DOI:
10.1039/D3LC00804E
(Paper)
Lab Chip, 2024,
24, 561-571
Dielectrophoretic enrichment of live chemo-resistant circulating-like pancreatic cancer cells from media of drug-treated adherent cultures of solid tumors†
Received
22nd September 2023
, Accepted 19th December 2023
First published on 19th December 2023
Abstract
Due to low numbers of circulating tumor cells (CTCs) in liquid biopsies, there is much interest in enrichment of alternative circulating-like mesenchymal cancer cell subpopulations from in vitro tumor cultures for utilization within molecular profiling and drug screening. Viable cancer cells that are released into the media of drug-treated adherent cancer cell cultures exhibit anoikis resistance or anchorage-independent survival away from their extracellular matrix with nutrient sources and waste sinks, which serves as a pre-requisite for metastasis. The enrichment of these cell subpopulations from tumor cultures can potentially serve as an in vitro source of circulating-like cancer cells with greater potential for scale-up in comparison with CTCs. However, these live circulating-like cancer cell subpopulations exhibit size overlaps with necrotic and apoptotic cells in the culture media, which makes it challenging to selectively enrich them, while maintaining them in their suspended state. We present optimization of a flowthrough high frequency (1 MHz) positive dielectrophoresis (pDEP) device with sequential 3D field non-uniformities that enables enrichment of the live chemo-resistant circulating cancer cell subpopulation from an in vitro culture of metastatic patient-derived pancreatic tumor cells. Central to this strategy is the utilization of single-cell impedance cytometry with gates set by supervised machine learning, to optimize the frequency for pDEP, so that live circulating cells are selected based on multiple biophysical metrics, including membrane physiology, cytoplasmic conductivity and cell size, which is not possible using deterministic lateral displacement that is solely based on cell size. Using typical drug-treated samples with low levels of live circulating cells (<3%), we present pDEP enrichment of the target subpopulation to ∼44% levels within 20 minutes, while rejecting >90% of dead cells. This strategy of utilizing single-cell impedance cytometry to guide the optimization of dielectrophoresis has implications for other complex biological samples.
Introduction
An overwhelming majority of cancer fatalities are attributed to metastasis,1,2 motivating the interest in developing therapeutics that target this mechanism.3 This need is especially critical for pancreatic cancer arising from pancreatic ductal adenocarcinoma (PDAC), which is the third leading cause of cancer deaths4 and has the shortest survival duration, due to its propensity for tumor metastasis.5–7 However, due to intra-tumoral heterogeneity, only a subpopulation of the parent cancer cells exhibits the ability for metastasis.8 Identifying this subpopulation across each of the multiple steps in the metastatic cascade can be challenging. An important first step in this cascade is the ability of cancer cells from an adherent solid tumor to survive as suspended cells that can enter the circulatory system,9 away from their extracellular matrix that provides them nutrient sources and waste sinks. This so-called anoikis resistance or anchorage-independent survival characteristic of chemo-resistant cancer cells after drug treatment serves as a pre-requisite for metastasis.10 Currently, circulating tumor cells (CTCs) serve as the leading model for such subpopulations,11 but the lack of reliable markers for their identification12 and their occurrence at ultra-low concentrations within liquid biopsies of PDAC patients13 has led to much interest in alternative sources of circulating-like cancer cell subpopulations for utilization within drug screening.14,15
Recent studies suggested that live cancer cells released into the suspension of drug-treated adherent cancer cell cultures exhibit chemo-resistance and tumor initiating properties.16–18 Furthermore, systematic alterations to their culture conditions, such as shear flow,19,20 hypoxia,21,22 and nutrients,23,24 can be used to generate greater numbers of live suspended cells that exhibit mesenchymal phenotypes. These cultures can serve as an in vitro source of circulating-like cancer cells with greater potential for scale-up, in comparison with CTCs. However, selective isolation of low levels of live circulating cells from the drug-treated culture media, while maintaining the cells in their suspended state, is challenging, due to their wide size distribution that overlaps with dead cells in the media.18 For this purpose, we present utilization of impedance cytometry for optimization of a flowthrough high frequency (1 MHz) dielectrophoresis (DEP) device with sequential 3D field non-uniformities to enrich the live chemo-resistant circulating cancer cell subpopulation from an in vitro culture of metastatic patient-derived PDAC cells.
Dielectrophoresis (DEP)25 has been widely utilized for phenotype-selective cell deflection to enable marker-free isolation of CTCs,26,27 while maintaining cells in their suspended state,28 including single-cell trapping approaches29 that enable the study of molecular signatures30 and biomechanical characteristics.31 However, planar electrode designs limit the throughput for cell deflection due to their limited spatial electric field gradient across the channel depth. While this may be alleviated by using higher voltages, it usually occurs at the cost of inducing irreversible pDEP trapping and cell electroporation. Recent approaches using 3D electrodes to pattern the electric field across the channel depth address this issue.32,33 Our prior work created a set of sequential lateral field non-uniformities orthogonal to the sample flow and extending over the device depth,34 to enable DEP deflection at 10–20 μL min−1 flow rates, along with an on-chip means for cell exchange into an optimal buffer prior to DEP.35 Since target CTCs have a more folded cell membrane structure and are larger in size on average than the background blood cells,36,37 CTCs can be selected over blood cells by positive DEP (pDEP) based on their lower DEP crossover frequency38,39 due to their larger size and higher membrane capacitance. However, the live chemo-resistant PDAC cell subpopulation suspended in the culture media exhibits a wide size distribution that overlaps with dead cells suspended in the media, which includes smaller apoptotic cells and larger necrotic cells from the adherent culture. To address the challenge of selecting small live suspended PDAC cells (<average in the size histogram) with lower DEP trapping force, while rejecting larger dead PDAC cells that exhibit a volumetric enhancement to their DEP trapping force, single-cell impedance cytometry40–42 is used to optimize pDEP deflection at high frequencies (1 MHz). This enables biophysical metrics of the cell membrane and cytoplasm to be used along with cell size as bases for pDEP selection of cells. Furthermore, by carrying out cell deflection by pDEP in the MHz range, rather than in the vicinity of its crossover from nDEP (<0.1 MHz),27,36 the likelihood of cell viability loss due to transmembrane potential drop that occurs for pDEP near the crossover is reduced.43 Single-cell impedance cytometry data gated by supervised machine learning to identify live vs. dead cells, despite their overlapping size distributions, are utilized to identify the DEP frequency range that enables pDEP enrichment of live circulating PDAC cells, as validated by live/dead fluorescence assays. This includes live cells of smaller size (<mean in the histogram) despite the lower volumetric contribution to their DEP trapping force. On the other hand, using size-based microfluidic isolation by deterministic lateral displacement (DLD), while most of the larger sized live cells can be selected into the displaced outlet, substantial numbers of larger-sized dead PDAC cells are also selected into the displaced outlet and smaller-sized live circulating PDAC cells are not selected into the displaced outlet due to their zigzag motion through the posts. In this manner, live chemo-resistant circulating-like pancreatic cancer cells can be enriched by pDEP deflection from the dilute fractions typically generated within in vitro cultures (<10% of total cell numbers). This sets the roadmap for future application to enrich CTC-like mesenchymal phenotypes from other in vitro cultures for emerging needs within molecular profiling, drug screening and subpopulation classification.
Results
Device for enriching suspended live PDAC cells
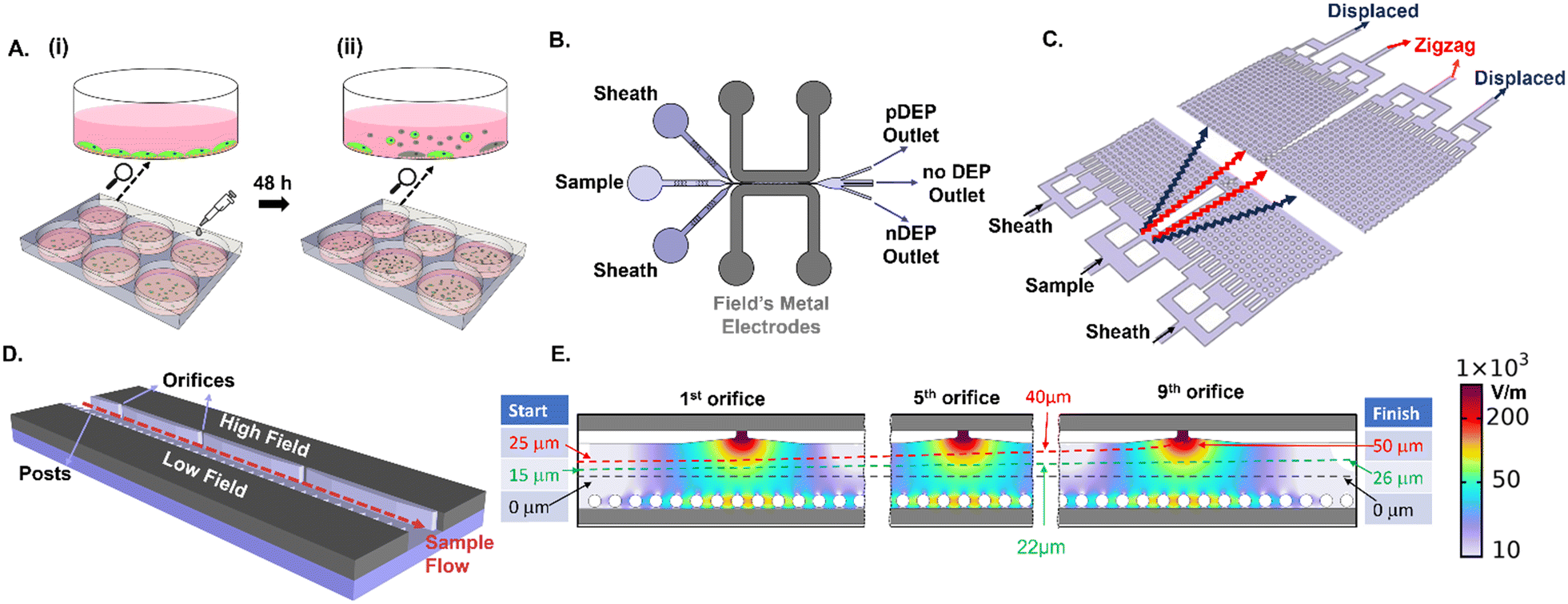
An overview of the device set-up and sample steps for enriching live PDAC cells from the suspension of adherent cultures is shown in Fig. 1. Adhered PDAC cultures (obtained from mouse xenograft models of metastatic tumors derived from patient T608) are subjected to 1 μg mL−1 gemcitabine treatment for 48 h (Fig. 1A), which is known to induce apoptosis within the majority of the adhered cells (>50%).44,45 The culture supernatant is composed chiefly of suspended dead cells released from the adhered culture, as well as limited numbers of live cells in the suspension (1–10% of total cell numbers depending on culture conditions) that exhibit anoikis resistance (Fig. 1A(ii)). Enrichment of the supernatant sample for live PDAC cells in the suspension is studied in a flowthrough microfluidic device by DEP (Fig. 1B) and DLD methods (Fig. 1C), using sheath flows to focus the cell streamlines in the device. For creating field non-uniformities to initiate DEP, Field's metal is introduced in the liquid state at 65 °C, which solidifies at room temperature to fill the electrode channels that adjoin the sample channel. Voltage application initiates spatial 3D field non-uniformities across the sample channel width, due to orifices on one side that create high electrical field points and posts on the other side that create a continuous metal layer of low electric field (Fig. 1D). Focusing of the cell sample streamline with respect to the electric field profiles in the device is a key feature of the design to achieve effective pDEP enrichment without cell viability loss due to pDEP entrapment at electrodes. Using sheath flows, cell streamlines from the sample are focused at the cross-sectional width of 15 μm from the channel center towards the high field region (green streamline of Fig. 1E). On the one hand, this position enables continuous pDEP deflection under the high spatial field extent from each of the nine successive orifices, which would not be possible if the cell streamline was focused right at the center of the channel cross-section (50 μm from the channel edge, as per the black streamline of Fig. 1E and S2†). On the other hand, this starting position (green streamline) ensures that after pDEP deflection towards the high field initiated by each of the nine sequential orifices, the net cell streamline leads to minimal entrapment and electroporation at any of the orifice tips, up to the last (or ninth) orifice (for instance, the red streamline of Fig. 1E shows entrapment). This is confirmed by particle tracing simulations and cell imaging experiments.
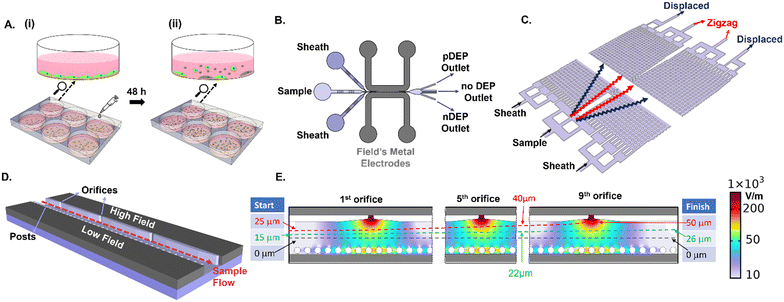 |
| | Fig. 1 Overview of the device and sample preparation steps. A. (i) Adherent PDAC cultures are drug treated at 1 μg mL−1 for 48 h to induce apoptosis and (ii) suspended cells released into the culture media are enriched for the live chemo-resistant population by: B. positive dielectrophoresis (pDEP) and C. deterministic lateral displacement (DLD). D. Schematic 3D view of the DEP device containing sequential field non-uniformities under voltage from fillable Field's metal electrodes in an adjoining channel, using orifices to create high field points and posts that create a continuous metal layer of low field. E. Simulated electric field profiles and flow focused streamline of the cell position at cross-sectional width after pDEP deflection at each orifice from the first orifice (start) up to the ninth orifice (finish). Optimal focusing of the start position (e.g., green streamline) enables successive pDEP deflection along device length without entrapment at the orifices, whereas a start position closer to the orifice (e.g., red streamline) exhibits entrapment due to pDEP at the ninth orifice, and a start position at the center of the channel cross-sectional width (e.g., black streamline) is too far away from the high field regions to exhibit significant pDEP deflection. | |
Metrics for analyzing live cells in suspension
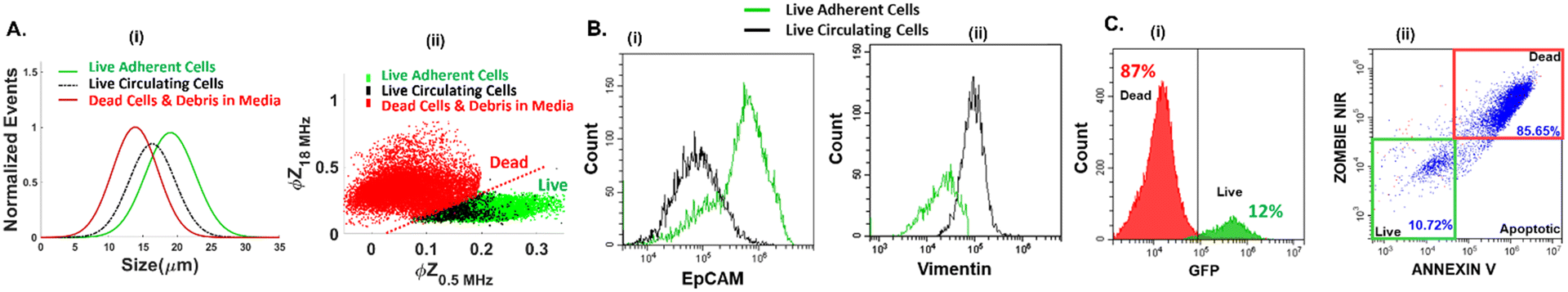
The phenotypes of live PDAC cells suspended in the media were compared to those of dead cells in the media, as well as to those of live adherent cells trypsinized from the culture. The metrics of cell size (Fig. 2A(i)), impedance phase (ϕZ in Fig. 2A(ii)), EpCAM (Fig. 2B(i)) and vimentin expression (Fig. 2B(ii)) were used for this purpose. Additional markers, including fibroblast activation protein, have been identified for isolation of the subpopulation of metastatic circulating cells.46,47 Using impedance cytometry, four frequencies (0.5 MHz, 2 MHz, 18 MHz, and 30 MHz) are simultaneously applied to compute impedance magnitude (|Z|) and phase (ϕZ) at each frequency for each cell. The electrical size of single-cell events can be quantified by normalization of |Z0.5MHz| to that of polystyrene beads of known size.42 This is consistent with forward scattering flow cytometry (ESI,† Fig. S7). The ϕZ value at low vs. high frequency (0.5 vs. 18 MHz in the scatter plot of Fig. 2A(ii)) can be used to gate live suspended cells.44 Using supervised learning based on the support vector machine (SVM) model to train impedance metrics with known samples of dead and live PDAC cells, the specific metrics that distinguish live vs. dead cells were identified to arise from comparison of the impedance phase (ϕZ) at low frequency (0.5 MHz) to that at high frequency (18 MHz or 30 MHz). This enabled computation of the hyperplane for label-free gating of live vs. dead cells (ESI,† section B1, Fig. S8 & Methods section). As per the sloped line in Fig. 2A(ii), live cells are roughly in the region of higher ϕZ0.5MHz levels due to effective electric field screening by their intact plasma membrane, and lower ϕZ18MHz levels due to lack of alteration in cytoplasmic conductivity by culture media penetration, as would be observed for dead cells with disrupted plasma membranes. In this manner, cell sizes in the heterogeneous sample can be attributed to live vs. dead circulating cells. Based on this, live circulating cells exhibit wide size distributions that overlap with dead cells in the suspension and trypsinized live adherent cells, with their average size falling between those of the dead cells and live adherent cells (Fig. 2A(i)). The remaining live PDAC cells after drug treatment in the adherent culture and in suspension are associated with chemo-resistant subpopulations.45 Interestingly, live circulating PDAC cells exhibit a high degree of phenotypic similarity to the chemo-resistant live adherent PDAC cell subpopulation across every measured impedance metric (ESI,† section A, Fig. S1). However, live circulating PDAC cells exhibit lower levels of EpCAM expression and higher levels of vimentin expression vs. live adherent PDAC cells (Fig. 2B(i) and (ii)), indicating the onset of mesenchymal characteristics for this chemo-resistant subpopulation, resembling those of CTCs, thereby highlighting the interest in enriching these circulating-like cancer cells. Due to the low proportion of live circulating cells, GFP expressing PDAC cells are used to optimize conditions for their microfluidic enrichment, since live PDAC cells retain their GFP signal level, while dead cells lose their GFP signal. Using GFP-expressing cancer cells, the GFP expression level (Fig. 2C(i)) corresponds to that from standard live/dead staining (Fig. 2C(ii)), using flow cytometry of a typical heterogeneous sample (12% live cell proportions based on the GFP level vs. 10.72% based on the live/dead assay under E-field OFF conditions). Results from live/dead staining of the pDEP enriched fraction under E-field ON conditions are shown in ESI,† Fig. S3, to validate pDEP enrichment based on the live/dead assay. While live/dead staining is the accurate live-cell quantification method, it requires extensive sample preparation and staining of the collected fraction at each outlet, which can lead to considerable cell loss. On the other hand, live-cell quantification by monitoring of GFP expression and impedance metrics requires no sample preparation. Hence, we utilize it in subsequent sections to optimize pDEP enrichment.
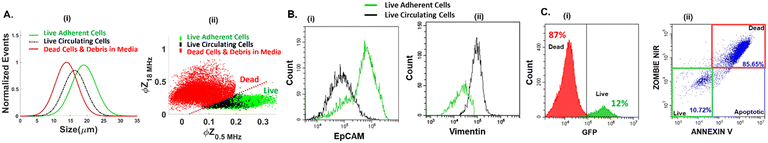 |
| | Fig. 2 A. (i) Overlap in electrical cell size distribution of respective cell types; (ii) live cells can be gated from dead cells using ϕZ metrics at multiple frequencies (based on ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 events in impedance cytometry – see ESI† Fig. S1 for other metrics and Fig. S8† for gate optimization by supervised learning). B. (i) EpCAM and (ii) vimentin expression (15000 events in flow cytometry). C. Comparison of the same heterogeneous cell sample for: (i) GFP expression and (ii) live/dead staining, after collection from the pDEP device under E-field OFF conditions (refer to Fig. S3† for collection under E-field ON conditions). 000 events in impedance cytometry – see ESI† Fig. S1 for other metrics and Fig. S8† for gate optimization by supervised learning). B. (i) EpCAM and (ii) vimentin expression (15000 events in flow cytometry). C. Comparison of the same heterogeneous cell sample for: (i) GFP expression and (ii) live/dead staining, after collection from the pDEP device under E-field OFF conditions (refer to Fig. S3† for collection under E-field ON conditions). | |
Optimizing field conditions for live PDAC cell enrichment
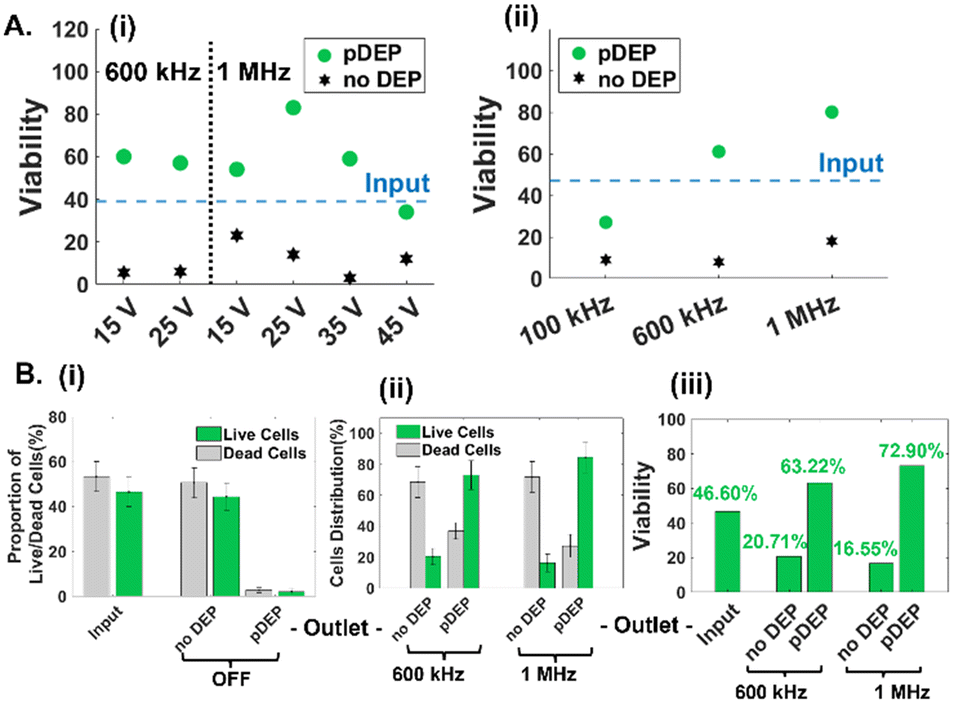
The low numbers and proportions of live circulating cancer cells in typical drug-treated samples (1–10% depending on culture conditions) vs. dead cells in the media, make it challenging to optimize pDEP conditions for enriching live circulating cells. Hence, the sample of suspended cells in the drug-treated media was mixed with the trypsinized adherent cell sample in equal parts (2 wells of suspended cells with 2 wells of lifted adherent cells from 6-well plates) for the results reported in Fig. 3 (input sample of ∼45% viability based on the GFP expression level) and in majority parts (3 wells of suspended cells with 1 well of lifted adherent cells from 6-well plates) for the results reported in Fig. 4 (input sample of ∼25% viability based on GFP expression) and finally, using only suspended cells for the results in Fig. 5 (input sample of ∼3% viability based on GFP expression). For quantifying pDEP collection from dilute input samples, the GFP expression level and impedance metrics were used to minimize sample preparation steps that could lead to cell loss. Following optimization of the cell streamline by flow focusing prior to flowthrough pDEP deflection (Fig. 1E), while ensuring that the deflection does not cause cell entrapment at any of the high field points up to the last orifice, we focused on optimization of field conditions of applied peak-to-peak AC voltage (15–45 Vpp) over 100 μm spacing and frequency (0.1–1 MHz). As per Fig. 3A(i), while the proportion of viable cells in the pDEP fraction increases from 15 to 25 Vpp levels due to greater DEP trapping force, it drops off at higher Vpp levels. This is likely due to cell deflection towards the vicinity of high field regions that lead to cell viability loss due to their entrapment and electroporation. Using 25 Vpp, pDEP deflection at 1 MHz led to higher proportions of viable cells in the pDEP fraction vs. that at lower frequencies (Fig. 3A(i) and (ii)). We did not explore frequencies beyond 1 MHz, due to decay in the frequency response of commercial amplifiers.48 Based on three independent runs under these optimized conditions (25 Vpp at 1 MHz) using an input PDAC sample of ∼45% live cells (Fig. 3B(i)), it is apparent that the entire sample (live and dead cells) passes undeflected into the “no DEP” outlet under field OFF conditions. This is apparent in a representative run (Fig. S4†), wherein >95% of the input sample (based on 650 live cell events collected in 15 minutes) is collected into the “no DEP” outlet. Live cells from the input sample are predominantly collected into the pDEP outlet at an ∼75% level at 600 kHz and ∼85% level at 1 MHz pDEP trapping frequency (Fig. 3B(ii)), with minimal loss into the “no DEP” outlet (18% at 600 kHz and 10% at 1 MHz). The selection purity, as measured by the viability of the collected sample, shows that the input sample of ∼45% live cells is enriched to ∼63% live cells at 0.6 MHz and to ∼73% live cells under 1 MHz conditions (Fig. 3B(iii)). For the representative run (ESI,† Fig. S4), under field ON conditions at 25 Vpp and 1 MHz, the entire live cell fraction (GFP+) gets deflected to the “pDEP” outlet (∼90%), with only limited live cell proportions passing through undeflected to the “no DEP” outlet (∼10%). These optimized field conditions (25 Vpp at 1 MHz) are subsequently explored at successively lower live cell proportions and using impedance cytometry to optimize DEP enrichment of dilute samples of live floating PDAC cells.
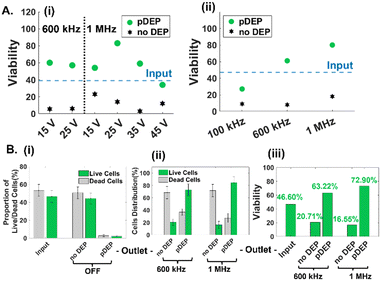 |
| | Fig. 3 A. Optimization of applied voltage (i) and frequency (at 25 Vpp) (ii), for maximizing enrichment of viable cells (GFP+) in the pDEP fraction using a sample input with ∼45% live PDAC cells. B. (i) Proportion of live vs. dead cells (GFP−) in the input at “no DEP” and “pDEP” outlets under field OFF conditions. (ii) Distribution of cells to the “no DEP” and “pDEP” outlets under field ON conditions (25 Vpp) at 600 kHz and 1 MHz pDEP trapping frequencies shows the collection efficiency of ∼85% for pDEP at 1 MHz. (iii) Selection purity determined based on viable cells % in each outlet at each DEP frequency. | |
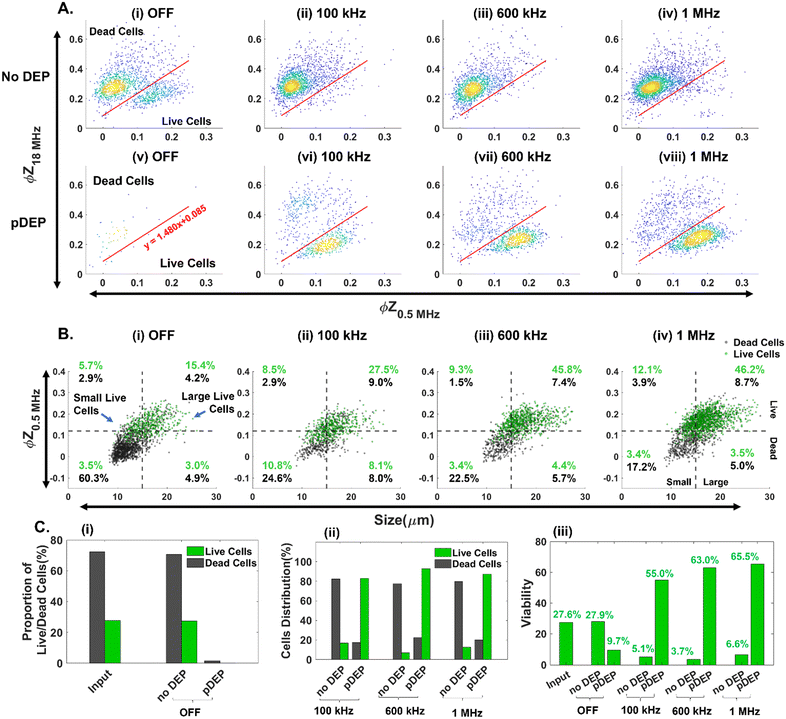 |
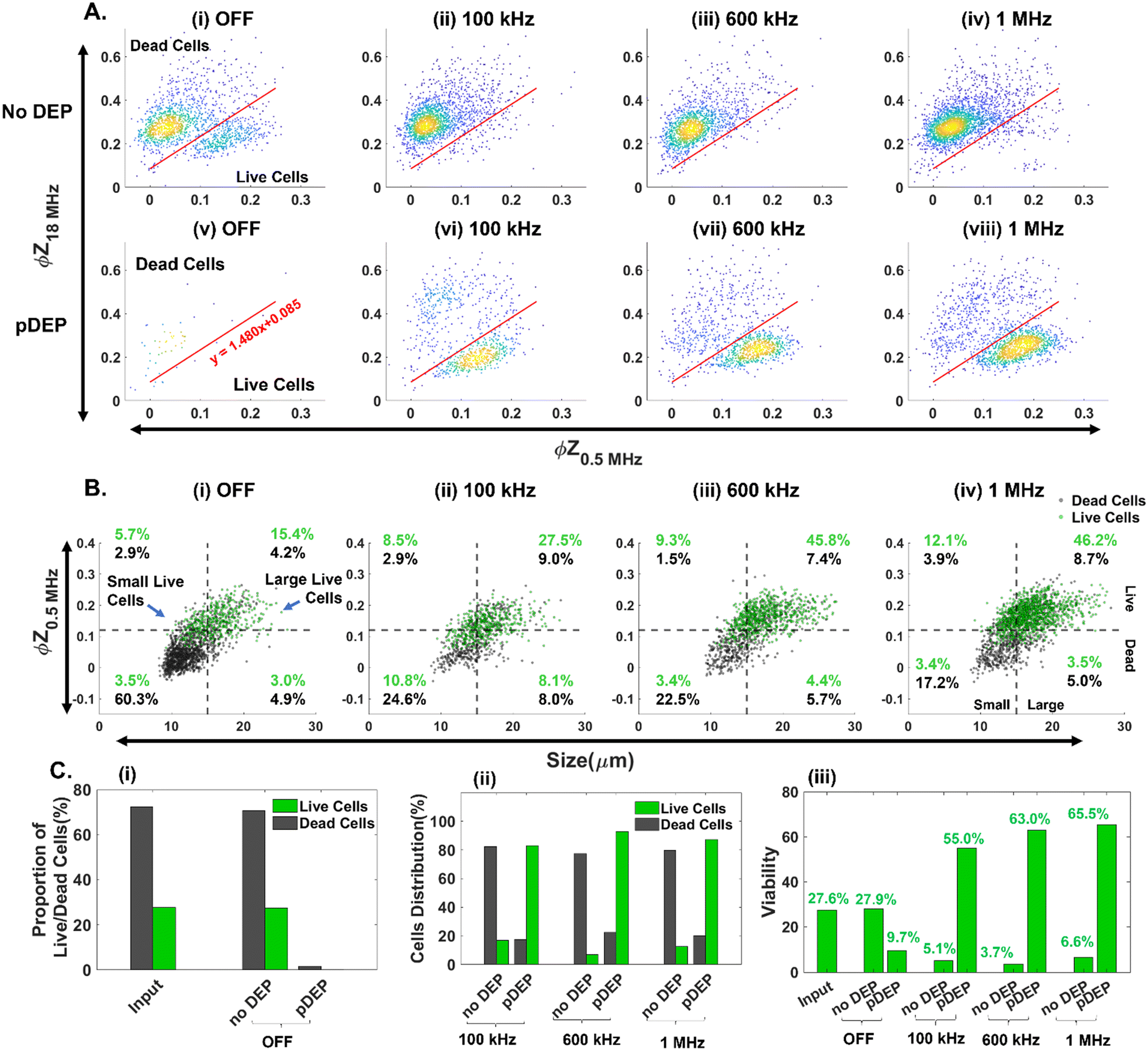
| | Fig. 4 Using impedance cytometry to optimize the frequency for pDEP enrichment (at 25 Vpp) of live cells over the entire size range of an input sample (∼27% live PDAC cells). Single-cell impedance scatter plots of: A) ϕZ18MHzvs. ϕZ0.5MHz for live cell gating of the sample from “pDEP” and “no DEP” outlets; and B) ϕZ0.5MHzvs. electrical size for size-stratified live vs. dead cells in the input and pDEP outlets post-DEP. C) Quantifying collection at the “pDEP” and “no DEP” outlets (25 Vpp at 0.1–1 MHz) based on: (i) proportion of live and dead cells; (ii) cell distribution in each outlet at different DEP trapping frequencies and (iii) viability of collected cells. All live vs. dead cell gates (lines in 2D plots) are obtained based on the optimized hyperplane from the SVM supervised learning model. | |
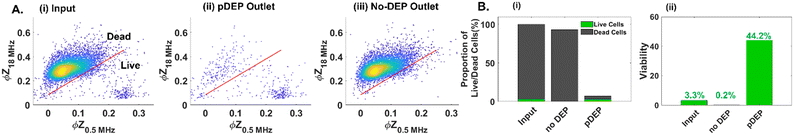 |
| | Fig. 5 DEP enrichment of samples with low levels (∼2–3%) of only live PDAC floating cells obtained from the cell culture supernatant after 48 h gemcitabine treatment (i.e., 100% floating cells). A. Impedance cytometry-based live cell gating of input and collected cells in the “pDEP” and “no DEP” outlets. B. Using impedance cytometry to quantify live cell proportions (i) and viability levels (ii) for cells collected in the “pDEP” and “no DEP” outlets. All live vs. dead cell gates (line in 2D plots) are obtained based on the SVM model (Fig. S8†). | |
Impedance cytometry to optimize live PDAC cell enrichment
In comparison with GFP expression for flow cytometry-based quantification of live vs. dead PDAC cells, impedance cytometry data allow for multiparametric label-free cell quantification, based on their electrical size 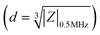 , apoptotic state44 (i.e., live vs. early apoptotic, late apoptotic and necrotic states based on ϕZ0.5MHzvs. ϕZ18MHz), cell membrane (|Z| and ϕZ in the 1–10 MHz range) and cell cytoplasmic properties (|Z| and ϕZ in the >10 MHz range). This allows for optimization of pDEP conditions by quantification of field-induced damage to the DEP enriched fractions, without needing to use GFP expressing PDAC cells. Live cell gating can be established based on the SVM model (optimized gate in Fig. 4A), with live cells roughly showing higher ϕZ0.5MHz levels (>0.125 at mean cell size) due to effective electric field screening by the intact plasma membrane, and lower ϕZ18MHz levels (<0.2 at mean cell size) due to the absence of any alteration to their cytoplasmic conductivity by penetration of the culture media that is observed for dead cells with disrupted plasma membranes. Using an input sample of ∼27% live PDAC cells, it is apparent that under field OFF conditions, cells pass undeflected into the “no DEP” outlet, with very few cell events in the pDEP outlet (Fig. 4A(i), (v) and C(i)). At successively higher frequencies of 0.1 MHz (Fig. 4A(ii)), 0.6 MHz (Fig. 4A(iii)) and 1 MHz (Fig. 4A(iv)), the number of events in the live cell gates successively increases. Next, we use the impedance scatter plot (Fig. 4B) of ϕZ0.5MHzvs. electrical size of cells to quantify the live cell #s (gated based on the SVM model) that are enriched in the pDEP outlet for each size range of interest. Using the intersection point of 15 μm for the histograms of live circulating cells vs. dead cells within the suspension of the culture media after gemcitabine treatment (Fig. 2A(i)), cell events of live circulating cells < 15 μm are called “small live cells” since they overlap with dead cells in the suspension, while those > 15 μm are called “large live cells”. Based on this, it is apparent that live cell events increase successively in the “large live cell” gate for pDEP collected fractions at 0.1 MHz and 0.6 MHz, while remaining substantially unchanged at 1 MHz. On the other hand, pDEP at 1 MHz is crucial for increasing the proportion of events in the “small live cell” gate of the pDEP collected fractions. The summary plot (Fig. 4C(i) and (ii)) confirms ∼80% collection of live cells into the pDEP outlet at 100 kHz and ∼90% collection at 600 kHz and 1 MHz DEP trapping frequencies, with 1 MHz enabling the enrichment of “small live PDAC cells”. Selection purity plots (Fig. 4C(iii)) show that the input PDAC sample of ∼27% live cells is enriched in the pDEP outlet to 55% live cells at 0.1 MHz, to 63% at 0.6 MHz and to 65.5% at 1 MHz.
, apoptotic state44 (i.e., live vs. early apoptotic, late apoptotic and necrotic states based on ϕZ0.5MHzvs. ϕZ18MHz), cell membrane (|Z| and ϕZ in the 1–10 MHz range) and cell cytoplasmic properties (|Z| and ϕZ in the >10 MHz range). This allows for optimization of pDEP conditions by quantification of field-induced damage to the DEP enriched fractions, without needing to use GFP expressing PDAC cells. Live cell gating can be established based on the SVM model (optimized gate in Fig. 4A), with live cells roughly showing higher ϕZ0.5MHz levels (>0.125 at mean cell size) due to effective electric field screening by the intact plasma membrane, and lower ϕZ18MHz levels (<0.2 at mean cell size) due to the absence of any alteration to their cytoplasmic conductivity by penetration of the culture media that is observed for dead cells with disrupted plasma membranes. Using an input sample of ∼27% live PDAC cells, it is apparent that under field OFF conditions, cells pass undeflected into the “no DEP” outlet, with very few cell events in the pDEP outlet (Fig. 4A(i), (v) and C(i)). At successively higher frequencies of 0.1 MHz (Fig. 4A(ii)), 0.6 MHz (Fig. 4A(iii)) and 1 MHz (Fig. 4A(iv)), the number of events in the live cell gates successively increases. Next, we use the impedance scatter plot (Fig. 4B) of ϕZ0.5MHzvs. electrical size of cells to quantify the live cell #s (gated based on the SVM model) that are enriched in the pDEP outlet for each size range of interest. Using the intersection point of 15 μm for the histograms of live circulating cells vs. dead cells within the suspension of the culture media after gemcitabine treatment (Fig. 2A(i)), cell events of live circulating cells < 15 μm are called “small live cells” since they overlap with dead cells in the suspension, while those > 15 μm are called “large live cells”. Based on this, it is apparent that live cell events increase successively in the “large live cell” gate for pDEP collected fractions at 0.1 MHz and 0.6 MHz, while remaining substantially unchanged at 1 MHz. On the other hand, pDEP at 1 MHz is crucial for increasing the proportion of events in the “small live cell” gate of the pDEP collected fractions. The summary plot (Fig. 4C(i) and (ii)) confirms ∼80% collection of live cells into the pDEP outlet at 100 kHz and ∼90% collection at 600 kHz and 1 MHz DEP trapping frequencies, with 1 MHz enabling the enrichment of “small live PDAC cells”. Selection purity plots (Fig. 4C(iii)) show that the input PDAC sample of ∼27% live cells is enriched in the pDEP outlet to 55% live cells at 0.1 MHz, to 63% at 0.6 MHz and to 65.5% at 1 MHz.
DEP enrichment from rare samples of live PDAC floating cells
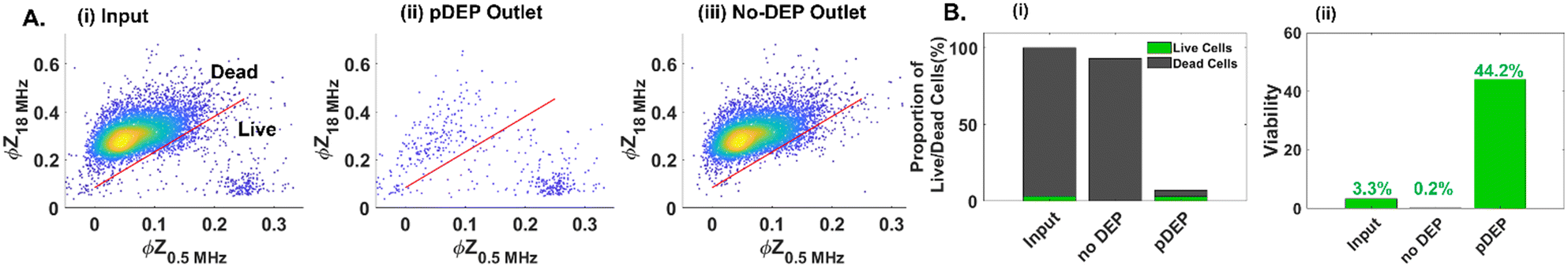
These optimized field parameters (25 Vpp at 1 MHz) and live-cell gating conditions (SVM model for ϕZ) are used to quantify pDEP enrichment of low levels of live PDAC circulating cell samples (2–3% live cells in a well plate after 48 h gemcitabine treatment, as per numbers and sizes in ESI,† Fig. S7). Based on the ϕZ gate from the SVM model (Fig. 5A), enrichment of low levels of live cells from the input sample (Fig. 5A(i)) into the “pDEP” outlet is apparent, with deflection of only minimal levels of dead cells (Fig. 5A(ii)), while the “no DEP” outlet is composed predominantly of dead cells (Fig. 5A(iii)). This is also apparent in the plots based on the ϕZ gate (Fig. 5B(i)) and GFP expression level (ESI,† Fig. S5A). The proportion of viable cells increases from ∼3% in the input sample to ∼44% collected in the pDEP outlet within 20 minutes (Fig. 5B(ii)), based on the ϕZ gate and based on the GFP expression level in ESI,† Fig. S5B. This demonstrates pDEP enrichment from dilute samples (∼3%) to ∼44%, at a collection efficiency of ∼60%, while rejecting >90% of dead cells.
DEP vs. DLD for live PDAC cell enrichment
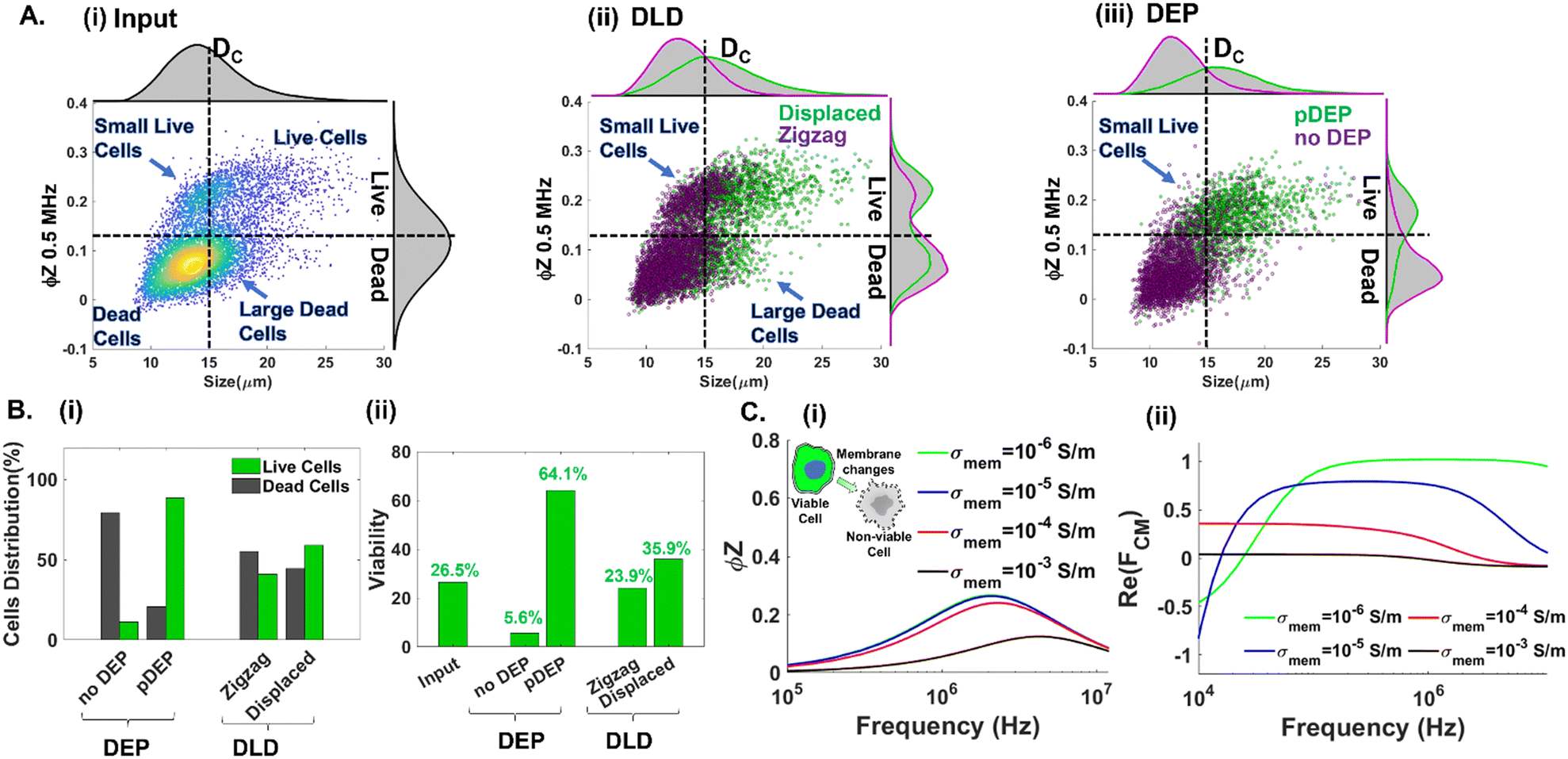
Next, we highlight the importance of utilizing multiple cellular biophysical metrics (size, plasma membrane and cytoplasmic properties) for enriching live circulating PDAC cells with wide and overlapping size distributions with other cell subpopulations in suspension. Impedance cytometry data (Fig. 4B(iv)) suggest that pDEP separation at 1 MHz selects live cells based on their membrane and cytoplasmic properties, with size included in the volumetric contribution to the trapping force. In contrast, microfluidic DLD only selects cells based on their size. Hence, we compare the separation metrics using DLD and pDEP for an input sample composed of ∼27% live PDAC cells (combining 3 wells of floating cells and 1 well of trypsinized adhered cells from a 6-well plate). As per Fig. 6A(i), while the input sample is composed predominantly of live cell events that are of greater cell size than dead cell events (top right quadrant vs. bottom left quadrant), there are substantial numbers of live cell events with cell size <15 μm (i.e., small live cells in the top left quadrant). Previously, we established that pDEP deflection at 1 MHz is needed to enrich small live cells <15 μm (Fig. 4B(iv)), whereas pDEP deflection at lower frequencies increases only the live cell populations of size >15 μm. It is also noteworthy that this input sample has a substantial number of events of size >15 μm in the gate of dead cells (right bottom quadrant of Fig. 6A(i)), which likely arises from necrotic cells released from the adherent culture under drug treatment. Using a DLD array designed for a cut-off size (DC) of 15 μm,49 we would expect that most of the live cells would be collected in the displaced outlet and most of the dead cells would be collected in the zigzag outlet. While this is indeed the case based on Fig. 6A(ii), wherein dead cells are collected in the zigzag outlet (left bottom quadrant) and live cells are collected in the displaced outlet (right top quadrant), the zigzag outlet also includes substantial live cell events for small cells (<15 μm in the left top quadrant) and the displaced outlet also includes substantial dead cell events arising from large cells (>15 μm in the bottom right quadrant). On the other hand, using pDEP deflection at 1 MHz, the subpopulation of small live cells (<15 μm) is collected in the pDEP outlet along with other live cells (top left and top right quadrants of Fig. 6A(iii)), while the large dead cells (>15 μm) remain undeflected and are collected in the “no DEP” outlet along with other dead cells (bottom left and bottom right quadrants of Fig. 6A(iii)). The summary plots of collected cell events at each outlet classified in Fig. 6B(i) for the proportion of live vs. dead cell events and in Fig. 6B(ii) for % viable cells show the poor separation purity of DLD vs. the high purity obtained with pDEP. Also refer to ESI† Fig. S6 for collected cell #s to compare live cell enrichment after pDEP vs. DLD. To illustrate the biophysical basis that allows pDEP to select small live cells despite the lower volumetric contribution to their net trapping force and reject large dead cells despite the higher volumetric contribution to their trapping force, we consider simulations of the frequency dispersion of the impedance phase (ϕZ) (Fig. 6C(i)) and the real part of the Clausius Mossotti factor (Re(fCM)) (Fig. 6C(ii)) for live cells with cell membranes of low conductivity (σmem ≤ 10−4 S m−1) and for dead cells with cell membranes of higher conductivity (σmem ≥ 10−3 S m−1). The ϕZ frequency dispersion is relatively stable for σmem levels ≤ 10−4 S m−1 (i.e., live cells), but sharply drops off for σmem levels ≥ 10−3 S m−1 (i.e., dead cells), especially in the 1–2 MHz range. As a result, live cells maintain interfacial polarization under DEP at 1 MHz due to sharp differences in cell vs. media dielectric properties that cause high positive Re(fCM) values at 1 MHz, whereas dead cells do not maintain sharp differences in cell vs. media dielectric properties, thereby exhibiting sharp drop-offs in Re(fCM) values in the 1 MHz region. Hence, despite the large volumetric contribution to the DEP trapping force from large dead cells (>15 μm), their low Re(fCM) value leads to minimal pDEP. Similarly, despite the small volumetric contribution to the DEP trapping force from small live cells (<15 μm), their high Re(fCM) value at 1 MHz causes them to be selected under pDEP deflection.
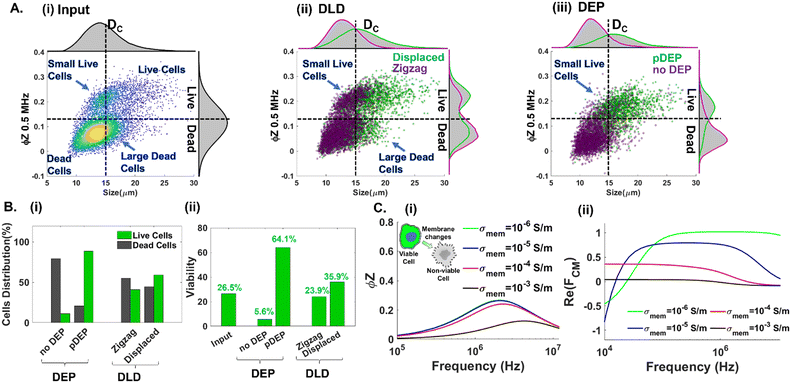 |
| | Fig. 6 Enrichment of live PDAC cells by DLD vs. DEP. A. Impedance scatter plots with respective histograms for: (i) the input sample (50% suspended cells and 50% trypsinized adhered cells), (ii) post-DLD enrichment and (iii) post-DEP enrichment. B. Summary plots of cells within the input and DEP enriched and DLD enriched outlets based on: (i) distribution of live/dead cells and (ii) % viable cells at each outlet. C. Frequency response of: (i) the impedance phase for viable (low membrane conductance) and non-viable cells (high membrane conductance) and (ii) Re(fCM) for viable and non-viable cells shows that Re(fCM) drops off to zero at ∼1 MHz for σmembrane > 10−4 S m−1, causing pDEP force to sharply drop off irrespective of cell size. | |
Conclusions
We present a flowthrough positive dielectrophoresis (pDEP) device to enrich live circulating cancer cells at rare levels (down to ∼3%) with chemo-resistant properties (i.e., high live cell numbers with drug treatment: ESI† Fig. S7A) and overlapping cell size distributions with apoptotic and necrotic cells in the culture suspension, by using impedance cytometry to optimize the pDEP frequency for selection of live cells over the entire size range (10–25 μm). A support vector machine (SVM) model is trained with live and dead cell samples to optimize gating of the impedance metrics of live vs. dead cells within the heterogeneous sample of broad cell size distributions, which is validated using cancer cells that lose GFP expression upon viability loss. While the subpopulation of large live circulating PDAC cells (>15 μm) can be enriched by pDEP at lower frequencies (0.1 and 0.6 MHz), pDEP at 1 MHz is essential for enriching the subpopulation of small live PDAC cells (<15 μm) which exhibits mesenchymal characteristics. This small live cell fraction cannot be enriched using size-based microfluidic DLD, due to size overlaps with apoptotic cells. Based on simulated frequency dispersions of the impedance phase and fCM, we show that live cells can maintain interfacial polarization under DEP conditions at 1 MHz due to sharp differences in cell vs. media dielectric properties that cause high positive fCM values. This enables enrichment despite lower volumetric contribution to DEP trapping force for cells of smaller size. On the other hand, dead cells (including smaller sized apoptotic and larger sized necrotic cells) are unable to maintain sharp differences in cell vs. media dielectric properties, thereby exhibiting sharp drop-offs in fCM values in the 1 MHz region causing minimal to no pDEP. As a result, low levels of live circulating PDAC cells with wide size distributions can be enriched from the drug-treated media of adherent cultures. Based on multiparametric information from impedance cytometry (cell size, viability, membrane capacitance, cytoplasmic conductivity, etc.) and its minimal sample preparation needs, we envision its broader utilization to optimize microfluidic enrichment using versatile sample types.
Methods
Cell samples
PDAC tumor samples were generated from specimens collected in collaboration with the University of Virginia Biorepository and Tissue Research Facility after approval of the University of Virginia Institutional Review Board for Health Sciences Research and after informed written consent from patients. Tumors were propagated in the pancreas of immunocompromised mice. Xenograft lines were established, transfected with GFP, and selected using puromycin and maintained in RPMI 1640 with 10% FBS and 2 mM glutamine.
Drug treatment
GFP transfected tumor cells were plated at 4 × 105 cells per ml in 6-well plates and allowed to adhere overnight in complete media (RPMI + 10% FBS). The cells were then treated with gemcitabine at a concentration of 1 μg ml−1 for 48 h. Floating cells were collected from the medium supernatant in the wells (centrifuged at 300g for 10 min). Adherent cells were washed in 1× PBS (Thermo Fisher) and then subjected to a 0.05% trypsin treatment (Thermo Fisher) for 10 min, and then complete media were added. The adherent cells were then centrifuged at 300g for 10 min. Cell pellets of each sample type were then resuspended in 1 mL of DEP buffer (sucrose and BSA, conductivity 45 μS cm−1) and counted using a hemocytometer. Floating cell samples were pDEP enriched, mixing in sample wells from adherent cells in some cases (Fig. 3 and 4), to increase live cell events for optimizing the DEP device.
Device design and fabrication
The DEP device as shown in Fig. 1B consists of two sheath inlets and one sample inlet. The active region of the device consists of a microfluidic channel flanked by two electrode channels that adjoins sequential orifices (20 μm) on one side and PDMS posts (25 μm diameter, 40 μm spacing) on the other side, to create spatial field non-uniformities across the channel cross-section orthogonal to the sample flow. The device was fabricated in a single layer to a depth of 50 μm using SU8 photolithography on a 4 inch silicon wafer. The silicon wafers were then silanized to allow for subsequent de-molding of PDMS. PDMS (Dow Chemicals) was poured onto the silicon master for micromolding at a ratio of 10:1 (base to crosslinker) and allowed to crosslink at 70 °C for 12 hours. Devices were then punched to create inlets and outlets for the electrodes, sample, and sheath channels. Devices were bonded to glass slides using oxygen plasma. Electrode channels were filled with liquefied Field's metal as described previously.34,35 Briefly, the device was immersed in a water bath at 65 °C and the liquefied Field's metal (RotoMetals) was introduced through a syringe using positive pressure. After complete filling of the electrode channel, the device was allowed to cool at room temperature resulting in solidification of the metal. DLD devices were fabricated as per prior reports.47
Device operation
Prior to introducing sample and sheath flows, the sample inlet and outlets were filled with 3% BSA (in 1× PBS) and left at room temperature for 30 min. The BSA solution was then removed. Sample and sheath fluid flows were introduced in their respective inlets using syringe pumps (Cetoni GmbH). Net flow rates of 2.64 μL min−1 were used for pDEP enrichment, based on a cell sample flow rate of 0.24 μL min−1, sheath flow near the orifice edge at 0.9 μL min−1 and sheath flow near the posts at 1.5 μL min−1. The chip was placed on a microscope stage equipped with a CMOS camera (Hamamatsu) for imaging cell streamlines. A function generator and amplifier were connected to the electrodes to deliver the studied voltage and frequency range.
Flow cytometry
After DEP enrichment, samples were stained using APC Annexin V (Biolegend) and Zombie NIR (Biolegend) to quantify viability and apoptotic levels using a Beckman Cytoflex flow cytometer and data were analyzed using Beckman CytExpert software. For analysis of EpCAM levels, samples were stained for 30 min with PE anti EpCAM (CD326) antibody (Biolegend). The samples were then centrifuged and washed twice before analysis. Cells were gated based on forward (FSC) and side scatter (SSC) to exclude debris. To exclude doublets, single cells were then gated based on side scatter – area vs. height. As a measure of viability, the intrinsic GFP signal was also measured and analyzed.
Impedance cytometry
Fractions at each outlet from the DEP device were centrifuged at 300g for 5 minutes and the resulting cell pellet was resuspended in 1× PBS along with 7 μm polystyrene reference beads for single-cell impedance cytometry using an impedance spectroscope (HF2IS, Zurich Instruments).50 The measured current was converted to voltage using a current amplifier (HF2TA, Zurich Instruments) at a gain factor of 1000 and a sampling rate of 115000 samples per s. Lock-in amplification was used to separate the real and imaginary signal components at each frequency, from which the impedance magnitude and phase were derived. Cells and beads (7 μm polystyrene, Sigma Aldrich) were co-flowed through a glass chip with top and bottom gold electrodes to acquire the impedance phase and magnitude at four frequencies (0.5 MHz, 2 MHz, 18 MHz, and 30 MHz) concurrently.
Data analysis
Impedance cytometry data were plotted and analyzed using MATLAB. The impedance signal of the cells was normalized by dividing it by the mean impedance of the reference polystyrene beads that show a frequency independent impedance response. Cells and beads were gated separately using impedance data at 30 MHz. The electrical diameter was calculated using the size of the reference beads and the normalized impedance magnitude at 0.5 MHz 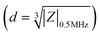 .
.
Machine learning
Machine learning (ML) algorithms were implemented using MATLAB. The data set composed of the phase and magnitude at 0.5, 2, 18 and 30 MHz, the electrical diameter and the impedance magnitude opacity was classified as live and dead subpopulations using SVM (support vector machine) models trained with a sample of untreated live cells and heat treated dead cells, using 70% data for testing and 30% for validation. Details are available in ESI† section B1, Fig. S8.
COMSOL simulation
To measure the spatial extent of the electric field from the high field point and its influence on particle deflection, the electric current module, flow module and particle tracing modules of COMSOL were used as described previously.34
Author contributions
A. Rane: conceptualization, investigation, formal analysis, writing; J. Jarmoshti: conceptualization, investigation, formal analysis, writing; A. Siddique & Sara Adair: methodology, investigation; K. Torres-Castro & C. Honrado: methodology, data curation; T. W. Bauer: resources, supervision; N. S. Swami: conceptualization, methodology, formal analysis, resources, writing, supervision, project administration, funding acquisition.
Conflicts of interest
The authors have no financial interests or other conflicts of interest related to this work.
Acknowledgements
This research was supported by the NCI Cancer Center Grant P30 CA44579, AFOSR grant FA2386-21-1-4070, NSF Award # #2222933, and the University of Virginia's Strategic Investment Fund to establish the Engineering in Medicine program and the University of Virginia's Cancer Center Trainee program.
Notes and references
- H. Dillekås, M. S. Rogers and O. Straume, Are 90% of deaths from cancer caused by metastases?, Cancer Med., 2019, 8(12), 5574–5576 CrossRef PubMed.
- C. L. Chaffer and R. A. Weinberg, A perspective on cancer cell metastasis, Science, 2011, 331(6024), 1559–1564 CrossRef CAS PubMed.
- M. Raudenská, K. Petrláková, T. Juriňáková, J. L. Fialová, M. Fojtů, M. Jakubek, D. Rösel, J. Brábek and M. Masařík, Engine shutdown: migrastatic strategies and prevention of metastases, Trends Cancer, 2023, 9(4), 293–308 CrossRef PubMed.
- L. Rahib, B. D. Smith, R. Aizenberg, A. B. Rosenzweig, J. M. Fleshman and L. M. Matrisian, Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States, Cancer Res., 2014, 74(11), 2913–2921 CrossRef CAS PubMed.
- J. M. Herman, M. J. Swartz, C. C. Hsu, J. Winter, T. M. Pawlik, E. Sugar, R. Robinson, D. A. Laheru, E. Jaffee and R. H. Hruban, Analysis of fluorouracil-based adjuvant chemotherapy and radiation after pancreaticoduodenectomy for ductal adenocarcinoma of the pancreas: results of a large, prospectively collected database at the Johns Hopkins Hospital, J. Clin. Oncol., 2008, 26(21), 3503 CrossRef PubMed.
- A. Jemal, R. Siegel, E. Ward, Y. Hao, J. Xu and M. J. Thun, Cancer statistics, 2009, Ca-Cancer J. Clin., 2009, 59(4), 225–249 CrossRef PubMed.
- J. P. Neoptolemos, D. D. Stocken, H. Friess, C. Bassi, J. A. Dunn, H. Hickey, H. Beger, L. Fernandez-Cruz, C. Dervenis and F. Lacaine, A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer, N. Engl. J. Med., 2004, 350(12), 1200–1210 CrossRef CAS PubMed.
- A. W. Lambert, D. R. Pattabiraman and R. A. Weinberg, Emerging biological principles of metastasis, Cell, 2017, 168(4), 670–691 CrossRef CAS PubMed.
- P. Paoli, E. Giannoni and P. Chiarugi, Anoikis molecular pathways and its role in cancer progression, Biochim. Biophys. Acta, Mol. Cell Res., 2013, 1833(12), 3481–3498 CrossRef CAS PubMed.
- Y.-N. Kim, K. H. Koo, J. Y. Sung, U.-J. Yun and H. Kim, Anoikis resistance: an essential prerequisite for tumor metastasis, Int. J. Cell Biol., 2012, 306879 Search PubMed.
- J. F. Edd, A. Mishra, K. C. Smith, R. Kapur, S. Maheswaran, D. A. Haber and M. Toner, Isolation of circulating tumor cells, iScience, 2022, 104696 CrossRef CAS PubMed.
- M. T. Gabriel, L. R. Calleja, A. Chalopin, B. Ory and D. Heymann, Circulating tumor cells: a review of non–EpCAM-based approaches for cell enrichment and isolation, Clin. Chem., 2016, 62(4), 571–581 CrossRef CAS PubMed.
- A. Semaan, V. Bernard, D. U. Kim, J. J. Lee, J. Huang, N. Kamyabi, B. M. Stephens, W. Qiao, G. R. Varadhachary and M. H. Katz, Characterisation of circulating tumour cell phenotypes identifies a partial-EMT sub-population for clinical stratification of pancreatic cancer, Br. J. Cancer, 2021, 124(12), 1970–1977 CrossRef CAS PubMed.
- M. Yu, A. Bardia, N. Aceto, F. Bersani, M. W. Madden, M. C. Donaldson, R. Desai, H. Zhu, V. Comaills and Z. Zheng, Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility, Science, 2014, 345(6193), 216–220 CrossRef CAS PubMed.
- A. A. Friedman, A. Letai, D. E. Fisher and K. T. Flaherty, Precision medicine for cancer with next-generation functional diagnostics, Nat. Rev. Cancer, 2015, 15(12), 747 CrossRef CAS PubMed.
- J. Y. Park, A. L. Jeong, H. J. Joo, S. Han, S.-H. Kim, H.-Y. Kim, J.-S. Lim, M.-S. Lee, H.-K. Choi and Y. Yang, Development of suspension cell culture model to mimic circulating tumor cells, Oncotarget, 2018, 9(1), 622 CrossRef PubMed.
- N. Sasaki, F. Gomi, F. Hasegawa, K. Hirano, M. Fujiwara, M. Toyoda and T. Ishiwata, Characterization of the metastatic potential of the floating cell component of MIA PaCa-2, a human pancreatic cancer cell line, Biochem. Biophys. Res. Commun., 2020, 522(4), 881–888 CrossRef CAS PubMed.
- J. Zhang, N. Chintalaramulu, R. Vadivelu, H. An, D. Yuan, J. Jin, C. H. Ooi, I. E. Cock, W. Li and N.-T. Nguyen, Inertial microfluidic purification of floating cancer cells for drug screening and three-dimensional tumor models, Anal. Chem., 2020, 92(17), 11558–11564 CrossRef CAS PubMed.
- Q. Huang, S. Li, X. Hu, M. Sun, Q. Wu, H. Dai, Y. Tan, F. Sun, C. Wang and X. Rong, Shear stress activates ATOH8 via autocrine VEGF promoting glycolysis dependent-survival of colorectal cancer cells in the circulation, J. Exp. Clin. Cancer Res., 2020, 39(1), 1–16 CrossRef PubMed.
- C. K. Ip, S.-S. Li, M. Y. Tang, S. K. Sy, Y. Ren, H. C. Shum and A. S. Wong, Stemness and chemoresistance in epithelial ovarian carcinoma cells under shear stress, Sci. Rep., 2016, 6(1), 1–11 CrossRef PubMed.
- J. Lan, H. Lu, D. Samanta, S. Salman, Y. Lu and G. L. Semenza, Hypoxia-inducible factor 1-dependent expression of adenosine receptor 2B promotes breast cancer stem cell enrichment, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(41), 9640–9648 CrossRef PubMed.
- M. Najafi, B. Farhood, K. Mortezaee, E. Kharazinejad, J. Majidpoor and R. Ahadi, Hypoxia in solid tumors: a key promoter of cancer stem cell (CSC) resistance, J. Cancer Res. Clin. Oncol., 2020, 146(1), 19–31 CrossRef PubMed.
- S.-H. Lin, T. Liu, X. Ming, Z. Tang, L. Fu, P. Schmitt-Kopplin, B. Kanawati, X.-Y. Guan and Z. Cai, Regulatory role of hexosamine biosynthetic pathway on hepatic cancer stem cell marker CD133 under low glucose conditions, Sci. Rep., 2016, 6(1), 1–10 CrossRef PubMed.
- K. Shibuya, M. Okada, S. Suzuki, M. Seino, S. Seino, H. Takeda and C. Kitanaka, Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells, Oncotarget, 2015, 6(2), 651 CrossRef PubMed.
-
H. Morgan and N. G. Green, AC electrokinetics: colloids and nanoparticles, Research Studies Press, 2003 Search PubMed.
- S. Shim, K. Stemke-Hale, A. M. Tsimberidou, J. Noshari, T. E. Anderson and P. R. Gascoyne, Antibody-independent isolation of circulating tumor cells by continuous-flow dielectrophoresis, Biomicrofluidics, 2013, 7(1), 011807 CrossRef PubMed.
- P. R. Gascoyne and S. Shim, Isolation of circulating tumor cells by dielectrophoresis, Cancers, 2014, 6(1), 545–579 CrossRef PubMed.
- P. G. Bonacci, G. Caruso, G. Scandura, C. Pandino, A. Romano, G. I. Russo, R. Pethig, M. Camarda and N. Musso, Impact of buffer composition on biochemical, morphological and mechanical parameters: A tare before dielectrophoretic cell separation and isolation, Transl. Oncol., 2023, 28, 101599 CrossRef CAS PubMed.
- M. Li and R. K. Anand, High-throughput selective capture of single circulating tumor cells by dielectrophoresis at a wireless electrode array, J. Am. Chem. Soc., 2017, 139(26), 8950–8959 CrossRef CAS PubMed.
- J. T. Banovetz, S. Manimaran, B. T. Schelske and R. K. Anand, Parallel Dielectrophoretic Capture, Isolation, and Electrical Lysis of Individual Breast Cancer Cells to Assess Variability in Enzymatic Activity, Anal. Chem., 2023, 95(20), 7880–7887 CrossRef CAS PubMed.
- H. Yang, T. Chen, Y. Hu, F. Niu, X. Zheng, H. Sun, L. Cheng and L. Sun, A microfluidic platform integrating dynamic cell culture and dielectrophoretic manipulation for in situ assessment of endothelial cell mechanics, Lab Chip, 2023, 23, 3581–3592 RSC.
- X. Xing, C. N. Ng, M. L. Chau and L. Yobas, Railing cells along 3D microelectrode tracks for continuous-flow dielectrophoretic sorting, Lab Chip, 2018, 18(24), 3760–3769 RSC.
- V. Varmazyari, H. Habibiyan, H. Ghafoorifard, M. Ebrahimi and S. Ghafouri-Fard, A dielectrophoresis-based microfluidic system having double-sided optimized 3D electrodes for label-free cancer cell separation with preserving cell viability, Sci. Rep., 2022, 12(1), 12100 CrossRef CAS PubMed.
- X. Huang, K. Torres-Castro, W. Varhue, A. Salahi, A. Rasin, C. Honrado, A. Brown, J. Guler and N. S. Swami, Self-aligned sequential lateral field non-uniformities over channel depth for high throughput dielectrophoretic cell deflection, Lab Chip, 2021, 21(5), 835–843 RSC.
- X. Huang, K. Torres-Castro, W. Varhue, A. Rane, A. Rasin and N. S. Swami, On-chip microfluidic buffer swap of biological samples in-line with downstream dielectrophoresis, Electrophoresis, 2022, 43, 1275–1282 CrossRef CAS PubMed.
- S. Shim, K. Stemke-Hale, J. Noshari, F. F. Becker and P. R. Gascoyne, Dielectrophoresis has broad applicability to marker-free isolation of tumor cells from blood by microfluidic systems, Biomicrofluidics, 2013, 7(1), 011808 CrossRef PubMed.
- C. Huang, J. P. Smith, T. N. Saha, A. D. Rhim and B. J. Kirby, Characterization of microfluidic shear-dependent epithelial cell adhesion molecule immunocapture and enrichment of pancreatic cancer cells from blood cells with dielectrophoresis, Biomicrofluidics, 2014, 8(4), 044107 CrossRef PubMed.
- F. S. Iliescu, W. J. Sim, H. Heidari, D. P. Poenar, J. Miao, H. K. Taylor and C. Iliescu, Highlighting the uniqueness in dielectrophoretic enrichment of circulating tumor cells, Electrophoresis, 2019, 40(10), 1457–1477 CrossRef CAS PubMed.
- G. I. Russo, N. Musso, A. Romano, G. Caruso, S. Petralia, L. Lanzanò, G. Broggi and M. Camarda, The role of dielectrophoresis for cancer diagnosis and prognosis, Cancers, 2021, 14(1), 198 CrossRef PubMed.
- K. C. Cheung, M. Di Berardino, G. Schade-Kampmann, M. Hebeisen, A. Pierzchalski, J. Bocsi, A. Mittag and A. Tárnok, Microfluidic impedance-based flow cytometry, Cytometry, Part A, 2010, 77(7), 648–666 CrossRef PubMed.
- T. Sun and H. Morgan, Single-cell microfluidic impedance cytometry: a review, Microfluid. Nanofluid., 2010, 8, 423–443 CrossRef CAS.
- C. Honrado, P. Bisegna, N. S. Swami and F. Caselli, Single-cell microfluidic impedance cytometry: From raw signals to cell phenotypes using data analytics, Lab Chip, 2021, 21(1), 22–54 RSC.
- J. Lu, C. A. Barrios, A. R. Dickson, J. L. Nourse, A. P. Lee and L. A. Flanagan, Advancing practical usage of microtechnology: a study of the functional consequences of dielectrophoresis on neural stem cells, Integr. Biol., 2012, 4(10), 1223–1236 CrossRef CAS PubMed.
- C. Honrado, A. Salahi, S. J. Adair, J. H. Moore, T. W. Bauer and N. S. Swami, Automated biophysical classification of apoptotic pancreatic cancer cell subpopulations by using machine learning approaches with impedance cytometry, Lab Chip, 2022, 22(19), 3708–3720 RSC.
- A. Salahi, C. Honrado, J. Moore, S. Adair, T. W. Bauer and N. S. Swami, Supervised learning on impedance cytometry data for label-free biophysical distinction of pancreatic cancer cells versus their associated fibroblasts under gemcitabine treatment, Biosens. Bioelectron., 2023, 231, 115262 CrossRef CAS PubMed.
- T. N. Pahattuge, I. M. Freed, M. L. Hupert, S. Vaidyanathan, K. Childers, M. A. Witek, K. Weerakoon-Ratnayake, D. Park, A. Kasi, M. F. Al-Kasspooles, M. C. Murphy and S. A. Soper, System modularity chip for analysis of rare targets (SMART-chip): Liquid biopsy samples, ACS Sens., 2021, 6(5), 1831–1839 CrossRef CAS PubMed.
- I. M. Freed, A. Kasi, O. Fateru, M. Hu, P. Gonzalez, N. Weatherington, H. Pathak, S. Hyter, W. Sun, R. Al-Rajabi, J. Baranda, M. L. Hupert, P. Chalise, A. K. Godwin, M. A. Witek and S. A. Soper, Circulating tumor cell subpopulations predict treatment outcome in Pancreatic ductal adenocarcinoma (PDAC) patients, Cells, 2021, 12(18), 2266 CrossRef PubMed.
- V. Farmehini, A. Rohani, Y.-H. Su and N. S. Swami, A wide-bandwidth power amplifier for frequency-selective insulator-based dielectrophoresis, Lab Chip, 2014, 14(21), 4183–4187 RSC.
- K. Torres-Castro, J. Jarmoshti, L. Xiao, A. Rane, A. Salahi, L. Jin, X. Li, F. Caselli, C. Honrado and N. S. Swami, Multichannel Impedance Cytometry Downstream of Cell Separation by Deterministic Lateral Displacement to Quantify Macrophage Enrichment in Heterogeneous Samples, Adv. Mater. Technol., 2023, 8(8), 2201463 CrossRef CAS PubMed.
- C. Honrado, N. Michel, J. H. Moore, A. Salahi, V. Porterfield, M. J. McConnell and N. S. Swami, Label-free quantification of cell cycle synchronicity of human neural progenitor cells based on electrophysiology phenotypes, ACS Sens., 2020, 6(1), 156–165 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Impedance biometrics of live adherent, live floating and dead cells, live/dead staining of samples in each outlet, GFP based quantification of enrichment of adherent and floating cells, movie showing inlets, the active region, and collection under E-field OFF and ON, SVM classification and dielectrophoretic shell modelling. See DOI: https://doi.org/10.1039/d3lc00804e |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Karina
Torres-Castro
c,
Karina
Torres-Castro
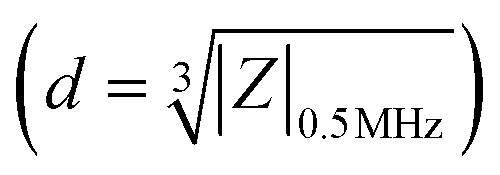 , apoptotic state44 (i.e., live vs. early apoptotic, late apoptotic and necrotic states based on ϕZ0.5MHzvs. ϕZ18MHz), cell membrane (|Z| and ϕZ in the 1–10 MHz range) and cell cytoplasmic properties (|Z| and ϕZ in the >10 MHz range). This allows for optimization of pDEP conditions by quantification of field-induced damage to the DEP enriched fractions, without needing to use GFP expressing PDAC cells. Live cell gating can be established based on the SVM model (optimized gate in Fig. 4A), with live cells roughly showing higher ϕZ0.5MHz levels (>0.125 at mean cell size) due to effective electric field screening by the intact plasma membrane, and lower ϕZ18MHz levels (<0.2 at mean cell size) due to the absence of any alteration to their cytoplasmic conductivity by penetration of the culture media that is observed for dead cells with disrupted plasma membranes. Using an input sample of ∼27% live PDAC cells, it is apparent that under field OFF conditions, cells pass undeflected into the “no DEP” outlet, with very few cell events in the pDEP outlet (Fig. 4A(i), (v) and C(i)). At successively higher frequencies of 0.1 MHz (Fig. 4A(ii)), 0.6 MHz (Fig. 4A(iii)) and 1 MHz (Fig. 4A(iv)), the number of events in the live cell gates successively increases. Next, we use the impedance scatter plot (Fig. 4B) of ϕZ0.5MHzvs. electrical size of cells to quantify the live cell #s (gated based on the SVM model) that are enriched in the pDEP outlet for each size range of interest. Using the intersection point of 15 μm for the histograms of live circulating cells vs. dead cells within the suspension of the culture media after gemcitabine treatment (Fig. 2A(i)), cell events of live circulating cells < 15 μm are called “small live cells” since they overlap with dead cells in the suspension, while those > 15 μm are called “large live cells”. Based on this, it is apparent that live cell events increase successively in the “large live cell” gate for pDEP collected fractions at 0.1 MHz and 0.6 MHz, while remaining substantially unchanged at 1 MHz. On the other hand, pDEP at 1 MHz is crucial for increasing the proportion of events in the “small live cell” gate of the pDEP collected fractions. The summary plot (Fig. 4C(i) and (ii)) confirms ∼80% collection of live cells into the pDEP outlet at 100 kHz and ∼90% collection at 600 kHz and 1 MHz DEP trapping frequencies, with 1 MHz enabling the enrichment of “small live PDAC cells”. Selection purity plots (Fig. 4C(iii)) show that the input PDAC sample of ∼27% live cells is enriched in the pDEP outlet to 55% live cells at 0.1 MHz, to 63% at 0.6 MHz and to 65.5% at 1 MHz.
, apoptotic state44 (i.e., live vs. early apoptotic, late apoptotic and necrotic states based on ϕZ0.5MHzvs. ϕZ18MHz), cell membrane (|Z| and ϕZ in the 1–10 MHz range) and cell cytoplasmic properties (|Z| and ϕZ in the >10 MHz range). This allows for optimization of pDEP conditions by quantification of field-induced damage to the DEP enriched fractions, without needing to use GFP expressing PDAC cells. Live cell gating can be established based on the SVM model (optimized gate in Fig. 4A), with live cells roughly showing higher ϕZ0.5MHz levels (>0.125 at mean cell size) due to effective electric field screening by the intact plasma membrane, and lower ϕZ18MHz levels (<0.2 at mean cell size) due to the absence of any alteration to their cytoplasmic conductivity by penetration of the culture media that is observed for dead cells with disrupted plasma membranes. Using an input sample of ∼27% live PDAC cells, it is apparent that under field OFF conditions, cells pass undeflected into the “no DEP” outlet, with very few cell events in the pDEP outlet (Fig. 4A(i), (v) and C(i)). At successively higher frequencies of 0.1 MHz (Fig. 4A(ii)), 0.6 MHz (Fig. 4A(iii)) and 1 MHz (Fig. 4A(iv)), the number of events in the live cell gates successively increases. Next, we use the impedance scatter plot (Fig. 4B) of ϕZ0.5MHzvs. electrical size of cells to quantify the live cell #s (gated based on the SVM model) that are enriched in the pDEP outlet for each size range of interest. Using the intersection point of 15 μm for the histograms of live circulating cells vs. dead cells within the suspension of the culture media after gemcitabine treatment (Fig. 2A(i)), cell events of live circulating cells < 15 μm are called “small live cells” since they overlap with dead cells in the suspension, while those > 15 μm are called “large live cells”. Based on this, it is apparent that live cell events increase successively in the “large live cell” gate for pDEP collected fractions at 0.1 MHz and 0.6 MHz, while remaining substantially unchanged at 1 MHz. On the other hand, pDEP at 1 MHz is crucial for increasing the proportion of events in the “small live cell” gate of the pDEP collected fractions. The summary plot (Fig. 4C(i) and (ii)) confirms ∼80% collection of live cells into the pDEP outlet at 100 kHz and ∼90% collection at 600 kHz and 1 MHz DEP trapping frequencies, with 1 MHz enabling the enrichment of “small live PDAC cells”. Selection purity plots (Fig. 4C(iii)) show that the input PDAC sample of ∼27% live cells is enriched in the pDEP outlet to 55% live cells at 0.1 MHz, to 63% at 0.6 MHz and to 65.5% at 1 MHz.
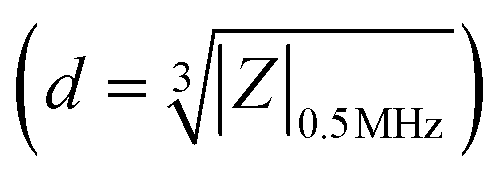 .
.