
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoporous oxide electrodes for energy conversion and storage devices
Jin Wook
Yang†
a,
Hee Ryeong
Kwon†
a,
Jin Ho
Seo†
a,
Sangwoo
Ryu
*b and
Ho Won
Jang
 *ac
*ac
aDepartment of Materials Science and Engineering, Research Institute of Advanced Materials, Seoul National University, Seoul 08826, Republic of Korea. E-mail: hwjang@snu.ac.kr
bDepartment of Advanced Materials Engineering, Kyonggi University, Suwon 16227, Republic of Korea. E-mail: sryu@kyonggi.ac.kr
cAdvanced Institute of Convergence Technology, Seoul National University, Suwon 16229, Republic of Korea
First published on 9th October 2023
Abstract
Nanoporous oxides have been established as key materials for constructing electrodes for energy conversion and storage devices, offering high surface area and a large number of active sites for electrochemical reactions. Herein, we mainly focus on the characteristics, synthesis, and application of various nanoporous oxide electrodes for energy conversion and storage devices. Features of various nanoporous oxides by dimensionality and their functionalities in electrodes are presented. The synthesis strategies for nanoporous oxide electrodes to control their morphology are introduced, including top-down and bottom-up methods. Recent advances in nanoporous oxide electrodes in energy conversion and storage devices, such as fuel cells, water splitting electrodes, solar cells, light-emitting diodes, batteries, and supercapacitors, are summarized. The roles of nanoporous oxides tailored to the specific requirement for high performance of each device are further discussed. This review provides valuable insights into the design of nanoporous oxide electrodes from a materials point of view, contributing to renewable energy technologies.
 Jin Wook Yang | Jin Wook Yang received his Ph.D. from the Department of Materials Science and Engineering at Seoul National University in 2023. He is currently working as a postdoc fellow under the supervision of Prof. Ho Won Jang. His research interests include materials synthesis and designing (photo)electrocatalysts for energy conversion devices. |
 Hee Ryeong Kwon | Hee Ryeong Kwon is currently a Ph.D. candidate under the supervision of Prof. Ho Won Jang at the Department of Materials Science and Engineering of Seoul National University. She received her B.S. degree from the Department of Materials Science and Engineering at Korea University in 2021. Her current research focuses on the synthesis and design of photoelectrochemical energy conversion devices. |
 Jin Ho Seo | Jin Ho Seo is currently a Ph.D. candidate under the supervision of Prof. Ho Won Jang at the Department of Materials Science and Engineering of Seoul National University. He received his B.S. degree from the Department of Materials Science and Engineering at the University of Seoul in 2023. His current research interests include materials synthesis and designing high entropy alloy materials for energy conversion devices. |
 Sangwoo Ryu | Sangwoo Ryu is an assistant professor at the Department of Advanced Materials Engineering of Kyonggi University. He received his Ph.D from the Department of Materials Science and Engineering at Pohang University of Science and Technology in 2010. He worked as a research associate at the University of Wisconsin-Madison from 2010 to 2016, and he worked as a research professor at KAIST from 2016 to 2018 before he joined Kyonggi University in 2018. His research interests are nanostructured (photo)electrochemical catalysts for hydrogen production and CO2 reduction, chemical and biosensors, and various ceramic thin film heterostructures. |
 Ho Won Jang | Ho Won Jang is a full professor at the Department of Materials Science and Engineering of Seoul National University. He received his Ph.D. from the Department of Materials Science and Engineering at Pohang University of Science and Technology in 2004. He worked as a research associate at the University of Wisconsin-Madison from 2006 to 2009. Before he joined Seoul National University in 2012, he had worked at the Korea Institute of Science and Technology (KIST) as a senior research scientist. His research interests include materials synthesis and device fabrication for (photo)electrocatalysis, chemical sensors, memristors, micro-LEDs, and thin film transistors. |
1. Introduction
Nanoporous oxides have gained significant attention as highly promising materials in the field of energy conversion and storage, revolutionizing the landscape of renewable energy technologies. With their tailored porosity and high surface area, nanoporous oxide electrodes offer unique structural and functional properties, enabling enhanced performance and efficiency in applications such as fuel cells, solar cells, batteries, supercapacitors, etc.1–4 As a result, various research studies have focused on the synthesis, characterization, and application of nanoporous oxide materials.The distinctive features of nanoporous oxide electrodes stem from their high surface area and ability to provide a large number of active sites for reactions, facilitating efficient charge transfer and ion diffusion processes. In electrochemical devices, the controlled porosity provides abundant active sites for electrochemical reactions and electrolyte penetration, improving stability in terms of accommodating volume changes upon oxidation–reduction reactions.5–7 Also, in energy conversion devices related to light energy, such as water splitting electrodes, solar cells, and light-emitting diodes (LEDs), complex nanostructures are advantageous for confining or emitting light, contributing to the high efficiency of devices.8–10
This review aims to provide a systematic overview of the recent advancements in nanoporous oxide electrodes for energy conversion and storage devices. First, we introduce nanoporous oxides by dimensionality and explain examples and characteristics of each structure. Then, we describe the advantages of nanoporous oxides in terms of charge and ion transport, light management, and active sites. Additionally, we delve into the various synthesis strategies employed to fabricate nanoporous oxide materials with precise control over their pore size, morphology, and composition. Furthermore, we explore the applications of nanoporous oxide electrodes in diverse energy conversion and storage devices. We highlight the performance enhancements achieved through the integration of nanoporous oxide electrodes and the key factors influencing their performance. Finally, we address the challenges and future perspectives in the field of nanoporous oxide electrodes. This covers strategies for improving their stability, scalability, and cost-effectiveness and expanding their applications in emerging energy technologies. By providing a comprehensive review of nanoporous oxide electrodes for energy conversion and storage devices, their potential expands the landscape of sustainable energy systems.
2. Nanoporous oxides
2.1. Structural features
Nanoporous oxides feature unique structures and properties by taking a porous structure and downsizing it to the nanoscale. These oxides possess a network of interconnected pores or voids with dimensions typically ranging from a few nanometers to a few hundred nanometers. Representative structures of nanoporous oxides can be classified into two categories. Firstly, some nanoporous oxides inherently possess internal pores within their substance.11 According to the International Union of Pure and Applied Chemistry (IUPAC) classification system, the common classification of porous materials is based on their pore sizes, which divides porous materials into microporous (with pore diameters of <2 nm), mesoporous (2–50 nm), and microporous (>50 nm) categories.12 These different-sized pores can be arranged either randomly or in an orderly manner to create numerous oxide structures with a high surface area. Secondly, nanoporous oxides also include structures with pores formed within the volume space of matrices due to nanostructured architectures, which encompass a wider range.13 These nanostructured oxides can exhibit various morphologies, such as nanoparticles,14 nanowires,15 nanotubes,16 or nanosheets.17 Furthermore, they can be diversified into distinct nanostructures that are advantageous in increasing the surface-to-volume ratio and accelerating the transport routes of mediators such as charge and light. In this review, we will cover numerous cases of nanoporous oxides by combining all the categories.In oxide materials, oxygen bonding plays a crucial role in forming strong bonds with other elements, leading to the formation of stable oxide structures. The specific bonding arrangements between metal and oxygen atoms determine the properties such as electrical conductivity, magnetic and optical behaviors, and catalytic properties.18 For example, basically, binary metal oxides include titania (TiO2),19 silica (SiO2),20,21 alumina (Al2O3), etc.22 Ternary oxides are exemplified by perovskite structures23 and intercalation compounds,24 and multinary oxides exist in various compositions. Besides that, graphene oxide (GO), a derivative of graphene, is a form of oxide material with oxygen functional groups attached to sp2 hybridized carbon atoms and the carbon lattice.25 By incorporating nanoporous structural strategies, these oxides with compositional diversity can be utilized as more effective electrodes for energy conversion and storage. As shown in Fig. 1, there are various nanoporous architectures that oxide materials can take, from zero-dimensional (0D) to three-dimensional (3D) structures. In this chapter, the structural characteristics of nanoporous oxides will be focused extensively on each dimensionality of the materials. In addition, we will discuss the strengths of nanoporous oxides, which play a major role in energy conversion and storage materials.
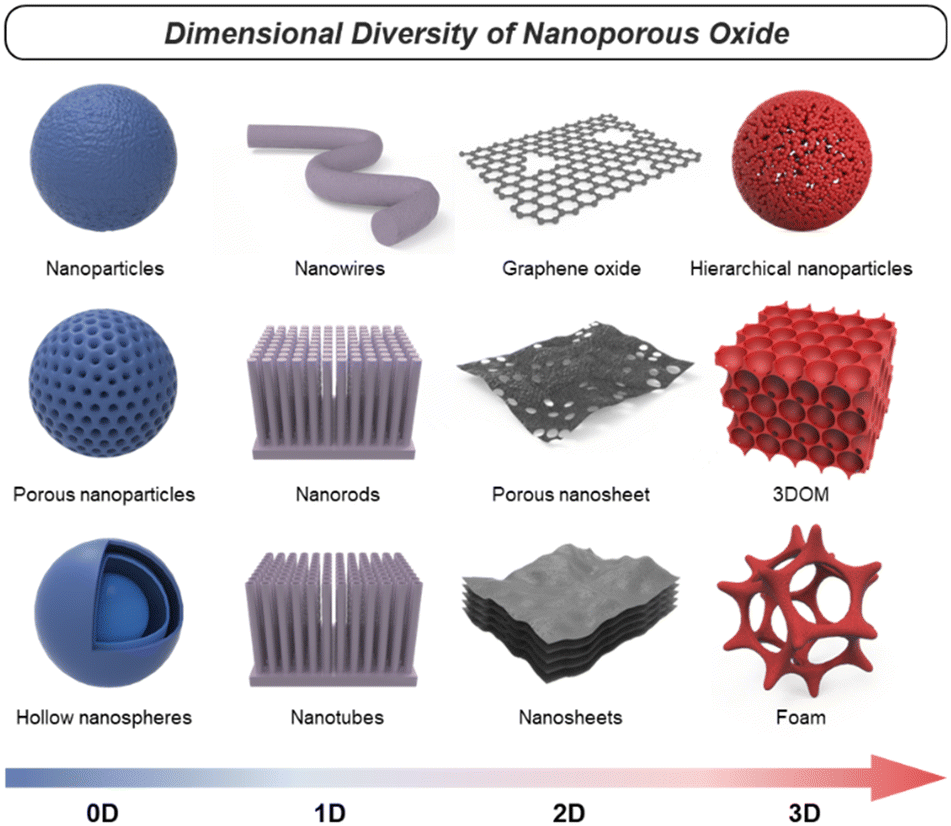 | ||
| Fig. 1 Schematics of various nanoporous oxides. | ||
2.2. Dimensionality
Essentially, since the particle size and the surface area of each particle are inversely proportional, synthesis techniques for smaller-sized solid nanoparticles have been developed. As mechanistic studies on the synthesis principle of solid-state or liquid-phase chemistry are supported, oxides with various compositions and precisely controlled sizes are inexhaustible.29 Among them, wide band gap semiconductor metal oxides, including ZnO,30 TiO2,31 SnO2,32 and NiO,33 have been studied with their unique advantages. Among the intense development efforts, Chen et al. succeeded in synthesizing black TiO2 nanocrystals through hydrogenation, broadening solar absorption for photocatalysis.34 The hydrogenated-black TiO2 nanocrystals were about 8 nm in diameter with a disordered outer layer surrounding a crystalline core of 1 nm thickness (Fig. 2a). This structural analysis, which discovered the lattice disorder of particles at the nanoscale, demonstrated that TiO2 nanoparticles' extended energy states enhance light absorption. In a recent study, a synthesis method has been proposed for TiO2 and antimony-doped tin oxide (ATO), utilizing calcination to achieve higher crystallinity while concurrently preventing the formation of nanoparticle agglomerates without sacrificial materials.35 This synthesis approach can be extended to a broader range of oxide materials, allowing for the formation of stable colloidal solutions with tailored crystallinity. On the other hand, in smaller 1.6 nm size WOx nanoparticles, the quantum confinement effect was confirmed in a limited width as the nanocrystal size became smaller.36 WOx quantum dots synthesized through the colloid process were identified to have excellent crystallinity and ultra-small crystal size through high-resolution transmission electron microscopy (HRTEM) images. Downsizing to 0D showed faster electrochemical kinetics compared to bulk materials, along with an increase in the band gap due to the strong quantum confinement. Metal oxide nanoparticles with narrower band gaps, such as Fe2O3 (ref. 37) and BiVO4,38 also make use of functions derived from the nanoscale geometry. Additionally, in spinel crystal structured-oxides with pseudocapacitive characteristics, a liquid-phase reaction synthesis has been devised.39 This synthesis enables sophisticated size control of MFe2O4 (M = Fe, Co, Mn, etc.) nanoparticles in the range of 3 to 20 nm. Based on the previous research, Ahn et al. synthesized monodispersed nanoparticles of Fe3O4 and conductive indium tin oxide (ITO) and applied them to pseudocapacitor electrodes.40Fig. 2b shows the high crystallinity of Fe3O4 nanoparticles and the uniformly controlled spherical size. Various oxides, such as those mentioned, have been investigated in the form of nanoparticles. Note that this research extends beyond simple geometric properties, aiming to achieve changes in electronic and optical characteristics as valuable energy materials. By modifying the electronic structure and controlling the crystalline properties, their impact has been significantly amplified.
 | ||
| Fig. 2 Dimensional diversity in the structure of porous oxide materials. (a) HRTEM image of hydrogenated TiO2 nanocrystals. A short dashed curve is applied to outline a portion of the interface between the crystalline core and the disordered outer layer. Reproduced from ref. 34. Copyright 2011 AAAS. (b) HRTEM images of Fe3O4 nanoparticles. The inset indicates the lattice fringe spacing. Reproduced from ref. 40. Copyright 2023 Elsevier. (c) HRTEM images of multi-shell hollow TiO2 nanoparticles. Reproduced from ref. 44. Copyright 2014 Wiley. (d) Cross-sectional SEM image of ultrathin corrugated TiO2 nanowire arrays. The inset indicates the HRTEM image of ultrathin corrugated TiO2 nanowire arrays. Reproduced from ref. 55. Copyright 2022 American Chemical Society. (e) SEM image of high entropy oxide nanofibers. The inset indicates the HRTEM image of high entropy oxide nanofibers. Reproduced from ref. 57. Copyright 2022 Wiley. (f) TEM images of MnO2 nanotubes (scale bar: 100 nm). Reproduced from ref. 59. Copyright 2015 Springer Nature. (g) HRTEM images of graphene oxide (GO) sheets after being etched for 1 h. Pores are highlighted in blue color. Reproduced from ref. 65. Copyright 2019 Springer Nature. (h) SEM image of WO3 nanosheets (scale bar: 200 nm). The inset indicates the HRTEM image of the crystal lattice structure of WO3 nanosheets (scale bar: 1 nm). Reproduced from ref. 70. Copyright 2014 Springer Nature. (i) TEM images of Y2O3 porous nanosheets (scale bars: 5 nm). The inset indicates the HRTEM image of the crystal lattice structure of Y2O3 nanosheets (scale bar: 2 nm). Reproduced from ref. 72. Copyright 2022 AAAS. (j) SEM image of CuO/ZnO nanorods. Reproduced from ref. 80. Copyright 2016 Elsevier. (k) SEM image of Ir/IrO2 of dealloyed Ir25Os75. Reproduced from ref. 86. Copyright 2017 Springer Nature. (l) SEM image of 3DOM La0.6Sr0.4MnO3 (LSMO) (scale bars: 200 nm). Reproduced from ref. 95. Copyright 2017 Springer Nature. | ||
Starting from solid nanospheres, nanoporous 0D materials can take on various structural modifications, transforming into hollow nanoparticles with empty interiors and core–shell or yolk–shell structures.41 Advances in nanotechnology have made it possible to precisely control the porosity, thickness, and number of shells and core and yolk materials. These advanced nanoparticles provide wide inner and outer surfaces, a fast charge transport path, and efficient light harvesting by multiple scattering. A study on Fe2O3 nanoparticles proved the advantage of the hollow shape by directly comparing solid and hollow nanoparticles with the performance of Li-ion batteries. The process of obtaining a hollow structure not only provides a larger reactive surface area on both sides but also leads to the formation of more cation vacancies, resulting in higher intercalation and conversion capacities.42 Specifically, the nanoporous geometry of hollow nanoparticles can be analyzed by N2 sorption isotherms based on the Brunauer–Emmett–Teller (BET) method, showing an increase in the BET surface area with decreasing shell thickness of hollow nanoparticles.43 As shown in Fig. 2c, the number of shells can be increased, and Hwang et al. introduced multi-shell TiO2 hollow nanoparticles with three shells to maximize the effect of multireflection.44 The synthesized multi-shell TiO2 hollow nanoparticles had a BET surface area of 171.3 m2 g−1, which is 2.7 times higher than the 63.6 m2 g−1 of the single shell. In addition, nanoporous characteristics were maximized with pores with a diameter of 4.09 nm in the shells themselves. Furthermore, oxide nanoparticles have been increasingly engineered into more complex structures for specific applications. Core–shell or yolk–shell oxide nanoparticles can also be formed with other materials, such as metals and organics.45,46 The structure of a porous CeO2 shell on a monometallic Pt yolk or bimetallic PtAg yolk is an example of an elaborate hetero-nanostructured oxide design.47 The yolk–shell structure allows for the encapsulation of different materials within the core, enabling the design of multifunctional nanoparticles with controlled release properties. Designing 0D structures with greater intricacy demonstrates the ongoing evolution of customized functionality. Therefore, it becomes evident that tailored interfaces using diverse oxides to harness the physicochemical properties can alter characteristics such as conductivity, catalytic activity, or optical behavior. Anticipating the synergy among individual components, the development of complex 0D nanoporous oxides becomes necessary.
When we delve into more specific examples of synthesized 1D structures, a prominent case is TiO2 1D semiconductors, which have made significant progress in the fields of photocatalysis and dye-sensitized solar cells (DSSCs).53 As the understanding of solution-based synthesis has improved, it has become possible to finely tune the length, diameter, and density of TiO2 nanorods or nanowires with specific crystal orientations and phases. Xing et al. synthesized well-separated single-crystal rutile TiO2 nanowire arrays with a length of 9 μm by a solvothermal method.54 The straight axial 1D geometry not only serves as an excellent charge separation and transport channel on its own but also provides a wide surface area, enabling effective noble metal decoration. Recently, the mono-micelle-directed assembly method has enabled the precise arrangement of extremely thin nanowires in TiO2 nanowire arrays (Fig. 2d).55 These nanoarrays consist of nanowires with a diameter of 8 nm, vertically aligned in a uniform spacing of 8 nm. Moreover, there are tightly packed concave structures with a diameter of 5 nm along the nanowire axis, forming a corrugated morphology. This design exploits the advantages of a nanoporous structure in a 1D configuration by reducing the radial distance and creating a direct axial pathway. In addition, other metal oxides such as SnO2, ZnO, Fe2O3, CuO, and Cu2O have been successfully synthesized in 1D nanoporous structures to obtain enhanced functionalities compared to their flat film structures. For the perovskite oxide materials, pioneering work by Hildebrandt et al. demonstrated the successful application of electrospinning to synthesize nanofibers of perovskite oxide materials, specifically Ba5Ta4O15 with its (111)-layered structure.56 In the synthesis process of Ba5Ta4O15 nanofibers, the BaCO3 intermediate stabilized and strengthened the fiber 1D morphology during polymer decomposition, providing high crystallinity with a length of several micrometers. Furthermore, it is possible to achieve 1D nanofibers of high entropy oxides, which are multicomponent oxides homogeneously mixed in a single crystal phase with five or more cation elements. The scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images in Fig. 2e present (Mg0.2Mn0.2Co0.2Ni0.2Zn0.2)Fe2O4 nanofibers with tens of micrometers in length and a diameter of 160 nm having abundant nanocrystals.57 As a result of N2 adsorption and desorption isotherm analysis, the synthesized high entropy nanofibers had a large specific surface area of 384.4 m2 g−1. The synergistic effect of multiple metal compositions and the 1D nanoporous structure leads to enhanced chemical adsorption abilities, making it a promising approach for various catalytic applications.
Nanotubes, on the other hand, are hollow cylindrical structures with nanoscale diameters. Anodizing metal foil is one of the representative methods to obtain metal oxide nanotube arrays. Geometrical modifications such as nano-bamboo and double-walled nanotubes can be achieved by controlling the anodization parameters.58 Another typical method is to obtain nanotube fibers through the calcination of nanofibers obtained by electrospinning. Niu et al. have developed an effective method for nanotube synthesis that can be applied to various materials, including inorganic single-metal oxides, binary-metal oxides, and multi-element oxides.59 An important mechanism to obtain porous oxide nanotubes in the electrospinning and pyrolysis processes is to set the gradient distribution of poly(vinyl alcohol) (PVA) while varying the molecular weight. Fig. 2f shows the synthesized MnO2 nanotubes with a diameter of approximately 50 nm. These nanotubes also exhibit abundant mesopores on their tube walls, ensuring a significantly enhanced specific surface area compared to nanowires. Besides, the electrospinning method, along with different post-treatments, has been applied to synthesize some interesting nanoporous 1D structures.60 For instance, a tube-in-tube structured spinel oxide including CoMn2O4, NiCo2O4, CoFe2O4, NiMn2O4, and ZnMn2O4 can be made into a 1D nanoporous structure by controlling the heating rate. In comparison to nanotubes and solid nanofibers, which have BET surface areas of 28.0 and 12.9 m2 g−1, the resulting spinel oxide nanotubes exhibit a significantly higher BET surface area of 47.3 m2 g−1.61 By carefully designing the fabrication process and parameters, researchers can tailor the size, morphology, and surface chemistry of the nanopores, allowing for precise control over the physicochemical properties. On the other hand, in terms of material development, it's essential to maintain structural stability while harnessing the advantages of 1D structures. The ultimate goal is to engineer materials with porous structures and desired properties under various challenging conditions, enabling successful applications across a wide range of industries. Therefore, understanding how stresses affect structural stability and developing solutions to enhance the robustness are also key aspects of 1D oxide development.
In 2D oxides, layered crystal oxides and non-layered crystal oxides are two different structural arrangements.63 Layered crystal oxides, also known as lamellar oxides, have a layered structure in which the oxygen atoms are arranged in stacked planes. The layers can be considered 2D sheets, and they are held together by weak van der Waals forces or other weak interlayer interactions. Examples of layered crystal oxides include graphite oxide, MnO2, V2O5, MoO3, and WO3. These layered oxides can be delaminated into 2D monolayer nanosheets via top-down exfoliation methods, including mechanical force exfoliation, mild chemical exfoliation, and electrochemical exfoliation. Intriguingly, GO, which has universal applicability in energy conversion and storage fields due to its unique functionality, is derived from a monolayer of graphite oxide.64Fig. 2g demonstrates the formation of sub-nanometer nanopores in a single layer of GO, indicating that the nanoporosity of GO can be increased through oxidative etching.65 In addition, GO nanosheets with a narrow pore size distribution centered around 0.4 nm and a high BET surface area of 611 m2 g−1 have been synthesized through calcination. The nanoporous GO electrodes have the benefits of abundant active sites based on their large surface area and nanopores in electrocatalytic reactions.66 For layered metal oxide 2D nanosheets, Yang et al. reported 2D MnO2 nanosheets intercalated with alkali metal cations by electrochemically exfoliating from bulk Mn metal. The synthesized nanosheets successfully achieved a mesoporous 2D geometry with pore sizes of 3–4 nm. In non-layered oxides, on the other hand, all atoms are linked by strong chemical bonds, leading to high-energy surfaces with surface dangling bonds. Some examples of non-layered oxides include WO3, TiO2, SnO2, and ZnO. Non-layered oxides without anisotropy make their exfoliation challenging, but a comprehensive knowledge of the surface energy and atomic bonding within the crystal structure has recently resulted in the successful synthesis of these materials with exfoliation.67,68 An example of such cases involves the synthesis of 2D nanocrystals with a thickness of 2 nm by insertion of K into bulk non-layered structures of wurtzite ZnO, β-MnO2, and anatase TiO2 through heat reaction under vacuum conditions.69
Nevertheless, the top-down approach has limitations in application depending on the crystal structure, low controllability, and low yield. As an alternative, a bottom-up synthesis has also been developed which can be applied more universally regardless of the crystal structure. More generalized bottom-up techniques have been investigated extensively in this flow, and one of them is the surfactant self-assembly method. This synthesis process includes forming inverse lamellar micelles using surfactants, incorporating hydrated inorganic oligomers, and subjecting them to hydrothermal or solvothermal treatment for improved organization and crystallization, resulting in well-crystallized nanosheets. It has been effectively used for certain transition metal oxides, including TiO2, ZnO, Co3O4, WO3, Fe3O4, and MnO2. The final product applied to WO3 exhibits a well-defined 2D nanosheet morphology with high crystallinity, as observed in the SEM and HRTEM images (Fig. 2h).70 Following such pioneering research, there has been active progress in the development of methods using 2D sacrificial templates and in situ analysis to elucidate the synthesis mechanism of 2D structured oxides.65,71 Yang et al. observed the 3D-to-2D transformation in rock salt structured cobalt oxide using in situ liquid-phase TEM. They revealed that the driving force of dimensional change is the variation in surface energy during the growth process, as supported by density functional theory calculations.65 In a recent study, a universally applicable synthesis method for nanoporous oxide nanosheets has been presented, which can be extended to rare-earth oxides, transition metal oxides, III-group metal oxides, II-group metal oxides, composite perovskite oxides, and high entropy oxides. The reported technique is a relatively simple and cost-efficient approach, utilizing a carbon template formed through the Maillard reaction of ammonium nitrate and glucose, along with a puffing process. The application of Y2O3 is shown in Fig. 2i, which reveals a dense arrangement of nanopores and an ultrathin 2D structure, highlighting its high surface area and excellent crystallinity.72 As such, new insights into the 2D oxide materials are being provided, and with adjustable porosity and structural stability, they are expected to be widely utilized in the field of energy nanotechnology.
In the construction of the nanoporous 3D oxides, the previously discussed 0D to 2D low-dimensional nanostructures serve as both the basic framework and building components. Branched oxide nanostructures with a tree-like structure, where nanorod branches are densely attached to nanorod trunks, effectively enhance nanoporosity. Identification of the growth mechanism and advances in synthesis technology have made it possible to synthesize structurally more complex hyperbranched nanoarrays.76 This design strategy has been widely reported with applications to various materials such as WO3,77 ZnO,78 and β-MnO2/α-Fe2O3,79 through controlling the length of the constituent units of branched oxides. For example, researchers introduced hierarchical ZnO/CuO electrodes. As shown in Fig. 2j, ZnO nanorod branches uniformly grow along the entire length of CuO nanorod arrays.80 Furthermore, hierarchical structures can be built by numerous assemblies with combinations of backbones and branches of lower-order materials. Ouyang et al. synthesized two types of hierarchical structure electrodes of NiCo2O4@NiO by growing ultrathin NiO nanosheets on 1D nanowires and 2D nanosheets of NiCo2O4, respectively.81 The BET analysis results to evaluate the nanoporous structure of the two types showed that the surface area of 1D nanowires NiCo2O4@NiO is higher at 81.3 m2 g−1 compared to 68.1 m2 g−1 when anchored on 2D nanosheets. In another study, LaNiO3@NiO nanoflowers were formed using a core–shell structure, where NiO nanosheets were wrapped around 0D LaNiO3 nanoparticles.82 Meanwhile, synthesis methods for 3D hierarchical structures have also been developed through the self-assembly of low-dimensional subunits.83 By controlling the additives in a hydrothermal synthesis process, different crystalline nuclei were formed, resulting in diverse hierarchical morphologies such as flowers, urchins, sheet-assembled spheres, and pyramid-assembled spheres.84 Jing et al. reported Bi2MoO6 microflowers consisting of extremely thin monolayers with a thickness of 0.8 nm.85 The utilization of hydrophobic chains in cetyltrimethylammonium ion additives allowed for the formation of a porous hierarchical structure to reduce surface free energy rather than a stacked monolayer.
3D hierarchical oxides can take the form of a foam or sponge providing rich 3D interconnected pore networks with a porous oxide framework. A typical example is a nanoporous structure composed of an Ir core and an IrO2 shell by dealloying of Ir25Os75 alloy as shown in Fig. 2k. Electrochemically active surface area (ECSA) tests validated the enhanced surface area of the nanoporous Ir/IrO2 structure as an oxygen evolution catalyst. The results showed a much higher ECSA, approximately 50 times greater than a flat surface.86 In another approach, an aliphatic ligand-based metal–organic framework (MOF) was thermally converted, resulting in nanoporous MgO and CeO2 with high crystallinities. The aliphatic ligand serves as a self-template to create nanopores, with their size determined by controlling the thermal conversion parameters. In the well-controlled nanoporous MgO, the BET surface area is 200 m2 g−1 with a pore hierarchy comprising micropores, mesopores, and macropores. A significant portion of the total pore volume is contributed by micropores, resulting in a volume of 0.37 cc g−1.87 Recently, there have been reports of synthesizing 3D foam structures by crosslinking 1D nanotubes or 2D nanoplates to create an abundant network of nanopores. On the one hand, Yan et al. introduced self-standing 3D hollow nanoporous SnO2-modified CuxO nanotubes for high energy density lithium-ion batteries.88 This distinctive design ensured electrical conductivity through metallic Cu nanotubes, which were then surrounded by CuxO and SnO2 layers to impart resistance to mechanical strain, resulting in an overall lamellar structure. On the other hand, through the synergistic effect of 2D nanosheets and wide accessible surface area, these 3D foam-inspired electrodes demonstrate high capacity and cyclic stability when applied to LiNi0.8Co0.15Al0.05O2 (ref. 89) and VO2 (ref. 90) battery cathode materials. At the same time, there have been advancements in visualizing complex and disordered 3D macrostructures and quantitatively assessing them using nanoscale X-ray computed tomography. These developments have greatly contributed to enhancing our understanding of intricate nanoporous 3D oxides.91 The recent advancement in 3D disordered structures, coupled with improved characterization techniques, will undoubtedly enhance our understanding of morphology, pore distribution, and surface chemistry. We expect that this approach will allow for more informed decision-making in electrode design and optimization. Ultimately, it is imperative that we strive for these interactive developments to better grasp and obtain feedback on the operation of electrodes in energy devices. In conjunction with highly advanced fabrication techniques, a sophisticated 3D ordered macroporous (3DOM) structure can give regularity to the network of pore arrays.92 The fabrication of the 3DOM structure involves self-assembling organic or inorganic sacrificial templates into a face-centered cubic lattice, filling metal oxide into the arranged sphere interstitials, and removing the templates. The ability to precisely control its pore architecture, like the pore size, shape, and arrangement, combined with its large surface area and tunable properties, makes it highly attractive for use in numerous fields. The structural benefits of the 3DOM morphology, which consists of periodic walls and open nanopores, have been applied to various materials such as V2O3 (ref. 93) and TiO2.94 In Fig. 2l, an SEM image of 3DOM La0.6Sr0.4MnO3 perovskite, 140 nm macropores are closely packed, and nano-sized channels form 2D hexagonal close-packed arrays. The structure is also an interconnection of hexapod-like building blocks with a body diameter of 80 nm and leg lengths of 15 nm.95 By optimizing these 3DOM architectures, it is possible to create robust and stable nanoporous structures that can withstand various environmental conditions and mechanical stresses.
2.3. Advantages
Nanoporous oxides possess various advantages that make them highly functional electrodes in energy conversion and storage applications. Specifically, nanoporous oxides provide functionalities such as enlargement of the specific surface area, fast charge separation and transport, and maximization of light utilization with their structural diversity in 0D to 3D discussed above (Fig. 3). We explore more detailed aspects of advantageous nanoporous oxides in this section. | ||
| Fig. 3 Three representative advantages of nanoporous oxides. | ||
Firstly, the enlarged specific surface area of nanoporous oxides assures an absolute exposed area where various chemical reactions can occur. In energy conversion and storage devices such as (photo)electrocatalysts, fuel cells, batteries, and supercapacitors, electrochemical reactions take place at the electrode–reactant interface. Nanoporous oxide electrodes extend an interface to interact with reactant species like ions or molecules, increasing the number of reaction sites. As a result, more reaction active sites led to higher reaction rates and improved overall performance.96 Additionally, in energy conversion and storage systems, efficient ion transport is crucial for high-performance operation. As the surface area increases, the ion diffusion path becomes shorter, reducing transport resistance and enhancing the power density and response time of the device.97 Aside from the major effect, the increased surface area can also contribute to high structural stability and cycling performance.98 Nanoporous structures provide greater resistance to volumetric changes in repeated charge and discharge processes, as the free volume of their interconnected pores serves as a buffer.
Secondly, nanoporous oxide electrodes facilitate rapid charge transport by providing shorter diffusion paths with reduced resistance for the movement of charge carriers. In particular, with a quantum confinement effect and an oriented charge transport channel, low-dimensional nanoporous oxides enhance carrier mobility by reducing charge carrier scattering. In the 3DOM structure, charge carriers move along the ultrathin skeleton, thereby effectively lowering the charge transport resistance of the electrode. As in the case of devices using photo-generated charge carriers, the nanoporous geometry allows photocarriers to travel much shorter distances to the electrode–reactant interface, reducing recombination and increasing charge separation efficiency.99 Nanoporosity can also overcome the limitation of the trade-off relationship where the depletion region and the carrier diffusion length are much shorter compared to the light penetration length in planar photoelectrodes.
Lastly, nanoporous oxides play a crucial role in increasing light absorption in solar energy conversion applications. Their rough or textured surfaces can scatter incident light multiple times, increasing the optical path length within the material. The extended path length enhances the chances of light being absorbed by the material, as it encounters more opportunities to interact with the nanostructure's surface. This light trapping effect results in a higher absorption coefficient compared to the film structure. In the fields of solar water splitting and solar cells, nanoporous photoelectrodes have been widely introduced to improve the non-directional scattering of incident light and suppress reflection.100 The light-trapping effect in nanoporous oxide electrodes offers opportunities for improving the performance and efficiency of optoelectronic devices.
3. Synthesis of nanoporous oxides
Porosity engineering has been worth investigating, yet there are still challenging sites due to fundamental difficulties with the formation of nanoscale level pores. There are two general approaches used to synthesize nanoporous oxide materials: bottom-up and top-down methods. The conventional top-down strategy involves the use of extra energy, such as chemical, electrochemical, thermal, and mechanical processes to break down bulk materials into nano-sized structures. Top-down approaches have an imprecise surface structure yet are intrinsically straightforward to synthesize massive products. Bottom-up engineering is a method of constructing a material from atoms and molecular species. This approach has the advantage of allowing for chemical reaction-based size and shape control. In the following subsections, we will cover various synthesis techniques for preparing nanoporous oxide materials.3.1. Bottom-up methods
The doctor blade method is one of the most utilized fabrication techniques for preparing nanoporous oxide electrodes for lithium-ion batteries,101 DSSCs,102–110 and electrochromic111–113 and (photo)electrochemical performances114–116 due to its ease of use and low price. Fig. 4a illustrates the standard doctor blade procedure for producing nanoporous oxide electrodes.117 Before coating, the surface of the substrate is cleaned, and a solution containing particles is spread on the substrate's surface. With the help of a blade, the solution is evenly dispersed throughout the surface, resulting reported a nanoporous WO3 film-coated ITO electrode prepared by the doctor-blade method for visible light-driven water splittingin a thin layer coated on the substrate. The residual solvent is then evaporated from the coated film while it is dried at the appropriate temperature to fix the coating. For example, Zhang et al. reported a nanoporous WO3 film-coated ITO electrode prepared by the doctor-blade method for visible light-driven water splitting.116 To create a nanoporous WO3 photoanode, a solution paste made of water, WO3 powder, polyethylene glycol, and marpolose was spread to an ITO glass surface that was cleaned with a UV-ozone treatment before being coated with a doctor blade and dried out. The final nanoporous structure on the ITO glass substrate, depicted in Fig. 4b and c, was formed by calcination at 550 °C. The synthesized nanoporous WO3 electrode produced a photoanodic current density of 1.8 mA cm−2 and displayed an onset potential of 0.67 V versus a reversible hydrogen electrode with an incident photon to current conversion efficiency (IPCE) of 45% at a light wavelength of 400 nm and an applied potential of 1.04 V versus Ag/AgCl. R. Gharbi's group presented nanoporous TiO2 for the performance of DSSCs, in which TiO2 paste was placed on a fluorine-doped tin oxide (FTO) glass surface with the use of adhesive tape to control the thickness and offer a non-coated space for electrical contact.107 The constructed DSSCs with nanoporous TiO2 photoanodes demonstrated better solar-to-current energy conversion efficiency with a short-circuit current density of 0.69 mA cm−2. Furthermore, Lionel Hirsch and colleagues produced nanoporous SnO2 films for DSSCs by applying commercial SnO2 colloidal suspensions and light-scattering SnO2 particles over conductive substrates with 3 M Magic Tape.109 Nanoporous SnO2 layers revealed excellent photovoltaic responses when used as electrodes for DSSCs, with an overall power conversion efficiency of 2.27% under AM 1.5G.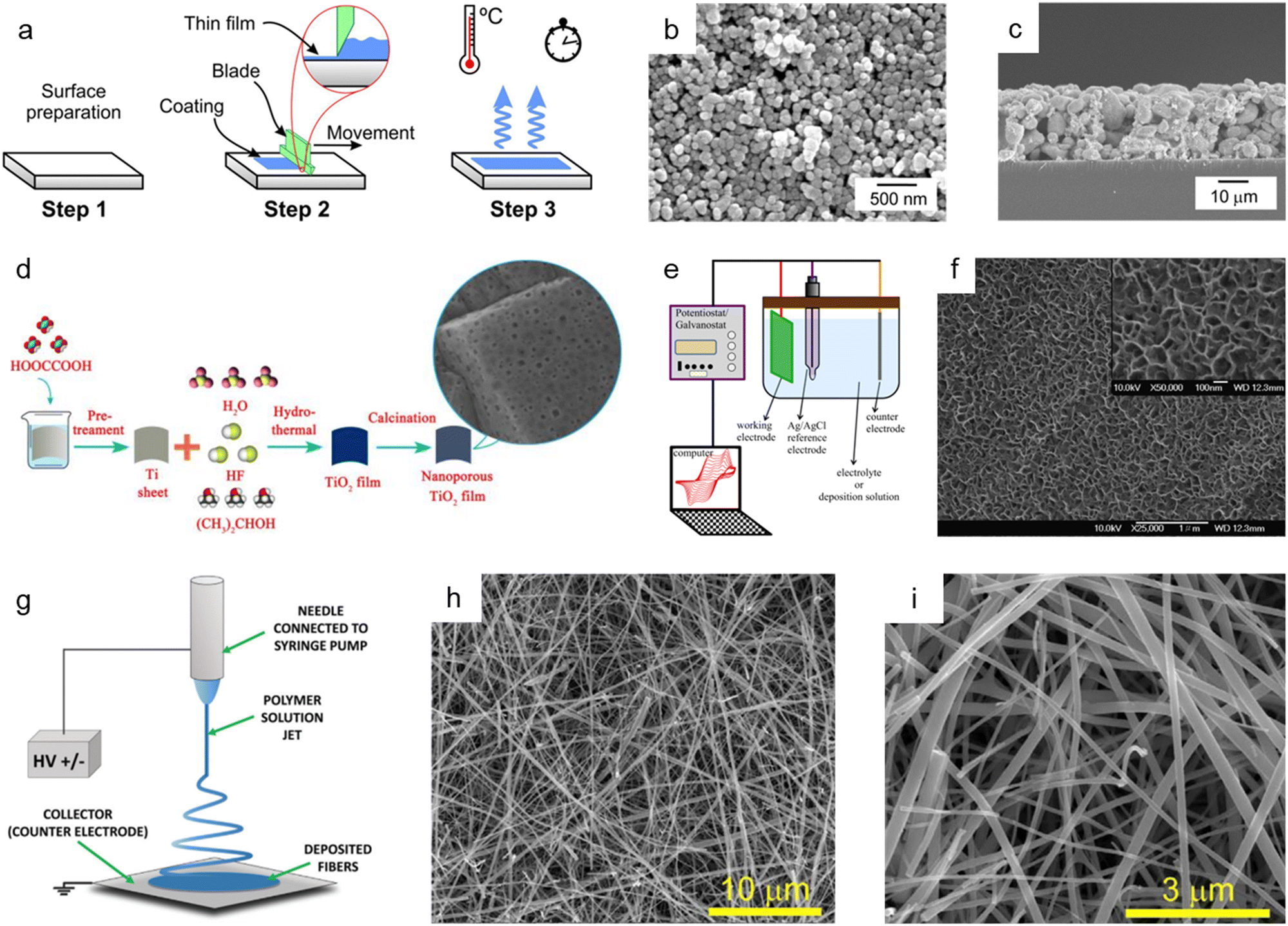 | ||
| Fig. 4 Bottom-up synthesis methods of nanoporous oxides. (a) Schematic diagram showing the mechanism of the doctor blade method. Reproduced from ref. 117. Copyright 2020 Springer Nature. (b) Top view and (c) cross-sectional SEM images of the nanoporous ITO/WO3 electrode. Reproduced from ref. 116. Copyright 2014 Elsevier. (d) A schematic illustration presenting the hydrothermal preparation of nanoporous TiO2 films. Reproduced from ref. 129. Copyright 2015 The Royal Society of Chemistry. (e) A schematic illustration of the typical electrodeposition system. Reproduced from ref. 131. Copyright 2016 Springer Nature. (f) SEM image of the electrodeposited nickel oxide film. Reproduced from ref. 132. Copyright 2007 Elsevier. (g) Schematics of the electrospinning setup with the standard nozzle configuration where the essential parts are underlined. Reproduced from ref. 143. Copyright 2022 Wiley. (h and i) SEM images of TiO2 nanofibers. Reproduced from ref. 149. Copyright 2017 Elsevier. | ||
The hydrothermal method has been commonly employed for synthesizing nanoporous oxides due to its simplicity, high yield, and superior crystallinity at a reasonable cost. In a typical hydrothermal approach, precursors are dissolved in an aqueous solution, sealed, and heated in a stainless steel autoclave. Without extra processing, high crystalline materials are produced by heterogeneous reactions that take place above the boiling point of water and atmospheric pressure. To successfully synthesize nanomaterials, it is essential to control the reaction parameters, such as the kind of precursor and solvent used, their concentration, stabilizing agents, reaction temperature, pressure, and duration. The reaction temperature, pressure, and time, in particular, determine the reaction rate, crystal growth, and characteristics. This method has been developed to synthesize a variety of nanoporous oxides for electrode materials that can be employed in Li-ion batteries,118 electrochemical systems,119–121 supercapacitors,122–127 DSSCs,128 and photocatalysts.129,130Fig. 4d depicts the fabrication process of nanoporous TiO2 films reported by Sekiguchi's group using a hydrothermal method for photocatalytic use.129 Before being immersed in HF aqueous solution with isopropanol in a Teflon-lined autoclave, a metallic Ti sheet was first etched with oxalic acid to remove the oxide layer. After the hydrothermal treatment, the Ti-sheet was washed with deionized water, dried in air, and then calcined at 600 °C. The as-made nanoporous TiO2 films with exposed {001} facets displayed superior UV photocatalytic activity. Cui et al. described the hydrothermally produced 3D nanoporous bamboo leaf-like copper oxide with a grain size of 50–80 nm for supercapacitive performance.125 The as-synthesized CuO electrode showed a specific capacitance of 269.6 F g−1 at a current density of 0.25 A g−1 and a capacitance retention ratio of 88.79% at a current density of 2 A g−1. Additionally, Bao et al. prepared nanoporous zinc oxide by a single hydrothermal step for the electrochemical reduction of CO2.119 The nanoporous ZnO demonstrated a substantially higher CO faradaic efficiency of 92.0%, indicating that the zinc oxide's nanopores boosted the electrocatalytic reduction of CO2.
Electrodeposition, which has the advantages of being straightforward and affordable and having increased interfacial interaction between substrates and coating materials, is another efficient technique for creating nanoporous oxide materials. The compositions, structures, and morphologies may be easily manipulated by modifying electrodeposition conditions. The conventional electrodeposition setup includes an anode, a cathode, an electrolyte solution in an electrochemical cell, and external power sources, as shown in Fig. 4e.131 The electrolyte solutions, in which an anode and a cathode are immersed, include metallic salts dissolved in water. Metallic ions are deposited as solid metals on the substrates by the electric current generated by external power sources through the electrolyte solution. By adjusting the voltage, pulse frequency, space between the components, deposition duration, and the use of nonconductive masks, porosity can be controlled through this technique. Electrodeposition has drawn attention in different applications (e.g., capacitors,132–134 batteries,135,136 DSSCs,137–139 electrochemistry133,140–142). Fig. 4f shows the SEM images of a nickel oxide film electrodeposited onto a stainless steel substrate for electrochemical capacitors using a plating bath of sodium acetate, nickel sulfate, and sodium sulfate mixture.132 The electrodeposition was performed in a standard three-electrode cell with an anodic current of 0.5 mA cm−2, and the deposited layer became highly porous after annealing at 300 °C. The synthesized nickel oxide film demonstrated outstanding performance with a specific capacitance of 167.3 F g−1 at 1 A g−1 and 156.6 F g−1 at 16.5 A g−1 charge/discharge. Luo et al. prepared a nanoporous ZnO thin film produced through cathodic electrodeposition for enhanced performance in DSSCs.139 The DSSCs made from nanoporous ZnO films achieved the highest solar-to-electric energy conversion efficiency of 5.08%, exceeding previous ZnO film-based DSSC efficiency levels. Likewise, Im et al. reported ultrathin nanoporous CuCo2O4 nanosheets produced via electrodeposition and air annealing as a bifunctional electrode for supercapacitors and water oxidation catalysis.133 The nanoporous CuCo2O4 electrode possessed a high specific capacitance of 1473 F g−1 at 1 A g−1 and an overpotential of 260 mV at 20 mA cm−2 with a Tafel slope of around 64 mV dec−1 in 1 M KOH solution.
Electrospinning is a versatile production technique used to produce continuous nanoporous oxide fibers with diameters less than 100 nm and a variety of compositions. Fig. 4g depicts an illustration of the fundamental electrospinning system, which consists of a high-voltage system, needles, and a collector.143 An exposed liquid droplet is electrified and subsequently extended toward the opposing collector when an electric field is generated between two electrodes. The qualities of the electrospun fibers can be adjusted using a variety of process parameters. For instance, the type of solvent or polymer used in the solution, the reaction temperature, the relative humidity, the electrical polarity applied to the nozzle, the distance between the nozzle and the collector, and the applied voltage, and all of these factors have a significant impact on the production of the final product. Molecular configurations and chemistry on the fiber's surface can be altered by changing a process parameter, which changes the fiber's properties to suit different applications, such as lithium-ion batteries,144–148 lithium-ion storage,149 DSSCs,150 and supercapacitors.145,151,152Fig. 4h and i present the SEM images of as-grown TiO2 nanofibers produced by two-step electrospinning for enhanced and rapid lithium-ion storage, as reported by Vilas G. Pol and co-workers.149 After wrapping reduced graphene oxide (rGO) onto anatase TiO2 nanofibers, the BET surface area increased from 54 m2 g−1 to 105 m2 g−1, resulting in a nanoporous structure with pore sizes ranging from 5 to 20 nm. The nanofiber anodes achieved a high reversible lithium-ion storage capacity of 200 mA h g−1 with a C/10 rate and good rate capability. Huang et al. presented electrospun ZnCo2O4 containing numerous nanopores (3 nm) as a high-performance lithium-ion battery anode material.144 The resultant ZnCo2O4 nanotubes had a high reversible capacity of 1454 mA h g−1 at 100 mA g−1 and reached 794 mA h g−1 at a current density of 2000 mA g−1 after 30 discharge/charge cycles. In addition, Binan Lu and colleagues developed new 3D nanoporous ZnWO4 nanoparticles using electrospinning for supercapacitors.151 The electrode fabricated with 3D nanoporous ZnWO4 nanoparticles could withstand a high current charge and discharge with a little capacitance fading, showing that the specific capacitance decreased by only 10% when the current density was increased from 40 A g−1 to 100 A g−1.
The hard-template method, also known as nanocasting, is widely applied in the production of nanoporous oxide materials. The precursor molecules are injected into stiff molds to create the final nanoporous oxide product. As observed in Fig. 5a,153 the rigid and porous template structure is impregnated with a mixture of metal precursors and chelating agents, which is then calcined and etched to remove the templates and produce the desired nanoporous oxide materials using acid or alkaline aqueous solution. Some specifications for templates are required to successfully synthesize desired products using nanocasting because the characteristics and quality of the templates and their interaction with the precursor determine the structure and morphology of the products. The templates should have stable, highly ordered porous structures and can be easily etched without affecting the properties of the product, so mesoporous silica (e.g., SBA-15,154–156 KIT-6,154,157,158 MSU-H,154 KCC-1,159 and MCM-41 (ref. 159)) or mesoporous carbon (e.g., CMK-1, and CMK-3) is commonly used as the porous hard template. Hard templating can be used on a variety of materials with high crystallinity due to the endurance of the stiff template to high temperatures and the ability to produce ordered and stable nanoporosity structures. However, the drawbacks of the hard template method include a reduced ability to adjust the pore size, resulting in a lower yield. Besides, the longer time necessary to thoroughly introduce metal precursors into the template and the difficulty in completely removing the hard template using acid or alkaline solution are still disadvantages. Despite these issues, there have been a few publications on the fabrication of nanoporous oxide materials employing hard templating. For instance, using mesoporous silica KIT-6 as a hard template, 3D highly ordered nanoporous CuO was produced for high-performance asymmetric supercapacitors, as shown in Fig. 5b.157 The highly ordered nanostructures are formed only when silica KIT-6 templates are present, according to the comparison of the TEM images of 3D highly ordered nanoporous CuO and bulk CuO shown in Fig. 5c–e. The electrode made from nanoporous CuO demonstrated outstanding electrochemical performance with a specific capacitance of 431 F g−1 at 3.5 mA cm−2, retaining over 70% of capacitance when operated at 70 mA cm−2. Im et al. reported the nanoporous TiO2 preparation with several mesoporous silica templates (SBA-15, KIT-6, and MSU-H) for improved photovoltaic characteristics of DSSCs.154 In comparison to nanocrystalline TiO2, the nanoporous TiO2 electrodes had a large surface area, a dye-adsorption capability that was 1.5 times higher, and greater photocurrent, fill factor, and solar energy conversion efficiency. Saeid Kadkhodazade and co-workers synthesized 3D-ordered nanocrystalline nanoporous NiMoO4 by nanocasting it from mesoporous silica KIT-6 for high-performance supercapacitor electrode materials.158 The nanoporous NiMoO4-based electrode showed a high area-specific capacitance of 4.25 F cm−2 at 3 mA cm−2, excellent rate capability of 2.18 F cm−2 at 120 mA cm−2, excellent cycling stability in 6000 continuous cycles at different current densities exhibiting only 8.4% loss after 3000 cycles at 7.5 mA cm−2, and high energy and power densities of 141.75 W h kg−1 at 0.6 kW kg−1 and 72.6 W h kg−1 at 24 kW kg−1.
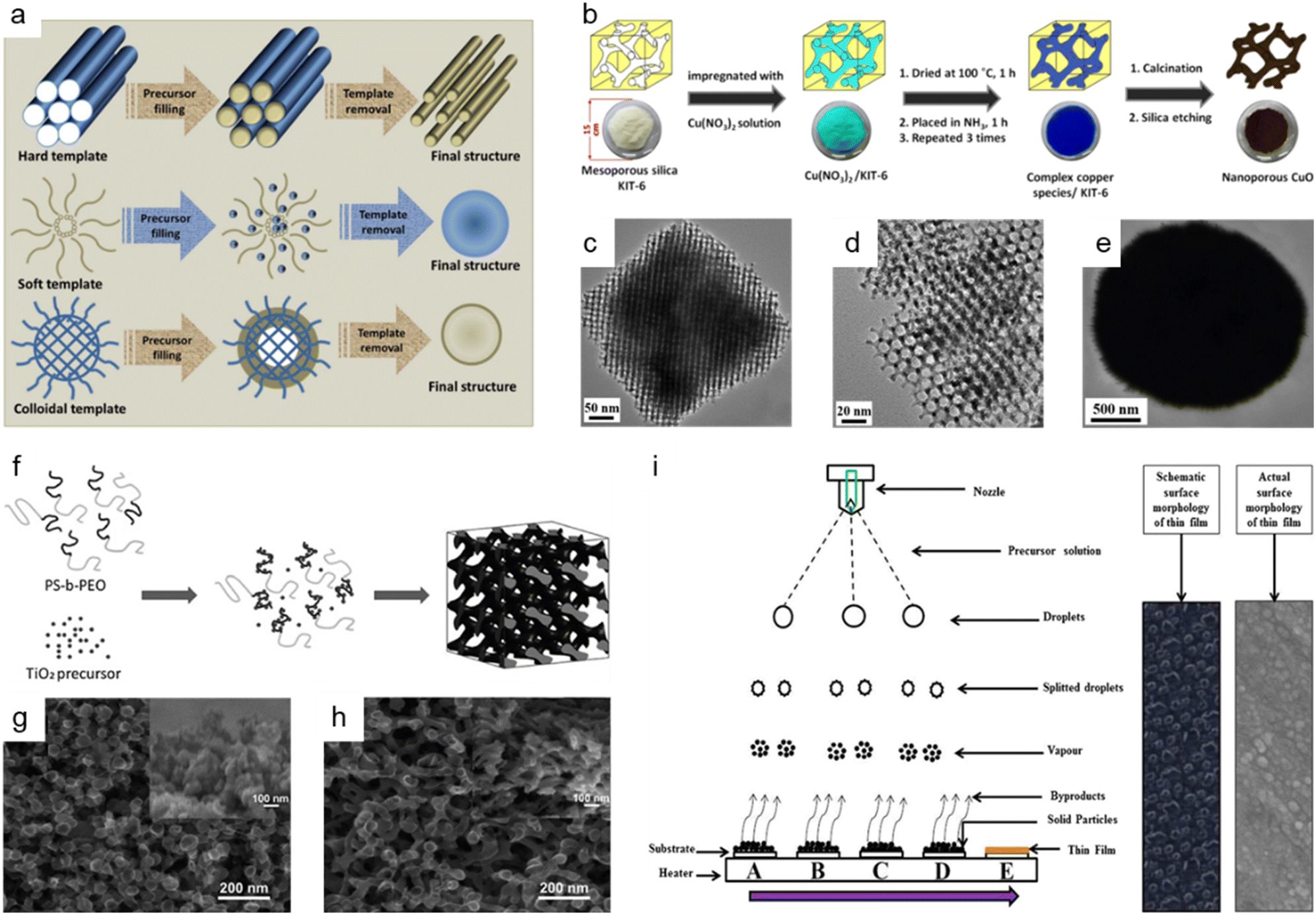 | ||
Fig. 5 Bottom-up synthesis methods of nanoporous oxides. (a) A schematic representation of the synthesis of materials using different types of templates. Reproduced from ref. 153. Copyright 2020 The Royal Society of Chemistry. (b) Schematic illustrations showing the synthesis of 3D highly ordered nanoporous CuO by nanocasting from KIT-6. (c and d) Typical TEM images of the 3D highly ordered nanoporous CuO. (e) Typical TEM images of the bulk CuO. (b–e) Reproduced from ref. 157. Copyright 2015 American Chemical Society. (f) Schematic diagram of the formation mechanism of nanostructured TiO2 using the PS-b-PEO block copolymer as a template. SEM images of nanostructured TiO2 films on FTO glass prepared with titania to polymer weight ratios of (g) 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and (h) 1:1. (f–h) Reproduced from ref. 161. Copyright 2010 The Royal Society of Chemistry. (i) Schematic of film formation by the spray pyrolysis technique. Reproduced from ref. 177. Copyright 2017 Springer Nature. :1 and (h) 1:1. (f–h) Reproduced from ref. 161. Copyright 2010 The Royal Society of Chemistry. (i) Schematic of film formation by the spray pyrolysis technique. Reproduced from ref. 177. Copyright 2017 Springer Nature. | ||
The use of soft templates has been widely described in the fabrication of nanoporous oxide materials, with the goal of co-assembly of surfactant molecules and inorganic species into ordered structures formed after removing the template, as illustrated in Fig. 5a.153 Surfactants for soft-templating are amphiphilic compounds that reduce surface and interfacial tension and have hydrophobic and hydrophilic equivalents. There are three types of surfactants used for soft templates: cationic surfactant (e.g., hexadecyltrimethylammonium bromide,160 cetylpyridinium chloride, etc.), anionic surfactant (e.g., sodium dodecyl sulfate, sodium dodecylbenzene sulphonate, etc.), and non-ionic surfactant (block co-polymer like PS-b-PEO,161 Pluronic PE10300,162 P-123,163–165 polyvinylpyrrolidone (PVP),166etc.). Soft matter, composed of surfactants, organic molecules, and block copolymers, interacts with the precursors via weak non-covalent bonding such as van der Waals forces or hydrogen bonding. Three general methodologies for the production of soft-template nanostructures have been reported: cooperative self-assembly, liquid crystal templating, and evaporation-induced self-assembly.153 The soft template method allows for the control of the morphology, size, and geometric structure, which is challenging with hard-template methods. Nonetheless, the fundamental obstacles in soft-templating are the limitation of crystal growth via heat treatment and the soft templates' low thermal stability. In addition, the complicated sol–gel processes and metal cation polymerization make detailed control of the porosity structure difficult. Fig. 5f shows the production process of the block copolymer PS-b-PEO templated nanoporous TiO2 for quantum-dot-sensitized solar cells as a soft template, and Fig. 5g and h show the corresponding SEM images of nanostructured TiO2 films on FTO glass.161 The solar cell could attain a higher IPCE value and greater photovoltaic performance by optimizing the soft template to titanium precursor ratio, with a peak IPCE value of 21% and a power conversion efficiency of 0.358% under AM 1.5G. Dai et al. prepared nanoporous Mn2O3 using Pluronic P123 as a template for increasing the high-rate zinc storage performance in Zn-ion batteries.164 The nanoporous Mn2O3 electrode exhibited a high reversible capacity of 233 mA h g−1 at 0.3 A g−1, a superior rate capability of 162 mA h g−1 at 3.08 A g−1, and exceptional cycling durability over 3000 cycles at a high current rate of 3.08 A g−1. Li et al. presented nanoporous SnO2 nanosheets as anode materials with PVP as a soft template for high-performance lithium-ion batteries.166 The nanoporous SnO2 nanosheets had an extremely high initial specific capacity of 2231 mA h g−1, a specific capacity of 688 mA h g−1 after 60 cycles at a current density of 0.2 A g−1, and an outstanding capacity retention of 224 mA h g−1 at 8 A g−1.
The colloidal-template strategy, which involves incorporating inorganic species into organic polymer (e.g., polystyrene (PS)167 and poly(methyl methacrylate) (PMMA)168,169) templates, is an effective and alternative way of synthesizing oxide materials with ordered nanopores (Fig. 5a153). The colloidal template methods have several advantages over the soft-template methods, including the ability to produce highly ordered nanoporous oxides and the ability to calcine at a relatively high temperature. Colloidal-templating offers the synergetic characteristics of both hard and soft templates since the inorganic components serve as a hard template to confine desired materials and increase stability, and the polymer template acts as a soft template to organize the inorganic particles through self-assembly. Colloidal templating permits the formation of ordered nanostructures with controlled porosity, good crystallinity, and structural integrity at a relatively high calcination temperature. For example, Ko et al. prepared high-efficiency perovskite solar cells based on vertically oriented nanoporous TiO2 employing PS-PMMA as a template.168 The best solar cells based on 1D nanoporous TiO2 nanorods showed an efficiency of 15.5% with an open-circuit voltage of 1.02 V, a short circuit current density of 20.0 mA cm−2, and a fill factor of 76.1%. Wang et al. reported a nanoporous manganese oxide fabricated using a polystyrene template for use in supercapacitor applications.169 The as-prepared nanoporous manganese oxide achieved a specific capacitance of 1018 F g−1 at a low current density of 500 mA g−1, revealing the prepared nanoporous manganese oxide as a viable candidate for supercapacitor applications. Even though colloidal templating has been extensively utilized for the synthesis of nanoporous oxide materials, several drawbacks limit its practical use. Because of the fragile properties of the material, the porous structure frequently collapses after the template removal, and the procedure is laborious and costly.
Spray pyrolysis is another adaptable synthesis process that is commonly used to produce nanoporous oxides (e.g., aluminum oxide,170 iron oxide,171–173 tin oxide,174,175 titanium oxide,176 ruthenium oxide,177 vanadium oxide,178 copper-bismuth oxide,179 cerium oxide180) based on thermal degradation of a liquid sample. In typical spray pyrolysis shown in Fig. 5i, the precursor solution is atomized into small droplets and delivered to the heated substrates as a form of vapor, creating thin films on the substrates.177 The spray pyrolysis approach allows for precise control of the properties of the product by adjusting the spray rate, air flow rate, solution concentration, droplet size, and distance from the nozzle to the substrate. Among these, the droplet size and deposition temperature have a major effect on the surface morphology and thickness of the film. Spray pyrolysis is an intriguing technology for producing nanoporous oxide materials since it allows for great thin-film adhesion on large substrates and homogeneity. Fig. 5i provides a schematic representation of the formation of thin films using the spray pyrolysis process together with both the schematic and actual surface micrographs of the thin films.177 The spray pyrolysis process was employed to load the nanoporous ruthenium oxide electrode on thin stainless steel substrates, which were then used as free-standing electrodes for supercharged capacitors. The electrodes made of nanoporous ruthenium oxide had an ultrahigh specific capacitance of 1429 F g−1 at a scan rate of 1 mV s−1, a higher energy density of 172 W h kg−1, a power density of 320 kW kg−1, a columbic efficiency of 94.74% in 1 M KOH, and a low charge transfer resistance of 0.89 Ω. U. J. Chavan and co-workers reported hematite α-Fe2O3 thin films made by a spray pyrolysis approach for the electrochemical supercapacitive performance.171 The α-Fe2O3 thin film electrodes had a maximum specific capacitance of 451 F g−1 within the potential window of −1.1 to 0.2 V in an aqueous 2 M KOH electrolyte and a 1.3 V voltage window with a specific energy and a specific power of 45 W h kg−1 and 1.25 kW kg−1 at 4 A g−1, respectively. R. M. Gamini Rajapakse et al. prepared fluoride-doped tin oxide on soda-lime glass films by using a specially designed atomized spray pyrolysis process for the operation of DSSCs.175 The synthesized FTO films provided an electronic conductivity of 1.17 × 103 S cm−1, an electron mobility of 10.89 cm2 V−1 s−1, a carrier density of 9.797 × 1020 cm−3, and a maximum light-to-electricity efficiency of 10.4% under AM 1.5 illumination for a cell active area of 0.25 cm2.
3.2. Top-down methods
Dealloying is a cutting-edge method that relies on the selective removal of sacrificial metal components from an alloy structure to produce nanoporous oxide materials under particular environmental conditions. As presented in Fig. 6a, dealloying can be achieved through chemical etching,3,181–186 electrochemical processing,181,187–190 immersion in liquid metal,181,191–193 and solid-state stripping,181,194–196 as well as by vapor-thermal processing.181,197–199 The metal atoms in the primary alloy are changed into metallic ions by localized oxidation when nanoporous oxide materials are produced using dealloying procedures. These metallic ions are then released in the dealloyed result. The morphology and surface properties of the dealloyed result can be determined by the atomic rearrangement and reaction kinetics, which are governed by input energy to deconstruct the metal alloy, reaction environment, and starting alloy composition. In particular, adjusting the conditions under which a sacrificial metal is removed, as well as the characteristics of the bulk alloy, can change the size distribution, volume, and interconnectivity of pores.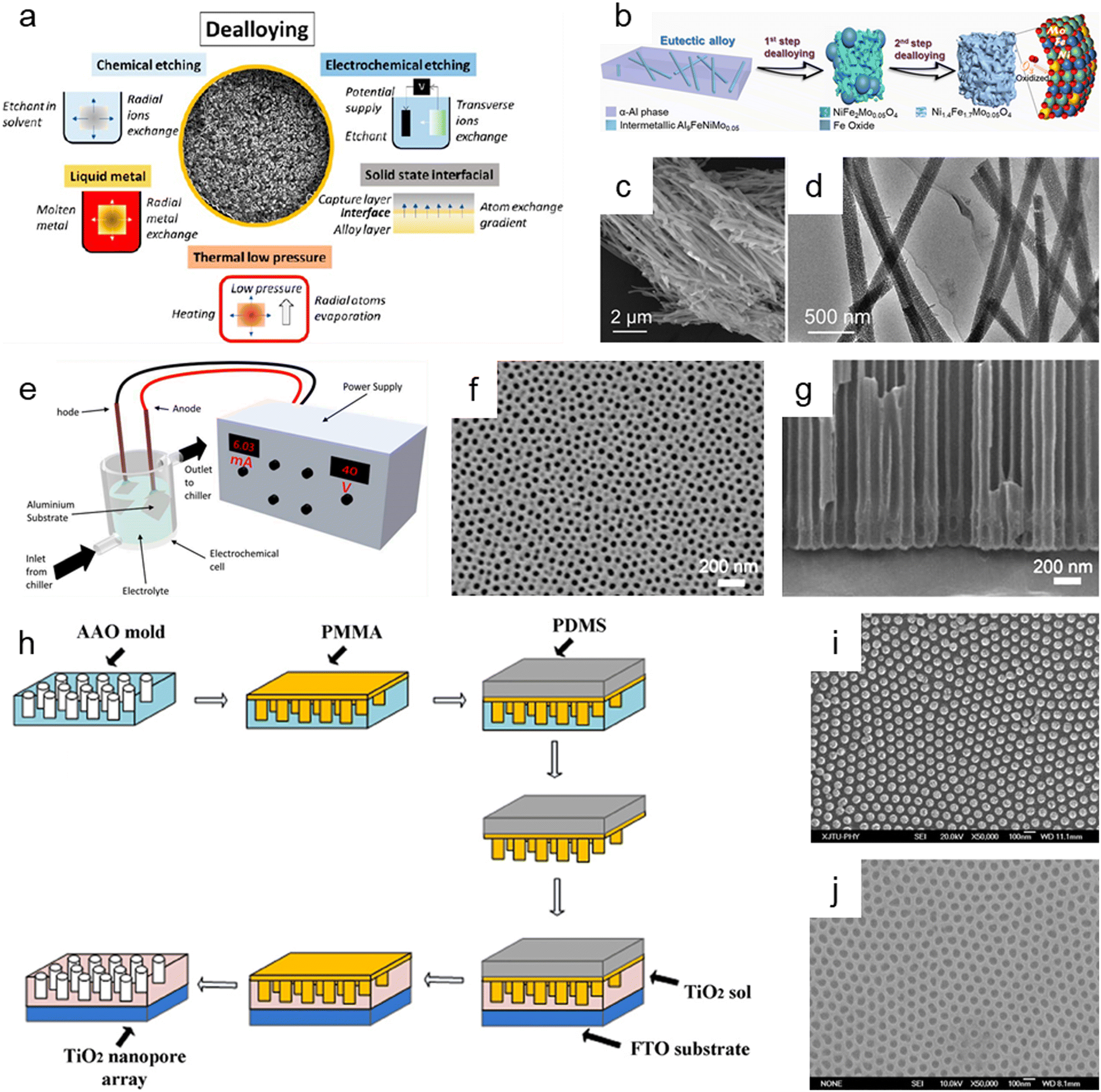 | ||
| Fig. 6 Top-down synthesis methods of nanoporous oxides. (a) Summary of the main dealloying pathways. Reproduced from ref. 181. Copyright 2023 American Chemical Society. (b) Schematic illustration showing the dealloying process of the AlNiFeMo alloy in corrosive solutions to fabricate Ni1.4Fe1.7Mo0.05O4 nanowires. (c) SEM and (d) TEM images of Ni1.4Fe1.7Mo0.05O4 nanowires. (b–d) Reproduced from ref. 182. Copyright 2023 American Chemical Society. (e) Typical experimental setup for the fabrication of nanoporous anodic alumina (NAA). Reproduced from ref. 212. Copyright 2022 MDPI. (f) Plane view and (g) cross-sectional SEM micrographs of AAO. Reproduced from ref. 200. Copyright 2011 Elsevier. (h) Schematics of the fabrication process of the nanoimprinted TiO2 nanopore arrays. (i) Top SEM image of the PMMA mold obtained from the AAO mold. (j) Top SEM image of the nanoimprinted TiO2 nanopore arrays with a diameter of 50 nm and an interpore distance of 100 nm. (h–j) Reproduced from ref. 213. Copyright 2011 Elsevier. | ||
Chemical dealloying is a procedure that involves a liquid-based redox reaction with the help of either an electrical supply or an acid or a base. On the other hand, electrochemical dealloying relies on selective etching using an electric field at a neutral pH environment. The primary distinction between the two etching methods is the kind of initial chemically dealloyed species, which is the atoms with fewer noble metal atoms in chemical dealloying versus atoms with lower corrosion potential in electrochemical dealloying. In liquid metal dealloying, the sacrificial metal is removed from the solid metal alloy while the alloy metals are still solid at high temperatures. Solid-state interfacial dealloying is accomplished by making contact between the dealloying agent and the alloy at a low temperature to promote interfacial diffusion and remove the sacrificial metal via solid-state transfer. Vapor-thermal dealloying is a process that requires selectively evaporating one of the sacred metal phases in a vacuum, resulting in the formation of vacancy sites. These vacancies offer routes for regional rearrangements and migrations of the noblest metal atoms still present in the system, which lowers the surface energy. Fig. 6b shows the preparation of nanoporous Ni1.4Fe1.7Mo0.05O4 nanowires via a two-step chemical dealloying procedure in which the AlNiFeMo alloy was immersed in corrosive solutions.182Fig. 6c and d illustrate the SEM and TEM images of the nanoporous Ni1.4Fe1.7Mo0.05O4 nanowires, which have an average diameter of 200 nm and a length of over 10 μm with an average pore/ligament size of around 4 nm. The 3D Mo-doped nanoporous NiFe oxide nanowires exhibited an efficient electrocatalytic oxygen evolution reaction performance with a low overpotential of 205 mV at 10 mV cm−2 and a small Tafel slope of 51.3 mV dec−1.
Anodization has emerged as one of the most popular synthesis techniques of various nanoporous oxides (e.g., aluminum oxide,200,201 titanium dioxide,202–204 tin oxide,205,206 tungsten oxide,207 tantalum pentoxide,208 vanadium pentoxide,209,210 and niobium oxide211) with well-controlled pores due to its distinctive electrochemical properties, large surface area, and high thermal stability. A typical electrochemical setup is required for anodizing the surface of a metal, which includes a chiller to maintain low-temperature conditions for the electrolyte, cathode metals, and high-purity metal foil for the anode, as well as an external power supply for galvanostatic or potentiostatic anodization, as shown in Fig. 6e.212 By adjusting the reaction parameters including the type, content, and concentration of electrolytes, applied potential/current, and temperature, the metal's surface is transformed in this electrolytic oxidation process into an oxide or hydroxide with the desired porosity. The porosity of the resulting oxide depends on the type of electrolyte used, and acidic electrolytes can produce nanoporous oxides. For example, C. T. Lee et al. used one-step anodization to create nanoporous anodic aluminum oxide films to evaluate the impacts of temperature and voltage mode on anodization processes.200 Al foil was anodized in one step using hybrid pulse anodization (HPA) and direct current anodization in 0.5 M oxalic acid at a temperature of 5–15 °C. As shown in Fig. 6f and g, which depict the SEM micrographs of the top-view and cross-section of the anodic aluminum oxide (AAO) formed by one-step HPA from 99.997% Al foil for 1 h at a temperature of 15 °C, a well-formed semicircle structure without cracks or voids at the interface between AAO and Al foil is achieved with pore diameters in the range of 45 ± 5 nm. For a supercapacitor electrode material, Han et al. described self-organized nanoporous tin oxide films made by anodizing a tin substrate in an aqueous electrolyte containing oxalic or phosphoric acid.206 The fabricated nanoporous tin oxide films had a maximum specific capacitance of 274 F g−1 and a long life in electrochemical charge/discharge cycles. Furthermore, Li et al. prepared oxygen-deficient Ta2O5 nanoporous films as self-supported electrodes for lithium microbatteries by electrochemical anodization of tantalum metal in an ammonium fluoride electrolyte and subsequent thermal annealing.208 The prepared nanoporous Ta2O5 films showed a high lithium capacity of about 480 mA h g−1 and exceptional cycling stability over 8000 cycles at a rate of 5C.
Imprinting, in which the structure and morphology of the material are pre-determined by the template containing nanopores, is another helpful method for synthesizing nanoporous oxide materials. By using the imprinting method, the nanostructure can be replicated by mechanical contact and 3D material displacement.213 In a typical imprinting procedure, a thin coating of polymer materials is applied to the template's surface to penetrate its pores and produce a stable structure that will help with the subsequent steps of mold construction. The flexible and highly elastic polymer materials are then injected onto the surface of the as-prepared sample to create a mold and replicate the desired patterns or structures in the following imprinting operations. After removing the template, the constructed mold is used to apply the necessary components to the sol while maintaining a constant applied pressure and temperature. The mold is subsequently removed, and nanoporous oxide structures are then calcined at a high temperature. X. Hu et al. reported a nanoimprinted TiO2 nanopore array for photovoltaic applications as an example of a nanoporous oxide material synthesized using an imprinting process.213Fig. 6h shows the typical fabrication process of the nanoporous TiO2 array, directly nanoimprinted on an FTO glass substrate using a PMMA/PDMS composite soft template, which is replicated from an AAO replica mold. Fig. 6i shows the top SEM images of the PMMA mold made from the AAO mold, demonstrating that the PMMA mold was highly ordered, and the sizes were accurately matched to the equivalent AAO molds. As shown in Fig. 6j, the replication from the PMMA/PDMS mold to the nanoporous TiO2 arrays was successful since the pore diameters of the nanoimprinted TiO2 nanopore arrays matched those of the respective AAO templates. The solar cells combined with the TiO2 nanopore arrays demonstrated that they might be more effective in quenching photoluminescence emission with a maximum efficiency of about 0.32% for a photovoltaic device. Kim et al. described highly ordered nanoimprinted TiO2 with nanopores for hybrid inverted bulk heterojunction solar cells.214 The as-synthesized solar cells showed a higher power conversion efficiency of 1.49% than on a flat titania of 1.18%.
All of the synthesis methods for nanoporous oxide materials discussed above have benefits, drawbacks, and limitations. Various synthetic strategies have been employed in previous studies to produce nanoporous oxides and examine their properties; however, utilizing these approaches completely to precisely regulate the structure and properties of nanoporous oxides remains a challenging task. Novel and simple strategies for thoroughly regulating nanoporous structures while ensuring structural uniformity and stability need yet to be developed for further practical applications of nanoporous oxide materials. For example, by properly integrating these distinct fabrication techniques, novel perspectives into the synthesis and control of intrinsic oxide nanoporous materials can be accomplished. Electrospinning can be used to produce nanoscale fibers, which can subsequently be applied to a dealloying process to generate a variety of metal-oxide nanoporous structures. These structures are intended to offer innovative solutions in a variety of applications through enhancing surface area, structural stability, and electrical properties. Current research on nanoporous oxide synthesis methods is expected to result in revolutionary advances in future nanotechnology and nanomaterial applications, as well as provide a new research direction in the field of nanoporous oxide synthesis and contribute to the development of innovative nanoporous oxide materials.
4. Applications of nanoporous oxide electrodes
4.1. Energy conversion devices
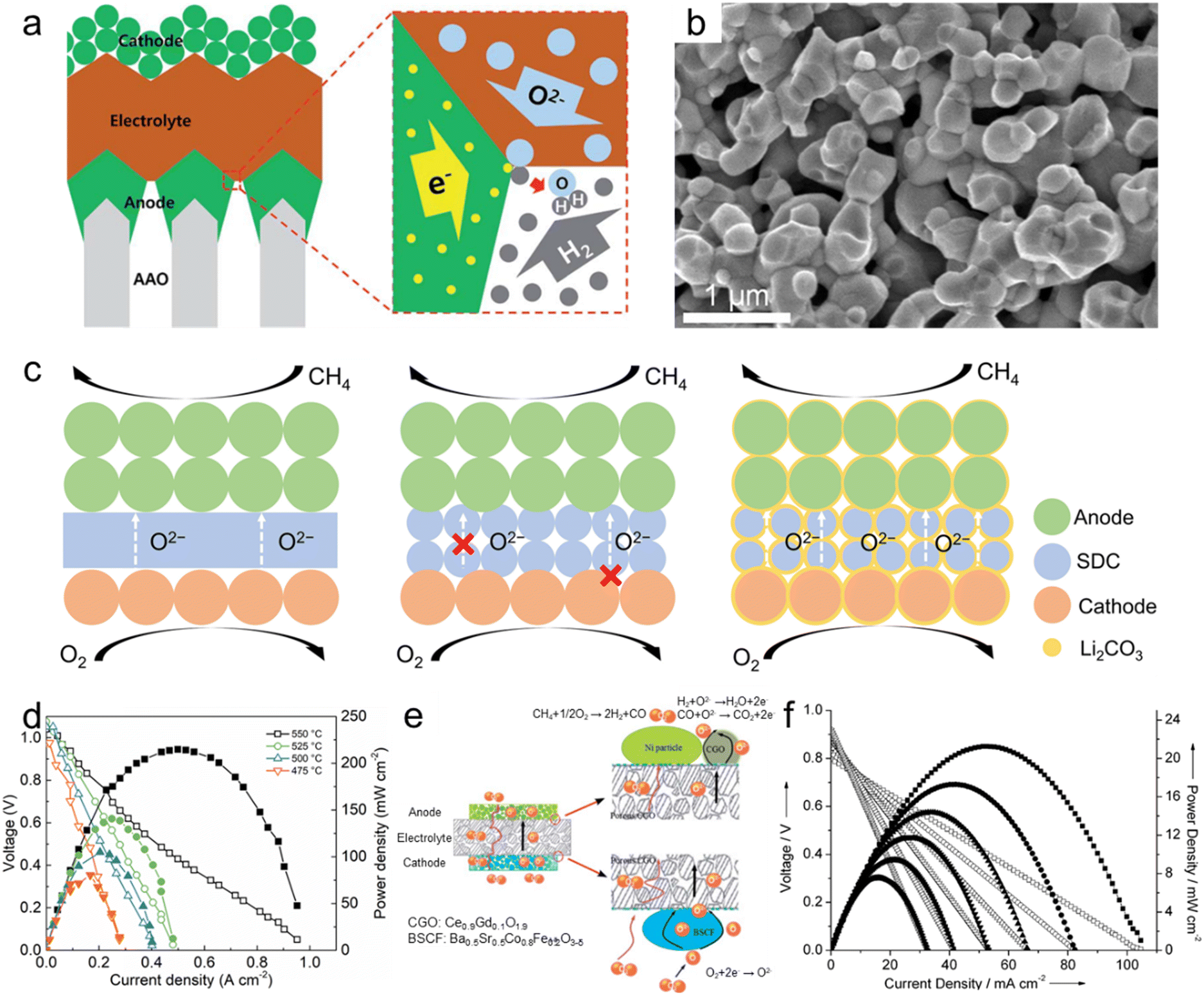 | ||
| Fig. 7 Nanoporous oxide electrodes for fuel cells. (a) Schematic showing the structure of a thin-film-based solid oxide fuel cell supported on an anodic aluminum oxide (AAO) template and the transport of gases, oxygen ions and electrons near the electrodes and the electrolyte. Reproduced from ref. 218. Copyright 2017 The Royal Society of Chemistry. (b) Cross-sectional SEM image of the LSTN-YSZ contact layer. Reproduced from ref. 219. Copyright 2023 Springer Nature. (c) Schematic of the SOFC, porous SOFC, and the CSSFC. (d) I–V and I–P curves of the CSSFC in CH4 at different temperatures. Reproduced from ref. 225. Copyright 2022 PNAS. (e) Schematic representation of the all-porous Gd0.1Ce0.9O1.9 (CGO)-supported fuel cell. (f) I–V and I–P curves of the all-porous fuel cell under a 4% CH4–96% He/4% O2–96% N2 atmosphere at different temperatures. Reproduced from ref. 226. Copyright 2013 The Royal Society of Chemistry. | ||
The second role of nanoporous oxides is as an electrolyte matrix. Various nanoporous oxides, such as yttria-stabilized zirconia (YSZ),220 gadolinium-doped ceria (GDC),221 scandia-stabilized zirconia (SSZ),222 and samarium-doped ceria (SDC),223 are commonly used as the electrolyte material in SOFCs. The nanoporous structure of these oxides provides a high surface area and interconnected pathways for oxygen ion transport. This enables efficient ion conduction through the electrolyte, facilitating the electrochemical reaction within the SOFC. Liu et al. developed a heterostructure electrolyte based on Gd0.15Ni0.05Ce0.8O2−δ (GNDC) and SnO2.224 The GNDC–SnO2 composite consisted of nanoscale particles, which offered sufficient grain boundaries and surface area. Based on the high ionic conductivity of 0.124–0.220 S cm−1, the fuel cell with the GNDC–SnO2 electrolyte recorded an OCV of 1.026 V and a high MPD of 879.4 mW cm−2 at 550 °C. Su et al. proposed a carbonate-superstructured solid fuel cell (CSSFC) with a tuned electrolyte as an advancement of the conventional SOFC.225 The in situ generation of the superstructured carbonate (Li2CO3) during the operation in SDC showed a high ionic conductivity of 0.17 S cm−1 at 550 °C. Fig. 7c illustrates the mechanism of the CSSFC. In nanoporous electrolytes of the SOFC, the specific area is larger than that of dense electrolytes, but it can lead to increased contact resistance between electrodes and require higher operating temperatures. On the other hand, incorporating molten carbonate into the nanoporous electrolyte improves oxygen ion conduction by creating a continuous interface between molten carbonate and solid ionic conductors. Additionally, it would establish a robust connection near the electrode–electrolyte interface. As shown in Fig. 7d, the CSSFC with the porous SDC electrolyte achieved a high OCV of 1.041 V and MPD of 215 mW cm−2 at 550 °C in dry methane fuel. Based on porous electrolytes and electrodes, Guo et al. suggested an all-porous SOFC.226 As shown in Fig. 7e, a porous Ce0.9Gd0.1O1.9 (CGO) electrolyte prepared by dry pressing was combined with a porous Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) cathode and a Ni particle anode. Oxygen flows from the cathode and is diffused by the porous electrolyte to the catalytic anode while methane is fed at the anode. The porous CGO electrolyte microstructure enhanced the ionic conductivity and increased the power density of the SOFC. As shown in Fig. 7f, the all-porous SOFC with the porous CGO electrolyte recorded an OCV of 0.8 V and MPD of 21.4 mW cm−2 at 750 °C. We summarized the materials, OCV, MPD, and operating temperatures of fuel cells in Table 1. Nanoporous oxide electrodes have significantly enhanced the electrochemical performance of SOFCs. Their high surface area and tailored pore structures could enable more efficient fuel oxidation and oxygen reduction reactions, resulting in higher power output and improved cell efficiency. Future research might focus on developing nanoporous oxide electrodes that enable SOFCs to operate at reduced temperatures. This advancement would lead to shorter start-up times, longer lifespan, and improved thermal management, making SOFCs more practical for a wider range of applications.
| Fuel cells | ||||
|---|---|---|---|---|
| Materials | OCV [V] | MPD [mW cm−2] | Temperature [°C] | Ref. |
| Ni-YSZ (AAO template) | 1.0 | 28 | 500 | 218 |
| Pt-YSZ (AAO template) | 1.02 | 350 | 500 | 2 |
| LSTN-YSZ (STS template) | 1.05 | 560 | 550 | 219 |
| NiO-YSZ | 1.04 | 187 | 600 | 220 |
| NiO-GDC | 1.0 | 178 | 800 | 221 |
| Ni-SSZ | 1.1 | 150 | 600 | 222 |
| NiO-BZCYYb | 1.047 | 640 | 650 | 223 |
| SnO2-GNDC | 1.026 | 879.4 | 550 | 224 |
| Ni-BZCYYb-SDC | 1.041 | 215 | 550 | 225 |
| NiO-CGO | 0.8 | 21.4 | 750 | 226 |
Hematite (α-Fe2O3), one of the most representative photoanode materials, has a low band gap of 2.1 eV, resulting in high theoretical photocurrent density. However, its actual efficiency is low due to a very short hole diffusion length of a few nanometers. Therefore, nanostructuring is essential in utilizing hematite-based photoanodes.234,235 Zhang et al. fabricated thick hematite films (∼1500 nm) constructed from highly ordered hematite mesocrystals (MCs) with solvothermal methods.236 As shown in Fig. 8a, the excellent penetration of the electrolyte into the mesoporous film and the increased specific surface area resulted in a larger depletion region compared to the Fe2O3 single crystal. The thin TiO2 layer formed on the surface of the MCs induced a steeper band banding, maximizing charge separation. Yoon et al. synthesized a Ge-doped porous Fe2O3 photoanode by immersing β-FeOOH based on the solution process in a Ge solvent followed by heat treatment.237 It exhibited higher porosity compared to Ge-doped hematite formed through a one-step process, and after coating a NiFeOx catalyst, it showed a high photocurrent density of 4.6 mA cm−2 at 1.23 VRHE. Jang et al. controlled the thickness and porosity of Fe2O3 through multiple regrowth processes using heat treatment of the synthesized FeOOH.234 It was first combined with a Si photocathode, and the tandem device achieved a solar-to-hydrogen (STH) conversion efficiency of 0.91%. By doping Ta into hematite nanorods through hydrothermal regrowth and hybrid microwave annealing, Zhang et al. obtained a high photocurrent density of 3.22 mA cm−2 at 1.23 VRHE.238
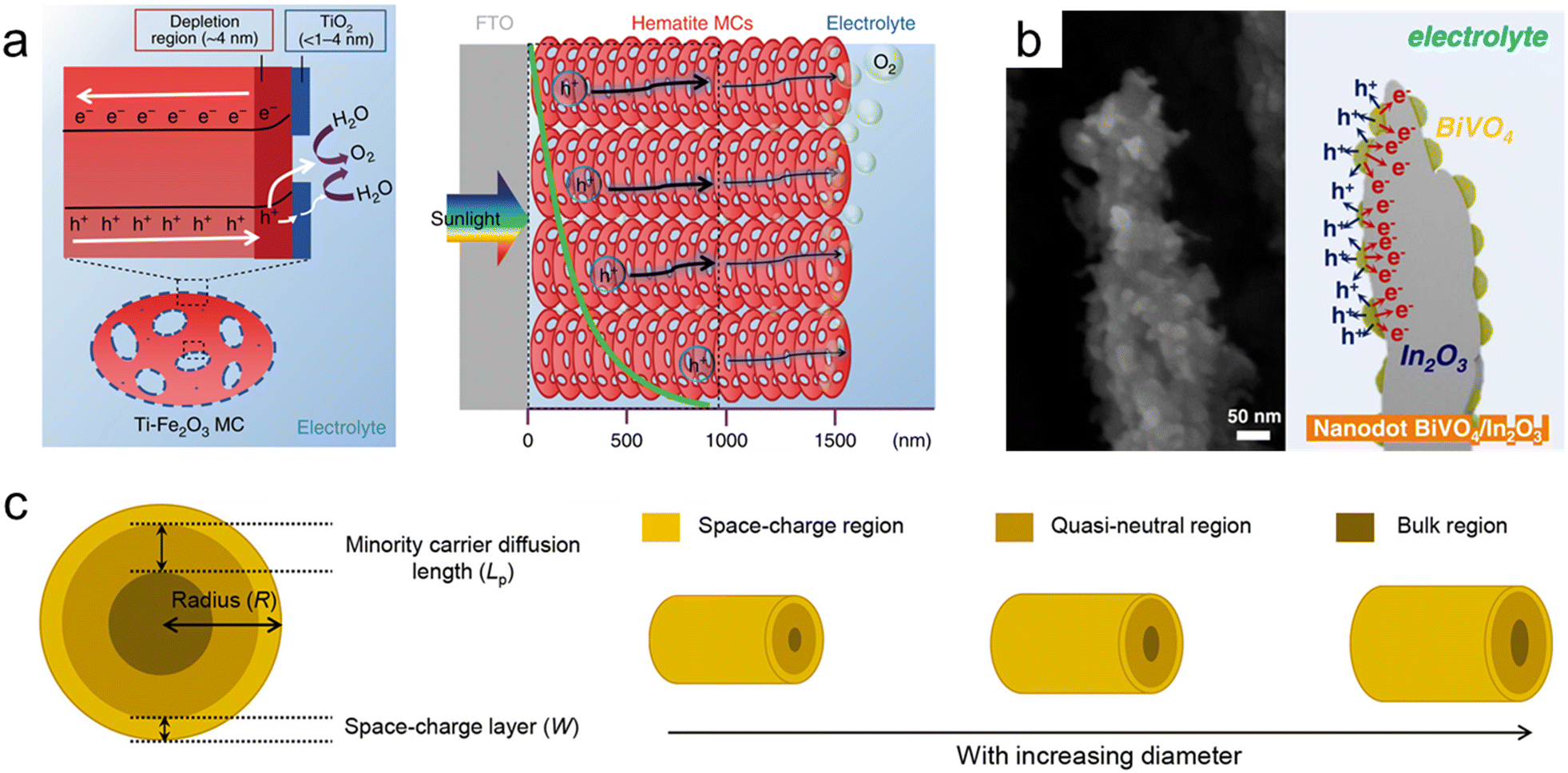 | ||
| Fig. 8 Nanoporous oxide electrodes for water splitting. (a) Illustration of charge carrier dynamics in a thick hematite mesocrystal (MC) film. Reproduced from ref. 236. Copyright 2019 Springer Nature. (b) Cross-sectional SEM image and illustration of nanodot BiVO4/In2O3 nanorods. Reproduced from ref. 247. Copyright 2023 Wiley. (c) Relationship between the minority carrier diffusion length (Lp) and the particle diameter. Reproduced from ref. 248. Copyright 2021 American Chemical Society. | ||
Bismuth vanadate (BiVO4) is a promising photoanode material with a band gap of 2.4 eV. However, its short hole diffusion length of 70 nm leads to degradation of the PEC performances due to charge recombination. To overcome this challenge, most studies have introduced nanoporous BiVO4.239,240 McDonald et al. synthesized nanoporous BiVO4 for the first time using electrodeposited BiOI.241 When the 2-dimensional plate-lick BiOI crystals were annealed together with a V-based precursor, they formed a few hundred-nanometer nanopores in BiVO4. Furthermore, Lee et al. obtained denser BiOI nanoplates by separating the electrodeposition process into nucleation and growth steps. Additionally, lactate buffer induced a uniform increase in local pH, resulting in a more uniform film formation and smaller nanopores in BiVO4.242 To further maximize the specific surface area of the existing nanoporous BiVO4, Yang et al. first introduced SnO2 nanorods underneath the BiVO4 layer.243 BiVO4 was vertically formed along the nanorods, and due to the coexistence of mesopores formed by SnO2 and nanopores of BiVO4, the BiVO4/SnO2 photoanode achieved a high photocurrent density of 6.3 mA cm−2 at 1.23 VRHE. Lee et al. synthesized various nanostructures of BiVO4 without BiOI precursors using a one-step synthesis method called pulsed electrodeposition.244–246 Also, In2O3 nanorods were first introduced as an electron transport layer for BiVO4 through glancing angle deposition.247 By adjusting the pulse cycle of electrodeposition, the coverage of BiVO4 was controlled, and a transition from a nanodot to a core–shell structure was observed at the specific cycle. As shown in the cross-sectional SEM image (Fig. 8b), BiVO4 nanodots have a larger specific surface area compared to the core–shell structure, increasing contact with the electrolyte. Also, nanodots below the diffusion length of BiVO4 resulted in the suppression of charge recombination and led to a high charge collection ability. Bera et al. conducted a more detailed analysis of the relationship between the minority carrier diffusion length (Lp) and particle diameter.248 As shown in Fig. 8c, as the radius of BiVO4 increases, the bulk region also increases, deteriorating the PEC performances since the probability of holes escaping bulk recombination in oxides is proportional to ex−/Lp. When the particle radius (R), except for the space charge layer (W) influenced by the drift, is smaller than the diffusion length, the bulk recombination is suppressed, causing high charge separation efficiency. Nanoporous oxide electrodes have enhanced light absorption in water splitting photoelectrodes. Their high surface area and tailored porosity allow for efficient light trapping and increased charge separation, improving solar-to-hydrogen conversion efficiency. Developing nanoporous oxide electrodes that exhibit enhanced stability under prolonged photoelectrochemical operation will be a key goal. These electrodes could resist degradation from factors like photocorrosion, ensuring longer-lasting and reliable water splitting performance.
Since the sensitizer is designed to absorb photons in the visible light region, semiconductor electrodes generally use wide band gap materials capable of absorbing the ultraviolet region in order to achieve efficient light harvesting.251 TiO2, which possesses a wide band gap (3.2 eV), high electron mobility, and stability, is the most representative material, and various TiO2 nanostructures based on solution processes have been developed for DSSCs.252 Choudhury et al. formed TiO2 nanowires through a hydrothermal reaction and synthesized hierarchical nanoforest structures of TiO2 using a second acid-assisted hydrothermal reaction.253 As shown in Fig. 9a, it is observed that TiO2 nanoarrays are densely formed along the Ti wire, maximizing the contact area with the electrolyte. Ag nanoparticles were synthesized on the TiO2 nanoforest by photon-assisted reduction, giving a 16.1% increase in efficiency compared to nanotree array (NTA)-based DSSCs. The device recorded a short circuit current density (Jsc) of 5.98 mA cm−2, which originated from the enhanced absorbance by the TiO2 nanostructures and the SPR effect of Ag nanoparticles. TiO2 NPs have numerous grain boundaries but low surface area. On the other hand, TiO2 nanospheres (SPs) have a high surface area but difficulty in charge transport at the SP interfaces. Therefore, the optimal size of SPs that combines the advantages of both structures is challenging.254,255 Yu et al. discovered the optimal size of porous TiO2 nanospheres (SPs) for achieving high power conversion efficiency (PCE) in DSSCs.256 Porous TiO2 SPs with sizes ranging from 100 to 200 nm were synthesized through the control of hydrolysis conditions during the sol–gel reaction. The DSSC consisting of SP100 with SP450 as a light scattering layer exhibited a high PCE of 10.66%. Kang et al. introduced hierarchical TiO2 (h-TiO2) with polyvinylpyrrolidone as a pore-forming agent to further enhance light absorption and the PCE of conventional mesoporous TiO2 (m-TiO2).257 It has been revealed that h-TiO2, which coexists with macropores, offers advantages over m-TiO2 in terms of diffusion and electrolyte injection. The optimized DSSCs consisting of h-TiO2 photoanodes with a long-persistence-phosphor layer showed the highest PCE of 8.05%.
 | ||
| Fig. 9 Nanoporous oxide electrodes for solar cells. (a) Scheme of the TiO2 nanoarray-based dye-sensitized solar cell (DSSC). (b) J–V curves of nano-tree TiO2 and Ag deposited nano-tree TiO2 DSSCs. Reproduced from ref. 253. Copyright 2021 Springer Nature. (c) FESEM image of ZnO hollow nanospheres. (d) J–V plots for DSSCs of SnO2 nanoparticles with different ZnO hollow nanospheres. Reproduced from ref. 261. Copyright 2018 American Chemical Society. (e) FESEM image of ZnO–TiO2 core–shell nanorods. (f) J–V characteristics of DSSCs applying ZnO nanorods and ZnO–TiO2 core–shell nanorods. Reproduced from ref. 262. Copyright 2020 MDPI. | ||
SnO2 and ZnO, which have wide band gaps, have also been extensively studied in DSSCs for UV absorption, although their electron injection rates are lower compared to that of TiO2.258–260 Banik et al. synthesized mesoporous hollow ZnO microspheres (HS) using the wet chemical process, as shown in Fig. 9c.261 It was used as the photoanode in DSSCs by forming composites with SnO2 NPs synthesized through a reflux process. ZnO HS exhibited excellent light scattering, and DSSCs recorded a high Jsc of 7.82 mA cm−2 by utilizing a synergistic effect at the optimal mixing ratio with SnO2 NPs (Fig. 9d). Zhang et al. introduced a core–shell structure of TiO2-coated ZnO nanorods deposited using sputtering.262 As shown in the SEM image (Fig. 9e), the TiO2 nanostructures deposited by mist chemical vapor deposition are observed to be formed around the ZnO nanorods, leading to increased roughness and surface area of the photoanode. NiO, which has a band gap of 3.6 eV, is generally used as a photocathode in DSSCs.263,264 Zannotti et al. introduced rGO to enhance the performances of NiO NPs formed by the sol–gel process.265 The rGO coated on the surface of NiO through the thermal reduction of GO not only increased the surface area and mesopore volume but also contributed to the improvement of electron transport. We summarized the materials, Voc, Jsc, and PCE of DSSCs in Table 1. Nanoporous oxide electrodes have enhanced light-harvesting capabilities in DSSCs. Their large surface area and well-defined porosity provided ample sites for dye molecule absorption, allowing for increased photon capture and improved overall PCE. Future research might focus on tailoring the pore structures of nanoporous oxide electrodes to optimize dye loading and electron transport. By precisely controlling the pore size and distribution, researchers can fine-tune the interaction between the dye molecules and the electrode, leading to improved charge separation and collection. Also, as DSSCs move closer to large-scale implementation, the development of scalable fabrication methods for nanoporous oxide electrodes will become crucial. Innovations in electrode production could help meet the growing demand for cost-effective and efficient solar energy solutions.
 | ||
| Fig. 10 Nanoporous oxide electrodes for light-emitting diodes. (a) Schematics of light trajectories in LEDs with the flat sapphire substrate (FSS) and patterned sapphire substrate (PSS). Reproduced from ref. 268. Copyright 2022 The Royal Society of Chemistry. (b) Cross-sectional ray-tracing images and schematic illustration of light trajectories of UV-LEDs on the FSS, PSS, and PSSA. Reproduced from ref. 269. Copyright 2020 Elsevier. (c) Cross-sectional SEM image of the nano–micro complex PSS (NMCPSS). Reproduced from ref. 273. Copyright 2016 American Chemical Society. (d) SEM image of the micro–nano PSS (MNPSS). Reproduced from ref. 274. Copyright 2013 IOP Science. (e) SEM image of the hybrid PSS. Reproduced from ref. 275. Copyright 2016 American Chemical Society. | ||
Various nanoporous substrates for LEDs have been developed as advancements from conventional patterned sapphire substrates.270–272 Zhou et al. introduced Ni nanodots on the PSS and fabricated nano–micro complex patterned sapphire substrates (NMCPSS) by inductively coupled plasma (ICP) etching.273 As shown in the SEM image (Fig. 10c), nanoscale cones derived from the Ni nanodots as the mask are densely formed on the microscale cones, making the substrate rougher. The light output power of the NMCPSS-based LED was 28.6% higher than that of the PSS-based LED. Through simulations, it was revealed that the enhancement was attributed to improved light extraction efficiency. Similarly, Cheng et al. fabricated a micro–nano patterned sapphire substrate (MNPSS) by using Ni nanoislands and ICP etching, as shown in Fig. 10d.274 3-Dimensional finite-difference time-domain simulation showed that the light extraction efficiency was 63% higher than that of the micropatterned sapphire substrate. Ke et al. prepared a hybrid patterned sapphire substrate (hybrid-PSS) by using an AAO etching mask.275 An Al–Ti film deposited by e-beam evaporation on the microscale PSS was anodized to form the AAO layer with nanopatterns. The PSS was etched by ICP with the nanopatterned AAO layer as the mask, and nanopatterns were intricately formed on the microcones (Fig. 10e). The increased contact area between the LED and hybrid-PSS induced light scattering and increased the LED's view angle of 15°, enhancing the injection current.
Nanoporous oxides can also facilitate the efficient transport and injection of charge carriers within the LEDs. The porous structure can provide a larger surface area for contact between the active area and the electrodes, enabling better charge injection and reducing energy losses.276 Chen et al. introduced NiOx as the hole injection layer for quantum dot LEDs (QLEDs).277 The NiOx film was easily fabricated by spin-coating and showed spongelike nanostructures. The larger surface area enhanced hole injection and lowered the turn-on voltage compared to the NiOx thin film. ZnO is the representative material in the electron transport layer for QLEDs because of its high electron mobility and transmittance. Kirkwood et al. introduced ZnO nanoparticles as the electron transport layer.278 Compared to conventional ZnO films, the ZnO nanoparticles exhibited enhanced electron mobility by tuning defect density. The QLEDs showed improved efficiency and operating time. In QLEDs, nanoporous oxides can also serve as a matrix for embedding quantum dots (QDs). They can provide a suitable host matrix for dispersing and aligning quantum dots, thereby improving the efficiency and color purity of QLEDs. Wang et al. introduced mesoporous silica as the host matrix for CsPbBr3 perovskite QDs.279 The CsPbBr3 QDs with a pore size of 12–14 nm were embedded in the mesoporous silica. The well-dispersed QDs prevent ion exchange and increase stability. Nanoporous oxides can also be applied as antireflection coatings on the surface of LEDs. These coatings help to minimize the reflection of light at the interface between the LED and the surrounding environment. By reducing unwanted reflections, more light can escape from the LED and contribute to the desired output, resulting in improved performance. Kim et al. introduced ITO nanorods by oblique-angle deposition and incorporated the ITO antireflection coating onto a GaInN LED.280 The GaInN LED with ITO antireflection exhibited a light extraction efficiency enhancement of 24.3% compared to the LED with conventional ITO.
4.2. Energy storage devices
Lui et al. developed 3DOM TiO2 electrodes as an anode in a Li-ion battery.287Fig. 11a and b show the comparison of lithiation pathways between the nanoparticle and 3DOM electrodes. In the nanoparticle electrodes, densely packed particles reduce the surface area contact with the electrolyte, and inside particles can be excluded from lithiation. Also, the movement of electrons in the nanoparticle electrode is limited to a specific path, and binding agents are required for high conductivity. On the other hand, in the 3DOM electrodes, the ordered pores offer high surface area contact with electrolytes, facilitating mass transport and complete lithiation. Also, the interconnected structures enhanced electron transport, and they can be manufactured without binding agents. The carbon-coated 3DOM TiO2 electrode from polystyrene exhibited high-rate performance with a capacity of 174 mA h g−1 at 2 A g−1. Also, the free volume in the 3DOM TiO2 electrode contributes to the highly stable performance (181 mA h g−1) with a capacity retention of 94.8% over 1000 cycles. Research on ternary nanoporous oxides for the electrodes of Li-ion batteries has also been conducted.288,289 Lou et al. prepared a 3DOM TiNbO7 anode with a high theoretical capacity of 387 mA h g−1.290 The honeycomb-like structure derived from the polystyrene crystal as a template offered a fast electron pathway and wide Li+ insertion/extraction region. The 3DOM-TiNb2O7 anode recorded not only a remarkable rate capability of 120 mA h g−1 at 50 C but also durable long-term cyclability of 82% capacity retention over 1000 cycles at 10 C, as shown in Fig. 11c. Pikul et al. synthesized a 3DOM LiMnO2 cathode on porous Ni and fabricated a Li-ion microbattery.136 The 3DOM Ni framework was formed by electrodeposition onto polystyrene particles and etching of polystyrene. MnO2 was electrodeposited on the 3DOM Ni framework and lithiated in molten salt. As shown in the cross-sectional SEM images (Fig. 11d), the NiSn anode and LiMnO2 cathode were uniformly formed into the 3DOM structure following the Ni frameworks. The Li-ion microbattery delivered an energy density of 2.5 μW h cm−2 μm−1 at 0.5 C, showing a high power density of 7.4 mW cm−2 μm−1, which is a comparable performance with supercapacitors. Similarly, Wang et al. fabricated a 3DOM Fe2O3 anode by using a Ni inverse opal as the support.291 Due to the short electrode–electrolyte distances for electron and ion transport, the 3DOM Fe2O3 electrode exhibited excellent cycling and rate performances. Based on the enhanced kinetics, the electrode showed a specific capacity of 450 mA h g−1 and a reduced voltage hysteresis of 0.62 V at 0.1 A g−1.
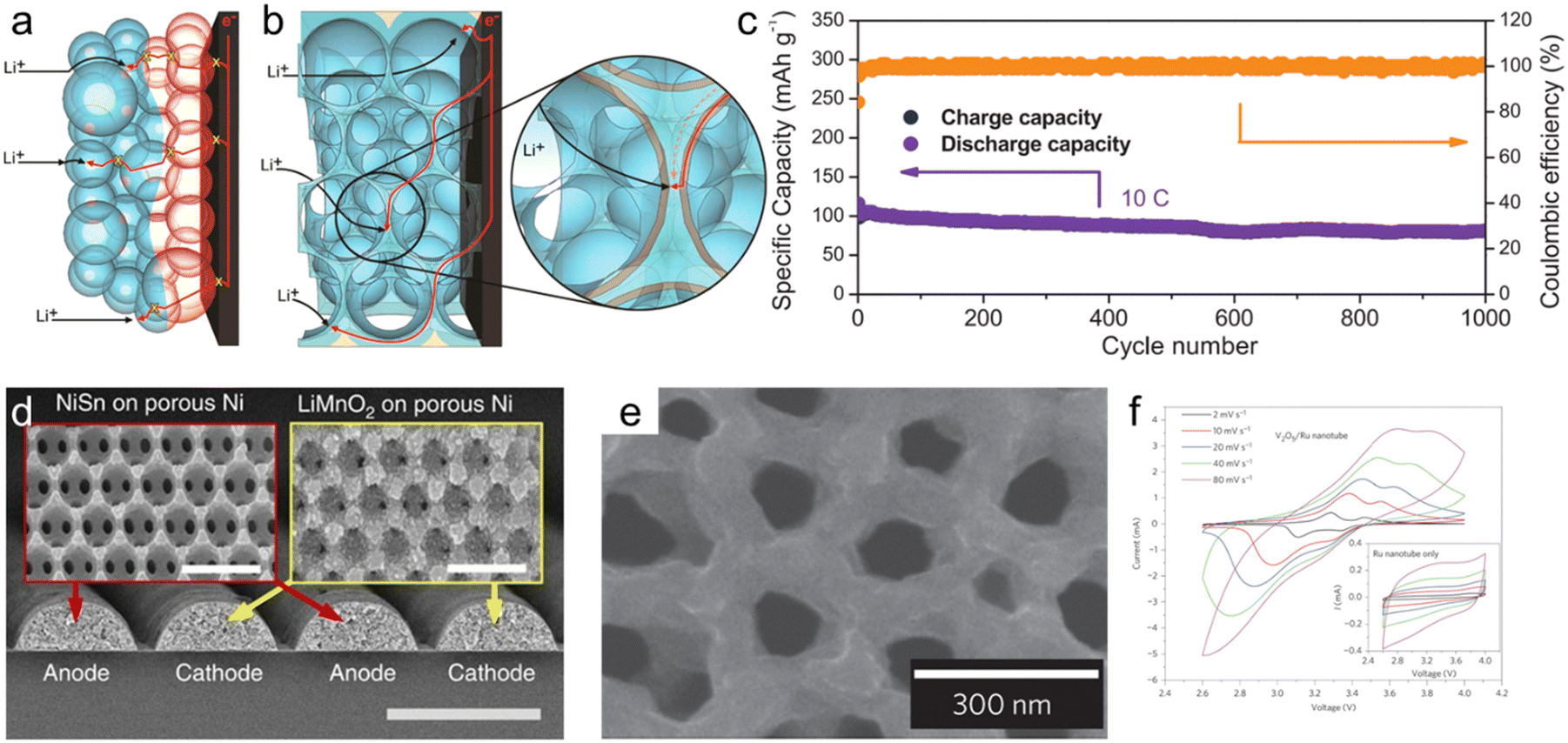 | ||
| Fig. 11 Nanoporous oxide electrodes for batteries. Schematic comparing lithiation pathways for (a) nanoparticle and (b) 3DOM electrodes. Reproduced from ref. 287. Copyright 2016 Elsevier. (c) Cycling performance and coulombic efficiency of the TiNb2O7 anode at 10 C after 7 cycles at 0.1 C. Reproduced from ref. 290. Copyright 2017 Elsevier. d) Cross-sectional SEM images of the interdigitated electrodes spanning two periods. The insets show NiSn on the porous Ni anode and LiMnO2 on the porous Ni cathode. Scale bars, 50 mm and 1 mm in the insets. Reproduced from ref. 136. Copyright 2013 Springer Nature. (e) SEM image of the device (top view), showing AAO pores remaining open after Ru and V2O5 ALD. (f) CVs of V2O5/Ru nanotubes and Ru nanotubes showing characteristic oxidation and reduction reaction peaks with scan rates up to 80 mV s−1. Reproduced from ref. 292. Copyright 2014 Springer Nature. | ||
Liu et al. developed an all-in-one nanopore battery array derived from a self-assembled AAO template.292 Each nanoelectrode was composed of a Ru nanotube current collector and V2O5 nanotube storage material, constructing a symmetric full nanopore battery array. The battery was asymmetrically cycled by forming the lithiated V2O5 anode and the pristine V2O5 cathode, as shown in the SEM image (Fig. 11e). In the CVs of the V2O5/Ru nanotubes (Fig. 11f), most of the reaction charge was used in the faradaic reaction process, indicating oxidation and reduction peaks. This means that the insertion of Li-ions took place quickly in the faradaic reaction, and these nanotube structures make the insertion of Li-ions comparable with fast double-layer charging. The nanopore battery array exhibited a high capacity of 80 mA h g−1 at 150 C with 80% retained capacity after 1000 cycles. We summarized the materials, capacities, and duration cycles of batteries in Table 2. While nanoporous oxide electrodes have shown potential in lithium-ion batteries, their application could extend beyond this. Research may focus on alternative energy storage systems, such as sodium-ion or potassium-ion batteries, where nanoporous oxide electrodes could exhibit superior performance and stability.
| Batteries | |||||
|---|---|---|---|---|---|
| Materials | Capacity [mA h g−1] | Specific current | Cycling capacity [mA h g−1] | Cycling [cls] | Ref. |
| 3DOM TiO2 | 174 | 2 A g−1 | 181 | 1000 | 287 |
| ZnCo2O4 | 932 | 1 A g−1 | 932 | 50 | 289 |
| 3DOM TiNbO7 | 387 | 1 C | 120 | 1000 | 290 |
| 3DOM LiMnO2 | — | — | — | 1000 | 136 |
| 3DOM Fe2O3 | 1000 | 0.2 A g−1 | 400 | 100 | 291 |
| V2O5/Ru NTs | 80 | 150 C | 80 | 1000 | 292 |
| Supercapacitors | ||||
|---|---|---|---|---|
| Materials | Specific capacitance [F g−1] | Specific power density [W kg−1] | Specific energy density [W h kg−1] | Ref. |
| TArGO | 372.1 | — | — | 302 |
| RuO2 | 50 | — | — | 305 |
| RuO2/CNT | 644 | 17000 |
4 | 5 |
| MnO2 | 144.1 | 1800 | 7.9 | 308 |
| MnO2/CNTs/MnO2 | 341.5 | — | — | 309 |
| Fe2O3/NPCTT | 1846 | 7.48 × 10−4 | 1.76 × 10−4 | 310 |
| Ni(OH)2–MnO2–rGO | 1985 | 392 | 54 | 314 |
| Co3O4 | 504 | 8000 | 36 | 315 |
000 cycle tests. The high EDLC was attributed to the high porosity of TArGO.
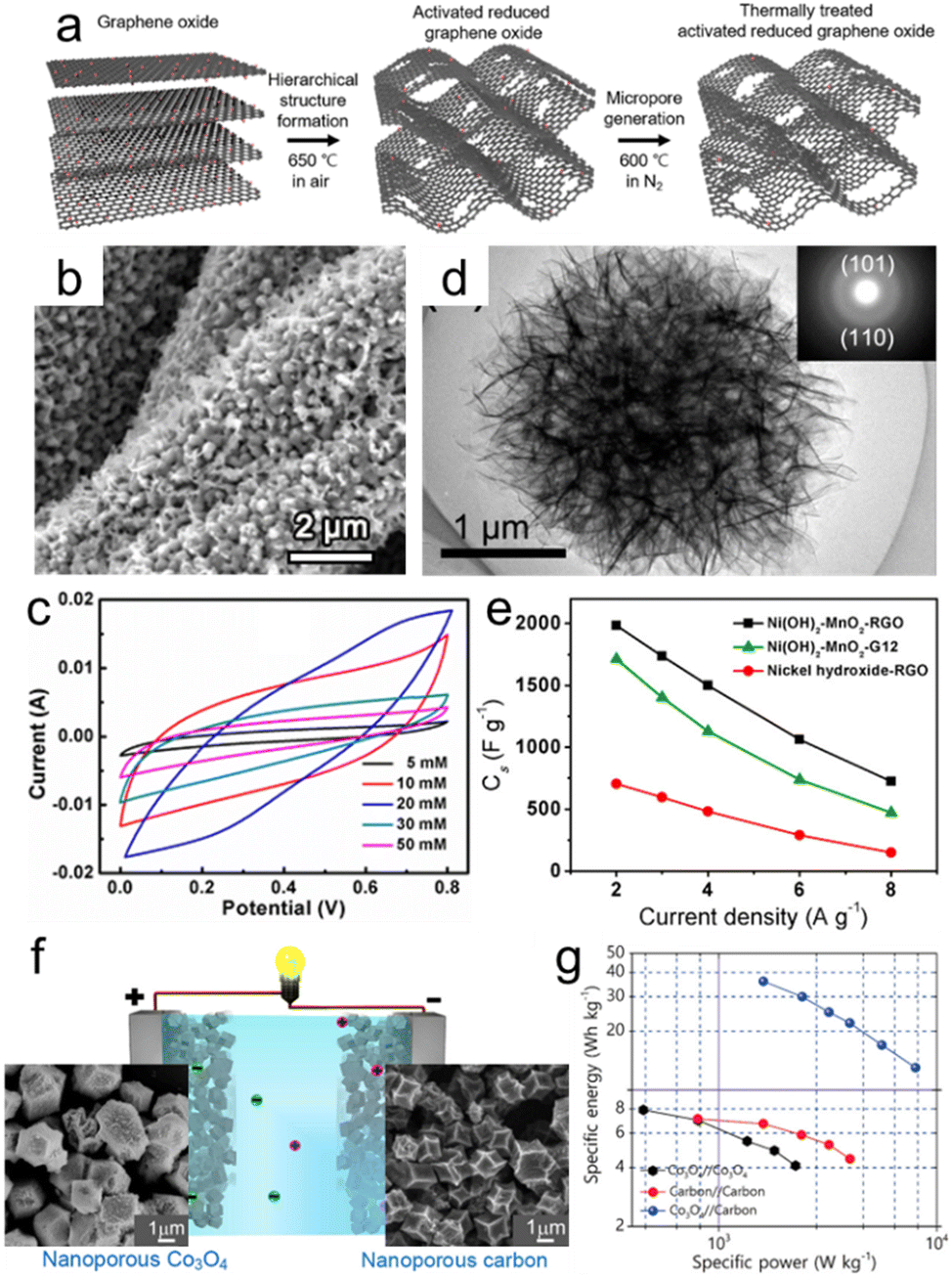 | ||
| Fig. 12 Nanoporous oxide electrodes for supercapacitors. (a) Schematic for the preparation of thermally treated activated reduced graphene oxide (TArGO) powder. Reproduced from ref. 302. Copyright 2021 Springer Nature. (b) SEM image of the MnO/nanoporous carbon tube textile (NPCTT) product. (c) CV curves of MnO/NPCTT electrodes with different CFe3+ at a scan rate of 10 mV s−1. Reproduced from ref. 310. Copyright 2020 Springer Nature. (d) TEM image and (e) specific capacitance of the Ni(OH)2–MnO2–rGO hybrid sphere electrode. Reproduced from ref. 314. Copyright 2014 American Chemical Society. (f) Schematics of asymmetric supercapacitors (ASC) with nanoporous Co3O4 and nanoporous carbon as the positive and negative electrodes, respectively. (g) Ragone plots of SSCs and ASC based on nanoporous carbon and nanoporous Co3O4 electrodes. Reproduced from ref. 315. Copyright 2015 American Chemical Society. | ||
RuO2 is one of the most efficient pseudocapacitive electrode materials.304 It exhibits high specific capacitance and fast charge/discharge rates. Patake et al. synthesized nanoporous RuO2 thin films for supercapacitors by chemical bath deposition. The large surface area of RuO2 contributed to the high specific capacitance of 50 F g−1.305 Warren et al. coated highly active RuO2 through atomic layer deposition on vertically aligned carbon nanotubes (CNTs) with extremely high porosity.5 A further electrochemical oxidation process was carried out to achieve a specific capacitance of 644 F g−1 that is close to the theoretical value of RuO2. MnO2, which is cost-effective and exhibits high specific capacitance, is an excellent alternative to RuO2.306,307 Kumar et al. chemically synthesized hierarchical nanostructured MnO2 for supercapacitors.308 Also, Wang et al. encapsulated MnO2 into porous CNTs, and the optimal MnO2/CNT/MnO2 nanocomposites showed an excellent specific capacitance of 341.5 F g−1 and good rate performance.309 This could be derived from the synergistic effect of MnO2 with high-capacity storage and CNTs with high electrical conductivity. Ding et al. developed a facile synthesis method of non-noble metal oxides onto a nanoporous carbon tube textile (NPCTT) toward flexible supercapacitors.310 The formation of nanostructures on the NPCTT was made possible through alkaline activation and infiltration of catalytic elements. As shown in Fig. 12b, MnO nanosheets were fabricated on the NPCTT without any aggregation, forming a porous and rough surface. This method was applied to the synthesis of hematite nanobelts, and the Fe2O3/NPCTT electrodes exhibited excellent flexibility with a high areal specific capacitance of 1846 mF cm−1 at 1 mA cm−2, as shown in Fig. 12c.
There are various attempts to achieve excellent supercapacitor performances through the combination of metal oxides and carbon-based oxides.311–313 Chen et al. reported Ni(OH)2–MnO2–rGO ternary hybrid sphere powders using a one-step hydrothermal method, as shown in Fig. 12d.314 Although Ni(OH)2 offers high capacitance and good cyclic stability, its relatively low electrical conductivity limits its performance. MnO2 and rGO complement each other, and the synergistic effect and high specific surface area resulting from the nanoporous structure provide excellent characteristics that can replace noble metal oxides. The Ni(OH)2–MnO2–rGO ternary hybrid spheres showed a significantly improved specific capacitance of 1985 F g−1 and energy density of 54 W h kg−1 at 2 A g−1 compared to other composites, as shown in Fig. 12e. Salunkhe et al. synthesized two supercapacitor materials, nanoporous carbon, and nanoporous Co3O4, by varying the thermal treatment conditions from a single zeolite imidazolate framework (ZIF-67) precursor.315 As shown in the SEM images (Fig. 12f), Co3O4 with a granular morphology offered a high surface area and good accessibility for ions, and nanoporous carbon also provided good electrical conductivity and a high surface area. Symmetric (SSC) and asymmetric supercapacitors (ASC) were fabricated using nanoporous carbon and Co3O4. The ASC recorded a high specific energy density of 36 W h kg−1 and a specific power density of 8000 W kg−1, which were higher than those of the SSCs. We summarized the materials, specific capacitances, power densities, and energy densities in Table 2. Nanoporous oxide electrodes have played a crucial role in advancing energy storage solutions. Their high surface area and porous nature provided ample room for ion storage, leading to improved energy density and faster charging/discharging rates in supercapacitors. The application of nanoporous oxide electrodes could extend beyond this. Future developments might lead to the integration of nanoporous oxide electrodes into multifunctional energy devices. For instance, combining energy storage with energy conversion (like in hybrid supercapacitor–solar cell systems) could offer self-sustaining power solutions for various applications.
5. Summary and outlooks
Herein, we have systematically discussed the advances in structures, synthesis techniques, and applications of nanoporous oxides. Nanoporous oxide electrodes have demonstrated tremendous potential in various energy conversion and storage applications. Their dimensional diversity, from 0D to 3D, enables the construction of infinite nanoporous structures by controlling the pore size and morphology with the effective utilization of graphene oxides and binary, ternary, and multinary metal oxides. Various synthesis strategies, including bottom-up and top-down approaches, have allowed for the creation of elaborate nanoporous oxide electrodes. By exploiting the advantages derived from the structural characteristics of nanoporous oxides, such as enlarged surface area, efficient charge separation and transport, and the light trapping effect, they have exhibited superior properties in the field of energy conversion and storage. From fuel cells to supercapacitors, nanoporous oxides have been used as multifunctional electrodes, successfully overcoming major challenges in energy nanotechnologies. Although significant advances in the construction and applications of nanoporous oxide electrodes have been achieved in recent decades, there are still several challenges that need to be addressed to fully realize their potential.First, it is important to carefully consider the performance degradation and stability issues associated with the increased surface area due to nanoporous structures. As mentioned above, the interconnected nanopore network enhances ion transport and accelerates the reaction rate; however, insufficient understanding of the flow dynamics within the nanoporous structure can adversely affect the reaction kinetics. In addition, even though the reaction site is extended by increasing the surface area, it can also introduce detrimental surface trap sites, which can degrade performance and exacerbate stability issues. Therefore, to effectively harness the nanoporous geometry, it is essential to focus on real-time operating conditions. Specifically, computational modeling techniques, such as molecular dynamics simulations and computational fluid dynamics, and in situ analysis during operation are imperative approaches. This can provide rapid feedback to mitigate adverse effects in the actual reaction kinetics and contribute to the design of optimized nanoporous oxide structures tailored to real operational scenarios. Furthermore, to ensure stable and high-efficiency operation, a strategic approach is also needed in the synthesis aspect. A specific example of this strategy is the development of synthesis methods for oxide functional coatings that can conformally coat the complex surface of nanoporous oxides. Many previous studies have shown the effectiveness of such coatings in deactivating trap sites, and this approach should be further developed into a more universal method for conformally coating a wider range of functional oxides.
Second, the fabrication processes of nanoporous oxide electrodes need to be optimized to scale up the synthesis while maintaining cost-effectiveness. Synthesis methods that are difficult to obtain high yields and require complex steps are challenging to use practically. The problem of synthesizing nanoporous oxides uniformly while controlling the desired composition, shape, and surface conditions also poses a major obstacle. Therefore, synthesis strategies need to be improved in a direction that enhances high productivity and uniformity, suitable for the targeted applications. Specifically, it is necessary to approach the development of synthesis methods through computational-based predictions. To identify patterns and predict optimal synthesis conditions, machine learning models can be actively utilized using a dataset of both successful and unsuccessful attempts. Additionally, there are methodologies that involve implementing generative models like GANs (generative adversarial networks). These models can generate new synthesis recipes based on known data and iteratively optimize them. By implementing these specific solutions, researchers can harness the power of computational-based predictions to optimize the fabrication processes of nanoporous oxide electrodes more effectively, reducing the need for extensive trial-and-error experimentation.
Third, nanoporous oxides can enhance their value as effective electrodes in multiscale devices that integrate various energy nanotechnologies, going beyond the development of a single device. By effectively harnessing the electrical and chemical properties of oxide materials and incorporating nanoporous structures, performance and stability have reached a certain level. Following the development of a single device, effective integration of energy conversion and storage in hybrid devices will ultimately have a significant impact on the future energy industry. Nanoporous oxides, with their unique structural characteristics and tunable properties, hold high potential for widespread utilization in the design of such hybrid devices.
We present design strategies for the cyclic process of feedback and optimization, along with the current system and future directions (Fig. 13). To date, we have accumulated empirical data on numerous nanoporous oxide materials, spanning from their synthesis to material characterization, followed by functionality assessment. Our development strategy is anticipated to be grounded in the active utilization of in situ analysis and computational techniques. In situ analysis allows for meticulous observation of the operation of designed nanoporous oxide electrodes, enabling the acquisition of the most precise and reliable feedback. Furthermore, the proactive use of computational techniques can expand our understanding of materials, from the synthesis to operational conditions, encompassing insights from the material level to the system level. In a more advanced phase, we envision the prospects of designing devices that integrate energy conversion and storage, based on nanoporous oxide electrodes. Although research at the system level holds the promise of significantly enhancing energy cycles, the complexity of interconnecting multiple systems currently presents delays. In the future, we anticipate that the considerable advancements in individual applications of nanoporous oxide electrodes will converge to create opportunities for their utilization in integrated devices. These system-level studies, built upon the substantial progress in various applications of nanoporous oxide electrodes, are expected to pave the way for the development of novel devices with improved efficiency and enhanced capabilities.
 | ||
| Fig. 13 Design strategies of nanoporous oxide electrodes. | ||
Author contributions
Jin Wook Yang: conceptualization, investigation, visualization, writing (original draft, review & editing). Hee Ryeong Kwon: conceptualization, investigation, visualization, writing (original draft, review & editing). Jin Ho Seo: conceptualization, investigation, visualization, writing (original draft, review & editing). Sangwoo Ryu: supervision. Ho Won Jang: conceptualization, writing (review & editing), supervision, funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) funded by the Korea government Ministry of Science and ICT (MSIT) (2021R1A2B5B03001851, 2021M3H4A1A03057403). This work was also supported by the KRISS (Korea Research Institute of Standards and Science) MPI Lab. Program. Jin Wook Yang acknowledges the NRF funded by the Korea government MSIT (RS-2023-00213786). The Inter-University Semiconductor Research Center and Institute of Engineering Research at Seoul National University provided research facilities for this work.Notes and references
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925–13931 CrossRef CAS.
- C. W. Kwon, J. W. Son, J. H. Lee, H. M. Kim, H. W. Lee and K. B. Kim, Adv. Funct. Mater., 2011, 21, 1154–1159 CrossRef CAS.
- N. Liu, H. Ma, L. Wang, Y. Zhao, Z. Bakenov and X. Wang, J. Mater. Sci. Technol., 2021, 84, 124–132 CrossRef CAS.
- D. Lei, D. C. Lee, E. Zhao, A. Magasinski, H. R. Jung, G. Berdichevsky, D. Steingart and G. Yushin, Nano Energy, 2018, 48, 170–179 CrossRef CAS.
- R. Warren, F. Sammoura, F. Tounsi, M. Sanghadasa and L. Lin, J. Mater. Chem. A, 2015, 3, 15568–15575 RSC.
- N. Ogihara, Y. Itou, T. Sasaki and Y. Takeuchi, J. Phys. Chem. C, 2015, 119, 4612–4619 CrossRef CAS.
- J. An, Y. B. Kim, J. Park, T. M. Gür and F. B. Prinz, Nano Lett., 2013, 13, 4551–4555 CrossRef CAS.
- H. Lee, J. W. Yang, J. Tan, J. Park, S. G. Shim, Y. S. Park, J. Yun, K. Kim, H. W. Jang and J. Moon, Adv. Sci., 2021, 8, 2102458 CrossRef CAS.
- S. M. H. Hejazi, J. Aghazadeh Mohandesi and M. Javanbakht, Sol. Energy, 2017, 144, 699–706 CrossRef CAS.
- J. H. Son, J. U. Kim, Y. H. Song, B. J. Kim, C. J. Ryu and J. L. Lee, Adv. Mater., 2012, 24, 2259–2262 CrossRef CAS.
- H. Yu, W. Wang, M. Liu, T. Zhao, R. Lin, M. Hou, Y. Kou, L. Chen, A. A. Elzatahry, F. Zhang, D. Zhao and X. Li, Sci. Adv., 2022, 8, eabq2356 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- J. Li, X. Jin, R. Li, Y. Zhao, X. Wang, X. Liu and H. Jiao, Appl. Catal., B, 2019, 240, 1–8 CrossRef CAS.
- T. A. Knecht and J. E. Hutchison, Chem. Mater., 2023, 35, 3151–3161 CrossRef CAS.
- M. Košiček, J. Zavašnik, O. Baranov, B. Šetina Batič and U. Cvelbar, Cryst. Growth Des., 2022, 22, 6656–6666 CrossRef.
- N. Du, H. Zhang, B. Chen, M. Xiangyang, Z. Liu, J. Wu and D. Yang, Adv. Mater., 2007, 19, 1641–1645 CrossRef CAS.
- J. Zhu, Z. Yin, H. Li, H. Tan, C. L. Chow, H. Zhang, H. H. Hng, J. Ma and Q. Yan, Small, 2011, 7, 3458–3464 CrossRef CAS.
- X. Yu, T. J. Marks and A. Facchetti, Nat. Mater., 2016, 15, 383–396 CrossRef CAS.
- Z. Yu, H. Liu, M. Zhu, Y. Li and W. Li, Small, 2021, 17, 1903378 CrossRef CAS.
- T. Liu, Y. Qu, J. Liu, L. Zhang, B. Cheng and J. Yu, Small, 2021, 17, 2103673 CrossRef CAS PubMed.
- G. Y. Yoo, N. Nurrosyid, S. Lee, Y. Jeong, I. Yoon, C. Kim, W. Kim, S. Y. Jang and Y. R. Do, ACS Appl. Mater. Interfaces, 2020, 12, 10626–10636 CrossRef CAS.
- C. W. Chen, H. W. Tsai, Y. C. Wang, Y. C. Shih, T. Y. Su, C. H. Yang, W. S. Lin, C. H. Shen, J. M. Shieh and Y. L. Chueh, Adv. Funct. Mater., 2019, 29, 1905040 CrossRef CAS.
- C. Sun, J. A. Alonso and J. Bian, Adv. Energy Mater., 2021, 11, 2000459 CrossRef CAS.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- W. Yu, L. Sisi, Y. Haiyan and L. Jie, RSC Adv., 2020, 10, 15328–15345 RSC.
- T. Hirano, S. Kaseda, C. Kiet Le Anh, F. Iskandar, E. Tanabe and T. Ogi, ACS Appl. Nano Mater., 2022, 5, 15449–15456 CrossRef CAS.
- M. Liu, H. Xia, W. Yang, X. Liu, J. Xiang, X. Wang, L. Hu and F. Lu, Appl. Catal., B, 2022, 301, 120765 CrossRef CAS.
- L. Li, J. Yan, T. Wang, Z. J. Zhao, J. Zhang, J. Gong and N. Guan, Nat. Commun., 2015, 6, 5881 CrossRef.
- J. Park, J. Joo, G. K. Soon, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 4630–4660 CrossRef CAS.
- P. Wainer, O. Kendall, A. Lamb, S. J. Barrow, A. Tricoli, D. E. Gómez, J. Van Embden and E. Della Gaspera, Chem. Mater., 2019, 31, 9604–9613 CrossRef CAS.
- L. Sang, Y. Zhao and C. Burda, Chem. Rev., 2014, 114, 9283–9318 CrossRef CAS.
- Z. Zhu, Y. Bai, X. Liu, C. C. Chueh, S. Yang and A. K. Y. Jen, Adv. Mater., 2016, 28, 6478–6484 CrossRef CAS.
- H. M. Jeong, K. M. Choi, T. Cheng, D. K. Lee, R. Zhou, I. W. Ock, D. J. Milliron, W. A. Goddard and J. K. Kang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7914–7919 CrossRef CAS.
- X. Chen, L. L. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- J. Klein, M. Philippi, F. Alarslan, T. Jähnichen, D. Enke, M. Steinhart and M. Haase, Small, 2023, 19, 2207674 CrossRef CAS PubMed.
- S. Cong, Y. Tian, Q. Li, Z. Zhao and F. Geng, Adv. Mater., 2014, 26, 4260–4267 CrossRef CAS PubMed.
- H. Xia, C. Hong, B. Li, B. Zhao, Z. Lin, M. Zheng, S. V. Savilov and S. M. Aldoshin, Adv. Funct. Mater., 2015, 25, 627–635 CrossRef CAS.
- M. Ye, Z. Zhao, Z. Hu, L. Liu, H. Ji, Z. Shen and T. Ma, Angew. Chem., 2017, 129, 8527–8531 CrossRef.
- S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, J. Am. Chem. Soc., 2004, 126, 273–279 CrossRef CAS.
- J. Ahn, Y. Song, Y. J. Kim, D. Nam, T. Kim, K. Kwak, C. Hoon Kwon, Y. Ko, S. J. Lee and J. Cho, Chem. Eng. J., 2023, 455, 140742 CrossRef CAS.
- M. Xiao, Z. Wang, M. Lyu, B. Luo, S. Wang, G. Liu, H. M. Cheng and L. Wang, Adv. Mater., 2019, 31, 1801369 CrossRef.
- B. Koo, H. Xiong, M. D. Slater, V. B. Prakapenka, M. Balasubramanian, P. Podsiadlo, C. S. Johnson, T. Rajh and E. V. Shevchenko, Nano Lett., 2012, 12, 2429–2435 CrossRef CAS PubMed.
- Z. Yan, W. Huang, X. Jiang, J. Gao, Y. Hu, H. Zhang and Q. Shi, Microporous Mesoporous Mater., 2021, 323, 111228 CrossRef CAS.
- S. H. Hwang, J. Yun and J. Jang, Adv. Funct. Mater., 2014, 24, 7619–7626 CrossRef CAS.
- S. Li, J. Niu, Y. C. Zhao, K. P. So, C. Wang, C. A. Wang and J. Li, Nat. Commun., 2015, 6, 7872 CrossRef CAS PubMed.
- M. Zhou, T. Wang, Z. He, Y. Xu, W. Yu, B. Shi and K. Huang, ACS Sustainable Chem. Eng., 2019, 7, 2924–2932 CrossRef CAS.
- N. Jiang, D. Li, L. Liang, Q. Xu, L. Shao, S. Bin Wang, A. Chen and J. Wang, Nano Res., 2020, 13, 1354–1362 CrossRef CAS.
- Q. Wei, F. Xiong, S. Tan, L. Huang, E. H. Lan, B. Dunn and L. Mai, Adv. Mater., 2017, 29, 1602300 CrossRef PubMed.
- I. Hussain, T. Mak and K. Zhang, ACS Appl. Nano Mater., 2021, 4, 129–141 CrossRef CAS.
- R. Venkatesan, R. Bauri and K. K. Mayuranathan, Energy Fuels, 2022, 36, 7854–7864 CrossRef CAS.
- H. Jin Han, G. Rac Lee, Y. Xie, H. Jang, D. J. Hynek, E. N. Cho, Y. Ji Kim, Y. S. Jung and J. J. Cha, Sci. Adv., 2021, 7, eabh2012 CrossRef.
- X. Li and J. Wang, InfoMat, 2020, 2, 3–32 CrossRef CAS.
- B. Liu and E. S. Aydil, J. Am. Chem. Soc., 2009, 131, 3985–3990 CrossRef CAS PubMed.
- Y. Xing, X. Sheng, H. Zhou, D. Wang, X. Chen and X. Feng, J. Phys. Chem. C, 2022, 126, 1966–1971 CrossRef CAS.
- P. Zhang, Z. Tian, Y. Kang, B. He, Z. Zhao, C. Te Hung, L. Duan, W. Chen, Y. Tang, J. Yu, L. Mai, Y. F. Li, W. Li and D. Zhao, J. Am. Chem. Soc., 2022, 144, 20964–20974 CrossRef CAS PubMed.
- N. C. Hildebrandt, J. Soldat and R. Marschall, Small, 2015, 11, 2051–2057 CrossRef CAS PubMed.
- L. Tian, Z. Zhang, S. Liu, G. Li and X. Gao, Energy Environ. Mater., 2022, 5, 645–654 CrossRef CAS.
- P. Roy, S. Berger and P. Schmuki, Angew. Chem., Int. Ed., 2011, 50, 2904–2939 CrossRef CAS PubMed.
- C. Niu, J. Meng, X. Wang, C. Han, M. Yan, K. Zhao, X. Xu, W. Ren, Y. Zhao, L. Xu, Q. Zhang, D. Zhao and L. Mai, Nat. Commun., 2015, 6, 7402 CrossRef PubMed.
- Y. Lei, Q. Wang, S. Peng, S. Ramakrishna, D. Zhang and K. Zhou, Adv. Energy Mater., 2020, 10, 1902115 CrossRef CAS.
- S. Peng, L. Li, Y. Hu, M. Srinivasan, F. Cheng, J. Chen and S. Ramakrishna, ACS Nano, 2015, 9, 1945–1954 CrossRef CAS PubMed.
- H. Wang, X. Liu, P. Niu, S. Wang, J. Shi and L. Li, Matter, 2020, 2, 1377–1413 CrossRef.
- H. Xie, Z. Li, L. Cheng, A. A. Haidry, J. Tao, Y. Xu, K. Xu and J. Z. Ou, iScience, 2022, 25, 103598 CrossRef CAS.
- Y. Tian, Z. Yu, L. Cao, X. L. Zhang, C. Sun and D. W. Wang, J. Energy Chem., 2021, 55, 323–344 CrossRef CAS.
- L. Mao, H. Park, R. A. Soler-Crespo, H. D. Espinosa, T. H. Han, S. B. T. Nguyen and J. Huang, Nat. Commun., 2019, 10, 3677 CrossRef.
- J. Li, J. Lv, Y. C. Hao, L. W. Chen, Y. Zuo, Y. Liu, S. Li, F. Zhang, F. Deng, A. X. Yin, J. Zhou, P. Li and B. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 47478–47487 CrossRef CAS PubMed.
- H. Kaur and J. N. Coleman, Adv. Mater., 2022, 34, 2202164 CrossRef CAS.
- A. Puthirath Balan, S. Radhakrishnan, C. F. Woellner, S. K. Sinha, L. Deng, C. D. L. Reyes, B. M. Rao, M. Paulose, R. Neupane, A. Apte, V. Kochat, R. Vajtai, A. R. Harutyunyan, C. W. Chu, G. Costin, D. S. Galvao, A. A. Martí, P. A. Van Aken, O. K. Varghese, C. S. Tiwary, A. M. M. R. Iyer and P. M. Ajayan, Nat. Nanotechnol., 2018, 13, 602–609 CrossRef CAS.
- L. Lin, N. Xu, C. Wu, J. Huang, A. Nattestad, X. Zheng, G. G. Wallace, S. Zhang and J. Chen, Matter, 2021, 4, 955–968 CrossRef CAS.
- Z. Sun, T. Liao, Y. Dou, S. M. Hwang, M. S. Park, L. Jiang, J. H. Kim and S. X. Dou, Nat. Commun., 2014, 5, 3813 CrossRef CAS PubMed.
- L. Peng, P. Xiong, L. Ma, Y. Yuan, Y. Zhu, D. Chen, X. Luo, J. Lu, K. Amine and G. Yu, Nat. Commun., 2017, 8, 15139 CrossRef PubMed.
- K. Liu, H. Jin, L. Huang, Y. Luo, Z. Zhu, S. Dai, X. Zhuang, Z. Wang, L. Huang and J. Zhou, Sci. Adv., 2022, 8, eabn2030 CrossRef CAS.
- W. Q. Wu, Y. F. Xu, J. F. Liao, L. Wang and D. Bin Kuang, Nano Energy, 2019, 62, 791–809 CrossRef CAS.
- H. Liao, X. Guo, Y. Hou, H. Liang, Z. Zhou and H. Yang, Small, 2020, 16, 1905223 CrossRef CAS PubMed.
- C. O'Dwyer, D. Navas, V. Lavayen, E. Benavente, M. A. Santa Ana, G. González, S. B. Newcomb and C. M. Sotomayor Torres, Chem. Mater., 2006, 18, 3016–3022 CrossRef.
- W. Q. Wu, H. L. Feng, H. S. Rao, Y. F. Xu, D. Bin Kuang and C. Y. Su, Nat. Commun., 2014, 5, 3968 CrossRef CAS PubMed.
- J. H. Kim, D. H. Kim, J. W. Yoon, Z. Dai and J. H. Lee, ACS Appl. Energy Mater., 2019, 2, 4535–4543 CrossRef CAS.
- S. H. Ko, D. Lee, H. W. Kang, K. H. Nam, J. Y. Yeo, S. J. Hong, C. P. Grigoropoulos and H. J. Sung, Nano Lett., 2011, 11, 666–671 CrossRef CAS PubMed.
- X. Gu, L. Chen, Z. Ju, H. Xu, J. Yang and Y. Qian, Adv. Funct. Mater., 2013, 23, 4049–4056 CrossRef CAS.
- U. Shaislamov, K. Krishnamoorthy, S. J. Kim, A. Abidov, B. Allabergenov, S. Kim, S. Choi, R. Suresh, W. M. Ahmed and H. J. Lee, Int. J. Hydrogen Energy, 2016, 41, 2253–2262 CrossRef CAS.
- Y. Ouyang, R. Huang, X. Xia, H. Ye, X. Jiao, L. Wang, W. Lei and Q. Hao, Chem. Eng. J., 2019, 355, 416–427 CrossRef CAS.
- Z. Hao, Z. Meng, X. Li, X. Sun, J. Xu, H. Nan, W. Shi, G. Qi, X. Hu and H. Tian, J. Colloid Interface Sci., 2022, 617, 430–441 CrossRef CAS PubMed.
- Z. Sun, J. H. Kim, Y. Zhao, F. Bijarbooneh, V. Malgras, Y. Lee, Y. M. Kang and S. X. Dou, J. Am. Chem. Soc., 2011, 133, 19314–19317 CrossRef CAS PubMed.
- Y. C. Tsai, N. Nhat Huy, J. Lee, Y. F. Lin and K. Y. A. Lin, Chem. Eng. J., 2020, 395, 124939 CrossRef CAS.
- K. Jing, W. Ma, Y. Ren, J. Xiong, B. Guo, Y. Song, S. Liang and L. Wu, Appl. Catal., B, 2019, 243, 10–18 CrossRef CAS.
- Y. T. Kim, P. P. Lopes, S. A. Park, A. Y. Lee, J. Lim, H. Lee, S. Back, Y. Jung, N. Danilovic, V. Stamenkovic, J. Erlebacher, J. Snyder and N. M. Markovic, Nat. Commun., 2017, 8, 1449 CrossRef PubMed.
- T. K. Kim, K. J. Lee, J. Y. Cheon, J. H. Lee, S. H. Joo and H. R. Moon, J. Am. Chem. Soc., 2013, 135, 8940–8946 CrossRef CAS PubMed.
- X. Yan, W. Liu, H. Kang, S. Zhang and S. Shi, Adv. Funct. Mater., 2023, 33, 2212654 CrossRef CAS.
- X. Tan, L. Guo, S. Liu, J. Wu, T. Zhao, J. Ren, Y. Liu, X. Kang, H. Wang, L. Sun and W. Chu, Adv. Funct. Mater., 2019, 29, 1903003 CrossRef.
- C. Wang, M. Wang, L. Liu and Y. Huang, ACS Appl. Energy Mater., 2021, 4, 1833–1839 CrossRef CAS.
- J. Hwang, C. Jo, K. Hur, J. Lim, S. Kim and J. Lee, J. Am. Chem. Soc., 2014, 136, 16066–16072 CrossRef CAS.
- J. E. S. Van Der Hoeven, A. V. Shneidman, N. J. Nicolas and J. Aizenberg, Acc. Chem. Res., 2022, 55, 1809–1820 CrossRef CAS PubMed.
- B. T. Liu, X. M. Shi, X. Y. Lang, L. Gu, Z. Wen, M. Zhao and Q. Jiang, Nat. Commun., 2018, 9, 1375 CrossRef PubMed.
- D. McNulty, E. Carroll and C. O'Dwyer, Adv. Energy Mater., 2017, 7, 1602291 CrossRef.
- Y. Wang, H. Arandiyan, H. A. Tahini, J. Scott, X. Tan, H. Dai, J. D. Gale, A. L. Rohl, S. C. Smith and R. Amal, Nat. Commun., 2017, 8, 15553 CrossRef CAS PubMed.
- X. Fan, Y. Liu, S. Chen, J. Shi, J. Wang, A. Fan, W. Zan, S. Li, W. A. Goddard and X. M. Zhang, Nat. Commun., 2018, 9, 1809 CrossRef.
- H. Zhang, X. Yu and P. V. Braun, Nat. Nanotechnol., 2011, 6, 277–281 CrossRef CAS PubMed.
- Y. Jiang, J. L. Yue, Q. Guo, Q. Xia, C. Zhou, T. Feng, J. Xu and H. Xia, Small, 2018, 14, 1704296 CrossRef PubMed.
- J. Deng, Y. Su, D. Liu, P. Yang, B. Liu and C. Liu, Chem. Rev., 2019, 119, 9221–9259 CrossRef CAS PubMed.
- J. Cai and L. Qi, Mater. Horiz., 2015, 2, 37–53 RSC.
- E. Larson, L. Williams, C. Jin, X. Chen, J. DiCesare, O. Sheppard, S. Xu and J. Wu, J. Mater. Res., 2022, 37, 2204–2215 CrossRef CAS.
- T. K. Das, P. Ilaiyaraja and C. Sudakar, Sol. Energy, 2018, 159, 920–929 CrossRef CAS.
- M. H. Habibi, A. H. Habibi, M. Zendehdel and M. Habibi, Spectrochim. Acta, Part A, 2013, 110, 226–232 CrossRef CAS PubMed.
- M. H. Habibi, E. Askari, M. Habibi and M. Zendehdel, Spectrochim. Acta, Part A, 2013, 104, 197–202 CrossRef PubMed.
- Y. J. Kim, M. H. Lee, H. J. Kim, G. Lim, Y. S. Choi, N. G. Park, K. Kim and W. I. Lee, Adv. Mater., 2009, 21, 3668–3673 CrossRef CAS.
- T. Azizi, A. E. Touihri, M. Ben Karoui and R. Gharbi, Optik, 2016, 127, 4400–4404 CrossRef CAS.
- M. Ben Karoui, S. Saadaoui, A. Torchani and R. Gharbi, J. Electron. Mater., 2021, 50, 4797–4805 CrossRef CAS.
- I. G. Yu, Y. J. Kim, H. J. Kim, C. Lee and W. I. Lee, J. Mater. Chem., 2011, 21, 532–538 RSC.
- Z. Tebby, T. Uddin, Y. Nicolas, C. Olivier, T. Toupance, C. Labrugère and L. Hirsch, ACS Appl. Mater. Interfaces, 2011, 3, 1485–1491 CrossRef CAS PubMed.
- P. K. Singh, B. Bhattacharya and R. K. Nagarale, J. Appl. Polym. Sci., 2010, 118, 2976–2980 CrossRef CAS.
- H. J. Kim, J. K. Seo, Y. J. Kim, H. K. Jeong, G. Il Lim, Y. S. Choi and W. I. Lee, Sol. Energy Mater. Sol. Cells, 2009, 93, 2108–2112 CrossRef CAS.
- Z. Tebby, O. Babot, T. Toupance, D. H. Park, G. Campet and M. H. Delville, Chem. Mater., 2008, 20, 7260–7267 CrossRef CAS.
- N. Nang Dinh, N. Minh Quyen, D. N. Chung, M. Zikova and V. Van Truong, Sol. Energy Mater. Sol. Cells, 2011, 95, 618–623 CrossRef CAS.
- S. Sharma, A. M. Volosin, D. Schmitt and D. K. Seo, J. Mater. Chem. A, 2013, 1, 699–706 RSC.
- H. V. Le, P. T. Pham, L. T. Le, A. D. Nguyen, N. Q. Tran and P. D. Tran, Int. J. Hydrogen Energy, 2021, 46, 22852–22863 CrossRef CAS.
- X. Zhang, D. Chandra, M. Kajita, H. Takahashi, L. Dong, A. Shoji, K. Saito, T. Yui and M. Yagi, Int. J. Hydrogen Energy, 2014, 39, 20736–20743 CrossRef CAS.
- D. Frederichi, M. H. N. O. Scaliante and R. Bergamasco, Environ. Sci. Pollut. Res., 2021, 28, 23610–23633 CrossRef CAS PubMed.
- J. Zhao, Z. Tao, J. Liang and J. Chen, Cryst. Growth Des., 2008, 8, 2799–2805 CrossRef CAS.
- X. Jiang, F. Cai, D. Gao, J. Dong, S. Miao, G. Wang and X. Bao, Electrochem. Commun., 2016, 68, 67–70 CrossRef CAS.
- K. Nguyen, N. D. Hoa, C. M. Hung, D. T. Thanh Le, N. Van Duy and N. Van Hieu, RSC Adv., 2018, 8, 19449–19455 RSC.
- P. S. Bassi, S. Y. Chiam, Gurudayal, J. Barber and L. H. Wong, ACS Appl. Mater. Interfaces, 2014, 6, 22490–22495 CrossRef CAS PubMed.
- J. Lv and T. Liang, Chem. Phys. Lett., 2016, 659, 61–65 CrossRef CAS.
- G. Wang, X. Shen, J. Horvat, B. Wang, H. Liu, D. Wexler and J. Yao, J. Phys. Chem. C, 2009, 113, 4357–4361 CrossRef CAS.
- Q. Abbas, S. H. Siyal, A. Mateen, N. U. Hassan, A. Idrees, Z. U. Rehman, E. M. T. El Din, M. A. Bajaber and M. S. Javed, Materials, 2022, 15, 4499 CrossRef CAS PubMed.
- F. Yang, X. Zhang, Y. Yang, S. Hao and L. Cui, Chem. Phys. Lett., 2018, 691, 366–372 CrossRef CAS.
- S. K. Meher, P. Justin and G. R. Rao, Electrochim. Acta, 2010, 55, 8388–8396 CrossRef CAS.
- R. Ahmad and M. A. Shah, Ceram. Int., 2023, 49, 6470–6478 CrossRef CAS.
- G. Rajamanickam, S. Narendhiran, S. P. Muthu, S. Mukhopadhyay and R. Perumalsamy, Chem. Phys. Lett., 2017, 689, 19–25 CrossRef CAS.
- T. Xu, H. Zheng, P. Zhang, W. Lin and Y. Sekiguchi, J. Mater. Chem. A, 2015, 3, 19115–19122 RSC.
- A. Manimekalai, P. Vivek, M. Manupriya, M. Umadevi and R. Parimaladevi, Surf. Interfaces, 2023, 41, 103176 CrossRef.
- Y. E. Firat and A. Peksoz, J. Mater. Sci., 2017, 28, 3515–3522 CAS.
- M. S. Wu, Y. A. Huang, C. H. Yang and J. J. Jow, Int. J. Hydrogen Energy, 2007, 32, 4153–4159 CrossRef CAS.
- S. M. Pawar, B. S. Pawar, P. T. Babar, A. T. A. Ahmed, H. S. Chavan, Y. Jo, S. Cho, J. Kim, B. Hou, A. I. Inamdar, S. N. Cha, J. H. Kim, T. G. Kim, H. Kim and H. Im, Appl. Surf. Sci., 2019, 470, 360–367 CrossRef CAS.
- H. Shen, X. Yang, J. Song, H. Gao, Z. Wu, J. Yu, W. Lei, J. Yang, G. He and Q. Hao, J. Solid State Electrochem., 2022, 26, 353–363 CrossRef CAS.
- X. He, Z. He, Q. Zou and L. Wu, Int. J. Energy Res., 2020, 44, 2100–2109 CrossRef CAS.
- J. H. Pikul, H. Gang Zhang, J. Cho, P. V. Braun and W. P. King, Nat. Commun., 2013, 4, 1732 CrossRef PubMed.
- C. Dunkel, M. Wark, T. Oekermann, R. Ostermann and B. M. Smarsly, Electrochim. Acta, 2013, 90, 375–381 CrossRef CAS.
- A. Y. El-Etre and S. M. Reda, Appl. Surf. Sci., 2010, 256, 6601–6606 CrossRef CAS.
- Z. Chen, Y. Tang, L. Zhang and L. Luo, Electrochim. Acta, 2006, 51, 5870–5875 CrossRef CAS.
- G. P. Wheeler and K. S. Choi, ACS Energy Lett., 2017, 2, 2378–2382 CrossRef CAS.
- H. Razmi and R. Mohammad-Rezaei, Electrochim. Acta, 2011, 56, 7220–7223 CrossRef CAS.
- K. R. Tolod, S. Hernández, M. Castellino, F. A. Deorsola, E. Davarpanah and N. Russo, Int. J. Hydrogen Energy, 2020, 45, 605–618 CrossRef CAS.
- D. P. Ura and U. Stachewicz, Macromol. Mater. Eng., 2022, 307, 2100843 CrossRef CAS.
- W. Luo, X. Hu, Y. Sun and Y. Huang, J. Mater. Chem., 2012, 22, 8916–8921 RSC.
- M. Kundu and L. Liu, J. Nanosci. Lett., 2015, 5, 11 Search PubMed.
- Z. Yang, G. Du, C. Feng, S. Li, Z. Chen, P. Zhang, Z. Guo, X. Yu, G. Chen, S. Huang and H. Liu, Electrochim. Acta, 2010, 55, 5485–5491 CrossRef CAS.
- L. Qiao, X. Wang, X. Sun, X. Li, Y. Zheng and D. He, Nanoscale, 2013, 5, 3037–3042 RSC.
- X. Zhao, L. Jiang, C. Ma, L. Cheng, C. Wang, G. Chen, H. Yue and D. Zhang, J. Power Sources, 2021, 490, 229534 CrossRef CAS.
- L. Thirugunanam, S. Kaveri, V. Etacheri, S. Ramaprabhu, M. Dutta and V. G. Pol, Mater. Charact., 2017, 131, 64–71 CrossRef CAS.
- Z. Zhou, W. Xiao, X. Shi, B. Ding, Q. Wang, Y. Zhan, H. Deng and Y. Du, J. Colloid Interface Sci., 2017, 490, 74–83 CrossRef CAS PubMed.
- Y. Yang, J. Zhu, W. Shi, J. Zhou, D. Gong, S. Gu, L. Wang, Z. Xu and B. Lu, Mater. Lett., 2016, 177, 34–38 CrossRef CAS.
- Y. Lu, Y. Liu, J. Mo, B. Deng, J. Wang, Y. Zhu, X. Xiao and G. Xu, J. Alloys Compd., 2021, 853, 157271 CrossRef CAS.
- R. R. Poolakkandy and M. M. Menamparambath, Nanoscale Adv., 2020, 2, 5015–5045 RSC.
- K. J. Hwang, D. W. Cho, J. W. Lee and C. Im, New J. Chem., 2012, 36, 2094–2100 RSC.
- Y. Lee and M. Kang, Mater. Chem. Phys., 2010, 122, 284–289 CrossRef CAS.
- S. E. Moosavifard, J. Shamsi and M. Ayazpour, Ceram. Int., 2015, 41, 1831–1837 CrossRef CAS.
- S. E. Moosavifard, M. F. El-Kady, M. S. Rahmanifar, R. B. Kaner and M. F. Mousavi, ACS Appl. Mater. Interfaces, 2015, 7, 4851–4860 CrossRef CAS PubMed.
- S. E. Moosavifard, J. Shamsi, S. Fani and S. Kadkhodazade, RSC Adv., 2014, 4, 52555–52561 RSC.
- A. Fadhli, D. Erika, S. Mardiana, C. B. Rasrendra, M. Khalil and G. T. M. Kadja, Chem. Phys. Lett., 2022, 803, 139809 CrossRef.
- N. J. Carroll, P. F. Crowder, S. Pylypenko, W. Patterson, D. R. Ratnaweera, D. Perahia, P. Atanassov and D. N. Petsev, ACS Appl. Mater. Interfaces, 2013, 5, 3524–3529 CrossRef CAS PubMed.
- Z. Liu, Y. Li, Z. Zhao, Y. Cui, K. Hara and M. Miyauchi, J. Mater. Chem., 2010, 20, 492–497 RSC.
- R. Z. Hou, P. Ferreira and P. M. Vilarinho, Chem. Mater., 2009, 21, 3536–3541 CrossRef CAS.
- W. Jiang, C. Jiang, X. Gong and Z. Zhang, J. Sol-Gel Sci. Technol., 2009, 52, 8–14 CrossRef CAS.
- D. Feng, T. N. Gao, L. Zhang, B. Guo, S. Song, Z. A. Qiao and S. Dai, Nano-Micro Lett., 2020, 12, 14 CrossRef CAS PubMed.
- F. Jonas, B. Lebeau, S. Siffert, L. Michelin, C. Poupin, R. Cousin, L. Josien, L. Vidal, M. Mallet, P. Gaudin and J. L. Blin, ACS Appl. Nano Mater., 2021, 4, 1786–1797 CrossRef CAS.
- W. Wei, P. Du, D. Liu, H. Wang and P. Liu, J. Colloid Interface Sci., 2017, 503, 205–213 CrossRef CAS PubMed.
- M. R. Krishnan, V. Rajendran and E. Alsharaeh, J. Non-Cryst. Solids, 2023, 606, 122198 CrossRef CAS.
- M. S. Seo, I. Jeong, J. S. Park, J. Lee, I. K. Han, W. I. Lee, H. J. Son, B. H. Sohn and M. J. Ko, Nanoscale, 2016, 8, 11472–11479 RSC.
- Y. Zhang, J. Li, F. Kang, F. Gao and X. Wang, Int. J. Hydrogen Energy, 2012, 37, 860–866 CrossRef CAS.
- S. H. Kim, B. Y. H. Liu and M. R. Zachariah, Chem. Mater., 2002, 14, 2889–2899 CrossRef CAS.
- A. A. Yadav, T. B. Deshmukh, R. V. Deshmukh, D. D. Patil and U. J. Chavan, Thin Solid Films, 2016, 616, 351–358 CrossRef CAS.
- S. Kumari, A. P. Singh, D. Sonal, R. Deva, S. Dass Shrivastav and V. R. Satsangi, Int. J. Hydrogen Energy, 2010, 35, 3985–3990 CrossRef CAS.
- T. Ishihara, H. Kim, Y. Inoishi and J. Matsuda, J. Am. Ceram. Soc., 2022, 105, 6718–6731 CrossRef CAS.
- J. Suffner, S. Kaserer, H. Hahn, C. Roth and F. Ettingshausen, Adv. Energy Mater., 2011, 1, 648–654 CrossRef CAS.
- G. R. A. Kumara, C. S. K. Ranasinghe, E. N. Jayaweera, H. M. N. Bandara, M. Okuya and R. M. G. Rajapakse, J. Phys. Chem. C, 2014, 118, 16479–16485 CrossRef CAS.
- E. L. Unger, F. Spadavecchia, K. Nonomura, P. Palmgren, G. Cappelletti, A. Hagfeldt, E. M. J. Johansson and G. Boschloo, ACS Appl. Mater. Interfaces, 2012, 4, 5997–6004 CrossRef CAS PubMed.
- R. S. Ingole, B. Y. Fugare and B. J. Lokhande, J. Mater. Sci., 2017, 28, 16374–16383 CAS.
- R. S. Ingole and B. J. Lokhande, Mater. Lett., 2016, 168, 95–98 CrossRef CAS.
- H. Xiu, T. Gao, N. An, Y. Wang, Y. Zhou, X. Qi, D. Liu and Y. Kuang, ACS Appl. Energy Mater., 2022, 5, 5127–5135 CrossRef CAS.
- S. F. Xue, Y. J. Li, F. H. Zheng, X. Bian, W. Y. Wu and C. H. Yang, Rare Met., 2021, 40, 31–39 CrossRef CAS.
- G. Scandura, P. Kumari, G. Palmisano, G. N. Karanikolos, J. Orwa and L. F. Dumée, Ind. Eng. Chem. Res., 2023, 62, 1736–1763 CrossRef CAS.
- Q. Zhang, H. Wu, S. Huang, X. Zhao, C. Hou, X. Zhuang, M. Wang, J. Han, Q. Chen and P. Liu, ACS Appl. Energy Mater., 2023, 6, 5435–5445 CrossRef CAS.
- S. L. Zhu, J. L. He, X. J. Yang, Z. D. Cui and L. L. Pi, Electrochem. Commun., 2011, 13, 250–253 CrossRef CAS.
- M. Niu, W. Xu, S. Zhu, Y. Liang, Z. Cui, X. Yang and A. Inoue, J. Power Sources, 2017, 362, 10–19 CrossRef CAS.
- A. Stepanovich, K. Sliozberg, W. Schuhmann and A. Ludwig, Int. J. Hydrogen Energy, 2012, 37, 11618–11624 CrossRef CAS.
- Z. Jin, J. Lyu, K. Hu, Z. Chen, G. Xie, X. Liu, X. Lin and H. J. Qiu, Small, 2021, 18, 2107207 CrossRef PubMed.
- K. Liang, X. Tang, B. Wei and W. Hu, Mater. Res. Bull., 2013, 48, 3829–3833 CrossRef CAS.
- M. Mirzaee and C. Dehghanian, J. Solid State Electrochem., 2018, 22, 3639–3645 CrossRef CAS.
- R. Li, N. Wu, J. Liu, Y. Jin, X. B. Chen and T. Zhang, Corros. Sci., 2017, 119, 23–32 CrossRef CAS.
- E. Hengge, J. Ihrenberger, E. M. Steyskal, R. Buzolin, M. Luckabauer, C. Sommitsch and R. Würschum, Nanoscale Adv., 2023, 5, 393–404 RSC.
- S. H. Joo and H. Kato, J. Alloys Compd., 2020, 831, 154733 CrossRef CAS.
- R. Song, J. Han, M. Okugawa, R. Belosludov, T. Wada, J. Jiang, D. Wei, A. Kudo, Y. Tian, M. Chen and H. Kato, Nat. Commun., 2022, 13, 5157 CrossRef CAS PubMed.
- C. Zhao, T. Wada, V. De Andrade, G. J. Williams, J. Gelb, L. Li, J. Thieme, H. Kato and Y. C. K. Chen-Wiegart, ACS Appl. Mater. Interfaces, 2017, 9, 34172–34184 CrossRef CAS PubMed.
- T. Wada, K. Yubuta and H. Kato, Scr. Mater., 2016, 118, 33–36 CrossRef CAS.
- C. Zhao, K. Kisslinger, X. Huang, J. Bai, X. Liu, C. H. Lin, L. C. Yu, M. Lu, X. Tong, H. Zhong, A. Pattammattel, H. Yan, Y. Chu, S. Ghose, M. Liu and Y. C. K. Chen-Wiegart, Nanoscale, 2021, 13, 17725–17736 RSC.
- C. Zhao, K. Kisslinger, X. Huang, M. Lu, F. Camino, C. H. Lin, H. Yan, E. Nazaretski, Y. Chu, B. Ravel, M. Liu and Y. C. K. Chen-Wiegart, Mater. Horiz., 2019, 6, 1991–2002 RSC.
- Y. Li, Q. Zhang, X. Zhao, H. Wu, X. Wang, Y. Zeng, Q. Chen, M. Chen and P. Liu, Adv. Funct. Mater., 2023, 33, 2214124 CrossRef CAS.
- Z. Lu, F. Zhang, D. Wei, J. Han, Y. Xia, J. Jiang, M. Zhong, A. Hirata, K. Watanabe, A. Karma, J. Erlebacher and M. Chen, Acta Mater., 2021, 212, 116916 CrossRef CAS.
- J. Han, C. Li, Z. Lu, H. Wang, Z. Wang, K. Watanabe and M. Chen, Acta Mater., 2019, 163, 161–172 CrossRef CAS.
- C. K. Chung, M. W. Liao, H. C. Chang and C. T. Lee, Thin Solid Films, 2011, 520, 1554–1558 CrossRef CAS.
- Y. Li, N. Peng, Y. Wen and L. Liang, Microporous Mesoporous Mater., 2020, 306, 110412 CrossRef CAS.
- T. Dikici, M. Erol, M. Toparli and E. Celik, Ceram. Int., 2014, 40, 1587–1591 CrossRef CAS.
- J. Kapusta-Kołodziej, A. Chudecka and G. D. Sulka, J. Electroanal. Chem., 2018, 823, 221–233 CrossRef.
- M. Erol, T. Dikici, M. Toparli and E. Celik, J. Alloys Compd., 2014, 604, 66–72 CrossRef CAS.
- L. Zaraska, D. Gilek, K. Gawlak, M. Jaskuła and G. D. Sulka, Appl. Surf. Sci., 2016, 390, 31–37 CrossRef CAS.
- D. V. Shinde, D. Y. Lee, S. A. Patil, I. Lim, S. S. Bhande, W. Lee, M. M. Sung, R. S. Mane, N. K. Shrestha and S. H. Han, RSC Adv., 2013, 3, 9431–9435 RSC.
- T. Zhang, M. Paulose, R. Neupane, L. A. Schaffer, D. B. Rana, J. Su, L. Guo and O. K. Varghese, Sol. Energy Mater. Sol. Cells, 2020, 209, 110472 CrossRef CAS.
- S. Xia, J. Ni, S. V. Savilov and L. Li, Nano Energy, 2018, 45, 407–412 CrossRef CAS.
- H. Lee, V. S. Kumbhar, J. Lee, Y. Choi and K. Lee, Electrochim. Acta, 2020, 334, 135618 CrossRef CAS.
- D. Lee, H. Lee, Y. T. Kim, K. Lee and J. Choi, Electrochim. Acta, 2020, 330, 135192 CrossRef CAS.
- J. E. Yoo, J. Park, G. Cha and J. Choi, Thin Solid Films, 2013, 531, 583–587 CrossRef CAS.
- K. S. Choudhari, C. H. Choi, S. Chidangil and S. D. George, Nanomaterials, 2022, 12, 444 CrossRef CAS PubMed.
- P. Zhong, W. Que and X. Hu, Appl. Surf. Sci., 2011, 257, 9872–9878 CrossRef CAS.
- W. H. Baek, I. Seo, T. S. Yoon, H. H. Lee, C. M. Yun and Y. S. Kim, Sol. Energy Mater. Sol. Cells, 2009, 93, 1587–1591 CrossRef CAS.
- V. M. Janardhanan, V. Heuveline and O. Deutschmann, J. Power Sources, 2008, 178, 368–372 CrossRef CAS.
- X. Lu, T. M. M. Heenan, J. J. Bailey, T. Li, K. Li, D. J. L. Brett and P. R. Shearing, J. Power Sources, 2017, 365, 210–219 CrossRef CAS.
- J. G. Rix, B. Mo, A. Y. Nikiforov, U. B. Pal, S. Gopalan and S. N. Basu, J. Electrochem. Soc., 2021, 168, 114507 CrossRef CAS.
- S. Ji, H. G. Seo, S. Lee, J. Seo, Y. Lee, W. H. Tanveer, S. W. Cha and W. Jung, RSC Adv., 2017, 7, 23600–23606 RSC.
- K. J. Kim, B. H. Park, S. J. Kim, Y. Lee, H. Bae and G. M. Choi, Sci. Rep., 2016, 6, 22443 CrossRef CAS PubMed.
- M. Liu, D. Dong, R. Peng, J. Gao, J. Diwu, X. Liu and G. Meng, J. Power Sources, 2008, 180, 215–220 CrossRef CAS.
- M. Singh, D. Zappa and E. Comini, Mater. Adv., 2022, 3, 5922–5929 RSC.
- C. Jin, Y. Mao, N. Zhang and K. Sun, Int. J. Hydrogen Energy, 2015, 40, 8433–8441 CrossRef CAS.
- M. Liu, D. Ding, Y. Bai, T. He and M. Liu, J. Electrochem. Soc., 2012, 159, B661–B665 CrossRef CAS.
- J. Liu, F. Yang, Z. Jiang, Y. Zhang, E. Hu, H. Wang and X. Yang, ACS Appl. Energy Mater., 2021, 4, 13492–13503 CrossRef CAS.
- H. Su, W. Zhang and Y. H. Hu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2208750119 CrossRef CAS PubMed.
- Y. Guo, M. Bessaa, S. Aguado, M. C. Steil, D. Rembelski, M. Rieu, J. P. Viricelle, N. Benameur, C. Guizard, C. Tardivat, P. Vernoux and D. Farrusseng, Energy Environ. Sci., 2013, 6, 2119–2123 RSC.
- J. W. Yang, S. H. Ahn and H. W. Jang, Curr. Opin. Green Sustainable Chem., 2021, 29, 100454 CrossRef CAS.
- B. R. Lee, S. Choi, W. S. Cheon, J. W. Yang, M. G. Lee, S. H. Park and H. W. Jang, Electron. Mater. Lett., 2022, 18, 391–399 CrossRef CAS.
- M. G. Lee, J. W. Yang, H. R. Kwon and H. W. Jang, CrystEngComm, 2022, 24, 5838–5864 RSC.
- J. Park, T. H. Lee, C. Kim, S. A. Lee, M. J. Choi, H. Kim, J. W. Yang, J. Lim and H. W. Jang, Appl. Catal., B, 2021, 295, 120276 CrossRef CAS.
- J. W. Yang, Y. J. Ahn, D. K. Cho, J. Y. Kim and H. W. Jang, Inorg. Chem. Front., 2023, 10, 3781–3807 RSC.
- S. Bera, S. A. Lee, C. M. Kim, H. Khan, H. W. Jang and S. H. Kwon, Chem. Mater., 2018, 30, 8501–8509 CrossRef CAS.
- M. J. Choi, T. L. Kim, K. S. Choi, W. Sohn, T. H. Lee, S. A. Lee, H. Park, S. Y. Jeong, J. W. Yang, S. Lee and H. W. Jang, ACS Appl. Mater. Interfaces, 2022, 14, 7788–7795 CrossRef CAS PubMed.
- J. W. Jang, C. Du, Y. Ye, Y. Lin, X. Yao, J. Thorne, E. Liu, G. McMahon, J. Zhu, A. Javey, J. Guo and D. Wang, Nat. Commun., 2015, 6, 7447 CrossRef PubMed.
- Y. Fu, Y. R. Lu, F. Ren, Z. Xing, J. Chen, P. Guo, W. F. Pong, C. L. Dong, L. Zhao and S. Shen, Sol. RRL, 2020, 4, 1900349 CrossRef CAS.
- Z. Zhang, I. Karimata, H. Nagashima, S. Muto, K. Ohara, K. Sugimoto and T. Tachikawa, Nat. Commun., 2019, 10, 4832 CrossRef PubMed.
- K. Y. Yoon, J. Park, M. Jung, S. G. Ji, H. Lee, J. H. Seo, M. J. Kwak, S. Il Seok, J. H. Lee and J. H. Jang, Nat. Commun., 2021, 12, 4309 CrossRef CAS PubMed.
- H. Zhang, D. Li, W. J. Byun, X. Wang, T. J. Shin, H. Y. Jeong, H. Han, C. Li and J. S. Lee, Nat. Commun., 2020, 11, 4622 CrossRef PubMed.
- K. H. Ye, H. Li, D. Huang, S. Xiao, W. Qiu, M. Li, Y. Hu, W. Mai, H. Ji and S. Yang, Nat. Commun., 2019, 10, 3687 CrossRef PubMed.
- K. Zhang, B. Jin, C. Park, Y. Cho, X. Song, X. Shi, S. Zhang, W. Kim, H. Zeng and J. H. Park, Nat. Commun., 2019, 10, 2001 CrossRef PubMed.
- K. J. McDonald and K. S. Choi, Energy Environ. Sci., 2012, 5, 8553–8557 RSC.
- D. K. Lee and K. S. Choi, Nat. Energy, 2018, 3, 53–60 CrossRef CAS.
- J. W. Yang, I. J. Park, S. A. Lee, M. G. Lee, T. H. Lee, H. Park, C. Kim, J. Park, J. Moon, J. Y. Kim and H. W. Jang, Appl. Catal., B, 2021, 293, 120217 CrossRef CAS.
- M. G. Lee, D. H. Kim, W. Sohn, C. W. Moon, H. Park, S. Lee and H. W. Jang, Nano Energy, 2016, 28, 250–260 CrossRef CAS.
- M. G. Lee, K. Jin, K. C. Kwon, W. Sohn, H. Park, K. S. Choi, Y. K. Go, H. Seo, J. S. Hong, K. T. Nam and H. W. Jang, Adv. Sci., 2018, 5, 1800727 CrossRef PubMed.
- M. G. Lee, J. W. Yang, H. Park, C. W. Moon, D. M. Andoshe, J. Park, C. K. Moon, T. H. Lee, K. S. Choi, W. S. Cheon, J. J. Kim and H. W. Jang, Nano-Micro Lett., 2022, 14, 48 CrossRef CAS PubMed.
- M. G. Lee, J. W. Yang, I. J. Park, T. H. Lee, H. Park, W. S. Cheon, S. A. Lee, H. Lee, S. G. Ji, J. M. Suh, J. Moon, J. Y. Kim and H. W. Jang, Carbon Energy, 2023, 5, e321 CrossRef CAS.
- S. Bera, S. A. Lee, W. J. Lee, J. H. Kim, C. Kim, H. G. Kim, H. Khan, S. Jana, H. W. Jang and S. H. Kwon, ACS Appl. Mater. Interfaces, 2021, 13, 14291–14301 CrossRef CAS PubMed.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- D. Kishore Kumar, J. Kříž, N. Bennett, B. Chen, H. Upadhayaya, K. R. Reddy and V. Sadhu, Mater. Sci. Energy Technol., 2020, 3, 472–481 CAS.
- M. Kokkonen, P. Talebi, J. Zhou, S. Asgari, S. A. Soomro, F. Elsehrawy, J. Halme, S. Ahmad, A. Hagfeldt and S. G. Hashmi, J. Mater. Chem. A, 2021, 9, 10527–10545 RSC.
- X. Hou, K. Aitola and P. D. Lund, Energy Sci. Eng., 2021, 9, 921–937 CrossRef CAS.
- B. D. Choudhury, C. Lin, S. M. A. Z. Shawon, J. Soliz-Martinez, H. Huq and M. J. Uddin, Sci. Rep., 2021, 11, 7552 CrossRef CAS PubMed.
- Y. Ding, L. Zhou, L. Mo, L. Jiang, L. Hu, Z. Li, S. Chen and S. Dai, Adv. Funct. Mater., 2015, 25, 5946–5953 CrossRef CAS.
- D. Chen, L. Cao, F. Huang, P. Imperial, Y. B. Cheng and R. A. Caruso, J. Am. Chem. Soc., 2010, 132, 4438–4444 CrossRef CAS PubMed.
- S. Yu, J. S. You, I. S. Yang, P. Kang, S. B. Rawal, S. Do Sung and W. I. Lee, J. Power Sources, 2016, 325, 7–14 CrossRef CAS.
- S. Q. Kang, E. Z. Chen, J. Di Cheng, X. Y. Gu, K. Wei, G. Z. Sun, X. P. Gao, X. J. Pan, J. Y. Zhou and E. Q. Xie, J. Alloys Compd., 2022, 905, 164295 CrossRef CAS.
- A. Listorti, B. O'Regan and J. R. Durrant, Chem. Mater., 2011, 23, 3381–3399 CrossRef CAS.
- S. Il Cho, H. K. Sung, S. J. Lee, W. H. Kim, D. H. Kim and Y. S. Han, Nanomaterials, 2019, 9, 1645 CrossRef PubMed.
- J. Yue, Y. Xiao, Y. Li, G. Han, Y. Zhang and W. Hou, Org. Electron., 2017, 43, 121–129 CrossRef CAS.
- A. Banik, M. S. Ansari and M. Qureshi, ACS Omega, 2018, 3, 14482–14493 CrossRef CAS PubMed.
- Q. Zhang, S. Hou and C. Li, Nanomaterials, 2020, 10, 1598 CrossRef CAS PubMed.
- H. F. Etefa, V. Kumar, F. B. Dejene, M. T. Efa and L. T. Jule, ACS Omega, 2023, 8, 15249–15258 CrossRef CAS PubMed.
- N. Li, E. A. Gibson, P. Qin, G. Boschloo, M. Gorlov, A. Hagfeldt and L. Sun, Adv. Mater., 2010, 22, 1759–1762 CrossRef PubMed.
- M. Zannotti, E. Benazzi, L. A. Stevens, M. Minicucci, L. Bruce, C. E. Snape, E. A. Gibson and R. Giovannetti, ACS Appl. Energy Mater., 2019, 2, 7345–7353 CrossRef CAS.
- G. G. Wang, Z. Q. Lin, D. D. Zhao and J. C. Han, Langmuir, 2018, 34, 8898–8903 CrossRef CAS PubMed.
- T. B. Tran, F. AlQatari and Q. H. Luc, Sci. Rep., 2021, 11, 4981 CrossRef CAS PubMed.
- S. Zhou, X. Zhao, P. Du, Z. Zhang, X. Liu, S. Liu and L. J. Guo, Nanoscale, 2022, 14, 4887–4907 RSC.
- H. Hu, B. Tang, H. Wan, H. Sun, S. Zhou, J. Dai, C. Chen, S. Liu and L. J. Guo, Nano Energy, 2020, 69, 104427 CrossRef CAS.
- Y. C. Lee, S. C. Yeh, Y. Y. Chou, P. J. Tsai, J. W. Pan, H. M. Chou, C. H. Hou, Y. Y. Chang, M. S. Chu, C. H. Wu and C. H. Ho, Microelectron. Eng., 2013, 105, 86–90 CrossRef CAS.
- C. Y. Chou, W. H. Lai, X. F. Li, C. Cheng, C. K. Huang and C. Y. Liu, Opt. Mater., 2021, 119, 111297 CrossRef CAS.
- S. H. Chao, L. H. Yeh, R. T. Wu, K. Kawagishi and S. C. Hsu, RSC Adv., 2020, 10, 16284–16290 RSC.
- Q. Zhou, M. Xu, Q. Li and H. Wang, IEEE Photonics Technol. Lett., 2017, 29, 983–986 CAS.
- Y. Cheng, L. Wang, Y. Zhang, H. Zheng, J. Ma, X. Yi, G. Wang and J. Li, ECS Solid State Lett., 2013, 2, 93–97 CrossRef.
- W. C. Ke, F. W. Lee, C. Y. Chiang, Z. Y. Liang, W. K. Chen and T. Y. Seong, ACS Appl. Mater. Interfaces, 2016, 8, 34520–34529 CrossRef CAS.
- V. Wood, M. J. Panzer, J. E. Halpert, J. M. Caruge, M. G. Bawendi and V. Bulović, ACS Nano, 2009, 3, 3581–3586 CrossRef CAS.
- W. S. Chen, S. H. Yang, W. C. Tseng, W. W. S. Chen and Y. C. Lu, ACS Omega, 2021, 6, 13447–13455 CrossRef CAS.
- N. Kirkwood, B. Singh and P. Mulvaney, Adv. Mater. Interfaces, 2016, 3, 1600868 CrossRef.
- H.-C. Wang, S.-Y. Lin, A.-C. Tang, B. P. Singh, H.-C. Tong, C.-Y. Chen, Y.-C. Lee, T.-L. Tsai and R.-S. Liu, Angew. Chem., 2016, 128, 8056–8061 CrossRef.
- J. K. Kim, S. Chhajed, M. F. Schubert, E. F. Schubert, A. J. Fischer, M. H. Crawford, J. Cho, H. Kim and C. Sone, Adv. Mater., 2008, 20, 801–804 CrossRef CAS.
- Y. An, Y. Tian, Q. Man, H. Shen, C. Liu, Y. Qian, S. Xiong, J. Feng and Y. Qian, ACS Nano, 2022, 16, 6755–6770 CrossRef CAS PubMed.
- Z. Liu, X. Yuan, S. Zhang, J. Wang, Q. Huang, N. Yu, Y. Zhu, L. Fu, F. Wang, Y. Chen and Y. Wu, NPG Asia Mater., 2019, 11, 12 CrossRef CAS.
- D. Liu, Z. Yang, P. Wang, F. Li, D. Wang and D. He, Nanoscale, 2013, 5, 1917–1921 RSC.
- S. Liu, L. Kang and S. C. Jun, Adv. Mater., 2021, 33, 2004689 CrossRef CAS.
- Z. Li, Y. Tan, X. Huang, W. Zhang, Y. Gao and B. Tang, Ceram. Int., 2016, 42, 18887–18893 CrossRef CAS.
- M. Nagasaki and K. Kanamura, ACS Appl. Energy Mater., 2019, 2, 3896–3903 CrossRef CAS.
- G. Lui, G. Li, X. Wang, G. Jiang, E. Lin, M. Fowler, A. Yu and Z. Chen, Nano Energy, 2016, 24, 72–77 CrossRef CAS.
- D. Tonti, M. J. Torralvo, E. Enciso, I. Sobrados and J. Sanz, Chem. Mater., 2008, 20, 4783–4790 CrossRef CAS.
- B. Qu, L. Hu, Q. Li, Y. Wang, L. Chen and T. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 731–736 CrossRef CAS.
- S. Lou, X. Cheng, Y. Zhao, A. Lushington, J. Gao, Q. Li, P. Zuo, B. Wang, Y. Gao, Y. Ma, C. Du, G. Yin and X. Sun, Nano Energy, 2017, 34, 15–25 CrossRef CAS.
- J. Wang, H. Zhou, J. Nanda and P. V. Braun, Chem. Mater., 2015, 27, 2803–2811 CrossRef CAS.
- C. Liu, E. I. Gillette, X. Chen, A. J. Pearse, A. C. Kozen, M. A. Schroeder, K. E. Gregorczyk, S. B. Lee and G. W. Rubloff, Nat. Nanotechnol., 2014, 9, 1031–1039 CrossRef CAS PubMed.
- K. Dai, X. Wang, Y. Yin, C. Hao and Z. You, Sci. Rep., 2016, 6, 38794 CrossRef CAS PubMed.
- P. K. Panda, A. Grigoriev, Y. K. Mishra and R. Ahuja, Nanoscale Adv., 2020, 2, 70–108 RSC.
- D. Nandi, V. B. Mohan, A. K. Bhowmick and D. Bhattacharyya, J. Mater. Sci., 2020, 55, 6375–6400 CrossRef CAS.
- S. Liu, Y. Yin, D. Ni, K. S. Hui, M. Ma, S. Park, K. N. Hui, C. Y. Ouyang and S. C. Jun, Energy Storage Mater., 2019, 22, 384–396 CrossRef.
- S. Liu, L. Kang, J. Hu, E. Jung, J. Henzie, A. Alowasheeir, J. Zhang, L. Miao, Y. Yamauchi and S. C. Jun, Small, 2022, 18, 2104507 CrossRef CAS.
- S. Liu, L. Kang, J. Zhang, E. Jung, S. Lee and S. C. Jun, Energy Storage Mater., 2020, 32, 167–177 CrossRef.
- L. Kang, M. Zhang, J. Zhang, S. Liu, N. Zhang, W. Yao, Y. Ye, C. Luo, Z. Gong, C. Wang, X. Zhou, X. Wu and S. C. Jun, J. Mater. Chem. A, 2020, 8, 24053–24064 RSC.
- S. Liu, L. Kang, J. Hu, E. Jung, J. Zhang, S. C. Jun and Y. Yamauchi, ACS Energy Lett., 2021, 6, 3011–3019 CrossRef CAS.
- C. Xiong, T. Li, Y. Zhu, T. Zhao, A. Dang, H. Li, X. Ji, Y. Shang and M. Khan, J. Alloys Compd., 2017, 695, 1248–1259 CrossRef CAS.
- J. Kim, J. H. Eum, J. Kang, O. Kwon, H. Kim and D. W. Kim, Sci. Rep., 2021, 11, 2063 CrossRef CAS.
- S. K. Singh, V. M. Dhavale, R. Boukherroub, S. Kurungot and S. Szunerits, Appl. Mater. Today, 2017, 8, 141–149 CrossRef.
- D. Majumdar, T. Maiyalagan and Z. Jiang, ChemElectroChem, 2019, 6, 4343–4372 CrossRef CAS.
- V. D. Patake and C. D. Lokhande, Appl. Surf. Sci., 2008, 254, 2820–2824 CrossRef CAS.
- R. Wang, Y. Ma, H. Wang, J. Key, D. Brett, S. Ji, S. Yin and P. K. Shen, J. Mater. Chem. A, 2016, 4, 5390–5394 RSC.
- J. Lee, J. Y. Seok, S. Son, M. Yang and B. Kang, J. Mater. Chem. A, 2017, 5, 24585–24593 RSC.
- A. Kumar, G. R. Dillip, A. Bharti, U. Maitra and A. J. Bhattacharyya, Electrochem. Sci. Adv., 2021, 2, e2100043 CrossRef.
- J. Wang, X. Guo, R. Cui, H. Huang, B. Liu, Y. Li, D. Wang, D. Zhao, J. Dong, S. Li and B. Sun, ACS Appl. Nano Mater., 2020, 3, 11152–11159 CrossRef CAS.
- Y. Ding, S. Tang, R. Han, S. Zhang, G. Pan and X. Meng, Sci. Rep., 2020, 10, 11023 CrossRef CAS PubMed.
- G. H. Jeong, S. Baek, S. Lee and S. W. Kim, Chem. – Asian J., 2016, 11, 949–964 CrossRef CAS PubMed.
- B. Lesbayev, M. Auyelkhankyzy, G. Ustayeva, M. Yeleuov, N. Rakhymzhan, Y. Maral and A. Tolynbekov, J. Compos. Sci., 2023, 7, 20 CrossRef CAS.
- X. Lang, A. Hirata, T. Fujita and M. Chen, Nat. Nanotechnol., 2011, 6, 232–236 CrossRef CAS.
- H. Chen, S. Zhou and L. Wu, ACS Appl. Mater. Interfaces, 2014, 6, 8621–8630 CrossRef CAS PubMed.
- R. R. Salunkhe, J. Tang, Y. Kamachi, T. Nakato, J. H. Kim and Y. Yamauchi, ACS Nano, 2015, 9, 6288–6296 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |