
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Path-dependent morphology of CH4 hydrates and their dissociation studied with high-pressure microfluidics†
Jidong
Zhang
 a,
Zhenyuan
Yin
*a,
Saif A.
Khan
b,
Shuxia
Li
c,
Qingping
Li
d,
Xiaohui
Liu
a and
Praveen
Linga
b
a,
Zhenyuan
Yin
*a,
Saif A.
Khan
b,
Shuxia
Li
c,
Qingping
Li
d,
Xiaohui
Liu
a and
Praveen
Linga
b
aInstitute for Ocean Engineering, Shenzhen International Graduate School, Tsinghua University, Shenzhen 518055, China. E-mail: zyyin@sz.tsinghua.edu.cn
bDepartment of Chemical and Biomolecular Engineering, National University of Singapore, Singapore 117582, Singapore
cSchool of Petroleum Engineering, China University of Petroleum (East China), Qingdao 266580, China
dState Key Laboratory of Natural Gas Hydrates, Technology Research Department CNOOC Research, Beijing 100192, China
First published on 30th January 2024
Abstract
Methane hydrates (MHs) have been considered a promising future energy source due to their vast resource volume and high energy density. Understanding the behavior of MH formation and dissociation at the pore-scale and the effect of MH distribution on the gas–liquid two phase flow is of critical importance for designing effective production strategies from natural gas hydrate (NGH) reservoirs. In this study, we devised a novel high-pressure microfluidic chip apparatus that is capable of direct observation of MH formation and dissociation behavior at the pore-scale. MH nucleation and growth behavior at 10.0 MPa and dissociation via thermal stimulation with gas bubble generation and evolution were examined. Our experimental results reveal that two different MH formation mechanisms co-exist in pores: (a) porous-type MH with a rough surface formed from CH4 gas bubbles at the gas–liquid interface and (b) crystal-type MH formed from dissolved CH4 gas. The growth and movement of crystal-type MH can trigger the sudden nucleation of porous-type MH. Spatially, MHs preferentially grow along the gas–liquid interface in pores. MH dissociation under thermal stimulation practically generates gas bubbles with diameters of 20.0–200.0 μm. Based on a custom-designed image analysis technique, three distinct stages of gas bubble evolution were identified during MH dissociation via thermal stimulation: (a) single gas bubble growth with an expanding water layer at an initial slow dissociation rate, (b) rapid generation of clusters of gas bubbles at a fast dissociation rate, and (c) gas bubble coalescence with uniform distribution in the pore space. The novel apparatus designed and the image analysis technique developed in this study allow us to directly capture the dynamic evolution of the gas–liquid interface during MH formation and dissociation at the pore-scale. The results provide direct first-hand visual evidence of the growth of MHs in pores and valuable insights into gas–liquid two-phase flow behavior during fluid production from NGHs.
1. Introduction
Methane hydrates (MHs) are ice-like crystalline materials consisting of water and CH4 molecules, which are formed at low temperatures (T) and high pressures (P).1 MHs have been considered a promising future source of energy for the transition to a low-carbon world due to their abundant reserves (∼3000 trillion cubic meters) and high energy storage capacity (∼170 vCH4/vH2O).2 Production of CH4 from NGH reservoirs has drawn considerable research and industrial interest in the past decade in countries like the USA, China, Japan, Canada, India, etc. The challenge faced in the past field production trials is to sustain a consistently high rate of CH4 production effectively and economically.3 This warrants a fundamental understanding of the dynamic physical behavior of MH formation and dissociation in geological media.Energy recovery from MHs is a coupled thermal–hydraulic–mechanical–chemical (THMC) process with multiphase (aqueous, gas, and MH) and multi-component flow and transport.4 The phase change of MHs from a gas–liquid two-phase system, the evolution of the MH morphology, and the distribution of MHs at the pore-scale provide critical insights into understanding the underlying THMC process.5 Both X-ray computed tomography (CT) and nuclear magnetic resonance (NMR) techniques have been employed to investigate the hydrate morphology and spatial distribution at the pore-scale in MH core samples extracted from the field. However, the relatively low spatial resolution of magnetic resonance imaging (MRI) (usually in the range of the millimeter-scale)6 and the extended scanning period of both NMR and CT (in the range of tens of minutes)7 limit the fast examination of the gas–water–hydrate interface dynamic change and the associated evolution of gas bubbles that usually occurs within seconds with a spatial scale of micrometers in pores.8 In addition, it has been recently reported that gas bubbles can be generated during MH dissociation but with no quantitative analysis. This warrants the development of novel experimental techniques to provide direct first-hand visual evidence for the growth and dissociation of MHs and the evolution of gas bubbles in pores.
The morphology and distribution of MHs directly affect the pore structure of hydrate-bearing sediments and the resulting gas–liquid two phase distribution.9 MH formation is similar to multiphase crystallization and is thus described by nucleation and growth processes.10 MH formation involves the enculturation of CH4 as a guest molecule in cages of H2O molecules stabilized by hydrogen bonds at low temperature (4–10 °C) and high pressure (>10 MPa in natural MH reservoir). The formation of MHs usually involves the following processes: (a) the dissolution of CH4 in H2O, (b) the generation of the MH nucleus after the induction time, and (c) the subsequent growth of MHs along with the transport of CH4 in H2O. The MH formation and dissociation processes involve the flow and distribution of multiple phases (gas, liquid and solid hydrate or ice) in addition to the transport of multiple components (H2O and CH4) within these phases. In addition, the process usually involves the generation and the development of a complex gas–liquid interface in the presence of MHs in the pores of sediments.
A previous study has reported that MH formation from gas bubbles is preferred at the gas–liquid interface.11,12 The typical MH morphologies are lamellar, pinnacle, needle-like, and flocculent.13,14 However, the development of MH films and the subsequent conversion of the remaining gas bubbles to MHs still lack direct visualization. In addition, it should be noted that CH4 gas exists in the liquid phase in both gaseous and dissolved phases.15 Past laboratory investigations mainly focused on MH formation from gas bubbles16 but with no MH formed from dissolved CH4 gas. Moreover, the uniform MH distribution can be attributed to the MH formation behavior, i.e., MH film formation at the gas–liquid interface and subsequent growth without preference.17 However, the evolution and distribution of gas bubbles during MH formation and their effect on MH film formation are not clarified. Therefore, the morphology, distribution, and growth rate of MHs formed from both CH4 gas bubbles and dissolved CH4 should be investigated.
MH dissociation in porous media is a time-dependent process involving heat and mass transfer and dissociation kinetics.18 Previous molecular simulation studies have shown that CH4 molecules are released into the liquid phase to generate CH4-rich regions during MH dissociation.15 The generation of CH4 bubbles reduces the concentration of CH4 molecules in the liquid phase, thus promoting the rate of MH dissociation.19 The direct visualization of the MH interface and gas bubble evolution during MH dissociation is essential for addressing the rate of MH dissociation.20,21 Kou et al.17 conducted X-ray experiments that revealed that the water layer covering the MH grows during MH dissociation. These findings show that the thickness of the water film at the front of the MH dissociation interface and the evolution of gas bubbles increase the complexity of the mass transfer. However, the detailed gas–liquid–MH interface in complex pore space and throat during MH dissociation is poorly understood, and many pore-scale investigations in the laboratory are still needed.
The diameter and distribution of gas bubbles during MH dissociation are crucial to the porous media's multiphase flow and heat transfer.22,23 The main mechanisms during MH dissociation are the gas bubble transport caused by capillary forces and the coalescence of gas bubbles due to the rupture of the liquid film.24 The large density gas bubble distribution within the pores hinders pressure and temperature propagation.11 However, there is a current lack of a robust methodology for quantitative analysis of gas bubbles, and the evolution of the number and distribution of gas bubbles during MH dissociation is still unclear. In addition, the inter-bubble growth, aggregation, and coalescence further increase the complexity of gas bubble evolution.25 Therefore, a detailed quantitative analysis of gas bubble size and distribution in complex porous media is necessary. Direct visualization of such a process without the addition of porous media at the macro scale (centimeter to meter scale) has been common in recent years.26 However, significant challenges remain in the direct visualization of the in situ MH formation and dissociation processes in the pores of sandy porous media (at the scale of micrometers within seconds).
Microfluidics is defined as the science and technology of fluid flow in microstructures with dimensions of 1–1000 μm. Microfluidics exploits both its most obvious characteristic of small size and less obvious characteristics of fluids in the microchannels, applications in biology and medicine,27 and applications in energy,28 hydrate-based CO2 sequestration,29 and H2 storage.30 In addition, high-pressure microfluidic chips are extremely useful in characterizing the phase change behavior of MH formation and dissociation.31,32 However, high pressure (>10 MPa) and low temperature (4–10 °C) are the key factors limiting the use of microfluidics in the field of gas hydrates. As previous studies suggest,32–34 the application of gas hydrates in microfluidic chips can be achieved by keeping the chip at a small constant pressure difference. Microfluidic chips have much to offer in the study of hydrate formation and dissociation behavior.
In this study, we designed a novel microfluidic chip to visualize the MH formation and dissociation behavior in the pore space. MHs were directly formed from CH4 and H2O above the quadruple point based on the memory effect of the dissociated H2O.35 MH dissociation via thermal stimulation with gas bubble generation and evolution was examined. The evolution of the MH morphology formed from the CH4 gas bubbles and dissolved CH4, as well as the gas–liquid–MH interface, was directly captured during MH formation and dissociation. In addition, the evolution of gas bubbles was quantified during MH dissociation. The pressure, temperature, and images of the hydrate/bubble morphology were acquired and analyzed in detail during the process. The experimental results aim to enhance the understanding of the pore-scale MH formation and dissociation behavior and the evolution of gas bubbles.
2. Experiment and procedure
2.1. Experimental apparatus and materials
Fig. 1 shows the schematic of the newly set up experimental apparatus for pore-scale investigation on the microfluidic chip with a picture of the apparatus shown in Fig. S1 in the ESI.† The entire apparatus consists of four major components: (a) a confining pressure holder (Tuochuang Technology Co., Ltd, Pmax = 20.0 MPa) equipped with quartz glass for visualization, with tubing (D = 1.6 mm) connected to a syringe pump for the control of the confining pressure. The pressure difference between the pressure holder and the microfluidic chip was maintained at 0.5 MPa in this study to ensure sufficient sealing and to avoid rupture of the microfluidic device; (b) an etched microfluidic chip (Zhenjiang Highray Chip Technology Co., Ltd) installed in the holder for MH formation and dissociation; (c) a constant flow-rate syringe pump (Sanotac Technology Co., Ltd, 0.001–9.999 mL min−1) equipped with a piston container (Tuochuang Technology Co., Ltd, Pmax = 20.0 MPa) in the circulating water bath for the injection of high-pressure CH4 and H2O into the microfluidic chip at the desired P and T; (d) a high-resolution charge coupled device (CCD) camera (Magellan Instrument Co., Ltd, image resolution = 3840 × 2160, pixel size = 0.94 μm per pixel, 30fps) associated with a customized in-house made light source which enabled real-time observation and monitoring of the pore-scale behavior of the gas–liquid two-phase flow and CH4 hydrates on the microfluidic chip. The interface of the microfluidic chip and constant flow-rate syringe pump is shown in Fig. S2 in the ESI.†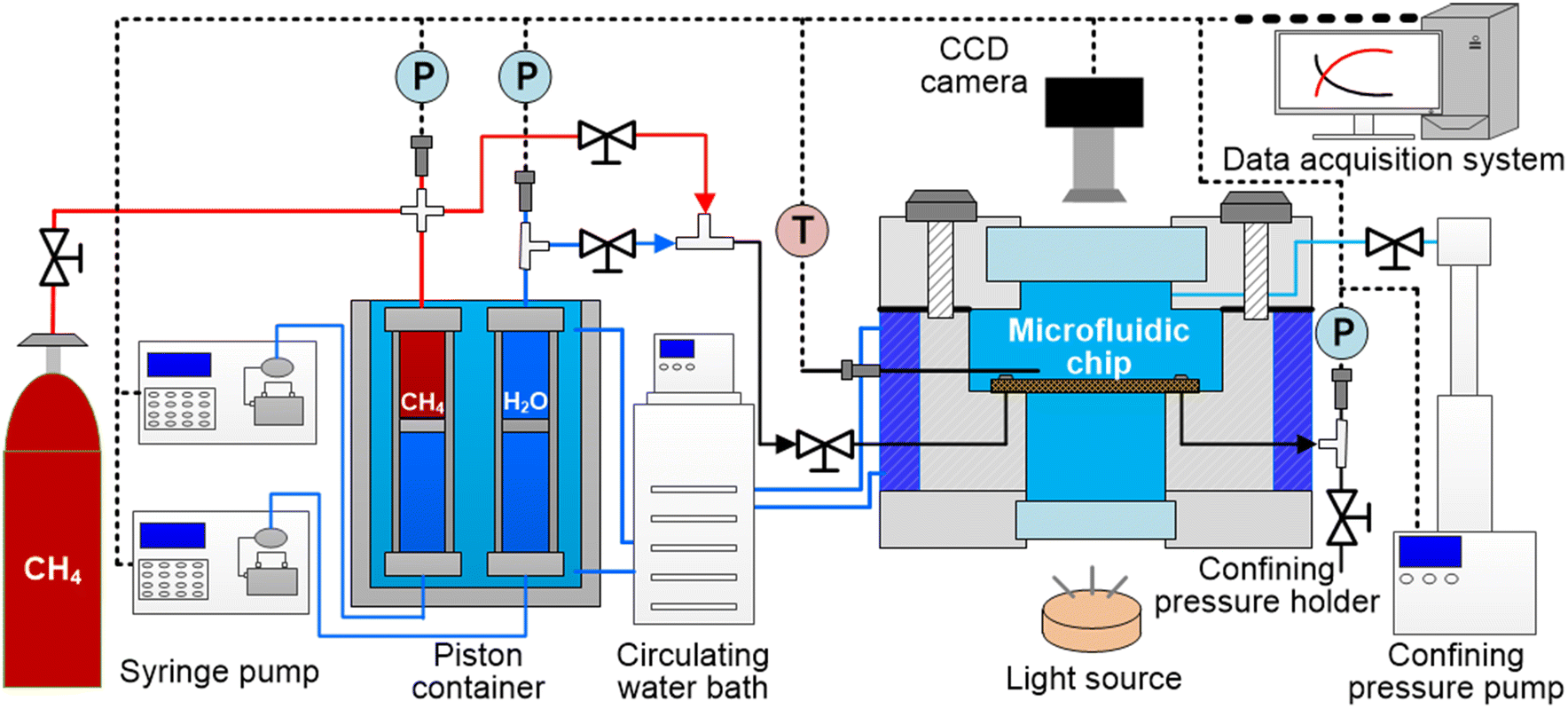 | ||
| Fig. 1 Schematic of the experimental apparatus consisting of the microfluidic chip for MH formation and dissociation. The confining pressure holder (Pmax = 20.0 MPa) was equipped with quartz glass and a CCD camera for pore-scale visualization and with cooling jackets circulating water for temperature control. A syringe pump was employed to control the confining pressure of the microfluidic chip to ensure a pressure difference of 0.5 MPa. A second syringe pump injected CH4 and H2O into the microfluidic chip to the desired pressure. P, T, and pore-scale images were recorded using a data acquisition system and stored. | ||
The temperature of the microfluidic chip was controlled by the cooling jacket surrounding the confining pressure holder. A PT-100 thermocouple (Omega Co., Ltd, accuracy ± 0.1 K) was positioned 2.0 cm above the microfluidic chip for temperature measurement. A pressure sensor (Maxonic Senex Co., Ltd, max. 20.0 MPa, accuracy ± 0.25%) was installed at the outlet of the microfluidic chip for pressure measurement. In this study, the effect of continuous air blowing for demisting at the upper and lower quartz glass surfaces coupled with the impact of ambient temperature was considered. In addition, we employed numerical simulation to calibrate the linear relationship between the thermocouple point and the temperature of the field of view (see Fig. 2c). The detailed process is provided in Fig. S3 in the ESI.† All the pressure, temperature, and pictures taken at the designated region of the microfluidic chip were recorded using a data acquisition device and stored on a personal computer.
 | ||
| Fig. 2 The microfluidic chip used in this study. (a) A picture of the microfluidic chip used in the study fabricated using laser etching (40.0 mm × 40.0 mm). The microfluidic chip exhibits a total pore volume of 1.75 mL with pore diameters ranging from 100.0 μm to 500.0 μm. (b) A representative cross-sectional profile of the microfluidic chip with a channel depth of 100.0 μm. (c) An image depicting local observation of the pore space (region 1 in panel a) showcasing CH4 gas bubbles and H2O distributed in the etched pores and the grains. The black line represents the gas–liquid interface, with multiple liquid drops unevenly distributed in gas bubbles due to the rupture of liquid films between adjacent bubbles. | ||
Fig. 2 shows the top and cross-section view of the etched microfluidic chip with detailed configuration and dimensions. The key steps for the fabrication of a microfluidic chip used in this study is explained in detail with a schematic shown in Fig. S4 in the ESI.† The material of the microfluidic chip was borosilicate glass, a hydrophilic material with a contact angle of 36.1° as shown in Fig. S5 in the ESI.† The entire microfluidic chip was 4.0 mm thick with an inlet and an outlet located at the top right corner and bottom left corner, as shown in Fig. 2a. The channel was fabricated at the center of the microfluidic chip using a combined technology of both laser etching followed by wet etching with dimensions of 40.0 × 40.0 mm. The pore space resulting from the etching ranged from 100.0 μm to 500.0 μm. The thickness of the etched flow channel was 100.0 μm with a trapezoidal cross-section (see Fig. 2b). Due to the limitation of the field of view of the CCD camera, only a section of the microfluidic chip can be observed at one time to capture the interface of gas–liquid and the evolution of CH4 hydrates at the pore-scale. For example, Fig. 2c presents an image captured using the apparatus, which shows the existence of CH4 bubbles located at the pore space and pore throats in the continuous H2O liquid phase in region 1 (3.6 mm × 2.0 mm) of the etched microfluidic chip. Pure CH4 > 99.9 mol% and L-Met (0.3 wt%, a kinetic promoter) synthesized MHs on the microfluidic chip.
2.2. Experimental procedure
Fig. 3 illustrates the P–T trajectory of the designed MH formation and subsequent MH dissociation under thermal stimulation in two cycles (path I and path II). It should be noted that in our trial experiments, the induction time of direct MH formation from CH4 and H2O was longer than 48 h above the quadruple point h in Fig. 3. However, the induction time can be significantly reduced during hydrate formation due to the memory effect of dissociated H2O.35 In addition, the ice-to-hydrate method can significantly reduce the induction time (tind).16 Thus, in path I, the temperature was lowered below the quadruple point to induce ice formation, after which MH formation was triggered from ice melting when the temperature was increased. In path II, MHs were directly formed after the thermal stimulation process of path I by lowering the MH equilibrium curve above the quadruple point. Due to the possible memory effect of the dissociated H2O, MH formation was successful with tind less than 2 h. The evolution of P and T during MH dissociation in path I is shown in Fig. S6 in the ESI.† The detailed morphology transition process of MH dissociation in path I is provided in Video S1 in the ESI.† In this study, pressure, temperature, and morphology images were acquired for both paths. However, the MH formation in the ice-to-hydrate method is rapid after ice melting, and the detailed formation process cannot be captured due to the CCD camera limitations. Thus, in this study, only the results from path II were analysed in detail and presented in the following Results and discussion section. | ||
| Fig. 3 Schematic of the P–T trajectory of the designed MH formation and MH dissociation under thermal stimulation to T = 287.8 K in two cycles. Path I refers to MH and ice formation below the quadruple point of CH4 + H2O and the subsequent MH formation above the quadruple point of CH4 + H2O with MH dissociation under thermal stimulation. Path II refers to the second cycle of MH formation above the quadruple point of CH4 + H2O with MH dissociation under thermal stimulation. A → B refers to CH4 injection to desired P = 9.0 MPa. B → C refers to the injection of 0.35 mL deionized water to P = 10.5 MPa. C → E refers to the process of ice-born MH formation. E → F refers to MH dissociation under thermal stimulation to T = 287.8 K. F → G refers to the 2nd cycle of MH formation starting 1.0 h after the 1st cycle under the memory effect. Pressure, temperature, and morphology images were acquired and analyzed during the entire process. | ||
The specific experimental procedure for path I (A → F) and path II (F → G) is as follows:
(a) The microfluidic chip was purged using CH4 gas to remove the residual air at P = 0.5 MPa, and CH4 gas was injected to desired P = 9.0 MPa (A → B in Fig. 3);
(b) After T stabilized at 288.2 K, a total of 0.35 mL of L-Met (0.3 wt%) was injected into the microfluidic chip at a constant flow rate of 0.02 mL min−1 (B → C in Fig. 3);
(c) The temperature of the microfluidic chip was decreased to 267.2 K for ice formation (C → D in Fig. 3);
(d) The temperature of the microfluidic chip was raised to 274.2 K to trigger ice melting and MH formation (D → E in Fig. 3);
(e) MH dissociation was induced by thermal stimulation to 287.8 K in path I (E → F in Fig. 3);
(f) After complete dissociation for 1 h, the T of the microfluidic chip was decreased to 275.2 K to induce direct MH formation above the quadruple point (F → G in Fig. 3);
(g) MH dissociation was induced by thermal stimulation to 287.8 K in path I (G → F in Fig. 3).
3. Results and discussion
3.1. MH formation behavior at the pore-scale on the microfluidic chip
 | ||
| Fig. 4 (a) Evolution of P and T overtime during MH formation with a rapid pressure drop of 1.0 MPa within 20.0 min starting at time 150.0 min; (b) P–T trace in relation to the MH equilibrium curve showing a sub-cooling of 12.1 K for MH formation on the microfluidic chip; (c)–(h) refer to the morphology images taken on the microfluidic chip at the same position and correlate with the timepoint shown in figure panels (a) and (b) with (c) and (d) showing the initial gas–liquid distribution before MH nucleation and (e)–(h) showing MH growth on the microfluidic chip after the nucleation at 5.0 min, 10.0 min, 25.0 min, and 240.0 min. | ||
Fig. 4c–h show the images acquired at location 1 (see Fig. 2c) on the microfluidic chip during the MH formation process discussed above, with the figure panel letter denoting the time stamp shown in Fig. 4a and b. Fig. 4c depicts the initial gas–liquid distribution on the microfluidic chip, where relatively large gas bubbles (DG = ∼200.0 μm) occupy the pore space with a flock of small bubbles (DG = ∼50.0 μm) scattering in the pore throats. Such a discontinuous gas bubble distribution agrees well with the abundant H2O injected into the system compared with relatively less CH4. In addition, the distribution of large and small bubbles agrees well with the capillary pressure induced at the pore space and throats. Fig. 4d shows the image acquired at the start of MH nucleation, where the gas bubbles slightly shrank due to CH4 dissolution and cooling.
Fig. 4e shows the distribution of gas bubbles at location 1 on the microfluidic chip 5 min after MH nucleation. No MH particle was observed at this location but the gas bubbles expanded and started to aggregate with surrounding gas bubbles. This can be attributed to the MH nucleation and growth at other places on the microfluidic chip, which resulted in a pressure decrease (ΔP = 0.2 MPa) within the flow channel. Fig. 4f shows the image acquired 30 seconds after the initial MH nucleation at position 1. It can be seen that two large gas bubbles still exist at this point in time because of the continuous gas bubble coalescence from Fig. 4e. It can be observed that the gas bubble located at the bottom right in the pore space turns dark and grayish, which is a distinct indication of MH nucleation and growth at this location.
Fig. 4g shows the image acquired 25 min after the initial MH nucleation at position 1, where the gas bubble at the pore space continues to shrink and the large gas bubble at the left pore throat completely turns into porous-type MH. Fig. 4h shows the image acquired 240 min after the initial MH nucleation at position 1, where no gas bubble existed in the field of view, and two different types of MH structures were observed with the remaining pore space occupied by H2O. The two types of MHs can be differentiated by their morphology, where the first type was MH with a porous-type structure (the region with a scattered dark grey color), and the second type was crystal-type MH (the region with a clear background white color).
A video showing the entire MH formation process at position 1 on the microfluidic chip is provided in Video S2 in the ESI.† It can be observed during the process that the aggregation and the coalescence of gas bubbles induce perturbation to the gas and liquid distribution and provide a favorable place for MH nucleation and growth. In addition, it can be observed that the distribution of MHs in the pore space and pore throat correlates well with the initial gas bubble distribution. This indicates that MH formation from such a static microfluidic channel is initiated from the gas–liquid interface and can propagate into both the pore space and pore throat without preference.
Two primary reasons can be attributed to the final heterogeneous distribution of MHs: (a) rigid MH film formation at the gas–liquid interface, which prevents the migration of gas bubbles, and (b) gas bubble aggregation and coalescence due to the pressure drop. Previously published studies have also provided experimental evidence for the MH film formation and MH distribution. Wang et al.11 observed that a thin MH film formed at the gas–liquid interface, and subsequent MHs grew in the liquid phase with different morphologies. In addition, Li et al.16 conducted experiments and revealed that MH formation usually occurs at the gas–liquid interface. The final MH distribution and morphology correlate with the gas bubble size and distribution. Their findings suggest that a MH film initially formed at the gas–liquid interface during MH formation. However, this previous study did not find the aggregation and coalescence of gas bubbles during MH formation, and their effect on MH film nucleation and growth at the gas–liquid interface still needs to be clarified. These results will be confirmed in the following section.
3.1.2.1. MH formation from CH4 gas bubble. Fig. 5 depicts the detailed images acquired at location 1 (see Fig. 2c) on the microfluidic chip during MH formation from the CH4 gas bubble (MHBub) in path II. Panel a1 shows the image acquired 0.5 min after the MH nucleation in the right pore space (see Fig. 4f), where a few faceted MH crystals formed from dissolved CH4 gas in the pore throat. Panel a2 depicts the crystal-type MH growth from dissolved CH4 gas and subsequent MHBub formation from gas bubbles in the pore throat. Panel a3 shows the image acquired 12.4 min after the initial MH nucleation. It can be seen that the gas bubble shrinks along the pore throat, after which crystal-type MH migrates toward the shrinking gas bubble. This can be attributed to the MH growth at other places on the microfluidic chip, which resulted in a pressure decrease (ΔP = 0.2 MPa) within the flow channel.
 | ||
| Fig. 5 Microscopy images showing the evolution and the interaction between CH4 gas bubbles and MHs grown in the pore-space and pore-throat at the same position on the microfluidic chip after MH nucleation. Panels (a1)–(a3) show that a few faceted crystal-type MHs form from the dissolved CH4 gas in H2O and subsequently migrate toward the shrinking gas bubble; panels (b1)–(b3) show that crystal-type MH migrates along the water film on the surface of the grain and comes into contact with the gas bubble with a layer of MH film formed at the gas–liquid interface along the pore throat towards the gas bubble; panels (c1)–(c3) show that the gas bubble breaks up and the remaining gas is released into the pore space for the continuing growth of porous-type MH from gas bubbles into the pore space. | ||
Panels (b1)–(b3) show the MH film formed at the gas–liquid interface along the left pore throat of location 1 (see Fig. 2c). Panel b1 shows the image acquired 6.0 seconds after crystal-type MH migration in panel a3. A thin water film was observed on the surface of the grain, where crystal-type MH migrates and grows with the surrounding gas bubble. This can be attributed to the hydrophilic grain (contact angle = 36.1°) and capillary pressure induced at the pore throat. Panel b2 shows that crystal-type MH comes into contact with a gas bubble, which triggers a layer of 240.0 μm MH film formed at the gas–liquid interface within 2.0 seconds. Panel b3 depicts the image acquired 2.2 min after the start of MH film nucleation. It can be seen that the MH film continues to grow along the gas–liquid interface toward the gas bubble with wall-climbing behavior. This was consistent with the findings in our prior reactor-scale study.39 Notably, flocculent MH was observed within the gas bubble.
Panels (c1)–(c3) show the porous-type MHBub growth from the CH4 gas bubble after MH film formation at the gas–liquid interface. Panel c1 shows the image acquired 2.4 min after MH film nucleation in panel b2, where the liquid phase breaks through the MH film into a gas bubble for the continuing growth of MHBub. Panel c2 depicts the gas bubble breaking up the MH film and the release of the remaining gas into the pore space. It was estimated that the local pressure difference across the gas bubble with the MH film was around 3.2 MPa before the sudden rupture of the hydrate film with the release of gas into the liquid phase (see panel c2 in Fig. 5). Details of the estimation method is presented in the ESI.† The location with released CH4 gas quickly nucleates and provides a site for subsequent porous MH formation. Panel c3 shows the image acquired 13.5 min after MH film nucleation at the gas–liquid interface, where no gas bubble existed in the field of view, and a porous-type MH morphology was observed after densification of the co-existing gas–liquid–hydrate.
A video showing the entire MHBub formation process of Fig. 5 is provided in Video S2 in the ESI.† It can be observed that the MH film growth along the gas–liquid interface (Δt = 10.0 s) was faster than the MH film growth towards the gas bubble (Δt = 1.1 min). This can contribute to the MH film preventing the contact of gas and liquid phases and thus inhibiting the mass transfer of water. In addition, once the MH film of the gas bubble breaks through, the release of the remaining gas bubble continues to flow into the aqueous phase, thus establishing an additional gas–liquid interface. This phenomenon enhances the formation of gas–liquid–MH co-existing at the gas–liquid interface. The gas finger continues its movement until a combination of reduced driving pressure and a thickened MH film fully arrests its flow. The intrinsic kinetics dominates the densification of the co-existing gas–liquid–MH into porous-type MH. This indicates that MHBub formation from gas bubbles can be considered a combination of mass transfer and intrinsic kinetics in such a static microfluidic chip.
Previous studies have also found MH film formation at the gas–liquid interface and porous-type MH formation from CH4 gas bubbles. Such MH film rupture was corroborated by the study of Fu et al.,40 where it takes a threshold pressure across the rigid MH film to release the entrapped gas. This may be attributed to the gas finger causing the flock of gas bubbles in the liquid face, generating and driving the porous-type MHBub. However, the general absence of direct observation of MH film nucleation and growth behavior at the gas–liquid interface in previous studies has limited the comprehensive understanding. It was generally accepted that the Gibbs free energy of nucleation was lower at the gas–liquid interface with higher concentrations of CH4.1 In this study, MH film formation at the gas–liquid interface was divided into three stages: (a) faceted MH crystal formation from dissolved CH4 gas, (b) MH crystal migration and growth toward the shrinking gas bubble and along the water film of the grain, and (c) MH crystals coming into contact with the gas bubble and triggering MH nucleation at the gas–liquid interface.
3.1.2.2. MH formation from dissolved CH4 gas. Fig. 6 shows that the crystal-type MH continues to grow from dissolved CH4 gas after MH nucleation on the microfluidic chip. Panel a1 depicts the image acquired 13.0 min after initial MH nucleation, where an initially hexahedral MH crystal (L = 23.8 μm) was observed in the pore throat. Panel a2 shows faceted MH crystal growth in the pore throat toward the gas bubble in the pore space. In addition, it can be observed that the gas bubble located at the bottom left pore throat completely turns into porous-type MHBub, and the gas bubble located at the right pore throat migrated to the middle pore throat. Panel a3 shows the image acquired 126.7 min after initial MH nucleation, where crystal-type MH continues to grow toward the shrinking gas bubble, and growth propagates the interface gradually expanding near the gas bubble. This can be attributed to the higher dissolved CH4 concentration near the gas bubble.
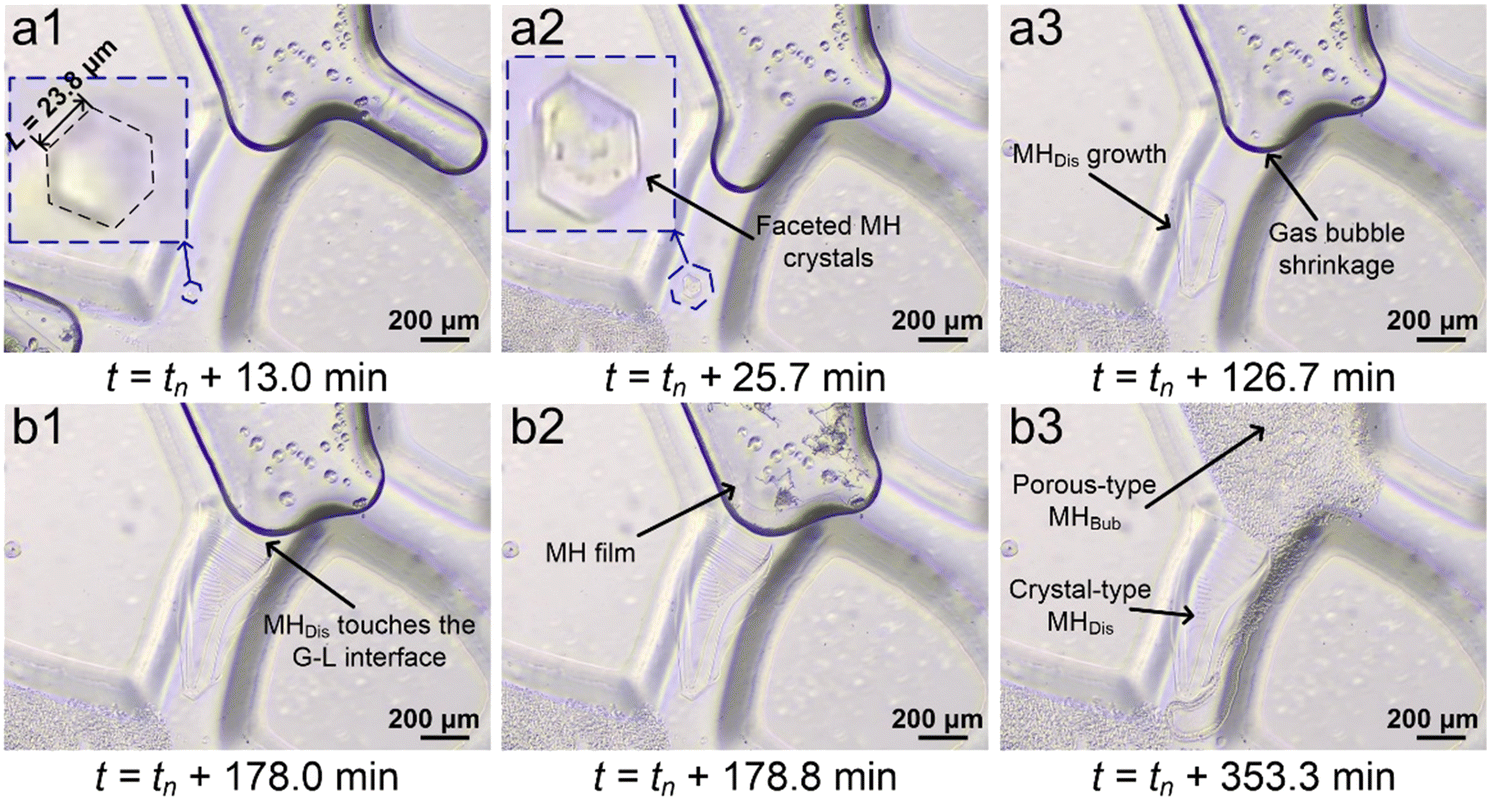 | ||
| Fig. 6 Microscopy images showing the evolution of significant MH crystal growth from dissolved CH4 gas in the pore-throat toward the pore-space at the same position on the microfluidic chip after MH nucleation. Panels (a1)–(a3) show that a hexahedral MH crystal (L = 23.8 μm) grows in the pore throat toward the shrinking gas bubble in the pore space; panels (b1)–(b3) show that the MH crystal formed from dissolved CH4 reaches the gas–liquid interface and triggers MH growth from CH4 gas bubbles (see Fig. 5) with two different MH morphologies co-existing on the microfluidic chip. | ||
Panel b1 shows the image acquired 178.0 min after the initial MH nucleation, where crystal-type MHDis comes into contact with shrinking gas bubbles in the pore space. It was interesting to note that crystal-type MHDis formation from dissolved gas in the pore throat was a slow and long process (Δt = 116.0 min) compared with porous-type MHBub formation (Δt = 13.5 min). This can be attributed to the low CH4 solubility (e.g., S = 0.0011 mol mol−1 CH4 in water, at 8.5 MPa and 273.45 K). Panel b2 shows the MH film formed at the gas–liquid interface after crystal-type MHDis touched the gas bubble, which was consistent with the porous-type MHBub formation process (see panels b1–b3 in Fig. 5). Panel b3 shows the image acquired 351.3 min after the initial MH nucleation, where two types of porous-type MHBub (region with a scattered dark grey color) and crystal-type MHDis (region with a clear background white color) were observed in the pore space and pore throat.
Comparing the MH formation behavior from the CH4 gas bubble and dissolved CH4 gas, one significant difference was the MH morphology of porous-type MHBub formed from the CH4 gas bubble and crystal-type MHDis formed from dissolved CH4 gas. Two major reasons may be attributed to the observed behavior: (a) the growth rate of crystal-type MH, which depends on the rate of mass transfer of CH4 molecules to the MH crystal surfaces, and (b) recrystallization due to Oswald ripening of porous-type MHBub. A video showing the recrystallization process of porous-type MHBub is provided in Video S3 in the ESI.† It can be seen that the broad particle size distribution of porous-type MHBub crystals was consistent with the findings in our prior reactor-scale study.41
3.2. MH dissociation behavior at the pore-scale on the microfluidic chip
 | ||
| Fig. 7 (a) Evolution of P and T over time and (b) P–T trace in relation to the CH4 hydrate equilibrium curve in the process of MH dissociation induced by thermal stimulation to T = 287.8 K. Panels (c1)–(c3) show the dissociation process of MHBub, which was initiated at the center of the pore space, and the generation of a gas bubble (DG = 72.7 μm in c1) growing outward with a layer of expanding water film (DL = 81.8 μm and DG = 379.6 μm in c3). Panels (d1)–(d3) show the dissociation of MHDis under thermal stimulation, which was initiated in the pore throat near the grain surface, and the generation of a gas bubble (DG = 101.9 μm in d1). The dissociating MH crystal shows a meniscus surface and is in good accordance with the growing gas bubbles. New gas bubbles are generated in the vicinity of the MHDis crystal with a flock of gas bubbles aggregating in the pore space. | ||
Fig. 7c shows the images acquired at the bottom right in the pore space on the microfluidic chip (see Fig. 2c) during the porous-type MHBub dissociation process. The figure panel letter denotes the time stamp in Fig. 7a and b. Panel c1 depicts the initial MHBub dissociation at location 1 (see Fig. 3), where a relatively small gas bubble (DG = 72.7 μm) occurs at the center of MHBub in the pore space. Panel c2 shows the gas–liquid–MH distribution acquired 82.0 min after MHBub dissociation. It can be observed that gas bubbles grow outward with a layer of expanding water film (DL = 62.1 μm and DG = 303.2 μm). In addition, the dissociating porous-type MHBub exhibits a circular surface and is in good accordance with the growing gas bubbles. Panel c3 depicts the image acquired 103.5 min after initial MH dissociation in the pore space. It can be seen that the water film continues expanding to 81.8 μm, accompanied by a new gas bubble (DG = 30.3 μm) generated on the surface of the MHBub dissociation interface.
Fig. 7d depicts the images acquired in the pore throats on the microfluidic chip during the crystal-type MHDis dissociation process. Panel d1 depicts the initial crystal-type MHDis dissociation in the pore throats, where a gas bubble (DG = 101.9 μm) was generated near the grain surface. It was interesting to note that the dissociation time of crystal-type MHDis is much lower than that of porous-type MHBub. Panel d2 depicts the image acquired 149.3 min after initial MH dissociation, where the dissociating MH crystal shows a meniscus surface shrinking into the pore throat and is in good accordance with the growing gas bubbles. Panel d3 shows the image acquired 158.8 min after initial MH dissociation. It can be observed that new gas bubbles are generated in the vicinity of crystal-type MHDis with a flock of gas bubbles aggregating in the pore space.
A video showing the entire MH dissociation process of Fig. 7 is provided in Video S4 in the ESI.† As can be seen, once gas bubbles were generated in the local MH dissociation, the MH dissociated interface in this area preferred to dissociate along the gas bubble direction until the gas bubbles were far from the MH dissociated interface. Subsequently, new gas bubbles were formed at the boundary of the MH without preference. As a result, the CH4 concentration in the liquid phase decreases, and the dissociation rate increases.19 In addition, it can be observed that the crystal-type MHDis directly dissociates in the liquid without CH4 gas bubble generation in Video S4 in the ESI.† This indicates that the liquid phase was not fully saturated with CH4 or the solubility of CH4 is increasing. The final large gas bubbles are mainly collected in the pore space with a flock of small bubbles scattering in the pore throats, consistent with the initial gas–liquid distribution. However, the gas phase saturation (gas bubble volume divided by pore volume) in the field of view after MH complete dissociation was lower than the initial gas phase saturation. Two major reasons can be attributed to the observed behavior: (a) the gas bubbles generated from the dissociation of MH would tend to be more uniformly distributed on the microfluidic chip; (b) the dissolved CH4 concentration increases after MH complete dissociation.
 | ||
| Fig. 8 Microscopy images showing the evolution of gas bubbles during CH4 hydrate dissociation under thermal stimulation to T = 287.8 K. Panels (a1)–(a3) show that a gas bubble grows continuously with a layer of water film around it occupying the pore space (ΔT = 0–1.5 K). Panels (b1)–(b3) show that a flock of gas bubbles is generated in the pore space during MH dissociation (ΔT = 1.8 K) and coalescence with adjacent gas bubbles forming one large gas bubble in the pore space. | ||
Fig. 8b shows a flock of gas bubbles and their coalescence with adjacent gas bubbles under the same higher temperature driving force (ΔT = 1.8 K). Panel b1 shows the generation and aggregation of a flock of gas bubbles in the pore space. As can be seen, the largest bubble was 145.1 μm, and the smallest bubble was only 31.5 μm. It should be noted that the aggregation and coalescence of these bubbles depend on the stability of the liquid film. Panels b2 and b3 shows the liquid film rupture of adjacent gas bubbles after aggregation. It was intriguing to note that a relatively large gas bubble coalesces with the surrounding gas bubbles. In addition, multiple liquid drops are unevenly distributed in gas bubbles due to the rupture of liquid films between adjacent bubbles. Such gas bubble coalescence agrees well with the difference of surface tension in the two bubbles caused by the Young–Laplace effect.42
By comparison of gas bubble evolution under different temperature driving forces, one significant difference was the gas bubble size during MH dissociation. Two major reasons can be attributed to the observed behavior: (a) the MH dissociation rate under different temperature driving forces and (b) the diffusion rate and solubility of CH4 in the liquid phase. Under higher temperature driving force, the high CH4 release rate from the MH dissociation interface was greater than the rate of CH4 diffusion into gas bubbles. This causes dissolved CH4 concentration to pass a threshold in the H2O film, thus generating a gas bubble. Such gas bubble evolution was corroborated by the balance between the MH dissociation rate and CH4 diffusion rate which determines the formation of gas bubbles.43
In addition, a video showing the entire gas bubble evolution on the microfluidic chip is provided in Video S4 in the ESI.† As can be seen, the gas bubble coalescence can be divided into four stages: (a) formation of the gas bubble; (b) gas bubble aggregation and thin film formation; (c) liquid film drainage; (d) liquid film rupture and formation of a large gas bubble in the pore space. In addition, the rupture of the liquid film between adjacent large gas bubbles produces a disturbance that can induce further gas bubble coalescence.24 It should be noted that the capillary force was a crucial factor influencing the gas bubble distribution on the 2D microfluidic chip compared with gravity.44 It was very likely that the capillary effects involving interfacial tension and the geometric structure of the pore space and pore throat determined the gas bubble size and distribution.
Fig. 9a shows the evolution of the number and average size of gas bubbles during MH dissociation (the initial hydrate dissociation on local observation using panel b in Fig. 2 is defined as the initial time, 0 min). The extraction method of gas bubble evolution is shown in Fig. S8 in the ESI.† As can be seen, the development of gas bubbles is divided into a single gas bubble growth stage (0–100.0 min), a fast accumulation stage of a flock of gas bubbles (100.0–140.0 min), and a gas bubble coalescence stage (140.0–250.0 min). A video showing the entire gas bubble evolution on the microfluidic chip is provided in Video S5 in the ESI.†Fig. 9b shows the evolution of gas bubble diameter distribution at the end of each stage. Initially, a single gas bubble grows continuously, ranging from 0 to a maximum of 237.7 μm. The number of gas bubbles was only 3 within the initial 100.0 min, of which 66.7% were located at 200.0–400.0 μm. It contributed to the lower temperature driving force (ΔT = 0–1.2 K) in the initial 100.0 min.
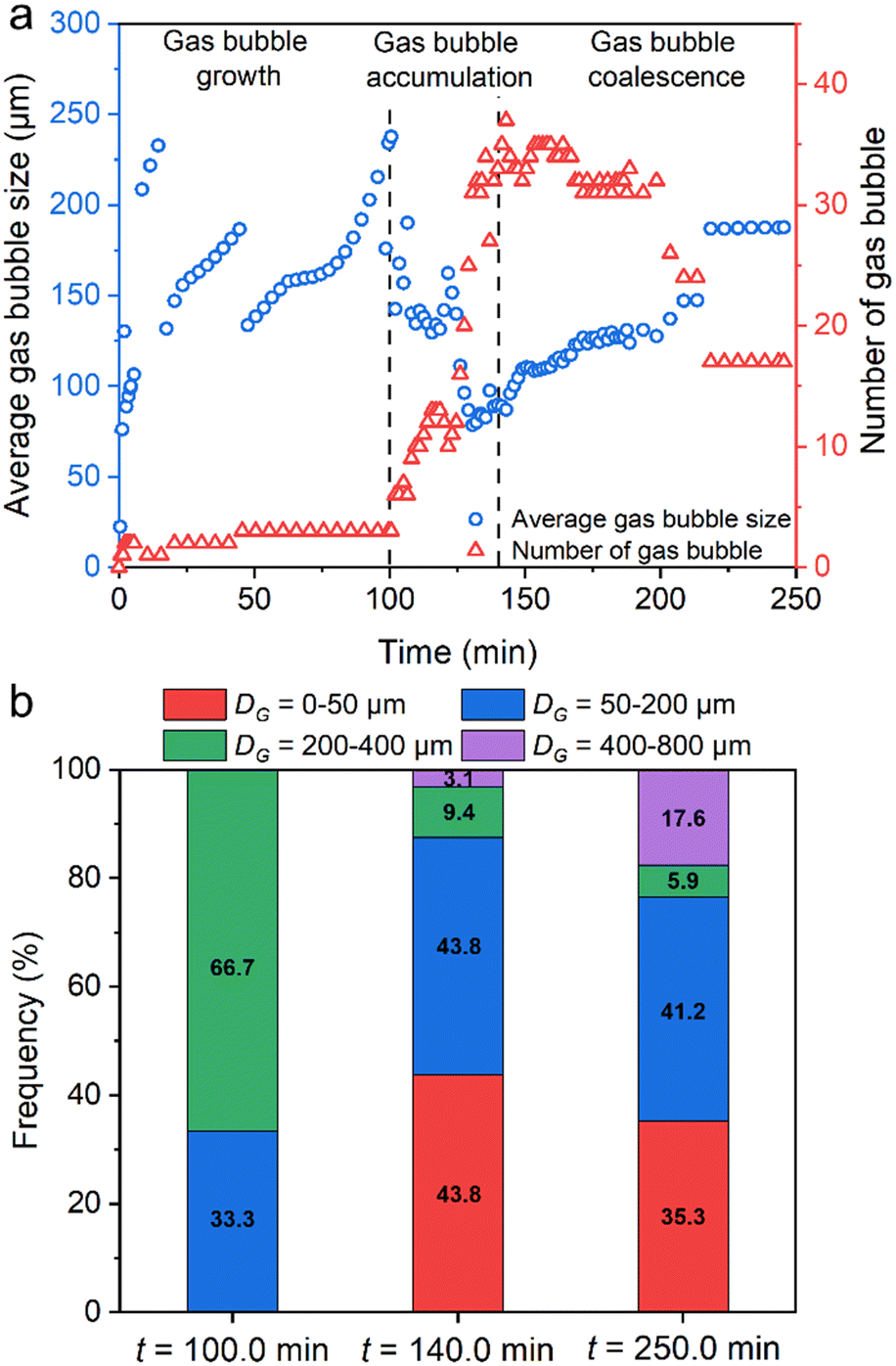 | ||
| Fig. 9 Evolution of the number of gas bubbles and the distribution of gas bubble diameter during MH dissociation based on local observation with panel b in Fig. 2 as a reference. Panel (a) shows three different stages of gas bubble evolution: single gas bubble growth stage (0–100.0 min), fast accumulation stage of flock of gas bubbles (100–140.0 min), and gas bubble coalescence stage. Panel (b) shows the evolution of gas bubble diameter distribution in each stage at td + 100.0 min, td + 140.0 min, and td + 250.0 min. | ||
In the gas bubble accumulation stage, a flock of gas bubbles were generated in the pore space (the peak number was 37), accompanied by a rapid drop in the average size of gas bubbles under high-temperature driving force (ΔT = 1.8 K). In addition, 43.8% of gas bubbles were located at 0–50.0 μm at 140.0 min. Moreover, the number of gas bubbles decreased slowly due to coalescence with adjacent gas bubbles. After the MH dissociation was completed, the total number of gas bubbles was 17, and the size of gas bubbles ranged from 20.1 μm to 769.7 μm with an average size of 187.7 μm on the microfluidic chip (35.3% with 6 bubbles in 0–50 μm, 41.2% with 7 bubbles in 50–200 μm, 23.5% with 4 bubbles in 200–800 μm). This result was consistent with the trend of the gas bubble evolution that has been mentioned in Fig. 8.
3.3. Two different MH formation mechanisms at the pore-scale
Fig. 10 shows the schematic of two different MH formation mechanisms and MH dissociation based on the above pore-scale observation on the microfluidic chip. Panels a1–a3 show the schematic of porous-type MHBub formation from CH4 gas bubbles. The initial gas–liquid distribution agrees well with the capillary effects involving interfacial tension, grain surface wettability, and porous media's geometric structure.45 MH formation from the CH4 gas bubble was preferred at the gas–liquid interface. In addition, the MH film surrounding the gas bubble serves as a mass transfer barrier of CH4 into H2O.46,47 However, sudden rupture of the MH film due to the local pressure difference inside and outside the gas bubble11 and the relatively low tensile strength of the MH film48 provides additional MH former gas. The release of remaining CH4 creates an additional gas–liquid interface and facilitates the continuous growth of co-existing gas–liquid–MH. The densification of the existing gas–liquid–MH completely turns into porous-type MH, dominated by the intrinsic kinetics and Ostwald ripening.10 | ||
| Fig. 10 Schematic of two different MH formation mechanisms and MH dissociation mechanisms. Panels (a1)–(a3) show the MHBub growth from the CH4 gas bubble in the pore space and throat. Panels (b1)–(b3) illustrate the MHDis growth from dissolved CH4 gas in the pore throat. Panels (c1)–(c3) depict the MH dissociation under thermal stimulation with different temperature driving forces. | ||
Panels b1–b3 depict the schematic of crystal-type MHDis formation from dissolved CH4 gas. Crystal-type MHDis formation from dissolved gas was a slow process controlled by the dissolution and diffusive mass transfer of CH4 molecules,35 essential for forming porous-type MHBub from isolated CH4 gas bubbles. A water film exists between the MH and solid particles due to the grain hydrophilic and pore geometric structures. The MH morphology difference between porous-type MHBub and crystal-type MHDis depends on the growth rate of MHs and the rate of mass transfer of CH4 molecules to the MH surfaces. In addition, the heterogeneous MH distribution in porous media was determined by the initial gas–liquid distribution, random nucleation sites, and CH4 gas dissolution.
Panels c1–c3 show the schematic of MH dissociation in the pore space and pore throat. Certain randomness exists in MH dissociation. Once MH dissociation begins, CH4 molecules escape from the broken hydrate cages and dissolve in the surrounding water phase.49 Gas bubbles form and grow when the concentration approaches the supersaturation limit.19 Additionally, the crystal-type MHDis formation from dissolved CH4 gas was more stable and required a longer time and higher driving force for dissociation. Two different evolution processes of gas bubbles consist of the growth of a single gas bubble and the coalescence of a gas bubble, which depend on the MH dissociation rate under different temperature driving forces.
4. Conclusions
Visualization of pore-scale MH formation and dissociation behavior and its effect on the pore structure and permeability are of significant interest to field-scale methane hydrate production tests. However, the challenges of in situ visualization techniques for the MH phase change limit further understanding. In this study, we developed a novel high-pressure microfluidic chip apparatus that is capable of direct observation of the MH formation behavior above the quadruple point. In addition, MH dissociation and gas bubble evolution under thermal stimulation were examined. The experimental results provide insights into the pore-scale MH formation and dissociation behavior, gas–liquid–MH interface, and gas bubble evolution. The high-quality images of gas–liquid–MH interfaces and gas bubble evolution can provide fundamental data for pore-scale CFD simulations of fluid flow and heat transfer. The following conclusions can be drawn:(a) Two different paths of MH formation were observed on the microfluidic chip, porous-type MHBub with a rough surface formed from CH4 gas bubbles and crystal-type MHDis formed from dissolved CH4 gas. The initial gas–liquid distribution, random nucleation sites, and CH4 gas dissolution determine the heterogeneous MH distribution in porous media;
(b) Porous-type MH formation from CH4 gas bubbles can be divided into four stages: (a) the MH film preferentially grows along the gas–liquid interface in pores; (b) the MH film collapses depending on the local pressure difference of the gas–liquid phase; (c) the release of remaining CH4 into the liquid phase facilitates the formation of co-existing gas–liquid–MH; (d) the densification of porous-type MH is dominated by the intrinsic kinetics and Ostwald ripening;
(c) Crystal-type MH formation from dissolved CH4 gas is a slow process controlled by the dissolution and diffusive mass transfer of CH4 molecules. Faceted MH crystals form and grow toward the shrinking gas bubble in the liquid phase. The growth and movement of crystal-type MH can trigger the sudden nucleation of porous-type MH;
(d) MH dissociation occurs at random sites in pores. The interface of MH exhibits a meniscus surface and is in good accordance with the growing gas bubbles. A layer of water film develops between the evolving gas bubble and dissociating bulk MH;
(e) Three distinct gas bubble evolution stages were observed under thermal stimulation: (a) single gas bubble growth with an expanding water layer at an initial slow dissociation rate; (b) quick generation of a flock of gas bubbles at a fast dissociate rate; (c) gas bubble coalescence with uniform distribution in the pore space with diameters ranging from 20.6–188.5 μm.
The experimental results provide valuable pore-scale understanding of MH formation and dissociation behavior in a static gas–liquid system in pores. It should be noted that depressurization is typically employed in the production of CH4 from NGH reservoirs.50 The evolution of the gas–liquid–MH interface and the generation of gas bubbles will be further examined in the depressurization scenario in our future studies. Future research also aims to integrate the high-pressure microfluidic technology with the in situ Raman testing technique to further investigate the micro-kinetics and the cage occupancy of the two different types of MHs observed in addition to CO2 hydrate51 and H2 hydrate52 for geological storage.
Author contributions
Jidong Zhang: conceptualization, formal analysis, data curation, writing – original draft. Zhenyuan Yin: conceptualization, methodology, formal analysis, writing – review & editing, supervision, funding acquisition. Saif A. Khan: investigation, formal analysis, writing – review & editing. Shuxia Li: investigation. Qingping Li: investigation. Xiaohui Liu: investigation, writing – original draft. Praveen Linga: investigation.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The financial support from the China National Science Foundation (52306254 and U21A20111), the Guangdong Basic and Applied Basic Research Foundation (2021A1515110755) and the Key Program of Marine Economy Development (Six Marine Industries) Special Foundation of Department of Natural Resources of Guangdong Province (GDOE[2022]43) are acknowledged. Z. Y. appreciates the financial support from the Ministry of Science and Technology of China (QN2022032009L), the Guangdong Pearl River Talent Program (2021QN02H836), and the Shenzhen Science and Technology Program (Grant No. RCBS20210609104537069), and the start-up fund from the Shenzhen High-level Talent Plan (QD2021011C). P. L. appreciates the Guangdong Foreign Talent Professor program.References
- E. D. Sloan Jr and C. A. Koh, Clathrate hydrates of natural gases, CRC press, 2007 Search PubMed.
- C. A. Koh and E. D. Sloan, AIChE J., 2007, 53, 1636–1643 CrossRef CAS.
- G. J. Moridis and E. D. Sloan, Energy Convers. Manage., 2007, 48, 1834–1849 CrossRef CAS.
- Z. Yin, Z. R. Chong, H. K. Tan and P. Linga, J. Nat. Gas Sci. Eng., 2016, 35, 1362–1387 CrossRef CAS.
- T. J. Kneafsey, Y. Seol, A. Gupta and L. Tomutsa, SPE J., 2010, 16, 78–94 CrossRef.
- G. Ersland, J. Husebø, A. Graue, B. Baldwin, J. Howard and J. Stevens, Chem. Eng. J., 2010, 158, 25–31 CrossRef CAS.
- M. Yang, Z. R. Chong, J. Zheng, Y. Song and P. Linga, Renewable Sustainable Energy Rev., 2017, 74, 1346–1360 CrossRef CAS.
- B. Pinho, Y. Liu, B. Rizkin and R. L. Hartman, Lab Chip, 2017, 17, 3883–3890 RSC.
- J. Zhang, X. Liu, D. Chen and Z. Yin, Int. J. Heat Mass Transfer, 2022, 192, 122901 CrossRef CAS.
- Z. Yin, M. Khurana, H. K. Tan and P. Linga, Chem. Eng. J., 2018, 342, 9–29 CrossRef CAS.
- S. Wang, Z. Cheng, Q. Liu, P. Lv, J. Lv, L. Jiang and Y. Song, Chem. Eng. J., 2021, 425, 130633 CrossRef CAS.
- B. Peng, A. Dandekar, C. Sun, H. Luo, Q. Ma, W. Pang and G. Chen, J. Phys. Chem. B, 2007, 111, 12485–12493 CrossRef CAS PubMed.
- Y. Li, J. Chen, A. M. Gambelli, X. Zhao, Y. Gao, F. Rossi and S. Mei, Chem. Eng. Sci., 2022, 248, 117230 CrossRef CAS.
- Q. Liu, Y. Liu, J. Xu, Y. Teng, Z. Ling, Y. Zhang, L. Jiang and Y. Song, Energy Rev., 2023, 2, 100011 CrossRef.
- S. A. Bagherzadeh, S. Alavi, J. Ripmeester and P. Englezos, J. Chem. Phys., 2015, 142, 214701 CrossRef PubMed.
- X. Li, C. Wang, Q. Li, W. Pang, G. Chen and C. Sun, Chem. Eng. Sci., 2022, 248, 117227 CrossRef CAS.
- X. Kou, J.-C. Feng, X.-S. Li, Y. Wang and Z.-Y. Chen, Energy, 2022, 239, 122230 CrossRef CAS.
- W. Liang, J. Wang and P. Li, Appl. Energy, 2022, 322, 119434 CrossRef CAS.
- T. Yagasaki, M. Matsumoto, Y. Andoh, S. Okazaki and H. Tanaka, J. Phys. Chem. B, 2014, 118, 1900–1906 CrossRef CAS PubMed.
- Q. Ouyang, J. S. Pandey and N. von Solms, Chem. Eng. J., 2023, 474, 145567 CrossRef CAS.
- J. S. Pandey, Ø. Strand, N. Von Solms, G. Ersland and S. Almenningen, Cryst. Growth Des., 2021, 21, 2793–2806 CrossRef CAS.
- Y. Ji, T. J. Kneafsey, J. Hou, J. Zhao, C. Liu, T. Guo, B. Wei, E. Zhao and Y. Bai, Fuel, 2022, 321, 124013 CrossRef CAS.
- A. Günther, S. A. Khan, M. Thalmann, F. Trachsel and K. F. Jensen, Lab Chip, 2004, 4, 278–286 RSC.
- D. Langevin, Curr. Opin. Colloid Interface Sci., 2015, 20, 92–97 CrossRef CAS.
- S. A. Khan, A. Günther, M. A. Schmidt and K. F. Jensen, Langmuir, 2004, 20, 8604–8611 CrossRef CAS PubMed.
- H. P. Veluswamy, A. J. H. Wong, P. Babu, R. Kumar, S. Kulprathipanja, P. Rangsunvigit and P. Linga, Chem. Eng. J., 2016, 290, 161–173 CrossRef CAS.
- R. Nosrati, M. Vollmer, L. Eamer, M. C. San Gabriel, K. Zeidan, A. Zini and D. Sinton, Lab Chip, 2014, 14, 1142–1150 RSC.
- D. Sinton, Lab Chip, 2014, 14, 3127–3134 RSC.
- S. Morais, A. Cario, N. Liu, D. Bernard, C. Lecoutre, Y. Garrabos, A. Ranchou-Peyruse, S. Dupraz, M. Azaroual and R. L. Hartman, React. Chem. Eng., 2020, 5, 1156–1185 RSC.
- M. Lysyy, N. Liu, C. M. Solstad, M. A. Fernø and G. Ersland, Int. J. Hydrogen Energy, 2023, 48, 31294–31304 CrossRef CAS.
- W. Chen and R. L. Hartman, Energy Fuels, 2018, 32, 11761–11771 CrossRef CAS.
- W. Chen, B. Pinho and R. L. Hartman, Lab Chip, 2017, 17, 3051–3060 RSC.
- S. Almenningen, J. Flatlandsmo, A. R. Kovscek, G. Ersland and M. A. Fernø, Lab Chip, 2017, 17, 4070–4076 RSC.
- X. Li, M. Liu, Q. Li, W. Pang, G. Chen and C. Sun, Fuel, 2023, 332, 125980 CrossRef CAS.
- X. Kou, J.-C. Feng, X.-S. Li, Y. Wang and Z.-Y. Chen, Appl. Energy, 2022, 306, 118015 CrossRef CAS.
- J. Zhang, Z. Yin, Q. Li, S. Li, Y. Wang and X.-S. Li, Energy, 2023, 128315 CrossRef CAS.
- J. Ren, Z. Yin, Q. Li, F. Wu, D. Chen and S. Li, Energy Fuels, 2022, 36, 14874–14887 CrossRef CAS.
- M. Li, S. Fan, Y. Wang, X. Lang and G. Li, AIChE J., 2023, e18069 CrossRef CAS.
- X. Liu, Y. Li, G.-J. Chen, D.-Y. Chen, B. Sun and Z. Yin, ACS Sustainable Chem. Eng., 2023, 11, 6057–6069 CrossRef CAS.
- X. Fu, J. Jimenez-Martinez, T. P. Nguyen, J. W. Carey, H. Viswanathan, L. Cueto-Felgueroso and R. Juanes, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 31660–31664 CrossRef CAS PubMed.
- J. Ren, S. Zeng, D. Chen, M. Yang, P. Linga and Z. Yin, Appl. Energy, 2023, 340, 120997 CrossRef CAS.
- P. Stevenson, Curr. Opin. Colloid Interface Sci., 2010, 15, 374–381 CrossRef CAS.
- S. A. Bagherzadeh, S. Alavi, J. Ripmeester and P. Englezos, J. Chem. Phys., 2015, 142, 214710 CrossRef PubMed.
- Z. Huang, M. Su, Q. Yang, Z. Li, S. Chen, Y. Li, X. Zhou, F. Li and Y. Song, Nat. Commun., 2017, 8, 14110 CrossRef CAS PubMed.
- E. Spangenberg, M. Priegnitz, K. Heeschen and J. M. Schicks, J. Chem. Eng. Data, 2015, 60, 258–268 CrossRef CAS.
- S. R. Davies, E. D. Sloan, A. K. Sum and C. A. Koh, J. Phys. Chem. C, 2010, 114, 1173–1180 CrossRef CAS.
- A. Touil, D. Broseta and A. Desmedt, Langmuir, 2019, 35, 12569–12581 CrossRef CAS PubMed.
- D. Atig, D. Broseta, J.-M. Pereira and R. Brown, Nat. Commun., 2020, 11, 3379 CrossRef CAS PubMed.
- S. A. Bagherzadeh, S. Alavi, J. A. Ripmeester and P. Englezos, Fluid Phase Equilib., 2013, 358, 114–120 CrossRef CAS.
- Z. R. Chong, Z. Yin, J. H. C. Tan and P. Linga, Appl. Energy, 2017, 204, 1513–1525 CrossRef CAS.
- X. Liu, J. Ren, D. Chen and Z. Yin, Chem. Eng. J., 2022, 438, 135504 CrossRef CAS.
- J. Zhang, Y. Li, Z. Yin, P. Linga, T. He and X. Y. Zheng, Chem. Eng. J., 2023, 467, 143459 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lc00950e |
| This journal is © The Royal Society of Chemistry 2024 |