
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A dual colorimetric-electrochemical microfluidic paper-based analytical device for point-of-care testing of ischemic strokes†
Silvia
Dortez‡
 a,
Marta
Pacheco‡
b,
Teresa
Gasull
c,
Agustín G.
Crevillen
*d and
Alberto
Escarpa
*ae
a,
Marta
Pacheco‡
b,
Teresa
Gasull
c,
Agustín G.
Crevillen
*d and
Alberto
Escarpa
*ae
aDepartment of Analytical Chemistry, Physical Chemistry and Chemical Engineering, University of Alcala, 28802, Alcala de Henares, Madrid, Spain. E-mail: alberto.escarpa@uah.es
bDepartment of Chemistry in Pharmaceutical Sciences, Analytical Chemistry, Faculty of Pharmacy, Complutense University of Madrid, 28040 Madrid, Spain
cCellular and Molecular Neurobiology Research Group, Department of Neurosciences, Germans Trias i Pujol Research Institute (IGTP), 08916, Badalona, Barcelona, Spain
dDepartment of Analytical Sciences, Faculty of Sciences, Universidad Nacional de Educación a Distancia (UNED), 28040, Madrid, Spain. E-mail: agustingcrevillen@ccia.uned.es
eChemical Research Institute “Andrés M. Del Río” (IQAR), University of Alcala, 28802, Alcala de Henares, Madrid, Spain
First published on 30th July 2024
Abstract
A novel microfluidic paper-based analytical device with dual colorimetric and electrochemical detection (dual μPAD) was developed for the assessment of transferrin saturation (TSAT) in samples from ischemic stroke patients. TSAT was calculated from the ratio between transferrin-bound iron, which was colorimetrically measured, and the total iron-binding capacity, which was electrochemically measured. To this end, a μPAD was smartly designed, which integrated both colorimetric and electrochemical detection reservoirs, communicating via a microchannel acting as a chemical reactor, and with preloading/storing capabilities (reagent-free device). This approach allowed the dual and simultaneous determination of both parameters, providing an improvement in the reliability of the results due to an independent signal principle and processing. The μPADs were validated by analyzing a certified reference material, showing excellent accuracy (Er ≤ 5%) and precision (RSD ≤ 2%). Then they were applied to the analysis of diagnosed serum samples from ischemic stroke patients. The results were compared to those provided by a free-interference method (urea-PAGE). Impressively, both methods exhibited a good correlation (r = 0.96, p < 0.05) and no significant differences were found between them (slope 1.0 ± 0.1 and the intercept 1 ± 4, p < 0.05), demonstrating the excellent accuracy of our approach during the analysis of complex samples from ischemic stroke patients, using just 90 μL of clinical samples and taking less than 90 min in comparison with the 18 hours required by the urea-PAGE approach. The developed fully integrated colorimetric-electrochemical μPAD is a promising ready to use reagent-free device for the point-of-care testing of TSAT, which can be used to assist physicians in the fast diagnosis and prognosis of ischemic strokes, where the decision-time is crucial for the patient's survival.
Introduction
Ischemic stroke is one of the leading causes of death and serious disability. According to the World Health Organization (WHO), annually, 15 million people worldwide suffer a stroke.1 Ischemic strokes are caused by vascular occlusion in the brain producing an inadequate blood supply to the brain tissues, causing loss of their functionality and ultimately a cerebral infarction. This ischemia generates neuronal death with the loss of electrical function and progresses through the generation of reactive oxygen species (ROS), destruction of cell membranes, and cell lysis. The progression of cerebral tissue towards an infarction depends on the magnitude and duration of the drop in cerebral blood flow and generally causes neuronal dysfunction leading to ischemic symptoms, typically motor function deficits such as loss of strength, sensitivity or tingling in parts of the body, difficulty speaking or altered mental status in patients.2The clinical diagnosis of patients who have suffered an ischemic stroke is a medical emergency that requires immediate treatment within a limited period. It is usually diagnosed rapidly by non-contrast computed tomography (CT) due to its wide availability and relatively short imaging time,3 magnetic resonance imaging (MRI) although it has generally limited availability, or by careful clinical assessment (clinical history and neurological examination).4 The problem is that the frequency of stroke mimics admissions in emergencies is usually very high. Moreover, tests such as simple CT are usually not informative, MRI gives many false negatives, and neurological examinations are sometimes impossible due to the consequences of brain damage induced by ischemia.5–7 For all these reasons, a differential diagnosis of ischemia versus other etiologies is required. In this context, biomarkers are valuable tools to support the clinical diagnosis of stroke, not only to guide treatment and prognosis, but also to identify patients at risk of the disease.
Most of the biomarkers used for the diagnosis and prognosis of ischemic stroke are proteins related to the pathophysiology of the disease, such as damage to brain tissue, inflammation, endothelium, and coagulation/thrombosis.8–11 The role of tissue iron stores appears crucial, as clinical studies have found evidence of ferroptosis-related brain damage in patients with acute ischemic stroke.12–14 Moreover, it was demonstrated that higher iron status was associated with an increased risk of stroke15 and with a detrimental effect on functional outcomes after ischemic stroke.16
Iron is an essential element for the growth and survival of almost all living organisms. It is involved in many biological processes due to its inherent redox properties between ferrous, i.e. Fe2+, and ferric, i.e. Fe3+, states.17 The iron form concentration must be tightly regulated by all organisms within the body to avoid the drastic consequences of iron deficiency or excess.18–20 In blood, most of the iron is bound to transferrin (Tf) (one Tf molecule can bind two Fe3+ ions), which transports iron in a soluble and non-toxic form throughout the body.21,22 Tf plays a vital role in iron metabolism in the body. Its main function is the transport of iron from sites of absorption, utilization, or storage to various tissues. In addition, it maintains Fe3+ in a soluble form under physiological conditions, which protects against the toxic side effects of ROS.21,23–26 Tf concentration in human serum is approximately 2–3 g L−1. Tf levels may also be related to body iron stores, since in the case of iron deficiency (anemia) an increase in the Tf level is observed. By contrast, a decrease in the Tf level is observed with iron overload.25–27
In this sense, transferrin saturation (TSAT) is considered an important biomarker of general body iron status since it can be used to diagnose iron deficiency and iron overload in combination with other serum biomarkers such as ferritin.22,28 Moreover, a high level of TSAT is related to an increased risk of stroke15 and brain damage induced by ischemia.29–34 In the latter scenario, a high level of TSAT (more than 30%) in patients during the critical first hours post-ischemic stroke onset means a worse ischemic stroke prognosis, as has been demonstrated in basic and clinical research.29
TSAT (%) is defined as the ratio between the amount of serum iron and the total iron-binding capacity (TIBC, the maximum amount of iron that can be captured by Tf):22
 | (1) |
| TIBC = [Tf] × conversion factor | (2) |
Therefore, the development of fast and reliable methods for determining TSAT is much needed, with special emphasis on simplicity, low-cost, and portability. These features would allow clinicians to use them for point-of-care testing (POCT), providing fast information and assisting in urgent decision-making such as the diagnosis/prognosis of ischemic stroke. In this context, paper-based analytical devices (PADs) offer several advantages such as portability, operational simplicity, miniaturization, biocompatibility, low-cost, ease of use, and availability, which make them perfect platforms for POCT.42,43 In addition, microfluidic paper-based analytical devices (μPADs) have other advantages inherent to microfluidic technology such as spatial control of fluids by using microchannels and chambers, and multiplexing by creating multiple detection reservoirs.44,45 Among detection modes employed in PADs, colorimetric and electrochemical detection methods are the most widely used. The colorimetric approach offers sensitivity, adaptability, and cost effectiveness, and it can be implemented on a smartphone, so it is attracting enormous attention from the scientific community for its simple quantitative analysis, portability, and connectivity.46,47 The electrochemical approach, commonly known as electrochemical paper-based analytical devices (ePADs), satisfies many of the requirements for in situ measurements due to portability and low power demands. These electrochemical sensors offer simple platforms with high sensitivity (greater than colorimetric detection), selectivity, accuracy, a wide linear range, low cost, inexpensive instrumentation, high compatibility with smart devices, and low sample volume requirements. Furthermore, in contrast to colorimetric assays, electrochemical methods offer a unique advantage: they are not affected by the color/turbidity of the sample matrix, which is often the cause of interference.48–50 Therefore, both types of detection, electrochemical and colorimetric, can complement each other by allowing the conversion of two independent signals, improving the reliability, sensitivity, and portability of future POCT tools for the analysis of relevant analytes.
In previous work by the authors, a PAD was developed to evaluate TSAT in serum samples, based on the colorimetric determination of Tf-bound iron using a smartphone-based color reader.33 The approach showed high accuracy (Er < 4%) and a good correlation (r = 0.93) with a reference method (urea-PAGE) for the analysis of serum samples from ischemic stroke patients. However, TIBC was measured by adding an excess amount of ferric iron to the serum sample (direct method), implying a multistep approach with intensive analyst intervention.
In this work, a strategy to measure the Tf-bound iron that has already been developed33 is combined with the electrochemical determination of Tf on a μPAD with colorimetric-electrochemical detection (dual μPAD) using anti-transferrin immunomagnetic beads (anti-Tf-MBs). This dual approach benefits from two independent signal conversions, simplifies the process, improves the sensitivity and selectivity, and stores several reagents on the μPAD (reagent-free device). The design of the μPAD was carried out using low-cost technologies such as tracing with hydrophobic ink on paper, allowing the desired flow of liquids, modification of electrodes through stencil-printing, and impregnation of chemicals for a reagent-free device. Several factors such as reagent volume, electrolyte, carbon ink, voltammetry parameters, among others were optimized to achieve the simultaneous quantification of Tf and Tf-bound iron, and therefore TSAT, with minimal analyst intervention. Finally, this method was validated using a certified reference material (human serum) and successfully compared with a well-established interference-free method (urea-PAGE) toward the analysis of complex samples from ischemic stroke patients.
Material and methods
Reagents, materials, and samples
Iron(III) chloride hexahydrate, hydroxylamine hydrochloride, ferrozine (3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine-p,p′-sulfonic acid monosodium salt hydrate), human transferrin (T3309), human serum, sodium hydroxide, acetic acid glacial, sodium acetate anhydrous, potassium hexacyanoferrate(II) trihydrate, and silver/silver chloride (60/40) paste were purchased from Merck (Darmstadt, Germany). Ortho-phosphoric acid, potassium chloride, and disodium hydrogen phosphate were purchased from Scharlau (Barcelona, Spain). Sodium dihydrogen phosphate and potassium hexacyanoferrate(III) were purchased from PanReac (Barcelona, Spain). Boric acid was acquired from Fluka (Darmstadt, Germany). Carbon paste (BG04) was purchased from SunChemical (New Jersey, USA). Carbon ink (LOCTITE EDAG PF407A) was acquired from Henkel (Düsseldorf, Germany). Disposable screen-printed carbon electrodes (SPCE DRP-110) were purchased from Metrohm (Madrid, Spain). Pierce™ Protein G Magnetic Beads and sodium chloride were acquired from Thermo Fisher Scientific (USA). Human serum (certified reference material, Spintrol H normal 1002121) was purchased from Spinreact (Girona, Spain). Anti-transferrin antibody (ab66952) was purchased from Abcam (Cambridge, UK).A stock solution of 2 M acetate buffer pH 4.8 was prepared by dissolving appropriate amounts of acetic acid and sodium acetate in Milli-Q water. A stock solution of 0.2 M Britton–Robinson (BR) buffer pH 3.0 was prepared by dissolving appropriate amounts of boric acid 0.04 M, ortho-phosphoric acid 0.04 M, and acetic acid 0.04 M in Milli-Q water adjusting the pH with sodium hydroxide 0.2 M. A stock solution of 0.1 M phosphate buffer saline (PBS) pH 7.4 was prepared by dissolving appropriate amounts of sodium dihydrogen phosphate, disodium hydrogen phosphate, sodium chloride, and potassium chloride in Milli-Q water. The solution of hydroxylamine was prepared by dissolving appropriate amounts in 2 M acetate buffer pH 4.8 to a concentration of 100 mg mL−1. The solution of ferrozine was prepared by dissolving appropriate amounts in 2 M acetate buffer pH 4.8 to a concentration of 49 mg mL−1. Iron(III) and Tf standard solutions were daily prepared by dissolving appropriate amounts in 0.2 M BR buffer solution of pH 3.0. All reagents and solvents were of analytical grade. All solutions were prepared in Milli-Q water (Merck Millipore, Darmstadt, Germany).
Whatman Chromatography Paper 1 CHR was purchased from Merck (Darmstadt, Germany). Invitrogen™ DynaMag™-2 Magnet was purchased from Thermo Fisher Scientific (USA). The waterproof marker pen Lumocolor® permanent CD/DVD/BD 310 was purchased from Staedtler (Nuremberg, Germany). Tesa 4024 clear packing tape, adhesive vinyl sheets from Tavolozza, and Samtian F40II Lightbox with LED were purchased from Amazon (Spain).
Pseudonymized serum samples of the multicenter, randomized, double-blind, placebo-controlled TANDEM-1 (ref. 29) (thrombolysis and deferoxamine in middle cerebral artery occlusion) study were used to evaluate TSAT by dual μPAD and compared with the urea-PAGE method used by Teresa Gasull's group.30 The serum samples used were specifically those collected at the Hospital Universitari Germans Trias i Pujol (HUGTP) in untreated patients. All samples were stored at −20 °C before use. TANDEM-1 study was approved by the Spanish Drug Agency (eudraCT 2007-0006731-31) and by local Ethics Committees, including the HUGTP Ethics Committee, and was registered on https://clinicalTrials.gov as NCT00777140.
Instrumentation
AutoCAD 2018 (Autodesk, student version) was used to design all the components of the μPAD. μPAD design was drawn on a sheet of filter paper (Whatman 1 CHR) using a desktop cutting plotter (Silhouette Cameo 3, Silhouette). The stencils of the Ag/AgCl connections and the reference electrode and of the carbon working and counter electrodes were cut on adhesive vinyl using a desktop cutting plotter (Silhouette Cameo 3, Silhouette). Photos were taken using a Realme X2 mobile and a lightbox (dimensions 44 × 44 × 44 cm, including 84 brightness LED light beads inside). Subsequent image analysis was performed by ImageJ software. Potentiostat Autolab PGSTAT101 (Methrom-Autolab, the Netherlands) was used for all the electrochemical measurements. Currents were recorded and displayed on a laptop by using the software Nova 2.1.5. An incubator (Eppendorf ThermoMixer C, Hamburg, Germany) was used for the immunopurification assays.Procedures
Fig. S1 (see the ESI†) shows the schematic procedure of the μPAD fabrication, which consists of three steps:48
(i) The dual μPAD patterns were drawn using AutoCAD software (Fig. S1A,† top). This pattern was transferred to a piece of filter paper using a cutter plot, in which the blade was replaced by a waterproof marker pen. Here, the waterproof marker pen ink penetrates the paper to form hydrophobic walls on the filter paper. In addition, it was painted with a thicker waterproof marker pen to mark the part that will delimit the electrochemical cell with its connectors, defining the testing area and confining the solution in the delimited area, thus avoiding its diffusion towards the electrical connections, and affecting the readout (Fig. S1B,† top).
(ii) Next, in the same way as in (i), the three-electrode pattern of Ag/AgCl and carbon was transferred to an adhesive vinyl sheet using a blade as an electrode cutter (Fig. S1A,† middle and bottom, respectively). So these Ag/AgCl and carbon adhesive vinyl patterns were stuck on the piece of filter paper where the μPADs were drawn (Fig. S1B,† middle and bottom, respectively). After, the three-electrode system was manually stencil-printed onto the hydrophilic semicircular electrochemical detection reservoir (Fig. S1C†) using a squeegee and two adhesive vinyl masks (one for Ag/AgCl pattern and the other for carbon pattern). Firstly, the adhesive vinyl mask with the Ag/AgCl pattern was pasted on the filter paper and a layer of Ag/AgCl ink was applied, with the help of a flat squeegee, to create the reference electrode and the electrical connections. The devices were allowed to cure at 90 °C for 30 minutes in an oven (Conterm J.P. Selecta, Barcelona, Spain) and after that time, the adhesive vinyl was removed with the help of a hot air dryer (Braun Cosmo 1000, Basingstoke, United Kingdom). Secondly, the adhesive vinyl mask with the carbon pattern was pasted on the filter paper and a layer of carbon ink was applied, with the help of a flat squeegee, to create the working and counter electrodes. The devices were allowed to cure at 120 °C for 30 minutes in an oven and after that time, the adhesive vinyl was removed with the help of a hot air dryer. Thermal curing was necessary to make the printed ink stable for the electrochemical measurements. Finally, dual μPADs were individually cut and the backside of the printing surface was covered with clear packing tape (without covering electrical connections) to prevent the solution from leaking out underneath the dual μPAD.
(iii) To produce a reagent-free μPAD, the colorimetric detection reservoir was impregnated with a hydroxylamine solution, as a reducing agent, and a ferrozine solution, as a colorimetric agent (Fig. S1D,† left). Hydroxylamine and ferrozine are commonly used for iron assays as a reducing agent and colorimetric agent, respectively.52–56 Three depositions of 10 μL (3 × 10 μL) of 100 mg mL−1 hydroxylamine in 2 M acetate buffer pH 4.8 and 0.5 μL of 49 mg mL−1 ferrozine in 2 M acetate buffer pH 4.8 (the optimal conditions of this chromogenic reagent were chosen based on our previous work57) were added to the colorimetric detection reservoir of μPAD, and it was dried completely in an oven at 60 °C for 2 min, after each deposition. The depositions were carried out carefully so as not to impregnate the electrochemical reservoir so that the reagents do not interfere with the electrochemical measurements. Finally, a magnet was pasted on the back of the μPAD at the height of the working electrode (Fig. S1D,† right). Then, the dual μPAD was ready to use.
The dual μPAD consists of a hydrophilic microfluidic channel (5 mm length × 5 mm width) that will connect an electrochemical semicircular detection reservoir (electrochemical cell of 13 mm diameter) with a colorimetric circular detection reservoir (10 mm diameter). The electrochemical cell consists of three 19 mm × 1 mm (length × width) rectangles separated by 1.5 mm that were used for electrical connections, the reference electrode that was an arc of 3 mm × 1 mm (length × width), the working electrode that was a circle of 4 mm diameter, and the counter electrode that was an arc of 12 mm × 1 mm (length × width). The protective rectangle for connections was 5 mm × 13 mm (length × width). The entire design of the dual μPAD is reported in Fig. S2.†
In a single sheet, 30 devices were drawn and stencil-printed. Then this sheet was cut to individually obtain the corresponding dual μPADs.
