
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembled and perfusable microvasculature-on-chip for modeling leukocyte trafficking†
Elisabeth
Hirth‡
a,
Wuji
Cao‡
 a,
Marina
Peltonen‡
a,
Edo
Kapetanovic
a,
Claudius
Dietsche
a,
Sara
Svanberg
a,
Maria
Filippova
b,
Sai
Reddy
a and
Petra S.
Dittrich
*a
a,
Marina
Peltonen‡
a,
Edo
Kapetanovic
a,
Claudius
Dietsche
a,
Sara
Svanberg
a,
Maria
Filippova
b,
Sai
Reddy
a and
Petra S.
Dittrich
*a
aDepartment of Biosystems Science and Engineering, ETH Zurich, 4056, Basel, Switzerland. E-mail: petra.dittrich@bsse.ethz.ch
bDepartment of Biomedicine, University of Basel, 4031 Basel, Switzerland
First published on 22nd November 2023
Abstract
Leukocyte recruitment from blood to tissue is a process that occurs at the level of capillary vessels during both physiological and pathological conditions. This process is also relevant for evaluating novel adoptive cell therapies, in which the trafficking of therapeutic cells such as chimeric antigen receptor (CAR)-T cells throughout the capillaries of solid tumors is important. Local variations in blood flow, mural cell concentration, and tissue stiffness contribute to the regulation of capillary vascular permeability and leukocyte trafficking throughout the capillary microvasculature. We developed a platform to mimic a biologically functional human arteriole–venule microcirculation system consisting of pericytes (PCs) and arterial and venous primary endothelial cells (ECs) embedded within a hydrogel, which self-assembles into a perfusable, heterogeneous microvasculature. Our device shows a preferential association of PCs with arterial ECs that drives the flow-dependent formation of microvasculature networks. We show that PCs stimulate basement membrane matrix synthesis, which affects both vessel diameter and permeability in a manner correlating with the ratio of ECs to PCs. Moreover, we demonstrate that hydrogel concentration can affect capillary morphology but has no observed effect on vascular permeability. The biological function of our capillary network was demonstrated using an inflammation model, where significantly higher expression of cytokines, chemokines, and adhesion molecules was observed after tumor necrosis factor-alpha (TNF-α) treatment. Accordingly, T cell adherence and transendothelial migration were significantly increased in the immune-activated state. Taken together, our platform allows the generation of a perfusable microvasculature that recapitulates the structure and function of an in vivo capillary bed that can be used as a model for developing potential immunotherapies.
Introduction
The capillary microvasculature is the smallest unit of the vascular system, connecting the terminal arterioles to the postcapillary venules.1 Exchange of solutes and gas between tissue and blood circulation occurs through this dense tight network through meticulous regulation of the microvascular permeability. The capillary is also highly regenerative; new blood vessel formation is essential to cure injuries and lesions but is also involved in diseases such as cancer where solid tumor growth is tied to enhanced vascularization.2,3 On the other hand, thorough vascularization enables drug administration or immune cell infiltration for clearance of the solid tumor.4 Thus, understanding the mechanisms regulating vessel permeability in health and disease can be beneficial for drug development.5Conventional 2D models often lack the complexity of a 3D environment, blood flow, cell interactions, and the proper extracellular matrix (ECM), all of which are crucial for vascular formation and functionality.6,7 Animal studies can provide some context but are limited due to species differences, making it challenging to accurately predict human physiological responses.8 Integrating these essential factors into microfluidics-based devices enables researchers to study human microvasculature under more physiologically relevant conditions. A specific application of microfluidics is the “vessel-on-a-chip”, which facilitates the creation of in vitro models of the microvasculature. These microfluidic systems often consist of a 3D hydrogel flanked by channels through which cells,9 drugs,10 angiogenic growth factors, or other compounds11 can be introduced. By applying various ECM and cell composition, vessel-on-chips were used to examine mechanical12,13 or chemical stimuli14 on endothelial cells (ECs) or vascularization of tissues such as tumors.9 Collagen15 and fibrin16 are among the most common hydrogels, the latter being of special relevance since it promotes self-assembly of the microvasculature from ECs and stromal cells embedded in the hydrogel by stimulating the secretion of basement membrane proteins.17–22 These 3D microfluidic models are particularly useful for studying the onset and progression of diseases affecting the vascular system, where traditional 2D in vitro systems struggle to replicate vessel function and architecture.23
Despite the advantages, current microfluidics-based platforms still lack important features to fully emulate the complexity of the microvasculature.19,24 For instance, many studies employ only one type of ECs, most commonly human umbilical cord vein ECs (HUVECs), which do not capture the endothelial subtype diversity in vivo. In addition, pericytes (PCs) are supportive mural cells that have been shown to affect vessel morphology and function, influencing vascular leakage and endothelial hyperplasia, both linked to various pathophysiologies.25,26 Many in vitro studies have focused solely on ECs, resulting in significant differences from in vivo blood vessels in terms of structure and function.19,27 Efforts to improve in vitro vascular systems have involved culturing perivascular cells and ECs together to better mimic in vivo conditions.28–30 Recent studies have highlighted the importance of PCs in stabilizing microvascular networks for long-term cultivation and protecting endothelial integrity.31 However, much remains to be explored about the relationship between EC–PC interactions, and evidence for this relationship is still inconclusive.
Recent studies suggest that fluid forces play a significant role as integrated signals for angiogenesis and vascular remodeling.32–34 For instance, interstitial flow (IF) has been found to play a critical role in both pathological and physiological angiogenesis, generating gradients of angiogenic factors that stimulate and guide vessel sprouting.32,33,35,36 Other studies have focused on a variety of parameters including mural cell types and ratios, matrix stiffness, and exogenous growth factors in the resulting morphology of self-assembled microvasculature.19,28,37–39 Nonetheless, a systematic and comprehensive study on various parameters such as ECM stiffness, varying flow conditions including interstitial flow and peristaltic flow, and PC cell density on the endothelial barrier properties of capillary microvasculature on-chip has not been described.
A key biological process involving the capillary microvasculature is the leukocyte recruitment cascade during tissue inflammatory response. The endothelium upregulates adhesion molecules in response to inflammatory cytokines released by tissue-resident macrophages such as tumor necrosis factor-alpha (TNF-α), and the mechanosignaling between circulating leukocytes and ECs ultimately results in the extravasation of leukocytes into the affected tissue space.40–42 In the context of cancer, trafficking of lymphocytes to tumors is critical for cancer immunotherapy with vaccines, immune checkpoint blockade, or adaptive T cell therapy. Thus, an in vitro model of a vascularized tumor for preclinical testing of the efficacy of cell therapies would be critical. Conventional in vitro models have employed one-dimensional linear or tubular models, where a gradient of inflammatory cytokines is applied across an ECM-mimicking hydrogel covered by a monolayer of ECs.43–45 While these models may excel at visualizing the leukocyte transmigration cascade of capture, arrest, rolling, and extravasation across an endothelial barrier, the simple linear or tubular structure does not fully recapitulate the complex 3D morphologies of in vivo capillaries. Additionally, these on-chip vasculature models lack the integration of PCs which is a key component of the capillary architecture and function.
To address these challenges, we have developed a microfluidic capillary microvasculature recapitulating several key cellular and functional aspects of the human capillary bed. By incorporating a heterogeneous mix of arterial and venous ECs, this study aims to investigate the specific interactions between PCs and ECs that drive the vasculogenic self-assembly of a microvascular network. Additionally, we aim to determine the effects of flow and hydrogel properties on vessel morphology and function by engineering microvascular networks from co-cultures of ECs and PCs. We hypothesize that different flow profiles, EC–PC cell ratios, and hydrogel stiffness properties will influence vessel morphology and endothelial barrier functionality. Lastly, this study evaluates the physiological function of the heterogeneous capillary microvasculature by mimicking the leukocyte recruitment and extravasation cascade. Together, this will inform future studies using vessel-on-a-chip platforms to consider these varying factors that can affect the biological properties of the vasculature.
Results and discussion
Flow-driven formation of pericyte-covered arteriole–venule capillary network
The microfluidic device consisting of three parallel channels separated by arrays of micropillars (Fig. 1A–C) was adapted from previously described methods and manufactured using PDMS.46 Hydrophobicity of the PDMS micropillar array surfaces allows for confinement of a cell-laden fibrin matrix in the central channel via surface tension.47 Fig. SI1† displays the workflow for chip seeding to form microvascular networks in vitro. To represent the cell composition of the microcirculation capillaries, GFP-HUAECs, RFP-HUVECs, and hPC-PL were mixed in a defined ratio with fibrinogen and thrombin and injected into the central channel. HUAECs and HUVECs were each seeded into one of the side channels, such that they line the walls of the two channels and fully cover the gel-channel interface, mimicking the larger arteriole and venule vessels that join the microvessels in the central gel channel into an interconnected network. High concentration of VEGF (50 ng ml−1) was supplemented to the endothelial growth medium during the first two days of chip culture to stimulate the vasculogenic self-organization of ECs and PCs, resulting in a perfusable capillary network forming after 4 days (Fig. 2A and brightfield images in Fig. SI2A–C†). Immunostaining with anti-human neuron-glial antigen 2 (NG2) revealed PCs closely associating with the capillary network, extending along the length of the microvessels and wrapping around, recapitulating the in vivo spatial organization (Fig. 2A and B). In addition, vascular endothelial (VE)-cadherin is present along the entire vessel indicating a proper endothelial barrier (Fig. SI2D and E†). | ||
| Fig. 1 Microvascular network formation on a microfluidic device. (A) Schematic illustration of blood vessels forming an intricate network of arteriole- and venule-like vessels formed by endothelial cells (HUAECs and HUVECs, respectively) with pericytes (PCs) residing along the vessels. T cells circulate in blood vessels and extravasate into the perivascular space upon being activated, i.e., in the presence of the protein ICAM-1 during the endothelial activation state. To emulate the microvasculature, we chose a microfluidic device with a central, fibrin-hydrogel filled chamber for cell co-culture and side channels, separated by an array of pillars, for culturing arteriole-like and venule-like vessels separately. The timeline of cell culture is depicted in the right illustration. (B) Photograph of the microfluidic device depicting the hydrogel region (filled with red dye) and side channels (blue dye). (C) Experimental setup during peristaltic flow condition. Syringes connected to the inlets deliver medium to the chip. Outlets are connected to a peristaltic pump which deposits the medium into tubes located outside of the pump. Up to 8 devices can be handled in parallel on one peristaltic pump. We used up to 16 devices in parallel. (D) Schematic overview of flow conditions used in the study: (1) peristaltic flow with either parallel flow or counterflow, (2) hydrostatic pressure-driven interstitial flow, and (3) static condition. Figure partially made with https://BioRender.com. | ||
 | ||
| Fig. 2 Peristaltic and interstitial flow promote the formation of microvascular networks. (A) Fluorescence image displaying NG2-stained PCs (red) residing on the endothelial barrier formed by HUAECs (green) and HUVECs (yellow) on day 6. Scale bar: 20 μm. (B) Cross-sectional micrographs and illustrative visualization capturing PC-enveloped vessels. Scale bar: 20 μm. (C) Fluorescence images (10× and 20× magnification) of the vessels consisting of HUAECs (green), HUVECs (yellow), and PCs (stained for actin) for different flow conditions. Scale bar: 50 μm. Fig. 2B partially made with https://BioRender.com. | ||
Although we included both arterial and venous ECs in our system, we did not observe any preferential association within the EC subtypes, and both types of ECs integrated evenly with each other in the resulting capillaries forming heterogeneous vessels consisting of both cell types (Fig. SI2E†). Nonetheless, we did not observe venous ECs migrating into the arteriole channel or arterial ECs migrating into the venule channel. Commercially available primary ECs were used for our study, but since EC identity can be fluid depending on their microenvironment, extensive transcriptomic profiling of the ECs prior to and after on-chip culture would be required to confirm their subtype identity and characterize any changes that may occur during 3D co-culture.
The significance of flow in the formation of microvascular networks has been shown in previous studies.33,35,36 Consequently, we were interested in exploring how diverse flow conditions affect the vascular structure in our heterogeneous vessel network. To systematically examine the influence of flow during in vitro vessel formation, microcapillary chips were cultured under three conditions: peristaltic flow applied at a constant rate of 60 μl h−1 to the arteriole/venule channels, 30 mmH2O hydrostatic pressure-driven interstitial flow across the hydrogel channel, and static culture, where medium was replenished every 24 hours (Fig. 1D). By day 6, when the vascular lumen network had become interconnected and capable of perfusion, distinct morphological variances were observed in maximum intensity projection images obtained through confocal microscopy (Fig. 2C). Both interstitial flow and peristaltic flow visibly promoted the formation of a dense microvascular network with interconnected lumen, while a sparse network with disconnected lumen was observed under static culture. To characterize the vessels and their structure, the average diameter of the networks was measured and compared to networks formed without flow. We observed that the presence of flow resulted in significantly larger vessel diameters in comparison to vessels formed under static conditions. This is generally consistent within varying conditions of PC-to-EC ratios and fibrinogen concentrations (see sections below and Fig. 4D and 5B). Our results corroborate with an earlier study showing that interstitial flow has significant effects in early vasculogenesis, forming vessels with higher interconnectivity and larger diameter.48 After a perfusable network is formed, the forces experienced by ECs in the system transition from interstitial flow to intraluminal flow, where the shear stress exerted on the vascular wall contributes to vessel modeling and increased diameter.
Perfusability serves as a crucial indicator when assessing the quality of engineered microvascular networks. To evaluate perfusability between the arteriole and venule channels, fluorescently tagged dextran and 1 μm FITC-labeled polystyrene beads were introduced into the venous channel by applying hydrostatic pressure between the inlet and outlet of the side channel (Fig. SI3†). In capillary networks cultured under both interstitial and peristaltic flow, the introduced beads were observed traversing through the network to the corresponding arteriole channel without entering the perivascular space (Fig. SI3C, Videos SI1 and SI2†). No perfusable networks were formed under static conditions, thus beads could not be perfused across the hydrogel channel.
Formation of the perfusable arteriole–capillary–venule network occurs through a multistep process: 1) vasculogenesis-like self-assembly of ECs and PCs in the hydrogel forming a capillary network, 2) angiogenic sprouting of ECs lining the arteriole/venule side channels into the capillary-containing hydrogel, and 3) anastomosis between the vasculogenic capillary and angiogenic sprouts forming an interconnected network.49 In all three flow conditions, a high concentration of VEGF (50 ng ml−1) was supplied to stimulate vasculogenesis in the hydrogel channel, but no VEGF gradient was applied across the side channels. While network-like structures in the hydrogel channel were observed in the static condition, the lack of interstitial flow resulted in minimal sprouting of ECs from the side channels and limited anastomosis with the capillary networks, leading to poor perfusability compared to the interstitial flow condition. This result suggests that interstitial flow alone can stimulate angiogenic sprouting independent of a VEGF gradient, as observed before.32 Interestingly, in the peristaltic flow condition, flow was applied parallelly in the arteriole and venule channels with equal pressure, thus no pressure difference was applied across the hydrogel channel. Nonetheless, we observed for this condition an equally interconnected network as in the interstitial flow condition.
In in vivo circulation, blood flows from the heart through arteries to the arterioles where it enters the capillaries, then passes through and joins the flow back to the heart in venules and veins, i.e., the arteriole and venule have a counterflow profile. Therefore, we hypothesized that applying a parallel- or counterflow profile in the side channels can induce distinct interstitial flow profiles within the hydrogel, which may influence the morphology of the microvasculature formed within. By employing a COMSOL Multiphysics® simulation model that mimics the fluid velocity and hydrogel properties in our device, we observed that a parallel flow profile in the side channels produced a slow flow within the hydrogel that is also parallel to the flow direction, whereas a counterflow profile generated opposing interstitial flows that intersects within the hydrogel, as well as increased flow rates along the hydrogel channel (Fig. SI5A and B†). We experimentally applied the two different flow profiles and imaged the network after 6 days of culture under perfusion. We expected differences in the average angle of vessel orientation between parallel- and counterflow conditions, e.g., aligning with the applied flow profile, but image analysis did not reveal a significant difference (Fig. SI5C and D†). However, an increase in the average vessel diameter could be observed in the counterflow condition with visibly higher cell density (Fig. SI5E†). The flow pattern with higher flow rates generated in the hydrogel under the counterflow condition drives EC migration and vascular remodeling in the well-perfused regions, which has been demonstrated in a variety of in vitro and in vivo models.50
The role of pericytes in the formation of the arteriol–venule capillary network
We then tested various cell combinations in the hydrogel channel to gain insight into each of their roles in the capillary network self-assembly process. We seeded only one of the arterial and venous ECs with PCs in a 12.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (AP, VP), as well as a 6.25:6.25:1 mixture of HUAECs, HUVECs and hPC-PLs (AVP). For all three conditions, we compared static with perfusion culture. The medium was spiked with VEGF (50 ng ml−1) in the first 2 days to induce vasculogenesis before reverting to the normal concentration of 5 ng ml−1 for the next 4 days.
:1 ratio (AP, VP), as well as a 6.25:6.25:1 mixture of HUAECs, HUVECs and hPC-PLs (AVP). For all three conditions, we compared static with perfusion culture. The medium was spiked with VEGF (50 ng ml−1) in the first 2 days to induce vasculogenesis before reverting to the normal concentration of 5 ng ml−1 for the next 4 days.
As seen in Fig. 3, HUVECs alone do not form vascular networks with PCs (VP) in static cultivation under the here-used conditions, though flow cultivation leads to small vessel sprouts emerging from the side channels. On the other hand, HUAECs in the presence of hPC-PL (AP) were able to form a visible network, with vessel density increasing when cultivated under flow. Interestingly, when both arterial and venous ECs are introduced with PCs (AVP) in the hydrogel, HUVECs participate in the self-organization process in the presence of HUAECs, resulting in the formation of a network comprised of both EC types. As before, flow application also leads to a denser network than the static culture condition. This suggests that there may be a preferential interaction between hPC-PLs and HUAECs that drives the process of self-organization of all EC types into a vascular network.
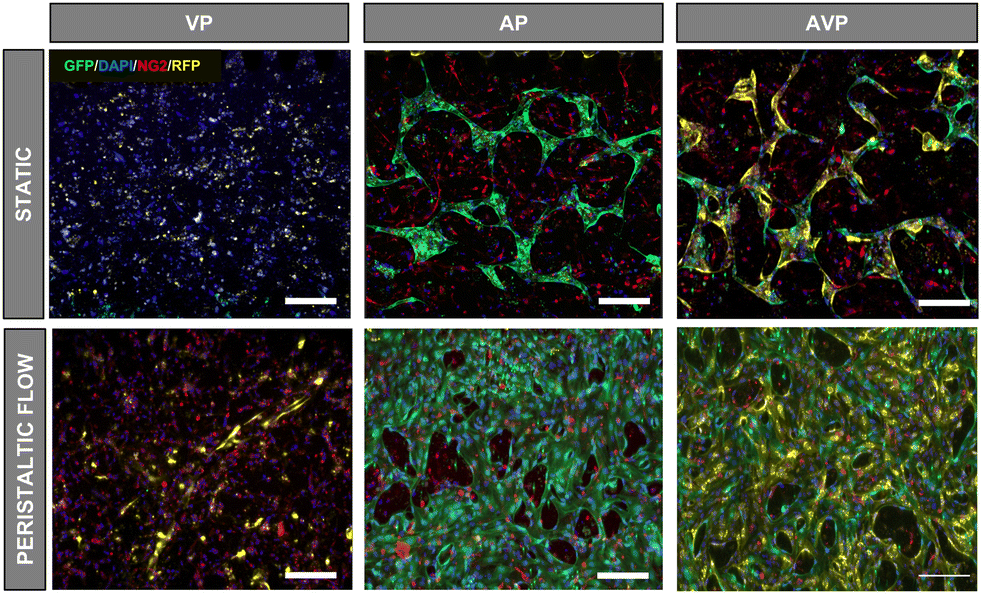 | ||
| Fig. 3 Devices with different EC combinations and PCs were set up and cultivated under flow or static conditions. Network formation was observed after fixation and immunostaining on day 6. The images indicate heterogeneous network formation when HUAECs are present, and a preferential interaction between hPC-PLs and HUAECs. Scale bar: 200 μm. VP = HUVEC (yellow) and hPC-PL (red); AP = HUAEC (green) and hPC-PL; AVP = HUVEC, HUAEC and hPC-PL. | ||
A previously confirmed interaction between PCs and ECs is the stimulation of basement membrane matrix synthesis and deposition, stabilizing the vessels and improving barrier function.51 Secretion of collagen IV, a major constituent of the basement membrane, was examined in devices containing all three cell types (AVP) and only ECs (AV), cultured in static and hydrostatic pressure/peristaltic pump-generated flow conditions. Indeed, while collagen IV secretion can be visualized in the EC-only AV devices by immunostaining, a significant increase was observed between the immunofluorescence intensity of collagen IV in the AVP networks (Fig. 4A).
 | ||
| Fig. 4 Influence of PCs on vessel formation, morphology, and functionality. (A) Collagen IV deposition in EC monocultures and networks of PCs and ECs at a ratio of 1:2 (N = 6 devices for each condition, ***p = 0.0002). Significance was calculated using an unpaired t-test. (B) Permeability coefficient for vessels treated with TNF-α compared to untreated controls. Vascular networks lacking PCs lead to a significant increase in permeability. Total acquisition time was 600 s. (C) Images of fluorescently labelled FITC–dextran supplied to EC monocultures and co-cultures of ECs and PCs in a PC-to-EC ratio of 1:5. Scale bar represents 30 μm. (D) Mean vessel diameter at different EC to PC ratios. Increased PC density resulted in narrower, non-perfusable vascular networks for all flow conditions (N = 5 devices per condition with average diameter calculated on n = 4 areas per device). A two-way ANOVA with Tukey correction for multiple comparisons was used to assess the significance between all groups. Data presented as mean ± SD. ns = not significant (p > 0.05), *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 and ****p ≤ 0.0001. (E) Fluorescence images of the network at different PC-to-EC ratio (HUAECs: green, HUVECs: yellow) for different flow conditions. Scale bar represents 50 μm. | ||
Next, we determined the permeability coefficient across the endothelial barrier using a previously established protocol.52 The average permeability coefficient of capillary networks formed with 1:12.5 PC-to-EC ratio was calculated to be 4.1 × 10−7 cm s−1, which is within the reported range of barrier permeabilities for 3D microvasculature network models,17 whereas the endothelial barrier in devices without PCs was three orders of magnitude higher at 10−4 cm s−1 (Fig. 4B and C). To evaluate the biological function of the microvascular barrier, we mimicked an inflammatory condition in the vascular networks by exposing the vessels to TNF-α, known to induce endothelial barrier dysfunction and vascular hyperpermeability. Vessel permeability increased to 4.6 × 10−6 cm s−1 upon exposure to TNF-α, indicating a disruption of the endothelium and increased vascular permeability (Fig. 4B and SI3B†).
To examine the effect of PCs on vessel morphology, we introduced a 1:1 ratio of HUVECs and HUAECs and increased the PC concentration. The introduction of PCs led to a significant reduction in vessel diameter regardless of flow conditions (Fig. 4D and E), which is in accordance with recent studies on vessels formed only by HUVECs and pericytes.31,53 Culturing microvascular networks with a 1:1 to 1:2 PC-to-EC ratio resulted in very small diameters and the networks were not perfusable anymore. As expected, collagen IV immunostaining also revealed increased deposition of the ECM protein in the basal lamina surrounding the microvessels with higher PC ratios (Fig. SI4†). In conclusion, PCs affect the arteriole–venule network formation and perfusability, as well as collagen IV concentration in the basement membrane matrix, ultimately improving the barrier properties by decreasing permeability. The ratio of PCs to ECs must be carefully chosen, with our data we conclude that a ratio of 1:12.5 is optimal and is used in the subsequent experiments.
Extracellular matrix concentration affects vessel morphology
Stiffness of tissues in human organs varies by a significant degree; the Young's modulus of native soft tissues ranges from 0.1–1 kPa in the brain to 10–30 kPa in muscle and nascent bone.54 Accurately mimicking these microenvironments is crucial for successfully replicating diverse tissue structures and disease models. We hypothesized that the various stiffnesses of in vivo tissue could be recapitulated by modeling a range of local ECM concentrations in our capillary-on-chip system.55 Fibrin hydrogel is a common choice of matrix used in developing in vitro microvasculature models. Due to its role in wound healing, fibrin promotes the cellular secretion of ECM and basement membrane proteins such as collagen IV and laminin.22 Additionally, its mechanical properties can be tuned by adjusting the fibrinogen concentration, making it an ideal choice for our study. The stiffness range of soft tissues (0.1–30 kPa) corresponds to fibrin hydrogel fabricated with 1 U ml−1 thrombin and fibrinogen at concentrations in the range of 2 to 20 mg ml−1.56,57 Here, we investigated how stiffness of the hydrogel impacts both vessel morphology and permeability. By manipulating the fibrinogen concentration in the hydrogel, we demonstrated that increasing fibrinogen concentrations led to changes in the average diameter of the vessels (Fig. 5A and B). Higher fibrinogen concentrations were associated with narrower vessels, even under peristaltic and interstitial flow conditions. Increasing fibrinogen concentrations did not have a significant effect on the permeability coefficients when comparing 5 mg ml−1 to 10 mg ml−1 within the same flow condition (Fig. 5C). Microvascular networks cultured with 15 mg ml−1 and 20 mg ml−1 fibrinogen failed to form perfusable networks, and we therefore could not further investigate the effect for high fibrinogen concentrations. | ||
| Fig. 5 Vessel morphology at increased fibrinogen concentration (mg ml−1). (A) Fluorescence images under varying fibrinogen concentrations and flow conditions. HUAECs: green, HUVECs: yellow, and PCs not stained. Scale bar: 50 μm. (B) Vessel diameters in dependence of the fibrinogen concentration. Reduced vessel diameters are linked to increased fibrinogen concentrations (N = 5 devices per condition with mean diameter ± SD calculated on n = 4 areas per device). A two-way ANOVA with Tukey correction for multiple comparisons was used to assess the significance between all groups. ns = not significant (p > 0.05), ***p ≤ 0.001, and ****p ≤ 0.0001. (C) Vascular permeability coefficient increased for two different fibrinogen concentrations and two flow conditions (N = 3 devices per condition). The increase was, however, not significant as determined using an unpaired t-test. Data presented as mean ± SD. | ||
Modeling inflammatory response and T cell adhesion and extravasation
One of the main functions of blood vessels is the transport of circulating cells. Under healthy conditions, the endothelium maintains a tight barrier that prevents cells from the bloodstream from passively transmigrating across. In the case of inflammation, however, inflammatory cytokines increase the permeability of the endothelial barrier, and the ECs express adhesion molecules for immune cells to attach and transmigrate into the perivascular space towards the diseased tissue.5 Here we use T cells, which are part of the adaptive immune system, to emulate the process of leukocyte trafficking within a capillary network. The initial adhesion to the activated microvasculature is facilitated via intercellular adhesion molecule-1 (ICAM-1)/CD54 expression on the endothelial side, with the T cells expressing the corresponding ligand (CD11a/CD18, LFA1) during inflammation.40 We mimicked the inflamed state by addition of TNF-α in the peristaltic pump perfusion reservoir and confirmed the upregulated ICAM-1 expression in the vasculature after 24 h by immunohistochemistry. As shown in Fig. 6A, ICAM-1 was present along the vessels after TNF-α exposure in contrast to the untreated vessels. | ||
| Fig. 6 T cells extravasate out of the lumen into the perivascular space when the endothelium is activated during inflammation. (A) ICAM-1 expression in the endothelium with and without TNF-α treatment, determined by immunostaining with human anti-ICAM-1. The graph depicts the fluorescence intensity per selected region of interest, ROI (*p ≤ 0.05, N = 1 for each condition, n ≥ 6). Scale bar: 50 μm. (B) IL-2-activated primary T cells were introduced to observe T cell adhesion, crawling and extravasation across the endothelial barrier (green and yellow: ECs, red: T cells). The white line indicates the movement of a T cell over 30 min, see also Video SI2†). The dashed line indicates cross section in the bottom micrographs. (C) Percentage of T cells perfused into the networks that have adhered to or transmigrated across the endothelial lumen after an overnight incubation for TNF-α-treated and untreated vessels (N = 6 and 8, ****p ≤ 0.0001). (D) Percentage of T cells perfused into the networks that have adhered to or transmigrated across the endothelial lumen. Data for TNF-α-treated and untreated vessels, and for TNF-α-treated vessels where T cells were pre-incubated with human antibodies against PSGL-1 and LFA1 (anti-PSGL-1 and anti-LFA1, respectively) (N = 11, N = 10 and N = 4, *p ≤ 0.05, ****p ≤ 0.0001). (E) Analysis of selected cytokines and chemokines in the cell culture supernatant, collected during the whole TNF-α treatment from the side channels (top) and the central channel (bottom), compared to controls without TNF-α treatment (N = 8, *p ≤ 0.05 and ***p ≤ 0.001). | ||
Upon activation by TNF-α, inflammatory cytokines and chemokines were secreted by ECs to activate and guide the immune cells toward the targeted tissue.58–60 To analyze the cytokine secretion in our system, we collected the outflow of our devices from both the side channels and the central, hydrogel-filled channel, for both the TNF-α-treated (TNF-α +) and untreated (TNF-α −) microvasculature during a 24 h period on peristaltic pump perfusion. Immunoassays targeting the following immune cell attracting chemokines and cytokines were performed:59 chemokine (C-C motif) ligand 2 (MCP-1), chemokine (C-X-C motif) ligand 1 (CINC-1), C-X-C motif chemokine 5 (ENA-78), C-X-C motif chemokine ligand 10 (CRG2), granulocyte-macrophage colony-stimulating factor (GM-CSF), macrophage colony-stimulating factor (M-CSF), chemokine (C-C motif) ligand 5 (RANTES), chemokine (C-X-C motif) ligand 2 (CINC-3), granulocyte colony-stimulating factor (G-CSF), interleukin 8 (CXCL8).59,61
Secretion levels of several chemokines and cytokines were significantly upregulated for TNF-α treated vessels, compared to those in the untreated condition (Fig. 6E). In particular, the levels of CXCL8, CINC-1, M-CSF, and MIG were significantly increased as compared to the control (TNF-α −), in both the supernatants from the central channel (gel) and the side channel outflows. CXCL-8 and CINC-1 are chemokines/cytokines that are upregulated when the tissue is inflamed to attract neutrophils out of the bloodstream towards the targeted tissue,62,63 while M-CSF attracts monocytes, which then differentiate into macrophages after transmigrating out of the blood.64 Additionally, MIG is a T cell-attracting and activating chemokine that is secreted during inflammation,65 and its secretion also significantly increased in TNF-α treated devices compared to the untreated. Overall, by inducing an inflammatory state in our capillary network, we observed a broad-range upregulation of signaling molecules relevant for immune cell recruitment.
Next, we introduced interleukin 2 (IL-2) activated primary T cells into TNF-α treated and untreated microvasculature via the venule channel to observe their perfusion and trafficking within our capillary networks. After 2 h of on-chip incubation, chips were perfused through the venule channel with fresh media to remove T cells that did not adhere or transmigrate through the vessel wall into the matrix space. During live imaging and perfusion, we were able to visualize the capture, arrest, and extravasation process of T cells (stained in red with CellTracker™ Deep Red, Fig. 6B, Video SI3†). By tracking individual cells through the capillary, we could also calculate T cell flow velocities, showing that T cells are trafficking the vessels at an approximate speed of 100 μm s−1. In vivo, T cells travel with an average speed of 200 μm s−1 in presence of TNF-α.66 Using confocal live imaging, we were able to follow the track of a single T cell that gets captured onto the endothelial lumen wall, crawls along the length of the vessel, and then extravasates into the perivascular space within 30 min. The cross-sectional views of the vessel depict the moment of extravasation and the movement away from the blood vessel (Fig. 6B). We then quantified the percentage of T cells that adhered or transmigrated across the capillary wall out of all perfused cells into the vessel during the 2 h incubation period and observed that this percentage is significantly higher in inflamed (TNF-α +) microvasculature than for non-inflamed (TNF-α −) (Fig. 6C). To confirm whether this observation is due to the biological process of leukocyte recruitment by inflamed endothelium or simply due to physical leakage through the capillary networks in our system, we conducted an inhibition assay by incubating the T cells with antibodies against P-selectin glycoprotein ligand 1 (PSGL-1), a ligand of the endothelial adhesion molecule P-selectin involved in the capture step, and lymphocyte function-associated antigen 1 (LFA1) which binds to endothelial ICAM-1 during arrest.40 The inhibition of EC biding ligands on the T cells resulted in less adhesion and transmigration compared to the positive control (TNF-α +) (Fig. 6D). The observed percentage of cells adhering or transmigrating was similar to that in the negative control (TNF-α −) at around 5%. From these findings we conclude that our capillary-on-chip allows us to mimic and influence key steps of T cell transmigration, facilitating the exploration of further key factors of this process in the future.
Conclusions
In conclusion, we have developed an in vitro capillary-on-chip model with in vivo similarity and morphology by co-culturing HUAECs and HUVECs with hPC-PLs. Our results demonstrated that flow, PC concentration and matrix stiffness significantly influences vessel morphology, perfusability and vessel permeability. Several observations of the heterogeneous network are similar to networks formed only by HUVECs, however, we observed that PCs preferentially associate with arteriole ECs during the self-assembly process. The limiting factor of our current study is elucidating the specific role of endothelial subtypes in the process of vessel formation. Thorough characterization of the primary HUAECs and HUVECs prior to and during on-chip co-culture should be examined in future studies. Additionally, quantitative measurements of mechanical and fluid forces in various flow conditions and patterns as well as local mechanical properties in the hydrogel need more attention.Our microfluidic model of the arteriole–venule and PC-enveloped capillary network enabled us to emulate key functional and mechanistic aspects of the human microvasculature during inflammation and leukocyte extravasation. We mimicked tissue inflammation by addition of TNF-α, which resulted in increased barrier permeability and upregulation of cytokines and chemokines associated with immune cell recruitment. We were able to observe in real-time the extravasation cascade of adhesion, rolling, and transmigration of T cells across a capillary network as it can be found in vivo. Inhibition of binding ligands involved in the cascade significantly decreased the adhesion/transmigration rate, demonstrating that we could recapitulate the true biological process of leukocyte trafficking.
Overall, our study contributes to the development of advanced models for drug screening, disease modeling and personalized medicine.
Materials and methods
Chip fabrication
A standard 4 inch silicon wafer (Si-Mat) was dehydrated for 10 min at 200 °C and cooled down prior to being spin coated with SU-8 2050 at 3250 rpm for 30 s. The photoresist layer was soft baked for 180 s at 65 °C and for 360 s at 95 °C before exposure to 160 mJ cm−2 at 365 nm through a transparency photomask (Micro Lithography Services) on an MA 6 mask aligner (Süss MicroTec). After a post-exposure bake for 60 s at 65 °C and 360 s at 95 °C, the wafer was developed using mr-Dev 600 developer for 5 min. The resulting feature height of the mold was 100 μm. As a final step, the wafer was placed in a 1H,1H,2H,2H-perfluorodecyltrichlorosilane atmosphere for 12 h at 100 mbar. This surface treatment facilitated the release of the polydimethylsiloxane (PDMS) cast from the wafer surface.Three-channeled microfluidic devices consisting of a central hydrogel channel flanked by lateral medium compartments were fabricated using conventional soft lithographic methods.46 To fabricate the PDMS part of the microfluidic chip, the oligomer and curing agent (Sylgard 183, Dow, MI, USA) were mixed together at a ratio of 10:1, degassed and poured onto the silicon wafer and cured overnight at 80 °C. Inlets and outlets were punched with a 1 mm outer diameter biopsy puncher (Miltex) and the devices bonded to a PDMS-coated #1.5 24 mm × 40 mm glass coverslip (Novoglas) with oxygen plasma treatment (Harrick Plasma, model no. PDC-32G) at 50 W for 60 s to enclose the microfluidic channels. Treated devices were put on an 80 °C heater prior to being cured overnight inside an 80 °C oven to recover hydrophobicity. Prior use, microfluidic chips were sterilized with UV light for 10 min and put in a desiccator for at least 1 h to remove any remaining air in the PDMS.
Cell culture
Red fluorescent protein (RFP)-expressing human umbilical vein endothelial cells and green fluorescent protein-expressing human umbilical artery endothelial cells (RFP-HUVEC/GFP-HUAEC, Angio Proteomie) were subcultured in flasks coated with 0.2% gelatin in vascular medium (Vasculife) with vascular endothelial growth factor (VEGF, Medium Complete Kit, iCell media supplement, CDI). Human pericytes isolated from placenta (hPC-PL, Promocell) were cultivated in pericyte growth medium (Promocell). Cells were maintained in a humidified incubator (37 °C, 95% humidity and 5% CO2) and cultivated to 90% confluency until passage number 8. Cell medium was replaced every 2 days and cells were detached using TrypLE Express (Gibco, cat no. 12605010).The cells were mixed in a ratio of 1:6.25:6.25 (hPC-PL:HUAEC:HUVEC) with both EC lines being 300000 cells per chip to achieve a final concentration of 320000 cells in total per chip. In AP (HUAEC + hPC-PL) and VP (HUVEC + hPC-PL) only systems, we used twice the number of ECs as for the AVP (HUAEC + HUVEC + hPC-PL) systems to be comparable. The pericyte-to-endothelial cell (PC-to-EC) ratio was further modified to explore the impact of PCs on vessel morphology. Specifically, PC-to-EC ratios of 1:12.5, 1:5, 1:2, and 1:1 were tested in comparison to EC monocultures where HUAECs and HUVECs were seeded in a 1:1 ratio, with 150000 cells per chip for each cell type.
Cells were detached and spun down at 300 rcf for 5 min and the cell pellet was resuspended in either Vasculife or PC growth medium before counting to achieve the right concentration of cells per chip. Mixed cell suspensions containing HUAEC and/or HUVEC with hPC-PL were spun down and the pellet resuspended in 10 μl of the hydrogel. The hydrogel was made by premixing 10 mg ml−1 fibrinogen from human plasma (Merck) and 0.15 U ml−1 aprotinin (Merck) dissolved in sterile PBS (−/−) (Gibco). Ice cold thrombin from human plasma (Merck) was diluted to 10 U ml−1 in high VEGF EGM-2 (50 ng ml−1 VEGF, Peprotech) and mixed into the cell suspension to a final concentration of 1 U ml−1. Subsequently, 10 μl of the cell-laden hydrogel mix was immediately injected into the central compartment of the device and let to polymerize for 15 min in an incubation chamber at 37 °C. Afterwards, 20 μl coating solution containing Vasculife medium supplemented with VEGF (50 ng ml−1), fibronectin from human plasma (0.1 mg ml−1, Merck) and collagen type I from rat tail (50 μg ml−1, Corning) was introduced to both side channels to promote endothelial cell adhesion. The chips were then incubated overnight in a humidified incubator (37 °C, 95% humidity and 5% CO2).
The next day, the side channels were washed with high-VEGF Vasculife medium and both HUAECs and HUVECs were seeded into the respective side channels at a cell density of 1000000 cells per ml (high VEGF Vasculife medium). To ensure that the cells were attached especially at the interface towards the central chamber, the device was tilted 90° or 270° for at least 2 h per side channel before seeding the other endothelial cell line. To induce an inflammatory response in the vessels, TNF-α (10 ng ml−1 in Vasculife medium, Thermo Fisher) was introduced to each side channel on day 6 and incubated overnight.
Flow application
Three different flow conditions were tested: 1) static flow, 2) interstitial flow and 3) peristaltic flow. Under static flow conditions, pipette tips containing 40 μl of medium were attached to both side channels and replaced every 24 h to replenish media. To establish interstitial flow across the gel, a hydrostatic pressure difference was created by adding pipette tips with 200 μl medium to both ports of one side channel and empty tips in the opposite side channel. The difference in height between the pipette tips drives interstitial flow through the gel region. The pressure gradient was alternated between the two side channels and restored every 24 h with fresh culture medium. The devices were kept in a humidified incubator with daily media changes for 6 days until perfusable microvascular networks were formed. The peristaltic flow was maintained by constantly perfusing the chips with a peristaltic pump (Ismatec® IPC-N, Cole Palmer) at a flow rate of 60 μl h−1. The same flow rate was also constantly applied during TNF-α (10 ng ml−1 in EGM-2) treatment as well as when T cells were supplied.To analyze the impact of flow direction on the vascular morphology, two flow profiles were set up during peristaltic flow. The parallel flow was created by attaching the tubing to the inlets positioned on the same side in parallel to the flow direction. The counterflow was established by attaching the tubing on the opposite sides of the side channels to produce an interstitial flow with opposing flows that intersect within the hydrogel. Confocal images were acquired and the average angle between the flow direction and vessel direction was measured using Image J software with the Orientation J plug-in.67,68
Immunohistochemistry
A list of antibodies used in this study is given in Table 1. Cells were fixed by addition of 4% paraformaldehyde (PFA, Thermo Fisher) for 30 min, followed by three washing steps of PBS (−/−) (Gibco). Permeabilization was done with 0.2% Triton-X (Merck) in PBS (−/−) for 30 min before blocking with 5% bovine serum albumin (BSA, Merck) and 0.1% Triton-X for at least 1 h. Primary antibodies (1:100 volume ratio diluted in PBS (−/−), 1.5% BSA and 0.1% Triton-X) and DAPI staining (NucBlue, 1:1000 dilution) were injected into the side channels of the device and incubated at 4 °C overnight followed by three washing steps of PBS (−/−) before imaging (Table 1). Actin filaments (F-actin) were stained using a 1:100 dilution of Alexa Fluor® conjugated phalloidin (Thermo Fisher Scientific), following the same incubation and washing steps as the primary antibodies. Confocal images were acquired using a Nikon Eclipse Ti2 Spinning Disk (Yokogawa) confocal microscope with a 10× and 20× objective. Z-stack images were captured with a 2 μm step size unless otherwise stated. The z-stack images and fluorescence intensity values in selected regions of interest (ROIs) were processed using ImageJ and Imaris software version 9.9.1.
| Antibody | Label | Distributor | Cat. no. |
|---|---|---|---|
| Anti-human VE-cadherin/CD144 | Alexa Fluor® 488 | Thermo Fisher Scientific | MA5-44146 |
| Anti-human NG2/MCSP | Alexa Fluor® 647 | Abcam | ab279348 |
| Anti/human ICAM-1/CD54 | Alexa Fluor® 647 | Novus Biologicals | 17-0549-42 |
| Anti-human collagen IV | Alexa Fluor® 647 | Thermo Fisher Scientific | 51-9871-82 |
| Phalloidin | Alexa Fluor® 647 | Thermo Fisher Scientific | A22287 |
Dextran-based permeability assay and vessel diameter measurement
A solution of 10 kDa Cascade Blue dextran and 70 kDa CF®633-labeled dextran (100 μl ml−1, Thermo Fisher) in Vasculife medium was prepared separately. Devices were placed inside an incubation chamber (37 °C, 95% humidity, 5% CO2) on a fluorescence microscope (Nikon Eclipse Ti2) with a spinning disk unit (Visitron Systems GmbH). The dextran solution was introduced via one side channel to assess the permeability of the endothelial barrier. By closing the corresponding outlet of this introductory channel, the dextran solution is forced into the microvasculature to get into the other side channel. Time-sequential images were acquired every 20 s for 10 min to create a 3D stack of the microvasculature. Vascular networks with a distinct boundary between the vessel wall and gel regions were chosen as ROIs. Similarly, fluorescent beads (diluted 1:1000) were injected through one of the side channels to assess perfusability of the vascular network between the side channels.
The permeability coefficient for different conditions was calculated using ImageJ software plug-ins ‘Trainable Weka Segmentation 3D’ and ‘Macro permeability’ in accordance with previously published protocols.52 Briefly, confocal z-stack files of dextran-perfused vessels were captured with a 6 μm step size at 0 and 10 min from which morphological parameters (volume of vessels, volume of the surrounding matrix, surface area of the vascular network) and the permeability coefficient were calculated according to a template. Several ROIs for each device were included in the analysis.
The vessel diameter was calculated using ImageJ software ‘Vessel diameter’ plug-in which provided automated measurements of vessel diameter based on ROI selection. In addition to the automated measurements, manual estimations were also performed. ROIs were manually drawn around the vessels of interest to obtain further diameter measurements.
T cell perfusion and live imaging
Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood of healthy human donors (Blutspendezentrum SRK beider Basel, Universitätspital Basel) via Lymphoprep (Stemcell Technologies). Human CD4+ and CD8+ T cells were extracted by magnetic negative selection using an EasySep Human Pan T Cell Isolation kit (STEMCELL Technologies). Primary T cells were cultured in XVivo-15 medium (Lonza) with 5% fetal bovine serum (FBS) and 50 μM 2-mercaptoethanol with freshly added 200 IU of recombinant human IL-2 (Peprotech), 100 μg mL−1 Normocin (Invivogen). On the day of thawing and magnetic selection, T cells were activated with anti-CD3/anti-CD28 Dynabeads (Thermo Fisher). After 3 days, beads were magnetically removed, and cells were cultured for up to 7 more days before used in the experiments.For the final activation, 100 ng ml−1 interleukin-2 was added. T cells were pre-stained with CellTracker™ Deep Red (Thermo Fisher) according to the provided protocol. A suspension of 1000000 cells per ml were flushed into one side channel and incubated for 1 h at 37 °C, after which the volume passing through the microvessel network to the other side channel was measured to determine total number of cells introduced into the microvasculature. Chips were then reconnected to the pumps for overnight perfusion to wash out non-adherent or extravasating T cells, then fixed with 4% PFA. T cells adhered to the vessel wall or extravasated into the perivascular space were imaged and counted utilizing Imaris software version 9.9.1. and ImageJ.
For live imaging of T cell extravasation, T cells were flushed in through one endothelial cell channel and placed back onto the microscope (Zeiss LSM 980). Z stacks were acquired at 37 °C, 5% CO2 and 95% humidity on multiple spots over a period of 40 min.
Inhibition assay
Primary T cells were stained with CellTracker™ Deep Red according to the manufacturer protocol (Invitrogen). Subsequently, anti-human PSGL-1/CD162 Alexa Fluor® 647 (R&D Systems) and anti-human CD11a PerCP/Cyanine5.5 (Biolegend) in 1:100 dilution were added to the T cells and incubated for 2 h at 37 °C, 5% CO2 and 95% humidity. After washing away unbound antibodies with PBS (−/−), T cells were put back into cultivation media (see T cell perfusion) to achieve a final concentration of 1000000 cells per ml. Normal T cell perfusion and counting were conducted (see T cell perfusion).
Luminex® bead-based immunoassay for cytokine and chemokine measurements
Cell culture supernatants were collected during the TNF-α treatment. Supernatants from non-treated devices were used as a negative control. A personalized Luminex® (Bio-Techne Corporation) panel was designed and used according to the manufacturer's protocol. The resulting well plate was measured with MAGPIX® (Luminex).Statistical analysis
All data were tested for normality and analyzed using Student's t-test for comparison between two independent samples and two-way ANOVA to determine the significance level by comparing means of three or more independent groups with two independent variables. Non-normal data were tested using the Mann–Whitney U test.Data are plotted as mean ± standard deviation (SD) unless otherwise stated. Statistical tests were performed using GraphPad Prism version 9.5.1 for Windows (GraphPad Software, San Diego, California, USA). At least three devices (≥3 regions per device) for each condition within an experiment were used for imaging and data analysis. Statistical significance was defined as p ≤ 0.05.
Author contributions
E. H., W. C., and P. S. D. conceived the project. E. H., W. C., M. P. designed and conducted the experiments and analyzed the data. E. H., W. C., M. P., and P. S. D. wrote the manuscript. S. S. assisted with flow experiments and discussion. E. K. and S. R. provided the T cells and discussion. C. D. conducted the immunoassays. M. F. provided endothelial cells and discussion; all authors reviewed and approved the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests.Acknowledgements
The authors thank the Automation and Single Cell Facility at BSSE for their technical support, Raphael Dezauzier for discussions and assistance with COMSOL modeling. This work was supported by the Swiss National Science Foundation as part of the NCCR Molecular Systems Engineering Project No. 51NF40-205608 and the Personalized Medicine Basel (PMB) Grant No. PMB-02-19.Notes and references
- J. Kreuger and M. Phillipson, Nat. Rev. Drug Discovery, 2016, 15, 125–142 CrossRef CAS PubMed.
- D. R. Myers and W. A. Lam, Annu. Rev. Biomed. Eng., 2021, 23, 1–26 CrossRef PubMed.
- A. S. Chung, J. Lee and N. Ferrara, Nat. Rev. Cancer, 2010, 10, 505–514 CrossRef CAS PubMed.
- Y. Zhao, X. Yu and J. Li, Acta Pharm. Sin. B, 2020, 10, 2018–2036 CrossRef CAS PubMed.
- L. Claesson-Welsh, E. Dejana and D. M. McDonald, Trends Mol. Med., 2021, 27, 314–331 CrossRef CAS PubMed.
- J. T. Daub and R. M. H. Merks, Bull. Math. Biol., 2013, 75, 1377–1399 CrossRef CAS PubMed.
- C. F. Buchanan, S. S. Verbridge, P. P. Vlachos and M. N. Rylander, Cell Adhes. Migr., 2014, 8, 517–524 CrossRef PubMed.
- D. E. Ingber, Adv. Sci., 2020, 7, 2002030 CrossRef CAS PubMed.
- Y. Nashimoto, T. Hayashi, I. Kunita, A. Nakamasu, Y. Torisawa, M. Nakayama, H. Takigawa-Imamura, H. Kotera, K. Nishiyama, T. Miura and R. Yokokawa, Integr. Biol., 2017, 9, 506–518 CrossRef PubMed.
- C. Kim, J. Kasuya, J. Jeon, S. Chung and R. D. Kamm, Lab Chip, 2014, 15, 301–310 RSC.
- V. van Duinen, W. Stam, E. Mulder, F. Famili, A. Reijerkerk, P. Vulto, T. Hankemeier and A. J. van Zonneveld, Int. J. Mol. Sci., 2020, 21, 4804 CrossRef CAS PubMed.
- J. D. Humphrey and M. A. Schwartz, Annu. Rev. Biomed. Eng., 2021, 23, 1–27 CrossRef CAS PubMed.
- P. A. Galie, D.-H. T. Nguyen, C. K. Choi, D. M. Cohen, P. A. Janmey and C. S. Chen, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7968–7973 CrossRef CAS PubMed.
- Q. Ma, H. Ma, F. Xu, X. Wang and W. Sun, Microsyst. Nanoeng., 2021, 7, 19 CrossRef CAS PubMed.
- C. F. Buchanan, E. E. Voigt, C. S. Szot, J. W. Freeman, P. P. Vlachos and M. N. Rylander, Tissue Eng., Part C, 2014, 20, 64–75 CrossRef CAS PubMed.
- L. Knezevic, M. Schaupper, S. Mühleder, K. Schimek, T. Hasenberg, U. Marx, E. Priglinger, H. Redl and W. Holnthoner, Front. Bioeng. Biotechnol., 2017, 5, 25 Search PubMed.
- M. B. Chen, J. A. Whisler, J. Fröse, C. Yu, Y. Shin and R. D. Kamm, Nat. Protoc., 2017, 12, 865–880 CrossRef CAS PubMed.
- J. Paek, S. E. Park, Q. Lu, K.-T. Park, M. Cho, J. M. Oh, K. W. Kwon, Y. Yi, J. W. Song, H. I. Edelstein, J. Ishibashi, W. Yang, J. W. Myerson, R. Y. Kiseleva, P. Aprelev, E. D. Hood, D. Stambolian, P. Seale, V. R. Muzykantov and D. Huh, ACS Nano, 2019, 13, 7627–7643 CrossRef CAS PubMed.
- S. Kim, H. Lee, M. Chung and N. L. Jeon, Lab Chip, 2013, 13, 1489–1500 RSC.
- X. Zhang, N. O. Abutaleb, E. Salmon and G. A. Truskey, Methods Mol. Biol., 2021, 2375, 77–90 CrossRef PubMed.
- M. Campisi, Y. Shin, T. Osaki, C. Hajal, V. Chiono and R. D. Kamm, Biomaterials, 2018, 180, 117–129 CrossRef CAS PubMed.
- E. D. Grassl, T. R. Oegema and R. T. Tranquillo, J. Biomed. Mater. Res., 2002, 60, 607–612 CrossRef CAS PubMed.
- E. Akbari, G. B. Spychalski and J. W. Song, Microcirculation, 2017, 24, e12363 CrossRef PubMed.
- S. R. Moses, J. J. Adorno, A. F. Palmer and J. W. Song, Am. J. Physiol., 2021, 320, C92–C105 Search PubMed.
- M. A. Mäe, L. He, S. Nordling, E. Vazquez-Liebanas, K. Nahar, B. Jung, X. Li, B. C. Tan, J. C. Foo, A. Cazenave-Gassiot, M. R. Wenk, Y. Zarb, B. Lavina, S. E. Quaggin, M. Jeansson, C. Gu, D. L. Silver, M. Vanlandewijck, E. C. Butcher, A. Keller and C. Betsholtz, Circ. Res., 2021, 128, e46–e62 CrossRef PubMed.
- M. Hellström, H. Gerhardt, M. Kalén, X. Li, U. Eriksson, H. Wolburg and C. Betsholtz, J. Cell Biol., 2001, 153, 543–554 CrossRef PubMed.
- L. L. Bischel, E. W. K. Young, B. R. Mader and D. J. Beebe, Biomaterials, 2013, 34, 1471–1477 CrossRef CAS PubMed.
- K. Yamamoto, K. Tanimura, M. Watanabe, H. Sano, H. Uwamori, Y. Mabuchi, Y. Matsuzaki, S. Chung, R. D. Kamm, K. Tanishita and R. Sudo, Tissue Eng., Part A, 2019, 25, 499–510 CrossRef CAS PubMed.
- J. P. Waters, M. S. Kluger, M. Graham, W. G. Chang, J. R. Bradley and J. S. Pober, J. Vasc. Res., 2013, 50, 324–331 CrossRef CAS PubMed.
- J. Kim, M. Chung, S. Kim, D. H. Jo, J. H. Kim and N. L. Jeon, PLoS One, 2015, 10, e0133880 CrossRef PubMed.
- M. Dibble, S. D. Cio', P. Luo, F. Balkwill and J. E. Gautrot, Sci. Rep., 2023, 13, 5729 CrossRef CAS PubMed.
- J. W. Song and L. L. Munn, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15342–15347 CrossRef CAS PubMed.
- S. Kim, M. Chung, J. Ahn, S. Lee and N. L. Jeon, Lab Chip, 2016, 16, 4189–4199 RSC.
- P. Campinho, A. Vilfan and J. Vermot, Front. Physiol., 2020, 11, 552 CrossRef PubMed.
- Y. Liu, J. Li, J. Zhou, X. Liu, H. Li, Y. Lu, B. Lin, X. Li and T. Liu, Micromachines, 2022, 13, 225 CrossRef PubMed.
- S. Zhang, Z. Wan, G. Pavlou, A. X. Zhong, L. Xu and R. D. Kamm, Adv. Funct. Mater., 2022, 32, 2206767 CrossRef CAS PubMed.
- J. S. Jeon, S. Bersini, M. Gilardi, G. Dubini, J. L. Charest, M. Moretti and R. D. Kamm, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 214–219 CrossRef CAS PubMed.
- J. S. Jeon, S. Bersini, J. A. Whisler, M. B. Chen, G. Dubini, J. L. Charest, M. Moretti and R. D. Kamm, Integr. Biol., 2014, 6, 555–563 CrossRef CAS PubMed.
- S. Nagaraju, D. Truong, G. Mouneimne and M. Nikkhah, Adv. Healthcare Mater., 2018, 7, e1701257 CrossRef PubMed.
- K. Ley, C. Laudanna, M. I. Cybulsky and S. Nourshargh, Nat. Rev. Immunol., 2007, 7, 678–689 CrossRef CAS PubMed.
- Z. Shulman, S. J. Cohen, B. Roediger, V. Kalchenko, R. Jain, V. Grabovsky, E. Klein, V. Shinder, L. Stoler-Barak, S. W. Feigelson, T. Meshel, S. M. Nurmi, I. Goldstein, O. Hartley, C. G. Gahmberg, A. Etzioni, W. Weninger, A. Ben-Baruch and R. Alon, Nat. Immunol., 2012, 13, 67–76 CrossRef CAS PubMed.
- D. J. Fowell and M. Kim, Nat. Rev. Immunol., 2021, 21, 582–596 CrossRef CAS PubMed.
- J. Lee, C. B. Breuer and E. Lee, Biochem. Soc. Trans., 2021, 49, 693–704 CrossRef CAS PubMed.
- L. de Haan, J. Suijker, R. van Roey, N. Berges, E. Petrova, K. Queiroz, W. Strijker, T. Olivier, O. Poeschke, S. Garg and L. J. van den Broek, Int. J. Mol. Sci., 2021, 22, 8234 CrossRef CAS PubMed.
- J. Lee, S.-E. Kim, D. Moon and J. Doh, Lab Chip, 2021, 21, 2142–2152 RSC.
- Y. Shin, S. Han, J. S. Jeon, K. Yamamoto, I. K. Zervantonakis, R. Sudo, R. D. Kamm and S. Chung, Nat. Protoc., 2012, 7, 1247–1259 CrossRef CAS PubMed.
- C. P. Huang, J. Lu, H. Seon, A. P. Lee, L. A. Flanagan, H.-Y. Kim, A. J. Putnam and N. L. Jeon, Lab Chip, 2009, 9, 1740–1748 RSC.
- K. Haase, F. Piatti, M. Marcano, Y. Shin, R. Visone, A. Redaelli, M. Rasponi and R. D. Kamm, Biomaterials, 2022, 280, 121248 CrossRef CAS PubMed.
- X. Wang, D. T. T. Phan, A. Sobrino, S. C. George, C. C. W. Hughes and A. P. Lee, Lab Chip, 2015, 16, 282–290 RSC.
- C. A. Franco, M. L. Jones, M. O. Bernabeu, I. Geudens, T. Mathivet, A. Rosa, F. M. Lopes, A. P. Lima, A. Ragab, R. T. Collins, L.-K. Phng, P. V. Coveney and H. Gerhardt, PLoS Biol., 2015, 13, e1002125 CrossRef PubMed.
- A. N. Stratman and G. E. Davis, Microsc. Microanal., 2011, 18, 68–80 CrossRef PubMed.
- C. Hajal, G. S. Offeddu, Y. Shin, S. Zhang, O. Morozova, D. Hickman, C. G. Knutson and R. D. Kamm, Nat. Protoc., 2022, 17, 95–128 CrossRef CAS PubMed.
- K. Fujimoto, S. Erickson, M. Nakayama, H. Ihara, K. Sugihara, Y. Nashimoto, K. Nishiyama, T. Miura and R. Yokokawa, Lab Chip, 2022, 23, 306–317 RSC.
- J. Liu, H. Zheng, P. S. P. Poh, H.-G. Machens and A. F. Schilling, Int. J. Mol. Sci., 2015, 16, 15997–16016 CrossRef CAS PubMed.
- M. Frye, A. Taddei, C. Dierkes, I. Martinez-Corral, M. Fielden, H. Ortsäter, J. Kazenwadel, D. P. Calado, P. Ostergaard, M. Salminen, L. He, N. L. Harvey, F. Kiefer and T. Mäkinen, Nat. Commun., 2018, 9, 1511 CrossRef PubMed.
- H. Duong, B. Wu and B. Tawil, Tissue Eng., Part A, 2009, 15, 1865–1876 CrossRef CAS PubMed.
- B. Bachmann, S. Spitz, B. Schädl, A. H. Teuschl, H. Redl, S. Nürnberger and P. Ertl, Front. Bioeng. Biotechnol., 2020, 8, 373 CrossRef PubMed.
- H. Lo, T. Lai, C. Li and W. Wu, Acta Pharmacol. Sin., 2014, 35, 339–350 CrossRef CAS PubMed.
- G. K. Griffin, G. Newton, M. L. Tarrio, D. Bu, E. Maganto-Garcia, V. Azcutia, P. Alcaide, N. Grabie, F. W. Luscinskas, K. J. Croce and A. H. Lichtman, J. Immunol., 2012, 188, 6287–6299 CrossRef CAS PubMed.
- P. Newton, G. O'Boyle, Y. Jenkins, S. Ali and J. A. Kirby, Mol. Immunol., 2009, 47, 485–492 CrossRef CAS PubMed.
- R. Ramjeesingh, R. Leung and C.-H. Siu, FASEB J., 2003, 17, 1292–1294 CrossRef CAS PubMed.
- R. C. Russo, C. C. Garcia, M. M. Teixeira and F. A. Amaral, Expert Rev. Clin. Immunol., 2014, 10, 593–619 CrossRef CAS PubMed.
- N. Sheikh, K. Tron, J. Dudas and G. Ramadori, Lab. Invest., 2006, 86, 800–814 CrossRef CAS PubMed.
- I. Ushach and A. Zlotnik, J. Leukocyte Biol., 2016, 100, 481–489 CrossRef CAS PubMed.
- R. Tokunaga, W. Zhang, M. Naseem, A. Puccini, M. D. Berger, S. Soni, M. McSkane, H. Baba and H.-J. Lenz, Cancer Treat. Rev., 2018, 63, 40–47 CrossRef CAS PubMed.
- B. J. Ballermann, A. Dardik, E. Eng and A. Liu, Kidney Int., 1998, 54, S100–S108 CrossRef PubMed.
- Z. Püspöki, M. Storath, D. Sage and M. Unser, Adv. Anat., Embryol. Cell Biol., 2016, 219, 69–93 Search PubMed.
- C. T. Rueden, J. Schindelin, M. C. Hiner, B. E. DeZonia, A. E. Walter, E. T. Arena and K. W. Eliceiri, BMC Bioinf., 2017, 18, 529 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lc00719g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |