
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Retracted Article: Gravity-perfused airway-on-a-chip optimized for quantitative BSL-3 studies of SARS-CoV-2 infection: barrier permeability, cytokine production, immunohistochemistry, and viral load assays†
Shannon L.
Faley
ab,
Niloufar A.
Boghdeh
c,
David K.
Schaffer
ad,
Eric C.
Spivey
 ab,
Farhang
Alem
c,
Aarthi
Narayanan
ce,
John P.
Wikswo
*abdf and
Jacquelyn A.
Brown
ad
ab,
Farhang
Alem
c,
Aarthi
Narayanan
ce,
John P.
Wikswo
*abdf and
Jacquelyn A.
Brown
ad
aVanderbilt Institute for Integrative Biosystems Research and Education, Vanderbilt University, Nashville, TN 37235, USA
bDepartment of Biomedical Engineering, Vanderbilt University, Nashville, TN 37235, USA. E-mail: john.wikswo@vanderbilt.edu
cBiomedical Research Laboratory, Institute of Biohealth Innovation, George Mason University, Manassas, VA 20110, USA
dDepartment of Physics and Astronomy, Vanderbilt University, Nashville, TN 37235, USA
eCollege of Science, Department of Biology, George Mason University, Fairfax, VA 22030, USA
fDepartment of Molecular Physiology and Biophysics, Vanderbilt University, Nashville, TN 37232, USA
First published on 8th February 2024
Abstract
Human microphysiological systems, such as organs on chips, are an emerging technology for modeling human physiology in a preclinical setting to understand the mechanism of action of drugs, to evaluate the efficacy of treatment options for human disease and impairment, and to assess drug toxicity. By using human cells co-cultured in three-dimensional constructs, organ chips can provide greater fidelity to the human cellular condition than their two-dimensional predecessors. However, with the rise of SARS-CoV-2 and the global COVID-19 pandemic, it became clear that many microphysiological systems were not compatible with or optimized for studies of infectious disease and operation in a Biosafety Level 3 (BSL-3) environment. Given that one of the early sites of SARS-CoV-2 infection is the airway, we created a human airway organ chip that could operate in a BSL-3 space with high throughput and minimal manipulation, while retaining the necessary physical and physiological components to recapitulate tissue response to infectious agents and the immune response to infection.
1 Introduction
With the highly contagious respiratory infection of SARS-CoV-2 sweeping the globe more than once and respiratory disease being one of the most common causes of morbidity and mortality worldwide,1 we can well understand why interest in modeling the human respiratory tract has reached an all-time high. Essential features that must be recapitulated include human cells, an air–liquid interface, co-culture of multiple cell types mimicking native airway architecture, and immune cell activation and recruitment, as well as compatibility with Biosafety Level 3 (BSL-3) containment practices and ease of translation into labs that study infectious disease.2 Clearly, the cells used for any infectious model are critical, as we were once again reminded in the case of SARS-CoV-2, where several promising drug candidates were advanced based on their efficacy in Vero cells. Unfortunately, these candidates showed no efficacy in human clinical trials, with the result that precious time and money were spent on compounds that had no future.3–5 Animals may respond to a specific viral infection quite differently than humans, if at all,6 and BSL-3 animal studies are particularly difficult and time consuming, and hence expensive. This in particular highlights the need for robust, early-stage in vitro cell models for candidate drug screening.Microphysiological systems (MPS) have the potential to meet this need in several ways. The ability to use primary human cells from the relevant tissue type generates a model that better mimics the actual infection process in humans.2 Another critical feature that MPS platforms can provide is the opportunity for an air–liquid interface that cannot be achieved with cellular monolayers on plastic.7 Although traditional cell culture of human airway epithelial cells can allow human cell–virus interaction, these cells fail to develop normally when cultured submerged in media.8 Thus, to achieve a mature and more physiologically relevant airway epithelium requires the culture to have an air–liquid interface, so that the air provides the necessary cues for development and there is still a feeding mechanism to maintain the cells. In addition, with an air–liquid interface, the epithelial cells are not in contact with cell media but instead exchange nutrients and metabolites across a porous membrane whose opposite side supports a layer of endothelial cells that are in direct contact with flowing cell culture media. The continuous perfusion of the basal (stromal) side of a cellular barrier may wash away secreted inhibitors that would be active in a non-perfused Transwell®.9 Hence, the MPS airway model requires not only an air–liquid interface but also perfusion and multiple cell types to provide the appropriate nourishment of the airway epithelium.10,11 With an increased focus on response to infectious agents, MPS platforms could also include immune cell components so that the tissue would be able to respond directly and locally to the infectious agent, particularly with regard to cytokine production. An appropriately designed and implemented MPS model would create the microenvironment into which a candidate therapeutic could be introduced not only to human cells but also to a living epithelial/endothelial barrier well in advance of human clinical trials. Lastly, the design must be easy to use in the laboratory. In the case of infectious disease studies, there is a particular need for model systems that are compatible with the restrictions placed on a BSL-3 environment, the most pressing of which are the need for the system to be totally enclosed, so that no aerosols are released in the lab environment, and a hardware design that can withstand the rigors of decontamination for component reuse or repair.12–16 These restrictions can be readily met with a compact, self-contained chip that needs neither extensive hardware nor external connections to transport fluids or air. Moreover, it should be straightforward to translate the platform into labs with expertise in infectious disease, but not necessarily in the use of microphysiological systems, so as to enable rapid drug screening and identification of therapeutic candidates that can be moved to more complex systems or animal models after passing these initial hurdles.
Although there are several excellent pulmonary MPS platforms that are readily used in a BSL-2 environment17–20 and have been applied to infectious disease studies,21 few meet all of our criteria necessary for a standalone airway MPS that can be used in BSL-3 laboratories, particularly without tubing and external reservoirs or pumps. Even the platforms that come closest22–25 struggle with secondary containment issues, bulky equipment, and ease of use with regard to tubing and setup that will prevent their widespread adoption in many BSL-3 spaces. Optimization for BSL-3 use includes 1) compact design, preferably without any tubing or vertical platforms that would prevent complete secondary containment, since open reservoirs to the incubator space are not permissible and separated chips, reservoirs, and pumps connected with tubing increase the risk of leaks and accidental spills; 2) no bulky powered equipment, such as a rocker plate or pump perfusion, as this would take up valuable cell culture space and would both necessitate running power cords into the incubator, which is particularly undesirable in a higher containment facility, and also complicate decontamination of equipment for repair or at the end of an experiment; and 3) ease of use, for rapid adoption – the COVID-19 pandemic taught us that for infectious disease experts to adopt a new model or platform rapidly, it cannot have a steep learning curve if it is going to be widely deployed. Since microfluidic platforms already existed that were well suited for imaging studies, and because we were interested in transcriptomics, Western blots, ELISA, RNAseq, and mass spectrometry for gene expression, cytokine detection, proteomics, and metabolomics, we focused our airway chip design on achieving greater biomass and conditioned media volumes than are possible with MPS models whose cells are contained in a small microfluidic channel. Also critical to our design was creating a simple, easy-to-use platform that was highly compatible with the restrictions of a BSL-3 environment and would allow greater throughput than is seen with many MPS platforms.
We have previously described our development of a microfluidic chip used to model the neurovascular unit (NVU),26–28 consisting of two cellular chambers separated by a semipermeable membrane, with flow in both chambers driven by external syringe pumps that could support the high flow rates required for shear-force polarization of the brain microvascular endothelial cells. At the outset of the COVID-19 pandemic, we concluded that our pump-perfused platform and other organ chips that use pressurized reservoirs, pumps, and gravity feed were ill-suited for BSL-3 use, with its strict rules regarding secondary containment and rigorous decontamination. In contrast to pseudovirus studies in microfluidic devices to quantify the binding of antiviral therapeutics to the spike protein,20 which can be performed in BSL-2 facilities, study of the active SARS-CoV-2 pathogen requires BSL-3. Furthermore, no existing standalone gravity-fed devices can support for a full day the flow rates and hence shear forces required to polarize microvascular endothelial cells.
We now report a novel microfluidic barrier chip capable of gravity-driven perfusion at a high rate of flow that is readily used in either BSL-2 or BSL-3 without the need for external pumps, reservoirs, or tubing. We designed our gravity-flow chip with large enough reservoirs that we could maintain the requisite flow rates for at least 24 hours between the addition of new media and removal of the old, or longer if smaller channels were used to restrict the flow rate. This in turn required the development of deep, large area reservoirs that could be replica cast with PDMS, a prerequisite for bonding to both the channels and chambers of our previous barrier chip. In Fig. 2 and the associated text, we describe in complete detail the novel design considerations and protocols to cast these reservoirs with smooth surfaces for bonding to PDMS microfluidics and without bubbles trapped in the PDMS in the bottom or sides of the reservoirs.
We describe the application of this system for modeling the response of an airway epithelium to a SARS-CoV-2 infection; elsewhere we present results obtained when the chips were used to create a gravity-perfused NVU that was used in BSL-3 facilities to study alphavirus infections.29 A key feature of our design is that these chips can be seeded and matured at the BSL-2 level and, when the chips demonstrate the appropriate physiological performance, they can be moved from BSL-2 into BSL-3 without the challenges of maintaining continuous perfusion during transfer, or the risks of biohazard release or the introduction of bubbles while disconnecting the chips from one perfusion system in BSL-2 and reconnecting the chips to another in BSL-3. Our approach thereby limits the time and space within BSL-3 to that required for infection and observation of the response, and it avoids either open handling of the chips or the transfer into BSL-3 of expensive hardware that cannot be readily removed for service or disposal.
2 Methods
2.1 Cell culture
All cells were maintained in BSL-2 facilities and incubated at 37 °C with 95% humidity and 5% CO2.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per ml and 1 × 106 cells per ml, respectively) to achieve >80% confluence and maintained in B-ALI basal media in both apical and basal chambers. Once the monolayer reached >95% confluence (within 24–48 hours of seeding), basal media was removed from both chambers, and replaced with B-ALI differentiation media supplemented with 2 μl ml−1 freshly thawed B-ALI inducer in the basal chamber only to achieve air-lift culture conditions. Basal media was exchanged every 48 hours with B-ALI differentiation media supplemented with inducer for the remainder of the experiment. The apical chamber was rinsed with sterile 1× PBS (Gibco, Cat#: 10010023) every 3–4 days to thin mucous and remove cellular debris.
000 cells per ml and 1 × 106 cells per ml, respectively) to achieve >80% confluence and maintained in B-ALI basal media in both apical and basal chambers. Once the monolayer reached >95% confluence (within 24–48 hours of seeding), basal media was removed from both chambers, and replaced with B-ALI differentiation media supplemented with 2 μl ml−1 freshly thawed B-ALI inducer in the basal chamber only to achieve air-lift culture conditions. Basal media was exchanged every 48 hours with B-ALI differentiation media supplemented with inducer for the remainder of the experiment. The apical chamber was rinsed with sterile 1× PBS (Gibco, Cat#: 10010023) every 3–4 days to thin mucous and remove cellular debris.
HMVEC-L – human lung microvascular endothelial cells (Lonza, Cat#: CC-2527) were cultured in BSL-2 according to manufacturer's recommendations using EGM™-2 MV Microvascular Endothelial Cell Growth Medium-2 BulletKit™ (Lonza, Cat#: CC-3202). Cells were initially seeded at a density of 10000 cells per cm2 in cell culture-treated T-25 flasks (ThermoFisher Scientific, Cat#: 156367) with B-ALI basal media (Lonza, Cat#: 00193514), and media was exchanged every 48 hours. Cells were passaged upon reaching 90% confluency using ReagentPack™ subculture reagents (Lonza, Cat#: CC-5034). Initial stocks were frozen in EGM™-2 MV Basal media with 10% DMSO (Sigma, Cat#: D2650) at P3 to ensure experimental consistency.
:1:5 ratio by volume, respectively, was applied to the apical and basal chambers of 1 μm Transwell® inserts in 24-well plates (Corning, Cat#: 3527) and incubated at 37 °C overnight to coat. Transwell membranes were then washed three times with 1× PBS prior to inversion of the inserts within the well plates. HMVEC-L cells were seeded on the basal side of the inserts at a concentration of 100000 cells per ml in EBM-2 media and allowed to incubate at 37 °C overnight to facilitate attachment. The inserts were then returned to an upright position with 500 μl EBM-2 media in the basal chamber. NHBE cells were seeded at a density of 500000 cells per ml of B-ALI basal media in the apical chamber and returned to a 37 °C incubator overnight. Once NHBE cells achieved >95% confluence in the apical chamber, all media was removed and replaced with 500 μl B-ALI differentiation media supplemented with 2 μl ml−1 inducer in the basal chamber.
2.2 Generation of SARS-CoV-2 for infection
The SARS-Related Coronavirus 2 isolate Italy-INMI1 (NR-52284) was obtained from BEI Resources. The virus was propagated in BSL-3 according to the manufacturer's recommendation. Briefly, Vero cells were grown to about 70–80% confluency in a cell culture flask and infected with the stock virus (obtained from BEI Resources) at a multiplicity of infection (MOI) of 0.1. Once about 30–40% of the cells were exhibiting cytopathic effect (CPE), which was observed as cell rounding and sloughing, the supernatant was collected and titered by traditional plaque assay in Vero E6 cells (ATCC, Cat#: CRL-1586). The final viral titer was determined to be 1 × 106 PFU ml−1.2.3 Viral infection of airway chips
The infection was carried out in BSL-3 by introducing the virus through the basal side port of the airway chip after the virus had been added to the perfusion medium to reach an MOI of 0.1. The virus-containing medium was continuously perfused into the airway chips overnight at 37 °C, 5% CO2 conditions. For control chips, the usual medium was used on each chamber. Twenty-four hours after the introduction of the virus into the unit, supernatants were collected from both outlet chambers of infected and control chips. Next, 1 ml of the medium was added to the inlet chambers. This process was repeated over the course of the study, and all airway chips were maintained at 37 °C, 5% CO2 in BSL-3 until the termination of the study. Supernatants were collected at 24, 48, 72, 120, and 168 hours post infection from both compartments of all (control and infected) airway chips.2.4 Plaque assay for viral infection
In BSL-3, quantification of infectious virus titer by plaque assay was performed in Vero E6-TMPRSS2 cells (kindly provided by Dr. Shirit Einav, Stanford University). TMPRSS2 cells were seeded in 12-well plates at 1.5 × 105 cells per well. Culture supernatants were diluted in Eagle's minimal essential medium (EMEM) (ATCC, Cat#: 30-2003) containing 10% fetal bovine serum (Cat#: 35-010-CV, Corning) and 2% Geneticin (Cat#: 10131027, Life Technologies Corporation), from 101 to 106, and the virus-containing medium was overlaid on the cells for 1-hour infection. At 1 hour post-infection, a 1 ml overlay of a 1:1 solution of 0.6% agarose in distilled H2O with 2× Eagle's minimal essential medium (Quality Biological, Cat#: 115-073-101) was added to the wells and allowed to solidify at room temperature. The plates were subsequently transferred to 37 °C, 5% CO2 culture conditions, and left for up to 72 hours. After 72 hours, the plates were fixed with 10% formaldehyde at room temperature. Approximately 24 hours after fixation, the agar plugs were removed and fixed cells were stained with 1% crystal violet in 20% methanol solution for 15 minutes. The plaques were counted for each plate, and we calculated plaque-forming units per ml (PFU ml−1) for each sample. The mean and standard deviation were determined using an average of three replicates for each sample.
2.5 Statistical analysis and data processing
All quantifications were performed by incorporating data obtained from triplicate samples unless indicated otherwise. Error bars in all figures indicate standard deviations. Plaque assay and proinflammatory cytokine data calculations were performed using Microsoft Excel. Graphs and P-values were designed and calculated using GraphPad Prism version 10.0.0 for MacOS. Significance values are from two-way ANOVA with Dunnett's post-test or unpaired two-tailed t-test and are indicated using asterisks: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.2.6 Airway chip
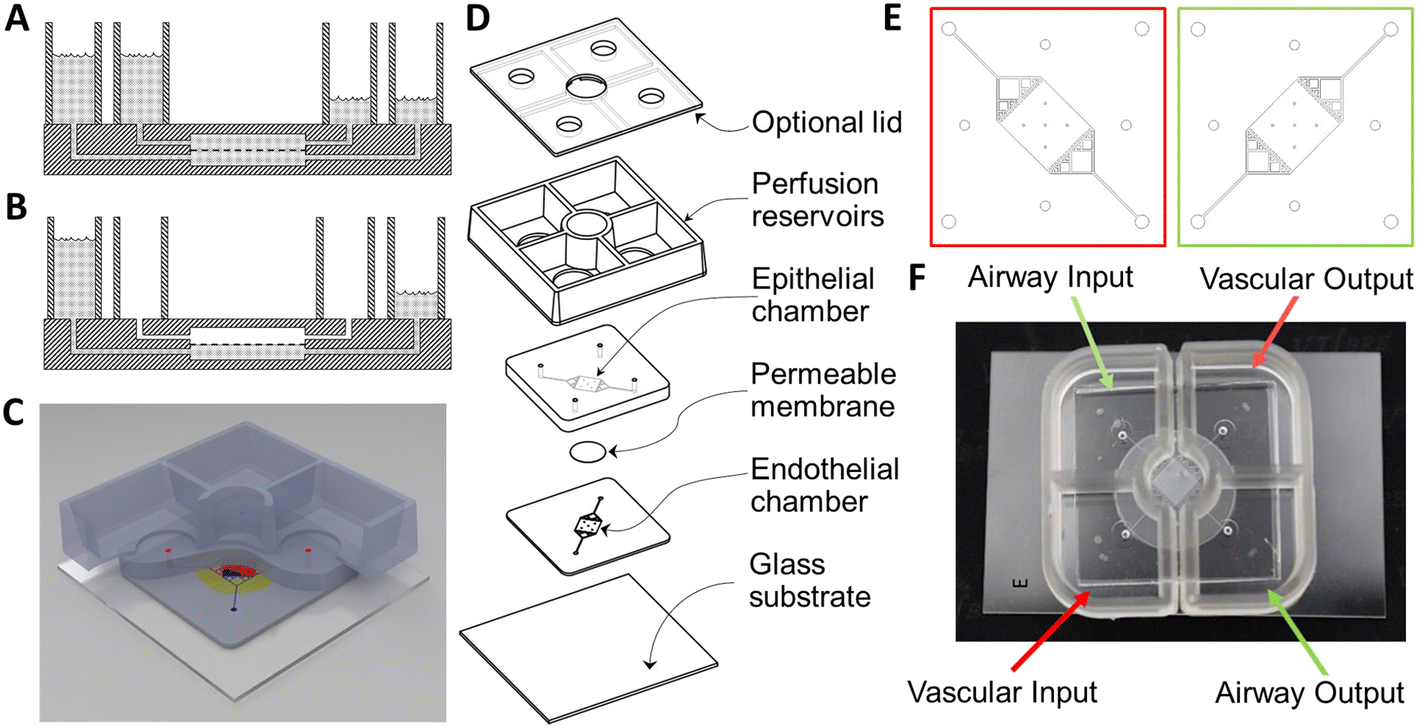 | ||
| Fig. 1 Layout and design of airway chip for BSL-3 use. A) A schematic representation of a gravity-perfused airway chip with two chambers separated by a PET membrane (dashed line) and bonded inside the PDMS (hatched), with both chambers perfused by the difference in height of the media in the supply and collection chambers. B) An airway chip with the epithelial cells on the upper surface of the barrier membrane providing an air–liquid interface. C) An illustration of the complete chip. D) An exploded drawing of the device. E) The mask layouts for the vascular (red) and airway (green) chambers and perfusion channels. F) A photograph of a completed airway chip with gravity reservoirs for use in a BSL-3 space. | ||
Unlike the pump-perfused NVU,27,28 the growth area of this airway chip and the gravity NVU29 were designed to be square, so that both sides of the membrane could be exposed to shear flow. This symmetry enables the gravity-driven flow to be more equal on both sides of the membrane, due to the fact that the fluidic resistance of the two flow chambers is nominally identical. This further means that reservoirs of comparable volume and dimension can be used to generate equivalent flow rates during the initial cell-seeding phase of cell growth, before the epithelial side is transitioned to airlift culture. All of these features contribute to the relative simplicity of the device. The geometry of the flow chamber was modeled and optimized in conjunction with reservoir geometry in order to have the fluidic resistance necessary to provide ∼2 μl min−1 mean volumetric flow rate, which has proven effective at inducing shear-driven tight junction formation in endothelial cells in the NVU.
Manufacturing of the passive-perfusion reservoirs began with a custom mold set consisting of a cavity mold and a flat lid, separated from each other with 1 mm-thick spacers (Fig. 2A). The cavities that form the reservoir walls were machined into 12 mm-thick poly(methyl methacrylate) (PMMA) stock (McMaster Carr, Atlanta, GA, P/N 4615T54) with a tapered end mill (McMaster Carr, P/N 8936A69) to a depth of 10 mm, with the 5° taper selected to ensure easy unmolding of the thick polydimethylsiloxane (PDMS) chamber from a deep mold. Demolding ports (Fig. 2B) were machined into the bottom of the cavity mold to aid in the removal of cured reservoirs from the mold. A second piece of PMMA was laser-cut and bonded to the top of the cavity mold to form a rim around its edge to contain any overflow during the casting process. A third sheet of PMMA was laser-cut to serve as the lid for the cavity mold. A handle was affixed to the lid to assist in the casting process. Finally, PMMA spacers were cut, sanded to precise thickness, and bonded to the underside of the lid. Sylgard 184 PDMS (Dow Corning, Midland, MI) was mixed in a 10:1 ratio of base to curing agent using a Thinky planetary conditioning mixer (Thinky, Tokyo, Japan, model AR-100). After covering the demolding ports with self-adhesive office tape, the liquid PDMS was poured into the cavity mold and placed under vacuum for 10 minutes to remove any air bubbles. The lid was then lowered onto the surface strategically so as to prevent the capture of air between the mold parts (Fig. 2C). The PDMS was allowed to cure at room temperature for a minimum of 48 hours before demolding so that it would not cure into a thermally expanded mold, which would lead to the device being larger than desired upon cooling. Note the complete absence of bubbles in the cast chambers.
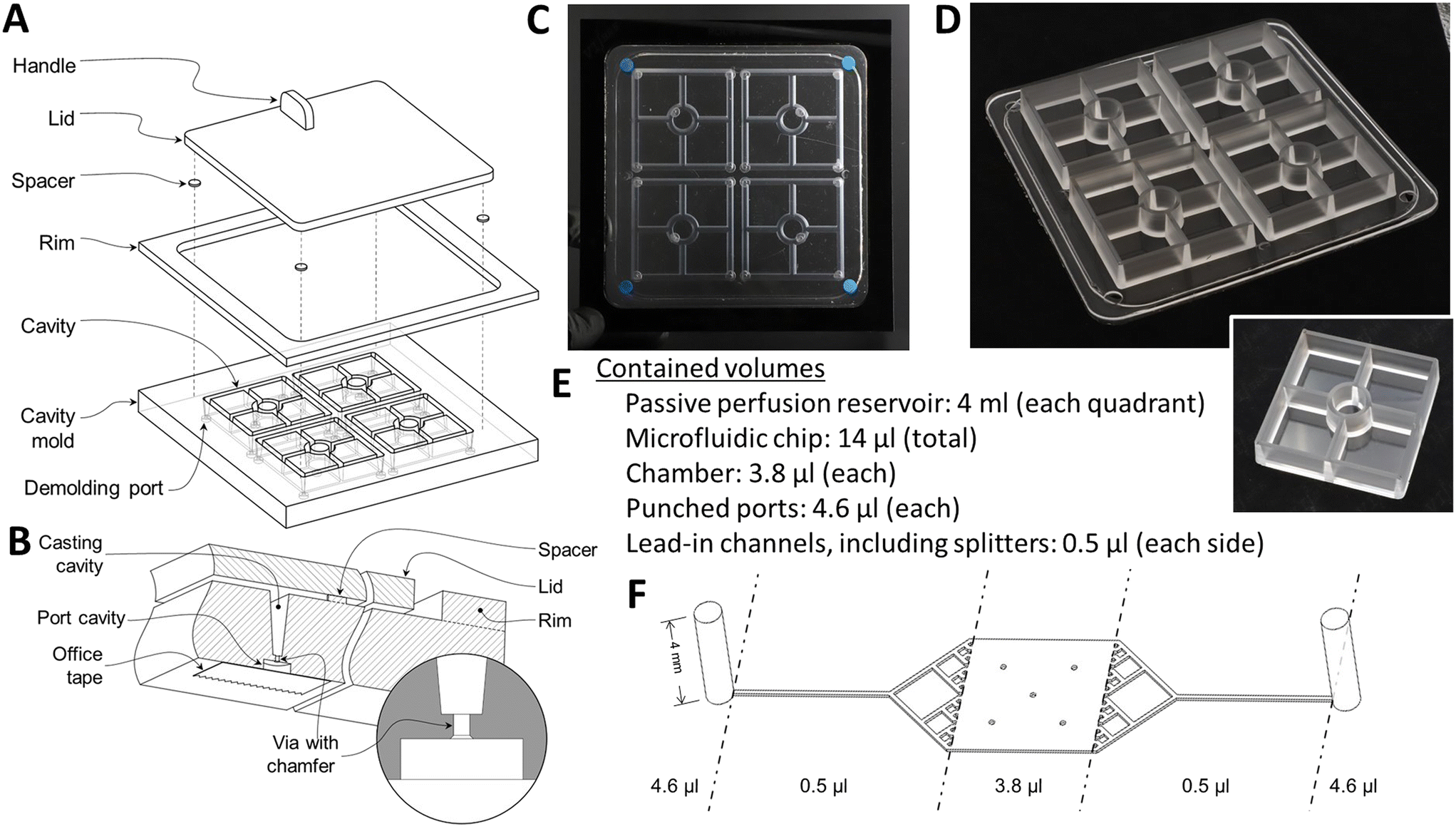 | ||
| Fig. 2 Fabrication of the gravity-perfused airway chip. A) An exploded drawing of the mold used to cast the deep supply and collection reservoirs. B) Details of the cavities on the bottom of the mold that, upon removal of the small PDMS plug, allow pneumatic or hydraulic pressure to simplify extraction of the PDMS reservoir casting to be removed from the deep, tapered mold channels that form the sides of the reservoirs. C) A photograph of the PDMS-filled mold prior to parting. D) The cast PDMS piece removed from the mold. Note that the four sets of reservoirs share a common, 1 mm thick bottom. The small, raised ring of PDMS near the edge is the overflow after an accurately measured volume of PDMS is poured into the open mold, degassed, and then displaced by the gradual lowering of the lid, with the excess PDMS being displaced into the gap between the lid and the rim. E) Device volumes. F) Port, channel, splitter, and chamber dimensions. | ||
Forceps were used to remove the larger, cured PDMS plug filling the demolding port cavity (Fig. 2B), with the chamfer on the outer end of the via ensuring that the larger plug and the smaller one that connects it to the reservoir wall would separate as a unit that would be torn from the reservoir wall by the other, sharp via edge. Hence the plugs can be separated from the molded reservoir wall without leaving a protruding fragment on the wall. Compressed air was injected through the demolding ports to part the reservoir casting from the mold. After demolding the reservoirs (Fig. 2D), flash was trimmed and a 12 mm diameter hole was punched from the floor of each quadrant using a biopsy punch (Difa Cooper, Milan, Italy).
The locations of the holes in the floors of the reservoirs match the access ports in the airway chip, which was cast using a similar molding approach, except that the channel and chamber pattern (Fig. 2F) was created using a modified photolithography technique. Briefly, a 100 μm thick dry film photoresist (SUEX 100, DJ Microlaminates, Sudbury, MA) was bonded to either a standard silicon wafer or a 22 gauge, #8 polished stainless steel wafer cut to fit a 120 mm square Petri dish (Stainless Supply, Monroe, NC). The pattern for the chip was exposed onto the dry film photoresist through a chrome on soda-lime glass photomask (Advanced Reproductions Corp., North Andover, MA). Once molded, two halves of the airway chip were bonded together with a corona treatment system (Electro-Technic Products, Chicago, IL, model BD-20AC).
To complete fabrication, the top surface of each airway chip and the bottom surface of each reservoir were also activated with corona treatment for 30 seconds, brought into contact with each other, and baked at 65 °C for a minimum of 4 hours. This process is very similar to the one used to create the gravity-flow neurovascular unit chips described previously.23
000 cells per ml EBM-2 media via syringe assembly. The airway chips were inverted within a T-150 resealable flask housing and incubated overnight at 37 °C to allow attachment of the HMVEC cells to the basal side of the membrane. They were then returned to an upright position and 5 ml EBM-2 media was added to the vascular inlet reservoir to initiate vascular flow. 100 μl of normal human bronchial epithelial cells (NHBE) (Lonza, #CC-25410S) at a concentration of 1 × 106 cells per ml B-ALI basal media (Lonza, 00193516) were subsequently loaded via syringe assembly into the airway compartment and incubated overnight at 37 °C to facilitate attachment. 5 ml B-ALI basal media was then added to the airway chamber inlet reservoir to initiate flow. If the NHBE monolayer failed to achieve 95% confluency within 48 hours of initial seeding, the NHBE loading step was repeated.
:10 in 1× PBS for five minutes. Cells were blocked with 500 μl 1× PBS containing 2% goat serum (Thermo-Fisher Scientific, Cat#: PCN5000) and incubated overnight at 4 °C.
:10 in 1× PBS via syringe assembly. Inlet chambers were filled with 5 ml Cytoperm solution and allowed to flow via gravity at room temperature for at least 15 minutes. Cells were blocked by removing the Cytoperm solution, then filling the inlet chambers with 5 ml 1× PBS with 2% goat serum and incubating overnight at 4 °C. Cells were then labeled with antibodies diluted in 1× PBS with 2% goat serum and allowed to incubate overnight at 4 °C (Fig. S1†), followed by a final wash with 1× PBS before imaging.
3 Results
The following results have allowed us to assess both the maturation of our airway chips in a BSL-2 environment and their ability to recapitulate in a BSL-3 environment the infection of the human airway with the SARS-CoV-2 virus, thereby demonstrating that gravity-perfused airway chips can be used to study viral infections over the course of several days with minimal complications in translating our MPS technology from BSL-2 to BSL-3 environments.3.1 NHBE maturation under static, Transwell® conditions
We performed initial validation experiments to verify the ability to generate an airway model comprising vascular and airway epithelial components under static conditions by culturing NHBE and HMVEC-L cells on the apical and basolateral surfaces, respectively, and assessing cellular markers and functional indicators of maturation over time.Maturation of a pseudostratified, polarized epithelium typified by mucin-producing goblet cells and ciliated columnar cells was analyzed through antibody staining of cellular markers cadherin-26 (CDH26, Fig. 3A), Vangl-1 (Fig. 3B), MU5AC (mucin, Fig. 3C), and alpha-tubulin (Fig. 3D) on days 0, 14, and 28 after airlift. In each case, staining profiles intensified over time with maximal expression observed after four weeks of culture. Next, we assessed permeability of the co-cultures to 330 Da sodium fluorescein over time. Fig. 3E shows that the barrier strengthened from 1.85 × 10−5 cm s−1 on day 0 to 7.83 × 10−6 cm s−1 on day 28 after airlift, providing evidence of increasing robustness of intercellular junctional integrity with maturation. Finally, we compared monolayer permeability to sodium fluorescein to co-culture assemblies after 28 days (NHBE and co-culture samples were maintained under airlift conditions). Fig. 3F shows that co-culture conditions resulted in markedly decreased permeability as compared to HMVEC-L (1.36 × 10−5 cm s−1) and NHBE (2.35 × 10−5 cm s−1) monolayers, emphasizing the importance of the synergism between vascular and epithelial layers in the development of the airway model.
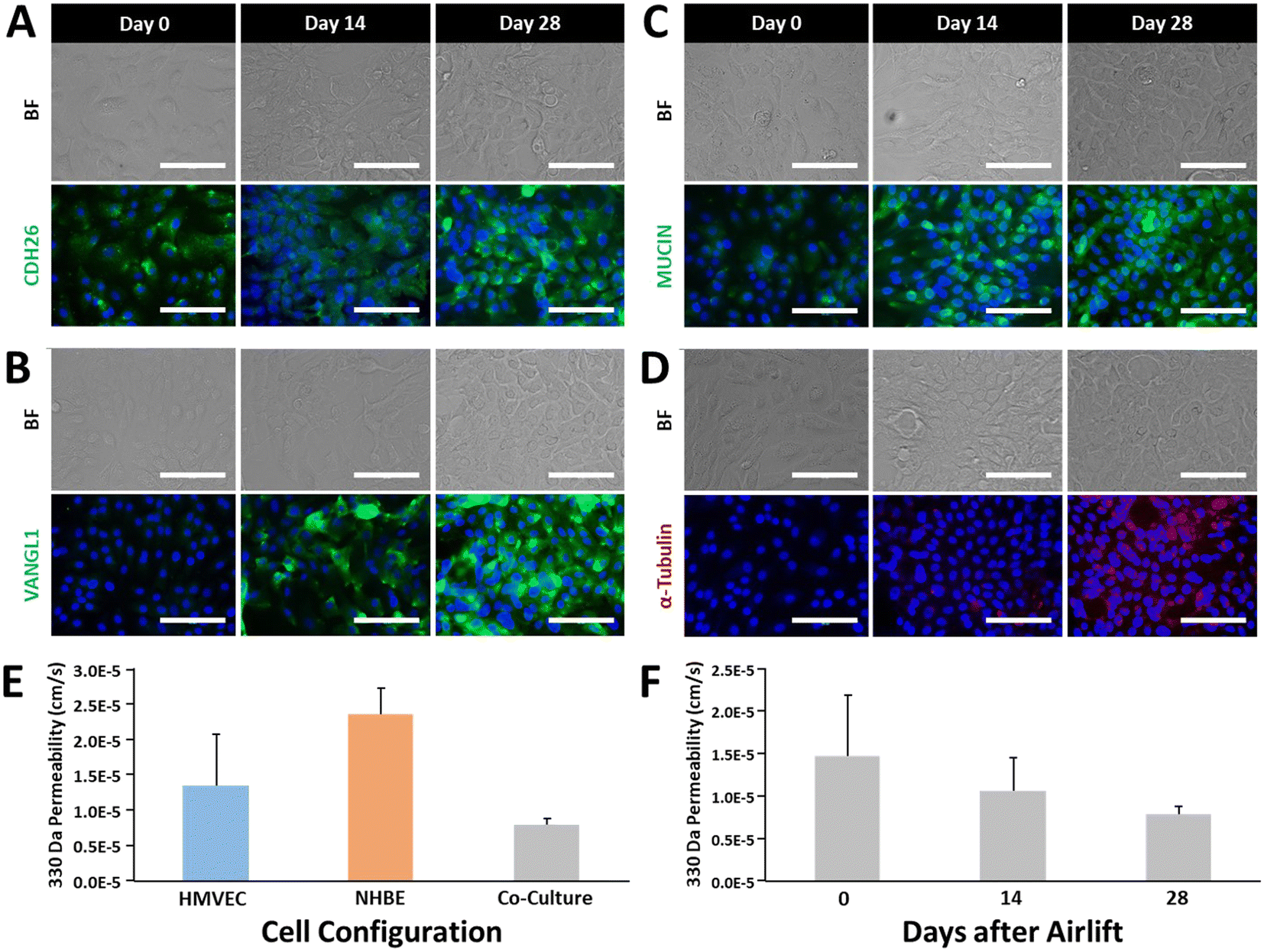 | ||
| Fig. 3 Maturation markers of airway epithelial development. A) Cadherin-26 expression, which is associated with cell–cell junctions, epithelial integrity, and maturation, shows an increase in expression over time. B) VANGL1 is a planar cell polarity marker associated with barrier maturity that increases in expression over time as the airway epithelium develops. C) Mucins are produced by goblet cells of surface epithelium and are associated with cilia formation and a mature epithelium. D) α-Tubulin is associated with cilia development and would indicate a well-differentiated epithelium within the bronchial area. E) Barrier permeability of airway cells in a Transwell in mono- and co-culture after 28 days in culture. F) Airway barrier formation over time in a Transwell containing both endothelial and epithelial airway cells. | ||
3.2 Co-culture in airway chips
We sought to demonstrate the ability to reproduce the static Transwell® airway model within a microphysiological platform subjected to continuous gravity-driven perfusion. Fig. 4A shows the permeability of airway chips to sodium fluorescein on days 0 and 30 after airlift, decreasing from 4.9 × 10−6 cm s−1 to 1.4 × 10−6 cm s−1, as compared to 7.83 × 10−6 cm s−1 in Transwells®, showing that the perfused chips have a seven-fold tighter barrier than that of Transwell® cultures, and that our observed barrier strength increased during four weeks of maturation. Notably, the permeability values are markedly lower with the 3D fluidic environment compared to static cultures at all time points. We also evaluated ACE-2 expression over time, as it has been well established that SARS-CoV-2 leverages ACE-2R to gain entry into cells.31,32Fig. 4B shows the concentration of soluble ACE-2 in the vascular and airway compartment effluent samples on days 15, 30, 45, and 60 after airlift, revealing a modest increase in ACE-2 secretion beginning after 45 days of culture. Although the platform configuration presents challenges in terms of imaging, we attempted to perform immunofluorescence assays of cellular markers associated with vascular integrity and pseudostratification of epithelial layers. In Fig. 4C, images show that the vascular (HMVEC-L) cells, which represent the endothelial layer of the airway chip, exhibit robust ZO-1 expression after 30 days post-airlift, demonstrating intact tight junctions. Likewise, epithelial layers (NHBE cells) positively stain MUCA5 (Mucin) and VANGL-1 30 days after airlift (Fig. 4D).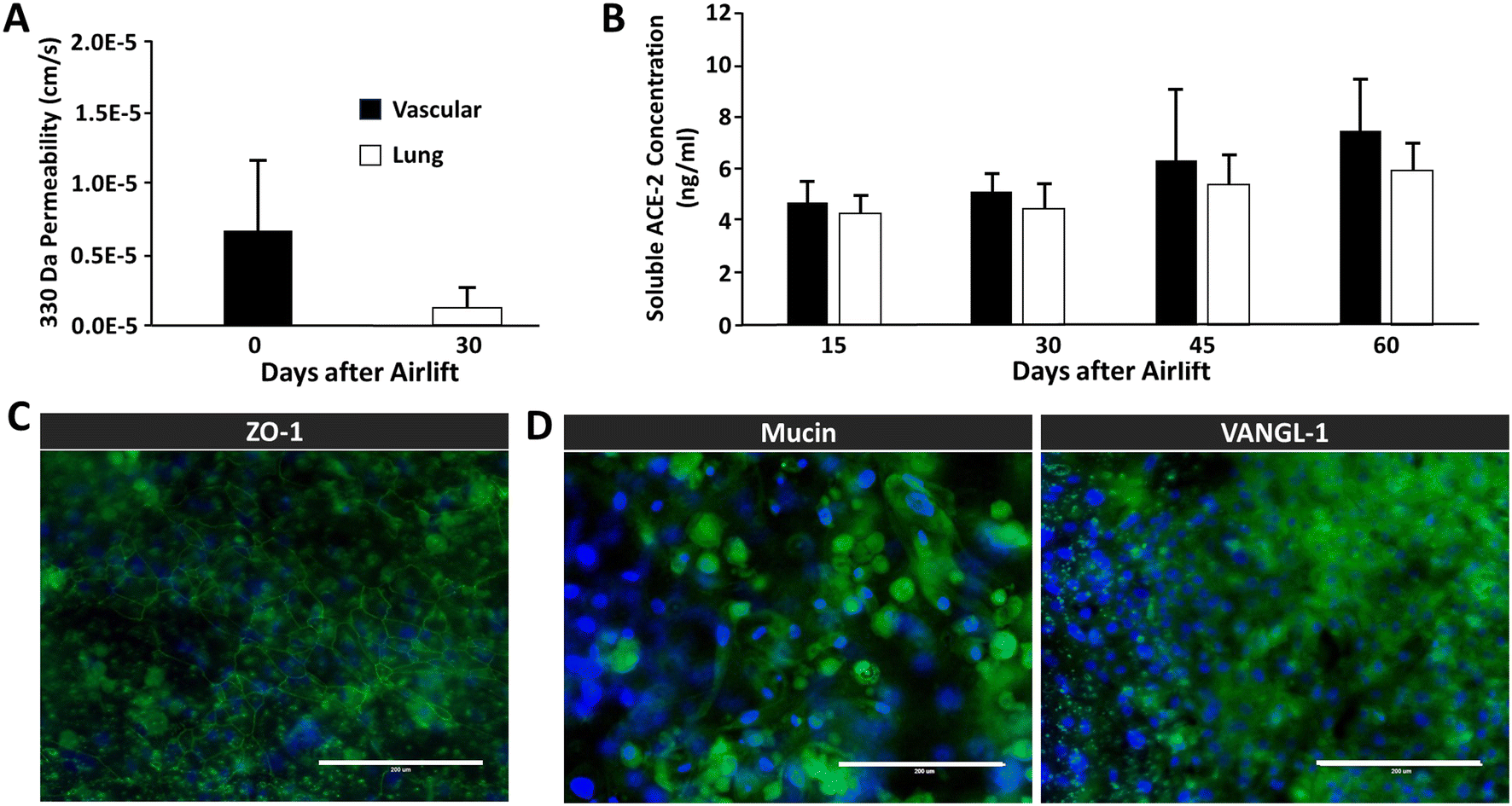 | ||
| Fig. 4 Maturation of the co-culture airway chip. A) After 30 days, there is a functioning airway epithelial-to-vascular barrier, as shown by a significant decrease in permeability to small fluorescein (N = 9, p = .001). B) Detection of soluble ACE-2 is present after 15 days in culture in both vascular and epithelial effluent (N = 9). C) Maturation of the airway vascular endothelium shows junctional protein expression of Z0-1. D) Airway epithelium on the chip was also shown to have maturation markers previously described, including VANGL1 and mucin. | ||
3.3 Assessing ACE-2 expression
Given the critical importance of ACE-2 to SARS-CoV-2 infection and the relatively low concentration of soluble ACE-2 detected in previous experiments, we sought to determine whether cytokine stimulation of airway epithelial cell monolayers would increase ACE-2 levels. Two cytokine candidates, IL-33 (ref. 33) and IFN-gamma,34 were identified from the literature as associated with, or likely to directly influence, elevated ACE-2 levels. Airway epithelial cells were cultured for 45 days on a Transwell® insert under airlift conditions, then exposed to IL-33, IFN-gamma, or a combination of both (see Fig. 5C) diluted in B-ALI differentiation media for 24 hours. Fig. 5A shows that the greatest increase in ACE-2 concentration was associated with the IFNg-Hi (100 ng ml−1 IFN-gamma), IL33/IFN-Lo (100 ng ml−1 IL-33 + 20 ng ml−1 IFN-gamma), and IL33/IFN-Hi (1000 ng ml−1 IL-33 + 100 ng ml−1 IFN-gamma), resulting in a 49%, 40%, and 34% change in ACE-2 concentration as determined by ELISA. We also assessed the impact of cytokine exposure upon barrier permeability to ensure that treatment did not disrupt cellular functioning. Fig. 5B shows that the IF-33-Hi (1000 ng ml−1 IL-33) and IL33/IFN-Hi cohorts were associated with a marked increase in barrier permeability, indicating potential cellular toxicity. We could also show that introduction of activated immune cells could affect blood–brain barrier permeability activation as assessed by cytokine production (Fig. S2†).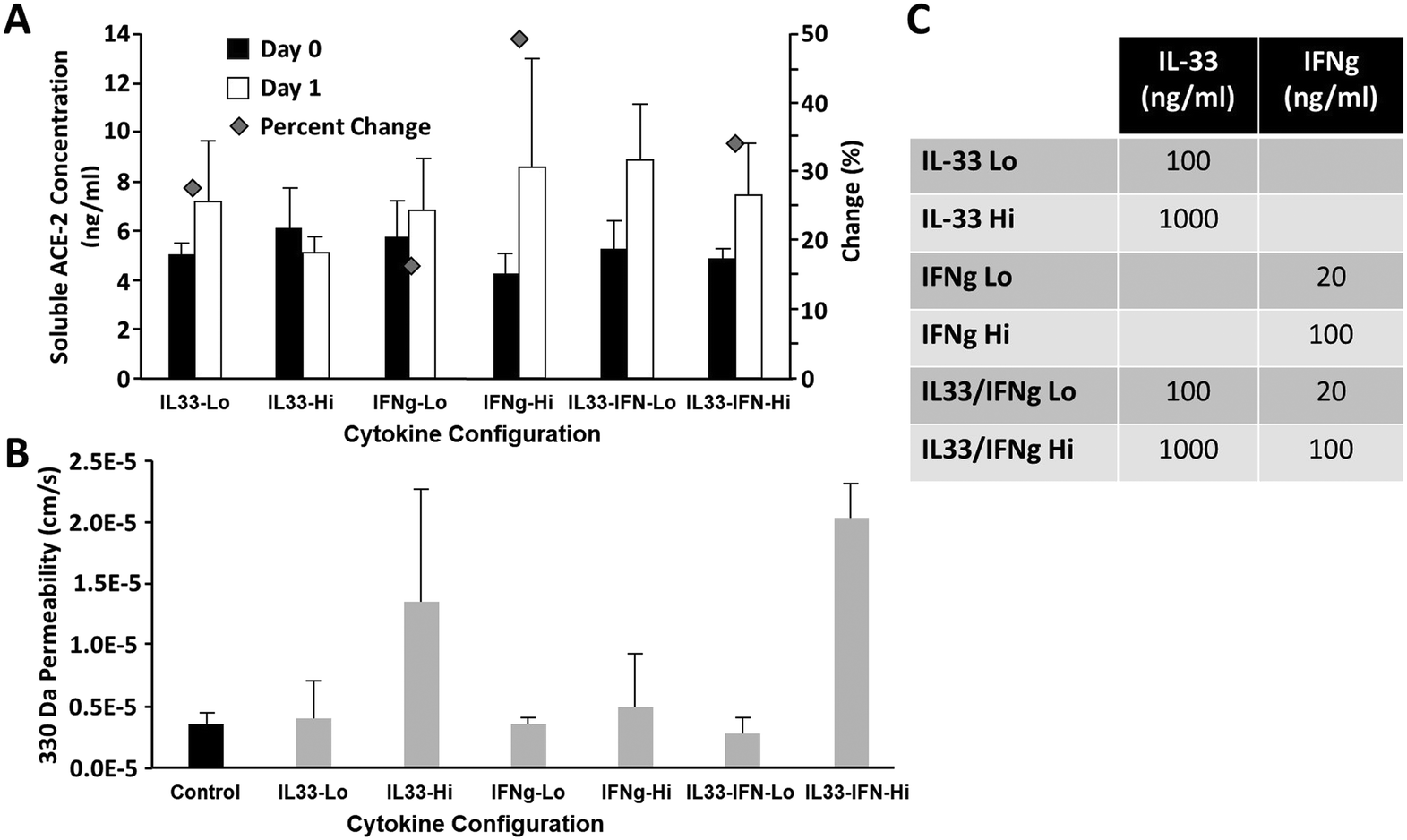 | ||
| Fig. 5 Effects of Il-33 and INF cytokine exposure on ACE-2 expression and airway chip permeability. A) Effects of IL-33 and INF on ACE-2 expression were most pronounced for Hi INF, Low IL-33 + INF, and Hi IL-33 + INF (N = 4). B) Effects of IL-33 and INF on airway chip permeability, the condition which most improved ACE-2 expression without loss of barrier function, was low IL-33 + INF (N = 4). C) Table overview of low and high concentrations of cytokines. | ||
3.4 Infection with SARS-CoV-2
Having validated the airway model for both physiological relevance for human bronchial airway and suitability for infectious disease testing, we were ready to study infection of the airway model with SARS-CoV-2 in a BSL-3 facility. Chips were allowed to mature for 38 days in BSL-2 before infection in BSL-3. By three days post initial infection, barrier disruption can be seen (N = 3; see Fig. 6A), and plaque assays show successful infection of both the airway and vascular side as early as 24 hours and continuing out to five days (Fig. 6B). Using microscale ELISA from MDS, a panel of 10 cytokines was evaluated over the course of the five-day infection (Table 1). From these data, three distinct response groups are evident (Fig. 7): one group can be identified by an early response seen only on the basal side, which was the site of infection (Fig. 7A) and is represented by the cytokines TNF alpha and interleukin 10. The second response group is one where only the basal side showed strong production (Fig. 7B), represented by interleukin 4, 13, and 12p70. The third is the group of cytokines that showed upregulation for both the basal and the vascular sides, indicating a response to the progression of the infection (Fig. 7C) and represented by interleukin 6, 8, 1β, and INF. Of the 10 cytokines investigated, only interleukin 2 showed relatively no response; however, this cytokine is more strongly associated with T cell activation and T cells were not included in this model, which may explain the lack of response in interleukin 2. Overall, these proinflammatory cytokines show a pronounced response to SARS-CoV-2 infection, demonstrating that the airway model successfully recapitulates the cytokine storm that has been associated with this infectious disease.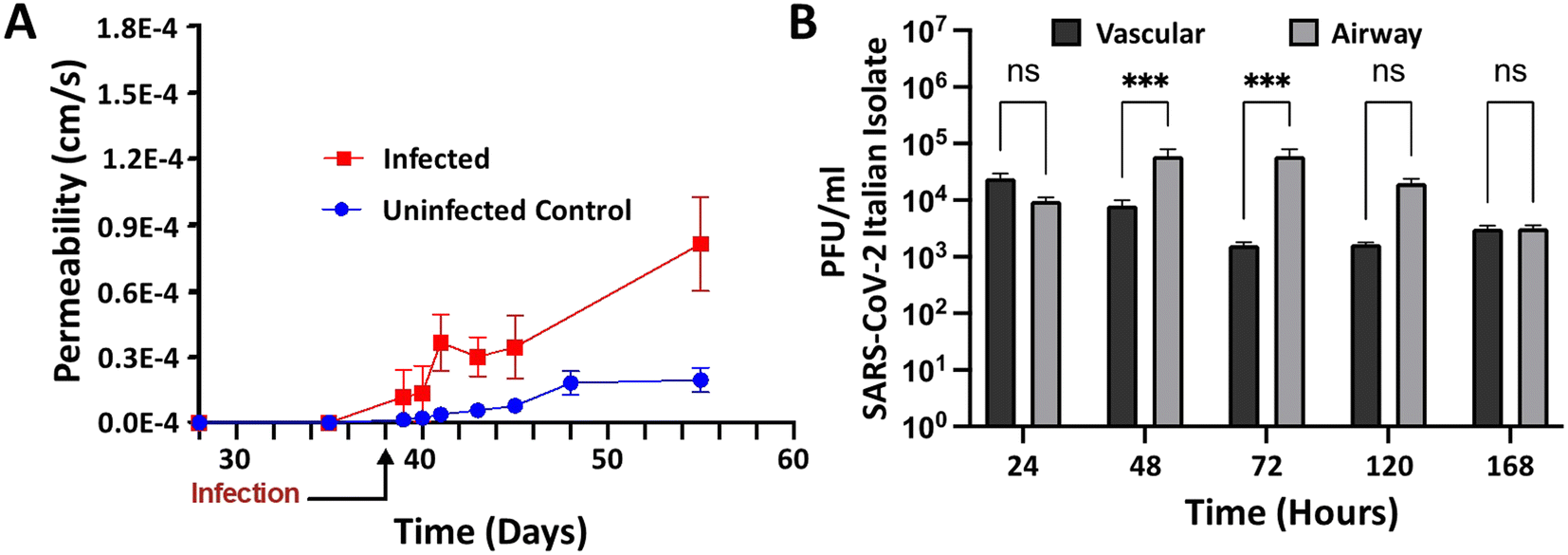 | ||
| Fig. 6 Airway chip permeability and viral load in the basal and vascular compartments in the context of SARS-CoV-2 infection. A) Permeability of the gravity airway chip was assessed by quantification of FITC-dextran levels in the basal and vascular chambers. Units were either infected with SARS-CoV-2 Italian isolate, or uninfected controls. Data are obtained as averages from N = 3 each for infected and for control samples. B) Longitudinal quantification of viral load in basal and vascular sides of the infected and control units. Data are obtained as averages from N = 3 each for all samples, and infectious titers are represented as PFU ml−1. Statistical analysis was performed using two-way ANOVA with Dunnett's post-test (***p < 0.001, ns = not significant). | ||
| Vascular | Airway | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time (HPI) | 24 | 48 | 72 | 120 | 168 | 24 | 48 | 72 | 120 | 168 |
| * = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001. | ||||||||||
| INF-γ | ns | * | * | ns | ns | *** | ** | ** | ns | ns |
| IL-1β | *** | * | * | * | ns | *** | *** | ** | * | * |
| IL-2 | ns | ns | ns | ns | ns | ns | ns | * | ns | ns |
| IL-4 | ns | ns | * | ns | ns | *** | *** | ** | * | ** |
| IL-6 | ** | ** | ** | ** | * | *** | **** | **** | **** | *** |
| IL-8 | *** | *** | *** | *** | ** | **** | **** | **** | **** | ns |
| IL-10 | ns | ns | ns | ns | ns | * | * | ns | ns | ns |
| IL-12p70 | ns | ns | ns | ns | ns | *** | ** | ** | * | ** |
| IL-13 | ns | ns | * | ns | ns | **** | *** | *** | ** | ** |
| TNF-α | ns | ns | ns | ns | ns | * | * | ns | ns | ns |
 | ||
| Fig. 7 Representative expression of three different patterns of cytokine response to SARS-CoV-2 infection in airway chips. The perfused media from the basal and the vascular sides of the units (infected [N = 3], control, [N = 3]) were collected at 24, 48, 72, 120, and 168 hours post infection. A) An early response on the basal/infected side that reduces over time. B) Robust cytokine response that is restricted to the basal side alone but maintained throughout the exposure. C) Robust cytokine response on both the basal and the vascular side. Statistical analysis was carried out using t-test *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. | ||
4 Discussion
The rapid emergence the COVID-19 pandemic and the world-wide spread of SARS-CoV-2 variants presented a breadth of challenges in diagnosis of the infection, identification of the specific pathogen, characterization of the sequence of events following infection, and determination of the mechanism of action of both the infection and possible therapeutic and prophylactic treatments. In vitro assays for any of these steps that involve active SARS-CoV-2 virus must be performed in a Biosafety Level 3 (BSL-3) facility.It is generally recognized that co-culture and barrier MPS models can recapitulate human physiology and pathology more accurately than mono-culture, mono-layer biology on plastic,7 and we explicitly demonstrated that co-cultures in Transwells® still had significantly reduced barrier function as compared to the MPS chips with the same cells. However, with an increase in the technical demands of the assay there is often a decrease in throughput. The vast majority of MPS studies to date have been conducted in BSL-2 level facilities, which require the assay operator to wear gloves and a lab coat, and only open the assay to air in a HEPA-filtered cell culture biosafety hood, i.e., the same as required for culture of any mammalian cells. The addition of syringe or peristaltic pumps, pressurized reservoirs, or static or dynamic reservoir height differences to drive media flow past cells enclosed in a microfluidic device is more of an inconvenience than a barrier to the use of MPS devices in standard BSL-2 laboratories, which are essentially designed to protect the cells from the investigator studying them.
One of the accepted challenges of using a syringe pump or pressurized reservoirs to perfuse a microfluidic organ-on-chip device is the need to avoid the introduction of bubbles in the feed lines when connecting a pump or pressurized reservoir to a chip that is already seeded with cells, in that a bubble can effectively scrub cells otherwise adherent to the microfluidic channel. This problem occurs in BSL-2 laboratories and is aggravated in BSL-3 because of the awkwardness of additional layers of personal protective gear and cramped space. In contrast, in a gravity-perfused device such as ours, the addition of media to a large, open supply reservoir or removal from collection reservoir can be done without the introduction of bubbles, and without the complexity of tethering an organ chip to a perfusion system.
In contrast to conventional BSL-2 studies, the study of active microbial and viral agents that can infect humans and for which there are no effective prophylactic or therapeutic treatments must be conducted in a BSL-3 facility, which protects not only the cells but also the investigators. In addition to the BSL-2 requirements, in BSL-3 investigators must be double-gloved and wear masks and more effective eye protection, and all biowaste and used laboratory supplies must be decontaminated, typically by incineration.12–16 Equipment maintenance and repair cannot involve simply removing the equipment from the BSL-3 space, repairing it, and later returning it to BSL-3. Equipment that is inoperable, obsolete, or simply no longer needed must be incinerated or otherwise thoroughly decontaminated. These requirements and the typically severe space constraints within a BSL-3 laboratory often discourage the introduction of complex and expensive instruments. Finally, BSL-3 facilities have stringent restrictions on the handling of fluids that could have been exposed to infective agents. For example, a typical cell culture flask must at all times reside within a secondary containment vessel such that spills or splashes cannot contaminate the culture hoods, incubators, bench tops, instruments, floors, or personnel. Actively perfused MPS systems can present unique containment, maintenance, and transport challenges in BSL-3.
Currently, infectious disease studies that require a safety level of BSL-3 or higher must overcome several challenges, such as getting bulky equipment into and possibly out of the facility. In addition, because of sterilization requirements, the usual methods for pumped perfusion are not ideal in this space, nor are the rocker plates or the graduated ring setups currently used for gravity-perfused devices, as many of these will not meet the requirements of secondary containment, or if they do will take up a significant space within the facility and reduce throughput. Although studies demonstrate that these methods can be used in a BSL-3 facility,21,22,25 they are not ideal and will retard the adoption of MPS within the BSL-3 space. In designing our device, we sought not only to make something possible, but also practical, robust, and reliable. Our design of a gravity-powered MPS enables both the study of SARS-CoV-2 and Venezuelan equine encephalitis virus (VEEV), and it can be rapidly adapted for new, emerging diseases in the future, thus preventing the time lag that affected research during the recent pandemic. In Fig. 2 and the associated text, we provide a simple yet detailed recipe that should enable other investigators to modify their devices for prolonged studies in a BSL-3 environment.
At present there are few lung organ chips that model the bronchial tracheal area of the respiratory system, which is critical for micro-organ modeling of airborne infectious disease. In addition, very few organ chip systems are highly compatible with the restrictions necessary for use in a BSL-3 environment. We acknowledge those that have been used in the space,21,22,25 but these chips are not practical for widescale deployment because they retain issues with secondary containment, space, and ease of use. For our airway chip to contribute something new, it needed to require no scaffolding, no powered rocking machine, and no tubing so that we could maintain a high level of secondary containment and throughput, which are limited by bulkier approaches, and streamline the ease of use so that laboratories that are not experienced with microfluidic systems could benefit from the technology and use their expertise in infectious disease or critical requirements for our chip design. After designing a BSL-3-compatible, gravity-perfused airway chip and completing perfusion testing, our first goal in validation was to demonstrate the ability to culture and generate a fully differentiated, polarized airway epithelial layer. To do so, we utilized multiple cell-surface staining markers as indicators of maturation,11 including cadherin-26, which has been shown to localize to the apical surface near ciliary membranes and support polarity,35 and Vangl-1, which is strongly associated with a planar-polarized ciliated epithelium.36,37 The presence of mucin-producing goblet cells was confirmed with mucin 5 AC staining.38 We were also able to show evidence of ciliation through positive staining with acetylated alpha tubulin.39 Taken together, these results strongly support the development of mature epithelium within the airway chip.
As we intended these organ chips for use in the fight against COVID-19, we also understood the importance of the expression of ACE-2, which is particularly relevant since it had been shown that although Vero cells not expressing ACE-2 could become infected with SARS-CoV-2, this different mechanism of infection provided poor concurrence with human drug trials.3 Through both immunohistochemistry and ELISA, we were able to demonstrate the expression of ACE-2 and to show how modulation of cytokine activity could affect the expression of ACE-2 with or without changes to vascular permeability. The final stage of validation showed that we could maintain physiologically relevant permeability40,41 over at least 30 days in culture.
Having succeeded in validating our novel airway chip, we were ready to test its reliability for modeling infectious disease by infecting it with SARS-CoV-2 in an actual BSL-3 space with live virus. While some groups have conducted studies using pseudoviruses or BSL-2-appropriate coronaviruses,42–44 these options obviously come with limitations with regard to providing direct information on SARS-CoV-2 itself. By utilizing the Italian strain of SARS-CoV-2 and demonstrating the ability to transfer the chips into a BSL-3 space, we were able to establish both infection of the airway epithelium and a transfer of infection to the airway endothelium, resulting in a marked increase in proinflammatory cytokines from both spaces within the airway chip and thus recapitulating key features of COVID-19 in a human cell-based airway chip. Early infection of the basal airway epithelium was characterized by upregulation of IL-10 and TNF alpha. TNF alpha has been implicated in assisting viral infection,45 and IL-10 has been associated with the early phase of COVID-19 infection;45 thus the airway chip is accurately mimicking the human response. In the case of the cytokinesis, upregulation was limited to the airway epithelial side. The cytokines IL-4 and 13 have been implicated in lung remodeling in response to COVID-19,45 as well as in the cytokine storm that includes IL-12p20.45 Finally, there are four cytokines that show significant upregulation, and both the airway epithelial and the vascular endothelial of the airway chip and serum concentrations of IL-8, IL-6, and IL-1β have all been shown to have correlations with disease progression or severity, thus indicating their presence in the vascular tissue.45,46 Given both the spatial and temporal expression of specific cytokine signatures that closely parallel those found in patients suffering from COVID-19 infections, we believe that this platform shows real potential for use not only in better understanding the consequences of SARS-CoV-2 infection, but also as a platform for novel therapeutics moving forward, not only for SARS-CoV-2 but of other viral agents that can enter the body through the airways, including those that have been weaponized for biological warfare, or by other means. This has been partially realized with our success in using this organ chip platform to study VEEV infection.47
5 Conclusions
We have demonstrated the ability to produce a microphysiological airway model consisting of a vascular component and a mature epithelial component, with the ability to introduce circulating immune components. We have validated this model's physiological function and its suitability for studies of SARS-CoV-2 infection by demonstrating the expression of both a permeability maturation marker and ACE-2. We have also shown that this airway model is capable of establishing a SARS-CoV-2 infection that leads to the production of a wave of cytokines reminiscent of the cytokine storm seen in human subjects. Furthermore, we have filled the need for a model system that is compatible with the restrictions placed on a BSL-3 environment, yet can be used easily and interchangeably in any other cell culture environment. By creating a human cell-based airway chip and demonstrating its ability to recapitulate key features of COVID-19 infection, we hope that this will be a useful platform in drug and intervention testing as novel strains of this and other respiratory pathogens continue to challenge vaccines.Author contributions
Shannon L. Faley: investigation, validation, formal analysis, data curation, writing – original draft, writing – review & editing. Niloufar A. Boghdeh: investigation, validation, formal analysis, data curation, writing – original draft, writing – review & editing. David K. Schaffer: conceptualization, resources, data curation, writing – review & editing. Eric C. Spivey: conceptualization, resources, writing – review & editing. Farhang Alem: conceptualization, methodology, validation, writing – review & editing, supervision. Aarthi Narayanan: conceptualization, methodology, validation, writing – review & editing, supervision, project administration, funding acquisition. John P. Wikswo: conceptualization, methodology, validation, writing – review & editing, supervision, project administration, funding acquisition. Jacquelyn A. Brown: conceptualization, investigation, methodology, validation, formal analysis, data curation, writing – original draft, writing – review & editing, supervision, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported in part by the Defense Threat Reduction Agency (HDTRA11810040 and HDTRA1-23-1-0003 to AN, and CBMXCEL-XL1-2-001 through a subaward from Los Alamos National Laboratory to JPW), the National Institutes of Health National Center for Advancing Translational Sciences (U01TR002383 to JPW) and the National Center for Advancing Translational Sciences, the National Institute of Neurological Disorders and Stroke, and Eunice Kennedy Shriver National Institute of Child Health and Human Development (UH3TR002097 to JPW). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Defense Threat Reduction Agency. We are indebted to Monika Judge and Clayton Britt at Vanderbilt for their assistance in the fabrication of the microfluidic, gravity-perfused airway chips, Dr. Shirit Einav of Stanford University for providing the Vero E6-TMPRSS2 cells, and Robin Trundy at Vanderbilt for her clarification of BSL-2 and BSL-3 requirements.References
- M. Xie, X. S. Liu, X. P. Cao, M. Z. Guo and X. C. Li, Respir. Res., 2020, 21, 49 CrossRef PubMed.
- D. A. Lagowala, S. Kwon, V. K. Sidhaye and D. H. Kim, Am. J. Physiol., 2021, 321, L1072–L1088 CAS.
- N. S. Ogando, T. J. Dalebout, J. C. Zevenhoven-Dobbe, R. Limpens, Y. van der Meer, L. Caly, J. Druce, J. J. C. de Vries, M. Kikkert, M. Barcena, I. Sidorov and E. J. Snijder, J. Gen. Virol., 2020, 101, 925–940 CrossRef CAS PubMed.
- B. K. Park, D. Kim, S. Park, S. Maharjan, J. Kim, J. K. Choi, M. Akauliya, Y. Lee and H. J. Kwon, Biomol. Ther., 2021, 29, 273–281 CrossRef CAS PubMed.
- S. Ramirez, C. Fernandez-Antunez, A. Galli, A. Underwood, L. V. Pham, L. A. Ryberg, S. Feng, M. S. Pedersen, L. S. Mikkelsen, S. Belouzard, J. Dubuisson, C. Solund, N. Weis, J. M. Gottwein, U. Fahnoe and J. Bukh, Antimicrob. Agents Chemother., 2021, 65, e0009721 CrossRef PubMed.
- M. R. Alexander, C. T. Schoeder, J. A. Brown, C. D. Smart, C. Moth, J. P. Wikswo, J. A. Capra, J. Meiler, W. B. Chen and M. S. Madhur, FASEB J., 2020, 34, 15946–15960 CrossRef CAS PubMed.
- D. E. Watson, R. Hunziker and J. P. Wikswo, Exp. Biol. Med., 2017, 242, 1559–1572 CrossRef CAS PubMed.
- A. N. Michi and D. Proud, Am. J. Physiol., 2021, 321, L263–L279 CAS.
- W. Shin, C. D. Hinojosa, D. E. Ingber and H. J. Kim, iScience, 2019, 15, 391–406 CrossRef CAS PubMed.
- T. Pasman, D. Baptista, S. van Riet, R. K. Truckenmuller, P. S. Hiemstra, R. J. Rottier, N. M. Hamelmann, J. M. J. Paulusse, D. Stamatialis and A. A. Poot, Membranes, 2021, 11, 197 CrossRef CAS PubMed.
- E. K. Vladar, J. V. Nayak, C. E. Milla and J. D. Axelrod, JCI Insight, 2016, 1, e88027 CrossRef PubMed.
- World Health Organization, Laboratory biosafety manual, 4th edn, 2020, https://www.who.int/publications/i/item/9789240011311 Search PubMed.
- Centers for Disease Control and Prevention, Biosafety in Microbiological and Biomedical Laboratories (BMBL), 6th edn, 2020, https://www.cdc.gov/labs/BMBL.html Search PubMed.
- L. Bane, Inspection Checklist for BSL-3 Laboratories: CDC/NIH, 2020, https://www.selectagents.gov/compliance/docs/checklists/BSL-3.pdf, https://www.selectagents.gov/compliance/preparing.htm, (accessed 2023-08-11).
- NIH Design Requirements, 2019, [updated 2-14-2019], https://orf.od.nih.gov/TechnicalResources/Documents/DRM/DRMDeskGuide.pdf, (accessed Aug. 11, 2023).
- NIH Design Requirements Manual: NIH, 2020 [updated 3-5-2020], https://orf.od.nih.gov/TechnicalResources/Documents/DRM/DRM1.503262020.pdf, (accessed Aug. 11, 2023).
- D. Huh, H. Fujioka, Y. C. Tung, N. Futai, R. Paine, J. B. Grotberg and S. Takayama, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18886–18891 CrossRef CAS PubMed.
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin and D. E. Ingber, Science, 2010, 328, 1662–1668 CrossRef CAS PubMed.
- A. Jain, R. Barrile, A. D. van der Meer, A. Mammoto, T. Mammoto, K. De Ceunynck, O. Aisiku, M. A. Otieno, C. S. Louden, G. A. Hamilton, R. Flaumenhaft and D. Ingber, Clin. Pharmacol. Ther., 2018, 103, 332–340 CrossRef CAS PubMed.
- L. L. Si, H. Q. Bai, M. Rodas, W. J. Cao, C. Y. Oh, A. D. Jiang, R. Moller, D. Hoagland, K. Oishi, S. Horiuchi, S. Uhl, D. Blanco-Melo, R. A. Albrecht, W. C. Liu, T. Jordan, B. E. Nilsson-Payant, I. Golynker, J. Frere, J. Logue, R. Haupt, M. McGrath, S. Weston, T. Zhang, R. Plebani, M. Soong, A. Nurani, S. M. Kim, D. Y. Zhu, K. H. Benam, G. Goyal, S. E. Gilpin, R. Prantil-Baun, S. P. Gygi, R. K. Powers, K. E. Carlson, M. Frieman, B. R. TenOever and D. E. Ingber, Nat. Biomed. Eng., 2021, 5, 815–829 CrossRef CAS PubMed.
- M. T. Raimondi, F. Donnaloja, B. Barzaghini, A. Bocconi, C. Conci, V. Parodi, E. Jacchetti and S. Carelli, Theranostics, 2020, 10, 7034–7052 CrossRef CAS PubMed.
- M. Zhang, P. Wang, R. H. Luo, Y. Q. Wang, Z. Y. Li, Y. Q. Guo, Y. L. Yao, M. H. Li, T. T. Tao, W. W. Chen, J. B. Han, H. T. Liu, K. L. Cui, X. Zhang, Y. T. Zheng and J. H. Qin, Adv. Sci., 2021, 8, 2002928 CrossRef CAS PubMed.
- Y. I. Wang and M. L. Shuler, Lab Chip, 2018, 18, 2563–2574 RSC.
- M. Gerigk, H. Bulstrode, H. H. Shi, F. Tönisen, C. Cerutti, G. Morrison, D. Rowitch and Y. Y. S. Huang, Lab Chip, 2021, 21, 2343–2358 RSC.
- P. Wang, L. Jin, M. Zhang, Y. S. Wu, Z. L. Duan, Y. Q. Guo, C. M. Wang, Y. Q. Guo, W. W. Chen, Z. Y. Liao, Y. Q. Wang, R. Lai, L. P. Lee and J. H. Qin, Nat. Biomed. Eng., 2023 DOI:10.1038/s41551-023-01054-w.
- J. A. Brown, V. Pensabene, D. A. Markov, V. Allwardt, M. D. Neely, M. Shi, C. M. Britt, O. S. Hoilett, Q. Yang, B. M. Brewer, P. C. Samson, L. J. M. McCawley, J. M. May, D. J. Webb, D. Li, A. B. Bowman, R. S. Reiserer and J. P. Wikswo, Biomicrofluidics, 2015, 9, 054124 CrossRef PubMed.
- J. A. Brown, S. G. Codreanu, M. Shi, S. D. Sherrod, D. A. Markov, M. D. Neely, C. M. Britt, O. S. Hoilett, R. S. Reiserer, P. C. Samson, L. J. McCawley, D. J. Webb, A. B. Bowman, J. A. McLean and J. P. Wikswo, J. Neuroinflammation, 2016, 13, 306 CrossRef PubMed.
- J. A. Brown, S. L. Faley, Y. Shi, K. M. Hillgren, G. A. Sawada, T. K. Baker, J. P. Wikswo and E. S. Lippmann, Fluids Barriers CNS, 2020, 17, 38 CrossRef CAS PubMed.
- N. A. Boghdeh, K. H. Risner, M. D. Barrera, C. M. Britt, D. K. Schaffer, F. Alem, J. A. Brown, J. P. Wikswo and A. Narayanan, Viruses, 2022, 14, 2799 CrossRef CAS PubMed.
- M. Fiorentino, M. M. Levine, M. B. Sztein and A. Fasano, PLoS One, 2014, 9, e85211 CrossRef PubMed.
- S. Lukassen, R. L. Chua, T. Trefzer, N. C. Kahn, M. A. Schneider, T. Muley, H. Winter, M. Meister, C. Veith, A. W. Boots, B. P. Hennig, M. Kreuter, C. Conrad and R. Eils, EMBO J., 2020, 39, e105114 CrossRef CAS PubMed.
- P. Verdecchia, C. Cavallini, A. Spanevello and F. Angeli, Eur. J. Intern. Med., 2020, 76, 14–20 CrossRef CAS PubMed.
- E. C. Lin and C. H. Hong, Biomedicines, 2022, 10, 1183 CrossRef CAS PubMed.
- J. Heuberger, J. Trimpert, D. Vladimirova, C. Goosmann, M. Q. Lin, R. Schmuck, H. J. Mollenkopf, V. Brinkmann, F. Tacke, N. Osterrieder and M. Sigal, EMBO Mol. Med., 2021, 13, e13191 CrossRef CAS PubMed.
- M. E. Lachowicz-Scroggins, E. D. Gordon, A. Wesolowska-Andersen, N. D. Jackson, H. J. MacLeod, L. Z. Sharp, M. Sun, M. A. Seibold and J. V. Fahy, Cell Discovery, 2018, 4, 7 CrossRef PubMed.
- M. T. Butler and J. B. Wallingford, Development, 2015, 142, 3429–3439 CAS.
- D. Devenport, J. Cell Biol., 2014, 207, 171–179 CrossRef CAS PubMed.
- K. A. Knoop and R. D. Newberry, Mucosal Immunol., 2018, 11, 1551–1557 CrossRef CAS PubMed.
- P. Ziegler, Y. R. Tian, Y. L. Bai, S. Abrahamsson, A. Backerholm, A. S. Reznik, A. Green, J. A. Moore, S. E. Lee, M. M. Myerburg, H. J. Park, K. W. Tang and K. H. Y. Shair, PLoS Pathog., 2021, 17, e1009041 CrossRef CAS PubMed.
- H. X. Lin, H. Li, H. J. Cho, S. Bian, H. J. Roh, M. K. Lee, J. S. Kim, S. J. Chung, C. K. Shim and D. D. Kim, J. Pharm. Sci., 2007, 96, 341–350 CrossRef CAS PubMed.
- N. R. Mathias, J. Timoszyk, P. I. Stetsko, J. R. Megill, R. L. Smith and D. A. Wall, J. Drug Targeting, 2002, 10, 31–40 CrossRef CAS PubMed.
- A. J. McAuley, P. J. van Vuren, M. U. R. Mohammed, F. Faheem, S. Goldie, S. Riddell, N. J. Godde, I. K. Styles, M. P. Bruce, S. Chahal, S. Keating, K. R. Blasdell, M. Tachedjian, C. M. O'Brien, N. B. Singanallur, J. N. Viana, A. V. Vashi, C. M. Kirkpatrick, C. A. MacRaild, R. M. Shah, E. Vincan, E. Athan, D. J. Creek, N. L. Trevaskis, S. Murugesan, A. Kumar and S. S. Vasan, Viruses, 2022, 14, 2417 CrossRef CAS PubMed.
- J. F. Tan, Q. W. Guo, L. L. Tian, Z. D. Pei, D. F. Li, M. X. Wu, J. H. Zhang and X. H. Gao, Eur. J. Pharm. Sci., 2023, 180, 106329 CrossRef CAS PubMed.
- J. W. Yang, Y. R. Lin, Y. L. Chu, J. H. Y. Chung, H. E. Lu and G. Y. Chen, Commun. Biol., 2022, 5, 70 CrossRef CAS PubMed.
- S. Haga, N. Yamamoto, C. Nakai-Murakami, Y. Osawa, K. Tokunaga, T. Sata, N. Yamamoto, T. Sasazuki and Y. Ishizaka, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7809–7814 CrossRef CAS PubMed.
- Y. M. Chang, M. R. Bai and Q. H. You, BioMed Res. Int., 2022, 2022, 2755246 Search PubMed.
- S. Saul, K. E. Huie, C. Tindle, M. Sibai, C. J. Ye, A. M. Khalil, K. Chiem, L. Martinez-Sobrido, J. M. Dye, B. A. Pinsky, P. Ghosh, S. Das, D. E. Solow-Cordero, J. Jin, J. P. Wikswo, D. Jochmans, J. Neyts, S. De Jonghe, A. Narayanan, S. Einav, M. Karim, L. Ghita, P. T. Huang, W. Chiu, V. Duran, C. W. Lo, S. Kumar, N. Bhalla, P. Leyssen, F. Alem, N. A. Boghdeh, D. Tran, C. A. Cohen and J. A. Brown, J. Clin. Invest., 2023, 133, e169510 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lc00894k |
| This journal is © The Royal Society of Chemistry 2024 |