 | (3) |
Results and discussion
Analytical design and development of the dual μPAD for TSAT assessment
As discussed in the introduction section, the assessment of the TSAT biomarker is essential in the clinical field, where both clinical parameters (Tf-bound iron and TIBC) are detected in independent and non-simplified manners. Here we proposed a μPAD simplifying and integrating the dual/simultaneous determination of both parameters.Fig. 1 shows the μPAD designed to integrate this dual determination approach, calculating the ratio between Tf-bound iron (colorimetric) and TIBC through the Tf concentration (electrochemical) measurements (eqn (3)) to obtain the TSAT clinical parameter in a fast and reliable manner. Indeed, this strategy could provide an improvement in the reliability of the analysis benefiting from the independent signal principle and processing. To this end, the sample (with Tf isolated and preconcentrated from the sample using anti-Tf-MBs to ensure that only Tf-bound iron was measured) was intentionally deposited in the electrochemical reservoir to immediately perform the TIBC assessment by exploiting the inherent electroactivity of Tf, allowing the sample to flow through the cellulosic reactor-microfluid channel (which connects both the detection modes with additionally stored hydroxylamine), where the reduction reaction from Fe3+ to Fe2+ in channel occurs and finally the sample reaches the colorimetric detection reservoir (with stored ferrozine), where the formation of the Fe2+–ferrozine complex occurs (adapted from ref. 57) (see Fig. 1). Subsequently, this μPAD was ready to be used and become a useful reagent-free device very suitable for point-of-care (POC) applications.
 | ||
| Fig. 1 Dual strategy for the determination of TSAT using anti-Tf-MBs, the electrochemical sensor, and the smartphone–μPAD sensor. | ||
Analytical performance of the dual μPAD
To obtain the best analytical performance of the dual μPAD and a reagent-free device, different electrochemical and colorimetric parameters were optimized (see ESI† for full optimization details). Table 1 summarizes all the optimizations performed and the best result obtained in each detection principle optimization.| Electrochemical parameters | ||
|---|---|---|
| Parameter | Studied interval | Optimal value |
| Commercial carbon ink | SunChemical | SunChemical (Fig. S3A and B†) |
| Henkel | ||
| Number of Ag/AgCl and carbon ink layers | 1 layer of Ag/AgCl + 1 layer of carbon ink | 1 layer of Ag/AgCl + 1 layer of carbon ink (Fig. S3C†) |
| 1 layer of Ag/AgCl + 2 layers of carbon ink | ||
| 2 layers of Ag/AgCl + 1 layer of carbon ink | ||
| 2 layers of Ag/AgCl + 2 layers of carbon ink | ||
| Volume of electrochemical cell | 20–50 μL | 30 μL (Fig. S4A†) |
| Electrochemical technique | SWV | SWV (Fig. S4B†) |
| DPV | ||
| Frequency of SWV | 50–400 Hz | 200 Hz (Fig. S4C†) |
| Acidic medium | 2 M acetate buffer pH 4.8 | 0.2 M BR buffer pH 3.0 (Fig. S5A†) |
| 0.2 M BR buffer pH 3.0 | ||
| Colorimetric parameters | ||
|---|---|---|
| Parameter | Studied interval | Optimal value |
| Volume of hydroxylamine | 1–30 μL | 3 × 10 μL (30 μL) (Fig. S6†) |
After optimizing the conditions, the analytical performance of the dual μPAD was evaluated. Linear calibration plots were constructed independently for the analysis of Tf and Fe3+ standard solutions (in 0.2 M BR buffer pH 3.0), by electrochemical and colorimetric detection, respectively (see Fig. 2). Each point on the calibration plots corresponds to the use of three independent dual μPADs.
 | ||
| Fig. 2 (A) Calibration plot of current intensity versus the concentration of Tf. Inset: Voltammograms corresponding to 30 μL of 0.2 M BR buffer pH 3.0 (black line), 0.25 g L−1 Tf (red line), 1 g L−1 Tf (blue line), 2.5 g L−1 Tf (green line), 5 g L−1 Tf (purple line), 7.5 g L−1 (orange line), and 10 g L−1 Tf (cyan line). SWV parameters: start potential +0.3 V, end potential +1.4 V, frequency 200 Hz, amplitude 0.05 V, and step potential 0.005 V (the background signal was linearized) (n = 3). (B) Calibration plot of color intensity versus the concentration of Fe3+. Inset: Image of dual μPADs used for calibration. Experimental conditions: 30 μL of Fe3+ standard solution from 3 μg mL−1 to 70 μg mL−1 in 0.2 M BR buffer pH 3.0. 2 min reaction time (n = 3). | ||
For electrochemical detection, the calibration curves were built by SWV using the signal generated by the accessible electroactive amino acids present in Tf such as cysteine, tryptophan, and tyrosine60–62 (at +0.8 V, without any interference of Fe3+, see Fig. S5B†) from 0.25 g L−1 to 10 g L−1 (Fig. 2A), obtaining an excellent linear correlation coefficient (r = 0.997). The calibration slope was 1.27 ± 0.05 μA L g−1 and the intercept was 1.1 ± 0.3 μA. The reproducibility of dual μPADs in terms of calibration slopes was very good (RSD = 4%). The limit of detection (LOD) was 0.061 g L−1 (calculated as 3 S/N criteria, using the standard deviation of the point of lowest concentration). In this sense, the LOD was satisfactory for Tf detection in human serum samples, because the normal range for Tf in human blood is between 2 and 3 g L−1.63,64
For colorimetric detection, the color tone gradually varied from colorless to purple with increasing iron concentration, as shown in the inset of Fig. 2B. The calibration curve for iron was linear in the range of 3–70 μg mL−1, showing an excellent correlation coefficient (r = 0.993). The calibration slope was 0.64 ± 0.03 px mL μg−1 and the intercept was 2 ± 1 px. The reproducibility of dual μPADs in terms of calibration slopes was also good (RSD = 5%). However, LOD was 2 μg mL−1 (calculated as 3 S/N criteria, using the standard deviation of the point of lowest concentration), which is slightly higher than the levels of iron present in human serum (normal range from 0.7 to 1.7 μg mL−1).65 But this was not a problem when the serum samples were analyzed because the Tf immunopurification step not only allowed the removal of the potential interferences but also preconcentrated the sample 3-fold (90 μL serum is concentrated in a 30 μL solution), thus allowing its quantification and improving the accuracy of TSAT assessment (see section Immunopurification step for Tf isolation and Tf-bound iron release in serum samples). So, the LOD of the overall method was reduced from 2 to 0.67 μg mL−1.
When analyzing serum samples, an immunopurification process is necessary to isolate Tf from the rest of the components of the sample. However, antibodies are glycoproteins and can be electroactive, so it is necessary to perform electrochemical measurements by SWV to several controls (MBs in 0.2 M BR buffer pH 3.0, 7.5 μg mL−1 anti-Tf antibody in 0.2 M BR buffer pH 3.0, and anti-Tf-MBs in 0.2 M BR buffer pH 3.0) and a blank (0.2 M BR buffer pH 3.0). As can be seen in Fig. S7,† the signal obtained for the anti-Tf antibody (Fig. S7,† purple line) is negligible compared to the peak at +0.8 V of Tf in the commercial serum sample after the immunopurification step without a preconcentration step (Fig. S7,† green line). In addition, the antibody concentration used was in excess but after incubation with the MBs, the excess was eliminated by washing. As can be seen in the controls using MBs and anti-Tf-MBs (Fig. S7,† red line and blue line, respectively) there was no anodic peak for the controls analyzed. Therefore, it is possible to use these anti-Tf and MBs in the immunopurification process. It was necessary to place a magnet on the back of the dual μPAD on the working electrode since MBs are brown and must be retained on the working electrode so that they do not mask the colorimetric signal obtained in the Fe3+ assay.
Then the dual μPAD-based approach was validated for both parameters (Tf-bound iron and Tf) by the analysis of a certified reference material (human serum) using the external calibration method (n = 3). The results are collected in Table 2.
| Parameter | Certified value | Dual μPAD value | E r (%) | |
|---|---|---|---|---|
![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) ± s ± s |
RSD (%) | |||
| Serum iron | 1.13 μg mL−1 | 1.08 ± 0.01 μg mL−1 | 1 | 4 |
| Tf | 1.84 g L−1 | 1.75 ± 0.04 g L−1 | 2 | 5 |
Based on these results, our approach showed excellent accuracy and precision. Furthermore, both parameters were measured on the same dual μPAD, demonstrating its multiplexing capacity and highlighting its robustness combined with the low-cost manufacturing process of the dual μPADs.
The stability of the reagent-free μPAD was also evaluated. Several dual μPADs were prepared and stored in plastic zipper bags at room temperature in the dark, to limit the oxidation of silver and degradation of the preloaded reagents on the dual μPAD due to light exposure. The stability of the electrochemical detector was evaluated over time by measuring the anodic peak intensity of a 5 mM ferro/ferri system solution by cyclic voltammetry (with three different dual μPADs). Impressively, after one year, there were no significant differences in the electrochemical signal concerning the anodic peak intensity obtained on the first day (t-test, α = 0.05, two sides), demonstrating its long stability over time (see Fig. S8†). However, the colorimetric assay was a limiting factor since it was stable for 40 days,33 being also a valuable one. This limitation is not a problem per se, for commercial purposes, because both reagents can be added to the μPAD when performing the colorimetric assay.
Assessment of TSAT in serum samples from ischemic stroke patients by using the dual μPAD
Once the good analytical performance of the dual μPAD for the simultaneous determination of Tf and Fe3+ was demonstrated, under optimal conditions, the reagent-free dual μPAD was evaluated for TSAT determination in 9 pseudonymized serum samples from ischemic stroke patients of the TANDEM-1. The results obtained for the TSAT values are shown in Fig. 3A where they were also compared with those obtained by the urea-PAGE method30 (see also Table S1†). Impressively, both methods, exhibited a good correlation coefficient (r = 0.96) and no significant differences were found since the values obtained for the slope (1.0 ± 0.1) and the intercept (1 ± 4), included 1 and 0, respectively (p < 0.05); consequently, revealing an excellent accuracy since the urea-PAGE method is a free-interference method (see a visual example in Fig. 3B).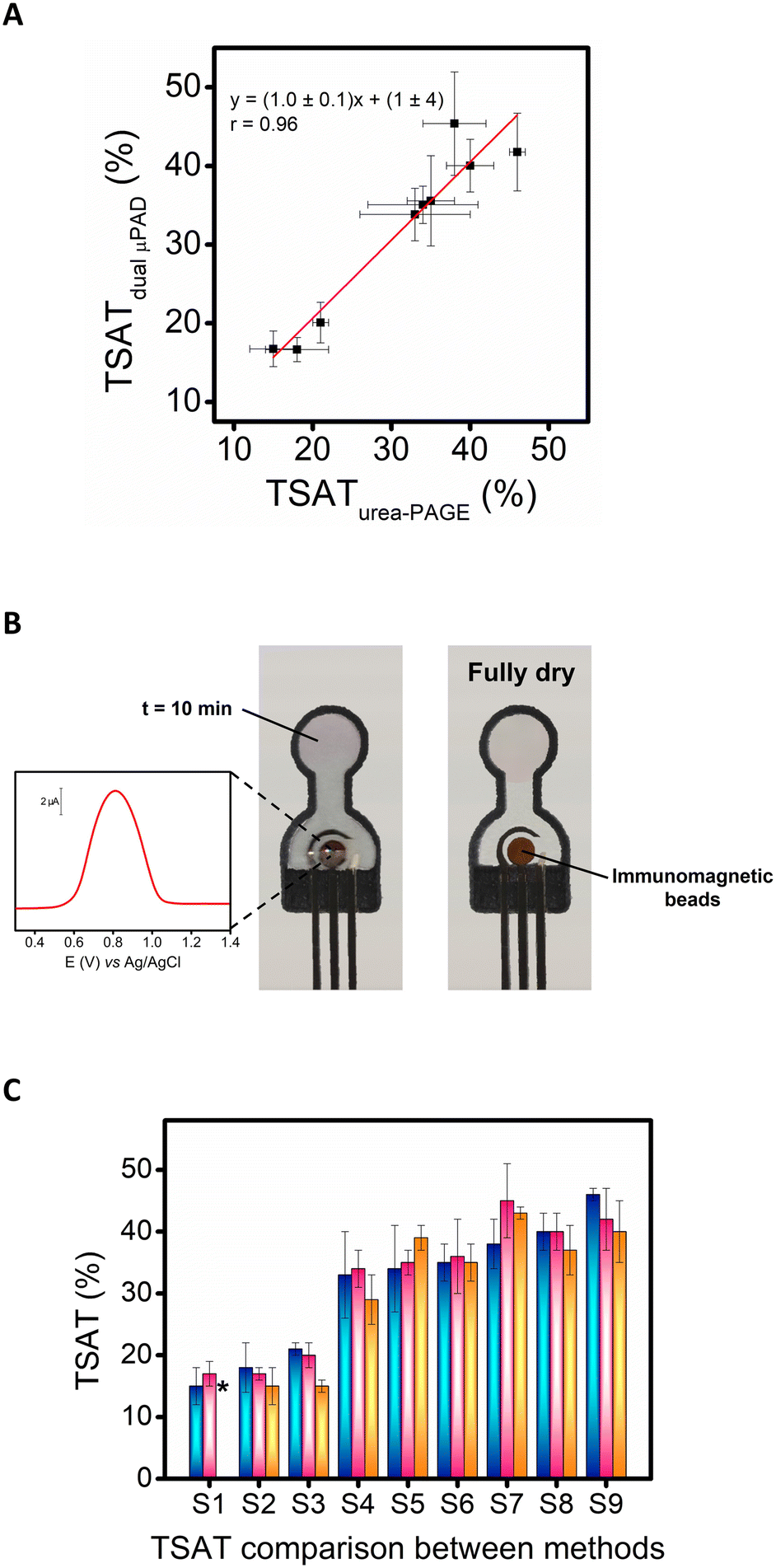 | ||
| Fig. 3 (A) Correlation between TSAT values obtained by dual μPAD and by urea-PAGE30 (n = 3). (B) Image of a dual μPAD used for sample S5 for the TSAT assessment using its two parameters: SWV for Tf (electrochemical detection) and Fe2+–ferrozine complex formation at 10 min (colorimetric detection) (left), and image of the same dual μPAD when it dries completely and the MBs are observed fully retained on the working electrode (brown circle) (right). Experimental condition: see section Procedures (the background signal was linearized) (n = 3). (C) Comparison in TSAT analysis between the urea-PAGE method30 (blue bar), our approach based on a dual μPAD (pink bar), and the previously reported method based on a colorimetric PAD (orange bar).33 * Sample S1 was not available in the colorimetric PAD.33 | ||
In addition, these results were compared with those obtained by our previously reported method based on a colorimetric PAD for the detection of TSAT33 (Fig. 3C), showing great similarities in the analysis of the same samples. Interestingly, the values provided by the dual μPAD are closer to the reference values (urea-PAGE) than those provided by our previous colorimetric μPAD for most of the cases. This means that the dual approach improved the method accuracy thanks to the electrochemical detection, which offers higher accuracy than colorimetric detection for the measurement of TIBC. These results demonstrated the suitability of the developed dual μPAD as a reliable microfluidic sensor for the assessment of TSAT using an easy procedure and a cost-effective device. Our approach turned out to be highly competitive compared to current methods since they use expensive benchtop equipment and are located in clinical laboratories or medical centers,29,36 and the dual μPAD shown here is cheap, portable, single-use (avoiding cross-contamination), easy-to-use, sensitive, specific, reagent-free, and deliverable to end-users, as well as the fact that it requires a very low clinical sample volume (90 μL). All these features make our dual detection device a potential candidate for a future POCT for ischemic stroke prognosis. On the other hand, the urea-PAGE method has the advantage of separating and detecting the different forms of Tf-bound iron (apo-Tf, monoferric-Tf, and diferric-Tf) but it is very slow (more than 18 h). Our approach takes less than 90 min and has the advantage of being independent of the color of the sample, thanks to the use of anti-Tf-MBs, so serum, blood, or hemolyzed samples may be analyzed.
In recent years, other authors have explored the extraordinary advantages of dual PADs combining colorimetry and electrochemical detection for detecting analytes of clinical relevance. The general characteristics of these devices are summarized in Table S2.†
In most cases, dual sensing devices integrating colorimetric and electrochemical detection have benefited from the excellent sensitivity and selectivity of electrochemical detection along with the simplicity of colorimetric techniques, despite their limited sensitivity. This approach has been used to develop devices with a hybrid sensing mode for the same analyte with the specific objective of expanding the working range in the practical detection in clinical applications.66–69 However, more interesting approaches involve analyzing different analytes simultaneously within a single integrated device to obtain a more comprehensive response, providing more information for clinical evaluation. For example, a colorimetric and electrochemical dual μPAD for the simultaneous detection of several biomarkers of periodontitis disease in saliva,70 or the one developed in this work for determining the TSAT (%) parameter through the selective and sensitive electrochemical detection of Tf (TIBC) and the colorimetric detection of Fe3+ (Tf-bond iron) in real human serum samples from patients who have suffered an ischemic stroke.
Conclusions
A novel dual μPAD was successfully developed for the assessment of TSAT in serum samples by the ratio between Tf-bound iron, which was assayed by a colorimetric approach, and TIBC, which was evaluated by an electrochemical approach. The use of electrochemical detection provided higher simplicity and accuracy to the approach since TIBC was directly measured by exploiting the inherent Tf electroactivity. Moreover, all reagents needed for the colorimetric approach were stored on the μPAD, yielding a reagent-free device, which is ready to be used. These dual μPADs were successfully applied to the analysis of serum samples from ischemic stroke patients, showing excellent accuracy in comparison to a free-interference method (urea-PAGE) and revealing key advantages such as low sample volumes and short analysis times required. Therefore, the developed dual μPAD may be a promising POCT device to assist physicians in the fast prognosis of ischemic stroke, where the decision-making time is crucial. In this sense, future research would be to perform the immunopurification process on the surface of the working electrode, creating a ready to use POCT device and simplifying the number of steps for analysis. Moreover, the concept of colorimetric and electrochemical μPAD could pave the way to new applications not only in clinical diagnostics and POC technologies but towards others belonging to the point-of-need in environmental and food as well as environmental forensic fields, because the possibility of performing two different detection modes provides higher flexibility and multiplexing capabilities since analysts can choose the best detection mode for each kind of target analyte.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
S. D.: conceptualization, investigation, methodology, formal analysis, visualization, validation, writing–original draft, writing–review & editing. M. P.: conceptualization, investigation, methodology, formal analysis, visualization, validation, writing–original draft, writing–review & editing. A. G. C.: conceptualization, investigation, formal analysis, supervision, writing–original draft, writing–review & editing. T. G.: conceptualization, resources, writing–review & editing. A. E.: conceptualization, investigation, project administration, funding acquisition, resources, formal analysis, supervision, writing–review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been financially supported by the TRANSNANOAVANSENS program from the Community of Madrid (P2018/NMT-4349) (A. E.), by the grant PID2020-118154GB-I00 funded by MCIN/AEI/10.13039/501100011033 (A. E.), by the NEURO-CHIP-CM program from the Community of Madrid (Y2020/NMT6312) (A. E.), by the RICORS RD21/0006/0024 (NextGeneration EU funding) and 2021SGR00925 (Agencia de gestio d'Ajuts Universitaris i de Recerca de Catalunya) (T. G.), and by the Spanish Ministry of Economy and Competitiveness (CTQ2017-86441-C2-1-R, FPI fellowship (S. D.)).References
- WHO EMRO|Stroke, Cerebrovascular accident|Health topics, https://www.emro.who.int/health-topics/stroke-cerebrovascular-accident/index.html.
- S. K. Feske, Am. J. Med., 2021, 134, 1457–1464 CrossRef.
- E. C. Jauch, J. L. Saver, H. P. Adams, A. Bruno, J. J. B. Connors, B. M. Demaerschalk, P. Khatri, P. W. McMullan, A. I. Qureshi, K. Rosenfield, P. A. Scott, D. R. Summers, D. Z. Wang, M. Wintermark and H. Yonas, Stroke, 2013, 44, 870–947 CrossRef PubMed.
- P. Vilela, Eur. J. Radiol., 2017, 96, 133–144 CrossRef PubMed.
- G. McClelland, H. Rodgers, D. Flynn and C. I. Price, Eur. J. Emerg. Med, 2019, 26, 2–8 CrossRef PubMed.
- S. J. Allder, A. R. Moody, A. L. Martel, P. S. Morgan, G. S. Delay, J. R. Gladman, P. Fentem and G. G. Lennox, Lancet, 1999, 354, 1523 CrossRef CAS PubMed.
- M. Pohl, D. Hesszenberger, K. Kapus, J. Meszaros, A. Feher, I. Varadi, G. Pusch, E. Fejes, A. Tibold and G. Feher, J. Clin. Neurosci., 2021, 93, 174–182 CrossRef PubMed.
- S. Sotgiu, B. Zanda, B. Marchetti, M. L. Fois, G. Arru, G. M. Pes, F. S. Salaris, A. Arru, A. Pirisi and G. Rosati, Eur. J. Neurol., 2006, 13, 505–513 CrossRef CAS.
- J. R. Lynch, R. Blessing, W. D. White, H. P. Grocott, M. F. Newman and D. T. Laskowitz, Stroke, 2004, 35, 57–63 CrossRef PubMed.
- M. A. Reynolds, H. J. Kirchick, J. R. Dahlen, J. M. Anderberg, P. H. McPherson, K. K. Nakamura, D. T. Laskowitz, G. E. Valkirs and K. F. Buechler, Clin. Chem., 2003, 49, 1733–1739 CrossRef CAS.
- G. C. Jickling and F. R. Sharp, Neurotherapeutics, 2011, 8, 349–360 CrossRef CAS.
- Q. Tan, Y. Fang and Q. Gu, Front. Pharmacol., 2021, 12, 1–15 Search PubMed.
- L. Magtanong and S. J. Dixon, Dev. Neurosci., 2019, 40, 382–395 CrossRef.
- Z. Geng, Z. Guo, R. Guo, R. Ye, W. Zhu and B. Yan, Brain Res. Bull., 2021, 172, 212–219 CrossRef PubMed.
- D. Gill, G. Monori, I. Tzoulaki and A. Dehghan, Stroke, 2018, 49, 2815–2821 CrossRef PubMed.
- Q. He, W. Wang, D. Xu, Y. Xiong, C. You, C. Tao and L. Ma, Stroke, 2024, 55, 423–431 CrossRef.
- C. Bolm, Nat. Chem., 2009, 1, 420 CrossRef CAS PubMed.
- A. N. Luck and A. B. Mason, Curr. Top. Membr., 2012, 69, 3–35 CAS.
- C. P. Anderson, M. Shen, R. S. Eisenstein and E. A. Leibold, Biochim. Biophys. Acta, Mol. Cell Res., 2012, 1823, 1468–1483 CrossRef CAS.
- D. M. Frazer and G. J. Anderson, BioFactors, 2014, 40, 206–214 CrossRef CAS.
- T. Kamińska-Gibas, J. Szczygieł, P. Jurecka and I. Irnazarow, Fish Shellfish Immunol., 2020, 102, 511–518 CrossRef PubMed.
- M. E. Elsayed, M. U. Sharif and A. G. Stack, Adv. Clin. Chem., 2016, 75, 71–97 CAS.
- H. Li and Z. M. Qian, Med. Res. Rev., 2002, 22, 225–250 CrossRef PubMed.
- J. O. Jeppsson, Structural studies on human transferrin, Dissertation, Umeå universitet, 1967 Search PubMed.
- N. C. Giri, Role of Transferrin in Iron Metabolism, Iron Metabolism - A Double-Edged Sword, 2022 Search PubMed.
- J. Parkkinen, L. von Bonsdorff, F. Ebeling and L. Sahlstedt, Vox Sang., 2002, 83(Suppl 1), 321–326 CrossRef.
- K. Gkouvatsos, G. Papanikolaou and K. Pantopoulos, Biochim. Biophys. Acta, Gen. Subj., 2012, 1820, 188–202 CrossRef PubMed.
- P. Cacoub, C. Vandewalle and K. Peoc'h, Crit. Rev. Clin. Lab. Sci., 2019, 56, 526–532 CrossRef PubMed.
- M. Millán, N. Degregorio-Rocasolano, N. P. de la Ossa, S. Reverté, J. Costa, P. Giner, Y. Silva, T. Sobrino, M. Rodríguez-Yáñez, F. Nombela, F. Campos, J. Serena, J. Vivancos, O. Martí-Sistac, J. Cortés, A. Dávalos and T. Gasull, Antioxidants, 2021, 10, 1270 CrossRef PubMed.
- N. DeGregorio-Rocasolano, O. Martí-Sistac, J. Ponce, M. Castelló-Ruiz, M. Millán, V. Guirao, I. García-Yébenes, J. B. Salom, P. Ramos-Cabrer, E. Alborch, I. Lizasoain, J. Castillo, A. Dávalos and T. Gasull, Redox Biol., 2018, 15, 143–158 CrossRef CAS.
- M. Castellanos, N. Puig, T. Carbonell, J. Castillo, J. M. Martinez, R. Rama and A. Dávalos, Brain Res., 2002, 952, 1–6 CrossRef CAS PubMed.
- I. García-Yébenes, M. Sobrado, A. Moraga, J. G. Zarruk, V. G. Romera, J. M. Pradillo, N. Perez De La Ossa, M. A. Moro, A. Dávalos and I. Lizasoain, Neurochem. Int., 2012, 61, 1364–1369 CrossRef.
- S. Dortez, N. DeGregorio-Rocasolano, M. Millán, T. Gasull, A. G. Crevillen and A. Escarpa, Anal. Chem., 2023, 95, 12391–12397 CrossRef CAS.
- S. H. Mehta, R. C. Webb, A. Ergul, A. Tawak and A. M. Dorrance, Am. J. Physiol., 2004, 286, 283–288 CrossRef.
- H. Yamanishi, S. Kimura, S. Iyama, Y. Yamaguchi and T. Yanagihara, Clin. Chem., 1997, 43, 2413–2417 CrossRef CAS.
- K. Strzelak, N. Rybkowska, A. Wiśniewska and R. Koncki, Anal. Chim. Acta, 2017, 995, 43–51 CrossRef CAS.
- H. Yamanishi, S. Iyama, Y. Yamaguchi, Y. Kanakura and Y. Iwatani, Clin. Chem., 2003, 49, 175–178 CrossRef CAS.
- T. Eleftheriadis, V. Liakopoulos, G. Antoniadi and I. Stefanidis, Renal Failure, 2010, 32, 1022–1023 CrossRef PubMed.
- C. Frank, O. Rienitz, R. Jährling, D. Schiel and S. Zakel, Metallomics, 2012, 4, 1239–1244 CrossRef.
- N. Kitsati, D. Liakos, E. Ermeidi, M. D. Mantzaris, S. Vasakos, E. Kyratzopoulou, P. Eliadis, E. Andrikos, E. Kokkolou, G. Sferopoulos, A. Mamalaki, K. Siamopoulos and D. Galaris, Haematologica, 2015, 100, 80–83 CrossRef PubMed.
- R. Agarwal, Kidney Int., 2004, 66, 1139–1144 CrossRef PubMed.
- P. B. Luppa, C. Müller, A. Schlichtiger and H. Schlebusch, TrAC, Trends Anal. Chem., 2011, 30, 887–898 CrossRef CAS PubMed.
- W. Zheng, K. Wang, H. Xu, C. Zheng, B. Cao, Q. Qin, Q. Jin and D. Cui, Anal. Bioanal. Chem., 2021, 413, 2429–2445 CrossRef CAS.
- W. Alahmad, A. Sahragard and P. Varanusupakul, Biosens. Bioelectron., 2021, 194, 113574 CrossRef CAS PubMed.
- T. Ozer, C. McMahon and C. S. Henry, Annu. Rev. Anal. Chem., 2020, 13, 85–109 CrossRef.
- G. Chen, C. Fang, H. H. Chai, Y. Zhou, W. Yun Li and L. Yu, Sens. Actuators, B, 2019, 281, 253–261 CrossRef CAS.
- G. G. Morbioli, T. Mazzu-Nascimento, A. M. Stockton and E. Carrilho, Anal. Chim. Acta, 2017, 970, 1–22 CrossRef CAS PubMed.
- S. Dortez, A. G. Crevillen, A. Escarpa and S. Cinti, Sens. Actuators, B, 2024, 398, 134704 CrossRef.
- Y. Xu, H. Ning, S. Yu, S. Liu, Y. Zhang, C. Niu, Y. Zhang, S. S. Low and J. Liu, Micromachines, 2023, 14, 142 CrossRef.
- K. Mahato and J. Wang, Sens. Actuators, B, 2021, 344, 130178 CrossRef.
- E. Carrilho, A. W. Martinez and G. M. Whitesides, Anal. Chem., 2009, 81, 7091–7095 CrossRef.
- Z. Miao, X. Gu, S. Lu, M. L. Brusseau, N. Yan, Z. Qiu and Q. Sui, J. Hazard. Mater., 2015, 300, 530–537 CrossRef CAS PubMed.
- G. Bengtsson, S. Fronæus and L. Bengtsson-Kloo, J. Chem. Soc., Dalton Trans., 2002, 2548–2552 RSC.
- G. L. Smith, A. A. Reutovich, A. K. Srivastava, R. E. Reichard, C. H. Welsh, A. Melman and F. Bou-Abdallah, J. Inorg. Biochem., 2021, 220, 111460 CrossRef CAS PubMed.
- L. L. Stookey, Anal. Chem., 1970, 42, 779–781 CrossRef CAS.
- Z. Yang, Y. Yan, A. Yu, B. Pan and J. J. Pignatello, Chem. Eng. J., 2020, 391, 123592 CrossRef CAS.
- S. Dortez, A. G. Crevillen and A. Escarpa, Talanta, 2023, 253, 123914 CrossRef CAS PubMed.
- T. Sierra, C. S. Henry, A. G. Crevillén and A. Escarpa, Analysis Sensing, 2022, 2, 2–8 Search PubMed.
- T. Sierra, A. G. Crevillen and A. Escarpa, Biosens. Bioelectron., 2021, 179, 113098 CrossRef CAS PubMed.
- A. H. B. Dourado, F. C. Pastrián and S. I. C. De Torresi, An. Acad. Bras. Cienc., 2018, 90, 607–630 CrossRef CAS.
- E. V. Suprun, E. V. Karpova, S. P. Radko and A. A. Karyakin, Electrochim. Acta, 2020, 331, 135289 CrossRef CAS.
- K. Moulaee and G. Neri, Biosensors, 2021, 11, 1–54 CrossRef PubMed.
- H. Kawabata, Free Radical Biol. Med., 2019, 133, 46–54 CrossRef PubMed.
- P. T. Gomme and K. B. McCann, Drug Discovery Today, 2005, 10, 267–273 CrossRef PubMed.
- W. Breuer, C. Hershko and Z. I. Cabantchik, Transfus. Sci., 2000, 23, 185–192 CrossRef CAS PubMed.
- K. Pungjunun, A. Yakoh, S. Chaiyo, N. Praphairaksit, W. Siangproh, K. Kalcher and O. Chailapakul, Microchim. Acta, 2021, 188, 1–11 CrossRef.
- H. Wang, C. Zhou, X. Sun, Y. Jian, Q. Kong, K. Cui, S. Ge and J. Yu, Biosens. Bioelectron., 2018, 117, 651–658 CrossRef CAS.
- W. Xue, P. Jia, Y. Wu, P. Wang, J. Shi, Y. Chan and M. Liu, Adv. Agrochem, 2023, 2, 269–275 CrossRef CAS.
- J. Xu, J. Shen, B. Zhang, Y. Zhang, X. Lv and G. Zhu, Electrochim. Acta, 2024, 481, 143952 CrossRef CAS.
- L. R. Sousa, H. A. Silva-Neto, L. F. Castro, K. A. Oliveira, F. Figueredo, E. Cortón and W. K. T. Coltro, Anal. Bioanal. Chem., 2023, 415, 4391–4400 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc00398e |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |