
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
SARS-CoV-2-induced disruption of a vascular bed in a microphysiological system caused by type-I interferon from bronchial organoids†
Kazuya
Fujimoto
 a,
Yoshikazu
Kameda
a,
Yuta
Nagano
a,
Sayaka
Deguchi
b,
Takuya
Yamamoto
bcd,
Rafal P.
Krol
e,
Peter
Gee
f,
Yasufumi
Matsumura
g,
Toru
Okamoto
h,
Miki
Nagao
g,
Kazuo
Takayama
*b and
Ryuji
Yokokawa
*a
a,
Yoshikazu
Kameda
a,
Yuta
Nagano
a,
Sayaka
Deguchi
b,
Takuya
Yamamoto
bcd,
Rafal P.
Krol
e,
Peter
Gee
f,
Yasufumi
Matsumura
g,
Toru
Okamoto
h,
Miki
Nagao
g,
Kazuo
Takayama
*b and
Ryuji
Yokokawa
*a
aDepartment of Micro Engineering, Kyoto University, Kyoto daigaku-Katsura, Nishikyo-ku, Kyoto 615-8540, Japan. E-mail: yokokawa.ryuji.8c@kyoto-u.ac.jp
bCenter for iPS cell Research and Application (CiRA), Kyoto University, Shogoin-Kawahara-cho 53, Sakyo-ku, Kyoto, 606-8507, Japan. E-mail: kazuo.takayama@cira.kyoto-u.ac.jp
cInstitute for the Advanced Study of Human Biology (WPI-ASHBi), Kyoto University, Yoshida-Konoe-cho, Sakyo-ku, Kyoto, 606-8501, Japan
dMedical-risk Avoidance based on iPS Cells Team, RIKEN Center for Advanced Intelligence Project (AIP), Shogoin-Kawahara-cho 53, Sakyo-ku, Kyoto, 606-8507, Japan
eResearch and Development Center, CiRA Foundation, Shogoin-Kawahara-cho 53, Sakyo-ku, Kyoto, 606-8397, Japan
fMaxCyte Inc., Gaithersburg, MD 20878, USA
gDepartment of Clinical Laboratory medicine, Kyoto University Graduate School of Medicine, Shogoin-Kawahara-cho 53, Sakyo-ku, Kyoto, 606-8507, Japan
hDepartment of Microbiology, School of Medicine, Juntendo University, Hongo 2-1-1, Bunkyo-ku, Tokyo 113-8421, Japan
First published on 9th January 2024
Abstract
Blood vessels show various COVID-19-related conditions including thrombosis and cytokine propagation. Existing in vitro blood vessel models cannot represent the consequent changes in the vascular structure or determine the initial infection site, making it difficult to evaluate how epithelial and endothelial tissues are damaged. Here, we developed a microphysiological system (MPS) that co-culture the bronchial organoids and the vascular bed to analyze infection site and interactions. In this system, virus-infected organoids caused damage in vascular structure. However, vasculature was not damaged or infected when the virus was directly introduced to vascular bed. The knockout of interferon-related genes and inhibition of the JAK/STAT pathway reduced the vascular damage, indicating the protective effect of interferon response suppression. The results demonstrate selective infection of bronchial epithelial cells and vascular damage by cytokines and also indicate the applicability of MPS to investigate how the infection influences vascular structure and functions.
Introduction
The COVID-19 pandemic caused by SARS-CoV-2 began in late 2019 and has still not completely ceased. The respiratory system is the primary target of SARS-CoV-2 owing to the high expression of ACE2 and TMPRSS2. In severe cases of COVID-19, the disease progresses to multi-organ dysfunction.1,2 Among organs, blood vessels suffer from multiple conditions such as endothelialitis, microthrombosis, and cytokine storms.2–5 While in vitro studies have investigated COVID-19 disease progression after direct infection, cytokine activation, and antiviral responses, several questions remain unanswered regarding SARS-CoV-2 infections. For example, even though ACE2 expression and infection to endothelial cells are indicated in patients with severe cases of the disease,5,6 several in vitro studies using primary cell culture report endothelial cells do not express ACE2 nor are they infected by SARS-CoV-2.7–9 The activation of endothelial cells by cytokines released from adjacent epithelial tissues and the attraction of immune cells have been also studied as part of the disease mechanism from patient biopsies.10–12The loss of structure and function of blood vessels upon infection is key to understanding COVID-19. The kidney, heart, and skin are damaged by the virus or by paracrine signals from primarily infected tissues that migrate through the vasculature.13,14 Morphological changes in pulmonary capillaries have been reported in samples from patients with upregulated cytokines or growth factors.5 Although the paracrine effects of cytokines are correlated with blood vessel malfunction or malformation,15,16in vivo observation of the vessel under the disease was impossible.
In vitro experiments have been widely utilized for SARS-CoV-2 study. Cell lines such as Vero E6 or A549, which derived from cancer cells, are most widely used.17,18 However, these cell lines largely differ from in vivo condition, especially in susceptibility to virus and immune response against infection. Recently, primary and stem-cell-derived cells are also used as more physiologically relevant cell source. For example, pulmonary epithelial cells form differentiated layer under air–liquid interface (ALI) culture has been reported.19,20
Even with higher functionality of cell source, conventional cell culture still lacks an ability to investigate interaction between blood vessel and other tissues. To address this gap, in vitro experimental approaches that mimic the interface between epithelial tissues and vasculature are needed. To understand the response of surrounding tissues in vitro, defining the infection site and recapitulating the interactions between epithelial tissues and blood vessels are critical.21
Microphysiological systems (MPS) to study SARS-CoV-2, such as SARS-CoV-2 MPS, enable the selection of the infection site because of its accessibility to the target tissues. Lu et al. reported that endothelialized vasculature-on-a-chip exposed to SARS-CoV-2 spike protein enters a hyperinflammatory state when co-cultured with peripheral blood mononuclear cells, including monocytes.22 Interactions between different cell types, migration of the virus, and the recruitment of immune cells have been studied using SARS-CoV-2 MPS with alveolar,23,24 bronchial,25 and intestine epithelial cells.26
However, these studies still suffer from several problems. First, not a few studies did not use authentic SARS-CoV-2, making it difficult to compare them with in vivo studies. In addition, most of the studies utilized a bilayer format MPS, which lacks the ability to assess blood vessel symptoms caused by the virus infection.
In the current study, we developed a SARS-CoV-2 MPS to recapitulate interactions between the 3D bronchial organoids (BOs) and the vascular bed (VB). The BOs and the VB, consisting of primary epithelial and endothelial cells, respectively, were co-cultured in a 3D microfluidic device previously reported,27 in which endothelial cells autonomously form the VB in a micro fluidic channel.28–30 We used the SARS-CoV-2 MPS to selectively introduce the virus into the BOs and/or the VB and analyze virus entry into each tissue and change of the VB's 3D structure upon viral infection. This proposed MPS model also enables us to study the paracrine effects between the BOs and the VB utilizing accessibility to the specific cell type, gene modification, and inhibitor treatment to block the immune response pathways.
Results
Microfluidic devices enabled the co-culture of bronchial organoids (BOs) and the vascular bed (VB)
The interactions between bronchial tissues and blood vessels were recapitulated using a three-layer microfluidic device. The BOs were cultured in a well (BO-culture well), and the VB was cultured in a channel fabricated with soft lithography (VB-culture channel) (Fig. 1A and S1A–C†). The BO-culture well and VB-culture channel were in contact with one another via a connecting hole (Fig. S1A†). A pillar array defined the side of the VB-culture channel and contained an extracellular matrix (ECM) gel consisting of fibrin and collagen, in which human umbilical vein endothelial cells (HUVECs) were suspended (Fig. S1A†). A perfusable VB was formed 4 days after HUVEC seeding. Normal human bronchial epithelial cells (NHBE) in Matrigel were differentiated to form BOs in the BO-culture wells (Fig. 1B).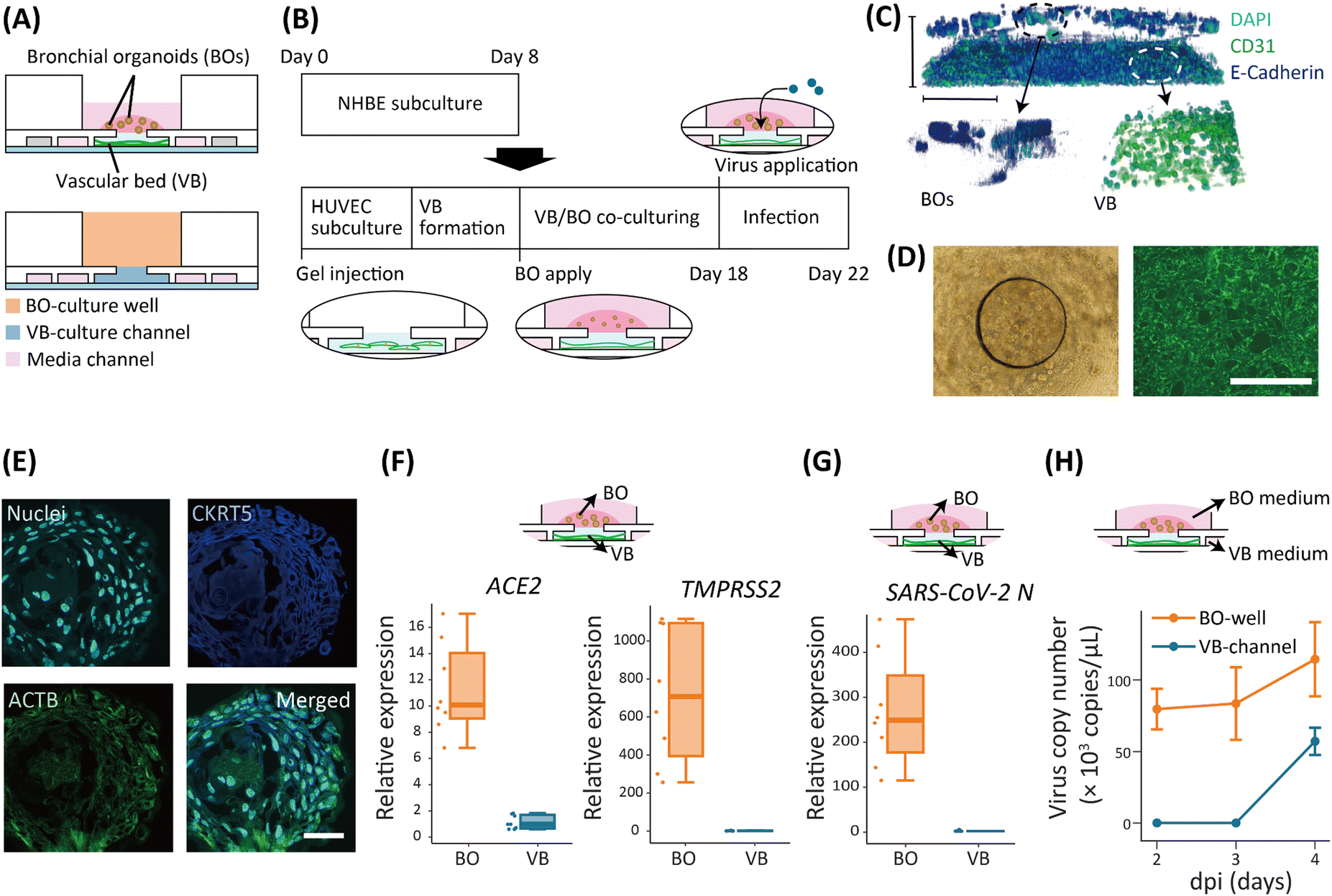 | ||
| Fig. 1 Co-culture of bronchial organoids and vascular beds in a microfluidic device. (A) Schematic of the co-culture device. The BOs were cultured in a culture well (BO-culture well) on top of a VB prepared in a microfluidic channel (VB-culture channel: blue). Culture media were supplied and exchanged through the channels neighboring the VB-culture channel (media channel: pink). (B) Protocol of the BO and VB culture and subsequent SARS-CoV-2 infection. (C) A 3D image of BOs and VB stained with DAPI (cyan), E-cadherin (blue), and CD31 (green) (top), enlarged view of BOs (left bottom), and VB (right bottom). Scale bar: 500 μm. (D) Microscope images of co-cultured BOs obtained. A phase contrast image of BOs in the BO-culture well (left) and fluorescent image of the VB (right). Scale bar: 1 mm. (E) Fluorescent image of a sectioned BO. Cyan: nuclei. Blue: cytokeratin 5. Green: acetylated α-tubulin. Scale bar: 20 μm. (F) Relative expression of ACE2 and TMPRSS2 between the BOs and the VB. N = 8 devices for the BOs and the VB each. (G) Expression of SARS-CoV-2 N gene in BOs and the VB at 4 dpi. N = 8 devices for each of the BOs and the VB. (H) Virus copy number in media taken from the BO-culture well and VB-culture channel until 4 dpi when the virus was introduced to the BO-culture well. N = 8 devices for each of the BO-culture well and VB-culture channel. | ||
VB formed 4 days after injection and VB perfusability was maintained with the flow generated by rocking with a shaker (Fig. S2A†). Without rocking, the perfusability of the VB was lost after 10 days of co-culture in all three devices tested (Fig. S2B†). The VB formed in the device remained perfusable for more than 20 days with the BOs, which was enough time to perform all the experiments in this study.
The layered co-culture of the BOs on the VB was visualized (Fig. 1C), in which globular shapes of the BOs and network shape of the VB were observed in the BO-culture well and VB-culture channel, respectively (Fig. 1D). The diffusion of virus particles was analyzed with numerical simulations of viral particle concentration to visualize how they travel in the ECM. When the virus was introduced into the BO-culture well, its particles permeated into the VB-culture channel within 24 hours (Fig. S3†).
We examined the differentiation of the BOs cultured in the device. First, basal cells and ciliated cells in the organoids were observed by staining cytokeratin 5 (KRT5) and acetylated α-tubulin (ACTB), respectively (Fig. 1E). The expression levels of ACE2, which codes for the virus receptor protein, and TMPRSS2, which codes for the protease necessary for virus entry into cells, were compared between the BOs cultured in the device and those cultured in a 24-well plate (Fig. S4†). We did not observe a significant difference in either gene expression between the device and the plate. Additionally, we investigated the expression levels of eight genes that are markers of bronchial epithelial cells (Fig. S4†). Three out of these eight markers, namely, MUC20, MCIDS, and PROM1, were significantly up-regulated on the device, and two markers, SCGB1A1 and NGFR, were significantly down-regulated. Expression differences between the plate and device were not significant for the remaining three genes (KLF5, MUC5B, and TUBA1A). Finally, the expression levels of all eight genes tested were within 50–200% of each other when comparing the expression levels of the genes on the device and the plate, consistent with a previous study.31
The expression of ACE2 and TMPRSS2 in the BOs and the VB was measured to investigate the susceptibility of these structures to the virus. We observed a 10-fold higher expression of ACE2 in the BOs than in the VB, while the expression of TMPRSS2 was 800-fold higher (Fig. 1F). These results suggest a lower sensitivity of the VB to virus infection.
SARS-CoV-2 showed higher replication in BOs than in the VB
We evaluated the infection of the BOs and the VB with SARS-CoV-2 by adding the virus to the BO-culture wells using 0.1 multiplicity of infection (MOI). At 4 days post-infection (4 dpi), Ct value of SARS-CoV-2 N in the BOs was 12.97 ± 0.93, which was comparable with previous studies using primary cells (Fig. S5†).20,31The expression of SARS-CoV-2 N was approximately 300-fold higher than in the VB (Fig. 1G), indicating that the virus replication occurred mostly in the BOs. The amount of virus was evaluated based on the virus copy number in the culture media taken from the BO-culture well and the VB-culture channel. The copy number in the BO-culture well at 2 dpi was over 100 × 103 copies per μL and remained at this level until 4 dpi (Fig. 1H). On the other hand, the copy number in the VB-culture channel was negligible until 3 dpi and increased to 50 × 103 copies per μL at 4 dpi.
The effect of viral application site on replication was next assessed in three ways: virus application to (i) both the BO-culture well and the VB-culture channel (BO+VB+); (ii) to the BO-culture well only (BO+VB−); (iii) and to the VB-culture channel only (BO−VB+). The virus copy number at 2 dpi in the BO-culture well and the VB-culture channel were high and low, respectively. In both BO+VB+ and BO+VB− (Fig. S6†), the virus replication occurred mainly in the BO-culture well regardless of whether the virus was introduced to the BO-culture well or the VB-culture channel. In contrast, the copy numbers were low for both the BO-culture well and the VB-culture channel in BO−VB+. Thus, we introduced the virus to the BO-culture well in subsequent experiments.
Bronchial cell markers were maintained on a chip and innate immunity-related genes were up-regulated by SARS-CoV-2 in both the BOs and the VB
Gene expression levels in the BOs were evaluated at 4 dpi with mock (no virus application) or SARS-CoV-2 introduced to the BO-culture well. The expression levels of three basal cell markers (TP63, KRT5, and NGFR), and two ciliated cell markers (MCIDAS and TUBA1A) were measured as bronchial markers (Fig. 2A). Only TP63 expression was significantly increased with infection, whereas MCIDAS expression was significantly decreased. Moreover, ACE2 expression was significantly higher in the BOs infected with SARS-CoV-2 than that in the mock, while TMPRSS2 expression was unchanged (Fig. 2B). Genes related to an innate immune response, IFNA1, IFNB1, MxA, ISG56, and ISG15, showed greatly increased expression in the BOs infected with the virus, indicating the induction of a strong innate immune response (Fig. 2C).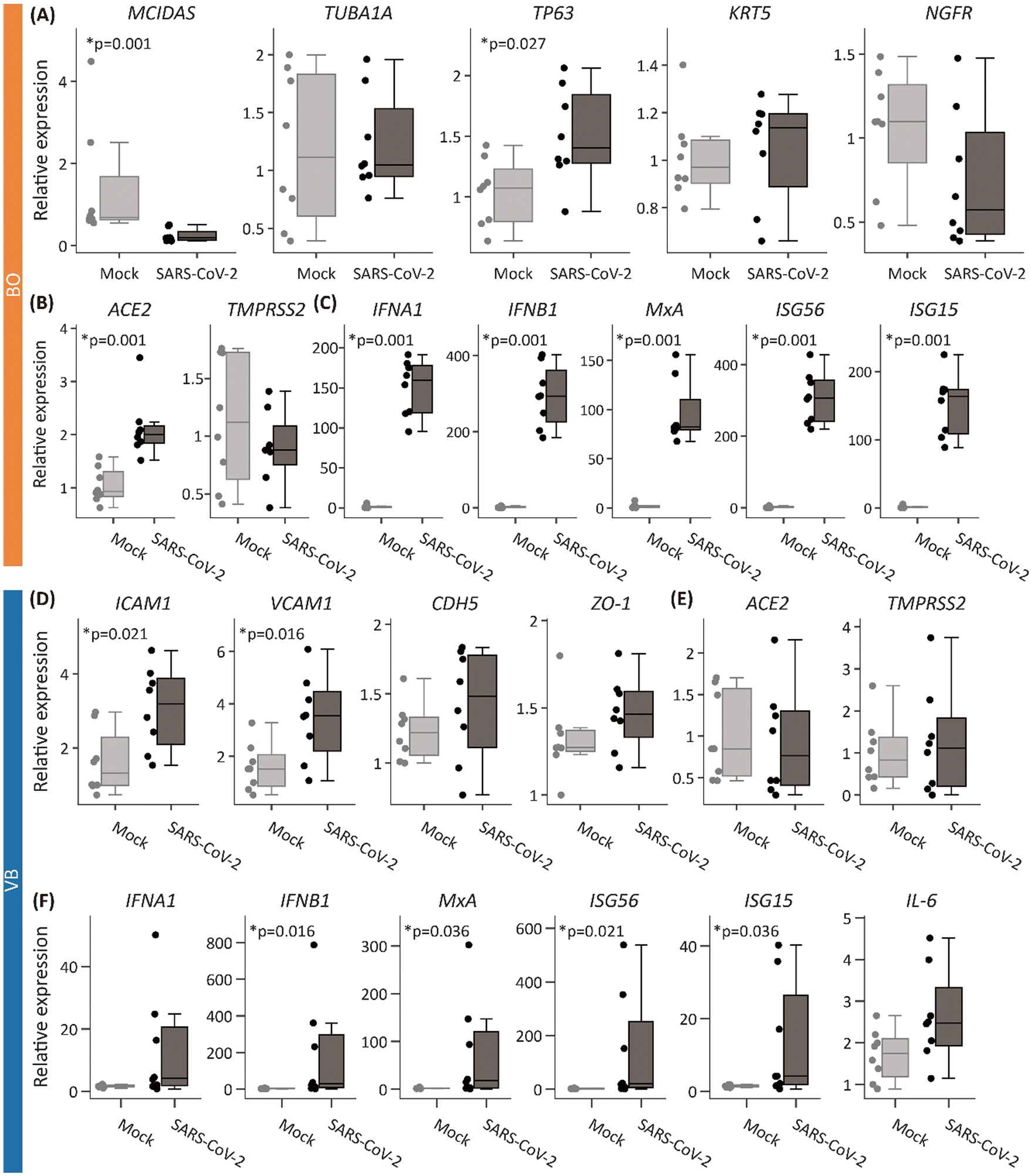 | ||
| Fig. 2 mRNA expression changes by SARS-CoV-2 infection in the BOs and the VB. Gene expression levels (A–C) in the BOs and (D–F) in the VB. (A) Differentiated bronchial markers. (B) Genes required for infection. (C) Genes related to innate immune response. (D) Endothelial cell genes. (E) Genes required for infection. (F) Genes related to innate immune response. Expression levels were normalized by the average in the mock device. p-Values were calculated by Wilcoxon's rank sum test. N = 8 devices. | ||
The expression of genes in the VB was also affected by the virus. We analyzed genes related to endothelial cell function, SARS-CoV-2 infection, and innate immunity. The expression of two cell adhesion molecule genes, ICAM1, and VCAM1, which are related to endothelialitis, was significantly increased with SARS-CoV-2, while two cell–cell adhesion genes, CDH5 and ZO-1, showed no significant change (Fig. 2D), similar to the unaltered levels of ACE2 and TMPRSS2 (Fig. 2E). Moreover, there was a significant increase in the innate immune response-related genes, IFNB1, MxA, ISG56, and ISG15 (p < 0.05), but no significant change was found for the expression of IFNA1 or IL6 (Fig. 2F).
RNA sequencing of the BOs reveals an up-regulation of immune response and down-regulation of cell proliferation
We performed BO transcriptome analysis to evaluate differential gene expression levels in the BOs with or without SARS-CoV-2. The BOs were collected at 4 dpi after co-culturing with the VB for whole RNA extraction and subsequent RNA sequencing. The innate immune response-related genes, RSAD2, HERC5, and IFI2, were up-regulated approximately 10-fold on a log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2 scale (Fig. 3A). Interestingly, PIN1, a prolyl isomerase that reportedly plays an important role in viral proliferation, was also significantly upregulated.32 The up-regulation of immune response pathways was detected by a gene ontology (GO) enrichment analysis, in which signals related to viral infection and interferon signaling were highly up-regulated (Fig. 3B). Most down-regulated pathways were related to cell proliferation. Moreover, the expression of genes from the GO term with the highest enrichment score, “defense response to the virus”, and the GO term with the lowest score, “mitotic sister chromatid segregation” are depicted in a heat map (Fig. 3C). The heat map of the genes included in the GO term “defense response to the virus” showed a consistent upregulation with SARS-CoV-2 among samples, while the genes included in the GO term “mitotic sister chromatid segregation” with SARS-CoV-2 were consistently downregulated among samples.
2 scale (Fig. 3A). Interestingly, PIN1, a prolyl isomerase that reportedly plays an important role in viral proliferation, was also significantly upregulated.32 The up-regulation of immune response pathways was detected by a gene ontology (GO) enrichment analysis, in which signals related to viral infection and interferon signaling were highly up-regulated (Fig. 3B). Most down-regulated pathways were related to cell proliferation. Moreover, the expression of genes from the GO term with the highest enrichment score, “defense response to the virus”, and the GO term with the lowest score, “mitotic sister chromatid segregation” are depicted in a heat map (Fig. 3C). The heat map of the genes included in the GO term “defense response to the virus” showed a consistent upregulation with SARS-CoV-2 among samples, while the genes included in the GO term “mitotic sister chromatid segregation” with SARS-CoV-2 were consistently downregulated among samples.
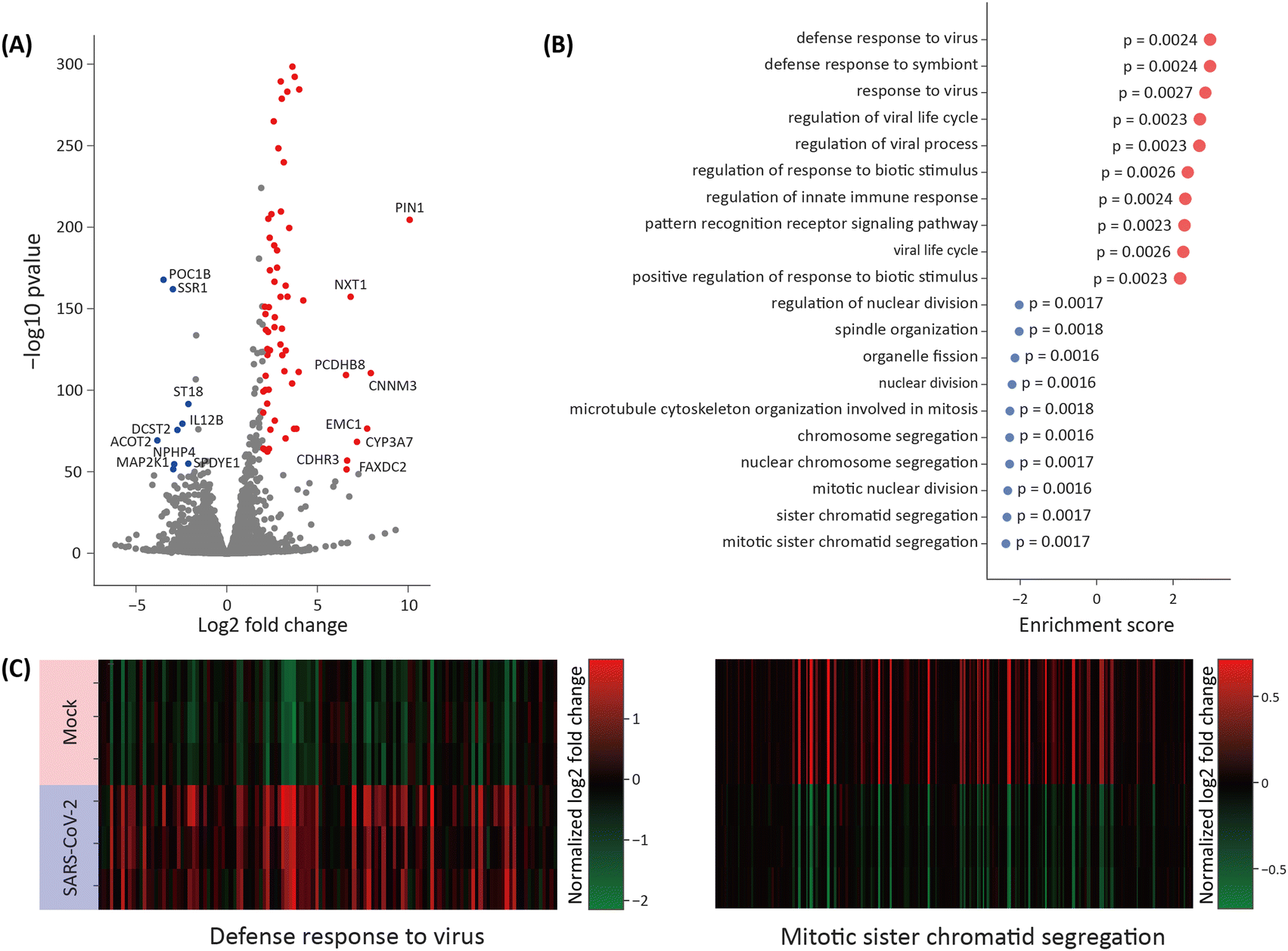 | ||
| Fig. 3 RNA sequencing results from the infected BOs. (A) Plot of the expression fold change (x-axis) and negative logp-value (y-axis). Gene expression levels that increased by more than 2 units on the x-axis and more than 50 units on the y-axis are colored in red. Those that decreased by 2 units on the x-axis and 50 units on the y-axis are colored in blue. (B) Normalized enrichment factor of the 10 top and bottom pathways analyzed in the GSEA. (C) Log2 fold change of genes in the GO term “defense response to virus” whose enrichment score was the largest in the GSEA (left) and in the GO term “mitotic sister chromatid segregation” whose enrichment score was the smallest. | ||
VB coverage and integrity were reduced by SARS-CoV-2 infection of the BOs
Morphological changes induced in the VB by the virus were observed by fluorescence microscopy. In the SARS-CoV-2-infected device, lumen structures in the VB thinned after the injection of the virus, whereas no significant change was observed in the mock device (Fig. 4A). VB images were quantitatively analyzed by calculating the ratio of the HUVEC-covered area to the entire area of the VB-culture channel. The ratio was significantly lower in the SARS-CoV-2-infected devices (0.64 ± 0.05; mean ± S.D.), compared to the ratio in the mock devices (0.87 ± 0.02) (Fig. 4B).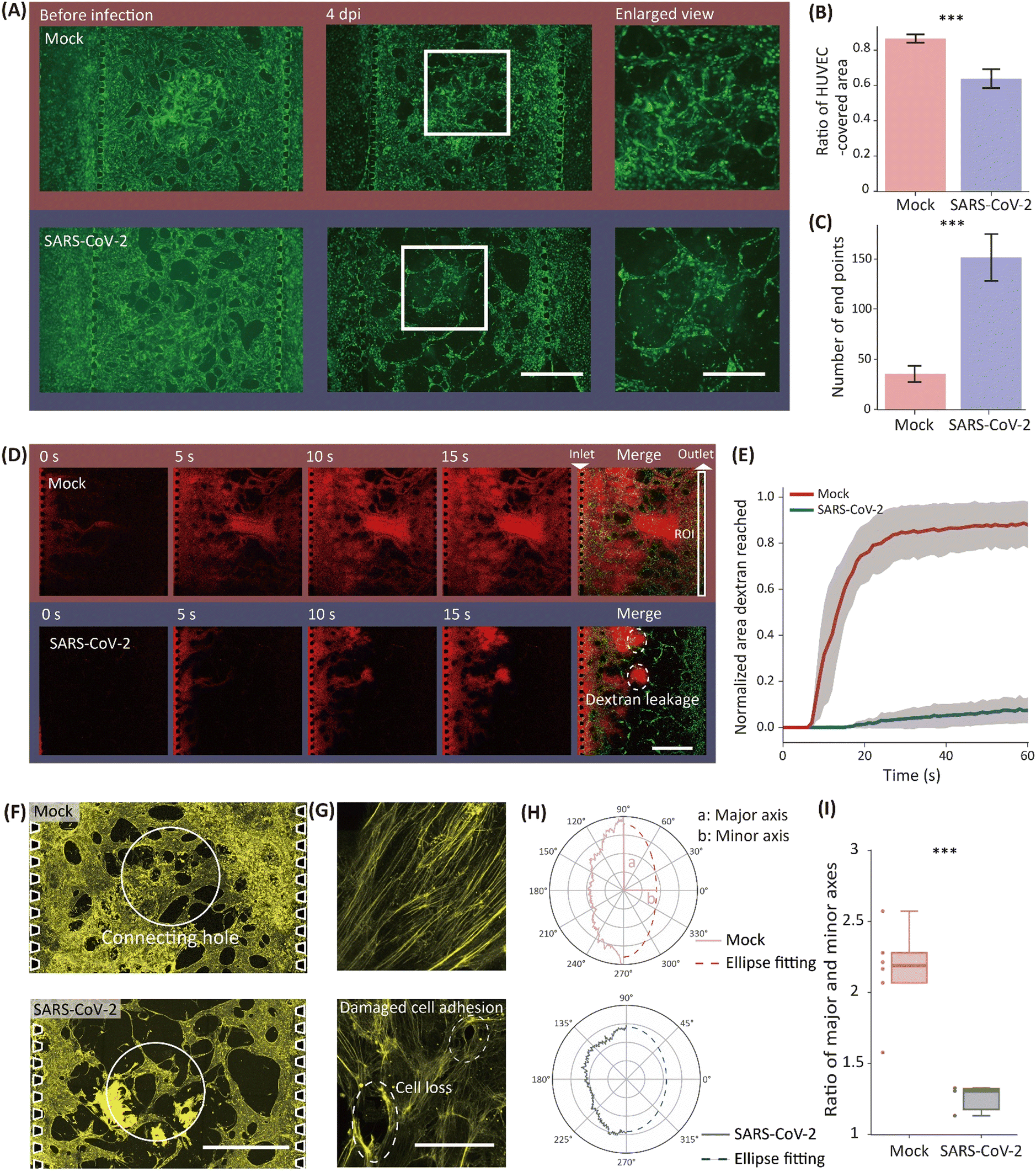 | ||
| Fig. 4 Morphological and functional effects of SARS-CoV-2 infection on the VB. (A) Fluorescent images of the VB before infection and 4 dpi. Scale bars: 1 mm. (B) The ratio of the HUVEC-covered area to the whole VB-culture channel area. (C) The number of end points per field of view. n = 3 biological replicates. ***p < 0.01 in (B) and (C) by Student's t-test. (D) Time-lapse images of rhodamine-labeled dextran solution flow (red) after injection from the left side. The rightmost images are overlaid by the VB (green) at 15 s. The white square in the merged image of the mock device corresponds to the ROI where the pixel intensity was evaluated. Dashed circles indicate the dextran leakage to the ECM from the VB due to the loss of integrity. Scale bar: 1 mm. (E) Time course change of the dextran area in the mock (red) and the SARS-CoV-2-infected device (blue). (F) Fluorescent image of the VB (actin filaments: yellow). White circles enclose the connecting hole. Scale bar: 1 mm. (G) Enlarged view of the actin filaments (yellow) in HUVECs. Dashed circles indicate the loss of cells and reduced cell–cell connection in the SARS-CoV-2-infected device (bottom). Scale bar: 100 μm. (H) Radar chart of the power spectrum against the angle of an actin filament following a 2D FFT. Raw spectra are plotted as solid lines. The ellipse fitting of the raw spectra are plotted as dashed lines. (I) The ratio of the major and minor axes values from the ellipse fitting of the orientation spectra in SARS-CoV-2 and mock devices. n > 3 ROIs from multiple replicates; ***p < 0.01 by Student's t-test. | ||
Moreover, the vascular network was also degraded in the SARS-CoV-2-infected devices, but not in the mock devices. The connectivity of the VB was evaluated by the number of end points, where a vessel terminates without connecting to any other vessel. We observed a significantly larger end point number (p < 0.01) in infected devices (151.7 ± 23.3 per device) than in mock devices (35.3 ± 8.1 per device), which quantitatively indicates the shrinkage and loss of the network connectivity in the SARS-CoV-2-infected device (Fig. 4C).
Vascular integrity was tested by introducing rhodamine B-conjugated dextran (rhodamine–dextran) solution into one side of the VB-culture channel through the media channel. The difference in the water head induced a fluidic force that led the solution from one end of the channel in which the dextran was applied to the other end. In the mock device, the dextran solution flowed through the VB and reached the opposite side of the VB-culture channel within 5 s (Fig. 4D, top). In contrast, the solution did not reach the opposite side in the SARS-CoV-2-infected devices even after 15 s (Fig. 4D, bottom). In addition, the dextran solution flowed out to the ECM through a damaged vascular wall or an end point in the VB-culture channel in the SARS-CoV-2-infected devices (Fig. 4D).
The loss of VB integrity was quantified by analyzing the flow of the labeled dextran solution. The ratio of the area where the VB was filled with dextran in a region of interest (ROI). The ROI was located at one end of the VB channel opposite to the side from which the dextran solution was inlet (Fig. 4D). In the mock devices, the ratio was approximately 0.9 in 20 s (N = 3 devices) and reached a plateau thereafter. The ratio of the SARS-CoV-2-infected devices was around 0.1 even after 60 s, indicating a loss of VB integrity (Fig. 4E).
The microscopic structure of VB was damaged by SARS-CoV-2 infection in the BOs
Structural damage to the VB was evaluated from images of fluorescently labeled actin filaments. Thinned vessels were visualized in the images with lower magnification (Fig. 4A and F). High-magnification images allowed the visualization of the individual actin filaments in the cells (Fig. 4G), whose orientations were less uniform in the infected device than in the mock device, owing to the loss of cells or cell–cell adhesions.The damage by SARS-CoV-2 to the VB structure was quantitatively measured by the orientation of actin filaments. A power spectrum was obtained from the 2D Fourier transformation of filament images taken from mock and infected devices. The sum of the power spectrum along the frequency (Fig. 4H) shows the degree of the orientation uniformity of the actin filaments. The ratio of the major and minor axes of an ellipse fitting was about twice as large in the mock device as in the infected device, indicating that the actin filament orientation was less uniform after virus application (Fig. 4I). This actin filament dysregulation is quantitative evidence of the damage of the VB structure by SARS-CoV-2.
Knocking out the type-I interferon pathway reduced vascular damage
Knocking out interferon alpha and beta receptor subunit 2 (IFNAR2) in HUVECs reduced the VB damage and the amount of virus detected in the VB-culture channel. A mutated VB was created using IFNAR2 KO-HUVECs (KO-VB), and the virus was introduced to the BO-culture well. The vascular structure of KO-VB remained intact after the infection (Fig. 5A), in contrast to the VB in the SARS-CoV-2-infected devices with wild-type HUVECs (WT-VB) (Fig. S7†). The virus copy number in the BO-culture well with the KO-VB (30.0 ± 0.4 × 103 copies per μL) was comparable with WT-VB (21.1 ± 1.3 × 103 copies per μL). However, in the VB-culture channel, the copy number with the KO-VB was significantly lower (1.1 ± 0.02 × 103 copies per μL) than with WT-VB (13.6 ± 1.3 × 103 copies per μL) (Fig. 5B).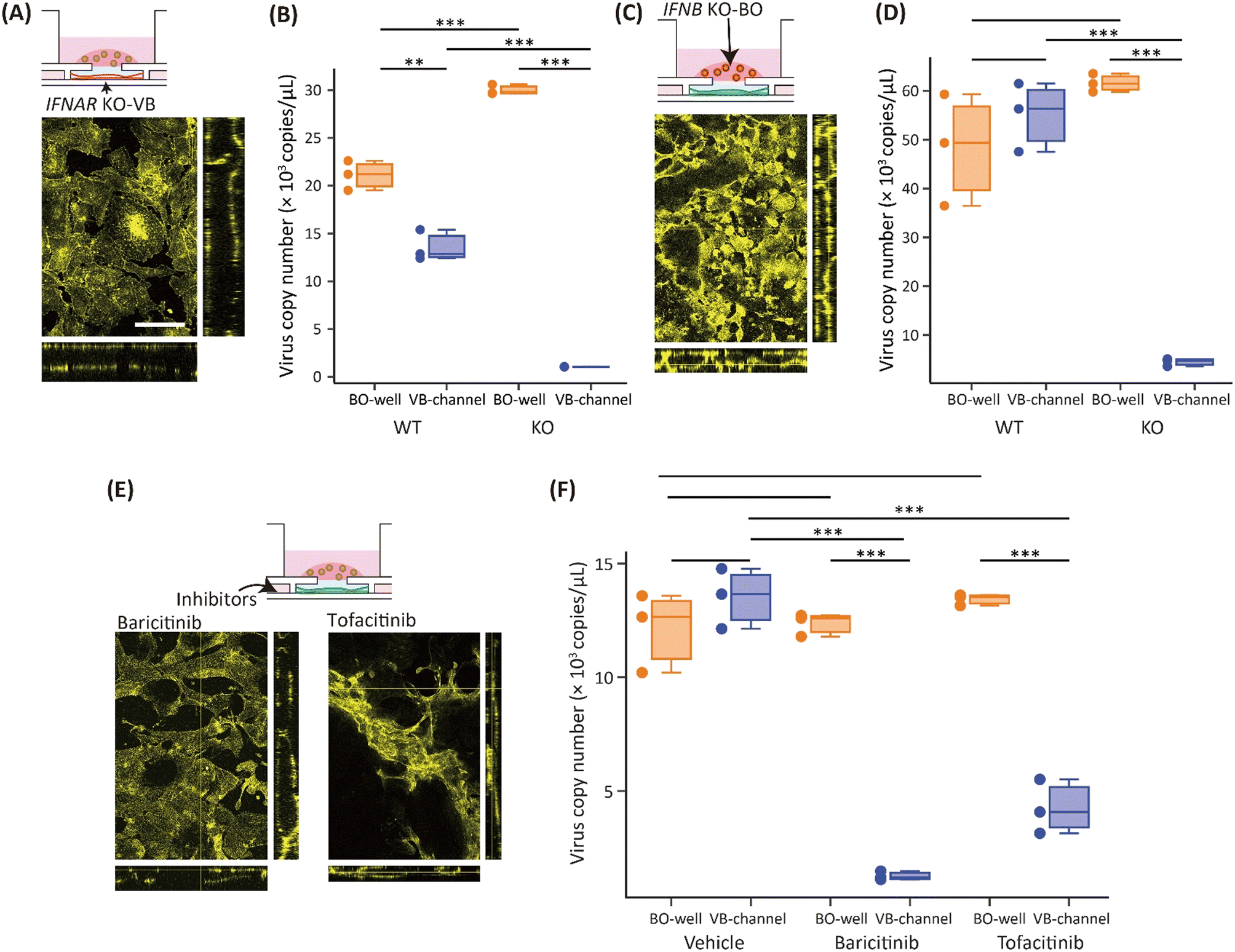 | ||
| Fig. 5 Reduction of vascular damage by suppression of the interferon pathway. (A) Orthogonal view of actin filaments (yellow) in KO-VB in the SARS-CoV-2-infected device. Scale bar: 100 μm. (B) The virus copy numbers at 4 dpi in the BO-culture well and VB-culture channel in the SARS-CoV-2-infected device culturing WT-BO and WT-VB or KO-VB. (C) Orthogonal view of actin filaments (yellow) in WT-VB in the SARS-CoV-2-infected device culturing KO-BO. (D) The virus copy numbers at 4 dpi in the BO-culture well and VB-culture channel in the SARS-CoV-2-infected device with WT-BO or KO-BO and WT-VB. (E) Orthogonal view of actin filaments (yellow) in WT-VB in SARS-CoV-2-infected devices with the administration of baricitinib or tofacitinib. (F) The virus copy number at 4 dpi in the BO-culture well and VB-culture channel in the SARS-CoV-2-infected devices treated with DMSO (vehicle), baricitinib, or tofacitinib. n = 3 replication of the devices for each condition, *p < 0.05, **p < 0.01, ***p < 0.005 by Student's t-test. | ||
Knocking out of interferon beta (IFNB1) in NHBEs also reduced the VB damage and viral gene expression. The BOs were prepared with IFNB1 KO-NHBEs (KO-BO) and co-cultured on WT-VB (Fig. 5C), and the virus copy number was measured. The copy number in the BO-culture well using the wild-type BO (WT-BO) (48.8 ± 9.3 × 103 copies per μL) was comparable with that using the KO-BO (62.0 ± 1.5 × 103 copies per μL) (Fig. 5D). In contrast, the copy number in the VB-culture channel with the KO-BO was an order of magnitude lower (4.9 ± 0.6 × 103 copies per μL) than with the WT-BO (55.5 ± 5.8 × 103 copies per μL). Thus, the use of KO-BO and KO-VB significantly reduced the virus copy number, indicating the crucial role of type-I interferon and its receptor pathway in vascular damage.
JAK/STAT inhibitors showed a protective effect on VB
The application of JAK/STAT inhibitors to SARS-CoV-2-infected devices resulted in the VB structure being maintained and reduced expression of virus genes in the VB-culture channels. Either baricitinib or tofacitinib, two JAK/STAT inhibitors, were introduced into the VB-culture channels in the infection experiments. VB treated with the inhibitors maintained its vascular structure (Fig. 5E). Additionally, the inhibitors also reduced virus copy number in the VB-culture channel. In the vehicle-treated devices, the copy number was 12.1 ± 1.4 × 103 copies per μL in the BO-culture well and 13.5 ± 1.1 × 103 copies per μL in the VB-culture channel. With baricitinib treatment, the virus copy number in the VB-culture channel was 1.3 ± 0.2 × 103 copies per μL, which was approximately 10-fold lower than in the vehicle condition, but the copy number in the BO-culture well was 12.4 ± 0.4 × 103 copies per μL, which was equivalent to the vehicle. The administration of tofacitinib gave similar results, with 4.2 ± 1.0 × 103 copies per μL in the VB-culture channel and 13.4 ± 0.2 × 103 copies per μL in the BO-culture well (Fig. 5F). These results suggest that both inhibitors suppressed the migration of the virus from the BO-culture well to the VB-culture channel.Discussion
In this study, we established the MPS model to evaluate the mutual interaction between the BOs and the VB in SARS-CoV-2 infections. The MPS based multi-layered-microfluidic-device enabled us to maintain VB and BOs with the help of media flow.The device design also makes it possible to define the initial infection site of SARS-CoV-2 while maintaining the diffusional transport of molecules and virus particles through the connecting hole. Multiple conditions were able to be tested with single batch of experiment, as approximately 30 devices were handled at one time. Replication of the virus in the BOs and transient damage in the VB were observed. The morphological damage to the VB caused by paracrine IFN-β was quantified. Additionally, the mechanisms of VB damage were examined using KO cells and JAK/STAT pathway inhibitor drugs.
The MPS model was examined by applying the virus to specific sites. Infection and replication in the BOs were confirmed by the detection of the virus gene in the BOs and the BO culture media at 2 dpi. (Fig. 1F and G). In contrast to the virus copy number observed in the BOs, a low virus copy number was seen in the VB (Fig. 1G). This finding is consistent with the much lower expression of ACE2 and TMPRSS2 in the HUVECs forming the VB than in NHBEs forming BOs (Fig. 1F).
Virus RNA copy number was negligible in VB culture media compared with BO culture media until 3 dpi, but became detectable level at 4 dpi (Fig. 1H). It can be explained by the two facts: one is the lower infection efficiency of the virus to endothelial cells until 3 dpi, because the copy number would drastically increase at earlier days if endothelial cells are infected. The other is that the virus released from the BOs were migrated to the VB-culture channel due to the loss of integrity of VB after 4 dpi.
Specificity of the virus infection to the BOs is also supported by high gene copy number in supernatants from the BO-culture well of the device where the virus was applied to the BO-culture well and VB-culture channel (BO+VB+) or only to the BO-culture well (BO+VB−). The copy number in the VB culture media in BO+VB− and BO−VB+ showed the same level, indicating that the infection of the VB was not significant. The BO-specific infection matched the higher expression of ACE2 and TMPRSS2 in the BOs than in the VB we observed here (Fig. 2B and D) and previously reported under in vitro conditions and from patients.7,8
When the virus was applied exclusively to the VB-culture channel (BO−VB+), a low gene copy number was observed both in the VB-culture channel and the BO-culture well. The low gene copy number observed in the VB-culture channel implies a lack of infection in the VB, consistent with the outcomes in BO+VB− and BO−VB+ conditions. Henceforth, predominant source of the observed virus in the BO-culture well can be deduced as the migration of the virus initially applied to the VB-culture channel.
The lack of infection in the BOs indicates the existence of a factor hindering virus migration from the VB-culture channel to the BO-culture well. Gravity emerges as a plausible factor that could restrict the diffusional transport of the virus from the VB-culture channel, situated at the bottom of the device to the BO-culture well. However, our theoretical analysis premising only gravitational and diffusional transport (Supplementary discussion and Table S1†) indicates that gravitational effect alone is insufficient to explain limited amount of the virus in the BO-culture well. An alternative hypothesis suggest that the VB structure can physically confine the virus, hindering its migration. This aligns with the results obtained in the experiments involving IFN-related gene KO cells. With KO cells, the VB remained intact post infection and a low virus count was observed, indicating confinement of the virus by the VB (Fig. 5A–D).
The gene expression changes in the BOs and the VB indicate an innate immune response caused by SARS-CoV-2 infection. Massive upregulation of innate immune response-related genes was observed in both the BOs and the VB (Fig. 2C and F). This trend was consistent with that seen in previous studies.19,33 All genes related to innate immune response were upregulated more than 100-fold in the BOs in SARS-CoV-2 infected devices (Fig. 2C). The transcriptome analysis showed a similar trend of up-regulation of the innate immune response in the BOs against virus infection (Fig. 3B). In the VB, although the expression levels of innate immune response-related genes in the VB were significantly up-regulated with the virus, the average up-regulation was lower than in the BOs (Fig. 2F). The difference in the expression suggests that the innate immune response is greater in the BOs, which is primarily infected with the virus.
Upon infection, the VB showed morphological changes (Fig. 4A–C) including shrinking and the loss of vascular integrity (Fig. 4D and E). The orientation of the cytoskeleton structure at the single-cell level was also significantly reduced (Fig. 4F–I). Considering the expression of innate immune response genes, the low susceptibility of the VB to the virus, and these structural damages, we hypothesized that the innate immune response in the BOs stimulated endothelial cells, resulting in morphological changes of the whole VB structure due to a loss of cell–cell connections, cell death, and detachment from the vascular structure. SARS-CoV-2 infection of the vasculature has been tested with capillary organoids.34 However, our system allowed us to also observe the paracrine effects of epithelial tissue mediated by SARS-CoV-2 infection.
A detailed mechanism of the VB damage and migration of the virus was further investigated by suppressing the innate immune response. The virus copy number in the VB-culture medium was reduced when using IFNAR KO-VB or IFNB KO-BO. VB damage was also mitigated (Fig. 5A and C). These results indicate that type-I interferon-related immune processes caused VB damage and the consequent migration of the virus from the BO-culture well to the VB-culture channel. Two JAK/STAT pathway inhibitors, baricitinib and tofacitinib, reduced the virus copy number (Fig. 5C). Because JAK/STAT activation is a major signaling pathway of type-I interferons, the reduction of the virus copy number by the inhibitors further indicates that VB damage is caused by a mechanism related to type-I interferon. The degree of the virus reduction was greater with baricitinib than with tofacitinib, which can be explained by the lower effectiveness of tofacitinib on JAK1.35 These results strongly suggest that type-I interferon responses induce vasculature damage and virus migration from the BOs to the VB. Thus, the suppression of downstream molecules may have a therapeutic effect in COVID-19 patients.36–38
Protective and detrimental aspects of type-I interferon in COVID-19 have been reported. In the initial phase of COVID-19, type-I interferon plays a primary and crucial role in the innate immune response. A higher risk of severe COVID-19 in patients with interferon gene abnormalities indicates the importance of this response in suppressing further disease aggravation.39–42 On the other hand, type-I interferons have also been reported to contribute to the cytokine storm in severe COVID-19 cases.43–46 The VB damage and migration of the virus demonstrated by our system are presumably associated with the detrimental effects of type-I interferon. Similarly, COVID-19 patients with vascular symptoms show an up-regulation of type-I interferon.2,47,48
Conclusion
We established a 3D co-culture model of epithelial tissues and the vasculature to explore the mechanisms of vascular dysfunction caused by SARS-CoV-2. One limitation of this model is that it does not include immune cells, which play a key role in suppressing viral replication and eliminating infected cells. Despite this deficiency, it enabled us to selectively define the initial infection site and to evaluate the 3D morphology of the vasculature. Obtained results revealed the endothelial damage caused by a paracrine type-I interferon response. The SARS-CoV-2 MPS developed here is thus advantageous for the application of the virus to specific cell types, assays of drug effect evaluations, and the study of genetic modifications, allowing us to study the direct and indirect effects of SARS-CoV-2 infection. Further development of this model, such as the integration with immune cells, will enable us to a deeper understanding of mechanisms underlying vascular dysfunctions in COVID-19.Materials and methods
Device design
The device was composed of three layers: two layers of polydimethylsiloxane (PDMS) and a bottom glass coverslip. The top PDMS layer was made of PDMS slab equipped with a 10 mm diameter well for the BO culture (BO-culture well) (Fig. S1A and B†). The middle PDMS layer contained microfluidic channels where HUVECs were cultured to form the VB (VB-culture channel) and two channels neighbouring the VB-culture channel (media channels). The media channels and the VB-culture channel were separated by an array of pillars to define the gels inside the VB-culture channel while maintaining a supply of nutrients and oxygen from the media to the VB through the pillar gaps (Fig. S1A†). The width of the VB-culture channel and side channels were 3 mm and 1 mm, respectively. Gel inlets with a diameter of 1 mm were located at both ends of the VB-culture channel and reservoirs with a diameter of 6 mm at the ends of the media channels. The BO-culture well and VB-culture channel were connected through a 1 mm hole punched at the center of the BO-culture well. Two channels located outside the media channels were filled with EGM2. Details of the device design were previously reported.27Device fabrication
The device was fabricated by bonding two layers made of PDMS and a glass bottom layer. The top PDMS layer was formed by casting the mixture of a PDMS prepolymer and curing agent (10:1 weight ratio) in a square plastic case. The middle PDMS layer was fabricated by the soft-lithography technique. First, SU-8 3050 (Nippon KAYAKU, Tokyo, Japan) was spin-coated onto a silicon substrate. The fluidic channel pattern was then exposed and developed using an SU-8 developer (Nippon KAYAKU). The height of the channel mold was 250 μm. A 10:1 mixture of PDMS prepolymer and curing agent (DuPont Toray Specialty Materials K.K. SILPOT 184, Tokyo, Japan) was spin-coated onto the mold to form a PDMS layer of approximately 500 μm thick and incubated in a vacuum chamber for 30 min to remove bubbles captured in the pillar array pattern (Fig. S1B-1†). The bottom PDMS layer was cured overnight at 80 °C.
The BO-culture well (10 mm in diameter) was punched into the top PDMS layer to a thickness of approximately 4 mm. The top layer was then bonded to the middle layer (Fig. S1B-2†) after activating their surfaces in a plasma processing machine (Femto Science, Covance MP, Hwasoeng, Korea). The bonded PDMS layers were peeled off from the mold (Fig. S1B-3†), and gel inlets and media reservoir were punched in with a 2 mm biopsy punch (KAI, BP20F, Tokyo, Japan) and a 6 mm biopsy punch, respectively. The center of the VB-culture channel was connected to the BO-culture well by a connecting hole (1 mm in diameter) punched by the biopsy punch. The bottom side of the two bonded PDMS layers was exposed to plasma and bonded with a 24 × 24 mm cover glass (Matsunami Glass Ind. Ltd., no. 1, Osaka, Japan) (Fig. S1B-4†). The connecting hole was sealed with a polyester membrane to maintain the gel inside the VB-culture channel. To fit the BO-culture well, a porous membrane (pore size: 0.4 μm, Sterlitech, Auburn, WA, USA) was cut to a circular shape with a diameter of 6 mm using a laser engraver. The membrane was then treated with 0.5% 2-mehacryloyl-oxyethylphophorylcholine polymer (Nichiyu, MPC polymer, Tokyo, Japan) in ethanol for 5 min and rinsed twice with deionized water. The membrane was dried and placed on the connecting hole in the BO-culture well before use. The device was disinfected using UV light for more than 1 hour in a 35 mm culture dish (Iwaki, Shizuoka, Japan) just before use. Further details of the fabrication process were previously reported.27
VB formation
The VB was formed by HUVECs in ECM gels. GFP-HUVECs (Angio-Proteomie, cAP-0001, Boston, MA, USA) at passage 5–6 were subcultured in a 100 mm culture dish for 4 days until they reached confluence with EGM2 (Lonza, CC-3162, Basel, Switzerland). All experiments including GFP-HUVECs were performed under approval from the Kyoto University Recombinant DNA Experiment Safety Committee (Approval no. 220171). On the day of the gel injection, the culture dishes were rinsed with PBS after aspiration of EGM2. The cells were collected by 0.05% trypsin treatment at 37 °C in a 5% CO2 incubator for 5 min followed by neutralization with twice the volume of Dulbecco's modified Eagle's medium (DMEM, Thermo Fisher Scientific, Waltham, MA, USA). Collected cells were centrifuged for 3 min at 150g and diluted to 2.0 × 107 cells per mL with EGM2. ECM gel was prepared to obtain a final composition of 5 mg mL−1 of fibrinogen (Sigma Aldrich, F8630, St. Louis, MO, USA), 0.2 mg mL−1 of collagen type 1 (Corning, 354236, Corning, NY, USA) neutralized with 1 N NaOH to pH 7.4, and 0.15 U mL−1 aprotinin (Sigma Aldrich, A6279) after mixing with the cell suspension. The ECM gel solution was then mixed with suspended HUVECs at a 1:1 volume ratio. After adding thrombin (0.5 U mL−1) to this, 15 μL of the mixture was immediately injected into the VB-culture channel from the gel inlet (Fig. S1†). Preparation of the ECM gel, mixing with the HUVEC suspension, and injection were performed on ice to prevent gelation before injection.
After injection, the device was incubated at 37 °C in a 5% CO2 incubator for 15 minutes to solidify the gels. Media channels were then filled with EGM2 supplemented with 50 ng mL−1 vascular endothelial growth factor A (VEGFA, Thermo Fisher Scientific, PHC9391) and 50 ng mL−1 basic fibroblast growth factor (bFGF, FujiFilm Wako, 064-04541, Osaka, Japan). The devices were incubated at 37 °C and 5% CO2. Two days after the injection, additional HUVECs (10 μL of 1.0 × 106 cells per mL) were introduced into the media channels, and the device was inclined with the injected side up for 20 minutes at 37 °C and 5% CO2 to allow cells to adhere to the side of the solidified gel. The additional HUVECs connected with HUVECs in the VB-culture channel to make VB perfusable from the media channels. The media were exchanged every other day. After the HUVECs formed a vascular network, typically 4 days after the gel injection, BOs co-culture was then started as described below. During co-culture, devices were kept on rocker (Next Advance, Infinity Rocker Mini, New York, NY, USA), with 1 cycle per min to maintain VB by flowing EGM2. Up to 30 devices were handled in single batch.
BO culture
The BOs were cultured by embedding NHBEs (Lonza, CC-2540) in Matrigel growth factor reduced (GFR) basement membrane matrix (Corning, 354230). NHBEs were subcultured until they reached confluence in BEGM (Lonza, CC-3170; normally 8 days). On the start day of the organoid culture, subcultured NHBEs were collected by treatment with TrypLE Select (Thermo Fisher Scientific, 12563011) after rinsing with PBS. The cell density was adjusted to 2 × 106 cells per mL in bronchial organoid culture media (BO expansion media).31 Matrigel was mixed with the cell suspension at a 4:1 volume ratio, and 25 μL of the mixture was gently placed in the BO-culture well. The device was then incubated for 15 min at 37 °C and 5% CO2 to solidify the gel. The BO-culture well was filled with 250 μL of BO expansion medium, and half of the volume in the well was exchanged every other day. Ten ng mL−1 bFGF and 10 μM Y27632 were added to the media until day 5 after the Matrigel introduction. The diameter of the organoids reached approximately 100 μm after 10 days.
SARS-CoV-2 preparation
SARS-CoV-2 strain B.1.1.214 (EPI_ISL_2897162) was isolated using a nasopharyngeal swab sample from a COVID-19 patient. This study was approved by the research ethics committee of Kyoto University (Approval no. R2379-3). The virus was proliferated in TMPRSS2/Vero cells (JCRB Cell Bank, JCRB1818, Osaka, Japan) and stored at −80 °C.49 The cells were cultured with minimum essential media (MEM, Sigma-Aldrich) supplemented with 5% FBS and 1% penicillin/streptomycin. All experiments including virus infections were performed in a biosafety level 3 (BSL3) facility at Kyoto University strictly following regulations and guidelines for SARS-CoV-2 research prescribed by Ministry of Health, Labor and welfare and Ministry of Education, Culture, Sports, Science and Technology, Japan.SARS-CoV-2 infection experiments
The BOs and the VB were exposed to SARS-CoV-2 10 days after the organoid culture started (14 days from the start of the VB culture). After removing the media and rinsing with PBS, SARS-CoV-2 diluted to 0.1 MOI in MEM supplemented with 10% FBS was applied to the device in three conditions: both the BO-culture well and VB-culture channel (BO+VB+), only the BO-culture well (BO+VB−), and only the VB-culture channel (BO−VB+). The devices were then incubated for 2 hours. As a mock condition, the same process was implemented without the virus. Infected devices were cultured until day 4 after the infection and then used for the evaluation. All experiments with SARS-CoV-2 were carried out in the BSL3 facility at Kyoto University strictly following regulations.BO cryosection and immunostaining
Matrigel containing the BOs was removed from the device using a spatula after the infection experiments. The gel was stored in 30% sucrose in PBS and frozen in O.C.T compound (Sakura Finetek, Tissue-Tek, Tokyo, Japan). Cryosections of the BOs embedded in the compound were obtained at a thickness of 10 μm and then fixed on a slide glass. Fixed sections were blocked with 10% donkey serum and incubated with the primary antibody overnight (see Table S2† for the list of antibodies used). After fluorescently labeled secondary antibodies were applied and the samples were incubated for 1 hour at room temperature, the slide glass was washed with PBS three times. Finally, a drop of anti-fade reagent (Thermo Fisher Scientific, SlowFade Gold S36936) was added to the glass slide. The sample was covered by placing a coverslip on the glass slide and sealed with nail polish for observation under a confocal microscope (Evident, FV3000, Tokyo, Japan).RNA expression in BO and VB cells
A reverse transcription-quantitative polymerase chain reaction (RT-qPCR) was performed to evaluate gene expression levels in both BO and VB cells. The BOs and the VB were separated by removing the BO gel from the BO-culture well with a spatula. The BOs were then processed in a lysis buffer. HUVECs forming the VB were also lysed by adding lysis buffer to the inlet of the VB-culture channel. Total RNA was isolated from the cell lysates using ISOGENE (NIPPON GENE, Tokyo, Japan). cDNA was synthesized using total RNA with the Superscript VILO cDNA Synthesis Kit (Thermo Fisher Scientific). Real-time RT-PCR was performed using SYBR Green PCR Master Mix (Thermo Fisher Scientific) on a QuantStudio 1 Real-Time PCR System (Thermo Fisher Scientific). Differences in gene expression levels between the BOs and the VB were evaluated using the ratio of the degree of expression in the BOs and the VB in the two structures. Relative quantification of the target mRNA levels was performed using the 2−ΔΔCT method. The values were normalized to the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The PCR primer sequences are shown in Table S3.†Virus copy number in culture media
The viral copy number in culture media was measured using RT-qPCR. The culture media of the BOs and the VB were obtained from the BO-culture well and VB-culture channel inlets, respectively. The media were mixed with an equal volume of 2× RNA lysis buffer (distilled water containing 0.4 U μL−1 SUPERase·In Rnase Inhibitor (Thermo Fisher Scientific), 2% Triton X-100, 50 mM KCl, 100 mM Tris-HCl (pH 7.4), and 40% glycerol) and incubated at room temperature for 10 min. The mixture was diluted 10 times with distilled water. Viral RNA was quantified using the One Step TB Green PrimeScript PLUS RT-PCR Kit on the QuantStudio 1 Real-Time PCR System. Primers used in this experiment are shown in Table S3.† Standard curves were prepared using SARS-CoV-2 RNA (105 copies per μL) purchased from Nihon Gene Research Laboratories.RNA sequencing and analysis
RNA from the BOs was extracted as described above. RNA integrity was assessed using a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). The library was constructed using a TruSeq stranded mRNA sample prep kit (Illumina, San Diego, CA, USA) following the manufacturer's instructions and sequenced using Illumina NextSeq550. The fastq files were generated using bcl2fastq-2.20. Adapter sequences and low-quality bases were trimmed from the raw reads using Cutadapt ver 1.14 and mapped to human reference genome sequences (hg38) using STAR ver. 2.5.3a with the GENCODE (release 36, GRCh38.p13) gtf file.50 Raw counts were calculated using htseq-count ver. 0.12.4 using the GENCODE gtf file.51–53 Gene expression levels were calculated as transcripts per kilobase million values, and differentially expressed genes were determined using DESeq2 v1.16.1.54 A total of 154 up-regulated and 174 down-regulated genes were subjected to pathway analysis with cluster profiler using the GSEA algorithm and GO Bioprocess gene set.55 Raw data concerning this study were submitted under the Gene Expression Omnibus accession number GSE 216851.VB observation
The mock and SARS-CoV-2 devices were treated with 4% PFA to fix and passivate the virus. Fluorescent images in the GFP channel were captured using an epi-fluorescence microscope (Evident, CKX 53) equipped with a color CMOS camera (Evident, DP74). Time-lapse images of rhodamine–dextran (rhodamine B isothiocyanate–dextran: 70 kDa, Sigma-Aldrich) solution flow were captured using the FV3000. The recording was started immediately after the application of the dextran solution from one side of the device at 1 s intervals and was halted after 60 s.Immunostaining for the VB was performed after the infection experiments. The device was fixed with a 4% PFA solution to fix the proteins in the tissues and deactivate the virus. The fixed sample was washed with PBS at least three times. The device was then permeated with 0.1% Triton X-100 for 1 hour and washed with PBS five times. The VB was incubated with 10% donkey serum in PBS (blocking buffer) for 1 hour at 4 °C. DAPI and Alexa Fluor 647 labeled phalloidin (Thermo Fisher Scientific, A3010) were diluted in blocking buffer and incubated for over 1 hour at 4 °C. The VB was then thoroughly washed by rinsing with PBS five times. Fluorescent images were recorded using the FV3000.
VB morphology analysis
The area covered with the VB inside the VB-culture channel was calculated from a binarized image with a threshold value calculated as| th = avgbg + 3 × stdbg, | (1) |
The number of end points of the VB was calculated from the skeletons of the binarized VB images. For each pixel in the skeleton, eight neighboring pixels were examined if they were included in the skeleton. Pixels were judged as a blind end when only one of the neighboring pixels was in the skeleton, indicating that the pixel was located at the end of a branch of the skeleton. The number of end points was counted from three replicates of the mock and SARS-CoV-2-infected devices.
The capability of the VB to guide fluidic flow was analyzed by binarizing the fluorescent images of rhodamine–dextran. An ROI was set to enclose a pillar array defining the VB-culture channel on the opposite end to the end where the dextran solution was applied (Fig. 4D). The number of pixels whose intensity in the fluorescent dextran channel was higher than the threshold was counted as the area with dextran and normalized by the whole ROI area. The time course of the area normalized with dextran was obtained from time-lapse images for three devices per SARS-CoV-2 or mock condition.
The degree of orientation of the actin filaments was analyzed using Fourier transformation. The stained actin filament images were binarized. The degree of orientation was calculated by a 2D fast Fourier transformation (FFT) using FibrOri8s03.56 Representative images used for the orientation analysis are shown in Fig. 4G. The integral of the power spectrum along the radial axis was calculated to obtain the density of the spectrum along the angular axis. The density function along the angular axis was fitted with an ellipse, and the ratio of the major and minor axes was calculated as the actin filament orientation. This ratio was statistically tested using the data obtained from more than three SARS-CoV-2 and mock devices. All image analysis except the FFT was implemented with custom-written Python codes using Python 3.8 and scikit-image 0.18.
IFNAR2 KO HUVEC preparation
All experiments related to IFNAR2 KO HUVEC were performed under approval from the Kyoto University Recombinant DNA Experiment Safety Committee (Approval no. 230037). HUVECs (PromoCell, C-12206, Heidelberg, Germany) at passage four were harvested when they reached approximately 80% confluency. The cell pellet was washed with Opti-MEM (Thermo Fisher Scientific, 31985-062) and centrifuged at 220g for 3 min. The supernatant was aspirated, and the cell pellet was resuspended with Opti-MEM at a concentration of 1.1 × 108 cells per mL.To complex the CRISPR-Cas9 RNP, 223 pmol Alt-R S. p. Cas9 nuclease V3 (IDT 1081059, Coralville, IA, USA) and 440 pmol IFNAR2-targeting sgRNA (GCCTATGTCACCGTCCTAGA, PAM sequence AGG; IDT 105321533) were mixed (final volume of 8 μL) and incubated at room temperature for 10 min. Then, 7.2 × 106 cells (66 μL) were mixed with the CRISPR-Cas9 RNP complex and electroporated in an OC-100x2 processing assembly using the MaxCyte STX electroporation platform (MaxCyte, Gaithersburg, MD, USA) with the program Optimization 8.
After electroporation, the cells were inoculated on two 100 mm dishes in Endothelial Cell Growth Medium 2 (PromoCell) supplemented with CEPT cocktail (a combination of chroman 1 (Tocris Bioscience, Bristol, UK), emricasan (Selleck Chemicals, Munich, Germany), polyamine supplement (Sigma-Aldrich), and trans-ISRIB (Tocris Bioscience)) and cultured for 36 hours in a humidified incubator in 5% CO2 at 32 °C. On day two, the first medium change was performed, and the culture temperature was shifted to 37 °C. The medium was changed daily until day 5, when the cells were harvested.
IFNB1 KO NHBE preparation
All experiments related to IFNB1 KO NHBE were performed under approval from the Kyoto University Recombinant DNA Experiment Safety Committee (Approval no. 230037). NHBEs at passage three were harvested when they reached approximately 80% confluency. The cell pellet was washed with Opti-MEM and centrifuged at 220g for 5 min. The supernatant was aspirated, and the cell pellet was resuspended with Opti-MEM at a concentration of 1.1 × 108 cells per mL.The IFNB1-targeting CRISPR-Cas9 RNP was prepared in two steps. First, 2 nmol crRNA (IDT) and 2 nmol Alt-R CRISPR-Cas9 tracrRNA, ATTO 550 (IDT 1077024) were mixed and incubated for 10 min. Then, 550 pmol of the resulting sgRNA was mixed with 279 pmol Alt-R S. p. Cas9 nuclease V3 (final volume of 10 μL) and incubated for 10 min. Next, 1.0 × 107 (90 μL) of the resuspended cells were mixed with the CRISPR-Cas9 RNP complex, and the mixture was electroporated in an OC-100x2 processing assembly using the MaxCyte STX electroporation platform with the program Optimization 0–2. After the electroporation, the OC-100x2 processing assembly was incubated in 5% CO2 at 37 °C for 20 min for membrane recovery. Following the recovery, the electroporated cells were inoculated on two 100 mm dishes in BEGM-2 BulletKit Medium (Lonza) supplemented with CEPT, and cultured in a humidified incubator in 5% CO2 at 37 °C. On day 2, the first medium change was performed, and the medium was then changed daily until day 5 when the cells were harvested.
Insertion/deletion (indel) frequency analysis
Genomic DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany). To analyze the indel frequency in IFNAR2 KO HUVECs, targeted genomic DNA regions cleaved with CRISPR-Cas9 RNP were amplified by PCR using the following primers: (forward) CCCTCCTGGAGCAGACATTA and (reverse) ACCAAGGCTCAGAGAAGCAA. To analyze the indel frequency in IFNB1 KO NHBEs, targeted genomic DNA regions cleaved with CRISPR-Cas9 RNP were amplified by PCR using the following primers: (forward) TTTCGAAGCCTTTGCTCTGGCA and (reverse) GCCATCAGTCACTTAAACAGCATCT. PCR-amplified DNA was cleaned using ExoSAP-IT for PCR Product Clean-Up (Affymetrix, USA), and the PCR products were sequenced, and the indel frequency was evaluated with the ICE CRISPR Analysis Tool (Synthego, Menlo Park, CA, USA):57 the indel frequencies were 97% and 92% when genome editing was performed using IFNAR2-targeting sgRNA and IFNB1-targeting sgRNA, respectively.Inhibitor assay
Inhibitors were added to the device at the same time as the virus was introduced. Baricitinib (MedChemExpress, HY-15315, Monmouth Junction, NJ, USA) and tofacitinib (MedChemExpress, HY-40534) were dissolved in DMSO to stock concentrations of 10 and 2 mM, respectively. They were further diluted in EGM2 at a 1:2000 volume ratio to provide 5 and 1 μM final concentrations, respectively, immediately before the application. DMSO was added to EGM2 at a 1:2000 volume ratio as a vehicle to confirm that DMSO did not harm the co-culture system. The media in the device were exchanged with the inhibitor every other day.
Statistical analysis
Wilcoxon's rank sum test was carried out for qPCR data and Student's t-test for other data to investigate the significance of differences. All statistical tests were carried out using python 3.8 and scikit-learn 0.23.Author contributions
Conceptualization: KF, YK, KT, RY. Funding acquisition: RPK, KT, RY. Methodology: KF, YK, YN, SD, TO, KT, RY. Resources: KT, RY. Software: KF, YN. Investigation: KF, YK, YN, SD, TY, RPK, PG, YM, TO, MN, KT, RY. Visualization: KF, YK, YN. Supervision: KT, RY. Writing – original draft: KF. Writing – review & editing: KF, YK, RPK, PG, KT, RY.Conflicts of interest
P. G. is an employee of MaxCyte Inc. R. Y. is one of the founders and shareholders of Physios Biotech, Inc. The other authors have no conflicts of interest to declare. All co-authors have agreed to the publication of this manuscript.Acknowledgements
This research was supported by AMED under Grant Numbers JP21gm1610005 (TK, RY) and JP22bm0104001 (RPK), the iPS Cell Research Fund, and the COVID-19 Private Fund (to the Dr. Shinya Yamanaka laboratory, CiRA, Kyoto University). We thank our technical staff (Ms. Aki Kubo, Ms. Shiho Morimoto, and Ms. Mayumi Moriwake) for the device fabrication, cell culture, and maintenance, Dr. Yoshio Koyanagi, Dr. Takeshi Noda, Dr. Yukiko Muramoto, Ms. Naoko Misawa, and Dr. Kazuya Shimura (Kyoto University) for the setup and operation of the BSL-3 laboratory, Ms. Masami Yamashita (Kyoto University) for the genome editing, Ms. Kazusa Okita and Ms. Satoko Sakurai (Kyoto University) for technical assistance with the RNA-seq experiments, and Ms. Rina Hashimoto, Ms. Natsumi Mimura, and Ms. Ayaka Sakamoto (Kyoto University) for technical assistance with the organoid culture, qPCR analysis, and SARS-CoV-2 infection experiments. We also thank the Single-Cell Genome Information Analysis Core (SignAC) in ASHBi for the RNA sequence analysis.References
- M. Yoshida, K. B. Worlock, N. Huang, R. G. H. Lindeboom, C. R. Butler, N. Kumasaka, C. Dominguez Conde, L. Mamanova, L. Bolt, L. Richardson, K. Polanski, E. Madissoon, J. L. Barnes, J. Allen-Hyttinen, E. Kilich, B. C. Jones, A. de Wilton, A. Wilbrey-Clark, W. Sungnak, J. P. Pett, J. Weller, E. Prigmore, H. Yung, P. Mehta, A. Saleh, A. Saigal, V. Chu, J. M. Cohen, C. Cane, A. Iordanidou, S. Shibuya, A.-K. Reuschl, I. T. Herczeg, A. C. Argento, R. G. Wunderink, S. B. Smith, T. A. Poor, C. A. Gao, J. E. Dematte, NU SCRIPT Study Investigators, G. Reynolds, M. Haniffa, G. S. Bowyer, M. Coates, M. R. Clatworthy, F. J. Calero-Nieto, B. Göttgens, C. O'Callaghan, N. J. Sebire, C. Jolly, P. De Coppi, C. M. Smith, A. V. Misharin, S. M. Janes, S. A. Teichmann, M. Z. Nikolić and K. B. Meyer, Nature, 2022, 602, 321–327 CrossRef CAS PubMed.
- D. M. Smadja, S. J. Mentzer, M. Fontenay, M. A. Laffan, M. Ackermann, J. Helms, D. Jonigk, R. Chocron, G. B. Pier, N. Gendron, S. Pons, J.-L. Diehl, C. Margadant, C. Guerin, E. J. M. Huijbers, A. Philippe, N. Chapuis, P. Nowak-Sliwinska, C. Karagiannidis, O. Sanchez, P. Kümpers, D. Skurnik, A. M. Randi and A. W. Griffioen, Angiogenesis, 2021, 24, 755–788 CrossRef CAS PubMed.
- W. Chen and J. Y. Pan, Biol. Proced. Online, 2021, 23, 4 CrossRef CAS PubMed.
- A. Bonaventura, A. Vecchié, L. Dagna, K. Martinod, D. L. Dixon, B. W. Van Tassell, F. Dentali, F. Montecucco, S. Massberg, M. Levi and A. Abbate, Nat. Rev. Immunol., 2021, 21, 319–329 CrossRef CAS PubMed.
- M. Ackermann, S. E. Verleden, M. Kuehnel, A. Haverich, T. Welte, F. Laenger, A. Vanstapel, C. Werlein, H. Stark, A. Tzankov, W. W. Li, V. W. Li, S. J. Mentzer and D. Jonigk, N. Engl. J. Med., 2020, 383, 120–128 CrossRef CAS PubMed.
- I. Hamming, W. Timens, M. L. C. Bulthuis, A. T. Lely, G. J. Navis and H. van Goor, J. Pathol., 2004, 203, 631–637 CrossRef CAS PubMed.
- L. Schimmel, K. Y. Chew, C. J. Stocks, T. E. Yordanov, P. Essebier, A. Kulasinghe, J. Monkman, A. F. R. Dos Santos Miggiolaro, C. Cooper, L. de Noronha, K. Schroder, A. K. Lagendijk, L. I. Labzin, K. R. Short and E. J. Gordon, Clin. Transl. Immunol., 2021, 10, e1350 CrossRef CAS PubMed.
- I. R. McCracken, G. Saginc, L. He, A. Huseynov, A. Daniels, S. Fletcher, C. Peghaire, V. Kalna, M. Andaloussi-Mäe, L. Muhl, N. M. Craig, S. J. Griffiths, J. G. Haas, C. Tait-Burkard, U. Lendahl, G. M. Birdsey, C. Betsholtz, M. Noseda, A. H. Baker and A. M. Randi, Circulation, 2021, 143, 865–868 CrossRef CAS PubMed.
- J. Nascimento Conde, W. R. Schutt, E. E. Gorbunova and E. R. Mackow, MBio, 2020, 11, 1–7 CrossRef PubMed.
- P. K. Jadaun and S. Chatterjee, Cytokine Growth Factor Rev., 2022, 63, 69–77 CrossRef CAS PubMed.
- C. Maucourant, I. Filipovic, A. Ponzetta, S. Aleman, M. Cornillet, L. Hertwig, B. Strunz, A. Lentini, B. Reinius, D. Brownlie, A. Cuapio, E. H. Ask, R. M. Hull, A. Haroun-Izquierdo, M. Schaffer, J. Klingström, E. Folkesson, M. Buggert, J. K. Sandberg, L. I. Eriksson, O. Rooyackers, H.-G. Ljunggren, K.-J. Malmberg, J. Michaëlsson, N. Marquardt, Q. Hammer, K. Strålin, N. K. Björkström and Karolinska COVID-19 Study Group, Sci. Immunol., 2020, 5, abd6832 CrossRef PubMed.
- H. M. Al-Kuraishy, A. I. Al-Gareeb, H. A. Al-Hussaniy, N. A. H. Al-Harcan, A. Alexiou and G. E.-S. Batiha, Int. Immunopharmacol., 2022, 104, 108516 CrossRef CAS PubMed.
- Y. Zhang, X. Geng, Y. Tan, Q. Li, C. Xu, J. Xu, L. Hao, Z. Zeng, X. Luo, F. Liu and H. Wang, Biomed. Pharmacother., 2020, 127, 110195 CrossRef CAS PubMed.
- T. Hubiche, N. Cardot-Leccia, F. Le Duff, B. Seitz-Polski, P. Giordana, C. Chiaverini, V. Giordanengo, G. Gonfrier, V. Raimondi, O. Bausset, Z. Adjtoutah, M. Garnier, F. Burel-Vandenbos, B. Dadone-Montaudié, V. Fassbender, A. Palladini, J. Courjon, V. Mondain, J. Contenti, J. Dellamonica, G. Leftheriotis and T. Passeron, JAMA Dermatol., 2021, 157, 202–206 CrossRef PubMed.
- S. Meini, T. Giani and C. Tascini, Mol. Biol. Rep., 2020, 47, 8301–8304 CrossRef CAS PubMed.
- M. Ackermann, S. J. Mentzer, M. Kolb and D. Jonigk, Eur. Respir. J., 2020, 56, 03147 CrossRef PubMed.
- M. Essaidi-Laziosi, F. J. Perez Rodriguez, N. Hulo, F. Jacquerioz, L. Kaiser and I. Eckerle, Lancet Microbe, 2021, 2, e571 CrossRef CAS PubMed.
- L. Wang, X. Fan, G. Bonenfant, D. Cui, J. Hossain, N. Jiang, G. Larson, M. Currier, J. Liddell, M. Wilson, A. Tamin, J. Harcourt, J. Ciomperlik-Patton, H. Pang, N. Dybdahl-Sissoko, R. Campagnoli, P.-Y. Shi, J. Barnes, N. J. Thornburg, D. E. Wentworth and B. Zhou, Emerging Infect. Dis., 2021, 27, 1380–1392 CrossRef CAS PubMed.
- T. Yamada, S. Sato, Y. Sotoyama, Y. Orba, H. Sawa, H. Yamauchi, M. Sasaki and A. Takaoka, Nat. Immunol., 2021, 22, 820–828 CrossRef CAS PubMed.
- C. B. Morrison, C. E. Edwards, K. M. Shaffer, K. C. Araba, J. A. Wykoff, D. R. Williams, T. Asakura, H. Dang, L. C. Morton, R. C. Gilmore, W. K. O'Neal, R. C. Boucher, R. S. Baric and C. Ehre, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2119680119 CrossRef CAS PubMed.
- R. F. Nicosia, G. Ligresti, N. Caporarello, S. Akilesh and D. Ribatti, Am. J. Pathol., 2021, 191, 1374–1384 CrossRef CAS PubMed.
- R. X. Z. Lu, B. F. L. Lai, N. Rafatian, D. Gustafson, S. B. Campbell, A. Banerjee, R. Kozak, K. Mossman, S. Mubareka, K. L. Howe, J. E. Fish and M. Radisic, Lab Chip, 2022, 22, 1171–1186 RSC.
- V. V. Thacker, K. Sharma, N. Dhar, G.-F. Mancini, J. Sordet-Dessimoz and J. D. McKinney, EMBO Rep., 2021, 22, e52744 CrossRef CAS PubMed.
- M. Zhang, P. Wang, R. Luo, Y. Wang, Z. Li, Y. Guo, Y. Yao, M. Li, T. Tao, W. Chen, J. Han, H. Liu, K. Cui, X. Zhang, Y. Zheng and J. Qin, Adv. Sci., 2021, 8, 2002928 CrossRef CAS PubMed.
- L. Si, H. Bai, M. Rodas, W. Cao, C. Y. Oh, A. Jiang, R. Moller, D. Hoagland, K. Oishi, S. Horiuchi, S. Uhl, D. Blanco-Melo, R. A. Albrecht, W.-C. Liu, T. Jordan, B. E. Nilsson-Payant, I. Golynker, J. Frere, J. Logue, R. Haupt, M. McGrath, S. Weston, T. Zhang, R. Plebani, M. Soong, A. Nurani, S. M. Kim, D. Y. Zhu, K. H. Benam, G. Goyal, S. E. Gilpin, R. Prantil-Baun, S. P. Gygi, R. K. Powers, K. E. Carlson, M. Frieman, B. R. tenOever and D. E. Ingber, Nat. Biomed. Eng., 2021, 5, 815–829 CrossRef CAS PubMed.
- A. Bein, S. Kim, G. Goyal, W. Cao, C. Fadel, A. Naziripour, S. Sharma, B. Swenor, N. LoGrande, A. Nurani, V. N. Miao, A. W. Navia, C. G. K. Ziegler, J. O. Montañes, P. Prabhala, M. S. Kim, R. Prantil-Baun, M. Rodas, A. Jiang, L. O'Sullivan, G. Tillya, A. K. Shalek and D. E. Ingber, Front. Pharmacol., 2021, 12, 718484 CrossRef CAS PubMed.
- Y. Kameda, S. Chuaychob, M. Tanaka, Y. Liu, R. Okada, K. Fujimoto, T. Nakamura and R. Yokokawa, Lab Chip, 2022, 22, 641–651 RSC.
- Y. Nashimoto, T. Hayashi, I. Kunita, A. Nakamasu, Y.-S. Torisawa, M. Nakayama, H. Takigawa-Imamura, H. Kotera, K. Nishiyama, T. Miura and R. Yokokawa, Integr. Biol., 2017, 9, 506–518 CrossRef PubMed.
- J. Paek, S. E. Park, Q. Lu, K. T. Park, M. Cho, J. M. Oh, K. W. Kwon, Y. S. Yi, J. W. Song, H. I. Edelstein, J. Ishibashi, W. Yang, J. W. Myerson, R. Y. Kiseleva, P. Aprelev, E. D. Hood, D. Stambolian, P. Seale, V. R. Muzykantov and D. Huh, ACS Nano, 2019, 13, 7627–7643 CrossRef CAS PubMed.
- M. Campisi, Y. Shin, T. Osaki, C. Hajal, V. Chiono and R. D. Kamm, Biomaterials, 2018, 180, 117–129 CrossRef CAS PubMed.
- E. Sano, T. Suzuki, R. Hashimoto, Y. Itoh, A. Sakamoto, Y. Sakai, A. Saito, D. Okuzaki, D. Motooka, Y. Muramoto, T. Noda, T. Takasaki, J.-I. Sakuragi, S. Minami, T. Kobayashi, T. Yamamoto, Y. Matsumura, M. Nagao, T. Okamoto and K. Takayama, Commun. Biol., 2022, 5, 1–9 CrossRef PubMed.
- T. Yamamotoya, Y. Nakatsu, M. Kanna, S. Hasei, Y. Ohata, J. Encinas, H. Ito, T. Okabe, T. Asano and T. Sakaguchi, Sci. Rep., 2021, 11, 18581 CrossRef CAS PubMed.
- H. M. Al-Kuraishy, A. I. Al-Gareeb, H. A. Al-Hussaniy, N. A. H. Al-Harcan, A. Alexiou and G. E.-S. Batiha, Int. Immunopharmacol., 2022, 104, 108516 CrossRef CAS PubMed.
- V. Monteil, H. Kwon, P. Prado, A. Hagelkrüys, R. A. Wimmer, M. Stahl, A. Leopoldi, E. Garreta, C. Hurtado del Pozo, F. Prosper, J. P. Romero, G. Wirnsberger, H. Zhang, A. S. Slutsky, R. Conder, N. Montserrat, A. Mirazimi and J. M. Penninger, Cell, 2020, 181, 905–913.e7 CrossRef CAS PubMed.
- I. B. McInnes, N. L. Byers, R. E. Higgs, J. Lee, W. L. Macias, S. Na, R. A. Ortmann, G. Rocha, T. P. Rooney, T. Wehrman, X. Zhang, S. H. Zuckerman and P. C. Taylor, Arthritis Res. Ther., 2019, 21, 183 CrossRef PubMed.
- P. Richardson, I. Griffin, C. Tucker, D. Smith, O. Oechsle, A. Phelan, M. Rawling, E. Savory and J. Stebbing, Lancet, 2020, 395, e30–e31 CrossRef CAS PubMed.
- S. Satarker, A. A. Tom, R. A. Shaji, A. Alosious, M. Luvis and M. Nampoothiri, Postgrad. Med., 2021, 133, 489–507 CrossRef CAS PubMed.
- J. Stebbing, G. Sánchez Nievas, M. Falcone, S. Youhanna, P. Richardson, S. Ottaviani, J. X. Shen, C. Sommerauer, G. Tiseo, L. Ghiadoni, A. Virdis, F. Monzani, L. R. Rizos, F. Forfori, A. Avendaño Céspedes, S. De Marco, L. Carrozzi, F. Lena, P. M. Sánchez-Jurado, L. G. Lacerenza, N. Cesira, D. Caldevilla Bernardo, A. Perrella, L. Niccoli, L. S. Méndez, D. Matarrese, D. Goletti, Y.-J. Tan, V. Monteil, G. Dranitsaris, F. Cantini, A. Farcomeni, S. Dutta, S. K. Burley, H. Zhang, M. Pistello, W. Li, M. M. Romero, F. Andrés Pretel, R. S. Simón-Talero, R. García-Molina, C. Kutter, J. H. Felce, Z. F. Nizami, A. G. Miklosi, J. M. Penninger, F. Menichetti, A. Mirazimi, P. Abizanda and V. M. Lauschke, Sci. Adv., 2021, 7, eabe4724 CrossRef CAS PubMed.
- P. Bastard, L. B. Rosen, Q. Zhang, E. Michailidis, H.-H. Hoffmann, Y. Zhang, K. Dorgham, Q. Philippot, J. Rosain, V. Béziat, J. Manry, E. Shaw, L. Haljasmägi, P. Peterson, L. Lorenzo, L. Bizien, S. Trouillet-Assant, K. Dobbs, A. A. de Jesus, A. Belot, A. Kallaste, E. Catherinot, Y. Tandjaoui-Lambiotte, J. Le Pen, G. Kerner, B. Bigio, Y. Seeleuthner, R. Yang, A. Bolze, A. N. Spaan, O. M. Delmonte, M. S. Abers, A. Aiuti, G. Casari, V. Lampasona, L. Piemonti, F. Ciceri, K. Bilguvar, R. P. Lifton, M. Vasse, D. M. Smadja, M. Migaud, J. Hadjadj, B. Terrier, D. Duffy, L. Quintana-Murci, D. van de Beek, L. Roussel, D. C. Vinh, S. G. Tangye, F. Haerynck, D. Dalmau, J. Martinez-Picado, P. Brodin, M. C. Nussenzweig, S. Boisson-Dupuis, C. Rodríguez-Gallego, G. Vogt, T. H. Mogensen, A. J. Oler, J. Gu, P. D. Burbelo, J. I. Cohen, A. Biondi, L. R. Bettini, M. D'Angio, P. Bonfanti, P. Rossignol, J. Mayaux, F. Rieux-Laucat, E. S. Husebye, F. Fusco, M. V. Ursini, L. Imberti, A. Sottini, S. Paghera, E. Quiros-Roldan, C. Rossi, R. Castagnoli, D. Montagna, A. Licari, G. L. Marseglia, X. Duval, J. Ghosn, HGID Lab, NIAID-USUHS Immune Response to COVID Group, COVID Clinicians, COVID-STORM Clinicians, Imagine COVID Group, French COVID Cohort Study Group, Milieu Intérieur Consortium, CoV-Contact Cohort, Amsterdam UMC Covid-19 Biobank, COVID Human Genetic Effort, J. S. Tsang, R. Goldbach-Mansky, K. Kisand, M. S. Lionakis, A. Puel, S.-Y. Zhang, S. M. Holland, G. Gorochov, E. Jouanguy, C. M. Rice, A. Cobat, L. D. Notarangelo, L. Abel, H. C. Su and J.-L. Casanova, Science, 2020, 370, abd4585 CrossRef PubMed.
- Q. Zhang, P. Bastard, Z. Liu, J. Le Pen, M. Moncada-Velez, J. Chen, M. Ogishi, I. K. D. Sabli, S. Hodeib, C. Korol, J. Rosain, K. Bilguvar, J. Ye, A. Bolze, B. Bigio, R. Yang, A. A. Arias, Q. Zhou, Y. Zhang, F. Onodi, S. Korniotis, L. Karpf, Q. Philippot, M. Chbihi, L. Bonnet-Madin, K. Dorgham, N. Smith, W. M. Schneider, B. S. Razooky, H.-H. Hoffmann, E. Michailidis, L. Moens, J. E. Han, L. Lorenzo, L. Bizien, P. Meade, A.-L. Neehus, A. C. Ugurbil, A. Corneau, G. Kerner, P. Zhang, F. Rapaport, Y. Seeleuthner, J. Manry, C. Masson, Y. Schmitt, A. Schlüter, T. Le Voyer, T. Khan, J. Li, J. Fellay, L. Roussel, M. Shahrooei, M. F. Alosaimi, D. Mansouri, H. Al-Saud, F. Al-Mulla, F. Almourfi, S. Z. Al-Muhsen, F. Alsohime, S. Al Turki, R. Hasanato, D. van de Beek, A. Biondi, L. R. Bettini, M. D'Angio, P. Bonfanti, L. Imberti, A. Sottini, S. Paghera, E. Quiros-Roldan, C. Rossi, A. J. Oler, M. F. Tompkins, C. Alba, I. Vandernoot, J.-C. Goffard, G. Smits, I. Migeotte, F. Haerynck, P. Soler-Palacin, A. Martin-Nalda, R. Colobran, P.-E. Morange, S. Keles, F. Çölkesen, T. Ozcelik, K. K. Yasar, S. Senoglu, Ş. N. Karabela, C. Rodríguez-Gallego, G. Novelli, S. Hraiech, Y. Tandjaoui-Lambiotte, X. Duval, C. Laouénan, COVID-STORM Clinicians, COVID Clinicians, Imagine COVID Group, French COVID Cohort Study Group, CoV-Contact Cohort, Amsterdam UMC Covid-19 Biobank, COVID Human Genetic Effort, NIAID-USUHS/TAGC COVID Immunity Group, A. L. Snow, C. L. Dalgard, J. D. Milner, D. C. Vinh, T. H. Mogensen, N. Marr, A. N. Spaan, B. Boisson, S. Boisson-Dupuis, J. Bustamante, A. Puel, M. J. Ciancanelli, I. Meyts, T. Maniatis, V. Soumelis, A. Amara, M. Nussenzweig, A. García-Sastre, F. Krammer, A. Pujol, D. Duffy, R. P. Lifton, S.-Y. Zhang, G. Gorochov, V. Béziat, E. Jouanguy, V. Sancho-Shimizu, C. M. Rice, L. Abel, L. D. Notarangelo, A. Cobat, H. C. Su and J.-L. Casanova, Science, 2020, 370, abdl4340 Search PubMed.
- J. Hadjadj, N. Yatim, L. Barnabei, A. Corneau, J. Boussier, N. Smith, H. Péré, B. Charbit, V. Bondet, C. Chenevier-Gobeaux, P. Breillat, N. Carlier, R. Gauzit, C. Morbieu, F. Pène, N. Marin, N. Roche, T.-A. Szwebel, S. H. Merkling, J.-M. Treluyer, D. Veyer, L. Mouthon, C. Blanc, P.-L. Tharaux, F. Rozenberg, A. Fischer, D. Duffy, F. Rieux-Laucat, S. Kernéis and B. Terrier, Science, 2020, 369, 718–724 CrossRef CAS PubMed.
- P. Bastard, A. Gervais, T. Le Voyer, J. Rosain, Q. Philippot, J. Manry, E. Michailidis, H.-H. Hoffmann, S. Eto, M. Garcia-Prat, L. Bizien, A. Parra-Martínez, R. Yang, L. Haljasmägi, M. Migaud, K. Särekannu, J. Maslovskaja, N. de Prost, Y. Tandjaoui-Lambiotte, C.-E. Luyt, B. Amador-Borrero, A. Gaudet, J. Poissy, P. Morel, P. Richard, F. Cognasse, J. Troya, S. Trouillet-Assant, A. Belot, K. Saker, P. Garçon, J. G. Rivière, J.-C. Lagier, S. Gentile, L. B. Rosen, E. Shaw, T. Morio, J. Tanaka, D. Dalmau, P.-L. Tharaux, D. Sene, A. Stepanian, B. Megarbane, V. Triantafyllia, A. Fekkar, J. R. Heath, J. L. Franco, J.-M. Anaya, J. Solé-Violán, L. Imberti, A. Biondi, P. Bonfanti, R. Castagnoli, O. M. Delmonte, Y. Zhang, A. L. Snow, S. M. Holland, C. Biggs, M. Moncada-Vélez, A. A. Arias, L. Lorenzo, S. Boucherit, B. Coulibaly, D. Anglicheau, A. M. Planas, F. Haerynck, S. Duvlis, R. L. Nussbaum, T. Ozcelik, S. Keles, A. A. Bousfiha, J. El Bakkouri, C. Ramirez-Santana, S. Paul, Q. Pan-Hammarström, L. Hammarström, A. Dupont, A. Kurolap, C. N. Metz, A. Aiuti, G. Casari, V. Lampasona, F. Ciceri, L. A. Barreiros, E. Dominguez-Garrido, M. Vidigal, M. Zatz, D. van de Beek, S. Sahanic, I. Tancevski, Y. Stepanovskyy, O. Boyarchuk, Y. Nukui, M. Tsumura, L. Vidaur, S. G. Tangye, S. Burrel, D. Duffy, L. Quintana-Murci, A. Klocperk, N. Y. Kann, A. Shcherbina, Y.-L. Lau, D. Leung, M. Coulongeat, J. Marlet, R. Koning, L. F. Reyes, A. Chauvineau-Grenier, F. Venet, G. Monneret, M. C. Nussenzweig, R. Arrestier, I. Boudhabhay, H. Baris-Feldman, D. Hagin, J. Wauters, I. Meyts, A. H. Dyer, S. P. Kennelly, N. M. Bourke, R. Halwani, N. S. Sharif-Askari, K. Dorgham, J. Sallette, S. M. Sedkaoui, S. AlKhater, R. Rigo-Bonnin, F. Morandeira, L. Roussel, D. C. Vinh, S. R. Ostrowski, A. Condino-Neto, C. Prando, A. Bonradenko, A. N. Spaan, L. Gilardin, J. Fellay, S. Lyonnet, K. Bilguvar, R. P. Lifton, S. Mane, HGID Lab, COVID Clinicians, COVID-STORM Clinicians, NIAID Immune Response to COVID Group, NH-COVAIR Study Group, Danish CHGE, Danish Blood Donor Study, St. James's Hospital, SARS CoV2 Interest group, French COVID Cohort Study Group, Imagine COVID-Group, Milieu Intérieur Consortium, CoV-Contact Cohort, Amsterdam UMC Covid-19, Biobank Investigators, COVID Human Genetic Effort, CONSTANCES cohort, 3C-Dijon Study, Cerba Health-Care, Etablissement du Sang study group, M. S. Anderson, B. Boisson, V. Béziat, S.-Y. Zhang, E. Vandreakos, O. Hermine, A. Pujol, P. Peterson, T. H. Mogensen, L. Rowen, J. Mond, S. Debette, X. de Lamballerie, X. Duval, F. Mentré, M. Zins, P. Soler-Palacin, R. Colobran, G. Gorochov, X. Solanich, S. Susen, J. Martinez-Picado, D. Raoult, M. Vasse, P. K. Gregersen, L. Piemonti, C. Rodríguez-Gallego, L. D. Notarangelo, H. C. Su, K. Kisand, S. Okada, A. Puel, E. Jouanguy, C. M. Rice, P. Tiberghien, Q. Zhang, A. Cobat, L. Abel and J.-L. Casanova, Sci. Immunol., 2021, 6, abl4340 CrossRef PubMed.
- L. Frumholtz, J.-D. Bouaziz, M. Battistella, J. Hadjadj, R. Chocron, D. Bengoufa, H. Le Buanec, L. Barnabei, S. Meynier, O. Schwartz, L. Grzelak, N. Smith, B. Charbit, D. Duffy, N. Yatim, A. Calugareanu, A. Philippe, C. L. Guerin, B. Joly, V. Siguret, L. Jaume, H. Bachelez, M. Bagot, F. Rieux-Laucat, S. Maylin, J. Legoff, C. Delaugerre, N. Gendron, D. M. Smadja, C. Cassius and Saint-Louis CORE (COvid REsearch), Br. J. Dermatol., 2021, 185, 1176–1185 CrossRef CAS PubMed.
- M. A. Akamatsu, J. T. de Castro, C. Y. Takano and P. L. Ho, EBioMedicine, 2021, 73, 103642 CrossRef CAS PubMed.
- J. S. Lee and E.-C. Shin, Nat. Rev. Immunol., 2020, 20, 585–586 CrossRef CAS PubMed.
- Y.-M. Kim and E.-C. Shin, Exp. Mol. Med., 2021, 53, 750–760 CrossRef CAS PubMed.
- J. Di Domizio, M. F. Gulen, F. Saidoune, V. V. Thacker, A. Yatim, K. Sharma, T. Nass, E. Guenova, M. Schaller, C. Conrad, C. Goepfert, L. De Leval, C. von Garnier, S. Berezowska, A. Dubois, M. Gilliet and A. Ablasser, Nature, 2022, 621, 14151 Search PubMed.
- L. Perico, A. Benigni, F. Casiraghi, L. F. P. Ng, L. Renia and G. Remuzzi, Nat. Rev. Nephrol., 2021, 17, 46–64 CrossRef PubMed.
- S. Matsuyama, N. Nao, K. Shirato, M. Kawase, S. Saito, I. Takayama, N. Nagata, T. Sekizuka, H. Katoh, F. Kato, M. Sakata, M. Tahara, S. Kutsuna, N. Ohmagari, M. Kuroda, T. Suzuki, T. Kageyama and M. Takeda, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 7001–7003 CrossRef CAS PubMed.
- M. Martin, EMBnet J., 2011, 17, 10–12 CrossRef.
- A. Dobin, C. A. Davis, F. Schlesinger, J. Drenkow, C. Zaleski, S. Jha, P. Batut, M. Chaisson and T. R. Gingeras, Bioinformatics, 2013, 29, 15–21 CrossRef CAS PubMed.
- A. Frankish, M. Diekhans, I. Jungreis, J. Lagarde, J. E. Loveland, J. M. Mudge, C. Sisu, J. C. Wright, J. Armstrong, I. Barnes, A. Berry, A. Bignell, C. Boix, S. Carbonell Sala, F. Cunningham, T. Di Domenico, S. Donaldson, I. T. Fiddes, C. García Girón, J. M. Gonzalez, T. Grego, M. Hardy, T. Hourlier, K. L. Howe, T. Hunt, O. G. Izuogu, R. Johnson, F. J. Martin, L. Martínez, S. Mohanan, P. Muir, F. C. P. Navarro, A. Parker, B. Pei, F. Pozo, F. C. Riera, M. Ruffier, B. M. Schmitt, E. Stapleton, M.-M. Suner, I. Sycheva, B. Uszczynska-Ratajczak, M. Y. Wolf, J. Xu, Y. T. Yang, A. Yates, D. Zerbino, Y. Zhang, J. S. Choudhary, M. Gerstein, R. Guigó, T. J. P. Hubbard, M. Kellis, B. Paten, M. L. Tress and P. Flicek, Nucleic Acids Res., 2021, 49, D916–D923 CrossRef CAS PubMed.
- G. H. Putri, S. Anders, P. T. Pyl, J. E. Pimanda and F. Zanini, Bioinformatics, 2022, 38, 2943–2945 CrossRef CAS PubMed.
- M. I. Love, W. Huber and S. Anders, Genome Biol., 2014, 15, 550 CrossRef PubMed.
- T. Wu, E. Hu, S. Xu, M. Chen, P. Guo, Z. Dai, T. Feng, L. Zhou, W. Tang, L. Zhan, X. Fu, S. Liu, X. Bo and G. Yu, Innovation, 2021, 2, 100141 CAS.
- T. Enomae, Y.-H. Han and A. Isogai, Nord. Pulp Pap. Res. J., 2006, 21, 253–259 CrossRef CAS.
- D. Conant, T. Hsiau, N. Rossi, J. Oki, T. Maures, K. Waite, J. Yang, S. Joshi, R. Kelso, K. Holden, B. L. Enzmann and R. Stoner, CRISPR J., 2022, 5, 123–130 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lc00768e |
| This journal is © The Royal Society of Chemistry 2024 |