
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Extending the shelf life of HLM chips through freeze-drying of human liver microsomes immobilized onto thiol–ene micropillar arrays†
Iiro
Rautsola
 a,
Markus
Haapala
a,
Leo
Huttunen
a,
Ossi
Korhonen
b and
Tiina
Sikanen
*ac
a,
Markus
Haapala
a,
Leo
Huttunen
a,
Ossi
Korhonen
b and
Tiina
Sikanen
*ac
aFaculty of Pharmacy, Drug Research Program, University of Helsinki, Finland. E-mail: tiina.sikanen@helsinki.fi
bFaculty of Health Sciences, School of Pharmacy, University of Eastern Finland, Finland
cHelsinki Institute of Sustainability Science, University of Helsinki, Finland
First published on 31st July 2024
Abstract
Microfluidic flow reactors functionalized with immobilized human liver microsomes (HLM chips) represent a powerful tool for drug discovery and development by enabling mechanism-based enzyme inhibition studies under flow-through conditions. Additionally, HLM chips may be exploited in streamlined production of human drug metabolites for subsequent microfluidic in vitro organ models or as metabolite standards for drug safety assessment. However, the limited shelf life of the biofunctionalized microreactors generally poses a major barrier to their commercial adaptation in terms of both storage and shipping. The shelf life of the HLM chips in the wetted state is ca. 2–3 weeks only and requires cold storage at 4 °C. In this study, we developed a freeze-drying method for lyophilization of HLMs that are readily immobilized inside microfluidic pillar arrays made from off-stoichiometric thiol–ene polymer. The success of lyophilization was evaluated by monitoring the cytochrome P450 and UDP-glucuronosyltransferase enzyme activities of rehydrated HLMs for several months post-freeze-drying. By adapting the freeze-drying protocol, the HLM chips could be stored at room temperature (protected from light and moisture) for at least 9 months (n = 2 independent batches) and up to 16 months at best, with recovered enzyme activities within 60–120% of the non-freeze-dried control chips. This is a major improvement over the cold-storage requirement and the limited shelf life of the non-freeze-dried HLM chips, which can significantly ease the design of experiments, decrease energy consumption during storage, and reduce the shipping costs with a view to commercial adaptation.
1. Introduction
The majority (>70%) of human pharmaceuticals are eliminated from the body via metabolic reactions catalyzed by cytochrome P450 (CYP) and uridine-5′-diphospo-glucuronosyltransferase (UGT) enzyme systems.1 The CYP enzyme system alone catalyzes >75% of these reactions and is particularly prone to drug–drug interactions resulting from competitive, uncompetitive, or noncompetitive binding of several pharmaceuticals to the same enzyme isoform.2 Irreversible CYP inhibition can result in long-lasting (days) loss of the inhibited enzyme activity, which may elevate the plasma concentrations of any other pharmaceutical normally cleared via the inhibited metabolic pathway above the toxic range. In contrast, reversible CYP inhibitors affect the clearance of only concomitantly administered pharmaceuticals. CYP interaction screening in vitro is thus an essential part of preclinical drug development.The most severe drug–drug interactions arise from irreversible binding of the pharmaceutical or its metabolites to the critical CYP isoforms. However, it is still difficult to identify irreversible CYP inhibitors using static in vitro assays. Currently, reversible and irreversible CYP inhibitors can only be distinguished based on time- and resource-intensive dialysis assays.3 Instead, flow-through microreactors functionalized with human liver microsomes (HLMs) are an enabling technique for studying human drug metabolism and related drug–drug interactions in vitro.4–6 HLMs are heterogeneous vesicle-like artifacts re-formed from the endoplasmic reticulum of liver cells via centrifugation, and they readily embed membrane-bound drug metabolizing enzymes such as the CYPs and UGTs. Compared to static enzyme activity assays, flow-through assay setups exploiting immobilized HLMs offer significant advantages by enabling the establishment of time-dependent concentration gradients of the test substance over time (e.g., for determining half-maximal inhibitory concentrations, IC50) and even elimination of the test substance from the feed to easily distinguish between reversible and irreversible enzyme inhibition.4,6,7 Moreover, the HLM chips, same as other immobilized enzyme microreactors,8 are also well suitable for producing drug metabolites for subsequent flow-through assays, such as microfluidic organ-on-a-chip systems, to facilitate examination of also the metabolites' effects in a streamlined manner. However, the limited shelf life of the HLM chips, same as any kind of biofunctionalized microreactors, poses a major challenge to their more extensive exploitation towards routine use in drug safety and efficacy testing. Biofunctionalized reactors often need to be stored at 4 °C (refrigerator) until use to prevent enzyme inactivation. Nevertheless, the HLM chips, for example, have only a limited lifetime of ca. two weeks.7 Although this suffices well for on-demand fabrication of laboratory prototypes, the short shelf life is a considerable barrier with a view to technology transfer toward commercialization. The HLMs as such are commercially available and typically cryopreserved at −80 °C, which enables their long-term storage, for several years at best (according to the supplier). However, when immobilized onto polymer-based microfluidic devices, the HLMs can no longer be cryopreserved because the crosslinked polymers (the support platforms) tend to lose their viscoelasticity in cryogenic temperatures.9 This may further compromise their mechanical durability and the bonding strength of the assembled biofunctionalized chips.
In this study, we evaluated the feasibility of freeze-drying techniques to lyophilization of HLMs immobilized inside microfluidic flow reactors made of off-stoichiometric thiol–ene (OSTE) as described in a previous work.7 Freeze-drying is an established approach for stabilizing proteins, peptides, and organic small molecules.10,11 In recent years, freeze-drying techniques have also been increasingly applied to more complex biomaterials, such as tissues,12 liposomes,13,14 and cell-secreted extracellular vesicles15 as well as rodent liver S9 fractions16 and human recombinant CYP.17 The freeze-dried biomaterials can be stored for extended periods in a regular refrigerator or even at room temperature, thus greatly simplifying and reducing the costs of storage, handling, and logistics. However, extending the freeze-drying approach to biological material immobilized on microfluidic devices is much less explored, with only a few previous studies focusing on freeze-drying of antibodies,18 nucleic acids19 or soluble enzymes.20,21 In comparison, lyophilization of membrane-bound enzymes, such as the microsomal CYPs and UGTs, is generally more demanding, requiring careful optimization of the freeze-drying conditions.17 To our knowledge, there are no previous studies evaluating the feasibility of freeze-drying for preservation of HLMs, either as such or immobilized on solid supports. From the microfabrication materials' perspective, freeze-drying enables much shorter exposure times at extremely low temperatures, and thus less material stress, compared with cryopreservation, which by default ensures better stability of the polymer platform in the long term. Moreover, after freeze-drying, the biofunctionalized flow reactors can be stored at room temperature, thus reducing the energy consumption during storage as well as the shipping costs, and thereby overall increasing the technological readiness toward commercialization.18,22
In this study, we developed a method specifically for lyophilization of HLMs immobilized onto OSTE microfluidic chips to demonstrate the feasibility of freeze-drying for preserving the enzymatic activity of microsomal CYPs and UGTs of the HLM chip in the long term. After initial optimization of the freeze-drying protocol, the long-term stability of the freeze-dried HLM chips stored at room temperature was demonstrated with two independent batches (each ≥40 chips) over several months.
2. Materials and methods
2.1. HLM chip design and microfabrication
The HLM chips used in the present study comprise a microchannel (4 mm × 30 mm × 0.2 mm, width × length × height) featuring an array of approximately 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 diamond-shaped micropillars (Fig. 1a and b). The micropillar arrays were implemented using UV replica molding of off-stoichiometric thiol–enes (OSTEs), as described in Tähkä et al.,23 and have a total surface area of ca. 800 mm2 over a total internal volume of ca. 17 μL. The microfabrication protocol comprises four steps, including (i) fabrication of the SU-8 master (for both cover and bottom layers) by photolithography, (ii) soft lithography to prepare the respective negative polydimethylsiloxane (PDMS) molds, (iii) UV replica molding of the cover and bottom OSTE layers, and (iv) bonding (Fig. 1c).
000 diamond-shaped micropillars (Fig. 1a and b). The micropillar arrays were implemented using UV replica molding of off-stoichiometric thiol–enes (OSTEs), as described in Tähkä et al.,23 and have a total surface area of ca. 800 mm2 over a total internal volume of ca. 17 μL. The microfabrication protocol comprises four steps, including (i) fabrication of the SU-8 master (for both cover and bottom layers) by photolithography, (ii) soft lithography to prepare the respective negative polydimethylsiloxane (PDMS) molds, (iii) UV replica molding of the cover and bottom OSTE layers, and (iv) bonding (Fig. 1c).
 | ||
| Fig. 1 (a) Scanning electron micrograph of the micropillar array. (b) A photograph of the HLM chip. (c) Schematic presentation of the OSTE chip microfabrication protocol. (d) Schematic presentation of the functionalization protocol to immobilize pre-biotinylated HLMs onto the thiol-rich micropillars. | ||
The SU-8 masters (step i) were fabricated by spin coating SU-8100 (Kayaku Advanced Materials, Westborough, MA) on single side polished silicon wafers (Siegert Wafer, Aachen, Germany) at 1150 rpm for 30 s to achieve an about 200 μm thick layer of SU-8. The SU-8 resist was soft baked first at 65 °C for 25 min and then at 95 °C for 70 min on a hot plate, and then exposed to collimated UV light for 30 s though a plastic photomask (OAI LS 30/5, nominal intensity 40 mW cm−2). Next, the SU-8 resist was post exposure baked first at 65 °C for 10 min and then at 95 °C for 40 min, and developed in propylene glycol methyl ether acetate (PGMEA; Sigma Aldrich, Burlington, MA) with stirring for 30 min. Finally, the SU-8 masters were rinsed with isopropanol, dried with nitrogen, and baked at 95 °C for 30 min.
The PDMS molds (step ii) were fabricated using Sylgard 184 (Dow, Midland, MI) with an elastomer/curing agent ratio of 10:1 (w:w). The PDMS prepolymer mixture was degassed under vacuum, applied on the SU-8 master, and cured in the oven at 70 °C overnight, then finally detached from the SU-8 master after cooling to room temperature.
The OSTE micropillar arrays were fabricated by mixing tetrathiol (pentaerythritol tetrakis(3-mercaptopropionate), PETMP; Bruno Bock GmbH, Marschacht, Germany) and triallyl (1,3,5-triallyl-1,3,5-triazine-2,4,6(1H,3H,5H)-trione, TATATO; Sigma-Aldrich, Burlington, MA) monomers in a molar ratio of 110:100 in terms of the thiol and allyl functional groups, yielding 10 mol% excess of thiols. The mixture was then degassed, applied on the PDMS molds (the micropillar array and the cover layer) and cured under a UV flood exposure lamp for 5 min (Dymax 5000-EC; Dymax Light Curing Systems, Torrington, CT; nominal intensity 225 mW cm−2). The cured OSTE layers were detached from the mold, heated on a hot plate at 100 °C for 2 h and laminated against each other. To finalize the bonding, the stack was exposed to UV light for 2 min (Dymax 5000-EC).
2.2. HLM chip surface functionalization
Before use, the OSTE micropillar arrays were functionalized sequentially with biotin, streptavidin, and pre-biotinylated HLMs (Fig. 1d), as described earlier by Kiiski et al.4 Briefly, the OSTE chips were first filled with 0.1 mg mL−1 biotin-PEG4-alkyne dissolved in ethylene glycol (Sigma-Aldrich, Burlington, MA) using 1% m/V TPO-L as a photoinitiator (BASF, Ludwigshafen, Germany). Next, the chip was exposed to UV light (LED 365 nm) through the cover layer for 1 min and then washed sequentially with ca. 2 mL of MeOH and deionized water. Next, the chip was filled with 0.5 μg mL−1 streptavidin solution (Thermo Fisher Scientific, Waltham, MA) in 0.01 M phosphate-buffered saline (PBS, pH 7.4), incubated at room temperature for 40 min and rinsed with ca. 2 mL of PBS.The pre-biotinylated HLMs were prepared with the help of biotinylated, fusogenic liposomes according to a previously reported protocol.7 To prepare the fusogenic liposomes, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl) (biotin-DOPE), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Liss-Rhod PE) (all Avanti Polar Lipids, Inc., Alabaster, AL) were individually dissolved in chloroform and then mixed in a weight ratio of 1:1:0.1:0.05. Next, the bulk solvent was evaporated under a nitrogen flow followed by removal of residual solvent by 2 h vacuum desiccation. The lipids were then resolubilized in PBS to achieve a total lipid concentration of 2 mg mL−1. The so obtained multilamellar lipid vesicles were extruded ca. 50 times through a 0.1 μm polycarbonate membrane to obtain unilamellar vesicles (Avanti Mini-Extruder; Avanti Polar Lipids, Inc., Alabaster, AL). Next, the unilamellar, biotinylated liposomes were mixed with commercial HLMs (BD Gentest #452161, pool of 20 donors, 20 mg mL−1 microsomal total protein in 250 mM sucrose) in a 1:1 (V/V) ratio and incubated at 37 °C for 15 min to obtain the pre-biotinylated HLMs (b-HLM) with 10 mg mL−1microsomal total protein concentration.
Finally, the streptavidin-functionalized chips were filled with pre-biotinylated HLMs and incubated at 4 °C until freeze-dried or used for enzyme activity testing as such. For refrigeration, the chip inlets were sealed with sealing tape (PS14335025, Stokvis Tapes, Alblasserdam, the Netherlands) to prevent evaporation.
2.3. Freeze-drying of the biofunctionalized HLM chips
The biofunctionalized HLM chips were freeze-dried using an FTS LyoStar II freeze dryer (SP Industries, Inc., Stone Ridge, NY). Just before freeze-drying, the HLM chips, preincubated at 4 °C for 5–7 days, were rinsed and filled with 10% (m/V) sucrose solution (a cryoprotection reagent). The freeze-drying protocol comprised loading, cooling, primary drying and secondary drying steps. The appropriate freeze-drying temperature was preliminarily determined by using a freeze-drying microscope (Nikon LV100D, Nikon, Tokyo, Japan) equipped with a temperature- and pressure-controlled sample stage (THMS350 V; Linkam Scientific Ltd., Salfords, UK) (Fig. 2a and b). Next, the freeze-drying protocol was further optimized with respect to temperatures and holding times at each of the four steps using the FTS LyoStar II freeze dryer (Fig. 2c and d). | ||
| Fig. 2 (a) Photograph of the microscopic freeze-drying setup used for preliminary method development. (b) Illustration of the drying process taking place inside the chip, from inlet and outlet toward the middle. (c) Photograph of the sample stage of the FTS LyoStar II freeze dryer with the NIR probe and a batch of HLM chips. (d) Freeze-drying temperatures of the three different freeze-drying protocols used in the initial optimization study. | ||
The optimized freeze-drying conditions used in long-term storage stability studies (protocol 3, Fig. 2d) comprised loading at room temperature, cooling to −50 °C at 5 °C min−1 and holding at −50 °C for 2 h, primary drying at −40 °C (50 mtorr) for 116 h, then −37 °C for 46 h, and finally secondary drying from −37 °C to room temperature in ca. 10–15 min at 1 °C min−1. The total freeze-drying time was thus 166 h. Two independent batches of HLM chips (batch 1, n = 52; batch 2, n = 40) were freeze-dried under these conditions. After freeze-drying, the HLM chips were stored at room temperature for the desired period of time until enzyme activity testing and rehydrated with 0.01 M PBS (pH 7.4) before use.
The residual water or ice content during the drying process was qualitatively monitored using a built-in capacitance manometer (vacuum pressure) and Pirani (thermal conductivity) sensors of the freeze-dryer as well as with near-infrared spectroscopy (NIR). The NIR spectrometer consisted of a short-wavelength infrared (SWIR) hyperspectral camera (970–2500 nm; Specim Oy, Oulu, Finland) equipped with a multichannel fiber-optic input module (VTT, Espoo, Finland), a multichannel fiber-optic light source with a 65 W halogen lamp (VTT) and a fiber-optic noncontact diffusion reflectance probe head (VTT). During freeze-drying, the NIR probe with an illumination spot of 2 mm was placed on the top of the chip (Fig. 2c) and a hundred spectra were collected and averaged every 2.5 min for the first 170 min of the freeze-drying cycle. Thereafter, during the primary and secondary drying steps, the spectra were collected once per hour (see the ESI,1 Fig. S1). The obtained spectra (range 1206–2169 nm) were pre-processed, first by smoothing (Savitzky–Golay; polynomial order: quadric; smoothing points: 15) to reduce the background noise and enhance the differences at the absorption wavelengths of water, and then by correcting the data using standard normal variate (SNV) correction to obtain comparable intensities. The pre-processed spectra were then analyzed with principal component analysis (PCA) using SIMCA software, version 17 (Sartorius, Goettingen, Germany, Germany), similarly as described earlier.24
2.4. Determination of the microsomal enzymatic activities of the HLM chips
The microsomal enzyme activities of the HLM chips were determined with the help of selected marker substrates of CYP and UGT. For this purpose, the rehydrated freeze-dried chips, or the non-freeze-dried control HLM chips equilibrated with PBS, were connected to a Pump 11 Pico Plus Elite syringe pump (Harvard Apparatus, Holliston, MA) through nanoport fluidic connectors (Idex, Northbrook, IL) and PTFE capillaries. The chips were placed on a PID-controlled heating block at 37 °C and the feed solution containing the enzyme selective marker substrate and the co-substrate were infused through the chip at a constant flow rate of 5 μL min−1 (Fig. 3a). The marker substrates used in the present study were luciferin-H, a CYP2C9-specific preluminescent marker compound (200 μM in 0.01 M PBS, pH 7.4; Promega, Madison, WI), and 8-hydroxyquinoline (8-HQ), a nonselective prefluorescent UGT marker compound (50 μM in 0.1 M Tris with 5 mM MgCl2, pH 7.5; Sigma-Aldrich, Burlington, MA). The cosubstrates for CYP and UGT marker reactions were nicotinamide adenine dinucleotide phosphate (NADPH, 1 mM; Sigma-Aldrich, Burlington, MA) and uridine diphosphate glucuronic acid (UDPGA, 1 mM; Sigma-Aldrich, Burlington, MA), respectively. The chip effluent was collected every 10 min (CYP reactions, a 50 μL fraction) or every 20 min (UGT reactions, a 100 μL fraction) for a total of 2 h using a CMA 470 refrigerated fraction collector (CMA Microdialysis AB, Kista, Sweden). | ||
| Fig. 3 (a) Schematic of the enzyme activity determination with the help of the HLM chip. (b) An example of CYP (luciferin-H) and UGT (8-HQ) activity profiles over time during the flow-through assays (n = 5 replicate chips each). The enzyme activities were normalized to the second effluent fraction collected from the CYP (t = 20 min) and UGT (t = 40 min) assays. The average enzyme activities were calculated based on the effluent fractions depicted with horizontal (CYP, t20 − t70) or vertical lines (UGT, t20 − t120). (c) Comparison of the average CYP activities between round and diamond-shaped micropillar arrays (n = 3 replicate chips each). The statistical analysis was performed using two-sample t-test, assuming unequal variances, with p < 0.05 as the threshold for statistically significant difference. n.s. denotes statistically nonsignificant difference (p > 0.05). (d) Impact of storage time on the average CYP and UGT activities of non-freeze-dried HLM control chips (n ≥ 3 replicate chips in each case). | ||
Before analysis, the effluent aliquots were mixed with either 50 μL of luciferin detection reagent (CYP reactions) or 10 μL of 4 M perchloric acid (UGT reactions) and incubated for ca. 20 min. The fractions were then analyzed for luminescence arising from the luciferin metabolite produced in CYP reactions, or for fluorescence (ex/em 245/475 nm) arising from 8-hydroxyquinoline glucuronide metabolite produced in UGT reactions, using a Varioskan LUX microtiter plate reader (Thermo Scientific, Waltham, MA).
The enzyme activity of each collected fraction was determined by subtracting the background signal (feed solution) from the sample signal (effluent) and calculating the average activity rate for each individual HLM chip from six consecutive fractions collected between 10 and 70 min (CYP reactions) or between 20 and 120 min (UGT reactions). The enzyme activity (fmol min−1) was calculated by multiplying the metabolite concentration (nmol L−1) with the flow rate (μmol L−1) as described by Pihlaja et al.6 The activities of n ≥4 chips were averaged to calculate the mean ± standard deviation (SD), excluding any individual chips deviating by SD ≥1.25 from the mean as outliers. Statistical analyses were performed with OriginPro 2021b, version 9.8.5.212, using Shapiro–Wilk test for normality of sample sets, and t-test or one-way ANOVA for normally distributed datasets and Kruskal–Wallis test for non-normally distributed datasets.
3. Results and discussion
3.1. Optimization of the chip design and the storage time prior to freeze-drying
Before freeze-drying experiments, the micropillar array design was optimized to obtain maximal enzyme activity. In the present study, two different micropillar shapes were compared, each arranged in arrays with a hexagonal lattice. The first array comprised ca. 14750 round-shaped pillars of Ø 50 μm and interpillar center-to-center distance of 90 μm, and the second array comprised ca. 14000 diamond-shaped pillars with 50 μm width and 100 μm length and interpillar center-to-center distance of 100 μm (Fig. 1a). The nominal surface-to-volume (A/V) ratios were respectively 35 mm−1 (657 mm2/18.5 mm3) and 47 mm−1 (802 mm2/17.0 mm3) for the round-shaped and diamond-shaped pillars. Both micropillar arrays were functionalized with HLM in an identical manner and stored for 2–7 days prior to enzyme activity testing. The conceptualization of the flow-through enzyme kinetic4 and enzyme inhibition6 assays conducted with the help of the HLM chip is described in detail in previous literature. Here, the CYP and UGT enzyme activities were determined using a constant flow rate (5 μL min−1) of the substrate solution (luciferin-H for CYP and 8-HQ for UGT) and measuring the corresponding metabolite concentration (luciferin for CYP and 8-HQ-glucuronide for UGT) in the effluent over time. Similar to earlier studies,4,6 inherent enzyme activity decay over time was observed with CYP but not UGT assays (Fig. 3b). The impact of pillar shape was assessed with the help of CYP enzyme activity over the first 1 h period. Compared with the round-shaped pillars, the diamond-shaped pillar arrays provided ca. 22% increase in the total HLM binding area (i.e., the nominal surface area), but increased the enzyme activity by 68% (n = 3 chips each), as illustrated in Fig. 3c. When normalized to the A/V ratio, the observed increase in the enzymatic activity was 27%. Owing to the chip-to-chip variation, assumed to primarily arise from the intrinsic biological variation inherent to the HLMs, the difference was not statistically significant (p = 0.062). Nevertheless, the diamond shape was concluded better, since it enhances the fluid dynamics inside the chip so that the substrate molecules delivered by the feed solution interact more efficiently with the enzymes immobilized onto the micropillars. Thus, the diamond-shaped pillar arrays were used in all further experiments.
To determine the shelf life of the non-freeze-dried HLM chips, the on-chip CYP and UGT activities were determined after storing the functionalized HLM chips in the wetted state at 4 °C for 2–23 days. As a result, both CYP and UGT activities of the HLM chips were observed to increase along with pre-incubation time for up to 7–9 days of storage in the fridge (Fig. 3d). After 9 days of pre-incubation, the enzyme activity started to decline and eventually faded away in ca. 3 weeks of cold storage. Based on these results, it was concluded that the approximate shelf life of the non-freeze-dried HLM chips is ca. 2 weeks at maximum, indicating that long-term storage in the wetted state for extended periods (months) is not possible. On the other hand, the data indicated that optimum enzyme activity is not achieved immediately after HLM immobilization. A similar trend was observed for both CYP and UGT activities, suggesting that the rise and decline of enzyme activity is likely interlinked with the stability of the biotinylated HLM on the avidin-coated chip surface rather than enzyme-specific processes. Although the initial biotin–avidin interaction between the pre-biotinylated HLM and the avidin-coated chip surface is likely very fast, additional lipid rearrangements inside the chip may take a longer time until reaching equilibrium. Based on these observations, the HLM incubation time before freeze-drying was set to 5–7 days to allow enough time for HLM immobilization while avoiding initiation of the activity decline from day 9 onward (Fig. 3d).
3.2. Optimization of the freeze-drying conditions
Freeze-drying of biomolecules typically requires a lyoprotectant for preserving their structural and functional integrity.13,14,16,25 Saccharides, such as sucrose and trehalose, are commonly used lyoprotectants for CYPs17,26 and liposomes.14 In this study, we used 10% (m/V) sucrose solution as the lyoprotectant to replace the non-immobilized b-HLM solution inside the chip just before freeze-drying. The sucrose incubation time before freeze-drying can have an impact on the enzyme activity, but in this study its impact was found to be insignificant if the time was within 1–24 h. It should be noted, however, that different lyoprotectants, such as trehalose, may have a different kind of impact on the (recovered) CYP and UGT activities.In addition, the uniformity of the drying process can influence the recovered enzyme activities. In this study, the impact of the connecting channel width (of HLM chips; Fig. 1b) on the drying process was evaluated to ensure a consistent progress of the drying process inside the chip during freeze-drying. For this purpose, micropillar arrays with connecting channel widths of 200, 400, 600 and 800 μm were filled with the 10% (m/V) sucrose solution and freeze-dried and the progress of drying was monitored visually. Although progressive drying from the inlet and outlet toward the middle (Fig. 2b) was observed with all channel widths, the 800 μm channel width was concluded to provide the most consistent results qualitatively and was thus adapted to all further assays.
Next, the freeze-drying conditions were optimized in terms of the freeze-drying temperature profile by comparing the short-term storage stability (up to 3 months) between three batches of HLM chips freeze-dried using different freeze-drying programs (Fig. 2d, protocols 1–3). As a result, no statistically significant differences (p > 0.05) were observed between the three different batches (protocols) in the recovered CYP and UGT activities after freeze-drying and <1 week or 1 month storage at room temperature (Fig. 4a and b). However, after 3 months of storage, protocol 1 resulted in significantly lower CYP activity compared with protocols 2 (p = 0.013) and 3 (p = 0.003) (Fig. 4a). Similarly for recovered UGT activity, no statistically significant differences between protocols were observed after 1 week or 1 month of storage at room temperature, but after 3 months of storage, protocol 1 resulted in significantly lower UGT activity compared to protocol 2 (p = 0.045) and protocol 3 (p = 0.006). On the other hand, protocol 2 yielded much higher chip-to-chip variation than protocol 3 in terms of both CYP (Fig. 4a) and UGT (Fig. 4b) enzyme activities after 1 and 3 months of storage. Therefore, protocol 3 was selected for the long-term storage stability studies, as it also provided recovered enzyme activities within the range of the non-freeze-dried control chips still after 1 and 3 months of storage at room temperature.
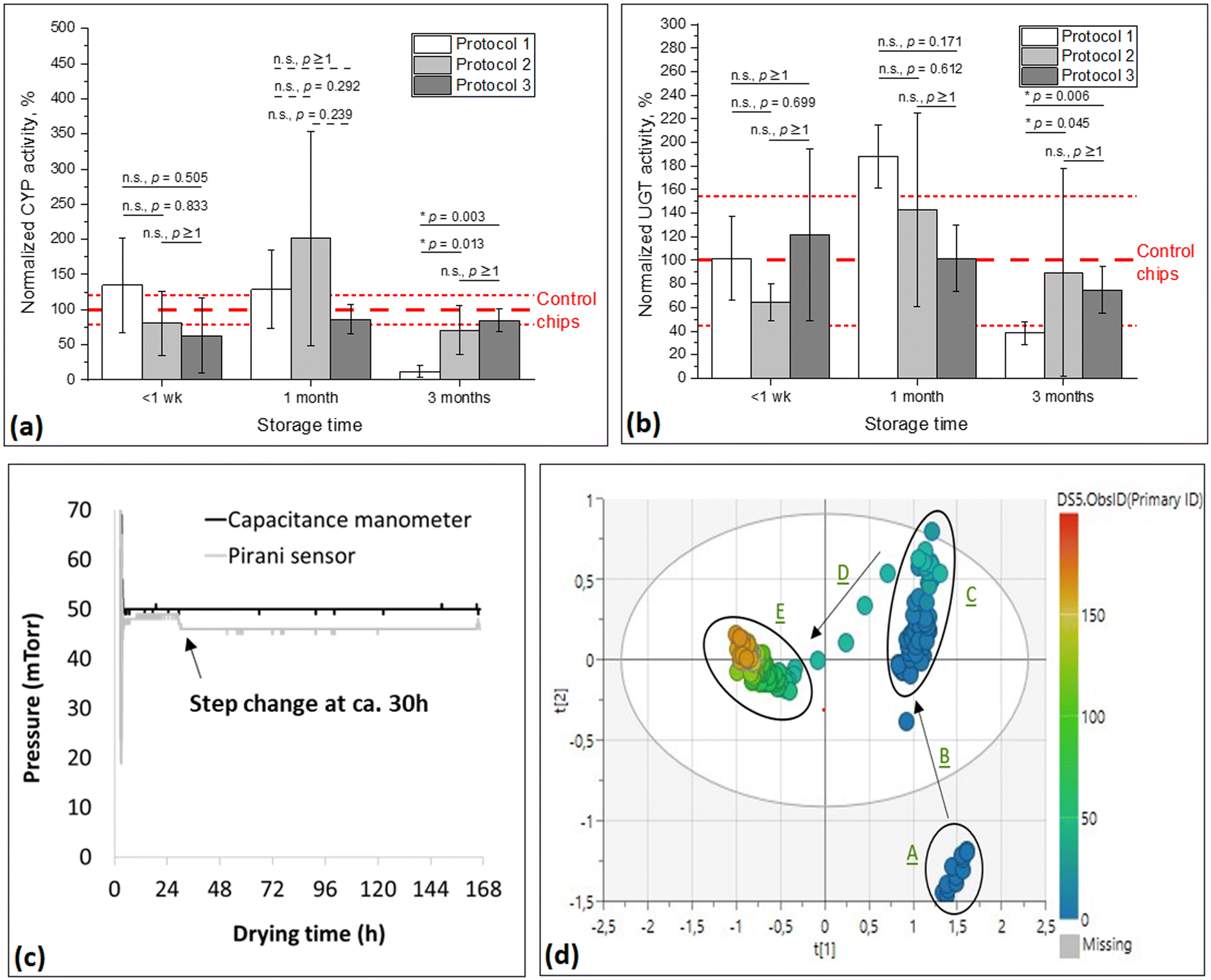 | ||
| Fig. 4 (a and b) The impact of freeze-drying (FD) process parameters on (a) the recovered CYP and (b) UGT activities of the HLM chips freeze-dried using the three different protocols given in Fig. 2d. The enzyme activities represent the average of n ≥3 chips in each case (n = 6 fractions each chip), normalized to the activity of the non-freeze-dried control chips. Before variance analysis, the datasets were tested for normality by Shapiro–Wilk test, after which the variance analysis was performed using one-way ANOVA with Bonferroni correction (for normally distributed datasets; solid line) or Kruskal–Wallis test with Dunn correction (for non-normally distributed datasets; dashed line), with p < 0.05 as the threshold for statistically significant difference. n.s. denotes statistically nonsignificant difference (p > 0.05) and * denotes statistically significant difference (p < 0.05). (c) The vacuum pressure (capacitance manometer) and thermal conductivity (Pirani sensor) collected during the freeze-drying protocol 1. (d) Principal component analysis (PCA) of the NIR spectra (1206–2169 nm) collected from an HLM chip during the freeze-drying protocol 1, using two principal components (r2 = 0.947 and Q2 = 0.947). The color bar indicates the process time from 0 h (blue) to 166 h (red). Clusters: liquid water (A), freezing (B), frozen water and sublimation (C), transition from frozen to dry state (D), and dry product (E). | ||
To confirm complete elimination of residual water during the freeze-drying process (protocol 3), the water or ice content over time was additionally monitored based on the gas composition using the Pirani sensor. During the primary drying step (Fig. 2d), the gas composition inside the drying chamber is primarily water vapor, which gives a higher Pirani reading (Fig. 4c). When all ice has sublimated from the chip, the gas composition inside the drying chamber changes to pure nitrogen gas, which is the inflow gas used to control the vacuum pressure. This can be observed as a step decrease in the Pirani reading at around 30 h after initiation of the drying process (Fig. 4c). Comparison of the process data (Pirani) with the NIR data (PCA, Fig. 4d) further confirmed that the transition from the frozen state to the dry state (end point of sublimation) occurs at around 30 h after initiation of the drying process. As visualized in Fig. 4d, the different freeze-drying process steps can be clearly seen in the PCA plot as clusters of liquid water (A), freezing (B), frozen water and sublimation (C), transition from frozen to dry state (D), and dry product (E).
3.3. Shelf life of the freeze-dried HLM chips
The long-term shelf-life stabilities of freeze-dried HLM chips were assessed using two independent batches of chips, pre-incubated with b-HLM for 5–7 days prior to freeze-drying with protocol 3 (Fig. 2d). After freeze-drying, these chips were stored at room temperature, in a sealed container with a silica gel moisture absorber and protected from light, for 16 months (batch 1) and 9 months (batch 2). To assess the recovered enzyme activities of the freeze-dried HLM chips, the CYP and UGT activities were determined immediately after drying (<1 week) as well as after 1 and 3 months and thereafter at 3 month intervals and normalized to those of the non-freeze-dried control chips. The control chips were biofunctionalized in the same manner and briefly incubated with 10% (m/V) sucrose solution, but not freeze-dried, prior to enzyme activity testing on the same day as the freeze-drying was initiated. The recovered enzyme activities of the first batch of freeze-dried HLM chips were categorically ≥60% of that of the control chips even after 16 months of storage (Fig. 5a and b). The second batch of freeze-dried chips yielded similar results, although with somewhat lower activity (between 40 and 60% of the control) after 6 months of storage. Based on these results, it could be concluded that the freeze-drying process itself is not detrimental to the immobilized HLMs or the membrane bound CYP and UGT enzymes therein. Although some variation in the recovered enzyme activities were observed in both batches over storage time, all freeze-dried HLM chips from both batches 1 and 2 had substantial enzyme activity at all timepoints, typically within 60–120% of the control activity, for as long as 16 and 9 months of storage, respectively. This kind of variation can arise from several factors, including most importantly the intrinsic heterogeneity of the HLM vesicle size, which can affect chip-to-chip variation, as well as the rehydration conditions, which were not specifically optimized in this study but were however kept identical between the different freeze-dried chip batches. Based on these results, the shelf life of the HLM chips could be extended from ca. 2–3 weeks (without freeze-drying at 4 °C) to anything between 3 and 16 months (with freeze-drying) depending on the threshold criteria. Considering the intrinsic chip-to-chip variation in enzyme activity (see control chips, Fig. 4a and b), which primarily results from the heterogenous size of the commercial HLM vesicles, our data suggest that freeze-drying is overall a feasible method for long-term preservation of microsomal enzyme activities of the HLM chips. | ||
| Fig. 5 (a) The recovered CYP and (b) UGT activities of the freeze-dried HLM chips assessed with two independent chip batches of n = 52 (batch 1) or n = 40 chips (batch 2). The enzyme activities of the freeze-dried chips present the average of n ≥3 replicate chips in each case and are normalized to those of the non-freeze-dried control chips (n = 6). | ||
Our results are also consistent with the shelf life stability data reported in previous literature, although similar long-term (several months) shelf-life stability studies are rare and most previous studies focused solely on comparison of the performance of biomolecules before and after freeze-drying at a single time point. In a previous study, however, freeze-drying of liver S9 fractions (non-immobilized), incorporating both membrane-bound microsomal and soluble cytosolic enzymes, enabled shelf lives of up to at least 6 weeks at room temperature (data for longer storage times were not reported).16 In another study, the catalytic activities of fructosyltransferases from Rhodotorula sp. LEB-V10, immobilized on niobium–graphite particles, could be retained for up to at least 6 months after freeze-drying.20 Here, notably, the recovered enzyme activity levels of the HLM chips (batch 1) were as high as 80% (CYP) and 60% (UGT) of that of the control chips even after 16 months of storage.
4. Conclusions
In this study, we present the proof-of-concept data on the feasibility of freeze-drying for long-term preservation of the enzyme activity of HLM immobilized on polymer-based micropillar arrays and demonstrate the recovery of selected CYP and UGT enzyme activities even after 16 months of storage at room temperature. Biofunctionalized flow-through HLM chips are an attractive approach for studying human drug metabolism in vitro, enabling likewise the production of human metabolite standards for subsequent flow-through drug safety and efficacy assays, such as organ-on-a-chip systems, and detailed mechanism-based drug–enzyme interaction studies in early stages of drug discovery and development. As a stand-alone in vitro model, the HLM chip is particularly well feasible for CYP enzyme inhibition assays, more precisely for facile discrimination between reversible and irreversible enzyme inhibitors among new and existing pharmaceuticals based on duration of their inhibitory impact toward the monitored enzyme system after the pharmaceutical exposure is discontinued. However, without freeze-drying, the HLM chips require cold storage and have very limited shelf life (ca. 2 weeks), which challenges the technology transfer from research laboratories to routine use. Through freeze-drying, the HLM chips can be stored at room temperature for several months, which decreases the energy consumption during storage as well as the cost of logistics substantially. Through further optimization of the freeze-drying conditions, including the selection of cryoprotectants and the rehydration conditions, freeze-drying could enable extending the shelf life of the HLM chips to a commercially viable range. At the same time, our proof-of-concept data of the lyophilization of immobilized HLM vesicles inside the OSTE microdevices suggest that freeze-drying could be a viable approach to lyophilization of also other types of lipid vesicles, such as extracellular vesicles and liposomes, or even intact cells, with a view to other applications based on biofunctionalized microfluidic devices.Data availability
The data supporting this article have been included as part of the ESI.1Author contributions
IR: investigation, methodology, conceptualization, data curation, formal analysis, writing – original draft. MH: methodology, writing – review & editing. LH: investigation, writing – review & editing. OK: investigation, methodology, formal analysis, writing – review & editing. TS: conceptualization, methodology, data curation, supervision, writing – review & editing, funding acquisition, project administration.Conflicts of interest
IR, MH, OK, and TS have patent PCT/EP2024/067617 pending to University of Helsinki.Acknowledgements
The work was financially supported by Business Finland (grant no. 6609/31/2019, R2B-EMiRA project), the Research Council of Finland (grants no. 309608 and 325222), and the NordForsk through the Nordic University Hub, NordicPOP (Patient Oriented Products, grant no. 85352). IR also acknowledges the Doctoral Programme in Chemistry and Molecular Sciences for personal funding. The Drug Discovery and Chemical Biology (DDCB) Core Facility, supported by the University of Helsinki and Biocenter Finland, and the Electron Microscopy Unit of the Institute of Biotechnology, University of Helsinki, are also acknowledged for providing access to the Varioskan LUX microplate reader and the scanning electron microscope, respectively.References
- L. C. Wienkers and T. G. Heath, Predicting in vivo drug interactions from in vitro drug discovery data, Nat. Rev. Drug Discovery, 2005, 4, 825–833 CrossRef CAS PubMed.
- U. M. Zanger and M. Schwab, Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation, Pharmacol. Ther., 2013, 138, 103–141 CrossRef CAS PubMed.
- S. Fowler and H. Zhang, In vitro evaluation of reversible and irreversible cytochrome P450 inhibition: Current status on methodologies and their utility for predicting drug-drug interactions, AAPS J., 2008, 10, 410–424 CrossRef CAS PubMed.
- I. Kiiski, E. Ollikainen, S. Artes, P. Järvinen, V. Jokinen and T. Sikanen, Drug glucuronidation assays on human liver microsomes immobilized on microfluidic flow-through reactors, Eur. J. Pharm. Sci., 2021, 158, 4–6 CrossRef PubMed.
- I. Kiiski, P. Järvinen, E. Ollikainen, V. Jokinen and T. Sikanen, The material-enabled oxygen control in thiol-ene microfluidic channels and its feasibility for subcellular drug metabolism assays under hypoxia: In vitro, Lab Chip, 2021, 21(9), 1820–1831 RSC.
- T. Pihlaja, I. Kiiski and T. Sikanen, HLM chip – A microfluidic approach to study the mechanistic basis of cytochrome P450 inhibition using immobilized human liver microsomes, Eur. J. Pharm. Sci., 2024, 197, 106773 CrossRef CAS PubMed.
- I. M. A. Kiiski, T. Pihlaja, L. Urvas, J. Witos, S. K. Wiedmer and V. P. Jokinen, et al., Overcoming the Pitfalls of Cytochrome P450 Immobilization through the Use of Fusogenic Liposomes, Adv. Biosyst., 2019, 3(1), 1–6 Search PubMed.
- B. Doyle, L. A. Madden, N. Pamme and H. S. Jones, Immobilised-enzyme microreactors for the identification and synthesis of conjugated drug metabolites, RSC Adv., 2023, 13(40), 27696–27704 RSC.
- M. Anthamatten, S. W. O'Neill, D. Liu, T. M. Wheler, R. S. Vallery and D. W. Gidley, Tunability of Free Volume and Viscoelastic Damping of Thiol-Ene Networks Deep in the Glassy State, Macromolecules, 2018, 51(7), 2564–2571 CrossRef CAS.
- Ľ. Krausková, J. Procházková, M. Klašková, L. Filipová, R. Chaloupková and S. Malý, et al., Suppression of protein inactivation during freezing by minimizing pH changes using ionic cryoprotectants, Int. J. Pharm., 2016, 509(1–2), 41–49 CrossRef PubMed.
- R. Pisano, V. Rasetto, A. A. Barresi, F. Kuntz, D. Aoude-Werner and L. Rey, Freeze-drying of enzymes in case of water-binding and non-water-binding substrates, Eur. J. Pharm. Biopharm., 2013, 85(3 PART B), 974–983 CrossRef CAS PubMed.
- A. Molnar, T. Lakat, A. Hosszu, B. Szebeni, A. Balogh and L. Orfi, et al., Lyophilization and homogenization of biological samples improves reproducibility and reduces standard deviation in molecular biology techniques, Amino Acids, 2021, 53(6), 917–928 CrossRef CAS PubMed.
- G. F. Boafo, K. T. Magar, M. D. Ekpo, W. Qian, S. Tan and C. Chen, The Role of Cryoprotective Agents in Liposome Stabilization and Preservation, Int. J. Mol. Sci., 2022, 23, 12487 CrossRef CAS PubMed.
- D. Guimarães, J. Noro, C. Silva, A. Cavaco-Paulo and E. Nogueira, Protective Effect of Saccharides on Freeze-Dried Liposomes Encapsulating Drugs, Front. Bioeng. Biotechnol., 2019, 7, 424 CrossRef PubMed.
- E. Trenkenschuh, M. Richter, E. Heinrich, M. Koch, G. Fuhrmann and W. Friess, Enhancing the Stabilization Potential of Lyophilization for Extracellular Vesicles, Adv. Healthcare Mater., 2022, 11(5), e2100538, DOI:10.1002/adhm.202100538.
- S. Buchinger, E. Campen, E. Helmers, V. Morosow, M. Krefft and G. Reifferscheid, Development of a freeze-drying protocol for the long-term storage of S9-fraction at ambient temperatures, Cryobiology, 2009, 58(2), 139–144 CrossRef CAS PubMed.
- A. Chefson, J. Zhao and K. Auclair, Sugar-mediated lyoprotection of purified human CYP3A4 and CYP2D6, J. Biotechnol., 2007, 130(4), 436–440 CrossRef CAS PubMed.
- S. Moon, Extending the Shelf-Life of Immunoassay-Based Microfluidic Chips through Freeze-Drying Sublimation Techniques, Sensors, 2023, 23, 8524 CrossRef CAS PubMed.
- T. Tonooka, Microfluidic device with an integrated freeze-dried cell-free protein synthesis system for small-volume biosensing, Micromachines, 2021, 12(1), 1–10 Search PubMed.
- E. Aguiar-Oliveira and F. Maugeri, Effects of lyophilization on catalytic properties of immobilized fructosyltransferase from Rhodotorula sp. LEB-V10, Food Bioprod. Process., 2013, 91(4), 609–616 CrossRef CAS.
- A. Ahlford, B. Kjeldsen, J. Reimers, A. Lundmark, M. Romani and A. Wolff, et al., Dried reagents for multiplex genotyping by tag-array minisequencing to be used in microfluidic devices, Analyst, 2010, 135(9), 2377–2385 RSC.
- A. Merivaara, J. Zini, E. Koivunotko, S. Valkonen, O. Korhonen and F. M. Fernandes, et al., Preservation of biomaterials and cells by freeze-drying: Change of paradigm, J. Controlled Release, 2021, 336, 480–498 CrossRef CAS PubMed.
- S. M. Tähkä, A. Bonabi, M. E. Nordberg, M. Kanerva, V. P. Jokinen and T. M. Sikanen, Thiol-ene microfluidic devices for microchip electrophoresis: Effects of curing conditions and monomer composition on surface properties, J. Chromatogr. A, 2015, 1426, 233–240 CrossRef PubMed.
- A. Merivaara, J. Kekkonen, J. Monola, E. Koivunotko, M. Savolainen and T. Silvast, et al., Near-infrared analysis of nanofibrillated cellulose aerogel manufacturing, Int. J. Pharm., 2022, 617, 121581 CrossRef CAS PubMed.
- E. Trenkenschuh, M. Richter, E. Heinrich, M. Koch, G. Fuhrmann and W. Friess, Enhancing the Stabilization Potential of Lyophilization for Extracellular Vesicles, Adv. Healthcare Mater., 2022, 11(5), 2100538 CrossRef CAS PubMed.
- H. L. van Beek, N. Beyer, D. B. Janssen and M. W. Fraaije, Lyophilization conditions for the storage of monooxygenases, J. Biotechnol., 2015, 203, 41–44 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc00429a |
| This journal is © The Royal Society of Chemistry 2024 |