
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA miniaturized microplasma excitation source coupled with photochemically induced volatile species generation as a cost-effective tool for in situ mercury pollution analyses †
Tymoteusz
Klis
,
Pawel
Pohl
,
Anna
Dzimitrowicz
and
Piotr
Jamroz
 *
*
Department of Analytical Chemistry and Chemical Metallurgy, Faculty of Chemistry, Wroclaw University of Science and Technology, Wybrzeze Stanislawa Wyspianskiego 27, Wroclaw 50-370, Poland. E-mail: piotr.jamroz@pwr.edu.pl; Fax: +48 71 320 38 07; Tel: +48 71 320 38 07
First published on 22nd October 2024
Abstract
A new portable miniaturized atmospheric microplasma discharge (μAPD) system coupled with the optical emission spectrometry (OES) detection for the determination of Hg in water samples was developed. The device was built from cheap, easily replaceable, and commercially available parts and was able to work with the photo-induced chemical vapor generation (PCVG). To optimize the photochemical volatile Hg generation process, a wide range of low molecular weight organic compounds (LMWOCs), namely formic, oxalic, acetic, propionic, and malonic acids, in addition to methanol, ethanol, glycerin, ethylene glycol, formaldehyde, and acetaldehyde, were tested to establish their influence on the signal of Hg. To assess the excitation and atomization potential of the newly developed μAPD system and its impact on the analytical performance of this microplasma excitation source, the plasma temperatures and the electron number density were evaluated. The analytical figures of merit were determined for the coupled PCVG-μAPD system. Additionally, the usability of the method was tested in reference to the analysis of selected environmental samples, i.e., tap, well, and river water spiked with Hg. A recovery test was also performed to evaluate the accuracy of the method. The examined analytical system allowed to detect Hg in water at a level lower than 0.33 μg L−1 when operating it at relatively low sample flow rates (2 mL min−1) and Ar supporting and plasma forming gas rates (20 mL min−1). The precision of measurements was better than 5% for formic acid. We believe that the presented system might be an attractive, cheaper alternative to commercial, highly expensive systems, e.g. based on inductively coupled plasma optical emission spectrometry (ICP OES).
1. Introduction
Mercury (Hg) is a toxic element known for its mobility and persistence in the natural environment.1 It is well known that Hg exhibits the intense negative chemical and biological activity and enters the environment through natural processes or improper human activities.1 As a consequence, the concentration of Hg in different parts of the environment is crucial to be monitored, particularly in the case of water consumed by living organisms. The negative impact of Hg on humans is associated with the exposure to this element through different routes, e.g., inhalation, skin contact, or ingestion. For this reason, determining both the presence and concentration of Hg in the natural environment is of great concern. Currently, the most common techniques used to determine Hg in liquid samples are atomic fluorescence spectrometry (AFS) and the chemical vapour generation (CVG) associated with atomic absorption spectrometry (AAS), inductively coupled plasma optical emission spectrometry (ICP OES), microwave-induced plasma optical emission spectrometry (MIP OES) and inductively coupled plasma mass spectrometry (ICP MS).2,3 Although reliable and precise, the mentioned analytical techniques involve instruments that are large, heavy, and restricted to use under stable environmental conditions (such as properly equipped laboratories).4 For these reasons, it is desirable to develop an apparatus that would be relatively inexpensive and portable and work under a wider range of atmospheric conditions, making it useable in the field analyses with the highest possible stability and the lowest limits of detection (LODs).Due to the low transport efficiencies related to the size of the instruments used and the significant matrix interferences associated with the direct introduction of samples into excitation and ionization sources or atomization cells in the case of atomic spectrometry methods, it is necessary to develop an indirect approach for the introduction of analytes that could solve the above-mentioned issues.5 One such approach is the CVG, which is generally divided into two main branches.5,6 One of them is the generation of Hg vapour, including the chemical vapors of the selected group of elements, i.e., hydrides of As, Bi, Ge, Pb, Sb, and Se, with reducing agents such as NaBH4 or SnCl2 (only for Hg). The other is the photo-induced CVG (PCVG), in which the reaction is propagated by the use of the UV irradiation only and one or more additional organic compounds. As such, there is no need to use such reagents like NaBH4/KBH4/SnCl2, which are toxic and accompanied by the overproduction of H2 (the case of NaBH4/KBH4), deteriorating the performance of common excitation, ionization or atomization sources. In addition to those two modes of the volatile species generation of given analytes, it is necessary to mention that the cold atmospheric plasma-induced CVG7 is also researched for Hg as well as some other techniques, including the electrochemical hydride generation, the alkylation, the chelation, or the microwave-assisted oxidation.6
The coupling of the miniaturized plasma sources with CVG techniques has been successfully achieved by a few scientific groups so far. Rong et al.8 coupled atmospheric pressure glow discharge (APGD) optical emission spectrometry (OES) with the CVG of Hg, using NaBH4 as the reducing agent. The authors reported a relatively low LOD of Hg (0.26 μg L−1), achieved by applying a low-cost and low-power apparatus. Similarly, Yuan et al.9 reported promising results for the OES determination of Hg using a point discharge microplasma as the excitation source and the introduction of Hg in the form of vapors produced in the CVG reaction with KBH4. The LOD of Hg achievable with the developed system was 0.15 μg L−1. Zheng et al.10 presented an alternative to the abovementioned technique for the generation of the Hg vapors, specifically the PCVG in the presence of formic acid. The LOD of Hg obtained for this method was at an extreme level, reaching 0.003 μg L−1. Recently, researchers have also developed portable analytical systems based on the 3D printing technique. Accordingly, Yang et al.11 proposed a dual mode CVG/PCVG micro-point discharge (μPD) system for the OES determination of inorganic and organic Hg species. The LOD of inorganic Hg was 0.1 μg L−1 in this case. Similarly, Li et al.12 presented the same system for detecting inorganic Hg at a level above 0.1 μg L−1.
In this research, a new, inexpensive, and low-power high-voltage alternating current (HV-ac) microplasma discharge (μAPD) source combined with the PCVG technique was developed and proposed for the determination of Hg in water samples. After the optimization of the system, the analytical performance of the PCVG-μAPD-OES method was evaluated. The possible interference in the case of the PCVG process was studied and the effectiveness of the PCVG of Hg was assessed. Additionally, the plasma parameters, i.e., the optical microplasma discharge temperatures and the electron number density, were determined for μAPD and PCVG-μAPD systems and compared. The PCVG-μAPD-OES method was validated, while its applicability was tested in the analysis of environmental water samples for the Hg content as an alternative and cheaper technique for the Hg determination. To the best of our knowledge, this is the first work where such an approach has been proposed, optimized, and validated.
2. Experimental
2.1. Microplasma device and instrumentation
A scheme of the μAPD system is presented in Fig. 1. Briefly, the device consists of a small sized quartz reactor (OD of 10 mm and length 30 mm) with a sharpened solid tungsten electrode (ID of 1.0 mm) and a hollow tungsten electrode (OD of 3.0 mm and ID of 1.5 mm). The distance between the electrodes was adjusted to 1.0 mm for the highest plasma stability. The microplasma was maintained in an open-air atmosphere. The HV-ac power supply was a laboratory-made device based on a commercially available and cheap (∼10 USD) neon power supply (Hongba, China). The power supply was operated at a maximum voltage of 5 kV and an output current of 30 mA, and connected to a step-down input voltage converter. It should be noted that this HV-ac power supply consumed only up to ∼10 W and can be powered by a battery. As plasma-supporting and carrier gas of the generated volatile Hg species, argon (Ar 5.0 SIAD, Poland) was applied. Miniature flow rates of Ar were used, i.e., within 10–120 mL min−1 (sccm – standard cubic centimeters per min at STP), and controlled using a Tylan General system, including an RO-28 gas flow meter and an F-2900 flow controller. The μAPD system was coupled with a PCVG quartz photoreactor (see Fig. 1). Sample solutions were introduced into the PCVG reactor with a 3-chanell ISM4312 peristaltic micropump (Ismatec, USA) and UV-irradiated. As a source of the UV radiation, a commercially available PURITEC HNS S 9 W UVC lamp (Osram, Germany) was used. The UV radiation was isolated from the environment using an aluminum foil. The foil served a dual purpose: its primary function was to protect the operator from the harmful UV radiation, while also reflecting the radiation near the reactor, potentially enhancing the irradiation efficiency. The emission spectrum of the UV-radiation lamp was recorded and is presented in Fig. S1.† As can be seen from Fig. S1,† the most intense radiation of this lamp was the UV-C region (approximately 50% of the total UV radiation). Additionally, a strong atomic emission line of Hg (Hg I) was observed at 253 nm. After the UV irradiation of the sample solutions, they were transported into a gas–liquid phase separator (GLS) chamber, where a stream of Ar swept the volatile Hg species and separated them from the liquid medium. Then, the volatile Hg species were introduced in the stream of Ar into the μAPD zone via the hollow tungsten electrode, while the liquid wastes were drained from the system at a flow rate equal to the sample flow rate, so as to maintain a stable liquid level in the GLS chamber (the waste pump was started 1 min after the system started). The photoreactor and the GLS chamber were cleaned by using 5% (m m−1) HNO3.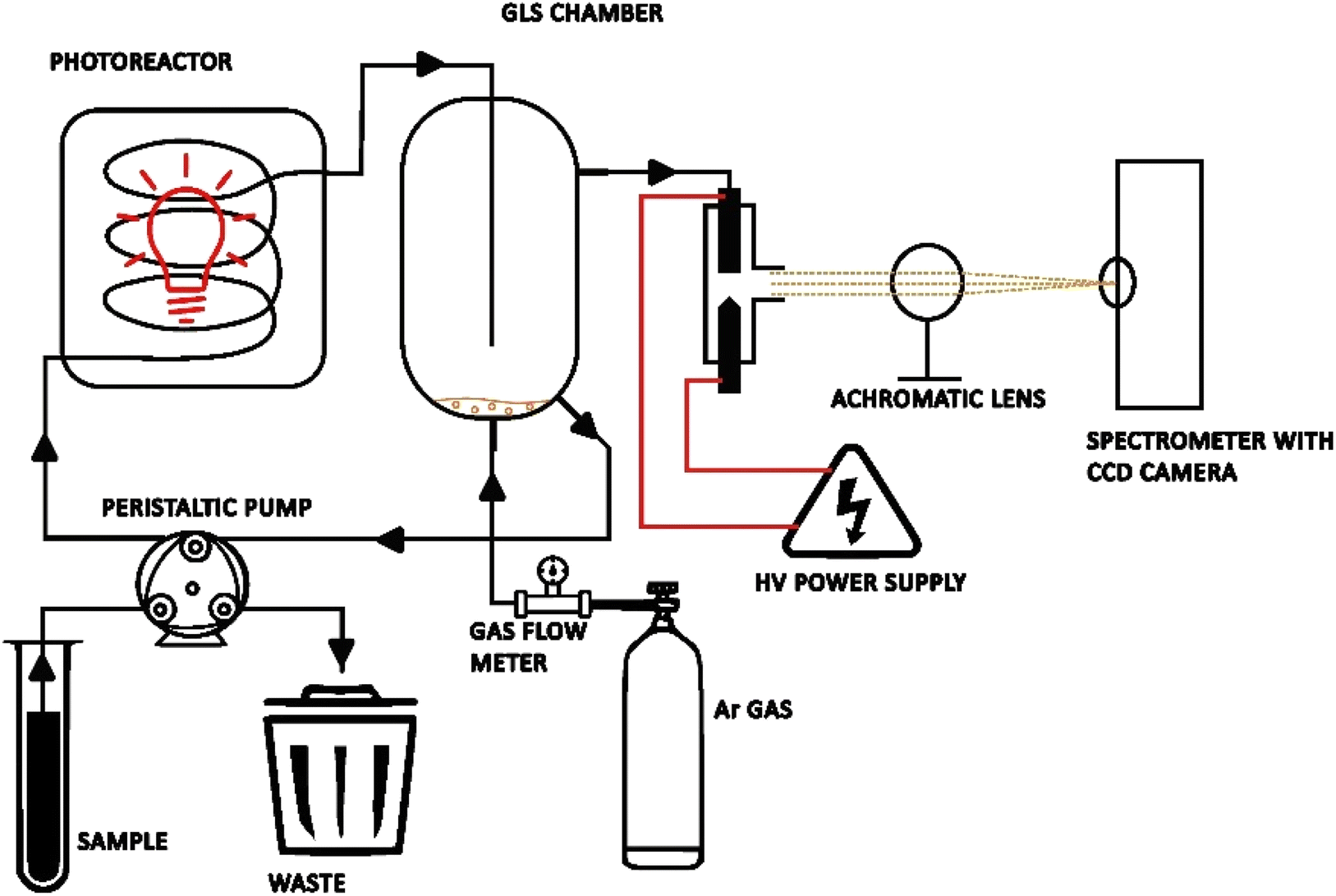 | ||
| Fig. 1 A representative scheme of the system (not in scale) of the μAPD coupled with the photoreactor (PCVG) system. | ||
The radiation emitted by the μAPD was collimated by using an achromatic UV lens (f = 60) and focused on an entrance slit (10 μm) of a Shamrock SR500 (Andor, UK) spectrograph. The spectrometer was equipment with two holographic gratings: 1800 and 1200 grooves per mm, for the spectral ranges 200–400 nm and 400–900 nm, respectively. For the detection of the radiation emitted by the μAPD source, a Newton DU-940 CCD camera (Andor, UK) with the pixel dimensions 1024 × 256 was applied. The CCD camera was operated in full vertical binning mode (FVB) with an integration time of 0.1 s and a 10-fold accumulation. The measured signals, being the intensities of the Hg I emission line at 253.7 nm, were background corrected, using a two-point correction method on one side of the recorded line profile.
2.2. Reagents and sample preparation
All chemical compounds used during the experiments were at least of analytical grade. Water solutions were prepared with deionized water (>20 MΩ cm) obtained with a DL3-400 water deionizing system (Polwater, Poland). A stock solution of Hg (1000 mg L−1) and an ICP multi-element solution containing Al, B, Ba, Be, Bi, Ca, Cd, Co, Cr, Cu, Fe, Ga, K, Li, Mg, Mn, Na, Ni, Pb, Se, Sr, Te, Ti, and Zn at concentrations of 100 mg L−1 were obtained from Supelco (Germany). Other reagents, low-molecular weight organic compounds (LMWOCs), including acetic acid (Sigma-Aldrich, Germany), formic acid, methanol and formaldehyde (POCH, Poland), as well as propionic acid, oxalic acid, malonic acid, glycerol, ethylene glycol, acetaldehyde and ethanol (Stanlab, Poland), were also applied for the PCVG reaction. The Hg and multi-element solutions were prepared by proper dilutions of the stock solutions. The following water samples, i.e., tap water (collected from Wroclaw city, Poland), river water (collected from the Odra river in Wroclaw, Poland), and well water (collected from Debica city, Poland), were analyzed. The pH of the analyzed samples was 7.5–8. The samples were collected into 0.5 L precleaned plastic containers. Next, the water samples were filtered through 0.45 μm PTFE syringe filters. The determination of Hg by the PCVG-μAPD-OES method in water samples was performed using the calibration curve with 9 standard solutions. For that purpose, HCOOH at a final concentration of 8% (m m−1) was added to the analyzed solutions of water samples and standards. Appropriate blank solutions were also prepared and analyzed. A spike-and-recovery study was also carried out. For that purpose, the analyzed water samples were spiked with Hg. For examining the hardness of the analyzed water samples, the complexometric titration method with EDTA and Eriochrome Black T was used.132.3. The determination of microplasma parameters
The rotational temperature (Trot) of OH and N2, i.e., Trot(OH) and Trot(N2), was estimated by fitting the experimental emission spectra of these molecules with their vibrational-rotational spectra simulated by using the Liftbase (in the case of OH) and Specair (in the case of N2) computer programs. For that purpose, the OH band spectra (for the A-X system and the (0–0) transition) in the range of 306–310 nm, with the band head at 309.0 nm, and the N2 band spectra (for the C–B system and the (0–2) transition) in the range of 376–380 nm, with the band head at 380.2 nm, were acquired and analyzed. A similar procedure was applied to determine the vibrational temperature of N2 – Tvib(N2); here, four bands of the N2 (C–B) system, corresponding to the (0–2), (1–3), (2–4), and (3–5) transitions, were taken into account. The excitation temperature of the H atoms (Texc(H)) was determined using the two emission line method for two prominent lines, i.e., Hα at 656.2 nm and Hβ at 486.1 nm. The electron number density (ne) was estimated on the basis of the Stark broadening of the Hβ line. For that purpose, the experimental profile of the Hβ line was fitted using the Voight line algorithm and only the Lorentz full width at half maximum (FWHM) was considered when calculating the Stark broadening. The van der Waals broadening of the Hβ line was included in the calculations. All necessary details on the determination of Trot(OH), Trot(N2), Tvib(N2), Texc(H) and ne are given in our earlier work.143. Results and discussion
3.1. Optical characteristics of atmospheric microplasma discharge
A typical optical emission spectrum in the range near the Hg I line at 253.7 nm (240–267 nm spectra zone for the CCD camera) for the μAPD excitation source without and with the PCVG system is presented in Fig. 2. For the PCVG, the concentration of Hg was 50 μg L−1. Moreover, the addition of 8% (m m−1) HCOOH was applied in the case of the PCVG reaction system. It should be noted that in the case of the PCVG-μAPD system, the Hg line with an excitation energy of 5.3 eV was observed at 253.7 nm due to the generation of the Hg volatile species in the PCVG reactor and their transport in the Ar (plasma-supporting and carrier gas) stream into the μAPD zone. Furthermore, multiple NO bands belonging to the γ-system of the (A2Σ+ − X2Π) transition were also observed in the spectra of both μAPD systems. The main NO bands of this system were for the following transitions: (1–3) at 244.0 and 244.7 nm, (0–2) at 247.1 and 247.8 nm, (1–4) at 255.0 and 255.9 nm, and (0–3) at 258.7 and 259.6 nm. Additionally, the OH band (2–0) at 262.2 nm was clearly observed in the system with the PCVG, resulting from the transport of the H2O vapour into the microplasma zone.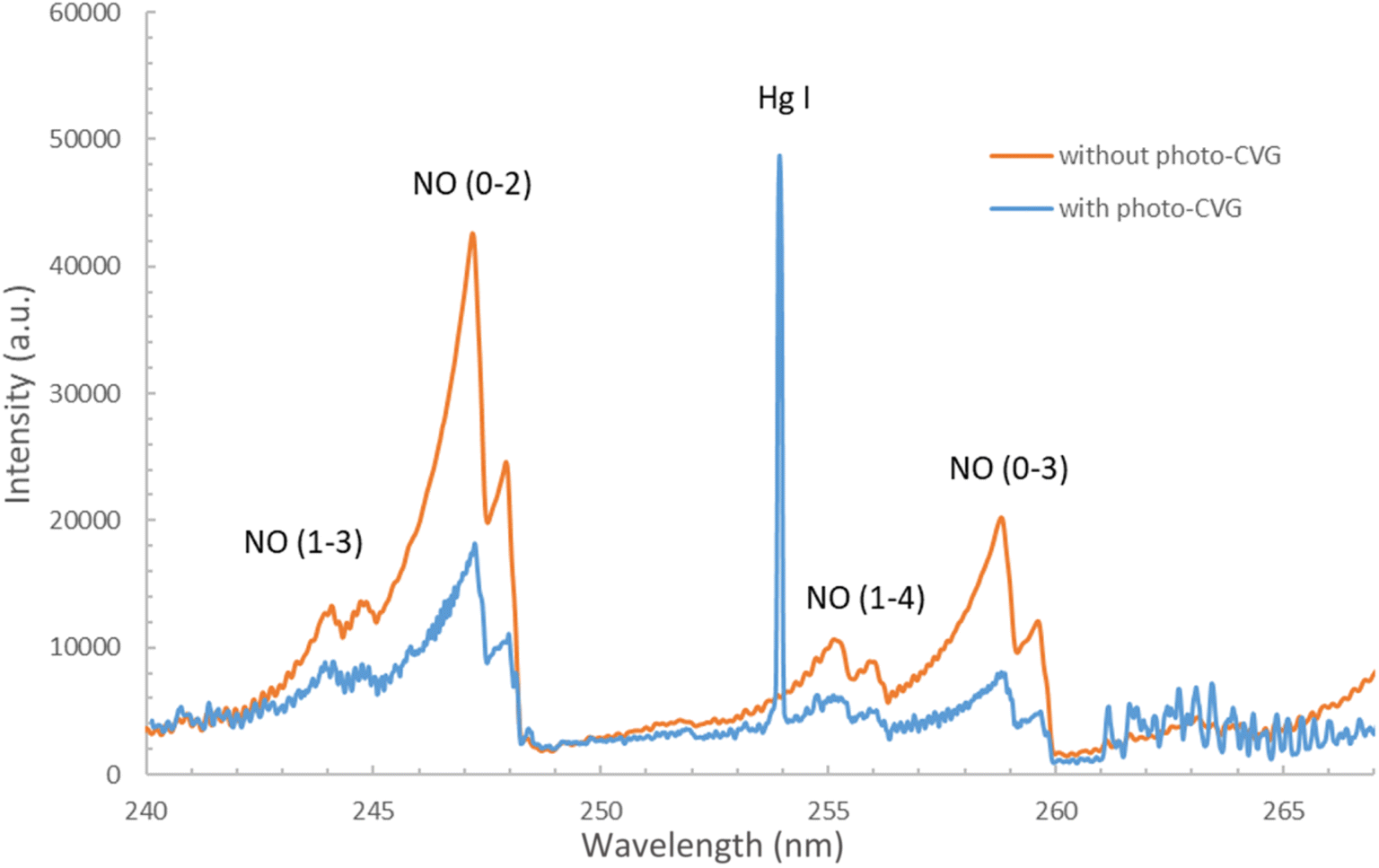 | ||
| Fig. 2 The optical emission spectrum of the microplasma discharge (μAPD) source with and without the photo-chemical vapour generation system. | ||
It was noted that the NO (A-X) bands were clearly visible in the emission spectrum of the μAPD system without the PCVG reactor. The NO molecule was likely formed from N2 and O2 present in the surrounding air atmosphere due to the following plasma chemical processes: N2 + O = NO + N and N + O2 = NO + O. The intensity of the abovementioned NO bands in the case of the μAPD system combined with the PCVG reactor was, however, almost 3-times lower than that in the case of the μAPD system. Since the presence of the NO bands near the Hg I line could be a possible source of the background signal fluctuation and the signal to background ratio lowering, the less intensive spectrum of the NO bands, particularly those for the (1–4) transition, was really promising and quite convenient. The decrease in the NO emission intensity was probably due to the quenching of the excited states of NO, i.e., NO(A), by the H2O vapor (when the liquid sample was introduced into the photoreactor) and/or H2 (product of the destruction of LMWOCs).14
The emission spectra were also collected in a wider spectral range, i.e., 200–900 nm, in order to identify the reactive species in the studied μAPD and PCVG-μAPD systems. Apparently, the NO (A-X) and N2 (C–B) emission bands were mainly identified in the 200–400 nm spectral region for both systems. The most intense bands of the N2 (C–B) system were noted at 337.1 nm (0–0), 357.7 nm (0–1) and 380.2 nm (0–2). Three strong bands of OH (A-X) with the bandheads at 262.2 nm (2–0), 289.3 nm (1–0) and 309.4 nm (0–0) were also easily excited in the case of the μAPD and PCVG-μAPD systems. Additionally, the atomic lines of H (H I at 486.1 nm and 656.2 nm) and O (O I at 777.4 and 844.6 nm), and multiple atomic lines of Ar (Ar I) in the range of 650–900 nm (with the excitation energy within 13–15 eV) were observed in both systems. It was found that the introduction of the volatile Hg species by using the PCVG reactor into the μAPD caused the growth of the intensity of the H I lines and the OH bands, probably due to the enhanced production of the H radicals in the case of the PCVG process and/or the transport of H2O vapor into the excitation source. In contrast, the intensity of the N2 and NO bands decreased, possibly due to the quenching of the excited states of N2 and/or NO by for example H2O and/or H2.14
3.2. Spectroscopic characterization of the atmospheric microplasma discharge
The selected spectroscopic parameters, i.e., Trot(OH), Trot(N2), Tvib(N2), Texc(H) and ne, were determined for the μAPD system with or without the PCVG system in order to evaluate the excitation and atomization potential of the newly developed excitation microsource. The results are presented in Table 1. Trot(N2) and Trot(OH) as well as Tvib(N2) were clearly higher in the case of the PCVG-μAPD system as compared to those assessed for the μAPD system. This could probably be a result of the transport of H2O vapor in the stream of Ar (plasma-supporting and cold vapour of Hg carrier gas) into the μAPD system. In contrast, Texc(H) and ne were higher in the case of the μAPD than those assessed for the system combined with the PCVG system. This pointed out that the electrons could be consumed by the H2O vapor and/or the possible products of the decomposition of the low-molecular weight organic compounds (LMWOCs) applied for the PCVG process.| T rot(N2), K | T rot(OH), K | T vib(N2), K | T exc(H), K | n e/1014, cm−3 | |
|---|---|---|---|---|---|
| PCVG-μAPD | 1200 ± 100 | 3100 ± 90 | 4970 ± 640 | 4800 ± 500 | 2.8 ± 0.2 |
| μAPD | 810 ± 100 | 2490 ± 140 | 3810 ± 360 | 5400 ± 500 | 3.3 ± 0.2 |
As can be seen from Table 1, for both examined μAPD systems Trot(OH) was higher than Trot(N2). The difference between these temperatures for a given system was about 1900 K (PCVG-μAPD) and 1700 K (μAPD). It could be presumed that the formation of the OH molecules, mainly coming from the H2O vapor, occurred in the plasma filament zone, while the excitation of the N2 molecules, coming from the open-air atmosphere, possibly occurred in the region of discharge outside this filament zone. Thus, Trot(OH) could represent the gas temperature of the filament zone, while Trot(N2) could relate to the mean gas temperature in the outside filament zone. Finally, for both μAPD systems examined here, the following relationship between the temperatures was observed: Trot(N2) < Trot(OH) < Tvib(N2) ≤ Texc(H). This inequality of the optical temperatures indicated a high nonequilibrium state of the studied μAPD systems. Moreover, estimated Trot in the filament zone of the microdischarge (3100 K for the PCVG-μAPD and 2490 K for the μAPD), Texc (∼4800–5400 K) and ne (∼3 × 1014 cm−3), though lower than those in ICP, i.e., 3500–4000 K, 6000–7800 K and ∼1015 cm−3, respectively,15 are sufficiently high to enable the efficient atomization and/or excitation of samples. The power density of both μAPD systems was estimated to be 12 kW cm−3 and was about 2- and 20-fold higher than the power density assessed for the APGD sustained in contact with a flowing liquid cathode (FLC) and a typical ICP system, respectively.16 The latter parameter was relatively high, primarily due to the small volume of the studied discharge.
3.3. Optimization of the μAPD system for the determination of Hg
The selected factors that influence the intensity of the Hg signal in the PCVG-μAPD-OES method were investigated to optimize the overall response of the analytical system. The sample and gas flow rates, along with the concentration of LMWOCs (alcohols, aldehydes and acids), were selected as the most important ones and examined accordingly. All experimental data were recorded using an Andor SR-500i spectrometer, monitoring the Hg I line at 253.7 nm. The intensity of this line was background-corrected, and the two-point correction method was applied. | ||
| Fig. 3 The normalized net intensity of the Hg I line as a function of (A) the Ar flow rate and (B) the sample flow rate detected for 8% m m−1 HCOOH. | ||
 | ||
| Fig. 4 The influence of the organic compounds and their concentration on the normalized net intensity of the signal of Hg in the PCVG-μAPD-OES method. | ||
It was found that for ethanol, methanol, acetaldehyde, and formaldehyde, the normalized net intensity of Hg I was lower than 10%, irrespectively of the concentration of the LMWOC used (2–10% m m−1). The signal of Hg rather slightly depended on the concentration of these compounds in the solution. This indicated that the reduction of the Hg(II) ions to the volatile Hg species in the presence of these compounds under the UV irradiation was insignificant. For other LMWOCs, such as propionic acid, malonic acid, formic acid, glycerin and ethylene glycol, the growth of their concentration in the range from 2 to 10% caused an increase in the normalized net intensity of the Hg I line from 16% to 100%, from 63 to 89%, from 86 to 100%, from 10 to 64%, and from 12 to 31%, respectively. In contrast, for oxalic acid and acetic acid, the analogous increase in the concentration of these LMWOCs resulted in a drop of the normalized intensity of the Hg I line from 60% and 37% to 42% and 19%, respectively. Finally, the highest signal of Hg was observed in the case of formic acid (at 8% m m−1), propionic acid (at 10% m m−1) and malonic acid (at 10% m m−1). It should be noted that the addition of certain LMWOCs, especially aldehydes (i.e., formaldehyde and acetaldehyde) and simple alcohols (i.e., methanol and ethanol), significantly reduced the stability of the μAPD system, leading to the higher fluctuations of the signal of Hg and the background intensity in the vicinity of the Hg I line. In the case of organic acids, the efficiency of the volatile Hg species formation during the UV irradiation seemed to be the highest among all the LMWOCs studied here. Additionally, the PCVG-μAPD system combined with the application of organic acids was distinguished by the high stability of the excitation source (RSD between 2 and 6%).
Formic acid (HCOOH) at a concentration of 8% (m m−1) was chosen for further experiments as the most convenient LMWOC for the PCVG of Hg. Similarly, this organic acid was previously reported in multiple PCVG techniques.5,10,19 Comparable results were also obtained with the use of propionic and malonic acids.
Considering that the PCVG efficiency was enhanced by increasing the addition of formic acid, two possible pathways could be responsible for that during the UV irradiation,10,18i.e., the formation of H radicals (HCOOH + hv → H˙ + ˙COOH → H2 + CO2) and/or CO radicals (HCOOH + hv → HCO˙ + ˙OH → CO + H2O). Both radicals generated under these conditions, i.e., H, CO, by the UV irradiation of HCOOH could reduce the Hg(II) ions present in the solution to the volatile Hg species.
3.4. Analytical performance and application
The analytical figures of merit were determined for the optimal parameters of the PCVG-μAPD system, i.e., flow rate of Ar: 20 mL min−1, sample flow rate: 2 mL min−1, and the presence of 8% (m m−1) HCOOH. A typical calibration curve was linear (R2 = 0.996) up to 100 μg L−1 (see Fig. 5). The precision of the measured intensity of the Hg I line (n = 9) was expressed as the relative standard deviation (RSD), and it was varied from 2.7% to 4.7% for different concentrations within the established linearity range. For other LMWOCs, the precision ranged from 4% to 12% (see Table 2). It is worth noting that similar linearity ranges of the calibration curves were observed for oxalic acid, acetic acid, propionic and malonic acid. To assess the usability of the applied LMWOC reagents, the LODs of Hg were calculated using the 3σ criterion.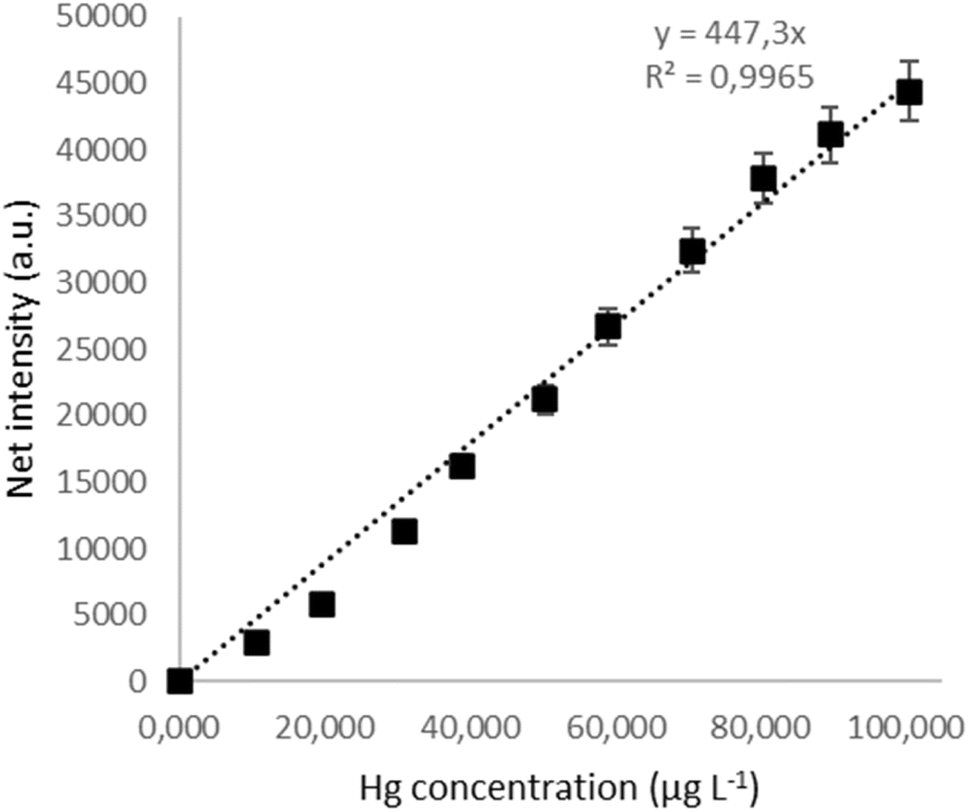 | ||
| Fig. 5 The typical calibration curve for 8% (m m−1) HCOOH. | ||
| LMWOCs | Concentration (m m−1) | LOD, μg L−1 | Precisiona (RSD), % |
|---|---|---|---|
| a Precision as RSD for 9 independent measurements. b Hg at a concentration of 10 μg L−1. c Hg at a concentration of 50 μg L−1. | |||
| Formic acid | 8% | 0.33 | 3.1b |
| Oxalic acid | 2% | 0.49 | 4.1b |
| Acetic acid | 2% | 0.74 | 6.2b |
| Propionic acid | 10% | 0.62 | 3.8b |
| Malonic acid | 10% | 0.34 | 4.0b |
| Methanol | 2% | 14.9 | 12.5c |
| Ethanol | 2% | 24.4 | 10.5c |
| Glycerin | 10% | 0.98 | 4.8c |
| Ethylene glycol | 10% | 1.80 | 5.5c |
| Formaldehyde | 10% | 19.3 | 11.5c |
| Acetaldehyde | 2% | 11.2 | 10.4c |
The determined LODs for the studied LMWOCs at their optimal concentrations are presented in Table 2. The lowest LOD of Hg (0.33 μg L−1) was noted for formic acid at a concentration of 8% (m m−1). The corresponding LOD values were also obtained for other organic acids, i.e., malonic acid, propionic acid, oxalic acid, and acetic acid, namely 0.34, 0.62, 0.49 and 0.74 μg L−1, respectively.
The obtained LOD of Hg as well as the precision for the PCVG-μAPD-OES method using 8% (m m−1) formic acid were comparable to those reported in the literature for other comparable techniques, as presented in Table 3. It should be noted that the LOD of Hg obtained here was comparable with ICP-OES coupled with PCVG and CVG techniques.6
| Sample introduction technique | Detection system | LOD, μg L−1 | Precision (RSD), % | Reference |
|---|---|---|---|---|
| a PCVG – photo-induced chemical vapor generation, CVG – chemical vapor generation, ICP – inductively coupled plasma, OES – optical emission spectrometry, AFS – atomic fluorescence spectrometry, APGD – atmospheric pressure glow discharge, HPLC – high-pressure liquid chromatography, and PD – point discharge. | ||||
| PCVG | ICP-OES | 1.20 | 9.5 | 20 |
| AFS | 0.02 | — | 18 | |
| AFS | 0.003 | 4.0 | 10 | |
| PD-OES | 0.10 | — | 21 | |
| μAPD-OES | 0.33 | 2.7–4.7 | This work | |
| CVG | ICP-OES | 0.1 | — | 6 |
| AFS | 0.08 | 4.8 | 22 | |
| μAPGD-OES | 2.3 | 4.1 | 23 | |
| APGD-OES | 0.26 | 0.7 | 8 | |
| PD-OES | 0.15 | 2.7 | 9 | |
| PD-OES | 0.10 | 4.5 | 10 | |
| PD-OES | 0.10 | 4.0 | 11 | |
| Plasma-assisted CVG | AFS | 0.03 | 6.2 | 24 |
| ICP-OES | 0.70 | 1.2 | 25 | |
| HPLC-AFS | 0.67 | 5.9 | 26 | |
| APGD-OES | 0.70 | 3.5 | 27 | |
To assess the potential analytical application of the method, at first the chemical interferences were studied. In this case, standard solutions of Hg (50 μg L−1), admixed with a multi-element standard solution containing elements such as: Al, B, Ba, Be, Bi, Ca, Cd, Co, Cr, Cu, Fe, Ga, K, Li, Mg, Mn, Na, Ni, Pb, Se, Sr, Te, Ti, and Zn, were prepared and analyzed by PCVG-μAPD-OES versus the calibration using the standard solutions not spiked with these elements. The concentration range of each interfering element was changed from 1 to 20 mg L−1. Measuring the Hg concentration in such sample solutions, it was found that for the solution containing 1 mg L−1 of each of the elements, the recovery of Hg was 103 ± 3.9%. However, when this concentration increased to 10 mg L−1 and 20 mg L−1, the recovery of Hg was lower, i.e., 96 ± 1.6% and 85 ± 3.0%, respectively. It was also established in the additional experiments that the determination of Hg by PCVG-μAPD-OES was not prone to the chemical interferences originating from the separately studied easily ionized elements (EIEs). Here, it was established that the following concentrations of the EIEs in the Hg solutions could be tolerated: up to 100 mg L−1 for Na, up to 20 mg L−1 for K, up to 200 mg L−1 for Ca, and up to 30 mg L−1 for Mg.
Finally, the PCVG-μAPD-OES method was employed for monitoring the Hg level in natural waters. The concentration of Hg was determined in three water samples: tap water, well water, and river water. In this case, formic acid (8% m m−1) was added for the efficient PCVG and the external calibration was used. Unfortunately, the concentration of Hg in these samples was below the LOD achievable with this method, i.e., 0.33 μg L−1. For that reason, the analyzed water samples were spiked with a standard Hg solution to a final concentration of 50 μg L−1, and the recoveries of Hg were evaluated. The results on the determination of Hg in the analyzed water samples are presented in Table 4 along with the hardness of these water samples. The recoveries obtained, being within 94–110%, confirmed the possible usability of the presented system for the in situ analysis of environmental water samples by the developed PCVG-μAPD system.
| Water origin (hardness) | Added (μg L−1) | Determined (μg L−1) | Recovery ± SD (%) |
|---|---|---|---|
| a dH: degree of hardness (1 dH represents 10 mg of CaO in 1 L of water, which corresponds to 7.1 mg L−1 of Ca or 4.3 mg L−1 Mg). | |||
| Tap (11.3 dH) | 0 | <0.33 | — |
| 50 | 47 ± 3 | 94 ± 6 | |
| Well (24.8 dH) | 0 | <0.33 | — |
| 50 | 48.5 ± 1.5 | 97 ± 3 | |
| River (15.9 dH) | 0 | <0.33 | — |
| 50 | 55 ± 2.5 | 110 ± 5 | |
As is presented in Table 4, the results of the Hg recoveries (94–110%) were acceptable independently of the hardness of water, which varied from 11 to 25 degrees of hardness, corresponding to 78–178 mg L−1 of Ca, and corroborated the previous findings about the lack of the chemical interferences in the PCVG-μAPD system from the EIEs. As such, it was confirmed that the typical composition of environmental water should not negatively influence the use of the examined method in its analysis.
4. Conclusions
This work presented an inexpensive and simple excitation microsource for optical emission spectrometry based on a μAPD coupled with the PCVG technique for the reliable determination of Hg. According to the World Health Organization (WHO) guidelines,28 the concentration of inorganic Hg in water consumed by humans should not exceed 6 μg L−1, which could mean that the excitation source presented here could be easily applied to monitor water and environmental pollution related to this toxic element. The LOD of Hg assessed with this method was 0.33 μg L−1; however, it could be expected that with sensitive optical fiber portable spectrometers, this LOD could be even better. Although the developed PCVG-μAPD-OES method provided a slightly worse LOD than that reported for CVG-ICP-OES, i.e., 0.1 μg L−1, it was considered to meet the green analytical chemistry standards and would be preferred over the CVG due to the absence of hazardous chemical compounds such as THB (NaBH4).The stable operation of the proposed μAPD excitation source under miniature flow rates of Ar (20 mL min−1) and a very low power consumption (∼10 W) makes the device inexpensive to maintain and operate. Spike-and-recovery and interference studies indicate that the apparatus could function as a highly specialized device for the trace Hg determination and/or as a handy tool for the rapid assessment of the potential Hg pollution in environmental water. Improving the stability of the μAPD and coupling it with a small portable spectrometer could be a very promising way to obtain a novel, fast, cheap, and easy-to-use apparatus capable of serving the role of an environmental pollution sensor.
Data availability
The authors declare that the data supporting the findings of this study are included within the paper and its ESI files.† Should any data files (e.g. raw data) be needed in another format, they are available from the corresponding author upon reasonable request. Source data are provided with this paper.Author contributions
Tymoteusz Klis: writing – original draft, methodology, investigation, writing – review & editing, visualization, data curation, conceptualization, formal analysis. Pawel Pohl: writing – review & editing and funding acquisition. Anna Dzimitrowicz: validation and writing – review & editing. Piotr Jamroz: writing – original draft, writing – review & editing, methodology, conceptualization, supervision, project administration, and formal analysis.Conflicts of interest
The authors declare no conflicts of interests.Acknowledgements
All studies reported here were funded by the National Science Centre (NCN), Poland, according to the Opus 17 project (UMO-2019/33/B/ST4/00356).References
- B. Gworek, W. Dmuchowski and A. H. Baczewska-Dabrowska, Environ. Sci. Eur., 2020, 32, 128 CrossRef CAS
.
- K. Leopold, M. Foulkes and P. Worsfold, Anal. Chim. Acta, 2010, 663, 127–138 CrossRef CAS PubMed
- Z. Cai and Z. Wang, Anal. Chim. Acta, 2022, 1203, 339724 CrossRef CAS PubMed
- Z. Cai and Z. Wang, Spectrochim. Acta, Part B, 2023, 199, 106578 CrossRef CAS
- Y. Yin, J. Liu and G. Jiang, TrAC, Trends Anal. Chem., 2011, 30, 1672–1684 CrossRef CAS
- P. Pohl, K. Greda, A. Dzimitrowicz, M. Welna, A. Szymczycha-Madeja, A. Lesniewicz and P. Jamroz, TrAC, Trends Anal. Chem., 2019, 113, 234–245 CrossRef CAS
- P. Pohl, P. Jamroz, K. Greda, M. Gorska, A. Dzimitrowicz, M. Welna and A. Szymczycha-Madeja, Anal. Chim. Acta, 2021, 1169, 338399 CrossRef CAS PubMed
- R. Rong, Z. Cai, X. Li and Z. Wang, J. Anal. At. Spectrom., 2022, 37, 2377–2382 RSC
- X. Yuan, K. Li, Y. Zhang, Y. Miao, Y. Xiang, Y. Sha, M. Zhang and K. Huang, Microchem. J., 2020, 155, 104695 CrossRef CAS
- C. Zheng, Y. Li, Y. He, Q. Ma and X. Hou, J. Anal. At. Spectrom., 2005, 20, 746–750 RSC
- J. Yang, Y. Lin, L. He, Y. Su, X. Hou, Y. Deng and Ch. Zheng, Anal. Chem., 2021, 93, 14923–14928 CrossRef CAS PubMed
- M. Li, K. Li, L. He, X. Zeng, X. Wu, X. Hou and X. Jiang, Anal. Chem., 2019, 91, 7001–7016 CrossRef CAS PubMed
-
S. Gupta and V. K. Jena, A Guide Book of Experiments in Applied Chemistry, LP Inc. Publisher North Carolina, USA, 2017 Search PubMed
- P. Jamroz, W. Zyrnicki and P. Pohl, Spectrochim. Acta, Part B, 2012, 73, 26–34 CrossRef CAS
- J. Borkowska-Burnecka, W. Zyrnicki, M. Wełna and P. Jamroz, Int. J. Spectrosc., 2016, 7521050, 1–9 Search PubMed
- P. Jamroz, P. Pohl and W. Zyrnicki, J. Anal. At. Spectrom., 2012, 27, 1032–1037 RSC
- Q. He, Y. Qiao, M. Zhao and J. Zhang, Spectrochim. Acta, Part B, 2022, 191, 106396 CrossRef CAS
- C. Han, C. Zheng, J. Wang, G. Cheng, Y. Lv and X. Hou, Anal. Bioanal. Chem., 2007, 388, 825–830 CrossRef CAS PubMed
- E. Covaci, M. Senila, C. Tanaselia, S. B. Angyus, M. Ponta, E. Darvasi, M. Frentiu and T. Frentiu, J. Anal. At. Spectrom., 2018, 33, 799–808 RSC
- B. B. A. Francisco, A. A. Rocha, P. Grinberg, R. E. Sturgeon and R. J. Cassella, J. Anal. At. Spectrom., 2016, 31, 751–758 RSC
- S. Zhang, H. Luo, M. Peng, Y. Tian, X. Hou, X. Jiang and Ch. Zheng, Anal. Chem., 2015, 87, 10712–10718 CrossRef CAS PubMed
- X. Wen, Y. Gao, P. Wu, Z. Tan, C. Zheng and X. Hou, J. Anal. At. Spectrom., 2016, 31, 415–422 RSC
- K. Greda, P. Jamroz and P. Pohl, J. Anal. At. Spectrom., 2014, 29, 893–909 RSC
- Z. Liu, Z. Zhu, Q. Wu, S. Hu and H. Zheng, Analyst, 2011, 136, 4539–4544 RSC
- Z. Zhu, G. C. Y. Chan, S. J. Ray, X. Zhang and G. M. Hieftje, Anal. Chem., 2008, 80, 7043–7050 CrossRef CAS PubMed
- Q. He, Z. Zhu, S. Hu and L. Jin, J. Chromat. A, 2011, 1218, 4462–4467 CrossRef CAS PubMed
- K. Greda, K. Swiderski, P. Jamroz and P. Pohl, Anal. Chem., 2016, 88, 8812–8820 CrossRef CAS PubMed
-
WHO/SDE/WSH/05.08/10, Mercury in Drinking-Water, World Health Organization 2005 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ja00306c |
| This journal is © The Royal Society of Chemistry 2024 |