
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Standalone cell culture microfluidic device-based microphysiological system for automated cell observation and application in nephrotoxicity tests†
Hiroshi
Kimura
 *a,
Hiroko
Nakamura
a,
Tomomi
Goto
a,
Wakana
Uchida
b,
Takayuki
Uozumi
b,
Daniel
Nishizawa
a,
Kenta
Shinha
a,
Junko
Sakagami
b and
Kotaro
Doi
c
*a,
Hiroko
Nakamura
a,
Tomomi
Goto
a,
Wakana
Uchida
b,
Takayuki
Uozumi
b,
Daniel
Nishizawa
a,
Kenta
Shinha
a,
Junko
Sakagami
b and
Kotaro
Doi
c
aMicro/Nano Technology Center, Tokai University, Kanagawa, Japan 259-1292. E-mail: hkimura@tokai-u.jp
bStem Cell Healthcare Business Unit, Nikon Corporation, Kanagawa, Japan
cInstitute of Industrial Science, The University of Tokyo, Tokyo, Japan 153-8505
First published on 22nd December 2023
Abstract
Microphysiological systems (MPS) offer an alternative method for culturing cells on microfluidic platforms to model organ functions in pharmaceutical and medical sciences. Although MPS hardware has been proposed to maintain physiological organ function through perfusion culture, no existing MPS can automatically assess cell morphology and conditions online to observe cellular dynamics in detail. Thus, with this study, we aimed to establish a practical strategy for automating cell observation and improving cell evaluation functions with low temporal resolution and throughput in MPS experiments. We developed a versatile standalone cell culture microfluidic device (SCCMD) that integrates microfluidic chips and their peripherals. This device is compliant with the ANSI/SLAS standards and has been seamlessly integrated into an existing automatic cell imaging system. This integration enables automatic cell observation with high temporal resolution in MPS experiments. Perfusion culture of human kidney proximal tubule epithelial cells using the SCCMD improves cell function. By combining the proximal tubule MPS with an existing cell imaging system, nephrotoxicity studies were successfully performed to automate morphological and material permeability evaluation. We believe that the concept of building the ANSI/SLAS-compliant-sized MPS device proposed herein and integrating it into an existing automatic cell imaging system for the online measurement of detailed cell dynamics information and improvement of throughput by automating observation operations is a novel potential research direction for MPS research.
Introduction
In the last decade, microphysiological systems (MPS) utilizing microfluidic technology have been proposed as an alternative to animal experiments, and their practical applications have been extensively studied in pharmaceutical and medical sciences.1 MPS, including organ-on-a-chip, are in vitro experimental systems that are employed for constructing physiological organ models by culturing cells using microfluidics.2 The recent declaration by the U.S. Environmental Protection Agency to essentially eliminate animal testing by 2035 and the U.S. Food and Drug Administration promulgation of the Modernization Act 2.0, which will not require animal testing before clinical trials by the end of 2022, have also accelerated the consideration of the practical applications of MPS for research.3 MPS have been proposed for various organs and tissues, including the liver, gut, kidney, and blood–brain barrier (BBB) to study their functions in relation to absorption, distribution, metabolism, and excretion, especially in the context of drug discovery.2,4–9The greatest advantage of microfluidic chip-based MPS is the maintenance of cultured cell function through fluidic shear stress (FSS) generated by the flow of the medium. The proximal tubules in the nephron—the smallest functional unit of the kidney—are among the tissues in which the effects of FSS have been extensively studied using MPS, as they are exposed to the flow of primary urine filtered by the glomerulus. In the early days of proximal tubule MPS (PT-MPS) research, Madin–Darby canine kidney (MDCK) cells and an immortalized proximal tubule epithelial cell line (HK-2) were cultured at the bottom of microchannels to investigate the effects of FSS on these cells.10–12 These studies showed that arbitrary FSS loading caused morphological changes, such as increased Na/K ATPase expression and enhanced cell ciliogenesis. A bilayer-type microfluidic chip incorporating a porous membrane was developed to model polar transport functions such as reabsorption and secretion in the proximal tubule.13,14 Jang et al. used a bilayer-type microfluidic chip to culture tubule-derived cells on a porous membrane, applied FSS to induce cell polarization, and observed a physiological response in which the administration of hormones, such as vasopressin or aldosterone, to the vascular channel alters osmolarity and sodium concentration in the tubule channel. Sakolish et al. cultured HK-2 cells in perfusion using a bilayer-type microfluidic chip to clarify the effect of the fluidic microenvironment on the development of renal disease;15 the results revealed that stress-related responses, such as the expression of α-smooth muscle actin and alkaline phosphatase activity, differed between static and fluidic culture conditions. Sadeghian et al. constructed a highly physiological PT-MPS by mixing tubular epithelial cells isolated from human-induced pluripotent stem cell (hiPSCs)-derived kidney organoids with human renal proximal tubule epithelial cells (RPTEC/TERT1 cells) and cultured them in perfusion using a bilayer-type microfluidic chip.16 Thus, several previous studies have demonstrated the usefulness of perfusion culture using MPS.
Many MPS have been developed as in vitro organ models for drug toxicity and pharmacokinetic evaluation in the drug discovery process.17,18 However, the low temporal resolution and the time and effort required to observe cell kinetics remain an issue in toxicity and kinetic studies of drugs under perfusion culture using MPS. To investigate the details of cell kinetics in toxicity and kinetic studies, observing cell conditions in the environment of drug exposure with high temporal resolution in minutes to hours and over the long term in hours to days is necessary. The MPS device for perfusion culture proposed thus far is in a format where microfluidic chips are connected to pumping systems, making it difficult to observe with a microscope or other measurement systems under perfusion culture conditions continuously. Therefore, in MPS experiments, the evaluation of cell conditions is limited to gene-level assays, optical cell observations such as bright-field imaging or immunostaining, and quantitative evaluation of compounds with low temporal resolution. To the best of our knowledge, no studies have achieved high temporal resolution and long-term cell observation in MPS under perfusion culture conditions.
Thus, in this study, we aimed to develop a standalone cell culture microfluidic device (SCCMD) that integrates a microfluidic chip and its peripherals into a versatile ANSI/SLAS-compliant-sized system for online cell observation in perfusion cell culture systems in MPS experiments. We automated cell observation in MPS experiments by integrating the developed SCCMD into an existing automated cell imaging system—BioStation CT (Nikon, Tokyo, Japan). We adapted this system to PT-MPS to validate the function of the cell observation system by combining the SCCMD and BioStation CT. First, perfusion cell culture of RPTEC/TERT1 cells with the SCCMD was shown to improve cell function. We used the proposed system to perform nephrotoxicity studies and successfully conducted online automated morphological and material permeability assessments of human RPTECs. We anticipate this concept of building the ANSI/SLAS-compliant-sized MPS device proposed herein and integrating it into existing automated cell imaging systems in order to enable the online measurement of detailed cell dynamics information and improve throughput by automating observation operations to pave the way for a novel direction in MPS research.
Materials and methods
Standalone cell culture microfluidic device (SCCMD)
We developed an ANSI/SLAS-compliant SCCMD with perfusion cell culture components for online cell observation under perfusion culture conditions using a microscope. The SCCMD, composed of microfluidic chips and a fluidic platform, was developed to operate independently at cell observation within an existing automatic cell imaging system [Fig. 1(a)].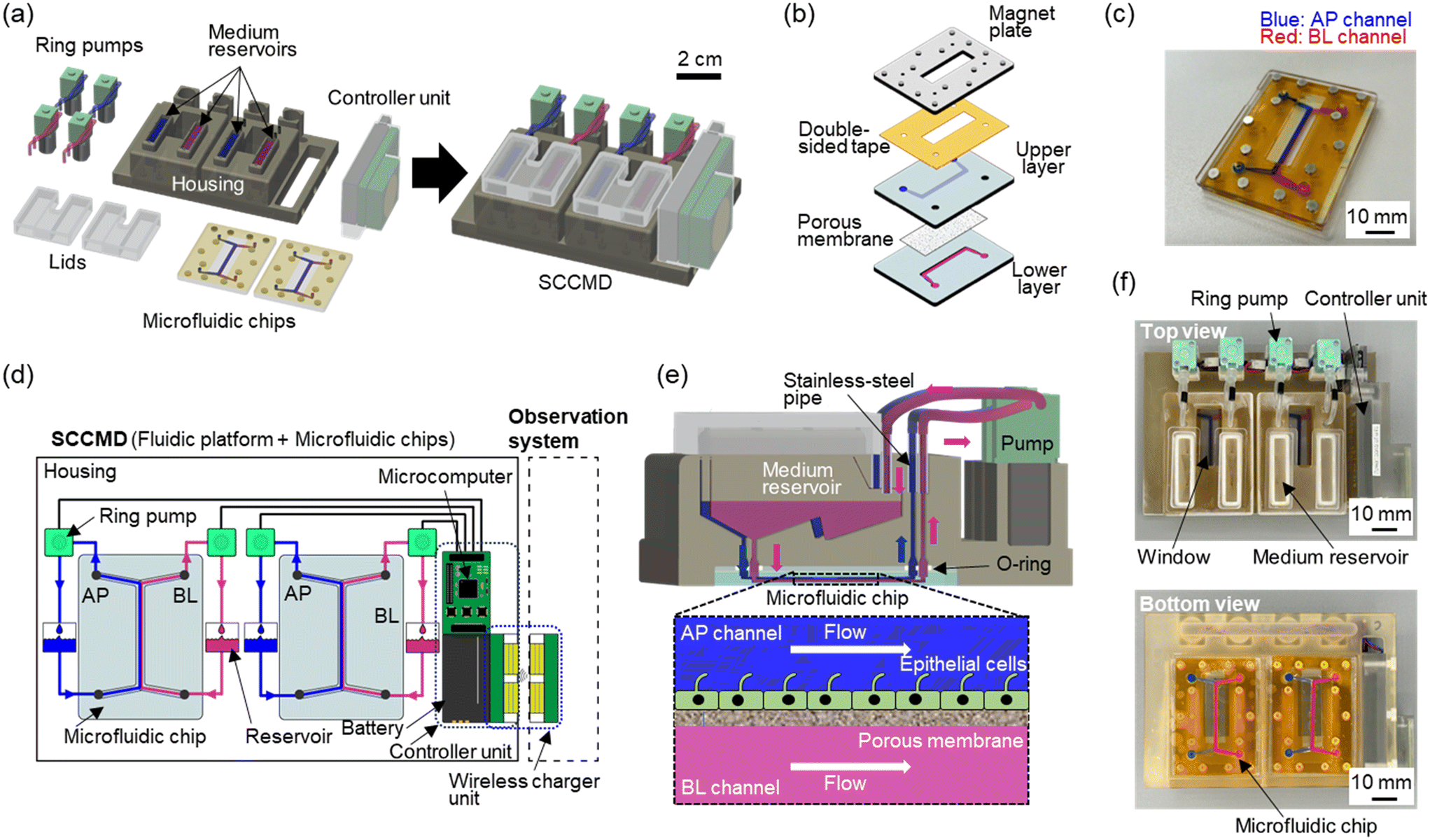 | ||
| Fig. 1 Standalone cell culture microfluidic device (SCCMD). (a) Schematic of the SCCMD components. The SCCMD integrates a controller unit, ring pumps, and microfluidic chips into a housing with medium reservoirs and plumbing. (b) Schematic of the exploded view of the bilayered microfluidic chip. The chip consisted of a porous membrane sandwiched between polydimethylsiloxane (PDMS) channel layers. The chip was further bound to the magnet plate using double-sided tape. (c) Photograph of the fabricated microfluidic chip. Blue: apical (AP) channel; red: basal (BL) side channel. (d) Fluidics and electrical schematic of the SCCMD. The fluidic circuits of the culture medium connected to each microchannel are independent. The controller unit consisted of a battery, microcomputer, and power receiving unit of a wireless charger unit. (e) Cross-sectional view of the SCCMD and cell culture sections of the microfluidic chip. The culture medium in the medium reservoir flows into the microchannel through a port at the bottom of the reservoir. It returns to the reservoir via a plumbing in the housing and ring pumps. Epithelial cells have a primary serial that acts as a mechanosensor for fluidic shear stress (FSS). (f) Top and bottom views of the SCCMD with microfluidic chips. The top side has four independent medium reservoirs, four ring pumps, a controller unit, and two windows for cell observation. Two microfluidic chips were attached using magnets on the underside. | ||
Microfluidic chip
A microfluidic chip for cell culture typically has bilayered microchannels separated by a porous membrane [Fig. 1(b)]. The microchannels were 0.6 mm high and 2.0 mm wide, with the upper and lower channels connected to the inlet and outlet ports 4 mm in diameter. The bottom area of the microchannel was approximately 1.0 cm2, and the culture area of the porous membrane was approximately 0.7 cm2. The microfluidic chip had a magnetic plate on the top surface to set it on the fluidic platform.Polydimethylsiloxane (PDMS; SILPOT184; Dow Toray, Tokyo, Japan) layers with microchannel structures were fabricated using conventional soft lithography.19 Polymethyl methacrylate (PMMA) molds for soft lithography were fabricated by machine cutting using a 3D modeling machine (MODELA MDX-50, Roland, Shizuoka, Japan). After machining, the surfaces of the PMMA molds were polished using an abrasive (12100; Nihon Maryo Kogyo, Tokyo, Japan) to obtain a smooth surface. A 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of unpolymerized PDMS and catalyst was poured into the PMMA mold and baked in an oven at 75 °C for 2 h to fabricate PDMS layers with a microchannel structure.
:1 mixture of unpolymerized PDMS and catalyst was poured into the PMMA mold and baked in an oven at 75 °C for 2 h to fabricate PDMS layers with a microchannel structure.
For permanent bonding to the PDMS layers, the surface of the polyethylene terephthalate porous membrane (pore size: 0.45 μm in diameter, 2000M12/640N453/A4, it4ip, Louvain-la-Neuve, Belgium) was coated with 6.7% (v/v) 3-aminopropyltriethoxysilane solution (KBE-903, Shin-Etsu Chemical, Tokyo, Japan) and 2.5% (v/v) glutaraldehyde solution (17026-32, Kanto Chemical, Tokyo, Japan) after plasma treatment using a plasma cleaner (PDC-32G, Harrick Plasma, Ithaca, NY, USA). Holes (4 mm in diameter) were made in the PDMS layer on the upper microchannel side using a biopsy punch (BP-40F, Kai Medical, Gifu, Japan) at the inlet and outlet ports of the upper- and lower-layer microchannels. The microfluidic chip was assembled by sandwiching a porous membrane between the plasma-treated PDMS layers. The microfluidic chip was then attached to a PMMA plate (thickness: 2 mm) with a neodymium magnet using double-sided tape (760H #25; Teraoka Seisakusho, Tokyo, Japan) [Fig. 1(c)].
Fluidic platform
The fluidic platform comprised a housing, four ring pumps (RP-Q1.5S-P45Z-DC3V, Aquatech, Osaka, Japan), and a controller unit integrated with a microcomputer for pump control, a lithium-ion battery, and a wireless charger [Fig. 1(d)]. All these components were organized within the ANSI/SLAS-compliant-sized housings embedding a plumbing and four independent medium reservoirs. Two microfluidic chips were mounted at the bottom of the fluidic platform using neodymium magnets for ease of setting. Hence, a single fluidic platform could independently control the upper and lower channels of the two microfluidic chips.The medium in the reservoir was perfused using ring pumps and discharged back into the reservoir through the plumbing in the housing, microfluidic chip, and ring pump [Fig. 1(e)]. Because the ring pump was DC motor-driven, the medium flow was controlled individually by changing the direct current (DC) power supply voltage or the pulse-width modulation setting of the microcontroller.
The housing of the fluidic platform was designed using 3D computer-aided design software program (AutoCAD, Autodesk, San Rafael, CA, USA) and fabricated via the 3D printing of an acrylic resin curable using ultraviolet (UV) rays. The 3D-printed housings were thoroughly cleaned with acetone, isopropanol, and ultrapure water using an ultrasonic cleaner to reduce material-induced cytotoxicity and autoclaved as a finishing treatment. After autoclaving, stainless steel pipes (O.D. 2 mm and I.D. 1 mm; SPLS2-15, Misumi, Tokyo, Japan) were attached to the ring pumps, neodymium magnets were attached using glue (235021, Henkel, Düsseldorf, Germany), and screws were attached to secure the microfluidic chips to the bottom of the housing.
The controller unit consisted of a microcontroller for pump control, lithium-ion battery, and power receiver for a wireless charger, and was custom-made by Maxell (Tokyo, Japan). A DC power supply (DCP3005, As One, Osaka, Japan) was used instead of a controller unit for the perfusion culture experiments without a cell observation system.
All functional components required for standalone perfusion cell culture were fully integrated into the housing of the ANSI/SLAS-compliant SCCMD [Fig. 1(f)]. The fluidic platform was sterilized with ethylene oxide gas (EOG) prior to cell culture experiments.
Cell preparation
RPTEC/TERT1 cells were obtained from the American Type Culture Collection (CRL-4031, ATCC, Manassas, VA, USA) and cultured in a hormonally defined, serum-free medium consisting of Dulbecco's modified Eagle's medium F12 (30-2006, ATCC, Manassas, VA, USA) supplemented with a growth kit (ACS-4007, ATCC, Manassas, VA, USA) and 0.1 mg mL−1 G418 sulfate (10131035; Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C and 5% CO2.The cells were cultured in tissue culture-treated dishes (100 mm diameter) prior to cell culture experiments. The culture medium was changed on alternate days and the cells were passaged at 70% confluence. No more than four passages were used for the culture experiments.
Cell culture in the SCCMD
Cell seeding was performed using only the microfluidic chip, without the fluidic platform. The microfluidic chip was sterilized using UV irradiation (20 W, NB-5, Nichiban, Osaka, Japan) for 20 min. The microchannels were coated with laminin 511 (892018; Nippi, Tokyo, Japan) at 37 °C for 1 h. The cells were seeded at a density of 2.0 × 105 cells per cm2 in the upper channel inlet of the microfluidic chip using a micropipette, and the lower channel was filled with the medium in the same manner. After seeding the cells, the microfluidic chip was placed in a 100 mm dish and incubated at 37 °C and 5% CO2 for approximately 4 d under static conditions. During static culture, the medium in the microfluidic channels was changed daily. After culturing the cells in this manner, the upper and lower microchannels were defined as apical (AP) and basal (BL) channels, respectively.After the cells formed a monolayer on the membrane under static conditions, the microfluidic chip was magnetically attached to the fluidic platform with a handmade 1.5 mm thick silicone O-ring at the ports. After 1.5–2.0 mL of medium was introduced into each reservoir, the cells were perfusion-cultured using ring pumps and maintained with medium changes on alternate days. The PT-MPS was constructed as an in vitro proximal tubule model using these operations (ESI† Appendix, Movie S1).
The FSS τ was calculated using the following equation:13,16
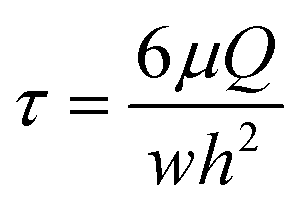 | (1) |
Real-time polymerase chain reaction (PCR)
Total RNA from the cells in the microchannels was collected using TRIzol (15596018, Thermo Fisher Scientific, Waltham, MA, USA) after 14 days of culture and isolated using Direct-zol RNA Microprep (R2062, Zymo Research, Irvine, CA, USA). After measuring the amount of RNA using a NanoDrop Lite spectrometer (ND-LITE; Thermo Fisher Scientific, Waltham, MA, USA), 1 μg of total RNA was reverse-transcribed using the iScript cDNA synthesis kit (1708891, Bio-Rad, Hercules, CA, USA) according to the manufacturer's instructions. Subsequently, complementary DNA was used for real-time PCR. All samples were tested with five (with-FSS) and three (without-FSS) biological replicates, and two technical replicates. The gene expression levels of MATE1, MATE2K, P-gp/MDR1, aquaporin-1, E-cadherin, and PPIA were evaluated using quantitative real-time PCR with TaqMan probes (ESI† Appendix, Table S1) and TaqMan Gene Expression Master Mix (4369016, Thermo Fisher Scientific, Waltham, MA, USA) on a CFX Connect real-time PCR detection system (Bio-Rad, Hercules, CA, USA). Gene expression levels were normalized with the expression level of PPIA (reference gene), and the calculated fold differences were compared with the gene expression levels in the cell culture without FSS.Immunofluorescence
The cells cultured in the microfluidic chips were fixed with 4% paraformaldehyde for 10 min at room temperature and rinsed gently with phosphate-buffered saline (PBS). After fixation, the cells were gently rinsed three times with PBS and permeabilized with 0.1% Triton X-100 in PBS for 5 min at room temperature. The cells were rinsed thrice with PBS and blocked with 1% bovine serum albumin (BSA) in PBS for 1 h. The primary antibodies, mouse anti-acetylated tubulin monoclonal antibody (1:200, T7451, Merck, Darmstadt, Germany), rabbit anti-ZO-1 polyclonal antibody (1:500, 21773-1-AP, Proteintech, Rosemont, IL, USA), mouse anti-OCT2 monoclonal antibody (1:100, MAB6547, R&D Systems, Minneapolis, MN, USA), rabbit anti-MATE1 polyclonal antibody (1:100, HPA021987, Merck, Darmstadt, Germany), and rabbit anti-aquaporin-1 monoclonal antibody (1:500, b219055, Abcam, Cambridge, UK), were incubated overnight at 4 °C in 1% BSA/PBS. The secondary antibodies, goat anti-mouse IgG conjugated with Alexa Fluor 488 (A-11029, Thermo Fisher Scientific, Waltham, MA, USA), goat anti-rabbit IgG conjugated to Cy3 (525-33371, Jackson ImmunoResearch, Philadelphia, PA, USA), and 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI; 340-07971, Dojindo, Kumamoto, Japan), were mixed in 1% BSA/PBS and incubated for 1 h at room temperature. After the final rinsing step with PBS, the membranes were cut from the microfluidic chips and mounted on glass slides using Vectashield (H-1700, Vector Laboratories, Newark, CA, USA).
After immunofluorescence staining, digital fluorescence images were acquired using a confocal microscope (A1R, Nikon, Tokyo, Japan) with NIS-Elements AR image analysis software (Nikon, Tokyo, Japan) at the Tokai University Imaging Center for Advanced Research. Z-Stack images were acquired with a confocal microscope in 0.3 μm steps and processed using maximum intensity projection. Because the fluorescence acquisition parameters of the microscope (such as laser power, detector sensitivity, and z-stack steps) were similar when observing the same protein, the fluorescence intensities of the acquired images under different experimental conditions (with and without FSS) could be compared qualitatively.
Automatic cell imaging system
BioStation CT (Nikon, Tokyo, Japan) was used as the base of the cell observation system to automate cell imaging using the MPS. BioStation CT is an incubator-based time-lapse system with a storage rack that can hold up to 30 ANSI/SLAS-compliant plates inside an incubator, a transport unit for transporting the plates, and an observation unit for microscopic observations. To operate the SCCMD, the volume contrast (VC) imaging module (Nikon, Tokyo, Japan) and five transmitters of the wireless charger unit (Maxell, Tokyo, Japan) were installed in the automatic cell imaging system [Fig. 2(a) and (b)]. | ||
| Fig. 2 BioStation CT-based cell imaging system. (a) Schematic representation of the system consisting of an incubator and observation parts. The power supply side of the wireless charger unit was installed inside the incubator. (b) Photographs showcasing the system and interior of the incubator. (c) Photograph of the standalone cell culture microfluidic device (SCCMD) with a holder for the system. (d) Photographs of the SCCMD, which is stored in storage racks, moved using the transport unit, and observed at the observation stage in the incubator. | ||
The transmitters were installed at a position aligned with the receiver of the SCCMD on the storage rack sidewall of the automatic cell imaging system. The transmitters were connected to a DC power supply and ready to supply power.
After preparing the cell culture, the SCCMD was mounted on a BioStation CT-specific holder and set on a storage rack [Fig. 2(c)]. Whenever the SCCMD was in the storage rack, the battery was charged wirelessly. The SCCMD was periodically transported from the storage rack to the observation stage by the transport unit at regular intervals [Fig. 2(d); ESI† Appendix, Movie S2]. The power supply from the battery drove the ring pump to keep the perfusion of the culture medium during imaging on the observation stage. This allowed for VC imaging of the cells and fluorescence observation inside the microchannel automatically under perfusion culture conditions.
VC imaging using the automatic cell imaging system
The VC imaging module produces a quantitative phase image from multiple bright-field Z-stack images20 and is expected to enable the observation of cells on porous membranes, which are usually difficult to observe using traditional light microscopy. To acquire VC images, 13 z-stack images of the cells were captured at 5 μm pitch with a 10× objective lens per observation point in the microchannel. The captured images were converted to VC images using NIS-Elements AR image analysis software. Cell numbers and intercellular spaces were quantified from VC images using the general analysis module of the NIS-Elements AR.Proximal tubule toxicity assay
The proximal tubules are among the most common targets of nephrotoxic drugs and chemicals. A toxicity assay using CDDP (P4394, Sigma-Aldrich, Burlington, MA, USA) was performed to evaluate the function of our system. RPTEC/TERT1 cells, which were precultured for approximately 4 d with the SCCMD, were exposed to CDDP concentrations of 0, 6.25, 12.5, 25, 50, and 100 μM in culture medium for 3 d under perfusion conditions. During CDDP exposure, cell toxicity was evaluated based on changes in cell morphology, with VC images obtained automatically every 3 h using the automatic cell imaging system.As another toxicity assessment method other than VC imaging, the paracellular marker lucifer yellow (LY, Wako, 128-06271, FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan) was used to evaluate monolayer permeability using time-lapse fluorescence images in microchannels obtained using the automatic cell imaging system. LY (200 μM) was introduced only into the medium on the upper channel side and perfused simultaneously with CDDP. During the 3 d of the toxicity assay, fluorescence images of the upper and lower channels were acquired every 3 h using the time-lapse function of the automatic cell imaging system. The fluorescence intensity of the images was quantified using CL-Quant (Nikon, Tokyo, Japan) image analysis software. LY concentration was determined from the fluorescence intensity using a calibration curve for LY obtained using the same experimental setup [ESI† Appendix, Fig. S1].
The values of the median effect concentration (EC50) were estimated using the following equation based on a four-parameter logistic model, which is an extension of the sigmoid Emax model presented by Schwinghammer et al. as a nonlinear model, using GraphPad Prism9 (GraphPad Software, San Diego, CA, USA).21
 | (2) |
Statistical analysis
All values are expressed as the mean ± standard deviation from multiple technical replicates, using at least three biologically independent experiments. Student's t-tests were performed for comparisons between the two groups in the FSS effect evaluation. Analysis of variance with Tukey's honest significant difference test was performed to compare three or more groups in the toxicity assay. Differences were considered statistically significant at p < 0.05.Results and discussion
SCCMD performance for perfusion cell culture
In this study, we developed an ANSI/SLAS-compliant sized SCCMD as a platform for automatic cell imaging under continuous medium perfusion conditions for MPS experiments. The medium perfusion circuit of the SCCMD consisted of microfluidic chips, housing embedding plumbing and medium reservoirs, and ring pumps. A magnetic crimp across the silicone O-ring connected the microfluidic chip port and housing plumbing. The housing plumbing port and silicone tubes of the ring pumps were connected via stainless steel pipes [Fig. 1(e)]. Owing to the use of this setup, no leakage of the culture medium occurred, and the culture medium could be perfused stably.The fluidic platform was sterilized using EOG. The microfluidic chip and O-rings were disposable, and the fluidic platform including the ring pumps was reused after washing and sterilizing with EOG. The SCCMD did not require tubing connection operation for medium change because all fluidic control functions, such as ring pumps and medium reservoirs, were integrated into the fluidic platform. The culture medium was easily replaced in the medium reservoir and sampled in the same manner as in conventional cell culture plates. In other words, the loading, removal, and sampling of culture media and reagents were simple. The integrated fluid control function simplified MPS handling and could significantly reduce the risk of contamination.
Microfluidic chips with cells were easily mounted and unmounted on the fluidic platform using magnets when the perfusion culture was started and finished. After the perfusion cell culture experiments, the microfluidic chips could be removed from the fluidic platform for immunostaining and cell sampling.
The SCCMD had the same ANSI/SLAS-compliant size as conventional multi-well plates; it was designed to be compatible with various existing observation and measurement systems such as microplate readers and microscope stage-top incubators. In this study, we operated the SCCMD using an automatic cell imaging system for observation of cells under continuous perfusion cell culture conditions for toxicity assessments (see below for detailed information).
Although some MPS platforms have already been proposed and marketed, standalone devices that integrate all fluid control functions in an ANSI/SLAS-compliant-sized system for online observation are yet to be developed. Typical MPS devices, such as OrganoPlate®,22 PhysioMimix®,23 and PD-MPS,24–26 have the same ANSI/SLAS-compliant-size as conventional multi-well plates for easy handling. However, these MPS devices require external devices such as shakers and pneumatic controls with tubing for perfusion cell culture. The MPS device of the Emulate system is not ANSI/SLAS-compliant, but its basic setup is similar to that of other MPS devices.27 Therefore, it is impossible to use these MPS devices to observe cells while perfusing the cell culture medium. In other words, online cell observation under perfusion culture conditions using existing observation equipment is not possible with these MPS devices, and it is difficult to automate the observation and improve the temporal resolution. The SCCMD was compatible with existing observation and analysis equipment designed for multi-well plates by integrating fluid control functions and the microfluidic chip in the ANSI/SLAS-compliant size, which not only enabled the automation of observation but also significantly reduced the need for dedicated control systems.
Validation of the FSS effect on RPTEC/TERT1 using the SCCMD
First, PT-MPS was established to investigate the effect of FSS on the human PRTEC line using only the SCCMD to evaluate the perfusion cell culture function of the SCCMD. RPTEC/TERT1 cells were cultured for 10 days under FSS conditions (w/FSS) with both the upper (AP) and lower (BL) channels with or without FSS conditions (w/o FSS) with only the lower channel flow using the SCCMD [Fig. 3(a)]. Cell function was evaluated from multiple perspectives using real-time PCR and immunostaining.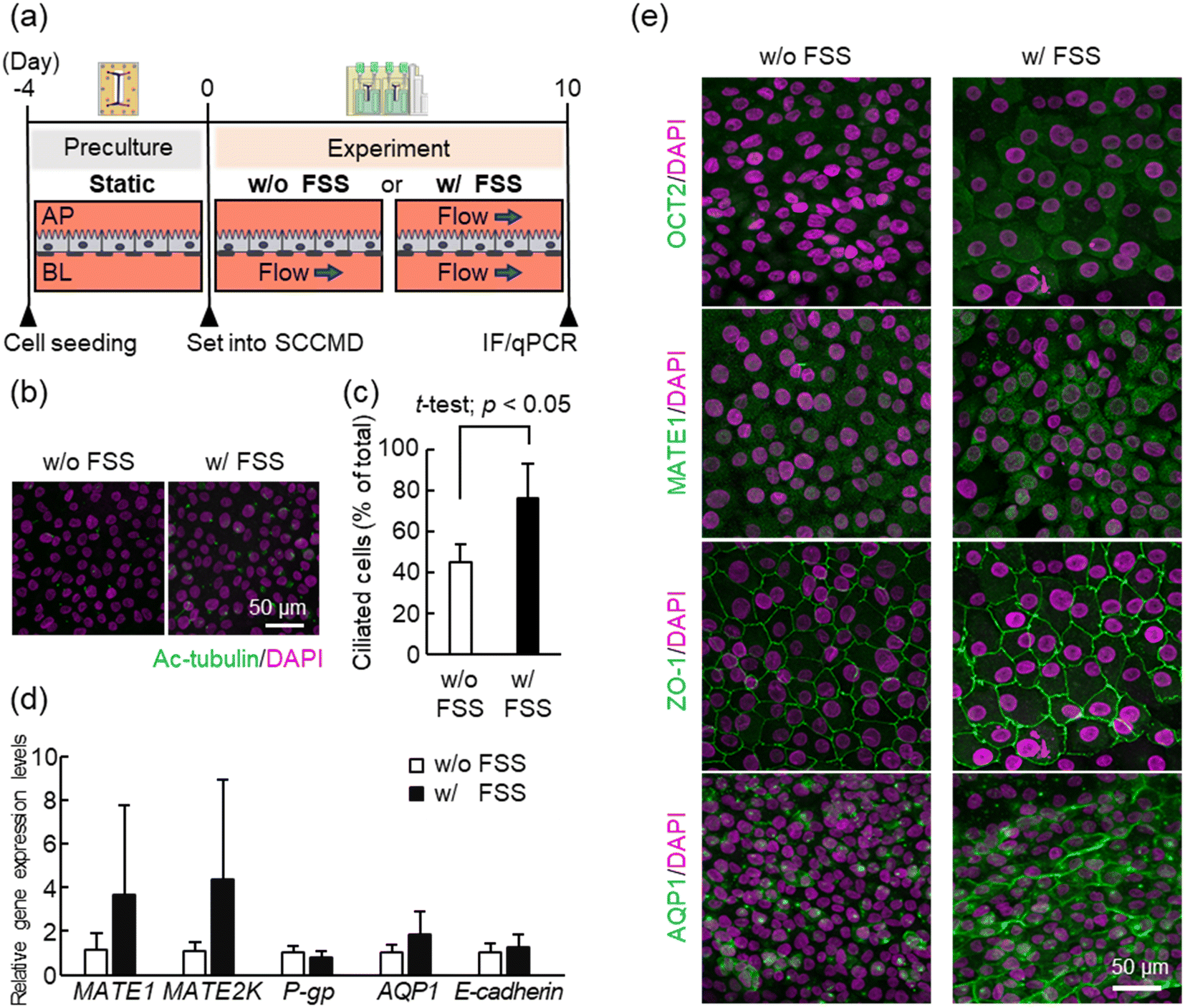 | ||
| Fig. 3 Fluidic shear stress (FSS) affects the functional expression of RPTEC/TERT1. (a) Protocol for proximal tubule microphysiological system (PT-MPS) preparation and FSS loading experiment. (b) Representative images of immunofluorescence (IF) staining of the primary cilia during culture w/FSS and w/o FSS. Green: Ac-tubulin; magenta: DAPI. (c) Comparison of the number of cells with primary cilia quantified from IF images using ImageJ (NIH, Bethesda, MD, USA). The number of ciliated cells was higher in the w/FSS group than in the w/o FSS group. (d) Comparison of the gene expression levels of representative functional proteins in RPTEC/TERT1 cells: MATE1 and MATE2K tended to be higher in w/FSS than in w/o FSS; however, there was little difference in the expression of other genes. (e) IF images of the functional proteins. Purple: DAPI. The expression of OCT2, MATE1, and ZO-1 was higher in w/FSS than in w/o FSS; aquaporin-1 (AQP1) differed in terms of the expression level and expression site between the conditions. | ||
Acetylated tubulin (Ac-tubulin) is a marker of primary cilia. The Ac-tubulin immunostaining results showed that the percentage of cells with a primary cilium under w/FSS culture conditions was significantly higher than that without FSS culture conditions [Fig. 3(b) and (c)]. This indicated that FSS loading promoted the expression of primary cilia in RPTEC/TERT1 cells.
The results of gene expression analysis using real-time PCR showed that the gene expression levels of MATE1, MATE2K, and aquaporin-1 in cells w/FSS culture conditions tended to be higher than those in cells w/o FSS culture conditions; however, the difference was not significant (MATE1 t-test, p = 0.23; MATE2K t-test, p = 0.21; and aquaporin-1 t-test, p = 0.19) [Fig. 3(d)]. Gene expression levels of P-gp and E-cadherin did not differ between the w/o FSS and w/FSS culture conditions. In summary, no apparent effect of FSS loading on RPTEC/TERT1 cells was detected using real-time PCR [Fig. 3(d)]. In contrast, immunostaining results for each functional protein showed that the fluorescence intensity of transporters OCT2 and MATE1, and the tight junction protein ZO-1 in cells cultured under w/FSS culture conditions was qualitatively higher than that in cells cultured w/o FSS [Fig. 3(e)]. ZO-1 was linearly distributed w/and w/o FSS, demonstrating the morphological characteristics of a healthy confluent epithelial monolayer. Aquaporin-1 changed in localization, being detected near the cytoplasm w/o FSS culture conditions, but in the intercellular spaces w/FSS culture conditions. These immunofluorescence staining results indicated that FSS loading affected the expression levels and localization of transporters such as OCT2, MATE1, ZO-1, and aquaporin-1, in RPTEC/TERT1 cells with no change in morphological soundness.
The primary cilia of proximal tubule epithelial cells (PTECs) function as mechanosensors that sense FSS.28 RPTEC/TERT1 cells also contain primary cilia.29 Although the biological mechanism is unknown, FSS loading may promote and maintain primary ciliary expression in the cells. Previous studies have reported that FSS-induced bending of primary cilia opens Ca2+ channels, and signaling through primary cilia increases cell volume and polar expression of aquaporin-1 and OCT2.30 The results of this experiment, showing changes in the expression and localization of various transporters in cells under FSS conditions, are consistent with those reported in previous studies. The results of E-cadherin gene expression analysis and ZO-1 immunofluorescence staining indicated that cell–cell adhesion functional proteins maintained a healthy morphology under FSS, consistent with the results of the previous study investigating detailed FSS effects on PTECs.31
In the PT-MPS studies reported to date, the effects of FSS have been investigated by comparing perfusion and nonperfusion conditions using single-layer microfluidic channels or by comparing static cell conditions using conventional culture plates.13 These studies had the limitation that the effect of differing levels of nutrients, such as glucose, arising from the difference in the amount of culture medium used for perfusion and static cultures could not be eliminated. A study using gut model cells showed that the effect of nutrient content on the increase or decrease in the ratio of medium volume per unit cell number is unexpectedly large compared with the effect of FSS.32 The disturbance caused by the introduction of cells into microfluidic chips may also be a major factor affecting cell function. In this study, to rigorously evaluate the effect of the presence or absence of FSS, cells were constantly perfused with culture medium on the lower channel side of the microfluidic chip to ensure nutrient supply and on the upper channel side only under FSS loading conditions. To our knowledge, this is the first study to validate the effects of shear stress on PT-MPS with only RPTEC/TERT1 cells under conditions that could eliminate the influence of nutrient supply differences and cell introduction into the microfluidic chip. Thus, we conclude that PT-MPS with RPTEC/TERT1 cells cultured under FSS conditions using the SCCMD is a useful model for studying renal proximal tubules.
Toxicity assessment by VC imaging using the automatic cell imaging system with the SCCMD
To validate the utility of the SCCMD in combination with a general-purpose automatic cell imaging system for toxicity assessment, the ability of the PT-MPS to quantitatively assess cell morphology in a nephrotoxicity assay using cisplatin (CDDP) was examined. The cells were cultured for approximately 4 d under w/FSS conditions (0.2 dyn cm−2) with both the upper and lower channels flowing before the experiment [Fig. 4(a)].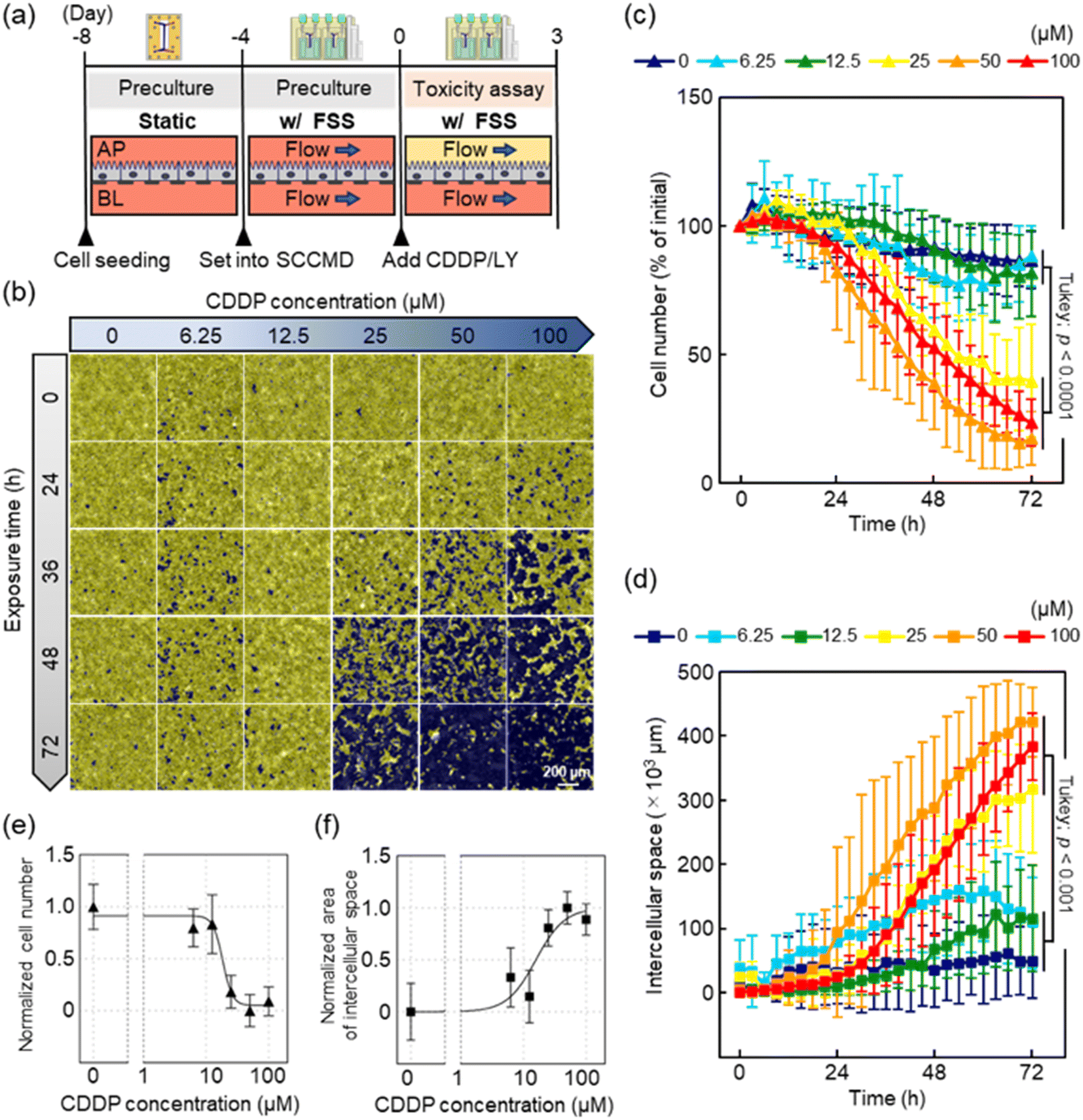 | ||
| Fig. 4 Cell morphology evaluation using the volume contrast (VC) method can be adapted for toxicity assessments. (a) Toxicity assessment protocol. (b) Examples of images obtained via automated segmentation of images generated using the VC method, in which images with different cisplatin (CDDP) concentrations and exposure times were arranged in a matrix. The yellow areas indicate automatically segmented cells, and the purple areas indicate intercellular spaces. (c) Cell number changes measured via the automatic segmentation of VC images, showing CDDP dose-dependent cell number changes. (d) Intercellular spaces measured via the automatic segmentation of VC images. Similar to the changes in cell number, CDDP dose-dependent temporal changes were observed in the intercellular space. (e) and (f) Dose–response curves for CDDP based on cell number and intercellular spaces at 72 h. Logistic regression analysis estimated the median effective concentration (EC50) to be 18.5 μM and 15.8 μM. | ||
VC images of cells exposed to CDDP were acquired by the automatic cell imaging system every 3 h during the 3 d experiment. Cell numbers and intercellular spaces were quantified from the data obtained from the automatic segmentation of VC images using NIS-Elements AR image analysis software. Graphs of the cell numbers exposed to CDDP were plotted with data normalized to the cell number measured from VC images at the beginning of the experiment. The differences between the intercellular spaces at each time point and the minimum intercellular spaces for each CDDP concentration were plotted as a graph.
The VC method facilitated the visualization of cells on a porous membrane, which is generally more difficult to observe in bright fields, as clearly as in the fluorescently stained cytoplasm images [Fig. 4(b)]. The accuracy of cell numbers measured through the automatic segmentation process of VC images was confirmed to be within 10% of the error rate, based on comparison with the cell counts obtained by counting images with Hoechst-stained nuclei [ESI† Appendix, Fig. S2].
The cell numbers remained virtually unchanged at a CDDP dose of ≤12.5 μM. At CDDP doses of ≥25 μM, cell numbers began to decrease with approximately 24 h of CDDP exposure; however, there was no difference in the downward trend in cell numbers between the groups. The cell numbers at 72 h of CDDP exposure were divided into two groups (0, 6.25, 12.5 and 25, 50, 100; Tukey's test, p < 0.0001) [Fig. 4(c)].
The intercellular spaces measured by the automatic segmentation of VC images showed a CDDP dose-dependent change. The intercellular spaces at CDDP doses of 6.25 μM and 12.5 μM did not significantly differ compared with those in the negative control (CDDP of 0 μM) (6.25 vs. 12.5, Tukey's test p = 0.71; 0 vs. 6.25, Tukey's test p = 1.00; and 0 vs. 12.5, Tukey's test p = 0.60). At a CDDP dose of ≥25 μM, the intercellular space increased with approximately 24 h of CDDP exposure. The toxicity assay endpoint, that is, the intercellular space at 72 h of CDDP exposure, was divided into two groups (0, 6.25, 12.5 and 25, 50, 100; Tukey's test, p < 0.001) [Fig. 4(d)].
Logistic regression analysis was performed to determine the relationship between cell numbers, intercellular spaces, and CDDP concentrations after 72 h of CDDP exposure [Fig. 4(e) and (f)]. The median effect concentration (EC50) values obtained from the cell numbers and intercellular spaces were 18.5 μM and 15.8 μM, respectively.
To assess CDDP toxicity, in this study, we measured changes in cell number and intercellular spaces using an automatic cell imaging system. These indices exhibited similar changes over time: a strong correlation was noted among cell number, intercellular spaces, and CDDP toxicity. The coefficient of determination R2 values for the fitting function obtained via logistic regression analysis were 0.81 for cell number and 0.68 for intercellular spaces. The results of the CDDP toxicity evaluation are discussed in detail in the next section. Nevertheless, the observation of multiple indicators over time has great potential for revealing phenomena that cannot be captured by conventional manual cell imaging or discrete observation by sampling.
In general, cells adhering to porous membranes are difficult to observe using phase-contrast imaging because of light scattering from the micrometer-sized pores. To address this issue, in this study, the automatic cell imaging system was equipped with a VC module. Several parameters of imaging and segmentation of RPTEC/TERT1 cells using VC were optimized in a preliminary experiment; the cell number quantification method thus assessed had sufficient accuracy, comparable to that of conventional fluorescent staining methods [ESI† Appendix, Fig. S2]. Although the cell area and cell aspect ratio were evaluated using automatically segmented VC imaging data, the evaluation of cytotoxicity using the cell area and cell aspect ratio did not have the same sensitivity as that using the cell number and intercellular spaces (data not shown). This may be attributed to the insufficient spatial resolution of the VC images owing to the limitations of the system optics and the optimization of the parameters used during segmentation to accurately identify the shapes of the individual cells. Accuracy owing to these parameters should be improved in future. However, if the optics, including the lens and camera of the automatic cell imaging system, and the parameters for VC and segmentation processing, are optimized, the cell area and cell shape (e.g., aspect ratio) can be used as indicators of cell status. In addition, the intensity of a VC image can be used as being indicative of refractive index and therefore be used to evaluate cell thickness and identify cell components. This would make it possible to identify cytotoxicity and morphological changes. Similarly, the automatic cell imaging system can be used in the same manner as in the PT-MPS evaluation shown in this study by optimizing various parameters, even when the cell type is changed.
The SCCMD was periodically moved by the transport unit from the storage rack to the observation area to perform cell observations; these observations took more than 20 min each time on the automated cell imaging system. Because of the dynamic and parallel nature of this operation, the SCCMD could not be wired for power supply and had to be run on battery power to enable medium perfusion to continue during cell observations. The concern that the lack of medium perfusion during observation might alter the conditions of material diffusion and FSS could thus be addressed.
Toxicity assessment via LY permeability measurement using the automatic cell imaging system with the SCCMD
To investigate functional methods other than morphological evaluation to evaluate cell condition, we examined, in a nephrotoxicity assay, the quantitative evaluation of layer permeability using the fluorescence observation function of the automatic cell imaging system with the SCCMD. The following phenomena were predicted: when cells are not present on the porous membrane, LY, a paracellular marker, will pass through the porous membrane from the AP channel to the lower BL channel via free diffusion [w/o cells in Fig. 5(a), left]. When the cells are on a porous membrane, LY will rarely leak from the AP channel to the BL channel because it is blocked by the cell monolayer [w/cells in Fig. 5(a), center]. If the drug damages the cell and the monolayer is disrupted, LY will leak from the AP channel to the BL channel [w/cells + CDDP in Fig. 5(a), right]. | ||
| Fig. 5 Fluorescence observation function enables the quantitative evaluation of the extent of monolayer permeation over time in toxicity assessment using our proposed system. (a) Diagram of lucifer yellow (LY) diffusion in the microfluidic channel. (b) Graph showing the fluorescence intensity ratio of LY in each microfluidic channel for each cisplatin (CDDP) concentration. Based on the CDDP dose, the leakage of LY was observed over time. (c) Graph of LY Papp quantified at 24, 48, and 72 h based on the observed fluorescence intensity. Compared to the negative control, a CDDP dose-dependent increase in LY Papp was observed after 24 h for a CDDP dose of ≥12.5 μM. (d) The dose–response curve of CDDP obtained from LY Papp at 72 h. Logistic regression analysis estimated the EC50 to be 17.1 μM. | ||
In this experiment, CDDP and 200 μM LY were added to the AP channel medium. Fluorescence images were captured in the AP and BL channels every 3 h, in addition to cell morphology observations. The fluorescence intensity within each channel was quantified from the fluorescence images using CL-Quant image analysis software. The intensity ratio within each channel at each time point was determined using the following equation:
 | (3) |
 | (4) |
Time-lapse fluorescence observations showed that when cells were not present on the porous membrane, LY passed through the porous membrane from the AP channel to the BL channel through free diffusion, reaching equilibrium after 72 h [w/o cells_AP and w/o cells_BL in Fig. 5(b)]. In contrast, no LY leakage from the AP channel to the BL channel was detected when cells were present on the porous membrane [0_AP and 0_BP in Fig. 5(b)]. At a CDDP dose of 6.25 μM, LY leakage from the AP channel to the BL channel was almost undetectable, similar to that observed at a CDDP dose of 0 μM (negative control). At a CDDP dose of 12.5 μM, a small amount of LY leakage was detected, as against that in the negative control, though this did not approach statistical significance. At a CDDP dose of 25 μM or more, significant LY leakage was detected after 20–40 h of CDDP exposure, albeit with a minor difference in the leakage trend. Two groups were differentiated based on the degree of LY leakage at the endpoint (0, 6.25, 12.5 and 25, 50, 100; Tukey's test, p < 0.05).
Based on the LY concentration determined from the measured fluorescence intensity using the function of the calibration curve, the apparent permeability (Papp) values of LY at 24, 48, and 72 h for each CDDP concentration were determined using the following equation33 [Fig. 5(c)]:
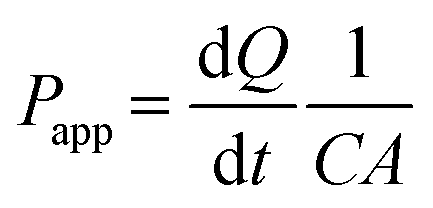 | (5) |
The LY Papp values with the negative control and CDDP dose of 6.25 μM were very low and remained almost unchanged for 72 h, respectively. The LY Papp value at a CDDP dose of 12.5 μM increased slightly compared to that of the negative control and CDDP dose of 6.25 μM. At a CDDP dose of ≥25 μM, the LY Papp increased after 24 h; however, the LY Papp at the endpoint was not significantly different. The EC50 concentration determined by logistic regression analysis from the Papp results was 17.1 μM, with an R2 value of 0.67 for that function [Fig. 5(d)].
In this experiment, the monolayer permeation phenomenon of LY observed over time generally exhibited the same behavior as that shown in Fig. 5(a). CDDP concentration-dependent leakage of LY was also observed. The Papp value of the negative control in this study was comparable to that obtained in previous studies on RPTECs,34 confirming the validity of the fluorescence observation method for quantification using the automatic cell imaging system. Although LY was used to assess toxicity in this study, this method can be adapted to various applications because several other fluorescent substances can serve as biomarkers. For example, rhodamine 123, a representative substrate of P-gp and a fluorescent substance, can be used to quantitatively evaluate P-gp function, making this method extremely versatile.5
In this study, cell numbers and intercellular spaces from VC images and LY leakage were measured using an automatic cell imaging system as indices of toxicity assessment. Interestingly, the data obtained over time showed a similar CDDP concentration-dependent behavior for all indices. At a CDDP dose of 25 μM or higher, a decrease in cell numbers, an increase in the number of intercellular spaces, and LY leakage started at approximately 24 h of exposure. However, an increase in LY Papp was observed in the LY measurements even at a CDDP dose of 12.5 μM. Although a detailed study is in the future, this result may indicate that the measurements of LY obtained through fluorescence observations exhibit higher sensitivity than the determination of cell numbers and intercellular spaces using VC images. For the endpoints, the experimental data for each CDDP concentration were divided into two groups for all the indices. These results demonstrated that the tubular toxicity of CDDP was not linear with the concentration and that toxicity is apparent at a certain threshold. Logistic regression equations were used to predict the threshold value, which was estimated as the EC50 value. The EC50 values obtained from the cell number, intercellular space, and LY Papp data were 18.5 μM, 15.8 μM, and 17.1 μM, respectively. Although there were slight differences, the EC50 values obtained from each measurement might be equivalent to each other given the general biological variability. These results prove that, in experiments using MPS, the multiple indices obtained using the different methods established in this study are useful for toxicity assessment.
CDDP is a useful anticancer drug for clinical use, but causes dose-dependent nephrotoxicity.35 Tubular cells take up CDDP into the cytoplasm via transporters. CDDP has multiple effects including gene regulation, direct cytotoxicity caused by reactive oxygen species, activation of mitogen-activated protein kinases, induction of apoptosis, and stimulation of inflammation and fibrogenesis. These effects of CDDP cause tubular damage and dysfunction in vivo.28
In this study, the EC50 values for CDDP in the culture insert (the conventional two-dimensional (2D) culture method; ESI† Appendix, Fig. S3) and the MPS were 29.3 μM and 15.8–18.5 μM, respectively, with the latter tending to be lower than the former. In clinical settings, CDDP is administered at various doses (10–100 mg m−2) depending on the type of cancer, leading to a wide range of CDDP blood concentrations (Cmax 5–35 μM).36 Considering the correlation between CDDP blood concentrations and nephrotoxicity, previous reports have indicated that patients receiving 80 mg m−2 CDDP with blood concentrations of up to 20 μM CDDP showed a decrease in the glomerular filtration rate, accompanied by elevated urinary levels of N-acetyl-β-D-glucosidase and β2-microglobulin.37 In contrast, patients receiving 60 mg m−2 CDDP did not develop acute kidney injury, as defined by a decrease in the glomerular filtration rate. On administration of a single dose of 50 mg m−2 CDDP, the maximum blood concentration of CDDP reached 13 μM, and only minor increases in the levels of biomarkers associated with proximal tubule damage, such as kidney injury molecule-1, neutrophil gelatinase-associated lipocalin, and albumin, were observed in urine.38 Considering these findings and associating the EC50 value of CDDP obtained in this study with the risk of renal injury in observed clinical practice, the EC50 value in the conventional method corresponds to a CDDP level that is indicative of severe renal injury, including pathologies other than proximal tubule injury. In contrast, the EC50 value of CDDP in MPS suggests clinically reversible renal injury. Hence, the proposed PT-MPS addresses the previous underestimation of CDDP nephrotoxicity by using conventional culture methods.
Previous studies have reported that CDDP is a substrate for the cation transporter OCT2 expressed in RPTECs.39 The results of the present and previous studies suggest that FSS loading leads to the expression of OCT2 in RPTECs.28 Thus, the toxicity assay using PT-MPS was predicted to be physiologically valid, with increased OCT2 expression in RPTECs and increased intracellular uptake of CDDP, resulting in higher toxicity than that by the conventional 2D culture method. Previous reports have shown that mechanostress from perfusion can alter the drug responsiveness of cell cultures, raising the issue of the inappropriateness of conventional static cell cultures as in vitro models to replace animal studies.40 In this study, we focused on the effects of FSS on PTECs and not only raised the issue of differential drug sensitivity due to physical stimuli in static cultures but also demonstrated differences in drug sensitivity due to pharmacological mechanisms.
The function of an MPS as an organ model depends not only on MPS hardware, but also on the activity and function of cells cultured in it. In this study, RPTEC/TERT1 cells were used as RPTECs. RPETC/TERT1 cells showed significantly lower activity of several key drug transporters than RPTECs in vivo. When assaying target drugs for drug transporters, cells with sufficient RPTEC activity are required. Ideally, primary RPTECs from humans used by Weber et al. would be easily available, but it is still difficult to obtain primary RPTECs in a stable manner.41 The large lot-to-lot variation in primary cells is also a major issue. hiPSC-derived RPTECs also exhibit immature differentiation and are difficult to isolate from organoids. In contrast, the method proposed by Sadeghian et al. for mixing RPTEC/TERT1 cells with RPTECs isolated from kidney organoids may solve the problem of cell supply in vitro in a realistic manner.16 Nevertheless, the development of RRTECs that are readily available and have high and stable functional activity is necessary to achieve PT-MPS more suitable for physiological settings in the future.
Conclusions
Herein, we propose an ANSI/ALAS-compliant-sized SCCMD with an integrated fluid control system for future automation, high-throughput drug efficacy, and toxicity studies using an MPS. The PT-MPS was constructed by perfusion-culture of RPTECs using the SCCMD and introduced into an automatic cell imaging system. In addition, we performed a CDDP toxicity assessment to validate the proposed method.Using the SCCMD, the effects of FSS loading on RPTECs were evaluated, and the results suggested that FSS loading not only preserves the primary cilia of the cells but also affects the expression level and localization of transporters. The ability of the system to measure cell number, intercellular space, and monolayer permeability over time was evaluated in the CDDP toxicity assessment using the automatic cell imaging system, demonstrating the validity of the observation method. The sensitivity of the PT-MPS in the determination of CDDP concentration was higher than that of the conventional 2D culture method, confirming that the toxicity response occurs by physiological chemical concentrations. An important point to emphasize in this study is that observing multiple indices over time using multiple methods has great potential for revealing phenomena that cannot be captured using conventional manual cell imaging or discrete observation by sampling. This can only be achieved using the experimental setup and the measurement method of the MPS proposed in this study.
Throughput is a bottleneck for drug discovery. However, most existing MPS lack throughput and functionality because there are trade-offs between these aspects in several cases. We developed an SCCMD that complies with existing standards and proposed a system that can be integrated into existing imaging systems. This system ensures functionality and improves throughput by automating the system, thereby demonstrating its usefulness. Although the SCCMD proposed in this study has only two microfluidic chips and has not yet achieved sufficient throughput, we infer that the MPS hardware is sufficient for validating this concept. We are currently developing an SCCMD that can accommodate six chips simultaneously. This advancement aims to enhance the throughput of MPS experiments. Given that the storage rack within the BioStation CT comprises 30 spaces, our system will facilitate the automated cell imaging of up to 180 chips in the future.
In this study, the SCCMD was utilized as a PT-MPS. However, with a typical bilayered microfluidic chip, it can be used as an MPS for various organs, such as the gut, lung, BBB, and glomerulus, by changing the cells being cultured. In other words, the concept of this study is not limited to the PT-MPS but can be adapted to various other organs. In addition, cell morphology evaluation methods and monolayer permeation tests using fluorescent substances, which were shown to be useful in this study, can be applied to toxicity, drug efficacy, and pharmacokinetic studies, and thus, can be used in all phases of drug discovery research using the MPS. Furthermore, this system is expected to contribute to biodigital transformation (BioDX) research through the use of a large number of images acquired by the system proposed in this study for machine learning to build artificial intelligence models for drug discovery applications.42 Considering the recent global situation, the MPS will further accelerate research and development as a powerful tool and method for drug discovery and as an alternative to animal experiments in life sciences. We expect that the concepts proposed in this study will become the standard for MPS.
Author contributions
Conceptualization: H. K. Methodology: H. K., H. N., T. G., W. U., T. U., D. N., and K. S. Software: H. N., T. G., W. U., T. U., and K. S. Validation: H. K., H. N., and T. G. Formal analysis: H. N., T. G., W. U., and T. U. Investigation: H. K., H. N., T. G., and K. D. Writing – original draft: H. K., H. N., T. G., and K. D. Visualization: H. N., T. G., and K. S. Supervision: H. K. Project administration: H. K. and J. S. Funding acquisition: H. K. All authors reviewed the manuscript.Conflicts of interest
W. U., T. U., and J. S. are employees of Nikon Corporation.Acknowledgements
We would like to thank Dr. Wataru Nihei and Prof. Norimoto Yanagawa for their discussions of this research, Miss. Arata Sato and Mr. Yuga Hara for their support with hardware fabrication, and Editage (https://www.editage.jp) for English language editing. This work was partially supported by JSPS KAKENHI (Grant numbers JP18H01849 and JP21H02824), and the Japanese Agency for Medical Research and Development (Grant number JP20be0304204). This work was also supported by the Tokai University Imaging Center for Advanced Research (TICAR).References
- U. Marx, T. Akabane, T. B. Andersson, E. Baker, M. Beilmann, S. Beken, S. Brendler-Schwaab, M. Cirit, R. David, E. M. Dehne, I. Durieux, L. Ewart, S. C. Fitzpatrick, O. Frey, F. Fuchs, L. G. Griffith, G. A. Hamilton, T. Hartung, J. Hoeng, H. Hogberg, D. J. Hughes, D. E. Ingber, A. Iskandar, T. Kanamori, H. Kojima, J. Kuehnl, M. Leist, B. Li, P. Loskill, D. L. Mendrick, T. Neumann, G. Pallocca, I. Rusyn, L. Smirnova, T. Steger-Hartmann, D. A. Tagle, A. Tonevitsky, S. Tsyb, M. Trapecar, B. Van de Water, J. Van den Eijnden-van Raaij, P. Vulto, K. Watanabe, A. Wolf, X. Zhou and A. Roth, ALTEX, 2020, 37, 365–394 Search PubMed.
- H. Kimura, Y. Sakai and T. Fujii, Drug Metab. Pharmacokinet., 2018, 33, 43–48 CrossRef CAS PubMed.
- L. Lehman-McKeeman and M. Davis, Med. Chem. Res., 2023, 32, 1235–1238 CrossRef CAS.
- S. Ishida, Front. Toxicol., 2021, 3, 657765 CrossRef PubMed.
- H. Kimura, T. Yamamoto, H. Sakai, Y. Sakai and T. Fujii, Lab Chip, 2008, 8, 741–746 RSC.
- K. Doi, H. Kimura, S. H. Kim, S. Kaneda, T. Wada, T. Tanaka, A. Shimizu, T. Sano, M. Chikamori, M. Shinohara, Y. T. Matsunaga, M. Nangaku and T. Fujii, Lab Chip, 2023, 23, 437–450 RSC.
- M. Chikamori, H. Kimura, R. Inagi, J. Zhou, M. Nangaku and T. Fujii, Biomicrofluidics, 2020, 14, 044102 CrossRef CAS PubMed.
- K. Shinha, W. Nihei, T. Ono, R. Nakazato and H. Kimura, Biomicrofluidics, 2020, 14, 044108 CrossRef CAS PubMed.
- C. M. Sakolish, M. B. Esch, J. J. Hickman, M. L. Shuler and G. J. Mahler, EBioMedicine, 2016, 5, 30–39 CrossRef PubMed.
- H. C. Huang, Y. J. Chang, W. C. Chen, H. I. C. Harn, M. J. Tang and C. C. Wu, Tissue Eng., Part A, 2013, 19, 2024–2034 CrossRef CAS PubMed.
- M. Zhou, H. Ma, H. Lin and J. Qin, Biomaterials, 2014, 35, 1390–1401 CrossRef CAS PubMed.
- E. M. Frohlich, X. Zhang and J. L. Charest, Integr. Biol., 2012, 4, 75–83 CrossRef CAS PubMed.
- K. J. Jang, A. P. Mehr, G. A. Hamilton, L. A. McPartlin, S. Chung, K. Y. Suh and D. E. Ingber, Integr. Biol., 2013, 5, 1119–1129 CrossRef CAS PubMed.
- K. J. Jang and K. Y. Suh, Lab Chip, 2010, 10, 36–42 RSC.
- C. M. Sakolish, B. Philip and G. J. Mahler, Biomicrofluidics, 2019, 13, 014107 CrossRef PubMed.
- R. B. Sadeghian, R. Ueno, Y. Takata, A. Kawakami, C. Ma, T. Araoka, M. Takasato and R. Yokokawa, Commun. Biol., 2023, 6, 483 CrossRef PubMed.
- A. Mahadeo, C. K. Yeung, J. Himmelfarb and E. J. Kelly, Curr. Opin. Toxicol., 2022, 30, 100341 CrossRef CAS PubMed.
- W. Y. Chen, E. A. Evangelista, J. Yang, E. J. Kelly and C. K. Yeung, Front. Pharmacol., 2021, 12, 695920 CrossRef CAS PubMed.
- H. Kimura, M. Nishikawa, N. Yanagawa, H. Nakamura, S. Miyamoto, M. Hamon, P. Hauser, L. Zhao, O. D. Jo, M. Komeya, T. Ogawa and N. Yanagawa, Biomicrofluidics, 2018, 12, 044107 CrossRef PubMed.
- Y. Park, C. Depeursinge and G. Popescu, Nat. Photonics, 2018, 12, 578–589 CrossRef CAS.
- T. L. Schwinghammer and P. D. Kroboth, J. Clin. Pharmacol., 1988, 28, 388–394 CrossRef CAS PubMed.
- S. J. Trietsch, G. D. Israëls, J. Joore, T. Hankemeier and P. Vulto, Lab Chip, 2013, 13, 3548–3554 RSC.
- N. Milani, N. Parrott, D. Ortiz Franyuti, P. Godoy, A. Galetin, M. Gertz and S. Fowler, Lab Chip, 2022, 22, 2853–2868 RSC.
- T. Satoh, S. Sugiura, K. Shinji, R. Nagasaki, S. Ishida, K. Kikuchi, M. Kakiki and T. Kanamori, Lab Chip, 2017, 18, 115–125 RSC.
- M. Shinohara, H. Arakawa, Y. Oda, N. Shiraki, S. Sugiura, T. Nishiuchi, T. Satoh, K. Iino, S. Leo, Y. Kato, K. Araya, T. Kawanishi, T. Nakatsuji, M. Mitsuta, K. Inamura, T. Goto, K. Shinha, W. Nihei, K. Komori, M. Nishikawa, S. Kume, Y. Kato, T. Kanamori, Y. Sakai and H. Kimura, Sci. Rep., 2021, 11, 5437 CrossRef CAS PubMed.
- H. Arakawa, S. Sugiura, T. Kawanishi, K. Shin, H. Toyoda, T. Satoh, Y. Sakai, T. Kanamori and Y. Kato, Lab Chip, 2020, 20, 537–547 RSC.
- K. S. Nitsche, I. Müller, S. Malcomber, P. L. Carmichael and H. Bouwmeester, Arch. Toxicol., 2022, 96, 711–741 CrossRef CAS PubMed.
- T. T. Nieskens and M. J. Wilmer, Eur. J. Pharmacol., 2016, 790, 46–56 CrossRef CAS PubMed.
- R. Fujii, S. Hasegawa, H. Maekawa, T. Inoue, K. Yoshioka, R. Uni, Y. Ikeda, M. Nangaku and R. Inagi, Am. J. Physiol., 2021, 321, F278–F292 CAS.
- J. A. Deane and S. D. Ricardo, Nephrology, 2007, 12, 559–564 CrossRef CAS PubMed.
- E. J. Ross, E. R. Gordon, H. Sothers, R. Darji, O. Baron, D. Haithcock, B. Prabhakarpandian, K. Pant, R. M. Myers, S. J. Cooper and N. J. Cox, Sci. Rep., 2021, 11, 14053 CrossRef CAS PubMed.
- W. Shin and H. J. Kim, Nat. Protoc., 2022, 17, 910–939 CrossRef CAS PubMed.
- J. D. Irvine, L. Takahashi, K. Lockhart, J. Cheong, J. W. Tolan, H. E. Selick and J. R. Grove, J. Pharm. Sci., 1999, 88, 28–33 CrossRef CAS PubMed.
- G. Specioso, D. Bovard, F. Zanetti, F. Maranzano, C. Merg, A. Sandoz, B. Titz, F. Dalcanale, J. Hoeng, K. Renggli and L. Suter-Dick, Bioengineering, 2022, 9, 516 CrossRef CAS PubMed.
- X. Yao, K. Panichpisal, N. Kurtzman and K. Nugent, Am. J. Med. Sci., 2007, 334, 115–124 CrossRef PubMed.
- P. Rajkumar, B. S. Mathew, S. Das, R. Isaiah, S. John, R. Prabha and D. H. Fleming, J. Clin. Diagn. Res., 2016, 10, XC01–XC04 CAS.
- N. Nagai, M. Kinoshita, H. Ogata, D. Tsujino, Y. Wada, K. Someya, T. Ohno, K. Masuhara, Y. Tanaka, K. Kato, H. Nagai, A. Yokoyama and Y. Kurita, Cancer Chemother. Pharmacol., 1996, 39, 131–137 CrossRef CAS PubMed.
- M. E. Ibrahim, C. Chang, Y. Hu, S. L. Hogan, N. Mercke, M. Gomez, C. L. O'Bryant, D. W. Bowles, B. George, X. Wen, B. Buckley, L. Aleksunes and M. S. Joy, Eur. J. Clin. Pharmacol., 2019, 75, 51–57 CrossRef CAS PubMed.
- P. F. Secker, L. Luks, N. Schlichenmaier and D. R. Dietrich, ALTEX, 2018, 35, 223–234 CrossRef PubMed.
- Y. A. Guerrero, D. Desai, C. Sullivan, E. Kindt, M. E. Spilker, T. S. Maurer, D. E. Solomon and D. W. Bartlett, AAPS J., 2020, 22, 53 CrossRef CAS PubMed.
- E. J. Weber, A. Chapron, B. D. Chapron, J. L. Voellinger, K. A. Lidberg, C. K. Yeung, Z. Wang, Y. Yamaura, D. W. Hailey, T. Neumann, D. D. Shen, K. E. Thummel, K. A. Muczynski, J. Himmelfarb and E. J. Kelly, Kidney Int., 2016, 90, 627–637 CrossRef PubMed.
- X. Chen, R. Roberts, W. Tong and Z. Liu, Toxicol. Sci., 2022, 186, 242–259 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lc00934c |
| This journal is © The Royal Society of Chemistry 2024 |