
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Automated centrifugal microfluidic system for the preparation of adaptor-ligated sequencing libraries†
Jimin
Guo‡
 a,
Daniel
Brassard‡
a,
Nadine
Adam‡
b,
Adrian J.
Verster
c,
Julie A.
Shay
c,
Caroline
Miville-Godin
a,
Mojra
Janta-Polczynski
a,
Jason
Ferreira
a,
Maxence
Mounier
a,
Ana V.
Pilar
b,
Kyle
Tapp
b,
Adam
Classen
b,
Matthew
Shiu
a,
Denis
Charlebois
d,
Nicholas
Petronella
c,
Kelly
Weedmark
b,
Nathalie
Corneau
*b and
Teodor
Veres
*a
a,
Daniel
Brassard‡
a,
Nadine
Adam‡
b,
Adrian J.
Verster
c,
Julie A.
Shay
c,
Caroline
Miville-Godin
a,
Mojra
Janta-Polczynski
a,
Jason
Ferreira
a,
Maxence
Mounier
a,
Ana V.
Pilar
b,
Kyle
Tapp
b,
Adam
Classen
b,
Matthew
Shiu
a,
Denis
Charlebois
d,
Nicholas
Petronella
c,
Kelly
Weedmark
b,
Nathalie
Corneau
*b and
Teodor
Veres
*a
aMedical Devices Research Center, Life Sciences Division, National Research Council of Canada, 75 de Mortagne Boulevard, Boucherville, QC J4B 6Y4, Canada. E-mail: teodor.veres@cnrc-nrc.gc.ca
bBureau of Microbial Hazards, Microbiology Research Division, Health Canada, 251 Sir Frederick Banting Driveway, Ottawa, ON K1A 0K9, Canada. E-mail: nathalie.corneau@hc-sc.gc.ca
cBureau of Food Surveillance and Science Integration, Bioinformatics High-Capacity Computing Laboratory, Health Canada, 251 Sir Frederick Banting Driveway, Ottawa, ON K1A 0K9, Canada
dCanadian Space Agency, 6767 Route de l'Aéroport, Saint-Hubert, QC J3Y 8Y9, Canada
First published on 4th December 2023
Abstract
The intensive workload associated with the preparation of high-quality DNA libraries remains a key obstacle toward widespread deployment of sequencing technologies in remote and resource-limited areas. We describe the development of single-use microfluidic devices driven by an advanced pneumatic centrifugal microfluidic platform, the PowerBlade, to automate the preparation of Illumina-compatible libraries based on adaptor ligation methodology. The developed on-chip workflow includes enzymatic DNA fragmentation coupled to end-repair, adaptor ligation, first DNA cleanup, PCR amplification, and second DNA cleanup. This complex workflow was successfully integrated into simple thermoplastic microfluidic devices that are amenable to mass production with injection molding. The system was validated by preparing, on chip, libraries from a mixture of genomic DNA extracted from three common foodborne pathogens (Listeria monocytogenes, Escherichia coli and Salmonella enterica serovar Typhimurium) and comparing them with libraries made via a manual procedure. The two types of libraries were found to exhibit similar quality control metrics (including genome coverage, assembly, and relative abundances) and led to nearly uniform coverage independent of GC content. This microfluidic technology offers a time-saving and cost-effective alternative to manual procedures and robotic-based automation, making it suitable for deployment in remote environments where technical expertise and resources might be scarce. Specifically, it facilitates field practices that involve mid- to low-throughput sequencing, such as tasks related to foodborne pathogen detection, characterization, and microbial profiling.
1. Introduction
Sequencing technologies are drastically changing microbiology practices in a wide range of applications, including food safety, environmental monitoring, and space genomics.1–4 These technologies enable increasingly thorough surveillance and monitoring by comprehensive pathogen identification and characterization. Indeed, the implementation of whole genome sequencing (WGS) by regulatory bodies has greatly augmented outbreak response, source tracking, and surveillance activities,5–9 with significant economic impact. For example, since 2013, the U.S. Food and Drug Administration's (FDA) GenomeTrakr database has been populated with WGS foodborne pathogen data from federal, state, and international laboratories. By increasing the effectiveness of public health response to outbreaks, this tool has saved an estimated $500 million annually in health benefits alone.10 Similarly, in 2017 PulseNet International, a global network of laboratories that includes a large Canadian cohort, transitioned away from traditional methodologies and began exclusively using WGS data to enhance outbreak detection, outbreak response and surveillance of foodborne pathogens.11 However, while WGS food safety applications are desired to identify pathogenic microorganisms present in food samples, a bottleneck limiting the widespread uptake of next generation sequencing technologies is the intensive sample workup required for sequencing, referred to as library preparation.1,4 Furthermore, while library preparation procedures can be automated by liquid handling workstations, these systems can be costly to procure and maintain, and do not provide a feasible solution for many settings where location, space constraints, technical expertise, and other environmental factors such as risk of cross-contamination, dust, dirt, or even microgravity preclude their use. Microfluidics-based automation of library preparation constitutes a promising alternative for compact, portable, miniaturized library preparation that is compatible with virtually any location with modest requirements.12Library preparation consists of sequential liquid handling and temperature incubation steps to convert a sample of nucleic acids into a format compatible with sequencing. The procedure required to prepare a library depends on various factors including sample type and purity, choice of sequencing platform, amount of genomic material available, and targeted information (e.g., general metagenomics versus de novo assembly, etc.). As such, procedures can range from a few simple steps to very complex assays requiring several hours with substantial hands-on time.
To prepare a library from a genomic DNA extract for sequencing on an Illumina instrument, the first step involves two common approaches: (i) DNA fragmentation (enzymatic or mechanical) followed by end preparation and adaptor ligation, or (ii) tagmentation, which involves simultaneous enzymatic fragmentation and adaptor insertion by the Tn5 transposase. Compared to protocols based on fragmentation, protocols based on tagmentation require significantly lower input, shorter incubation times, and fewer liquid transfer steps, making them ideal for microfluidics automation. As such, a plethora of microfluidic methods have implemented variations of tagmentation via centrifugal microfluidics,13 digital microfluidics,14 or classic pump-driven microfluidics at high throughput.15,16 However, tagmentation workflows have some performance shortcomings, as the resulting libraries tend to yield decreased coverage in GC-rich regions and have relatively short insert sizes,17–20 which compromises the quality and accuracy of genome assembly, single nucleotide variant identification,19,21,22 and metagenomic taxonomy.23 While fragmentation-based workflows are less impacted by these issues, they require more complex procedures, often comprising additional end preparation, adaptor ligation, PCR amplification, and DNA cleanup steps.24 The complexity of reliably performing sophisticated liquid transfer steps and effectively achieving thermal cycling make it challenging to implement fragmentation-based workflows on microfluidics chips.
In almost all library preparation workflows currently in use, DNA cleanup steps require the most liquid transfer operations, which is challenging to automate. The objective of DNA cleanup is to remove undesired DNA fragments such as primer dimers and adaptor dimers and resuspend the library in a buffer compatible with the subsequent steps of the protocol. Typically, DNA cleanup is performed through solid phase reversible immobilization (SPRI) by binding desired nucleic acid fragments to paramagnetic beads in the presence of salt and molecular crowding agents. The beads are then trapped with a magnet to remove the supernatant, washed with buffers containing a high concentration of ethanol, followed by elution of the nucleic acids. Because of the number of fluid transfer steps required by the SPRI process and the intrinsic difficulty of manipulating an ethanol-based wash buffer within microfluidics devices due to its low surface tension and low contact angle,25 DNA cleanup protocols must often be modified before integration into microfluidics-based systems. In one previous approach, a magnet was applied to pull DNA-bound paramagnetic beads from one reservoir to another through an oil-filled channel, thereby removing most of the supernatant in one movement with limited need for additional wash steps.26 Alternatively, magnetic force and electroosmotic flow in opposing directions have been used to separate DNA-bound paramagnetic beads from the supernatant.27 While these approaches can circumvent the limitations of some microfluidic platforms in handling complex liquid transfer steps, such modifications may require extensive optimization, must be fully compatible with other functions including PCR amplification, and ought to demonstrate robustness with pre-programmed settings.
Direct integration of conventional DNA cleanup protocols requires microfluidic technologies that can support complex liquid manipulations. Digital microfluidics (DMF) can perform DNA purification by manipulating micro- to nanoliter droplets over magnetically captured SPRI beads using electrical potentials applied on an array of electrodes.14,28,29 Another approach makes use of an array of integrated pneumatic valves in a multilayer polydimethylsiloxane (PDMS) device fabricated by soft lithography.16 Both approaches have successfully demonstrated automation of library preparation.12,14,28 However, they rely on the integration of numerous active elements in a single-use microfluidic device, which can impact both fabrication cost and device reliability since the failure of a single electrode or valve can affect the assay execution. These platforms also offer limited capacity to process raw, unprocessed samples, can face challenges with respect to reliably manipulating some reagents or samples, and can be impacted by the presence of air bubbles. Alternatively, centrifugal microfluidics has recently been demonstrated as a viable approach for bead cleanup and library preparation. Bead mixing, sedimentation, washing, and elution steps can be achieved in low-cost disposable cartridges by combining centrifugal force, pumping by thermal expansion of air, siphon valves, and capillary effects.13,30 The centrifugal force also provides additional means to prepare raw samples or manipulate beads and limits issues associated with air bubbles, which is a key advantage for field deployment. However, the number of independent steps that can be reliably integrated in classical centrifugal microfluidic platforms remains limited by the space available on the device and by the small number of independent forces available for assay execution. Therefore, integration of multiple rounds of DNA cleanups, as required in many library preparation workflows, remains a considerable challenge for centrifugal microfluidics. In summary, while centrifugal microfluidics is an ideal approach for developing single-use and automated library preparation workflows, its full potential has yet to be explored by developing technologies that permit integration with minimal modifications to standard library preparation procedures and original reagent compositions, compatibility with a wide range of raw samples, and low-cost consumables.
In this paper, we report on the design and validation of an automated on-chip fragmentation-based library preparation workflow for Illumina based on the PowerBlade platform, an advanced pneumatic centrifugal microfluidic technology published previously.25,31–35 In this technology, air pneumatic control lines are employed in addition to the centrifugal force, providing additional degrees of freedom to program intricate liquid displacements. We demonstrate the on-chip integration of a complex fragmentation-based library preparation procedure in simple thermoplastic microfluidic devices that do not contain any active element and are fabricated by a scalable injection molding process. We present optimization of the on-chip protocol, including the liquid manipulation steps required to perform two complete on-chip DNA cleanup steps, SPRI bead manipulation strategies, and PCR thermocycling. To validate the developed assay, libraries were prepared from a mixture of genomic DNA extracts from common foodborne bacterial pathogens to represent a low-diversity metagenomic sample. We show that both the quality control metrics and the sequencing data generated from libraries prepared on-chip are comparable with those prepared by a manual procedure. Both types of libraries allowed precise taxonomic identification and relative abundance quantification of bacterial populations in the metagenomic samples, demonstrating the utility of this microfluidic library preparation technology for applied food microbiology. The technology also opens the door for the integration of even more complex sample preparation workflows that could, as an example, include a DNA extraction step from a raw sample31 in addition to the actual library preparation.
2. Assay design and on-chip integration
2.1 Overview of fragmentation-based library preparation procedure
An overview of the library preparation procedure that was integrated for microfluidic on-chip automation is shown in Fig. 1 (refer to Materials and methods section for additional details). The initial sample consisted of 500 ng of mixed genomic DNA (gDNA) which, for the context of this study, was obtained by conventional DNA extraction from cultures of Listeria monocytogenes, Escherichia coli O157:H7 and S. enterica subsp. Enterica serovar Typhimurium. As a first step, the gDNA underwent enzymatic fragmentation, end preparation, and dA-tailing by mixing the sample with the fragmentation master mix and cycling temperature to 37 °C for 8 min and 65 °C for 30 min. After cooldown, ligation master mix was added, mixed, and incubated at 20 °C for 15 min to complete adaptor ligation. After a first round of DNA cleanup using SPRI beads at 0.8× reagent to sample volume ratio and ethanol-based wash buffer, the library was resuspended in a PCR master mix. The library was then amplified in the presence of the SPRI beads, followed by a second complete DNA cleanup and elution of the final library in a standard buffer. Fragment size analysis and DNA quantification of the prepared libraries was performed prior to sequencing.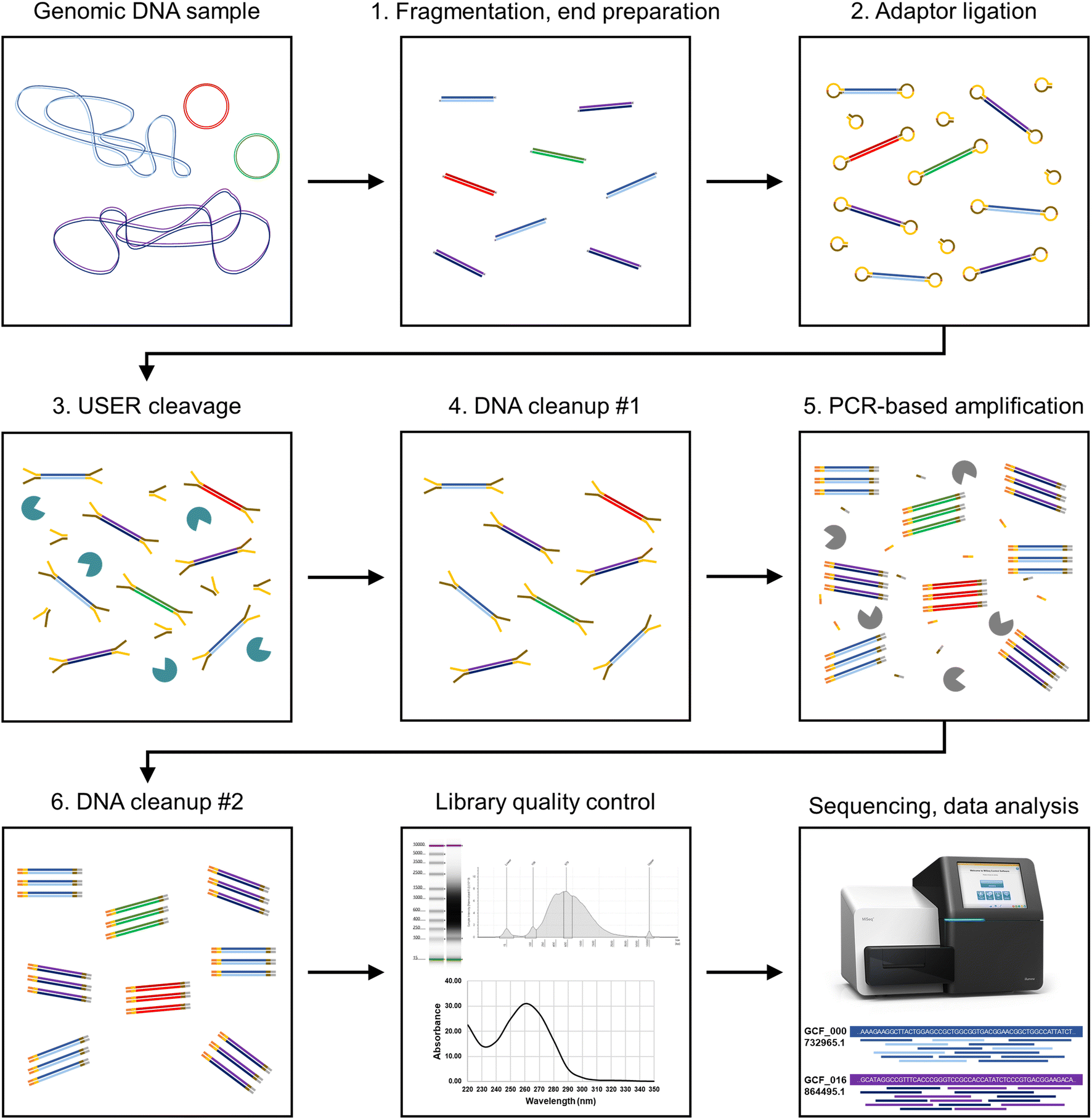 | ||
| Fig. 1 Overview of the main steps of the library preparation workflow adapted for microfluidic on-chip automation. In the workflow, genomic DNA underwent fragmentation and end preparation including dA-tailing (1), followed immediately by adaptor ligation (2). Adaptors were cleaved by the USER enzyme (3) and adaptor-ligated fragments were purified using SPRI bead cleanup (4). Following PCR amplification of isolated fragments (5), a second round of DNA cleanup was performed (6), resulting in a final DNA library. DNA library quality was assessed, after which samples were sequenced on an Illumina NGS platform. | ||
2.2 Centrifugal microfluidic platform
Fig. 2a shows a picture of the PowerBlade centrifugal microfluidic platform that was used for assay automation. The principles behind the operation of this platform have been published previously in detail.25,31 In summary, the platform is designed to provide precise control over the air pressure applied to two microfluidic devices located on opposite sides of the rotating stage using a series of pneumatic lines connected to manifolds (Fig. 2b). Each manifold contains eight (8) holes surrounded by O-rings that align with openings on the microfluidic device called “pressure ports” when the device is secured on the rotating platform (see inset of Fig. 2b). Each pressure line is connected to a three-way electromechanical valve located on the rotating platform. By switching these valves, the platform can control each pressure line to either act as a normal vent or to be connected to an air pump located in the platform. The air pressure from the pump is transferred to the rotating platform using a rotary feedthrough. | ||
| Fig. 2 (a) Picture of the pneumatic-centrifugal microfluidic “PowerBlade” platform used for the on-chip library preparation assays. (b) Close-up view of the rotating stage showing the pneumatic and thermal interfaces between the microfluidic device and the platform. The inset shows the design of the manifold that creates the pneumatic connections with the microfluidic device. (c) Schematics and (d) picture showing the main features of the 5 × 10 cm2 microfluidic device developed for library preparation and initial position of the reagents within the reservoirs. (e) 3D schematic exploded view presenting the assembly of the microfluidic device. | ||
Air pressure applied to one or more pneumatic lines during centrifugation is used to trigger various fluidic actions inside the microfluidic devices, including: transfer of fluid from one reservoir to another, pumping of liquid toward the center of rotation, generation of predefined liquid displacements by balancing centrifugal and pneumatic forces, mixing by generating air bubbles at the bottom of a reservoir, etc.25,31 The platform also includes four 2 × 2 mm2 zones (two per device) where the temperature can be actively controlled during centrifugation using thermoelectric elements. All the steps required for assay execution are controlled by a pre-programmed execution sequence running on custom-made LabVIEW software providing a graphical user interface. While not required for automated execution, a video of the assay can also be recorded through a camera and a flash synchronized with the rotation of the platform.
2.3 Design and fabrication of the microfluidic devices
The microfluidic devices developed for the automation and integration of the library preparation procedure are shown in Fig. 2c and d. Their design remains very simple compared to the complexity of the target library preparation protocol. It consists of: a main reaction chamber connected to six reagent chambers (R1 to R6) and a waste reservoir, all of which are connected to one of the eight pressure ports of the device. As the pressure ports are located close to the center of rotation, the centrifugal force displaces all liquids away from the pneumatic connections, preventing contamination of the platform by the sample or liquid reagents. The main reaction chamber, which is designed to host all the reactions required for library preparation, is aligned with one of the temperature-controlled zones of the platform (identified as thermal interface in Fig. 2c) to enable thermal incubation and cycling. Before the assay is started, reagent chambers R1 to R6 are each manually loaded with the respective reagent through the pressure ports, using a standard pipette. Fig. 2d illustrates the initial configuration of the reagents within the device. The loading of the initial genomic DNA sample and retrieval of the prepared library at the end of the automated sequence is performed using a pipette through a dedicated sample inlet/outlet located in the main reaction chamber. This configuration greatly limits risk of carry-over from one experiment to another due to potential contamination of the pneumatic manifold during pipetting.Since the microfluidic devices are single-use, a “design for manufacturing” process was followed to ensure compatibility with high volume production methods such as injection molding, and to minimize the complexity of the assembly procedure. As an example, the device thickness is kept as uniform as possible (about 2.5 mm) to minimize the risk of creating defects such as sink marks during the injection molding process. Channels, reservoirs, and access holes are also relatively large and have a low aspect ratio (the smallest feature is 500 μm), which facilitates high quality mold manufacturing with industry standard techniques. All the main features of the devices were fabricated by a one-step injection molding process using Zeonor 1060R thermoplastics. As shown in Fig. 2e, assembly was performed by inserting absorbent paper in the waste reservoir and laminating a 127 μm thick polycarbonate film on the bottom of the injected part using a silicone-based adhesive film. The role of the absorbent paper is to prevent the liquid located in the waste reservoir from flowing back into the main reaction chamber during retrieval of the prepared library at the end of the automated assay. Finally, once the cartridge was placed onto the platform and the DNA sample was loaded, another sheet of polycarbonate film was laminated to the top side of the device on a raised wall surrounding the region where the main reaction chamber is located. As described in more detail below, the role of this film is to thermally isolate the main reaction chamber and to close the sample loading port.
2.4 Control of liquid reagents
During the automated assay, the rotational speed of the platform is kept between 200 and 1000 rpm. To transfer a reagent from one of the six reagent reservoirs to the main chamber, positive air pressure is applied to one of the pressure ports of the platform. The air pressure creates a force that pushes the liquid through the outlet channel toward the main reaction chamber. The centrifugal force then further displaces the liquid toward the bottom of the main reaction chamber (i.e., away from the center of rotation). It is noteworthy that the two large reaction chambers R5 and R6 are placed further away from the center of rotation compared to the main reaction chamber. Therefore, depending on the rotation speed and liquid density, only a minimal air pressure is required to generate and maintain liquid transfer.25 These design characteristics can be used to precisely control the volume of reagent transferred to the main reaction chamber (e.g., by controlling the duration and intensity of an air pressure pulse) and allows for repeated transfer of multiple aliquots from the same reservoir during an assay. Liquid transfer from the main chamber to the waste chamber is performed by applying a negative pressure (i.e., lower than atmospheric pressure) to pressure port #7. Alternatively, if a positive pressure is applied to the waste chamber, air bubbles can be generated at the bottom of the main reaction chamber. This process, called “bubble mixing”, is one of the key advantages of the pneumatic centrifugal microfluidic PowerBlade technology. Due to the presence of strong centrifugal force associated with the rotation of the platform, the air bubbles generated by the pneumatic system are rapidly displaced toward the center of rotation before they become larger than the size of the reservoir. The fluid convection currents associated with the rise of the air bubbles provides effective mixing of various reagents and, depending on the intensity of the pressure pulse, can be used to resuspend SPRI beads sedimented at the bottom of the main reaction chamber. More details on the bubble mixing process are available elsewhere.252.5 On-chip manipulation of SPRI beads
Virtually all library preparation workflows contain at least one DNA cleanup step, which usually relies on paramagnetic SPRI beads and a chaotropic agent to purify DNA fragments and perform size selection. When performed manually, this step typically makes use of a magnet to immobilize beads and exchange buffers; when the magnet is removed, paramagnetic beads can be suspended in another solution. The developed centrifugal microfluidic platform programs the capture and release of SPRI beads using only centrifugal and pneumatic forces. Bead immobilization is achieved through sedimentation, which was found to require between 5 to 15 min at 1000 rpm (corresponding to an acceleration of about 120 g) depending on the viscosity of the buffer. The bottom edge of the main reaction chamber was designed as a circle arc aligned with the center of rotation of the platform to ensure uniform packing of the beads on the bottom wall following sedimentation. Naturally, bead sedimentation was also found to take place in reservoir R5 containing the SPRI bead reagent (see Fig. 2c and d). To ensure uniform dispersion of the beads in this reservoir, bubble mixing steps were performed in this reservoir every 2 to 3 minutes for the duration of the assay and just before transfer of SPRI bead reagent to the main reaction chamber.After bead sedimentation, supernatant removal is performed by applying negative pressure on the waste chamber (using pressure port #7) while the platform is rotating at 400 rpm to withdraw liquid from the main reaction chamber through the side exit channel. This channel is designed to leave a small amount of liquid in the main reaction chamber during supernatant removal to limit bead loss. To further avoid disturbing the sedimented beads, supernatant transfer is performed by slowly increasing the negative pressure from about −0.6 to −1.7 psig while recording the pump speed in real time (rotation speed of 400 rpm). The automated script is programmed to stop the pump when a sudden increase in pump speed is detected, indicating that air is being pulled toward the waste chamber instead of liquid. This slow liquid removal process is similar to that performed manually in tubes, where gentle pipetting is generally recommended to avoid disturbing the beads. These settings resulted in effective and repeatable liquid withdrawal from the beads with minimal bead loss. A video providing an example of on-chip DNA cleanup is available in Video S1.†
Another advantage of the developed system is that the beads can be dried at the end of the DNA cleanup steps to remove traces of ethanol left from the wash buffer. Ethanol contamination can indeed inhibit PCR amplification36 and can therefore be detrimental to the library preparation process. While air drying beads is routinely performed during manual DNA cleanup, it can be very challenging to integrate in many microfluidic platforms considering the closed architecture of the devices. In our system, we found that beads can be dried effectively by generating air flow in the microfluidic device using the pneumatic manifold. The drying process was performed for 3 minutes by creating a gentle air flow in the main reaction chamber through activation of pressure port #7 at a pressure of 0.5 psig at a rotation speed of 1000 rpm. More details about the developed on-chip air drying process are available in previous publications.31,35
During DNA cleanup, it is also critical to precisely control the volume of SPRI bead reagent that is transferred to the main reaction chamber as it affects the DNA fragment size selection process.37 In the microfluidic devices, the volume of reagent transferred is a function of device design, liquid properties, rotational speed, and air pressure pulse duration and intensity. Fig. S1a† shows a series of five SPRI bead reagent transfer tests performed to evaluate the reproducibility of the process (see also Video S2†). The system was found to perform precise and reproducible volume transfer which is required to ensure adequate control over the size selection process during DNA cleanup. Alternatively, by adjusting the duration of the applied air pressure pulse at a given rotation speed, the platform can be used to program the volume of SPRI bead reagent transferred, as demonstrated in Fig. S1b and c† (see also Video S3†). As discussed later, this provides the capacity to control the DNA fragment size selection process depending on the needs of the target assay or sequencing platform.
2.6 Temperature control and calibration
Successful integration of a library preparation workflow in microfluidics requires effective on-chip temperature control for nucleic acid amplification and enzymatic reactions. Various elements of device design have therefore been optimized to provide accurate and repeatable temperature control. To maximize heat transfer from the platform to the sample, the bottom cover of the device is thin (127 μm polycarbonate film with a 41 μm silicone adhesive layer) and a layer of thermal tape was applied between the bottom cover and the platform's heating element (Fig. 2d). The thermal tape ensures firm and consistent contact with the heating element despite thermal expansion,33 which can lead to deformation of the thin device cover. Additionally, since cold air flowing rapidly above the spinning device can reduce the temperature of the contained liquids, a raised edge was integrated above the main reaction chamber as part of the chip design. During operation, this raised edge is covered with a thin transparent film, creating an air pocket (Fig. 2e). To further isolate the device from the cold air, a holder with a transparent cover is placed above the device during operation (Fig. S2a†).To ensure accurate control over the temperature of the liquid reagents, temperature calibration was performed by inserting a thermocouple directly into the main reaction chamber filled with an aqueous buffer (Fig. S2a†). Measurements were made at the same rotational speed as the value defined in the execution sequence for that step (i.e., from 400 to 800 rpm). Temperature readings from the thermocouple were obtained through a pre-calibrated custom-made wireless thermocouple amplifier system attached to the rotating stage of the platform. The temperature setpoints in the execution sequence running on the platform were then adjusted until the target temperature was reached within the main reaction chamber. An example of the temperature recorded inside the main reaction chamber during the PCR amplification step is provided in Fig. S2b,† demonstrating the capacity to control the temperature to precisely reach the target setpoints of 55 °C, 65 °C and 95 °C at every cycle at a rotation speed of 400 rpm. To evaluate the performance of on-chip PCR amplification, we amplified a diluted Illumina-compatible library for 4 cycles using both a conventional thermal cycler and the developed centrifugal microfluidic platform. The resulting on-chip and manual amplicons displayed almost identical fragment size distributions, as shown in Fig. S3.† These results indicate that on-chip PCR does not introduce a noticeable bias to preferably amplify or eliminate specific DNA fragments.
3. Results and discussion
3.1 On-chip DNA cleanup by SPRI
Considering the key importance of DNA cleanup in library preparation, a set of experiments was performed to evaluate the capabilities of the on-chip DNA cleanup step compared to a standard manual workflow. DNA cleanup by SPRI beads determines library yield and fragment size profile. To assess on-chip SPRI DNA cleanup, the method was evaluated using a DNA ladder with SPRI beads. The input ladder contained 19 DNA fragments with sizes ranging from ∼100 bp to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 bp, all of which were recovered by both the on-chip and manual cleanup processes at comparable molecular concentrations (Fig. 3a). Similar results were obtained regardless of input amount (results not shown). These results demonstrate the capability of the microfluidic system to effectively carry out DNA cleanup without the need of a magnet.
000 bp, all of which were recovered by both the on-chip and manual cleanup processes at comparable molecular concentrations (Fig. 3a). Similar results were obtained regardless of input amount (results not shown). These results demonstrate the capability of the microfluidic system to effectively carry out DNA cleanup without the need of a magnet.
 | ||
| Fig. 3 (a) Molar concentrations of DNA fragments from a 1 kb+ DNA ladder (New England Biolabs N0550L) recovered by 1.0× volume ratio of SPRI bead reagent to sample using the manual procedure or the on-chip procedure (eluted in an equal volume). Samples were analyzed on an Agilent Bioanalyzer using the High Sensitivity DNA kit (Agilent Technologies). (b) Molar concentrations of DNA fragments from a 1 kb+ DNA ladder (Invitrogen 10787018) recovered by various volume ratios of SPRI bead reagent to sample using the on-chip procedure (eluted in an equal volume). Samples were analyzed with an Agilent TapeStation using the High Sensitivity D5000 Assay (Agilent Technologies). | ||
Furthermore, on-chip transfer of the SPRI bead reagent was highly reproducible. When 100 μL of SPRI beads was set to be transferred from a reservoir containing a stock of 200 μL, the difference between the targeted volume and the transferred volume was at most 3 μL (Fig. S1A†). For a successful library preparation, it is often necessary to adjust the volume ratio between the SPRI beads and the sample to selectively exclude short DNA fragments, which tend to be primer dimers, adaptor dimers, or fragments with inserts shorter than the length of reads. We therefore programmed the PowerBlade to transfer various volumes of SPRI beads from reservoir R5 filled with 200 μL to the main reaction chamber (Fig. S1B†). After the initial time duration to build up pressure prior to transfer, the transfer duration and the volume of the reagent approximately followed a linear relationship (Fig. S1C†). Using this knowledge, we varied the SPRI bead to sample volume ratios to 0.4×, 0.8×, and 1×, and observed progressive inclusion of the smaller fragments with increasing ratios (Fig. 3b). Aside from demonstrating programmable on-chip size selection, these results showcase a feature of the PowerBlade centrifugal microfluidic system to dispense any pre-defined volume of liquid from a stock, resembling the operation of a robotic system.
3.2 Automated library preparation
Fig. 4 provides an overview of the main steps executed on the centrifugal microfluidic platform during the automated library preparation process. Table S1† provides the detailed conditions associated with each step of the automated protocol.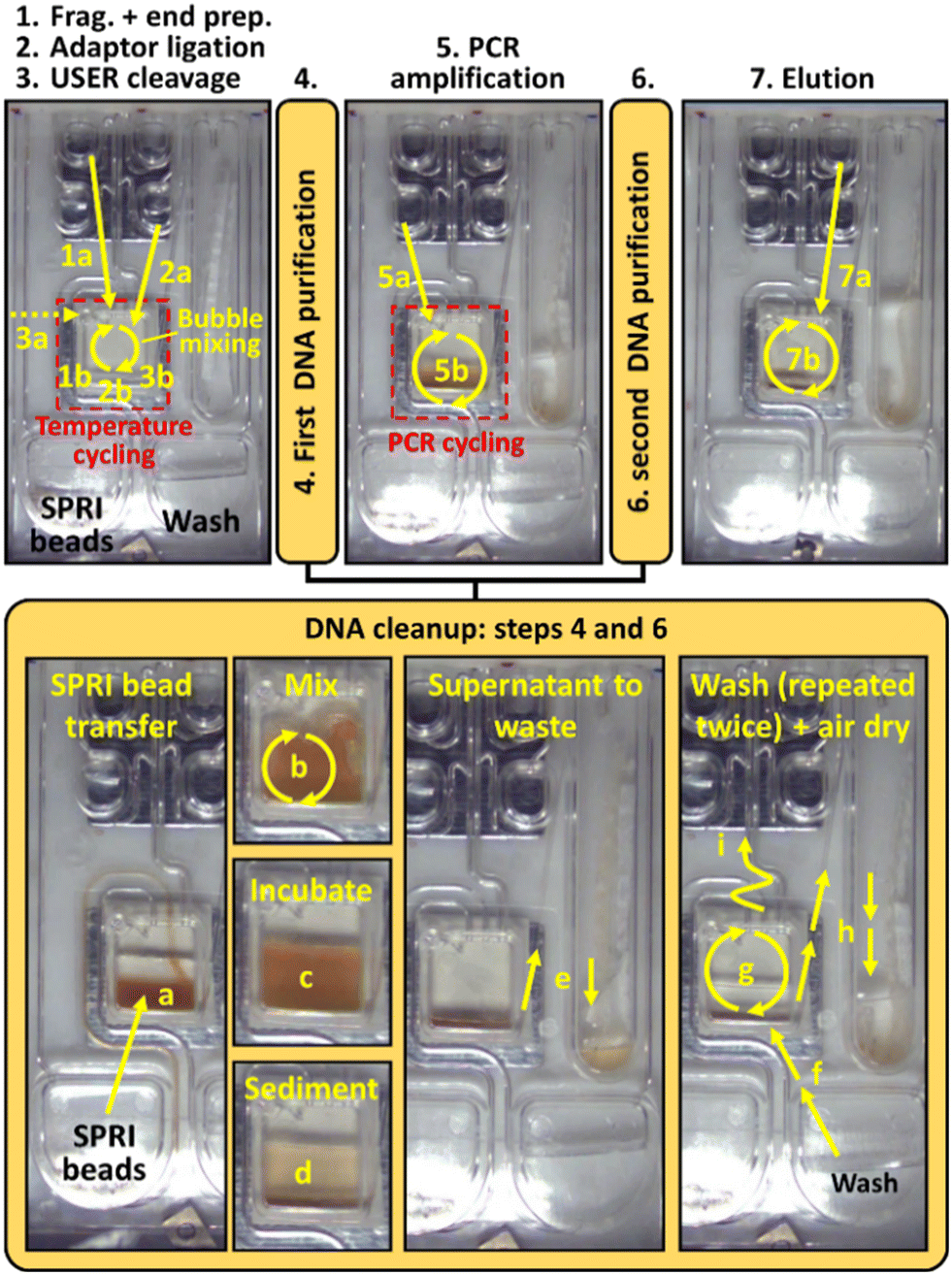 | ||
| Fig. 4 Sequence of images showing the main steps of the automated on-chip library preparation assay. The straight yellow arrows indicate the liquid transfer steps from one chamber to another and the curved yellow arrows indicate bubble mixing actions. Steps requiring local temperature control are highlighted with a red dash line. Numbers and letters respectively indicate processing steps and substeps (i.e., 1a, 1b, 2a, 2b, etc.). During the automated assay, the liquid manipulation steps are preformed under centrifugation (i.e., the rotational speed of the platform is kept between 200 and 1000 rpm) and are triggered by the activation of one specific pressure port of the platform at a pressure in the range of −1.7 to 3.0 psig. More details are available in Table S1 of ESI.† | ||
After manually filling the device with the liquid reagents according to the layout shown in Fig. 2d, the device is connected to the manifold of the platform and the pre-recorded script is started. The assay is then executed automatically by the PowerBlade platform, ensuring a high level of reproducibility of the fluidic manipulation and incubation steps. Initially, the platform is set to a rotational speed of 800 rpm, providing sufficient centrifugal force to displace all the liquids away from the center of rotation. The first step of the protocol is to transfer the fragmentation master mix to the main reaction chamber where the genomic DNA sample is located (step 1a). This is performed by activating pressure port #3 at a pressure of 2 psig for 0.5 s. The two liquids are then thoroughly mixed in the main reaction chamber by bubble mixing (step 1b) following the activation of pressure port #7 at a pressure of 2 psig for 5 s (rotational speed of 400 rpm). The thermoelectric heater of the platform is then activated to cycle the temperature of the solution to 37 °C for 10 min, 65 °C for 30 min and 25 °C for 5 min (step 1c).
The second step of the protocol, adaptor ligation, is performed at a rotation speed of 800 rpm through a similar set of operations, except that the ligation master mix is transferred by activating pressure port #6 and undergoes a different incubation protocol (refer to Table S1† for details). It is noteworthy that the ligation master mix loaded to reservoir R4 at the beginning of the assay also contains the adaptors. The adaptors and ligation enzymes are therefore in the same solution throughout fragmentation and end preparation, which can promote the formation of adaptor dimers. To minimise adaptor dimerization, the developed assay makes use of hairpin adaptors. Ligation of these hairpin adaptors to inserts (or to each other) caps the double-stranded DNA with hairpin structures on both ends. Enzymatic excision of the uracil nucleotide via a USER enzyme is then used to create a fork structure at each end of the insert, enabling PCR and strand-specific addition of DNA handles. Meanwhile, removing the uracil nucleotides from adaptor dimers destabilizes them due to their relatively short double-stranded regions. In the automated script, the rotation of the platform is stopped momentarily to allow manual loading of the USER enzyme mixture through the loading port of the main reaction chamber (step 3a). This manual step is performed while the device is still attached to the platform, and since it requires less than one minute to complete, adds negligible hands-on time compared to the overall duration of the 4.5 hour assay. Moreover, if a workflow without this manual intervention is deemed necessary in some settings, it could be eliminated by changing the chip loading protocol. For example, loading both the sample and fragmentation buffer directly into the main reaction chamber at the beginning of the assay would free up reservoir R1 for the USER enzyme. After USER enzyme loading, rotation of the platform is resumed at 800 rpm and bubble mixing is performed (step 3b), followed by incubation at 37 °C for 15 min (step 3c).
A first round of DNA cleanup is then performed as follows. First, an aliquot of SPRI bead reagent is transferred from reservoir R5 to the main reaction chamber (step 4a). The conditions of the transfer (port #1 at 3.0 psig for 5.37 s at 400 rpm rotation speed) were optimized to reach a volume ratio of 0.8× between the SPRI bead reagent and the volume of liquid in the main reaction chamber. After bubble mixing (step 4b), beads are incubated at a low rotational speed of 200 rpm for 10 min (step 4c) and sedimented at a high rotational speed of 1000 rpm for 15 min (step 4d). The supernatant is then slowly pulled to the waste chamber (step 4e) using the procedure described previously (see section 2.5). While a small fraction of beads may be transferred to the waste chamber during that step, this was found to not significantly affect the efficiency of the bead cleanup (as discussed in section 3.1). A first aliquot of about 200 μL of wash buffer is then transferred to the main reaction chamber (step 4f1, port #1 at 1.5 psig for 2.80 s at 400 rpm), gently mixed with the sedimented beads (step 4g1, port #7 at 0.6 psig for 4.0 s at 400 rpm), incubated for 12 min at 1000 rpm to sediment the beads (step 4g1), and slowly transferred to waste (step 4h1) using the procedure described previously (see section 2.5). A second wash step is then performed using a similar procedure (see steps 4f2 to 4h2 of Table S1†), except that the duration of the air pressure pulse to transfer a 200 μL aliquot of wash buffer is increased from 2.80 s to 3.20 s to compensate for the lower volume of wash buffer in reservoir R6. Finally, the beads are air dried by activating pressure port #7 at 0.5 psig for 3 min at a rotation speed of 1000 rpm (step 4i).
After the first DNA cleanup, the PCR master mix is transferred from reservoir R3 to the main reaction chamber (step 5a, port #2 at 1.5 psig for 5.0 s at 400 rpm) and mixed with the beads through bubble mixing (step 5b, port #7 activated twice at 2.5 psig for 5.0 s at 400 rpm), which enables elution of the DNA fragments from the beads. Thermal cycling is then performed while keeping the rotation speed at 400 rpm as detailed in Table S1† (step 5c): 95 °C initial denaturation followed by 6 cycles of 95 °C, 55 °C, and 65 °C. It is noteworthy that SPRI beads are still present in the main reaction chamber during PCR amplification. While SPRI beads can affect the PCR amplification process, we found that, in comparison to other commercially available PCR master mixes, HiFi HotStart ReadyMix is able to provide high quality PCR amplification despite the presence of the SPRI beads (data not shown).
After PCR amplification, a second round of DNA cleanup is performed (step 6) at 0.8× SPRI bead volume ratio using a similar procedure to that of the first round (see description of step 4 above and Table S1†). As detailed in Table S1,† durations of the air pressure pulses between steps 4 and 6 are slightly different to ensure transfer of the appropriate volume despite the lower liquid level in the reservoirs at this stage of the assay. Finally, the library is eluted by transferring the elution buffer to the main reaction chamber (step 7a, port #5 activated twice at 1.5 psig for 1.0 s at 800 rpm) and performing bubble mixing (step 7b, port #7 activated twice at 2.5 psig for 5.0 s at 800 rpm). At the end of the automated protocol, the rotation of the platform stops. The library and beads are then manually pipetted out of the reaction chamber, followed by removal of the magnetic beads using a magnet.
3.3 Library quality metrics and sequencing results
Once the library preparation procedure described in section 2.1 was adapted for automation on the PowerBlade, three replicate sets of libraries were prepared, with each set comprising an on-chip library and a manually prepared library. The input of the library preparation was a defined mixture of genomic DNA from three common, well characterized foodborne bacterial pathogens: L. monocytogenes, E. coli O157:H7, and S. enterica subsp. enterica serovar Typhimurium. The resulting libraries both displayed comparable DNA fragment sizes between 400 bp to 800 bp (Fig. S4†). For both types of libraries, some adaptor dimers around 100 bp in size were detectable, but their low relative concentration did not interfere with library quantification, pooling, or sequencing.For each of three separate replicate sets, on-chip and manual libraries were pooled at equal molar ratios and sequenced using paired-end Illumina technology. Sequencing data for the libraries prepared on-chip were evaluated by comparing standard performance metrics with sequence data generated via the manual method. The on-chip and manual libraries shared comparable quality scores when evaluated at the read level (Fig. 5a), and comparable coverage along the full lengths of the three input genomes (Fig. 5c). Nonetheless, for one replicate, the manual library data showed lower coverage than the on-chip library across the genome specifically for the E. coli and S. enterica assemblies (respectively: E. coli average fold coverage 105 vs. 123; S. enterica average fold coverage 54 vs. 77; data not shown). This might be attributed to imperfect pooling during preparation of input DNA samples, exacerbated in the lower-abundance organisms.
 | ||
| Fig. 5 (a) Distribution of reads by Q-score. The proportion (%) of paired-end reads from on-chip (blue) and manual (red) library datasets were binned according to Q-score. Error bars depict standard deviation for three technical replicates. (b) Organism abundance estimates for on-chip and manual library data. Reads were classified by organism using MetaPhlAn for three replicate datasets. Theoretical relative abundance values (i.e., L. monocytogenes (73.5%): E. coli (19.2%): S. enterica (7.2%) were compared to average abundances for on-chip and manual library data (error bars = standard deviation). (c) Representative comparison of read coverage plots against each of the reference genomes. Standardized reads were aligned to the reference genome, and the coverage depth was plotted for each dataset. Representative data from replicate 2 is shown. | ||
Furthermore, MetaPhlAn metagenomics estimates for the relative abundances of input genomes from L. monocytogenes, E. coli, and S. enterica were highly similar for both the on-chip (74.4 ± 4.7%, 16.0 ± 2.9% and 9.6 ± 2.0% respectively) and manual libraries (75.5 ± 1.2%, 15.5 ± 1.3%, and 9.07 ± 0.57% respectively), and were comparable to the theoretical ratios based on the starting gDNA molar ratios (73.5%, 19.2%, and 7.23% respectively) (Fig. 5b). Statistical analysis of the relative abundances via a two-way ANOVA revealed no significant differences between the two methodologies (p = 0.997). These results indicate that the qualities of the on-chip libraries were comparable to those of the manual counterparts.
GC content biases, which are often present in WGS, can have pervasive effects on downstream genomic data analysis, and can result in erroneous conclusions such as failure to identify functional motifs during bacterial serotyping, distortion of the composition of a metagenome, or misleading HLA genotyping.22,38,39 We therefore sought to test whether the on-chip prepared libraries showed any GC bias difference from the manually prepared libraries. The reference genomes were partitioned into regions of 1000 bases, and the regions were binned based on their % GC content. This was plotted against the fold coverage for each respective bin. We observed only weak correlations (correlation coefficient |r| ≤ 0.4) between bin coverage and GC content for all three input genomes in both the on-chip libraries and the manual libraries (Fig. 6). These results indicate that neither the manual nor on-chip processes introduce strong GC-content bias. In contrast, tagmentation-based library preparation workflows, which are often used in microfluidics due to their simplicity, can lead to prominent GC content biases.17–20 Collectively, reads from the on-chip libraries and the manual libraries contain virtually identical information to support mapping, de novo assembly, and other read count-based analyses.
 | ||
| Fig. 6 GC content bias scatterplot analysis. Coverages of the genomes divided into bins of 1000-base regions with various GC contents, for (a) L. monocytogenes, (b) E. coli, and (c) S. enterica. For each panel, the r scores show the Spearman correlation coefficient between GC content and fold coverage based on a sliding window of 1000 and a minimum coverage of 10. Representative data from replicate 2 is shown. | ||
4. Conclusion
We demonstrated the automation of a complete ligation-based library preparation workflow in simple thermoplastic microfluidic cartridges that contain no active elements and are amendable to low-cost mass manufacturing. The PowerBlade, a field-deployable centrifugal microfluidic platform, delivers programmed centrifugal force, pneumatic force, and temperature control to stepwise carry out the reactions within the device using a pre-programmed execution sequence. The developed microfluidic devices, while simple in design, have effectively and reliably executed complex liquid manipulation steps, including several rounds of DNA purification and PCR amplification. Compared to the libraries prepared with a manual procedure, on-chip libraries displayed comparable genome coverages, supported de novo assembly equally well, and revealed almost identical relative abundances of the input species. For both sets of libraries, there was minimal GC-content bias. Notably, while preparing a library manually demands a user to stand by for a number of hours, the automated procedure described herein requires considerably less hands-on time.While this paper reports on the preparation of Illumina-compatible ligation-based libraries, the developed liquid transfer, mixing, bead handling, and thermal cycling steps are generic and customizable. Similarly, as the buffers required by the assay are loaded on-chip in separate reservoirs before the assay, substitution with other reagents is very straightforward. Hence, the developed microfluidic cartridges and platform could also be used to prepare libraries that are compatible with other applications and NGS platforms such as those of Oxford Nanopore and Pacific Biosciences. Furthermore, if needed by specific workflows the chip design could also be modified (e.g., with additional reservoirs, etc.) with minimal impact on the main liquid manipulation strategies demonstrated herein.
One constraint of the microfluidic platform is the limited number of independent pressure lines available on the platform, which can ultimately limit the number of independent fluidic steps that can be reliably carried out on-chip. By increasing the number of pressure ports, additional important functionalities could be integrated within an automated on-chip workflow. As an example, we believe that the developed technology can support the integration of a DNA extraction step31 before library preparation, which would allow end users to automate the entire workflow from a raw sample (e.g. a bacterial culture) to the final prepared library.
The innovative approach to automated library preparation presented in this paper is amenable to deployment in areas where resources and technical expertise may be more limited, including remote settings such as distant regions and deep space environments. By eliminating the need for user expertise, hands-on time, and dedicated laboratory space, and by enabling deployable, automated, user-friendly NGS technologies, we believe this platform will facilitate rapid, on-site pathogen detection and characterization, with ultimate benefits to public health and food safety practices.
Materials and methods
Design, fabrication, and assembly of the microfluidic devices
The microfluidic devices consist of a 50 mm wide by 100 mm long body fabricated in Zeonor 1060R (Zeon Chemicals, Louisville, KY) through an injection molding process (Proto Labs, Maple Plains, MN) using a custom design. All the channels, reservoirs and access holes are patterned in a single step during the injection molding process. Cartridges were sealed by laminating a 41 μm thick silicone adhesive film (ARclear 93495; Adhesives Research, Glen Rock, PA) and a 127 μm thick polycarbonate film (McMaster-Carr, Aurora, OH) on both the bottom side of the device and above a raised edge located above the main reaction chamber. A piece of about 40 mg of absorbent paper (cleanroom wipes, Cleanroom World, Centennial, CO) was inserted into the waste chamber before closing the device.On-chip DNA cleanup by SPRI
To compare the efficacy of on-chip DNA cleanup to that of a standard manual DNA cleanup, the microfluidics platform was programmed to transfer 80 μL of AMPure XP SPRI reagent (Beckman Coulter, Montreal, Québec) to an 80 μL DNA suspension containing 125 ng of a 1 kb+ DNA ladder (New England Biolabs, Whitby, Ontario). The on-chip SPRI procedure is described in section 2.5, and the manual procedure was performed as per SPRI manufacturer instructions. The recovered DNA fragments were eluted in 50 μL of 10 mM Tris-HCl pH 8. One microlitre of the eluted samples was analyzed with an Agilent Bioanalyzer using the High Sensitivity DNA kit (Agilent Technologies, Germany). DNA fragment sizes were approximated to the values listed by the DNA ladder manufacturer.To demonstrate the effects of varying the volume ratios of SPRI bead reagent to DNA sample on-chip, the microfluidics platform was programmed to transfer either 40, 80, or 100 μL of SPRI beads into a 100 μL DNA suspension containing a different 1 kb+ DNA ladder (Invitrogen, Burlington, Ontario). This corresponds to bead-to-sample volume ratios of 0.4×, 0.8×, and 1.0× respectively. The recovered DNA fragments were eluted in 50 μL of 10 mM Tris-HCl pH 8, and were analyzed with an Agilent TapeStation using the High Sensitivity D5000 Assay (Agilent Technologies). DNA fragment sizes were approximated to the values listed by the DNA ladder manufacturer.
gDNA extraction, quantification, and size analysis
Strains of three common bacterial foodborne pathogens with closed genomes were chosen for sequencing: Listeria monocytogenes serovar 4b strain ATCC 13932, Escherichia coli O157:H7 strain EDL933, and Salmonella enterica subsp. Enterica serovar Typhimurium ATCC 14028 (NCBI genome accession numbers GCA_003031895.1, GCA_000732965.1, and GCA_016864495.1 respectively). From −80 °C stocks, strains were plated onto Brain–Heart Infusion (BHI) agar media (BD, Mississauga, Ontario) and incubated overnight at 37 °C. Single colonies were inoculated into BHI broth and incubated overnight at 37 °C; 1 mL aliquots of overnight culture were used to extract genomic DNA using the PureLink™ Genomic DNA Mini Kit (Invitrogen, Burlington, Ontario). For consistency, manufacturer instructions for Gram-positive bacterial cells were followed for all strains regardless of Gram status, with lysis performed in 180 μL of lysozyme digestion buffer (25 mM Tris-HCl pH 8.0; 2.5 mM EDTA; 1% Triton X-100; 20 mg mL−1 lysozyme) supplemented with 50 U mutanolysin for increased lysis efficiency. Purified DNA was eluted in 10 mM Tris-HCl, 0.1 mM EDTA, pH 8.0. DNA quantification was performed using the Qubit 1× dsDNA High Sensitivity assay (Invitrogen, Burlington, Ontario).Library preparation
For all libraries, gDNA extracts from L. monocytogenes, E. coli, and S. enterica ser. Typhimurium were combined at 73.5:19.2:7.2 molar ratios based on genome size (mass ratios of 6:3:1), for a total of 500 ng of input bacterial gDNA in 10 mM Tris-HCl, 1 mM EDTA, pH 8.0. Manual libraries were prepared using fragmentation, end preparation, dA-tailing, adaptor ligation, and USER-based adaptor cleavage reagents as per manufacturer instructions for the NEBNext® Ultra™ II FS DNA Library Prep Kit for Illumina (New England Biolabs, Whitby Ontario), in double-volume reactions. Following USER incubation, SPRI cleanup of the adaptor-ligated DNA was performed using AMPureXP SPRI beads (Beckman Coulter) at a 0.8× reagent to sample volume ratio. DNA fragments were eluted in 100 μL 1× KAPA HiFi HotStart ReadyMix (Roche, Mississauga, Ontario) containing NEBNext® Multiplex Oligos for Illumina® index primers (New England Biolabs), followed by PCR amplification (initial denaturation: 60 seconds at 95 °C; 4 cycles of: 20 seconds at 95 °C, 30 seconds at 50 °C, 2 minutes at 65 °C; final elongation: 5 minutes at 65 °C; hold at 4 °C) on an Eppendorf Nexus GSX1 Thermocycler (Eppendorf, Hamburg, Germany) without removal of SPRI beads. Following PCR, amplified fragments were put through a second round of SPRI cleanup as described above. The final library was eluted in 50 μL of 1 mM Tris, 0.01 mM EDTA, pH 8.0. DNA quantification was performed using the Qubit 1× dsDNA High Sensitivity assay, and DNA fragment analysis was performed using the TapeStation High Sensitivity D5000 assay (Agilent Technologies).
Automated, on-chip libraries were generated using a similar protocol as well as the same reagents and volumes as the manual method, as described in detail in section 3.1 and Table S1.† Minor differences between the on-chip and manual protocols include the number of PCR cycles (6 vs. 4) and fragmentation time (8 min vs. 5 min).
Sequencing and read processing
For each replicate, manual and on-chip libraries were pooled in equimolar ratios based on quantification and peak size results from DNA fragment analysis. Paired-end (2 × 250, v3 chemistry) sequencing was performed using a MiSeq instrument loaded with 15–17 pM pooled library, spiked with 3% phiX according to manufacturer instructions (Illumina Inc). Reads were trimmed and quality filtered using BBMap40 (v38.18; bbduk adaptor trimming, removal of bases with quality scores <12 from read ends, remove reads <20 bases after trimming; (https://sourceforge.net/projects/bbmap); seqtk (v1.3-r106, seed = 100; https://github.com/lh3/seqtk) was used to downsample reads such that both the on-chip and manual library datasets comprised the same number of reads (replicate 1: 18089048; replicate 2: 10747188; replicate 3: 15863936).
Sequence analysis: genome assembly, mapped coverage, and metagenomics
Processed reads were de novo assembled using metaSPAdes41 (v3.13.0), and reference mapped using bowtie (v2.1.4).42 Genome coverage was determined using samtools43 (v1.9) to create pileups to matched reference genomes. Custom Python scripts were used to summarize GC content and coverage from the pileup, and data was visualized using custom R scripts (v4.1.2; https://www.r-project.org) with ggplot2 (ref. 44) (v3.3.6) and plotly (v4.10.0; https://github.com/plotly/plotly.R/releases/tag/v4.10.0) (see https://github.com/ajverster/LabOnAChipAnalysis/). For metagenomic analysis, processed reads were analyzed using MetaPhlAn45 (v3.1.0; mpa_v31_CHOCOPhlAn_201901 database; taxa minimum abundance = 0.001). Relative abundance data was analyzed and visualized using RStudio (v1.4.1106) with readr (v2.1.2), dplyr (v1.0.8), matrixStats (v0.63.0), ggplot2 (v3.3.5) and scales (v1.1.1), and were analyzed with a two-way ANOVA followed by Tukey's multiple comparisons test using GraphPad Prism 9 (GraphPad Software Inc., Boston, MA).Author contributions
JG, DB, NA, AVP, DC, NC, and TV conceptualized the integrated library preparation assay. DB and JG designed the initial microfluidic devices which were then further optimized by MM, JF, and MS. MS adapted the design for compatibility with injection molding. JG, DB, MJP, NA and AC designed and optimized the library preparation protocol and its integration in microfluidics. JG, NA, DB, KW, AVP, NC, and TV designed the validation experiments. NA, CMG, MJP, MM, JF, JG, AVP, KT, AC, and KW collected data for assay validation. AV, JG, NA and NP conceptualized the bioinformatics pipeline. AV and JS performed the bioinformatics analysis. JG, DB, NA, AV, JS, and KW curated data, performed data visualization, and wrote the manuscript. All authors reviewed and approved the manuscript prior to submission.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Research Council, Health Canada and the Canadian Space Agency. We thank the financial support from the Genomics Research and Development Initiative and the NRC Pandemic Preparedness Program. We thank our colleagues Aaron Bessoff, Francois Normandin, Émilie Leblanc-Gaudreau, Marc Alexandre Chan, and Simon Geissbuehler for the technical work on the platform design, electronic circuits, and control software. Thanks to Dr. Annika Flint and Dr. Richard Harris for critical comments on the manuscript. The authors also wish to acknowledge the contributions of Health Canada support staff in the Rapid Diagnostics Laboratory and staff of the StemCore Laboratories at the Ottawa Hospital Research Institute whose efforts helped enable this research.References
- B. Jagadeesan, P. Gerner-Smidt, M. W. Allard, S. Leuillet, A. Winkler, Y. Xiao, S. Chaffron, J. Van Der Vossen, S. Tang, M. Katase, P. McClure, B. Kimura, L. Ching Chai, J. Chapman and K. Grant, Food Microbiol., 2019, 79, 96–115 CrossRef CAS PubMed.
- B. Imanian, J. Donaghy, T. Jackson, S. Gummalla, B. Ganesan, R. C. Baker, M. Henderson, E. K. Butler, Y. Hong, B. Ring, C. Thorp, R. Khaksar, M. Samadpour, K. A. Lawless, I. MacLaren-Lee, H. A. Carleton, R. Tian, W. Zhang and J. Wan, npj Sci. Food, 2022, 6, 1–6 CrossRef PubMed.
- S. Shokralla, J. L. Spall, J. F. Gibson and M. Hajibabaei, Mol. Ecol., 2012, 21, 1794–1805 CrossRef CAS PubMed.
- L. Rutter, R. Barker, D. Bezdan, H. Cope, S. V. Costes, L. Degoricija, K. M. Fisch, M. I. Gabitto, S. Gebre, S. Giacomello, S. Gilroy, S. J. Green, C. E. Mason, S. S. Reinsch, N. J. Szewczyk, D. M. Taylor, J. M. Galazka, R. Herranz and M. Muratani, Patterns, 2020, 1, 100148 CrossRef CAS.
- M. Hoffmann, Y. Luo, S. R. Monday, N. Gonzalez-Escalona, A. R. Ottesen, T. Muruvanda, C. Wang, G. Kastanis, C. Keys, D. Janies, I. F. Senturk, U. V. Catalyurek, H. Wang, T. S. Hammack, W. J. Wolfgang, D. Schoonmaker-Bopp, A. Chu, R. Myers, J. Haendiges, P. S. Evans, J. Meng, E. A. Strain, M. W. Allard and E. W. Brown, J. Infect. Dis., 2016, 213, 502–508 CrossRef PubMed.
- H. M. Blankenship, S. E. Dietrich, E. Burgess, J. Wholehan, M. Soehnlen and S. D. Manning, Microorganisms, 2023, 11, 1298 CrossRef CAS PubMed.
- M. W. Gilmour, M. Graham, G. Van Domselaar, S. Tyler, H. Kent, K. M. Trout-Yakel, O. Larios, V. Allen, B. Lee and C. Nadon, BMC Genomics, 2010, 11, 1–15 CrossRef PubMed.
- B. R. Jackson, C. Tarr, E. Strain, K. A. Jackson, A. Conrad, H. Carleton, L. S. Katz, S. Stroika, L. H. Gould, R. K. Mody, B. J. Silk, J. Beal, Y. Chen, R. Timme, M. Doyle, A. Fields, M. Wise, G. Tillman, S. Defibaugh-Chavez, Z. Kucerova, A. Sabol, K. Roache, E. Trees, M. Simmons, J. Wasilenko, K. Kubota, H. Pouseele, W. Klimke, J. Besser, E. Brown, M. Allard and P. Gerner-Smidt, Clin. Infect. Dis., 2016, 63, 380–386 CrossRef PubMed.
- S. Gillesberg Lassen, S. Ethelberg, J. T. Björkman, T. Jensen, G. Sørensen, A. Kvistholm Jensen, L. Müller, E. M. Nielsen and K. Mølbak, Clin. Microbiol. Infect., 2016, 22, 620–624 CrossRef CAS PubMed.
- B. Brown, M. Allard, M. C. Bazaco, J. Blankenship and T. Minor, PLoS One, 2021, 16, e0258262 CrossRef CAS PubMed.
- C. Nadon, I. Van Walle, P. Gerner-Smidt, J. Campos, I. Chinen, J. Concepcion-Acevedo, B. Gilpin, A. M. Smith, K. M. Kam, E. Perez, E. Trees, K. Kubota, J. Takkinen, E. M. Nielsen, H. Carleton and FWD-NEXT Expert Panel, Eurosurveillance, 2017, 22, 30544 CrossRef.
- J. F. Hess, T. A. Kohl, M. Kotrová, K. Rönsch, T. Paprotka, V. Mohr, T. Hutzenlaub, M. Brüggemann, R. Zengerle, S. Niemann and N. Paust, Biotechnol. Adv., 2020, 41, 107537 CrossRef CAS.
- J. F. Hess, M. E. Hess, R. Zengerle, N. Paust, M. Boerries and T. Hutzenlaub, Anal. Chim. Acta, 2021, 1182, 338954 CrossRef CAS.
- H. Kim, M. J. Jebrail, A. Sinha, Z. W. Bent, O. D. Solberg, K. P. Williams, S. A. Langevin, R. F. Renzi, J. L. Van De Vreugde, R. J. Meagher, J. S. Schoeniger, T. W. Lane, S. S. Branda, M. S. Bartsch and K. D. Patel, PLoS One, 2013, 8, e68988 CrossRef CAS.
- Q. Zhang, X. Xu, L. Lin, J. Yang, X. Na, X. Chen, L. Wu, J. Song and C. Yang, Lab Chip, 2022, 22, 1971–1979 RSC.
- S. Kim, J. De Jonghe, A. B. Kulesa, D. Feldman, T. Vatanen, R. P. Bhattacharyya, B. Berdy, J. Gomez, J. Nolan, S. Epstein and P. C. Blainey, Nat. Commun., 2017, 8, 13919 CrossRef CAS.
- S. Picelli, Å. K. Björklund, B. Reinius, S. Sagasser, G. Winberg and R. Sandberg, Genome Res., 2014, 24, 2033–2040 CrossRef CAS PubMed.
- A. Adey, H. G. Morrison, Asan, X. Xun, J. O. Kitzman, E. H. Turner, B. Stackhouse, A. P. MacKenzie, N. C. Caruccio, X. Zhang and J. Shendure, Genome Biol., 2010, 11, R119 CrossRef CAS PubMed.
- S. Gunasekera, S. Abraham, M. Stegger, S. Pang, P. Wang, S. Sahibzada and M. O'Dea, PLoS One, 2021, 16, e0253440 CrossRef CAS.
- J. H. Lan, Y. Yin, E. F. Reed, K. Moua, K. Thomas and Q. Zhang, Hum. Immunol., 2015, 76, 166–175 CrossRef CAS PubMed.
- A. D. Tyler, S. Christianson, N. C. Knox, P. Mabon, J. Wolfe, G. Van Domselaar, M. R. Graham and M. K. Sharma, PLoS One, 2016, 11, e0148676 CrossRef PubMed.
- L. Uelze, M. Borowiak, C. Deneke, I. Szabó, J. Fischer, S. H. Tausch and B. Malorny, Appl. Environ. Microbiol., 2020, 86, e02265-19 CrossRef.
- M. B. Jones, S. K. Highlander, E. L. Anderson, W. Li, M. Dayrit, N. Klitgord, M. M. Fabani, V. Seguritan, J. Green, D. T. Pride, S. Yooseph, W. Biggs, K. E. Nelson and J. Craig Venter, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 14024–14029 CrossRef CAS PubMed.
- D. R. Bentley, S. Balasubramanian, H. P. Swerdlow, G. P. Smith, J. Milton, C. G. Brown, K. P. Hall, D. J. Evers, C. L. Barnes, H. R. Bignell, J. M. Boutell, J. Bryant, R. J. Carter, R. Keira Cheetham, A. J. Cox, D. J. Ellis, M. R. Flatbush, N. A. Gormley, S. J. Humphray, L. J. Irving, M. S. Karbelashvili, S. M. Kirk, H. Li, X. Liu, K. S. Maisinger, L. J. Murray, B. Obradovic, T. Ost, M. L. Parkinson, M. R. Pratt, I. M. J. Rasolonjatovo, M. T. Reed, R. Rigatti, C. Rodighiero, M. T. Ross, A. Sabot, S. V. Sankar, A. Scally, G. P. Schroth, M. E. Smith, V. P. Smith, A. Spiridou, P. E. Torrance, S. S. Tzonev, E. H. Vermaas, K. Walter, X. Wu, L. Zhang, M. D. Alam, C. Anastasi, I. C. Aniebo, D. M. D. Bailey, I. R. Bancarz, S. Banerjee, S. G. Barbour, P. A. Baybayan, V. A. Benoit, K. F. Benson, C. Bevis, P. J. Black, A. Boodhun, J. S. Brennan, J. A. Bridgham, R. C. Brown, A. A. Brown, D. H. Buermann, A. A. Bundu, J. C. Burrows, N. P. Carter, N. Castillo, M. C. E. Catenazzi, S. Chang, R. N. Cooley, N. R. Crake, O. O. Dada, K. D. Diakoumakos, B. Dominguez-Fernandez, D. J. Earnshaw, U. C. Egbujor, D. W. Elmore, S. S. Etchin, M. R. Ewan, M. Fedurco, L. J. Fraser, K. V. F. Fajardo, W. S. Furey, D. George, K. J. Gietzen, C. P. Goddard, G. S. Golda, P. A. Granieri, D. E. Green, D. L. Gustafson, N. F. Hansen, K. Harnish, C. D. Haudenschild, N. I. Heyer, M. M. Hims, J. T. Ho, A. M. Horgan, K. Hoschler, S. Hurwitz, D. V. Ivanov, M. Q. Johnson, T. James, T. A. H. Jones, G. D. Kang, T. H. Kerelska, A. D. Kersey, I. Khrebtukova, A. P. Kindwall, Z. Kingsbury, P. I. Kokko-Gonzales, A. Kumar, M. A. Laurent, C. T. Lawley, S. E. Lee, X. Lee, A. K. Liao, J. A. Loch, M. Lok, S. Luo, R. M. Mammen, J. W. Martin, P. G. McCauley, P. McNitt, P. Mehta, K. W. Moon, J. W. Mullens, T. Newington, Z. Ning, B. L. Ng, S. M. Novo, M. J. O'Neill, M. A. Osborne, A. Osnowski, O. Ostadan, L. L. Paraschos, L. Pickering, A. C. Pike, A. C. Pike, D. C. Pinkard, D. P. Pliskin, J. Podhasky, V. J. Quijano, C. Raczy, V. H. Rae, S. R. Rawlings, A. C. Rodriguez, P. M. Roe, J. Rogers, M. C. R. Bacigalupo, N. Romanov, A. Romieu, R. K. Roth, N. J. Rourke, S. T. Ruediger, E. Rusman, R. M. Sanches-Kuiper, M. R. Schenker, J. M. Seoane, R. J. Shaw, M. K. Shiver, S. W. Short, N. L. Sizto, J. P. Sluis, M. A. Smith, J. E. S. Sohna, E. J. Spence, K. Stevens, N. Sutton, L. Szajkowski, C. L. Tregidgo, G. Turcatti, S. vandeVondele, Y. Verhovsky, S. M. Virk, S. Wakelin, G. C. Walcott, J. Wang, G. J. Worsley, J. Yan, L. Yau, M. Zuerlein, J. Rogers, J. C. Mullikin, M. E. Hurles, N. J. McCooke, J. S. West, F. L. Oaks, P. L. Lundberg, D. Klenerman, R. Durbin and A. J. Smith, Nature, 2008, 456, 53–59 CrossRef CAS PubMed.
- L. Clime, D. Brassard, M. Geissler and T. Veres, Lab Chip, 2015, 15, 2400–2411 RSC.
- A. Snider, M. Nilsson, M. Dupal, M. Toloue and A. Tripathi, SLAS Technol., 2019, 24, 196–208 CrossRef CAS PubMed.
- L. Schneider, F. Cui and A. Tripathi, Biomicrofluidics, 2021, 15, 024104 CrossRef CAS.
- H. Kim, M. S. Bartsch, R. F. Renzi, J. He, J. L. Van de Vreugde, M. R. Claudnic and K. D. Patel, J. Lab. Autom., 2011, 16, 405–414 CrossRef CAS PubMed.
- A. M. Foudeh, D. Brassard, M. Tabrizian and T. Veres, Lab Chip, 2015, 15, 1609–1618 RSC.
- J. F. Hess, M. Kotrová, S. Calabrese, N. Darzentas, T. Hutzenlaub, R. Zengerle, M. Brüggemann and N. Paust, Anal. Chem., 2020, 92, 12833–12841 CrossRef CAS PubMed.
- D. Brassard, M. Geissler, M. Descarreaux, D. Tremblay, J. Daoud, L. Clime, M. Mounier, D. Charlebois and T. Veres, Lab Chip, 2019, 19, 1941–1952 RSC.
- L. Clime, J. Daoud, D. Brassard, L. Malic, M. Geissler and T. Veres, Microfluid. Nanofluid., 2019, 23, 1–22 CrossRef.
- M. Geissler, D. Brassard, L. Clime, A. V. C. Pilar, L. Malic, J. Daoud, V. Barrère, C. Luebbert, B. W. Blais, N. Corneau and T. Veres, Analyst, 2020, 145, 6831–6845 RSC.
- B.-U. Moon, L. Clime, D. Brassard, A. Boutin, J. Daoud, K. Morton and T. Veres, Lab Chip, 2021, 21, 4060–4070 RSC.
- L. Malic, D. Brassard, D. Da Fonte, C. Nassif, M. Mounier, A. Ponton, M. Geissler, M. Shiu, K. J. Morton and T. Veres, Lab Chip, 2022, 22, 3157–3171 RSC.
- L. Rossen, P. Nørskov, K. Holmstrøm and O. F. Rasmussen, Int. J. Food Microbiol., 1992, 17, 37–45 CrossRef CAS PubMed.
- N. J. Lennon, R. E. Lintner, S. Anderson, P. Alvarez, A. Barry, W. Brockman, R. Daza, R. L. Erlich, G. Giannoukos, L. Green, A. Hollinger, C. A. Hoover, D. B. Jaffe, F. Juhn, D. McCarthy, D. Perrin, K. Ponchner, T. L. Powers, K. Rizzolo, D. Robbins, E. Ryan, C. Russ, T. Sparrow, J. Stalker, S. Steelman, M. Weiand, A. Zimmer, M. R. Henn, C. Nusbaum and R. Nicol, Genome Biol., 2010, 11, R15 CrossRef.
- J. Grützke, B. Malorny, J. A. Hammerl, A. Busch, S. H. Tausch, H. Tomaso and C. Deneke, Front. Microbiol., 2019, 10, 1805 CrossRef PubMed.
- J. H. Lan, Y. Yin, E. F. Reed, K. Moua, K. Thomas and Q. Zhang, Hum. Immunol., 2015, 76, 166–175 CrossRef CAS PubMed.
- B. Bushnell, BBMap: A Fast, Accurate, Splice-Aware Aligner, Lawrence Berkeley National Laboratory, 2014 Search PubMed.
- S. Nurk, D. Meleshko, A. Korobeynikov and P. A. Pevzner, Genome Res., 2017, 27, 824–834 CrossRef CAS PubMed.
- B. Langmead and S. L. Salzberg, Nat. Methods, 2012, 9, 357–359 CrossRef CAS.
- P. Danecek, J. K. Bonfield, J. Liddle, J. Marshall, V. Ohan, M. O. Pollard, A. Whitwham, T. Keane, S. A. McCarthy, R. M. Davies and H. Li, GigaScience, 2021, 10, giab008 CrossRef PubMed.
- H. Wickham, ggplot2: Elegant Graphics for Data Analysis, Springer Cham, Cham, 2016 Search PubMed.
- F. Beghini, L. J. McIver, A. Blanco-Míguez, L. Dubois, F. Asnicar, S. Maharjan, A. Mailyan, P. Manghi, M. Scholz, A. M. Thomas, M. Valles-Colomer, G. Weingart, Y. Zhang, M. Zolfo, C. Huttenhower, E. A. Franzosa and N. Segata, eLife, 2021, 10, e65088 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lc00781b |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |