
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA gravity-driven tissue chip to study the efficacy and toxicity of cancer therapeutics†
Pouria
Rafsanjani Nejad
 a,
Astha
Lamichhane
a,
Prasiddha
Guragain
a,
Gary
Luker
b and
Hossein
Tavana
*a
a,
Astha
Lamichhane
a,
Prasiddha
Guragain
a,
Gary
Luker
b and
Hossein
Tavana
*a
aDepartment of Biomedical Engineering, University of Akron, Akron, OH, USA. E-mail: tavana@uakron.edu
bDepartments of Radiology, Microbiology and Immunology, Biomedical Engineering, University of Michigan, Ann Arbor, MI, USA
First published on 24th October 2024
Abstract
Tissue chip and organs-on-chip technologies have emerged as promising tools in preclinical studies. In oncology, this is driven by the high failure rates of candidate drugs in clinical trials mainly due to inadequate efficacy or intolerable toxicity and the need for better predictive preclinical models than those traditionally used. However, the intricate design, fabrication, operation, and limited compatibility with automation limit the utility of tissue chips. To tackle these issues, we designed a novel 32-unit tissue chip in the format of standard 96-well plates to streamline automation, fabricated it using 3D printing, and leveraged gravity-driven flow to bypass the need for external flow devices. Each unit includes three interconnected tissue compartments that model liver, tumor, and bone marrow stroma. The focus on liver and bone marrow stroma was due to their respective roles in drug metabolism and disturbances to the bone marrow niche from off-target toxicity of chemotherapies. We analyzed flow patterns, mixing, and oxygen transport among and within the compartments through finite element simulations and demonstrated the utility of the tissue chip to study the efficacy of commonly-used cytotoxic cancer drugs against tumor cells and their toxicity toward liver and bone marrow cells. The ability to simultaneously study drug efficacy and toxicity in high throughput can help select promising therapeutics in early stages of drug discovery in preclinical studies.
Introduction
Around 90% of drug candidates in clinical trials fail to advance for use in patients. The rate of approval of drugs in the oncology space is only 5–7%.1–3 High failure rates contribute to soaring drug development costs that currently exceed one billion dollars per drug.4 Lack of clinical efficacy and unmanageable toxicity account for up to 80% of these failures.2 A major challenge to reliably predict the safety and efficacy of drug candidates in patients has been the inadequate preclinical models used in the process of drug development. Cell-based assays and animal models have been the standard preclinical models for many decades. Traditional two-dimensional cell cultures have been a useful drug discovery tool because they offer ease of use in standard cultureware and compatibility with robotic tools for high throughput phenotypic screening. However, they do not reproduce the intricate three-dimensional environments of human tissues in terms of composition and architecture of cellular and acellular components and the respective signaling events.5–7 On the other hand, animal models provide a truly physiological system to evaluate organ-level functions and responses to treatments and enable longitudinal studies. However, the lack of human tissue stroma and associated signaling with cells of interest and differences in drug metabolism and immune system functions from humans render animal models inaccurate for many drug efficacy and toxicity studies.8–10To address the limitations of existing models, tissue chip technologies have been developed to recreate, at least in part, the three-dimensionality, compositional and structural complexity, and interconnectivity of human tissues and organs and to improve preclinical drug development.11–13 In the oncology space, various tumor tissue chips were developed to model different types of cancer, such as those of the lung,14,15 brain,16,17 liver,18,19 colon,20,21 and breast.22–24 Because off-target toxicity is a major challenge with cancer treatments, it is critical to determine systemic effects of cancer drugs. This need has led to the development of the so-called organs-on-chip or body-on-chip systems, where the interconnectivity of human organs is mimicked by connecting different tissue compartments via fluidic channels.25–29 The interconnectivity is often achieved by connecting separate tissue chips using external tubing,28 or on the same device fabricated using techniques such as 3D printing,27 CNC cutting polymer sheets,26,29 and/or injection molding.26 These devices have shown the feasibility of determining the efficacy of drug compounds on a desired organ and their potential toxicities and inform potential benefits and risks of the treatments.
Although organs-on-chip devices have been successful in predicting physiological responses consistent with or even more accurate than those from animal models,30,31 their widespread adoption, particularly in drug discovery, remains limited.32 Complex design, tedious fabrication, and challenges with operation of these devices are main obstacles against scaling up for drug testing and screening. Tissue chips often incorporate multiple components, such as gaskets, tubing, and membranes, and require substantial expertise to maintain tissue compartments with dynamic flow. Due to these challenges and the vast number of potential drug candidates, pharmaceutical companies frequently default to the conventional 2D cell cultures for initial high throughput screening with robotic tools. However, due to the inaccuracies of 2D cultures, there is a pressing need for models that can capture systemic effects of drugs while offering a reasonable throughput.
To address these challenges, recent efforts have focused on improvements such as leveraging accessible manufacturing processes, e.g., 3D printing,27,33,34 implementing automated flow technologies,35 designing devices that house arrays of independent systems,36 and developing plate-format tissue chips.37–39 Complementing these efforts, this study introduces a multi-organ tissue chip platform that employs a standard 96-well format to seamlessly integrate with automated laboratory equipment for drug screening studies. By integrating multiple organ-specific tissues, it effectively simulates systemic interactions in a high throughput manner. This device has the potential to improve the efficiency of selecting promising compounds and preventing premature elimination of potentially valuable drug candidates.
Results
Design of the tissue chip
The conceptual design of the tissue chip is shown in Fig. 1. Three tissue compartments for liver, tumor, and bone marrow stroma and two side reservoirs form a single unit and are fluidically connected to allow exchange of media, cell secretome, and drug compounds among them through gravity-driven flow. The gravity-driven flow is sustained by tilting the chip on a custom-built plate rocker (Video S1†). Each tissue chip has 32 identical units. These units are arranged into two identical arrays, each consisting of 2 columns and 8 rows (Fig. 2a and S1†), with a diagonal offset between them to maximize the use of space and minimize the use of resin for printing. Due to this diagonal offset, aspirating and dispensing fluids with standard liquid handlers or multi-channels pipettes is done separately and in two steps for the two arrays. The units are connected with elevated cylindrical channels of 650 μm in diameter. Flow through the channels is facilitated by tilting the plate on the rocker. Additionally, the chip contains a series of independent reservoirs on either side of the matrices and at the top and bottom of the chip. During tissue culture, these reservoirs contain distilled water to minimize evaporation from the tissue compartments and the known edge effect in microwell plates. The spacing between wells in each array matches that of a standard 96-well plate to ensure compatibility with multi-channel pipettes and robotic liquid handling systems for automated processes to substantially improve experimental throughput and testing accuracy (Fig. S1 and Video S2†). We fabricated the plate of the tissue chip using 3D printing and bonded it against a polycarbonate film to ensure a leakage-free device (Fig. 2a). | ||
| Fig. 1 Conceptual design of the tissue chip representing breast tumor, liver, and bone marrow in three compartments that are fluidically connected and supported by gravity-driven convective flow from two side reservoirs. | ||
 | ||
| Fig. 2 (a) Design and fabrication of the tissue chip with 32 identical units. Food coloring dyes in the fabricated device (top right image) show 32 leak-free independent units. (b) Schematic representation of robotic formation and collagen encapsulation of breast cancer spheroids and transfer to the tissue chip. (c) Phase images of MDA-MB-231 cells micropatterned in an aqueous two-phase system forming a spheroid in 4 days and embedded in a collagen matrix (1 day after encapsulation). Scale bar is 200 μm. | ||
Establishing and maintaining cell culture in the tissue chip
Using our established cell printing technology,40 we formed breast cancer spheroids in standard microplates using the triple negative breast cancer MDA-MB-231 cell line. After spheroid formation, we aspirated and mixed them with a collagen solution and dispensed the spheroid–collagen mixture into the tumor compartment of each unit (Fig. 2b and c). Single cell suspensions of HepG2/C3A and HS-5 cells in collagen were used for the liver and bone marrow stroma compartments, respectively. HepG2/C3A cells were selected because they are immortalized, possess many functions of differentiated hepatocytes, and are not tumorigenic. HS-5 cells were also selected as immortalized surrogates for bone marrow mesenchymal stromal cells.Because liver cells use a different culture medium than breast cancer and bone marrow stroma cells, we studied the effects of different combinations of the media on the metabolic activities of the cells prior to seeding the cells in the tissue chip. We normalized the metabolic activities of each cell type to that of cells in their native medium (Fig. S2†). The metabolic activities of cells in the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of EMEM and DMEM were comparable to those in the native media for all three cell types, with average values of 123%, 97%, and 94% for the HepG2/C3A, MDA-MB-231, and HS-5 cells, respectively (Fig. S2†). Other ratios, especially when more than 80% EMEM was used, led to significant reduction in the metabolic activities of MDA-MB-231 and HS-5 cells. Based on this analysis, we used a 1:1 ratio of EMEM:DMEM to maintain the cells in the tissue chip.
:1 mixture of EMEM and DMEM were comparable to those in the native media for all three cell types, with average values of 123%, 97%, and 94% for the HepG2/C3A, MDA-MB-231, and HS-5 cells, respectively (Fig. S2†). Other ratios, especially when more than 80% EMEM was used, led to significant reduction in the metabolic activities of MDA-MB-231 and HS-5 cells. Based on this analysis, we used a 1:1 ratio of EMEM:DMEM to maintain the cells in the tissue chip.
Next, we examined the capability for on-chip analysis of cells using both bioluminescence and PrestoBlue assays, which are widely used to determine cell metabolic activity and viability. We developed collagen-embedded cultures in the tissue compartments with different cell densities. The measured signal intensity values from either assay showed a strong correlation with the seeded cell density with R2 values of 0.92 for bioluminescence and 0.96 for the PrestoBlue assays, similar to or better than the benchmark measurements in standard microplates (Fig. S3†). Additionally, both assays easily distinguished between blanks and the lowest cell density of 5 × 103 cells per well (p < 0.001). Furthermore, we evaluated the consistency between on-chip measurements of the PrestoBlue signal and measurements after transferring the supernatant media to a standard plate. Both methods gave similar results (Fig. S3†), validating the feasibility of performing high precision analysis of cells directly in the tissue chip.
To ensure the utility of the tissue chip for long-term cell cultures, we quantified cell viability within each compartment over an 8-day period by measuring cellular metabolic activity (Fig. 3). After 4 and 8 days, metabolic activities increased by 1.4- and 2.3-fold for breast cancer cells, 1.7- and 4.7-fold for liver cells, and 1.8- and 3.4-fold for bone marrow stromal cells. Furthermore, MDA-MB-231 cells significantly invaded into the surrounding collagen matrix, which is a key phenotype of this cell line, HepG2/C3A liver cells formed clusters, and HS-5 bone marrow stromal cells assumed a more elongated, mesenchymal phenotype. Additionally, confocal imaging of live and dead cells showed minimal cell death in the tissue compartments over 8 days (Fig. 3). Therefore, the tissue chip supported growth and normal phenotypes of the cells during culture.
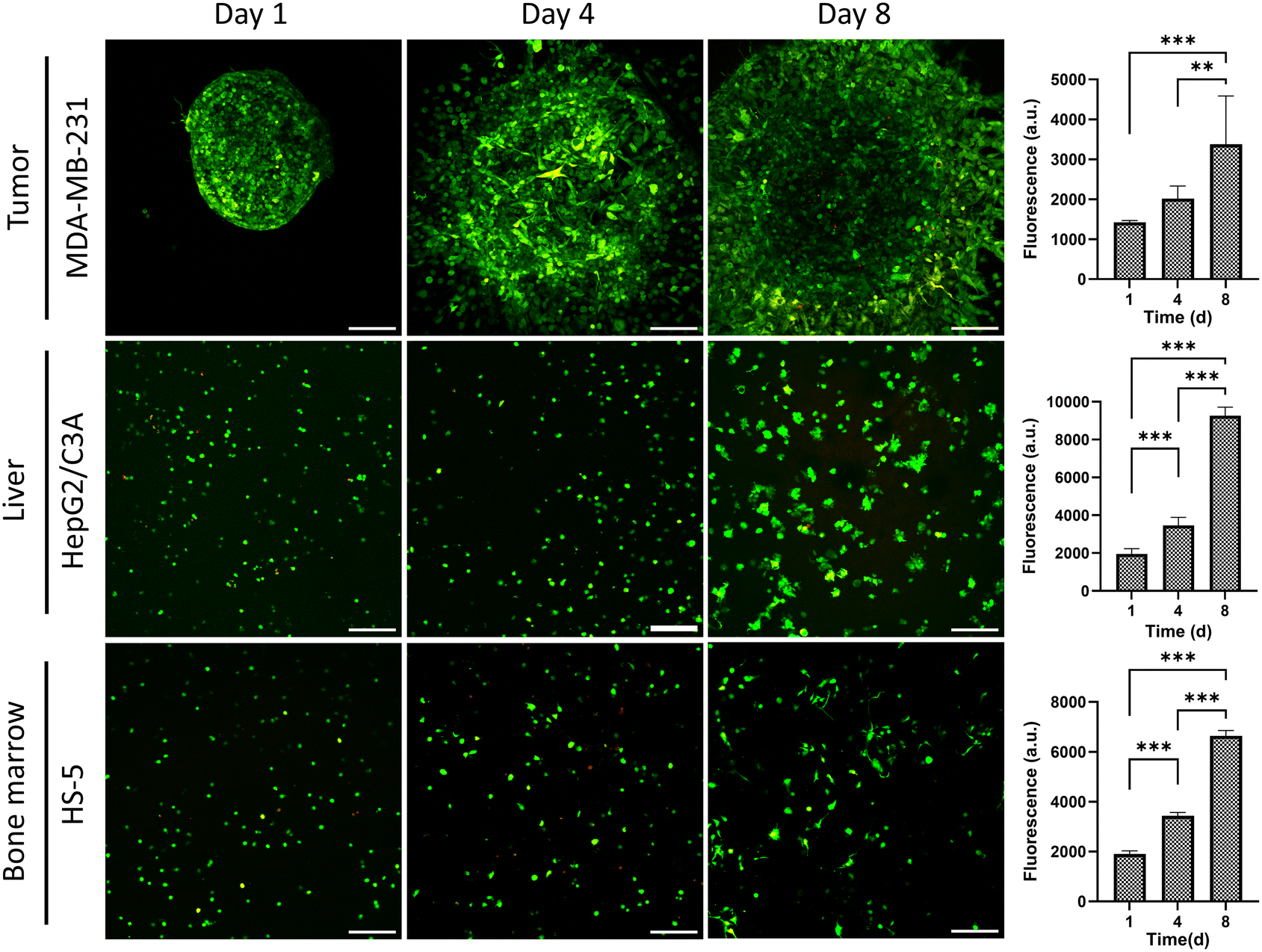 | ||
| Fig. 3 Confocal images of MDA-MB-231 spheroids, HepG2/C3A liver cells, and HS-5 bone marrow stromal cells in their respective compartments in the tissue chip over an 8-day culture. Bar graphs represent the viability of cells measured using a PrestoBlue assay. MDA-MB-231 cells also show significant invasion into the collagen matrix. Scale bar is 200 μm. **p < 0.01, ***p < 0.001. | ||
Transport studies in the tissue chip
We conducted three sets of finite element simulations to analyze fluid flow, chemical mixing, and oxygen distribution in the tissue chip. First, we characterized the gravity-driven flow using a moving mesh model (Fig. 4a–f, Video S3†). This simulation involved continuously altering the gravitational vector to emulate a tilting motion. Graphical representation of the simulation in Fig. 4a–c shows fluid height fluctuations of about 2.5 mm in the side reservoirs resulting from the gravity-driven flow among the compartments. Since the tissue compartments experienced both influx and efflux of culture medium, their fluid heights fluctuated by about 1.0 mm per cycle. | ||
| Fig. 4 (a–c) Numerical simulation shows that tilting of the tissue chip generates gravity-driven flow and changes of about 1 mm in the fluid height within the tissue compartments. (d) The tissue chip is tilted ±7° within a period of 40 s to maintain a dynamic fluid flow. (e) Volume flow rates and (f) net fluid volumes transferred through the channels connecting the side reservoirs and tissue compartments. Q1 and V1, Q2 and V2, Q3 and V3, and Q4 and V4 correspond to flow through the left, middle left, middle right, and right channels. V = ∫ − Qdt, limits of the integral are 0 and t. (g) Transport and distribution of a hypothetical small molecule from an initial source in the middle tissue compartment through the entire device due to the gravity-driven flow. The color bar shows concentration range in mM. (h) Concentrations of the hypothetical small molecule in the tissue compartments over a 20 min period. (i and j) Oxygen concentration profiles within tissue compartments in the presence and absence of flow and at different consumption rates of 2.5, 25, and 100 amol s−1 cell−1. The color bar shows percent oxygen concentration relative to that at the fluid–air interface. (k) Quantified oxygen concentrations at different consumption rates. → next to the y-axis in panels (b) and (c) represents 0.5 s before the presented time and indicates the last time point before the next tilt started. | ||
Additionally, we obtained volume flow rates between the compartments, labeled Q1–Q4, in each of the channels from left to right. We used a negative sign to indicate flow in the negative direction of the x-axis. The starting flow rates of 3–4 μL s−1 gradually declined within each 20-second tilt (Fig. 4d and e). Notably, the flow rates through the channels between the three tissue compartments (Q2 and Q3) remained within 75% of the initial values, whereas the flow rates in the channels leading to the elevated reservoir decreased to approximately 35% of its initial maximum (Q4 for 0–20 s and Q1 for 20–40 s). Next, we computed the volume of medium displaced through each channel, labeled V1–V4, by integrating the flow rates over time (Fig. 4f). The result showed that 55 μL is transferred within a single tilt in the tissue compartments (V3 at 20 s). Considering that there is a total medium volume of 650 μL in the entire tissue chip, about 8% of this volume is transported during each single tilt.
To assess mixing efficiency across and within the compartments, we conducted a static mesh simulation and used the flow rate Q2 between the tissue compartments as an imposed boundary condition. We introduced a hypothetical compound with a diffusion coefficient of 1 × 10−9 m2 s−1 into the middle compartment to approximate the diffusivity of small metabolites and drugs such as 5-FU.41 The result showed that the transport of the compound through compartments by the gravity-driven flow results in almost full mixing within 20 minutes (Fig. 4g and h and Video S4). At the 4-second mark, traces of the compound were present in the downstream compartment. At the 40-second mark, the compound was present in all compartments and there was a significant reduction in the concentration in the middle compartment. At 20 minutes, the compound was relatively evenly distributed with average concentrations in the three compartments (1 to 3, from left to middle) being 99 μM, 94 μM, and 149 μM, respectively, and the overall average in the tissue chip was 100 μM (Fig. 4h). To further validate this result, we used a food coloring dye to experimentally demonstrate uniform distribution of the dye in the tissue chip in 20 minutes (Video S5†).
To investigate oxygen transport in the chip, we assigned different oxygen consumption rates over a broad range to the cell culture regions of the three tissue compartments and conducted a static mesh simulation (Fig. 4i–k, Video S6 and S7†). We determined the volume average of oxygen concentration in the cellular region of each compartment and normalized it to the oxygen concentration at the fluid–air boundary. Our result showed that with a consumption rate of 2.5 amol s−1 cell−1, the average oxygen concentration within the cellular regions remained at or above 90% of the initial maximum concentration under both static (no tilting) and convective flow (due to tilting) conditions. At the higher oxygen consumption rate of 25 amol s−1 cell−1, the presence of flow improved oxygen concentration to 78.8% compared to 24.6% under a static condition. This effect was also clear at the highest consumption rate of 100 amol s−1 cell−1, where flow maintained an average oxygen concentration of 24.6%, but a static condition led to complete depletion of oxygen in the cellular regions. The concentration heatmap for this compartment showed that convection transports oxygen-rich media to the upper region of the compartment (Fig. 4i, R0 = 100 amol s−1 cell−1). Nonetheless, the flow velocity in the tissue culture region was significantly lower than that in the upper region (Fig. S4†) and oxygen distribution was predominantly due to diffusion.
Modeling systemic drug responses in the tissue chip
Next, we studied the feasibility of replicating systemic effects of cancer therapeutics in the tissue chip. Specifically, we tested whether the liver compartment could activate a prodrug, tegafur, and generate a toxic effect in the tumor and bone marrow stroma compartments. We used the chemotherapy drug 5-fluorouracil (5-FU) as a negative control. 5-FU is commonly used to treat various cancers,42 including the triple negative breast cancer.43 By inhibiting the enzyme thymidylate synthase, 5-FU prevents DNA replication and induces cell death.42 However, the effectiveness of 5-FU is limited due to its rapid breakdown by dihydropyrimidine dehydrogenase (DPD) in the liver.44 To address this issue, an oral cocktail of uracil and tegafur (UFT) with a molar ratio of 1:4 has been developed.45 In this combination, tegafur acts as a prodrug of 5-FU and is converted to the active form in the liver by the cytochrome P-450 2A6 enzyme, while uracil boosts the bioavailability of 5-FU by competitively inhibiting DPD.
We added 5-FU (50 μM) and UFT (200 μM tegafur and 800 μM uracil) one day after forming the tissue compartments. To reliably evaluate effects of UFT activation on the cells, we used 5-FU in the tissue chips containing all three tissue compartments and also prepared tissue chips without a liver compartment to serve as control conditions. Five days post-treatment, we quantified cell viability using a PrestoBlue assay and imaging of live and dead cells (Fig. 5). As expected, 5-FU generated significant toxicity in all three compartments and reduced the viability of breast cancer, liver, and bone marrow stromal cells to 35%, 20%, and 24%, respectively. In tissue chips with UFT added, the viability of breast cancer, liver, and bone marrow stromal cells reduced to 64%, 47%, and 37%, respectively, but in the absence of a liver compartment, there was only marginal toxicity from UFT and the viability of breast cancer and bone marrow stromal cells was 93% and 73%, respectively. The live/dead cell staining was also consistent with these measurements and showed the highest number of dead cells (red) in the 5-FU-treated condition. Furthermore, UFT treatment inhibited cancer cell invasion and increased the number of dead cells, especially in the presence of a liver compartment.
 | ||
| Fig. 5 Viability of MDA-MB-231 breast cancer, HepG2/C3A liver cancer, and HS-5 bone marrow stromal cells 5 days post-treatment: control indicates no treatment, UFT (− liver) indicates a tissue chip lacking a liver compartment but with UFT added, UFT (+ liver) indicates a complete tissue chip and UFT added, and 5-FU indicates a complete tissue chip and 5-FU added. Graphs represent quantified results of cell viability. All tissue compartments contained 3D cultures. Scale bar is 200 μm. *p < 0.5, **p < 0.01, ***p < 0.001. | ||
Drug screening studies with the tissue chip
We investigated the utility of the tissue chip to evaluate systemic and dose-dependent responses of cells to both chemotherapy and targeted therapy drugs. We selected 5-FU and doxorubicin as chemotherapeutics, and trametinib (a MEK inhibitor) as a targeted therapy drug, and administered them to the tissue chips (Fig. 6). Following the treatments, we used a four-parameter logistic regression model to obtain half-maximal inhibitory concentrations (IC50). We used individual 3D cultures in 384-well plates as a negative control (Fig. S5†). | ||
| Fig. 6 Dose responses of cells in different tissue compartments to drug treatments. The respective IC50 and AUC values for the treatments are also shown. Vertical dashed lines in the graphs represent IC50 concentrations. Each data point is an average of 2 replicates for 5-fluorouracil and 4 replicates for doxorubicin and trametinib. Error bars show standard errors. | ||
5-FU showed toxicity across all compartments, with IC50 values ranging from 1 to 10 μM (AUC = 0.61–0.78), and showed similar, but lower, toxicity to the cultures in 384-well plates. Doxorubicin caused significant toxicity in the breast cancer and bone marrow stroma compartments at low concentrations, with IC50 values of 5.5 nM and 4.6 nM (AUC = 0.47 and 0.45), respectively, while showing reduced toxicity to liver cells (IC50 = 231 nM, AUC = 0.72). Interestingly, doxorubicin was toxic to breast cancer cells in the tissue chip at much lower concentrations and had an IC50 value almost 70-fold lower than that in the conventional mono-culture model (cf.Fig. 6 and S5†). Additionally, it generated greater toxicity to bone marrow stromal cells in the tissue chip and gave an IC50 value almost 33-fold lower than that in the conventional mono-culture model than in the standard well-plate. Trametinib was much more selective toward breast cancer cells with an IC50 value of 0.06 nM (AUC = 0.63), compared to the liver and bone marrow stroma compartments that gave IC50 values of 12.2 nM (AUC = 0.75) and more than 1 μM (AUC = 0.98), respectively. Responses of cells to 5-FU and trametinib were relatively similar in the tissue chip and the conventional well-plate cultures.
Discussion
In early stages of drug discovery, efficiently screening large numbers of compounds is crucial to identify promising candidates and eliminate potentially toxic ones.32 Traditionally, potential drugs have been first selected using 2D cell cultures and then validated in animal models. However, results from animal studies often do not translate well to humans due to interspecies differences. Organs-on-chip platforms have shown great promise in enhancing the quality of preclinical data by replicating certain physiological conditions in humans. This capability provides a major opportunity in preclinical studies prior to using animal models, with the goal of ultimately replacing them,30 which was signed into law in 2022 as the FDA Modernization Act 2.0.46An area of need in developing tissue chips is increasing their throughput capabilities for the early stage drug discovery process, particularly during the hit-to-lead and lead optimization phases. Existing devices are often limited by their complex designs and fabrication, as well as reliance on external fluid actuation and control components. These factors collectively hinder the scalability of tissue chips for large-scale screening required in the initial stages of drug development.
To address these issues, we used 3D printing to fabricate the tissue chip because it is widely available and allows fast design iterations with significantly reduced labor and cost compared with techniques such as microfabrication.33 The growing availability of biocompatible resins and ongoing advances to enhance fabrication resolution and capabilities make this a viable approach to develop tissue chips.47 Additionally, gravity-driven flow has been successfully incorporated into many designs to avoid the complexities associated with external components required for flow control designs.27,48–51 Leveraging gravity-driven flow and a design that conforms to the conventional cultureware standards, we fabricated a tubeless and pumpless tissue chip compatible with automated liquid handling, imaging, and analysis tools that are conveniently available in biomedical research laboratories, core centers, an pharmaceutical companies. The compatibility with automation significantly enhances the precision and throughput of experiments.
We integrated three distinct tissue models into the chip: collagen-embedded tumor spheroids to study cell invasion;52,53 a 3D liver compartment to investigate drug metabolism and its effects on other tissues; and a 3D bone marrow stroma compartment to evaluate off-target toxic effects of cancer drugs. We incorporated a stromal environment in the bone marrow compartment because long-term myelosuppression induced by chemotherapies is often associated with disturbances to bone marrow stroma.54,55 Offering multiple fluidically-connected 3D tissue models is significant to inform possible off-target drug toxicity and efficacy.
Off-target toxicity of cancer drugs is a major challenge for patients undergoing treatments. In the past, several clinical trials failed due to unmanageable toxicity to patients. For example, significant toxicity due to simultaneous inhibition of commonly dysregulated PI3K/Akt and MEK/ERK pathways in several cancers led to frequent dose interruptions and reductions, or stopping of the trials altogether.56–59 Developing high throughput models that replicate systemic effects of drugs and their combinations in preclinical studies could greatly improve outcomes for patients.
Meeting the metabolic demand of cells in tissue chips is critical for maintaining long-term cultures for drug testing applications. Our finite element analysis established that the gravity-driven convective flow sufficiently mixed the media in different tissue compartments and enhanced oxygen transport. Our selected oxygen consumption rates of 2.5, 25, and 100 amol s−1 cell−1 covered a wide range reported for the following: 1) average cellular oxygen uptake in the human body of ∼2.5 amol s−1 cell−1 based on calorimetric studies,60 2) 16.8 and 32.5 amol s−1 cell−1 for cancer cells such as MDA-MB-231 and MCF7 cells, respectively, based on in vitro studies,60 and 3) 400 amol s−1 cell−1 or higher for more oxygen demanding cells such as primary hepatocytes.61 Our simulation showed that the convective flow through the compartments met the metabolic needs of cells and that the extra height of media over the cultures did not limit oxygen transport to cells. At the high rate of 100 amol s−1 cell−1, we observed oxygen depletion in the tissue compartments, potentially limiting the culture of primary cells or tissue fragments with greater oxygen demands. This limitation arises from reduced flow rates in the collagen-filled cellularized parts of the compartments, where oxygen distribution primarily occurs via diffusion. This limitation is even more pronounced in standard culture plates due to the lack of convective flow. Addressing this issue requires strategies such as increasing the effective surface area-to-volume ratio of the hydrogel.
The fluidic connections among the compartments in the tissue chip helped mimic metabolic activation of a chemotherapeutic prodrug and enabled quantifying its systemic toxicity in different tissues. We found that the toxicity from tegafur was lower than that with the active drug 5-FU. This is likely due to the lower metabolic activity of the HepG2/C3A cells than primary hepatocytes, and the inherent susceptibility of HepG2/C3A cells to tegafur, given their immortalized nature and sensitivity to chemotherapeutics. Additionally, the tissue chip captured dose-dependent toxicity from traditional chemotherapy drugs and the MEK inhibitor, trametinib. The MEK inhibitor showed greater specificity toward the breast tumour spheroid and less toxicity in other tissues. This effect is due to the constitutively active MAPK/MEK/ERK pathway in MDA-MB-231 cells,53,62 highlighting the benefits of therapies that target cancer-specific driver mutations to enhance specificity and reduce off-target toxicity.
Our tissue chip contains 32 identical units to generate sufficient data for dose–response studies. We achieved this using four concentrations (including control), each with four replicates, for each of the three tissue compartments. Dose responses of tissue cultures can also be obtained using independent tissue compartments in standard culture plates. We compared these approaches and found similar dose responses of cells to 5-FU and trametinib on the chip and in conventional culture plates. These drugs are in their active forms and do not require metabolic activation in the liver. However, a key advantage of fluidically-connected tissue compartments is the ability to capture effects of secondary metabolites in addition to the primary drug.63,64 We showed this capability with the UFT experiments. Additionally, we observed greater toxicity from doxorubicin in the tissue chip compared to standalone cultures. Although we did not investigate the underlying mechanism, doxorubicin is known to undergo extensive metabolism in the liver,65,66 and one of its major metabolites, doxorubicinol (DOXol), induces oxidative stress in cells. While doxorubicinol is less effective at intercalating into DNA,67 the resulting oxidative stress can be damaging to cells, which may explain the greater toxicity of doxorubicin in the tissue chip.
Transport of drugs, nutrients, and oxygen in the tissue chip occurs via flow from the top of the compartments and passive diffusion through the collagen hydrogel to the cells. However, in the body, the vascular system is responsible for transporting oxygen and biomolecules, with endothelial cells acting as the potential target in certain cancer therapies.68,69 Adding a vascular component into future designs will improve the physiological relevance of transport processes in the tissue chip. Lastly, incorporating more complex cultures with hydrogels that mimic the mechano-chemical properties of native tissues, along with primary cells and patient-derived tissue fragments, can further enhance the translational potential of the tissue chip.
Materials and methods
Design and fabrication of the tissue chip and rocker platform
The tissue chip was designed in SolidWorks software. It consisted of a plate and a bottom layer polycarbonate film. The plate contained 32 units, each with three tissue compartments and two fluid reservoirs. Each compartment or reservoir was designed as two stacked cylindrical wells with diameters of 5.0 mm at the top and 3.5 mm at the bottom. The cross-sectional area of the bottom cylinder is comparable to that of a well in a standard 384-well plate and holds about 30 μL of media or extracellular matrix hydrogels. The top cylinder is wider to accommodate more media and increase the media–air surface area to increase transport of oxygen and carbon dioxide. These top cylinders are interconnected by microchannels with a circular cross-section and a diameter of 650 μm, which represents the smallest printable dimension without the uncured resin clogging the channels. The plate was fabricated using a Formlabs 3B resin printer and a clear biocompatible resin (Formlabs BMCL-01), washed in isopropanol twice, and cleaned using pressurized air. To drain uncured resin, particularly from inside the channels connecting tissue compartments, each plate was positioned vertically during both print and wash steps.To enable inverted microscopy, each plate was printed without a bottom layer and subsequently attached to a transparent, food-grade polycarbonate sheet. To attach the bottom layer, the bottom surface of the plate was sanded down to a smooth surface, dipped in chloroform inside a chemical fume hood, and pressed against the polycarbonate sheet on a flat surface in a fume hood. Adequate time was allowed for the chloroform to evaporate. The assembly was post-cured in a UV chamber at 60 °C according to the resin manufacturer's specifications. To enhance bonding and prevent leaking, the polycarbonate layer was attached to the plate prior to the post-curing step. The assembly, i.e., the tissue chip, was sterilized using filtered 70% ethanol and UV light, and then rinsed three times with deionized, sterile water. Polycarbonate was used for the bottom layer due to its compatibility with autoclaving and its chemical resistance to ethanol used to sterilize the tissue chip.
An in-house plate rocker was used to generate gravity-driven flow. This rocker was constructed by CNC cutting of acrylic sheets and then assembling them with a cyanoacrylate-based adhesive. A servo motor, interfaced with an Arduino board, was used to adjust the tilt angle and frequency of rocking.
Finite element simulation
Three finite element analyses were carried out using COMSOL Multiphysics software, leveraging the laminar flow (spf) and transport of diluted species (tds) modules. The first simulation determined the flow profile through a moving mesh analysis, wherein the fluid in each compartment was considered a deformable geometry. A dynamic gravitational force was applied to the model and its direction over time was adjusted according to the tilt angle. The upper surfaces were defined as free surfaces with an external pressure of zero. The simulations were initiated from a horizontal position. The duration of the initial tilt was set to 10 seconds to cover a simulation period of −10 seconds to 0 seconds, i.e., half a cycle. This was followed by a full tilt cycle of 20 seconds for the ascent and 20 seconds for the descent. Volume flow rates between the compartments, Q1–Q4, were calculated by integrating the product of flow velocity and cross-sectional vector area of connecting channels in the x-axis direction. Displaced media volumes were found as the time integrals of the corresponding Q values. To avoid negative values, integration was started at t = 0 s and flow was considered in the negative x-axis direction.The second analysis involved investigating the transport of a hypothetical small molecule among the interconnected compartments. To reduce the computational load, a static mesh simulation was used to estimate the solution based on flow rates obtained from the previous moving mesh simulation. The top surfaces of the left and right fluid reservoirs were defined as the inlet and outlet, respectively. The inlet flow was adjusted to replicate the flow rates from the moving mesh simulation. This simulation was done for 20 minutes, i.e., for 30 tilting cycles, and the temporal concentration distribution was determined.
The third simulation was done with a static mesh and a constant flow rate of 2.5 μL s−1 through the compartments to evaluate the distribution of oxygen within the tissue chip. Boundary conditions were established by calculating the oxygen concentration at the top surface of each compartment using Henry's law and the walls being assigned a zero-flux condition. An oxygen consumption reaction was defined within the cell culture region, with the reaction rate calculated using the product of cell density and an average cellular consumption rate for each compartment. The consumption rates, ranging from 2.5 to 100 amol s−1 cell−1, were sourced from the literature. The analysis was carried out for 48 hours and the resulting oxygen concentration data was normalized relative to the surface concentration to facilitate a clear visualization of oxygen distribution.
Cell culture
Triple negative breast cancer cell line MDA-MB-231, human bone marrow mesenchymal stromal cell line HS-5, and human hepatocyte cell line HepG2/C3A were purchased from ATCC. MDA-MB-231 cells were previously transduced to produce GFP and click beetle green and HS-5 cells were transduced to produce click beetle red.70 MDA-MB-231 and HS-5 cells were cultured in high glucose DMEM (Sigma) with 10% fetal bovine serum (Sigma), 1% streptomycin/penicillin/amphotericin B (ThermoFisher), and 1% GlutaMAX (Gibco). HepG2/C3A cells were cultured in low glucose EMEM (Sigma), 10% fetal bovine serum (Sigma), and 1% streptomycin/penicillin (ThermoFisher). A 1:1 mixture of the two media was used for cell culture in the tissue chip. Cells were kept in a humidified incubator at 37 °C supplemented with 5% CO2 and subcultured when they were about 90% confluent.
Robotic tumor spheroid formation and cell culture in the tissue chip
Four days prior to seeding in the tissue chip, breast cancer spheroids were robotically formed using an aqueous two-phase system (ATPS) technique.40 Initially, solutions of 5% (w/v) polyethylene glycol (35 kDa PEG; Sigma-Aldrich) and 12.8% (w/v) dextran (500 kDa DEX; Pharmacosmos) were prepared in the complete growth medium. To improve spheroid formation of MDA-MB-231 cells, 0.24% (w/v) methyl cellulose (Fisher Scientific) and 2% rat tail collagen I (Corning) were added to these two solutions, respectively. Subsequently, a suspension of MDA-MB-231 cells at a density of 80 × 106 cells mL−1 was mixed with the DEX solution in a 1:1 volume ratio. Concurrently, 30 μL of the PEG solution was dispensed into each well of a 384-well ultralow attachment (ULA) round-bottom plate (Corning). Next, 250 nL of the cell suspension in DEX phase solution was robotically dispensed into each well of the ULA plate using an automated liquid handler (Bravo SRT, Agilent), resulting in each DEX phase drop containing 10 × 103 cells. The cultures were maintained with culture medium additions on days 1 and 3. After compact spheroids formed, they were robotically aspirated from the ULA plate, mixed with collagen to a final concentration of 3.5 mg mL−1, and added to the tumor compartment of the tissue chip. Then, bone marrow stroma and liver cells were suspended in 3.5 mg mL−1 collagen as single cells with a density of 600 × 103 cells mL−1. The compartments were then loaded by transferring 25 μL of the respective suspension to obtain a final density of 15 × 103 cells per compartment. To maintain collagen at 4 °C, we developed cooling plates for the liquid handler using thermoelectric (TEC) plates that dissipated heat into a circulating water system. The chip was then kept in a CO2 incubator for 1 hour to allow the collagen to gel followed by adding culture medium to maintain the cells in culture.
Drug treatments
The chemotherapy drugs 5-fluorouracil (5-FU) and doxorubicin (DOX), the prodrug tegafur, and uracil were purchased from Selleckchem. These compounds were dissolved in dimethyl sulfoxide (DMSO) to prepare stock solutions of 50 mM 5-FU and DOX, and 100 mM tegafur and uracil. Drug solutions were diluted in the tissue chip medium. A 50 μM concentration of 5-FU and a mixture of 200 μM tegafur and 800 μM uracil cocktail were used for single-dose experiments. One day after initiating the cultures, the medium was replaced with drug-containing medium in the entire chip and was subsequently replenished every two days. Similar concentrations of drugs were also used in 384-well plates populated with the same tissue models, i.e., collagen-embedded spheroids of MDA-MB-231 cells and suspensions of HepG2/C3A and HS-5 cells in 25 μL collagen. The viability of cells in each compartment was quantified using a PrestoBlue assay following 5 days of treatments.Cellular assays and viability measurements
To evaluate cellular metabolic activity, a resazurin-based assay (PrestoBlue™, Thermo Fisher Scientific) was used. The existing culture medium was removed, and each well was supplemented with 50 μL of medium containing 15% (V/V) of PrestoBlue reagent. This addition resulted in a final PrestoBlue concentration of 10% (V/V), accounting for the volume of the collagen solution. Following a 1-hour incubation, fluorescence measurements were conducted using a plate reader (Synergy H1M, BioTek Instruments) at an excitation wavelength of 560 nm and an emission wavelength of 590 nm. In addition, cell viability in the chips was investigated using bioluminescence measurements. The medium in each well was replaced with 30 μL of phenol red-free medium containing luciferin. Considering the volume of collagen, a final luciferin concentration of 500 μg mL−1 was used. Following a 10-minute incubation, luminescence measurements were done using a plate reader (Synergy H1M, BioTek Instruments).Imaging of live and dead cells
Live/dead staining was performed using calcein AM (CA/AM) and propidium iodide (PI). Following the removal of the culture medium from the compartments, a 50 μL staining solution containing 6 μM CA/AM and 15 μM PI was added to each compartment followed by incubation for 1 hour. After staining, z-stack images were obtained using a confocal microscope (Nikon A1), and then maximum intensity z-projection was used to collapse the images.Statistical analysis
For biochemical assays, each sample had four replicates unless otherwise noted. The statistical analyses of sample means were performed using GraphPad Prism software. One-way ANOVA, complemented by Tukey's pairwise comparisons, was used to evaluate differences between groups. A p-value less than 0.05 was considered statistically significant. The area under the curve (AUC) was computed using GraphPad Prism software. To assess the dose-dependent responses of cells to the treatments, a four-parameter logistic curve was fitted to the data.Conclusions
We introduced a multi-organ tissue chip that leverages gravity for convective flow among fluidically-connected tissue compartments, eliminating the need for external tubing and pumping and significantly simplifying both assembly and operation of the device. Furthermore, the compatibility of our design with the standard 96-well plate format ensured automated exchange of reagents and improved throughput and precision. Additionally, the standard format allowed for facile formation of 3D cultures representing liver, bone marrow, and breast tumor to quantitatively study systemic drug responses including both targeted and off-target toxicities. The 3D architecture of the tissue cultures offers the potential to incorporate stromal cells in the tumor and liver compartments to facilitate tumor–stromal interactions and help maintain hepatocytes in long-term cultures, respectively. In addition, the use of 3D printing to fabricate the tissue chip facilitates convenient modifications to the design in future studies such as optimizing the compartments for primary cell and tissue cultures. Overall, this tissue chip provides a scalable model to study inter-organ interactions and for early-stage drug discovery due to its medium throughput.Data availability
Details of methods and designs for the studies presented in the manuscript are available upon request from the corresponding author.Author contributions
Pouria Rafsanjani Nejad: investigation, methodology, conceptualization, formal analysis, visualization, writing. Astha Lamichhane: methodology. Prasiddha Guragain: methodology. Gary D. Luker: funding, resources, writing, editing. Hossein Tavana: conceptualization, supervision, funding, project administration, resources, writing, editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support for this project was provided by grant CA225549 from National Institutes of Health and grant W81XWH-22-1-0119 from the U.S. Department of Defense. Fig. 1 and several panels of Fig. 2 were created in https://Biorender.com.References
- H. Dowden and J. Munro, Nat. Rev. Drug Discovery, 2019, 18, 495–496 CrossRef CAS PubMed.
- M. Hay, D. W. Thomas, J. L. Craighead, C. Economides and J. Rosenthal, Nat. Biotechnol., 2014, 32, 40–51 CrossRef CAS PubMed.
- L. Hutchinson and R. Kirk, Nat. Rev. Clin. Oncol., 2011, 8, 189–190 CrossRef PubMed.
- O. J. Wouters, M. McKee and J. Luyten, JAMA, 2020, 323, 844–853 CrossRef PubMed.
- J. Jitcy Saji, M. Sibusiso Tebogo and N. Monde, in Cell Culture, ed. M. Radwa Ali, IntechOpen, Rijeka, ch. 2, 2018, DOI:10.5772/intechopen.81552.
- W. H. Abuwatfa, W. G. Pitt and G. A. Husseini, J. Biomed. Sci., 2024, 31, 7 CrossRef PubMed.
- C. Jensen and Y. Teng, Front. Mol. Biosci., 2020, 7, 33 CrossRef CAS PubMed.
- L. Meigs, L. Smirnova, C. Rovida, M. Leist and T. Hartung, ALTEX, 2018, 35, 275–305 CrossRef PubMed.
- G. A. V. Norman, JACC Basic Transl. Sci., 2019, 4, 845–854 CrossRef PubMed.
- N. T. Doncheva, O. Palasca, R. Yarani, T. Litman, C. Anthon, M. A. M. Groenen, P. F. Stadler, F. Pociot, L. J. Jensen and J. Gorodkin, Nucleic Acids Res., 2021, 49, 1859–1871 CrossRef CAS PubMed.
- D. E. Ingber, Nat. Rev. Genet., 2022, 23, 467–491 CrossRef CAS PubMed.
- G. Vunjak-Novakovic, K. Ronaldson-Bouchard and M. Radisic, Cell, 2021, 184, 4597–4611 CrossRef CAS PubMed.
- C. M. Leung, P. de Haan, K. Ronaldson-Bouchard, G.-A. Kim, J. Ko, H. S. Rho, Z. Chen, P. Habibovic, N. L. Jeon, S. Takayama, M. L. Shuler, G. Vunjak-Novakovic, O. Frey, E. Verpoorte and Y.-C. Toh, Nat. Rev. Methods Primers, 2022, 2, 33 CrossRef CAS.
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin and D. E. Ingber, Science, 2010, 328, 1662–1668 CrossRef CAS PubMed.
- D. J. Jung, T. H. Shin, M. Kim, C. O. Sung, S. J. Jang and G. S. Jeong, Lab Chip, 2019, 19, 2854–2865 RSC.
- A. Marino, M. Battaglini, A. Carmignani, F. Pignatelli, D. De Pasquale, O. Tricinci and G. Ciofani, APL Bioeng., 2023, 7, 036103 CrossRef CAS PubMed.
- G. Silvani, C. Basirun, H. Wu, C. Mehner, K. Poole, P. Bradbury and J. Chou, Adv. Ther., 2021, 4, 2100106 CrossRef.
- E. Moradi, S. Jalili-Firoozinezhad and M. Solati-Hashjin, Acta Biomater., 2020, 116, 67–83 CrossRef CAS PubMed.
- F. Sharifi, O. Yesil-Celiktas, A. Kazan, S. Maharjan, S. Saghazadeh, K. Firoozbakhsh, B. Firoozabadi and Y. S. Zhang, Bio-Des. Manuf., 2020, 3, 189–202 CrossRef CAS.
- A. Sontheimer-Phelps, D. B. Chou, A. Tovaglieri, T. C. Ferrante, T. Duckworth, C. Fadel, V. Frismantas, A. D. Sutherland, S. Jalili-Firoozinezhad, M. Kasendra, E. Stas, J. C. Weaver, C. A. Richmond, O. Levy, R. Prantil-Baun, D. T. Breault and D. E. Ingber, Cell. Mol. Gastroenterol. Hepatol., 2020, 9, 507–526 CrossRef PubMed.
- C. Strelez, S. Chilakala, K. Ghaffarian, R. Lau, E. Spiller, N. Ung, D. Hixon, A. Y. Yoon, R. X. Sun and H.-J. Lenz, iScience, 2021, 24, 102509 CrossRef CAS PubMed.
- Y. Chen, D. Gao, Y. Wang, S. Lin and Y. Jiang, Anal. Chim. Acta, 2018, 1036, 97–106 CrossRef CAS PubMed.
- I. Sigdel, A. Ofori-Kwafo, R. J. Heizelman, 3rd, A. Nestor-Kalinoski, B. Prabhakarpandian, A. K. Tiwari and Y. Tang, Front. Bioeng. Biotechnol., 2023, 11, 1227119 CrossRef PubMed.
- Y. Liu, R. Liu, H. Liu, T. Lyu, K. Chen, K. Jin and Y. Tian, Analyst, 2023, 148, 5822–5842 RSC.
- S. Schneider, M. Bubeck, J. Rogal, H. J. Weener, C. Rojas, M. Weiss, M. Heymann, A. D. van der Meer and P. Loskill, Lab Chip, 2021, 21, 3963–3978 RSC.
- K. Ronaldson-Bouchard, D. Teles, K. Yeager, D. N. Tavakol, Y. Zhao, A. Chramiec, S. Tagore, M. Summers, S. Stylianos, M. Tamargo, B. M. Lee, S. P. Halligan, E. H. Abaci, Z. Guo, J. Jacków, A. Pappalardo, J. Shih, R. K. Soni, S. Sonar, C. German, A. M. Christiano, A. Califano, K. K. Hirschi, C. S. Chen, A. Przekwas and G. Vunjak-Novakovic, Nat. Biomed. Eng., 2022, 6, 351–371 CrossRef CAS PubMed.
- M. B. Esch, H. Ueno, D. R. Applegate and M. L. Shuler, Lab Chip, 2016, 16, 2719–2729 RSC.
- A. Skardal, S. V. Murphy, M. Devarasetty, I. Mead, H.-W. Kang, Y.-J. Seol, Y. Shrike Zhang, S.-R. Shin, L. Zhao, J. Aleman, A. R. Hall, T. D. Shupe, A. Kleensang, M. R. Dokmeci, S. Jin Lee, J. D. Jackson, J. J. Yoo, T. Hartung, A. Khademhosseini, S. Soker, C. E. Bishop and A. Atala, Sci. Rep., 2017, 7, 8837 CrossRef PubMed.
- C. W. McAleer, C. J. Long, D. Elbrecht, T. Sasserath, L. R. Bridges, J. W. Rumsey, C. Martin, M. Schnepper, Y. Wang, F. Schuler, A. B. Roth, C. Funk, M. L. Shuler and J. J. Hickman, Sci. Transl. Med., 2019, 11, eaav1386 CrossRef CAS PubMed.
- D. E. Ingber, Adv. Sci., 2020, 7, 2002030 CrossRef CAS PubMed.
- L. Ewart, A. Apostolou, S. A. Briggs, C. V. Carman, J. T. Chaff, A. R. Heng, S. Jadalannagari, J. Janardhanan, K.-J. Jang, S. R. Joshipura, M. M. Kadam, M. Kanellias, V. J. Kujala, G. Kulkarni, C. Y. Le, C. Lucchesi, D. V. Manatakis, K. K. Maniar, M. E. Quinn, J. S. Ravan, A. C. Rizos, J. F. K. Sauld, J. D. Sliz, W. Tien-Street, D. R. Trinidad, J. Velez, M. Wendell, O. Irrechukwu, P. K. Mahalingaiah, D. E. Ingber, J. W. Scannell and D. Levner, Commun. Med., 2022, 2, 154 CrossRef PubMed.
- C. Probst, S. Schneider and P. Loskill, Curr. Opin. Biomed. Eng., 2018, 6, 33–41 CrossRef.
- L. A. Milton, M. S. Viglione, L. J. Y. Ong, G. P. Nordin and Y.-C. Toh, Lab Chip, 2023, 23, 3537–3560 RSC.
- H. Lee and D.-W. Cho, Lab Chip, 2016, 16, 2618–2625 RSC.
- A. Herland, B. M. Maoz, D. Das, M. R. Somayaji, R. Prantil-Baun, R. Novak, M. Cronce, T. Huffstater, S. S. F. Jeanty, M. Ingram, A. Chalkiadaki, D. Benson Chou, S. Marquez, A. Delahanty, S. Jalili-Firoozinezhad, Y. Milton, A. Sontheimer-Phelps, B. Swenor, O. Levy, K. K. Parker, A. Przekwas and D. E. Ingber, Nat. Biomed. Eng., 2020, 4, 421–436 CrossRef CAS PubMed.
- T. Satoh, S. Sugiura, K. Shin, R. Onuki-Nagasaki, S. Ishida, K. Kikuchi, M. Kakiki and T. Kanamori, Lab Chip, 2018, 18, 115–125 RSC.
- A. Junaid and T. Hankemeier, Bio-Protoc., 2021, 11, e4070 CAS.
- J. C. Mejías, M. R. Nelson, O. Liseth and K. Roy, Lab Chip, 2020, 20, 3601–3611 RSC.
- J. Parrish, K. S. Lim, K. Baer, G. J. Hooper and T. B. F. Woodfield, Lab Chip, 2018, 18, 2757–2775 RSC.
- E. Atefi, S. Lemmo, D. Fyffe, G. D. Luker and H. Tavana, Adv. Funct. Mater., 2014, 24, 6509–6515 CrossRef CAS PubMed.
- L. M. P. Verissimo, I. Cabral, A. M. T. D. P. V. Cabral, G. Utzeri, F. J. B. Veiga, A. J. M. Valente and A. C. F. Ribeiro, J. Chem. Thermodyn., 2021, 161, 106533 CrossRef CAS.
- K. Miura, M. Kinouchi, K. Ishida, W. Fujibuchi, T. Naitoh, H. Ogawa, T. Ando, N. Yazaki, K. Watanabe, S. Haneda, C. Shibata and I. Sasaki, Cancers, 2010, 2, 1717–1730 CrossRef CAS PubMed.
- L. Yin, J. J. Duan, X. W. Bian and S. C. Yu, Breast Cancer Res., 2020, 22, 61 CrossRef PubMed.
- M. Malet-Martino and R. Martino, Onco Targets Ther, 2002, 7, 288–323 CAS.
- S. Fujii, S. Kitano, K. Ikenaka and T. Shirasaka, Jpn. J. Cancer Res., 1979, 70, 209–214 CAS.
- Cancer Discovery, 2023, 13, 805 Search PubMed.
- M. S. Viglione, A. Saxton, D. Downs, A. T. Woolley, K. A. Christensen, P. M. Van Ry and G. P. Nordin, Lab Chip, 2024, 24, 2202–2207 RSC.
- J. H. Sung, C. Kam and M. L. Shuler, Lab Chip, 2010, 10, 446–455 RSC.
- C. Oleaga, C. Bernabini, A. S. Smith, B. Srinivasan, M. Jackson, W. McLamb, V. Platt, R. Bridges, Y. Cai, N. Santhanam, B. Berry, S. Najjar, N. Akanda, X. Guo, C. Martin, G. Ekman, M. B. Esch, J. Langer, G. Ouedraogo, J. Cotovio, L. Breton, M. L. Shuler and J. J. Hickman, Sci. Rep., 2016, 6, 20030 CrossRef CAS PubMed.
- P. G. Miller and M. L. Shuler, Biotechnol. Bioeng., 2016, 113, 2213–2227 CrossRef CAS PubMed.
- J. J. Tronolone, J. Lam, A. Agrawal and K. Sung, Biomed. Microdevices, 2021, 23, 25 CrossRef CAS PubMed.
- S. L. Ham, P. S. Thakuri, M. Plaster, J. Li, K. E. Luker, G. D. Luker and H. Tavana, Onco Targets Ther, 2018, 9, 249–267 Search PubMed.
- S. Singh, A. Lamichhane, P. Rafsanjani Nejad, J. Heiss, H. Baumann, R. Gudneppanavar, N. D. Leipzig, M. Konopka, G. D. Luker and H. Tavana, Mol. Cancer Res., 2022, 20, 1166–1177 CrossRef CAS PubMed.
- J. E. May, C. Donaldson, L. Gynn and H. R. Morse, Mutagenesis, 2018, 33, 241–251 CrossRef CAS PubMed.
- A. Mendelson and P. S. Frenette, Nat. Med., 2014, 20, 833–846 CrossRef CAS PubMed.
- P. L. Bedard, J. Tabernero, F. Janku, Z. A. Wainberg, L. Paz-Ares, J. Vansteenkiste, E. Van Cutsem, J. Pérez-García, A. Stathis, C. D. Britten, N. Le, K. Carter, D. Demanse, D. Csonka, M. Peters, A. Zubel, H. Nauwelaerts and C. Sessa, Clin. Cancer Res., 2015, 21, 730–738 CrossRef CAS PubMed.
- Z. A. Wainberg, M. Alsina, H. P. Soares, I. Braña, C. D. Britten, G. Del Conte, P. Ezeh, B. Houk, K. A. Kern, S. Leong, N. Pathan, K. J. Pierce, L. L. Siu, J. Vermette and J. Tabernero, Target. Oncol., 2017, 12, 775–785 CrossRef PubMed.
- T. Shimizu, A. W. Tolcher, K. P. Papadopoulos, M. Beeram, D. W. Rasco, L. S. Smith, S. Gunn, L. Smetzer, T. A. Mays, B. Kaiser, M. J. Wick, C. Alvarez, A. Cavazos, G. L. Mangold and A. Patnaik, Clin. Cancer Res., 2012, 18, 2316–2325 CrossRef CAS PubMed.
- K. S. Saini, S. Loi, E. de Azambuja, O. Metzger-Filho, M. L. Saini, M. Ignatiadis, J. E. Dancey and M. J. Piccart-Gebhart, Cancer Treat. Rev., 2013, 39, 935–946 CrossRef CAS PubMed.
- B. A. Wagner, S. Venkataraman and G. R. Buettner, Free Radical Biol. Med., 2011, 51, 700–712 CrossRef CAS PubMed.
- S. Kidambi, R. S. Yarmush, E. Novik, P. Chao, M. L. Yarmush and Y. Nahmias, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15714–15719 CrossRef CAS PubMed.
- K. P. Hoeflich, C. O'Brien, Z. Boyd, G. Cavet, S. Guerrero, K. Jung, T. Januario, H. Savage, E. Punnoose, T. Truong, W. Zhou, L. Berry, L. Murray, L. Amler, M. Belvin, L. S. Friedman and M. R. Lackner, Clin. Cancer Res., 2009, 15, 4649–4664 CrossRef CAS PubMed.
- H. Kimura, Y. Sakai and T. Fujii, Drug Metab. Pharmacokinet., 2018, 33, 43–48 CrossRef CAS PubMed.
- M. B. Esch, T. L. King and M. L. Shuler, Annu. Rev. Biomed. Eng., 2011, 13, 55–72 CrossRef CAS PubMed.
- M. G. Sturgill, D. A. August and D. E. Brenner, Cancer Invest., 2000, 18, 197–205 CrossRef CAS PubMed.
- M.-A. Le Bot, D. Glaise, D. Kernaleguen, D. Ratanasavanh, D. Carlhant, C. Riche and A. Guillouzo, Pharmacol. Res., 1991, 24, 243–252 CrossRef CAS PubMed.
- A. D. Heibein, B. Guo, J. A. Sprowl, D. A. MacLean and A. M. Parissenti, BMC Cancer, 2012, 12, 381 CrossRef CAS PubMed.
- S. K. Golombek, J.-N. May, B. Theek, L. Appold, N. Drude, F. Kiessling and T. Lammers, Adv. Drug Delivery Rev., 2018, 130, 17–38 CrossRef CAS PubMed.
- P. M. Glassman, J. W. Myerson, L. T. Ferguson, R. Y. Kiseleva, V. V. Shuvaev, J. S. Brenner and V. R. Muzykantov, Adv. Drug Delivery Rev., 2020, 157, 96–117 CrossRef CAS PubMed.
- S. P. Cavnar, A. D. Rickelmann, K. F. Meguiar, A. Xiao, J. Dosch, B. M. Leung, S. Cai Lesher-Perez, S. Chitta, K. E. Luker, S. Takayama and G. D. Luker, Neoplasia, 2015, 17, 625–633 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc00404c |
| This journal is © The Royal Society of Chemistry 2024 |