
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development and application of reference and routine analytical methods providing SI-traceable results for the determination of technology-critical elements in PCB from WEEE†
Giancarlo
D'Agostino
 *a,
Marcus
Oelze
b,
Jochen
Vogl
b,
Jean-Philippe
Ghestem
c,
Nicolas
Lafaurie
c,
Ole
Klein
d,
Daniel
Pröfrock
d,
Marco
Di Luzio
a,
Luigi
Bergamaschi
a,
Radojko
Jaćimović
e,
Caroline
Oster
f,
Johanna
Irrgeher
g,
Shaun T.
Lancaster
g,
Anna
Walch
g,
Anita
Röthke
h,
Lena
Michaliszyn
h,
Axel
Pramann
h,
Olaf
Rienitz
h,
Timo
Sara-Aho
i,
Oktay
Cankur
j,
Derya
Kutan
jl and
Johanna
Noireaux
k
*a,
Marcus
Oelze
b,
Jochen
Vogl
b,
Jean-Philippe
Ghestem
c,
Nicolas
Lafaurie
c,
Ole
Klein
d,
Daniel
Pröfrock
d,
Marco
Di Luzio
a,
Luigi
Bergamaschi
a,
Radojko
Jaćimović
e,
Caroline
Oster
f,
Johanna
Irrgeher
g,
Shaun T.
Lancaster
g,
Anna
Walch
g,
Anita
Röthke
h,
Lena
Michaliszyn
h,
Axel
Pramann
h,
Olaf
Rienitz
h,
Timo
Sara-Aho
i,
Oktay
Cankur
j,
Derya
Kutan
jl and
Johanna
Noireaux
k
aIstituto Nazionale di Ricerca Metrologica (INRIM), Unit of Radiochemistry and Spectroscopy c/o Department of Chemistry, University of Pavia, via Taramelli 12, Pavia, 27100, Italy. E-mail: g.dagostino@inrim.it; m.diluzio@inrim.it; l.bergamaschi@inrim.it
bBundesanstalt für Materialforschung und –prüfung (BAM), Inorganic Trace Analysis, Richard-Willstätter-Straße 11, Berlin, 12489, Germany. E-mail: marcus.oelze@bam.de; jochen.vogl@bam.de
cBureau de Recherches Géologiques et Minières (BRGM), Water, Environment, Processes and Analysis Department, 3 avenue Claude Guillemin, 45000 Orleans, France. E-mail: jp.ghestem@brgm.fr; n.lafaurie@brgm.fr
dHelmholtz-Zentrum Hereon (Hereon), Max-Planck-Straße 1, 21502 Geesthacht, Germany. E-mail: ole.klein@hereon.de; daniel.proefrock@hereon.de
eJožef Stefan Institute (JSI), Department of Environmental Sciences, Jamova cesta 39, Ljubljana, 1000, Slovenia. E-mail: radojko.jacimovic@ijs.si
fLaboratoire Nationale des Essais et de la métrologie (LNE), Department Environment and Climate change, 1 rue Gaston Boissier, Paris, 75015, France. E-mail: caroline.oster@lne.fr
gChair of General and Analytical Chemistry, Montanuniversität Leoben (MUL), Franz Josef-Straße 18, Leoben, 8700, Austria. E-mail: johanna.irrgeher@unileoben.ac.at; shaun.lancaster@unileoben.ac.at
hPhysikalisch-Technische Bundesanstalt (PTB), Bundesallee 100, Braunschweig, 38116, Germany. E-mail: anita.roethke@ptb.de; lena.michaliszyn@ptb.de; axel.pramann@ptb.de; olaf.rienitz@ptb.de
iFinnish Environment Institute (Syke), Research Infrastructure, Metrology, Mustialankatu 3, Helsinki, 00790, Finland. E-mail: timo.sara-aho@syke.fi
jNational Metrology Institute of Turkey (TUBITAK), Inorganic Chemistry Laboratory, Gebze Kocaeli, 41470, Turkey. E-mail: oktay.cankur@tubitak.gov.tr
kLaboratoire Nationale des Essais et de la métrologie (LNE), Division Chemistry-Biology, 1 rue Gaston Boissier, Paris, 75015, France. E-mail: johanna.noireaux@lne.fr
lDepartment of Physics, Gebze Technical University, Gebze, 41400, Kocaeli, Turkey. E-mail: derya_kutan_@hotmail.com
First published on 18th September 2024
Abstract
The recovery and reprocessing of technology-critical elements (TCE) present in printed circuit boards (PCB) from electrical and electronic waste is essential both for recycling valuable materials subject to supply risk and for reducing the environmental impact. Although the quantitative knowledge of TCE amounts in end-of-life PCB plays a key role, there are neither matrix certified reference materials nor harmonized analytical methods available to establish the traceability of the results to the International System of Units. To fill these gaps, we developed and applied five reference analytical methods based on ICP-MS standard addition calibrations and INAA k0- and relative calibrations suitable to certify reference materials. In addition, we developed and tested six analytical methods based on more commonly used ICP-MS external standard calibrations to provide industry with routine analysis methods. Twenty TCE (Ag, Au, Co, Cu, Dy, Ga, Gd, Ge, In, La, Li, Nd, Ni, Pd, Pr, Pt, Rh, Sm, Ta and Ti) were selected as target analytes and a batch of powdered PCB was used as measurement material. An overall mutual agreement was observed among data collected by reference methods at a few percent relative uncertainty levels. Moreover, all but one of the methods developed for routine analysis demonstrated their suitability in industrial applications by producing data within ± 20% of the values established with reference methods.
1. Introduction
The production of electrical and electronic waste (WEEE) is increasing exponentially as a result of the global use of high-tech equipment combined with shortening of their lifespan. According to the Global E-waste Monitor 2024,1 the total amount of WEEE generated worldwide in 2022 was 62 × 106 t and it is now anticipated to reach 82 × 106 t in 2030. On the other end, only 22.3% of the WEEE was collected and recycled in 2022 with all its consequences concerning loss of valuable materials and environmental impact.To address the issue of the increasing amount of WEEE, the European Union has introduced the directives WEEE 2012/19/EU and RoHS 2011/65/EU; the first sets collection and recycling targets for all WEEE types while the latter restricts the use of hazardous materials in the production of electronic and electrical goods. Recently, the Circular Economy Action Plan of the European Green Deal has given incentives to increase the collection and recycling of electrical and electronic products to contribute to the supply of materials for the digital and environmental transition.
Management costs of the current and future WEEE stream can be partially covered by recovery and reprocessing of noteworthy amounts of technology-critical elements (TCE) found in the waste, declared as “critical” due to their risk of supply shortage and vital importance for the production of new technologies.2
While the knowledge of TCE content is essential both to determine the economic value of the WEEE and to increase the recycling processes, no specific and harmonized analytical methods are available. In addition, there are no matrix certified reference materials (CRM) to validate or improve the analytical methods and/or establish traceability of the results to the International System of Units (SI). Only the BAM-M505a electronic scrap is available but, since it was obtained by melting with pyrite, it does not consist exclusively of WEEE. This has a direct impact in WEEE recycling, e.g., discrepancies in collected measurement data and difficulties in comparing the efficiency of recovery processes.3
One of the major problems that must be addressed in quantitative elemental analysis of WEEE performed with techniques requiring the chemical degradation of sample matrix in solution is the high heterogeneity of the materials, which are complex mixtures of metals, plastics and organic substances.
Printed circuit boards (PCBs) are a promising source of recycling as they can contain up to 60 elements, most of them TCE. So far, however, only the traditional valuable metals are being recycled such as copper, gold, silver and platinum group metals. For determining the elemental composition of PCB in selected smartphones, Bookhagen et al. developed and validated with the ERM®-EZ505 (electronic scrap with pyrite reference material, no longer available) a multipurpose analytical method based on inductively coupled plasma optical emission spectrometry (ICP-OES) and mass spectrometry (ICP-MS). Mass fractions of Ag, Al, As, Au, Ba, Bi, Ca, Cd, Ce, Co, Cr, Cu, Dy, Er, Eu, Fe, Ga, Ge, Hf, Ho, In, La, Li, Lu, Mg, Mn, Mo, Na, Nb, Nd, Ni, Pb, Pd, Pt, Rb, Sb, Sc, Si, Sm, Sn, Sr, Ta, Tb, Ti, Tm, V, W, Y, Yb, Zn and Zr were obtained, including measurement uncertainties.4
A preliminary study for the production and characterization of a reference material for PCB was recently carried out using ICP-OES and instrumental neutron activation analysis (INAA).5 Mass fractions of Ag, Al, As, Au, Ba, Cr, Cu, Fe, Ni, Pb, Sb, Si, Sn and Zn were reported, including measurement uncertainties but without stated evidence of SI traceability.6
In the present study, a similar approach was adopted by focusing on TCE qualified as critical and strategic by the European Commission, e.g. Ag, Au, Co, Cu, Dy, Ga, Gd, La, Li, Nd, Ni, Pd, Pr, Pt, Sm and Ta. We developed and applied reference analytical methods based on ICP-MS standard addition calibrations and INAA k0- and relative calibrations establishing traceability to the SI. In addition, we developed and tested analytical methods based on ICP-MS external standard calibrations, more commonly used in measurement laboratories and suitable as routine methods.
Equation models describing the operation of the developed reference methods are recalled. Sample preparation methods that allow for PCB digestion and measurement procedures are described. Finally, results obtained with ICP-MS based on external calibration are compared to results obtained with reference methods to test their suitability in routine measurements.
2. Description of the analytical methods and the calibration strategies
2.1 ICP-MS
In the field of ICP-MS, various calibration strategies have been established, ranging from simple external calibration to more elaborate methods such as standard addition and isotope dilution. In the light of the extraordinary complexity of the PCB matrix, the application of ICP-MS standard addition techniques following complete sample digestion is a reasonable choice to develop reference methods, because the calibration within the matrix itself eliminates virtually completely biases arising from different sensitivities of the analytes between the sample matrix and the calibration standards.7 The same applies to matrix-matched standard addition techniques when sample matrix effects are fully compensated for. In the case of elements with at least two isotopes, isotope dilution techniques are also possible but the higher experimental costs and efforts often prevent their use.Analytical methods based on external standard techniques without matrix matching might yield biased results when applied to PCB. However, because of their experimental simplicity, they are suitable to develop and optimize digestion protocols, and, in case results are in agreement with reference methods, they can be adopted for routine analysis in industrial research and development laboratories.
There are different calibration methods available for standard addition. Among them, the so-called gravimetric standard addition with natural internal standard requires a standard z with an SI traceable mass fraction of the analyte X, wz, and an element Y already present in the sample matrix acting as a “natural” internal standard y.
The measurement equation, linking the mass fraction of X in a sample, wx, to wz, and adopted in this study is8
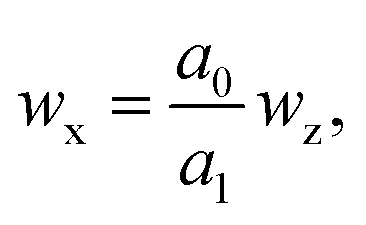 | (1) |
 | (2) |
A further measurement equation adopted in this study, linking the mass fraction of X in a sample, wx, to an SI traceable standard and developed for matrix-matched standard addition calibration is provided as well by the least squares fit of yi = a1xi + a0 to (yi, xi) data, where a0 is the intercept (signal intensity from the standard solution i which no standard is added to), and a1 is the slope of the line. Specifically:
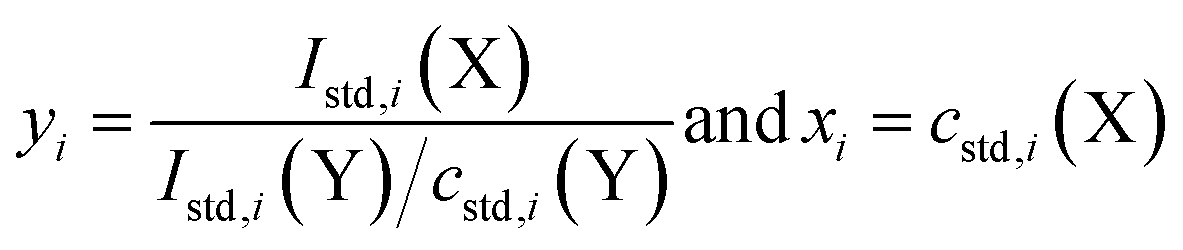 | (3) |
Then the mass fraction of the analyte in the sample can be calculated according to
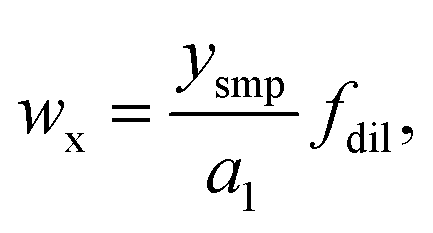 | (4) |
 with csmp(Y) the concentration of Y into the sample solution, and Ismp(X) and Ismp(Y) the signal intensities of X and Y from sample solution, respectively.
with csmp(Y) the concentration of Y into the sample solution, and Ismp(X) and Ismp(Y) the signal intensities of X and Y from sample solution, respectively.
Symbols adopted in the measurement equations of the ICP-MS gravimetric standard addition with natural internal standard and matrix-matched standard addition calibrations are summarized in Table 1.
| Symbol | Quantity |
|---|---|
| a Gravimetric standard addition with natural internal standard. b Matrix-matched standard addition. | |
| X | Analytea,b |
| Z | Added standarda |
| Y | Internal standarda,b |
| w x | Mass fraction of Xa,b |
| w z | Mass fraction of Za |
| a 0 | y-Intercepta,b |
| a 1 | Slopea,b |
| m i | Mass of the i-th solutiona |
| ρ i | Density of the i-th solutiona |
| m z,i | Mass of Z in the i-th solutiona |
| m smp,i | Mass of the sample in the i-th solutiona |
| I i (X) | Signal intensity of X from the i-th solutiona |
| I i (Y) | Signal intensity of Y from the i-th solutiona |
| c std,i(X) | Concentration of X in the i-th standard solutionb |
| c std,i(Y) | Concentration of Y in the i-th standard solutionb |
| c smp,i(Y) | Concentration of Y in the sample solutionb |
| I std,i(X) | Signal intensity of X from the i-th standard solutionb |
| I std,i(Y) | Signal intensity of Y from the i-th standard solutionb |
| I smp(X) | Signal intensity of X from the sample solutionb |
| I smp(Y) | Signal intensity of Y from the sample solutionb |
| f dil | Dilution factorb |
2.2 INAA
Despite the limited availability of laboratories equipped with the necessary facilities and relatively high experimental costs, INAA is a realistic choice along with ICP-MS standard addition to develop candidate reference methods because measurements can be performed in the solid state without difficulties associated with dissolving PCB samples. Measurement procedures are improved by adjusting neutron irradiation, radioactivity decay and gamma counting times to reduce or, in some cases, to cancel out matrix interferences.There are two calibration methods available: the single comparator, hereafter called k0-INAA,9 and the direct comparator, hereafter called relative-INAA.10 The first allows to measure any analyte X with a single element Y in a standard y, the latter requires a standard z for each analyte X to be measured.
The measurement equation adopted in this study for the k0-INAA is
 | (5) |
In case of relative-INAA calibration method, the single standard y is replaced by a direct standard z of the analyte X to be measured. The measurement equation is:
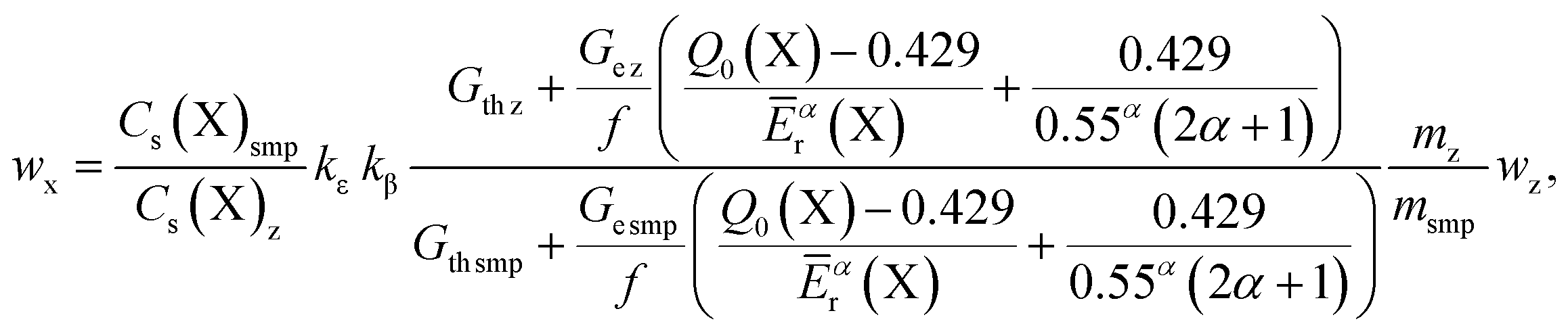 | (6) |
Symbols adopted in the measurement equations of the k0 and relative INAA calibrations are summarized in Table 2.
| Symbol | Quantity |
|---|---|
| a k 0. b Relative. | |
| X | Analytea,b |
| Y | Single standarda |
| Z | Direct standardb |
| w x | Mass fraction of Xa,b |
| w y | Mass fraction of Ya |
| w z | Mass fraction of Zb |
| m smp | Mass of the samplea,b |
| C s(X)smp | γ rays count rate at saturation of X in the samplea,b |
| C s(Y)y | γ rays count rate at saturation of Y in the single standarda |
| C s(X)z | γ rays count rate at saturation of X in the direct standardb |
k
0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Au(X) Au(X) |
k 0 value of Xa |
|
k
0Au(Y) |
k 0 value of Ya |
| k ε | γ efficiency ratio (Y/X)a,b |
| k β | Neutron flux gradient correctiona,b |
|
G
thsmp |
Thermal neutron self-shielding correction of the samplea,b |
|
G
thy |
Thermal neutron self-shielding correction of the single standarda |
|
G
thz |
Thermal neutron self-shielding correction of the direct standarda |
|
G
esmp |
Epithermal neutron self-shielding correction of the samplea,b |
|
G
ey |
Epithermal neutron self-shielding correction of the single standarda |
|
G
ez |
Epithermal neutron self-shielding correction of the direct standardb |
| f | Thermal to epithermal conventional flux ratioa,b |
| Q 0(X) | Resonance integral to thermal cross section ratio of Xa,b |
| Q 0(Y) | Resonance integral to thermal cross section ratio of Ya |
| Ē r(X) | Effective resonance energy of Xa,b |
| Ē r(Y) | Effective resonance energy of Ya |
| α | Deviation of the epithermal flux from the 1/E trenda,b |
3. Experimental
Ten laboratories with well-established experience in measuring trace elements in different matrices, hereafter called L1, L2, L3, L4, L5, L6, L7, L8, L9 and L10, participated in this study. The adopted techniques and calibration methods are summarized in Table 3. The target analytes were Ag, Au, Co, Cu, Dy, Ga, Gd, Ge, In, La, Li, Nd, Ni, Pd, Pr, Pt, Rh, Sm, Ta and Ti.| Laboratory | Technique | Calibration |
|---|---|---|
| a Matrix-matched to alkali flux blank. b With natural PCB material internal standard. c Matrix-matched to PCB material. | ||
| L1 | ICP-MS | External standard |
| L2 | ICP-MS | External standard |
| L3 | ICP-MS | External standard |
| L4 | ICP-MS | External standarda |
| L5 | ICP-MS | External standard |
| L6 | ICP-MS | Standard additionb |
| L7 | ICP-MS | Standard additionc |
| L8 | ICP-MS | Standard additionb and external standard |
| L9 | INAA | k 0 and relative |
| L10 | INAA | k 0 |
The PCB material was selected from small WEEE category (audio and video appliances, toys, personal care products, culinary equipment, etc.). Approximately 500 kg of waste was collected in an industrial sorting chain by the waste recovery company “Envie 2E Midi-Pyrenees” (France). The wastes were dismantled manually to extract the PCB. The resulting sample (485 kg) was first shredded with an industrial cutting mill (Bohmier Maschinen GmbH) to a particle size <30 mm. A quarter of the sample (122 kg) was then shredded to a particle size <10 mm and divided to obtain sub-sample masses of 4 kg. Three of these subsamples were used for testing processing methodologies from particle size <10 mm to (i) 2 mm, (ii) 750 μm and (iii) 200 μm, respectively. In detail, (i) consisted of 1 shredding step to 2 mm in a lab knife mill (Retsch SM2000), (ii) consisted of 3 shredding steps to 2 mm, 1 mm and 750 μm in the lab knife mill and (iii) consisted of 1 shredding step to 750 μm in the lab knife mill and 1 milling step to 200 μm in a universal grinder (FL1 Poittemille; Poittemille Company, Bethune, France) using ring holes. The losses of material were <1% in (i) and (ii) while in (iii) they are unknown. No particles were removed during the shredding steps. Sampling and processing methodologies are described in ref. 11 and 12.
The 200 μm particle size PCB powder obtained from the application of methodology (iii) was sent to the participating laboratories for the development of the analytical methods. A preliminary TCE survey of the material carried out by ICP-MS highlighted (i) Co at hundreds of μg g−1 level, (ii) Ga, Nd, Pr, La, Au and Pd at tens of μg g−1 level, (iii) Ge, Dy and Gd at μg g−1 level and (iv) Pt and Rh at tenths of μg g−1 level. In addition, the preliminary survey highlighted (i) Fe and Cu at tens of percent level, (ii) Al, Sn, Br and Zn at percent level, (iii) Ba, Cr, Mn, Ni, Pb, Sb and W at thousands of μg g−1 level, (iv) Ag, Mo and Sr at hundreds of μg g−1 level, and (v) As and Cd at tens of μg g−1 level. No homogeneity measurements were made.
For each laboratory, methods adopted to prepare PCB samples and SI standards, and measurement procedures are reported below.
3.1 Sample and standard preparation methods
L1 – The sample preparation process involved three separate aliquots of the PCB material, each weighing approximately 1 g. These aliquots were subjected to ashing, and the resulting reduction in mass was recorded. From each of these aliquots, three additional individual aliquots weighing 200 mg each were extracted. These aliquots underwent treatment with 5 mL aqua regia at 120 °C for a duration of 24 h. Subsequently, each of the nine resulting samples was measured three times using external calibration based on gravimetrically prepared SI traceable standard solutions.
L2 – Sample digestion was carried out using (i) microwave aqua regia (Anton Paar Mutiwave GO) or (ii) peroxide fusion. In (i), 500 mg of sample was mixed with 8 mL of aqua regia (3/1 HCl/HNO3) and heated to 175 °C for 30 min. In (ii), the sample was first calcinated at 550 °C, then it was fused with sodium peroxide by mixing 300 mg sample with 2 g of sodium peroxide in a zirconium crucible; the mixture was heated to 700 °C and the sample was taken up in 80 mL water acidified with 5 mL HNO3. SI traceable standard solutions were gravimetrically prepared for calibration.
L3 – All preparatory laboratory work was performed in a class 10000/1000 clean room. Type I reagent-grade water (18.2 MΩ cm) was obtained from a Milli-Q Integral water purification system equipped with a QPod-Element polishing system (Merck-Millipore, Darmstadt, Germany). Analytical grade HNO3 (w = 65%, Fisher Scientific, Schwerte, Germany) and analytical grade HCl (w = 30%, Carl Roth, Karlsruhe, Germany) were further purified by double sub-boiling in perfluoralkoxy-polymer (PFA)-subboling stills (DST-4000 & DST-1000, Savillex, Minnesota, USA) operated under clean room conditions. HBF4 (w = 38%, Chem-Lab, Zedelgem, Belgium) was used in ultra-pure quality for sample digestion without any further purification.
Standard solutions (all traceable to NIST standards) for calibration were prepared from either single elements as well as custom-made multi-element standards of different composition (Inorganic Ventures, Christiansburg, USA).
The aliquots (300 mg ± 5 mg) of the material were digested in triplicates with 5 mL HNO3, 2 mL HCl and 1 mL HBF4 for 300 min at 180 °C either with a MARS Xpress or a MARS 6 microwave (CEM Corp., Kamp Lintfort, Germany) in 55 mL pre-cleaned TFM digestion vessels following the protocol of Zimmermann et al.13 The reference material BAM M505a was treated similarly and digested in duplicates per digestion batch (12 samples in triplicates).
L4 – Alkali fusion after sample ashing was selected as the best sample digestion method resulting in a visually clear and residue-free solution. For dry ashing, 1 g to 3 g of powdered PCB was placed in a Thermolyne 48000 furnace at 600 °C in pre-cleaned quartz crucibles. For alkali fusion, 250 mg to 400 mg of dry ashed sample were mixed with 3 g to 4.5 g of sodium peroxide for a 1:10 mass ratio in 25 mL zirconium crucibles with lid. The crucibles were then placed in a furnace at 600 °C for 1 h. Due to the high content of organic compound (16% weight), the alkali fusion was performed only on ashed samples to avoid damaging the crucibles. Given the high salt content induced by the use of the alkali flux, the calibration standards were matrix-matched to a flux blank obtained by the exact same preparation as the sample.
L5 – Samples were firstly combusted in a furnace (Carbolite ELF 11/14B; Carbolite Gero, Sheffield, UK) at 550 °C for 5 h to remove the carbon matrix. Sample digestion was based on microwave assisted digestion (Multiwave Pro, Anton Paar, Graz, Austria) using dilute aqua regia5 with an addition of tetrafluoroboric acid (HBF4) as a substitute for hydrofluoric acid.13 In detail, 100 mg of combusted PCB material was digested in a solution containing 1.25 mL HNO3 (≥65%, p.a. grade; Carl Roth GmbH, Karlsruhe, Germany), 3.75 mL HCl (37%, p.a. grade; Carl Roth GmbH), 4 mL reagent grade I water (18.2 MΩ cm, MilliQ IQ 7000; Merck, Darmstadt, Germany), and 1 mL HBF4 (38%, ultra-pure; Chem-Lab, Zeldelgem, Belgium). The digestion was carried out using a temperature program with 10 min ramp to 200 °C and holding for 30 min, then leaving samples to cool to 50 °C. Samples were then filtered through 0.45 μm filter discs (Minisart regenerated cellulose; Sartorius, Göttingen, Germany) and diluted to 50 mL with reagent grade I water. Subsequent dilutions were carried out using dilute HNO3 (2% mass fraction). A separate HF-based digest was carried out for determinations of Ta following the procedure reported in ref. 4. Briefly, 5 mL HNO3, 2 mL HCl, 0.5 mL H2O2 and 1 mL HF were added to 100 mg of PCB sample. The sample was then digested using the same microwave procedure as for the HBF4 digestion. Following digestion, the sample was complexed with 9 mL of saturated (47 mg mL−1) boric acid solution to complex the HF and further diluted with dilute HNO3 (mass fraction = 2%) before analysis. Calibration standard solutions were prepared gravimetrically from SI traceable standards: ICP multi-element standard solution VI (Merck Certipur, Darmstadt, Germany), AHF-CAL-7 (Inorganic Ventures, US) and Calib. Std #2 Precious Metals (AccuStandard, Inc., US).
L6 – Sample aliquots of approximately 300 mg were weighed into 30 mL quartz vials and the samples were ashed at 500 °C in an MLS PYRO.lab oven for 5 hours. The material was then weighed into 90 mL TFM tubes. 10 mL 65% HNO3 (subboiled, Merck p.a.), 3 mL 48% HBF4 (Sigma-Aldrich), and 4 mL 30% H2O2 (suprapur, Merck) were used to digest the samples in an MLS ETHOS.lab microwave system within 2.5 h at 210 °C (30 min linear ramp from room temperature to 210 °C, 1 h constant at 210 °C). After cooling down, the solutions were evaporated to complete dryness within 5 h in an MLS ETHOS.lab evaporation system at 70 °C and approximately <450 mbar. The residues were re-dissolved in a mixture of 5 mL 25% HCl (subboiled, Merck p.a.), 5 mL 65% HNO3 (subboiled, Merck p.a.), and 2 mL 30% H2O2 (suprapur, Merck) and again digested in an MLS ETHOS.lab microwave system within 2.5 h at 210 °C as described above. After cooling down, the solutions were evaporated to complete dryness within 6 h in an MLS ETHOS.lab evaporation system at 70 °C and approximately <450 mbar. The residues were re-dissolved in 15 mL 0.15 mol kg−1 mol L−1 HNO3 (subboiled, Merck p.a.). Therefore, the tubes were sonicated in an ultra-sonic bath at 80 °C for 30 minutes. All sample solutions were then filtered into 25 mL pre-cleaned and pre-weighed Duran glass bottles through PET filter discs with 0.2 μm pore diameter. All TFM tubes were rinsed with another 4 mL 3 mol L−1 HNO3 (subboiled, Merck p.a.) filtered through the same filter discs into the according 25 mL Duran glass bottles. The filter discs were rinsed with another 1 mL of 3 mol L−1 HNO3. Each of the sample solutions was then subdivided into k = 4 aliquots of masses msln,i to apply gravimetric standard addition with natural internal standard calibration. Accordingly, in eqn (2), msmp,i = msmp(msln,i/msln), where msmp is the mass of the sample and msln is the mass of the total diluted solution. Appropriate masses mz,i of SI traceable standard solutions z were subsequently added to the aliquots to obtain the solutions i. This allowed to distinguish between inhomogeneities in the samples and insufficient reproducibility of the sample preparation (especially the digestion) on the one hand and the quality of the measurement itself on the other hand.
L7 – Samples were prepared by digesting and dissolving at least 3 replicate samples of 200 mg in Milestone Ethos SEL microwave digestion system. The digestion was carried out at 210 °C using the concentrated acid mixture HNO3 – HCl (1:3). For Ga and Li, HF was also added to the digestion vessel. Then, equal amounts from the digested sample solutions were combined in a test tube to represent the sample matrix which was used for the preparation of matrix-matched addition calibration solutions. Each calibration solution contained the same amount sample solution adjusted to have optimum analyte concentration for introduction to the instruments. The linear calibration plot was prepared by spiking the matrix containing solution by spiking varying amount of SI traceable standard calibration solutions. The concentration of each analyte in the sample is calculated using the least square method from the analytical calibration plot between the concentration of the analyte and the analytical signal. The internal standards (Tl and Sc) were also added to all solutions at approximately the same concentration in order to observe and correct the drifts on the instrument sensitivity during the measurement. When preparing the sample and the standard solutions, all dilutions and additions of standards were done gravimetrically using the analytical balance.
L8 – Samples were digested with aqua regia. A single reaction chamber microwave system (Ultrawave, Milestone Srl, Sorisole, Italy) was employed in the acid digestion of the samples. The chamber acts as the pressure vessel instead of individual sample vessels in traditional closed vessel microwave digestion units. Acid-leached glass vessels were used for digestion, and their weight was recorded before use. 400 mg sample was weighed and 10 mL of aqua regia (3:1 HCl:HNO3) added. A base load of 2% (v/v) hydrogen peroxide was placed in the chamber. The rack with the sample vessels was placed in the chamber, which was pressurized to 40 bar with nitrogen before heating. The sample vessels are immersed in the base load, which ensures an even temperature distribution throughout the samples. A temperature-controlled microwave program was used consisting of a 25 min ramp to 240 °C and holding for 15 min, followed by a cooling stage to 80 °C and depressurization. Samples were diluted to 40 mL with reagent grade I water in the digestion vessels. The final weight of the samples including the vessel was recorded. There was a grayish-brown residue, which was washed several times with grade I water, and the final aqueous suspension was screened using TXRF and found to be composed primarily of Ti, W and Zr, with smaller traces of V, Cr, Fe and Br. SI traceable standard solutions were gravimetrically prepared for calibration.
A summary of the digestion methods adopted for the dissolution of PCB samples is reported in Table 4.
| Laboratory | Acid mixture | Acid ratio | Temperature (°C) | Pre-ashing step | Code |
|---|---|---|---|---|---|
| a Sample to flux ratio. b Adopted only for Li and Ge. c Adopted only for Ta. | |||||
| L1 | HNO3, HCl | 1:3 |
120 | Yes | A |
| L7 | HNO3, HCl | 1:3 |
210 | No | A |
| L8 | HNO3, HCl | 1:3 |
240 | No | A |
| L2 | Alkali fusion | 1:7a |
700 | Yes | B |
| L4 | Alkali fusion | 1:10a |
600 | Yes | B |
| L3 | HNO3, HCl, HBF4 | 5:2:1 |
180 | No | C |
| L5 | HNO3, HCl, HBF4 | 1.25:3.75:1 |
200 | Yes | C |
| L6 | 1st HNO3, HBF4, H2O2 | 10:3:4 |
210 | Yes | D |
| 2nd HNO3, HCl, H2O2 | 5:2:2 |
||||
| L7 | HNO3, HCl, HF | 2:5:2 |
210 | No | Eb |
| L5 | HNO3, HCl, H2O2, HF | 10:4:1:2 |
200 | Yes | Fc |
L10 – Samples were sealed into polyethylene containers with a diameter of 8 mm and the height of the PCB samples varied from 3.5 mm to 6 mm. In total, 8 aliquots were prepared for the study, with sample masses varying from 210 mg to 300 mg. Four aliquots were used for short irradiation (30 s) and four for long irradiation (1 h). Before irradiation, the sample of each aliquot was measured on a calibrated analytical balance. To achieve SI traceability, the Al-0.1% Au alloy certified reference material ERM-EB530A produced by the Joint Research Centre (JRC, Belgium) was used to prepare Au standard discs with 7.2 mm diameter and 0.1 mm thickness.
3.2 Measurement procedures
L1 – The measurements were conducted using a Thermo Scientific iCAP quadrupole-ICP-MS instrument. The analyses were executed employing the standard sample introduction system, which includes a concentric nebulizer and a cyclonic spray chamber. Operational parameters of the ICP-MS instrument were tuned to optimize sensitivity, utilizing a tuning solution while closely monitoring the oxide formation rate, specifically based on the 140Ce16O/140Ce ratio to ensure consistent and reliable day-to-day performance.
External calibration was carried out using a freshly prepared multi-elemental standard solution based on Merck's multi-element standard IV (7Li, 59Co, 60Ni and 71Ga) and Merck's single-element standards (139La, 143Nd, 104Pd and 141Pr). Yttrium (89Y) served as the internal standard.
L2 – The analysis was carried out in triplicate using a TQ ICP-MS Agilent 8900 with external standard calibration. Three internal standards were used for quantification (Sc, Y and Ir). Solution blanks were systematically run. For most of elements, solid reference materials were used as quality controls (inter-laboratory test sample GEOPT35 and BAM M505A). The measurement procedure was developed to consider the specificity of the PCB matrix. Depending on elements and interferences, different gas modes were used on the TQ ICP-MS: gas-free, helium or oxygen (without or with mass transition). The three modes have been systematically used and the quantification isotopes have been selected based on comparison between the different modes and the potential interferences. For example, Ge has been measured at mass-shift 88 with O2 in order to prevent Nd interference.
L3 – A procedure based on external calibration was developed to measure Ag, Au, Co, Cu, Dy, Ga, Gd, Ge, La, Li, Nd, Ni, Pd, Pr, Pt, Ta and Ti via isotopes 107Ag, 197Au, 59Co, 65Cu, 163Dy, 69Ga, 157Gd, 72Ge, 139La, 7Li, 146Nd, 60Ni, 105Pd, 141Pr, 181Ta and 47Ti, respectively. Determination of elemental mass fractions in the digests was performed using an inductively coupled plasma tandem mass spectrometer (ICP-MS/MS) (Agilent 8800, Agilent Technologies, Tokyo, Japan) coupled to an ESI SC-4 DX FAST autosampler (Elemental Scientific, Omaha, Nebraska, USA).14 Detection modes can be found in Table S3.† The instrument was tuned daily using a tune solution containing Li, Co Y, Ce and Tl at a concentration of 10 μg L−1. Quantification was performed by external calibration covering a concentration range from 0 μg L−1 to 10000 μg L−1. Solutions and blanks were prepared on a daily basis from traceable, custom made multi-element standards (Inorganic Ventures, Christiansburg, USA). Wash blanks were measured after each sample triplicate to monitor and avoid potential carry-over effects.
Multi-element data were processed using MassHunter version 4.4 or higher (Agilent Technologies, Tokyo, Japan) and a custom written Excel© spreadsheet. The isobaric interference of 115Sn on 115In was corrected for by peak stripping as implemented in MassHunter using the signal of 118Sn and the isotopic abundances provided by IUPAC's Commission on Isotopic Abundances and Atomic Weights.15 Combined uncertainties were estimated using a Kragten spreadsheet approach16 taking into account reproducibility, repeatability and measurement precision for each sample. The significant number of digits of elemental mass fractions are given according to GUM and EURACHEM guidelines, whereby the uncertainty determines the significant number of digits to be presented with the value.17
L4 – An external standard calibration procedure with alkali fusion matrix-matching was used to measure Au, La, Li, Pd and Ta via isotopes 197Au, 139La, 7Li, 105Pd and 181Ta, respectively. The calibration standards were prepared in a diluted alkali fusion blank to take into account the matrix effect attributed to the high Na content of the flux.
L5 – The analysis was carried out using a NexION 5000 ICP-MS/MS (PerkinElmer, Waltham, MA, USA) with external calibration and nitrous oxide (medicinal grade; Linde Gas GmbH, Stadl-Paura, Austria) as a reaction gas.18,19 Operating parameters are listed in Table S5.† The gravimetrically prepared SI standard solutions were combined into one stock solution that was further diluted to obtain an 11-point calibration. For every sample digestion, the calibration standards were made fresh within 12 h of the measurements.
L6 – A reference (gravimetric) standard addition with natural internal standard calibration procedure was developed to measure Co, La and Li via isotopes 59Co, 7Li and 139La, respectively. The internal standard elements Y, Sb and Ba via isotopes 89Y, 121Sb and 127Ba, respectively, were chosen based on their relative signal intensity to be able to adjust signal intensity ratios Ri of the analyte element X over the internal standard Y between 0.1 and 10. The signal intensity ratios were measured using an HR-ICP-MS (Element XR, Thermo Fisher Scientific, Germany) operated in low resolution.
An ordinary least squares fit of a linear equation to (yi, xi) data obtained using eqn (2) yielded the slope a1 and the y-intercept a0 which in turn yielded the mass fraction wx of the analyte element in the original solid sample according to eqn (1). Uncertainty budgets consistent with the GUM20 were obtained on a sample-per-sample basis using the measurement eqn (1) as a comprehensive closed form equation.
L7 – A reference matrix-match standard addition procedure was developed to measure Au, Dy, Ga, La, Li, Nd, Pd, Pr, Pt and Sm via isotopes 197Au, 161Dy, 163Dy, 71Ga, 139La, 6Li, 7Li, 144Nd, 145Nd, 146Nd, 105Pd, 106Pd, 108Pd, 141Pr, 194Pt, 196Pt, 147Sm and 149Sm; 205Tl was used as internal standard.
The uncertainty for each analyte in the sample was calculated according to eqn (4) by using a commercial software GUM Workbench. GUM approach was used during the calculations. The contributions from each source were identified and quantified in the uncertainty budget.
L8 – The measurements were carried out using a Thermo iCAP TQ ICP-MS/MS (Thermo Scientific, Bremen, Germany) either with reference (gravimetric) standard addition with natural internal standard calibration or external calibration. Helium was used as a collision gas and oxygen as a reaction gas. Operating parameters are listed in Table S8.†
For the external calibration, four internal standards were used (Sc, Ge, Rh, Ir). The additions were made so that the contribution from the PCB sample was less than 1% of the total internal standard signal. The calibration solutions were prepared from NIST SRMs except for Ga, Pd and Pt for which commercial standard stock solutions from Inorganic Ventures (I.V. Labs, Inc., Christiansburg, Virginia, USA) were used. 4–5 calibration points were prepared, and the calibration range was selected to fit the PCB sample which was gravimetrically diluted 50× or 200×, depending on the analyte. The calibration standards were made fresh on each day of measurement and the acid matrix was matched to the digested samples. The reference (gravimetric) standard addition with natural internal standard calibration was used to measure Co, La, Pr and Au via isotopes 59Co, 139La, 141Pr and 197Au respectively. 59Co was measured on mass using ICP-MS/MS with helium as a collision gas, 139La, 141Pr and 197Au were measured using ICP-MS/MS with oxygen as a reaction gas, La and Pr as oxides and Au on mass. Internal standard isotopes 55Mn, 137Ba (for La and Pr) and 206Pb, respectively, were chosen based on their relative signal intensity and closeness in mass to the analytes. 55Mn was measured on mass with He, 137Ba and 206Pb as oxides with oxygen as the reaction gas. The signal intensity ratios Ri of the analyte element X over the internal standard Y varied between 0.6 and 3.2. Uncertainty budgets consistent with the GUM were obtained on three replicate samples using eqn (1) as measurement model.
Samples and standards were placed in cartridge cases and irradiated in the carousel facility of a 250 kW TRIGA Mark II research reactor with a thermal neutron flux of 1.0 × 1012 cm−2 s−1.21 The neutron irradiation lasted 1 h at nominal reactor power. Irradiated cartridge cases were collected after 2 days cooling due to the high Cu activity. Samples and standards were extracted and individually placed in gamma counting containers.
Gamma spectrometry was performed with a Ge detector ORTEC GEM50P4-83 (50% relative efficiency, 1.9 keV full-width half maximum at 1332.5 keV energy) connected to a multichannel analyzer ORTEC DSPEC 502 and controlled by a PC. The detector is placed inside a low-background graded lead shield located in an underground laboratory room with temperature controlled at 22 °C. The GEM50P4-83 detector is extensively characterized in terms of counting efficiency using a mix of single nuclide gamma sources with SI traceable activity measured at different distances from the end-cap. The gamma counting of samples and standards was carried out in two steps: the first started 2 days after neutron irradiation and lasted 5 days, the latter started 14 days after neutron irradiation in order to wait for the interfering Br decay and lasted 16 days.
Collected gamma spectra were elaborated with HyperLab program version 2014.1(ref. 22) to get the net peak areas of the radionuclides produced by the analytes. The output data were processed with the rel-INRIM software to quantify Au, La, Co and Ta, and the k0-INRIM software to quantify Ag and Cu.23 Both software are homemade developed to obtain uncertainty budgets of elemental mass fractions on a sample-per-sample basis using eqn (5) and (6) as measurement models for k0 and relative calibration, respectively, and according to the GUM.20
L10 – A k0-INAA procedure was developed to measure total content of elements via its radionuclides induced by neutron by short irradiation of 30 s (Cu, Dy, In and Ti) and by long irradiation of 1 h (Ag, Au, Co, Cu, La, Sm and Ta). Depending on the mass fraction of a particular element in the samples, an element can be detected in both irradiations, e.g. Cu. Samples and standards were fixed in sandwich form and irradiated in the carousel facility of a 250 kW TRIGA Mark II research reactor with a thermal neutron flux of 1.1 × 1012 cm−2 s−1.
After short irradiation, induced activity in the samples was measured after 15 min, 20 min, 120 min, 24 h and 20 days of cooling times on an absolutely calibrated HPGe detector (45% relative efficiency) connected to a multichannel analyzer ORTEC DSPEC PLUS™ and controlled by a PC. After long irradiation, induced activity in the samples was measured after 5, 11 and 30 days of cooling times on an absolutely calibrated HPGe detector (40% relative efficiency) connected to a multichannel analyzer CANBERRA Multiport II and controlled by a PC. For peak area evaluation, the HyperLab program version 2014.1 (ref. 22) was used. For elemental mass fractions and effective solid angle calculations, the software package Kayzero for Windows was applied using eqn (5) as measurement model.24
4. Results and discussion
4.1 Digestion methods
Several digestion methods have been tested for the complete decomposition of samples to make available a standardized protocol for ICP-MS methods. Significant outcomes obtained by laboratories are reported below.L4 – In general, the acid digestion in a microwave with no pre-ashing step led to a black residue left in solution. With a pre-ashing step and the addition of HF at high temperature (240 °C), a small opaque residue that is almost undetectable is left. With the alkali fusion step, no residues are visible. In addition to alkali fusion, two microwave-assisted acid digestions were tested with and without ashing step: a mixture of HNO3:HCl and the same mixture with the addition of HF. For Ta, using only aqua regia prevents the recovery of this element and HF is absolutely necessary. In addition, it seems preferable to add an ashing step for the recovery of Li.
L5 – Initial attempts at digesting the PCB material using H2O2 to remove the plastic matrix were unsuccessful, indicated not only by the large proportions of black residue observed following analysis, but poor comparability between of the TCE mass fractions obtained to other laboratories. Therefore, instead of using H2O2, a combustion step was employed prior to the microwave-assisted digestion in order to remove the plastic matrix. This eliminated the black residue, however the digestion was still found to be incomplete. Instead, an opaque mixture resulted, which required filtration before analysis. The resulting brown residue was screened using ED-XRF and found to be composed primarily of Ti and Zr. Analysis of the sample extract revealed good comparability of TCE mass fractions to other laboratories, with the exception of Ta (likely lost along with Ti and Zr in the residue) as the mass fraction determined was approximately half that of other laboratories. Therefore, a separate HF digestion needed to be performed specifically for this element.
L6 – Prior to the final digestion procedure and sample analysis, several digestion procedures have been applied and tested with the PCB material. Generally, for sample digestion, fusion techniques were not applied, because the latter are usually accompanied by large amounts of blank contaminating the analytes (here, Li, Co, and La). Therefore, microwave (MW) assisted acid digestion procedures were tested exclusively. For digestion, a triple blend consisting of HNO3, HCl, and HF was used first. According to a comprehensive study carried out by Zimmermann et al.,13 HF could be substituted by HBF4 mainly used for silicate Si–O decomposition in the matrix due to in situ HF generation during the digestion process. Initial tests used only a two-step digestion in (1st Step HNO3 + HCl blend in a high-pressure 40 bar MW-assisted run: turboWAVE™ MLS) followed by the 2nd step MW digestion (HNO3/HCl/HBF4) in an ETHOS.lab™ MLS instrument. The amount of non-dissolved residues mainly due to organic matrix components led to a triple digestion procedure, starting with a high-temperature ashing in a PYRO.lab™ MLS MW oven (sample mass ≈ 300 mg, heating duration: 5 h, T = 500 °C). After ashing, ≈ 25% of the initial sample mass was removed enabling larger surface areas for the successive MW acid digestion steps (1st HNO3/HCl turboWAVE™ MLS; 2nd HNO3/HCl/HBF4 ETHOS.lab™ MLS). Complete digestion was not achieved, and green opaque solid residues remained on the bottom of the vials. In order to facilitate the digestion procedure in standard labs, the final procedure was adapted to a two-step acid digestion protocol with an initial ashing step, which can be performed in a standard laboratory microwave without the need of high pressure (see Section 3.1.1).
4.2 TCE measurement results
The TCE mass fraction values (dry-mass basis, i.e. corrected for moisture content) obtained with reference methods by participating laboratories are reported in Table 5; number, n, and mass, msmp, of the measured samples, average mass fraction,![[w with combining macron]](https://www.rsc.org/images/entities/i_char_0077_0304.gif) x, uncertainty of the average mass fraction, u(x), and experimental standard deviation of the individual mass fraction value j, s(wx j) are given.
x, uncertainty of the average mass fraction, u(x), experimental standard deviation, s(wx j), and uncertainty of the average mass fraction including the contribution due to inhomogeneity and/or large data scattering, if present, u*(x), are given
x, uncertainty of the average mass fraction, u(x), and experimental standard deviation of the individual mass fraction value j, s(wx j) are given.
x, uncertainty of the average mass fraction, u(x), experimental standard deviation, s(wx j), and uncertainty of the average mass fraction including the contribution due to inhomogeneity and/or large data scattering, if present, u*(x), are given
| X | Lab (technique) | n | m smp/mg |
x/μg g−1 |
u(x)/μg g−1 |
s(wxj)/μg g−1 |
u*(x)/μg g−1 |
|---|---|---|---|---|---|---|---|
| a Relative. b k 0. c Standard addition with internal standard. d Matrix-matched standard addition. | |||||||
| Ag | L9 (INAA)b | 12 | 170 | 517 | 11 | 7.0 | 11 |
| L10 (INAA)b | 8 | 210–300 | 530 | 19 | 8 | 19 | |
| Au | L7 (ICP-MS)d | 5 | 200 | 38.7 | 1.3 | 0.6 | 1.4 |
| L8 (ICP-MS)c | 3 | 400 | 37.4 | 0.95 | 0.74 | 1.2 | |
| L9 (INAA)a | 12 | 170 | 38.26 | 0.20 | 1.12 | 0.41 | |
| L10 (INAA)b | 8 | 210–300 | 39.3 | 1.5 | 1.6 | 1.6 | |
| Co | L6 (ICP-MS)c | 4 | 300 | 788 | 2.8 | 13 | 10 |
| L8 (ICP-MS)c | 3 | 400 | 800 | 15 | 2.3 | 15.2 | |
| L9 (INAA)a | 12 | 170 | 786.5 | 6.0 | 29.5 | 11.2 | |
| L10 (INAA)b | 8 | 210–300 | 757 | 27 | 20 | 29 | |
| Cu | L9 (INAA)b | 12 | 170 | 95434 |
2582 | 1525 | 2628 |
| L10 (INAA)b | 8 | 210–300 | 105360 |
4115 | 5164 | 4648 | |
| Dy | L10 (INAA)b | 4 | 210 | 3.83 | 0.24 | 0.40 | 0.43 |
| L7 (ICP-MS)d | 5 | 200 | 2.73 | 0.08 | 0.05 | 0.09 | |
| Ga | L7 (ICP-MS)d | 5 | 200 | 10.46 | 0.4 | 0.3 | 0.44 |
| In | L10 (INAA)b | 4 | 210 | 2.296 | 0.087 | 0.065 | 0.103 |
| La | L6 (ICP-MS)c | 4 | 300 | 33.9 | 1.4 | 1.9 | 1.7 |
| L7 (ICP-MS)d | 3 | 200 | 36.6 | 1.5 | 1.4 | 1.8 | |
| L8 (ICP-MS)c | 3 | 400 | 39.3 | 0.75 | 0.84 | 1.1 | |
| L9 (INAA)a | 12 | 170 | 39.99 | 0.33 | 0.66 | 0.33 | |
| L10 (INAA)b | 8 | 210–300 | 38.3 | 1.4 | 1.4 | 1.6 | |
| Li | L6 (ICP-MS)c | 4 | 300 | 79.2 | 0.44 | 1.7 | 1.4 |
| L7 (ICP-MS)d | 3 | 200 | 76.2 | 2.9 | 0.7 | 3.0 | |
| Nd | L7 (ICP-MS)d | 5 | 200 | 83.9 | 3.3 | 2.8 | 3.7 |
| Pd | L7 (ICP-MS)d | 5 | 200 | 15.6 | 0.6 | 0.4 | 0.65 |
| Pr | L7 (ICP-MS)d | 3 | 200 | 11.8 | 0.4 | 0.1 | 0.41 |
| L8 (ICP-MS)c | 3 | 400 | 11.8 | 0.25 | 0.12 | 0.28 | |
| Pt | L7 (ICP-MS)d | 5 | 200 | 0.279 | 0.016 | 0.020 | 0.020 |
| Sm | L10 (INAA)b | 4 | 300 | 4.72 | 0.20 | 0.21 | 0.27 |
| L7(ICP-MS)d | 3 | 200 | 5.33 | 0.22 | 0.22 | 0.31 | |
| Ta | L9 (INAA)a | 12 | 170 | 281.6 | 2.0 | 7.1 | 3.0 |
| L10 (INAA)b | 8 | 210–300 | 303 | 11 | 9 | 12 | |
| Ti | L10 (INAA)b | 4 | 210 | 66403 |
2353 | 736 | 2438 |
To check the data for the presence of any kind of inhomogeneity and/or large data scattering, e.g. reproducibility of sample preparation (digestion and tableting) and analyte inhomogeneity in samples, we applied the concept of degree of equivalence
| dj = wxj − x, | (7) |
 | (8) |
In case of negligible correlation between wx j and x, the covariance term in eqn (8) is omitted. When a dj value is larger than its uncertainty, the corresponding mass fraction value wx j is not consistent with the overall average and a type A uncertainty is added to the measurement uncertainty according to:25,26
 x) is calculated with the approximated formula
x) is calculated with the approximated formula 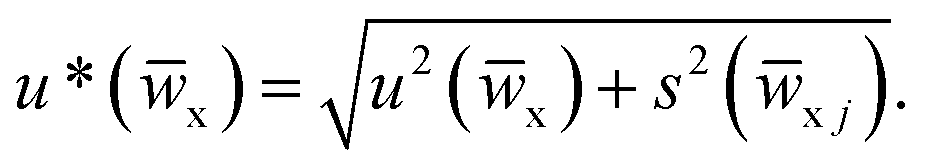 The u*(x) values obtained by each participating laboratory are reported in Table 5.
The u*(x) values obtained by each participating laboratory are reported in Table 5.
The TCE mass fraction values (dry-mass basis, i.e. corrected for moisture content) obtained with routine methods by participating laboratories are reported in Table 6; n, msmp, x and expanded uncertainty (k = 2) of the average mass fraction, u(x), are given.
x, and expanded uncertainty (k = 2) of the average mass fraction, U(x), are given
| X | Lab | n | m smp/mg |
x ± U(x)/μg g−1 |
X | Lab | n | m smp/mg |
x ± u(x)/μg g−1 |
|---|---|---|---|---|---|---|---|---|---|
| a Alkali fusion. | |||||||||
| Ag | L3 | 6 | 200 | 530 ± 80 | Li | L1 | 9 | 200 | 65 ± 18 |
| Au | L2 | 3 | 500 | 37.5 ± 4.0 | L3 | 6 | 200 | 61 ± 10 | |
| L3 | 6 | 200 | 55 ± 10 | L4a | 3 | 300–450 | 76.6 ± 6.0 | ||
| L4a | 3 | 300–450 | 34.7 ± 5.0 | L5 | 3 | 120 | 66.3 ± 2.6 | ||
| L5 | 3 | 120 | 38.2 ± 2.4 | Nd | L1 | 9 | 200 | 92 ± 14 | |
| L8 | 4 | 400 | 38.6 ± 3.0 | L2a | 3 | 300(ii) | 84.6 ± 8.0 | ||
| Co | L1 | 9 | 200 | 780 ± 94 | L3 | 5 | 200 | 49 ± 8 | |
| L2a | 3 | 300 | 840 ± 80 | L5 | 3 | 120 | 85.6 ± 9.2 | ||
| L3 | 6 | 200 | 760 ± 120 | L8 | 4 | 400 | 88.8 ± 9.8 | ||
| L5 | 3 | 120 | 768 ± 44 | Ni | L1 | 9 | 200 | 3680 ± 468 | |
| L8 | 4 | 400 | 775 ± 78 | L2a | 3 | 300 | 4120 ± 400 | ||
| Cu | L2a | 3 | 300 | 118000 ± 10000 |
L3 | 6 | 200 | 3920 ± 400 | |
| L3 | 6 | 200 | 112000 ± 16000 |
L8 | 4 | 400 | 3880 ± 430 | ||
| L8 | 4 | 400 | 103000 ± 7000 |
Pd | L1 | 9 | 200 | 17 ± 2 | |
| Dy | L2a | 3 | 300 | 2.83 ± 0.2 | L2a | 3 | 300 | 13.3 ± 1.3 | |
| L3 | 6 | 200 | 1.6 ± 1.6 | L3 | 5 | 200 | 13.3 ± 2.2 | ||
| L5 | 6 | 120 | 2.86 ± 0.18 | L4a | 3 | 300–450 | 13.8 ± 2.0 | ||
| L8 | 4 | 400 | 2.57 ± 0.26 | L5 | 3 | 120 | 14.6 ± 1.4 | ||
| Ga | L1 | 9 | 200 | 12 ± 2 | L8 | 4 | 400 | 13.9 ± 1.6 | |
| L2a | 3 | 300 | 10.2 ± 1.0 | Pr | L1 | 9 | 200 | 13 ± 2 | |
| L3 | 6 | 200 | 10.2 ± 2.4 | L2a | 3 | 300 | 10.9 ± 1.0 | ||
| L8 | 4 | 400 | 10.1 ± 1.2 | L3 | 6 | 200 | 6.4 ± 2.8 | ||
| Gd | L2a | 3 | 300 | 4.50 ± 0.40 | L5 | 3 | 120 | 10.4 ± 0.8 | |
| L3 | 6 | 200 | 2.8 ± 2 | L8 | 4 | 400 | 11.4 ± 1.1 | ||
| L5 | 3 | 120 | 4.61 ± 0.46 | Pt | L2 | 3 | 500 | 0.26 ± 0.03 | |
| L8 | 4 | 400 | 4.70 ± 0.44 | L3 | 6 | 200 | 0.7 ± 1.2 | ||
| Ge | L2a | 3 | 300 | 2.50 ± 0.20 | L5 | 3 | 120 | 0.249 ± 0.080 | |
| L3 | 6 | 200 | 2.5 ± 5.0 | L8 | 4 | 400 | 0.270 ± 0.064 | ||
| La | L1 | 9 | 200 | 39 ± 6 | Sm | L2a | 3 | 300 | 3.94 ± 0.40 |
| L2a | 3 | 300 | 36.8 ± 3.6 | Ta | L2a | 3 | 300 | 282 ± 28 | |
| L3 | 5 | 200 | 17.2 ± 3.6 | L3 | 6 | 200 | 280 ± 60 | ||
| L4a | 3 | 300–450 | 38.0 ± 5.6 | L4a | 3 | 300–450 | 262.9 ± 35 | ||
| L5 | 3 | 120 | 35.2 ± 3.2 | L5 | 3 | 120 | 285 ± 48 | ||
| L8 | 4 | 400 | 36.3 ± 3.8 | Ti | L3 | 6 | 200 | 6800 ± 3400 | |
Rh is the only element among the selected TCE that was not quantified. Mass fraction values obtained both with reference and routine methods are available for Ag, Au, Co, Cu, Dy, Ga, La, Li, Nd, Pd, Pr, Pt, Sm, Ta and Ti, while for In, and Ge and Ni, only values obtained with candidate reference and routine methods are available, respectively.
Experimental data are plotted for each element in the order of increasing mass fraction values in Fig. 1 and 2. The Ge, In and Ti graphs were excluded due to lack of data. Results obtained with reference methods are indicated with gray dots and used, when available, to calculate the reference mass fraction value as weighted average; u*(x) is used as weight. Results obtained with routine methods are indicated with black dots. The reference mass fraction value is represented by the (horizontal) dotted line (dash–dot) and the corresponding expanded uncertainties (k = 2) by the solid lines. In addition, the dotted lines (dot–dot) represent the reference mass fraction value ± 20%. For Gd and Ni, the reference mass fraction value is calculated in the same way but using data collected with routine methods; u(x) is used as weight instead of u*(x).
 | ||
| Fig. 1 Pt, Dy, Gd, Sm, Ga, Pr, Pd and Au results obtained by the participating laboratories. Gray and black dots indicate data obtained with candidate reference and routine methods, respectively. The (horizontal) dotted line (dash-dot) represents the reference mass fraction value and the solid lines the corresponding expanded uncertainties (k = 2). The dotted lines (dash–dash) show the reference mass fraction value ± 20%. Error bars indicate expanded uncertainties (k = 2). The right-hand y-axis shows the relative difference with respect to the reference value, Δrefw. | ||
Even if the measured PCB material was not previously characterized in terms of homogeneity, we can use the results to evaluate the analytical performance of the reference and routine methods. To this aim, the (maximum) mutual relative difference, Δmutw, and the (maximum) relative difference with respect to the reference value, Δrefw, are reported below.
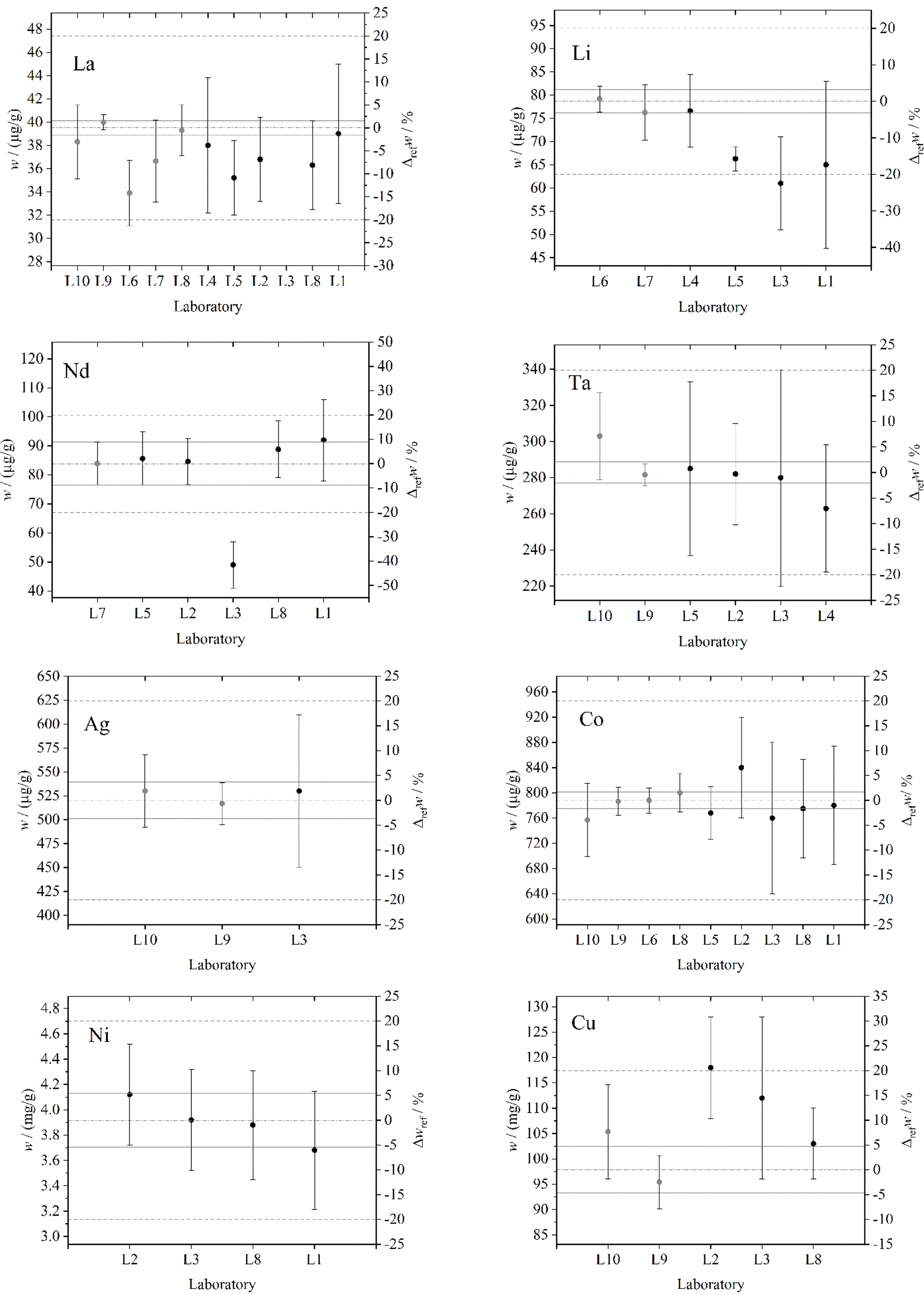 | ||
| Fig. 2 La, Li, Nd, Ta, Ag, Co, Ni and Cu results obtained by the participating laboratories. Gray and black dots indicate data obtained with candidate reference and routine methods, respectively. The horizontal dotted line (dash–dot) represents the reference mass fraction value and the horizontal solid lines the corresponding expanded uncertainties (k = 2). The horizontal dotted lines (dash–dash, where visible) show the reference mass fraction value ± 20%. Error bars indicate expanded uncertainties (k = 2). The right-hand y-axis shows the relative difference with respect to the reference value, Δrefw. | ||
For what concerns candidate reference methods, solid analysis using k0 and relative INAA produced data in mutual agreement for Ag (L9, L10; Δmutw = 2.5%), Au (L9, L10, Δmutw = 2.7%), Co (L9, L10; Δmutw = 3.8%), Cu (L9, L10; Δmutw = 10.2%), La (L9, L10; Δmutw = 4.3%) and Ta (L9, L10; Δmutw = 7.5%). Wet analysis carried out by L7, L8 and L6 using standard addition ICP-MS produced data in mutual agreement for Au (L7, L8; Δmutw = 3.4%), Co (L6, L8; Δmutw = 1.5%), La (L6, L7, L8; Δmutw = 13.7%), Li (L6, L7; Δmutw = 3.8%) and Pr (L7, L8; Δmutw = 0.2%). In addition, there is a mutual agreement between standard addition ICP-MS and INAA for Au (L7, L8, L9, L10; Δmutw = 5.0%), Co (L6, L8, L9, L10; Δmutw = 5.6%), La (L7, L8, L9, L10; Δmutw = 8.4%) and Sm (L7, L10; Δmutw = 12.3%), and a mutual disagreement for Dy (L7, L10; Δmutw = 39.6%); for La, L6 is in agreement with L10 (Δmutw = 8.4%) and in disagreement with L9 (Δmutw = 15.4%). In case of Dy, the noteworthy discrepancy observed between L7 (matrix-matched standard addition ICP-MS) and L10 (k0-INAA) could be due to the combined effect of a hidden systematic error and the large expanded uncertainty of L10 value (22.5%). Since the L7 value is in full agreement with the L5, L2 and L8 values, the origin of the systematic error might be due to the fact that Dy content determined by L10 was very close to the detection limit (about 3 μg g−1). The relative difference of values observed in case of La measured by L6 (standard addition with natural internal standard ICP-MS) and L9 (relative INAA) is less striking and below 20%, which we consider in this study as a threshold of evidence for discrepancy.
For what concerns candidate routine methods based on ICP-MS external calibration, data collected by L3 (Au, Dy, Gd, La, Li, Nd, Pr, Pt) and L2 (Cu, Sm) are outside the ± 20% of the reference mass fraction. Excluding such cases, data are in agreement with the reference value in case of Ag (L3; Δrefw = 1.9%), Au (L2, L4, L5, L8; Δrefw = −9.3%), Co (L1, L3, L5, L8; Δrefw = −3.6%), Cu (L3, L8; Δrefw = 14.5%), Dy (L2, L5, L8; Δrefw = −7.3%), Ga (L1, L2, L3, L8; Δrefw = 14.8%), Gd (L2, L5, L8; Δrefw = 2.9%), La (L1, L4, L8; Δrefw = 2.9%), Li (L1, L4; Δrefw = −17.4%), Nd (L1, L2, L5, L8; Δrefw = 9.7%), Ni (L1, L2, L3, L8; Δrefw = 5.9%), Pd (L1, L3, L4, L5, L8; Δrefw = −14.9%), Pr (L1, L2, L8; Δrefw = 10.2%), Pt (L4, L5, L8; Δrefw = −10.8%) and Ta (L2, L3, L4, L5; Δrefw = −7.1%); data are in disagreement with the reference value in case of Au (L3; Δrefw = 43.7%), Cu (L2; Δrefw = 20.6%), La (L5; Δrefw = −10.9%), Li (L3, L5; Δrefw = −22.5%), Nd (L3; Δrefw = −41.6%), Pd (L2; Δrefw = −14.9%), Pr (L3, L5; Δrefw = −45.7%) and Sm (L2; Δrefw = −20.9%); L2 was the only participating laboratory using alkali fusion digestion for Cu and Co quantification. A sound reason for the outstanding discrepancies observed in most of the L3 determinations has not yet been found. The remaining discrepancies are close or below the 20% threshold and negligible in most industrial applications requiring routine TCE measurements.
Mass fraction values for Ag, Au, Co, Cu, Dy, Ga, In, La, Li, Nd, Ni, Pd, Pt, Sm, Ta and Ti in PCB are also reported in ref. 4 or 6 with mass fraction ranges (0.07–0.88) mg g−1, (0.04–1.41) mg g−1, (0.13–0.54) mg g−1, (9–494) mg g−1, (21–189) μg g−1, (12–267) μg g−1, (10–144) μg g−1, (0.5–6.3) μg g−1, (0.8–40.5) μg g−1, (0.03–0.79) mg g−1, (1.8–82.9) mg g−1, (4–178) μg g−1, (1.1–25) μg g−1, (0.6–4.1) μg g−1, (0.07–2.8) mg g−1 and (0.26–7.3) mg g−1, respectively. Compared to these ranges, values for Ag, Cu, Nd, Ni, Pd and Ta obtained in this study are within, values for Co, La, Li, Sm and Ti are above and values for Au, Ga, In and Pt are below; values for Gd and Pr were not reported in either ref. 4 or 6.
5. Conclusion
Three ICP-MS reference methods adopting standard addition calibrations and two INAA reference methods adopting k0 and relative calibrations were independently developed and applied by five laboratories to measure selected TCE mass fractions in powdered samples of a PCB material. Six additional ICP-MS methods based on external standard calibrations were also independently developed and tested to provide measurement methods suitable for routine TCE measurements.Twenty TCE were selected as target analytes. All except Rh were quantified in the range within fractions of μg g−1 (Pt) and 0.1 g g−1 (Cu). In addition to the elements reported in ref. 4 and 5 we quantified Gd at the level of a few μg g−1.
Reference methods produced data in full mutual agreement for Ag, Au, Co, Cu, La, Li, Pr, Sm and Ta with relative expanded uncertainties ranging within 0.8% (La) and 7.2% (Ag), while some disagreement was observed for Dy and La. In addition, all but one of the routine methods produced data in agreement with the reference value or within ± 20% of it.
Accordingly, the developed ICP-MS standard addition and INAA k0 and relative methods are suitable to certify matrix reference materials made from end-of-life PCB. Moreover, analytical methods used in industries and research laboratory working in WEEE recycling can be harmonized starting from the ICP-MS external calibration methods here applied whose performances are suited to the purpose of determining the economic and strategic value of PCB through TCE quantification.
In conclusion, this paper presented for the first time fully validated and traceable protocols for the analysis of TCE in PCB. This advancement is particularly crucial given the current geopolitical shifts that are significantly impacting the availability of raw materials and precious metals. The rapid acceleration of technology deployment globally further exacerbates these challenges. In response, the MetroCycleEU consortium has developed comprehensive methods to address this complex matrix, thereby supporting recycling industries and measurement laboratories in adopting these essential techniques to benefit society.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project (20IND01 MetroCycleEU) has received funding from the EMPIR programme co-financed by the Participating States and from the European Union's Horizon 2020 research and innovation programme. The authors have no competing interests to declare that are relevant to the content of this article.References
- C. P. Baldé, et al., Global E-Waste Monitor 2024, International Telecommunication Union (ITU) and United Nations Institute for Training and Research (UNITAR), Geneva/Bonn, 2024 Search PubMed.
- D. H. Dang, M. Filella and D. Omanović, Technology-critical elements: an emerging and vital resource that requires more in-depth investigation, Arch. Environ. Contam. Toxicol., 2021, 81, 517–520, DOI:10.1007/s00244-021-00892-6.
- S. Das and Y. P. Ting, Evaluation of wet digestion methods for quantification of metal content in electronic scrap material, Resources, 2017, 6, 64, DOI:10.3390/resources6040064.
- B. Bookhagen, W. Obermaier, C. Opper, C. Koeberl, T. Hofmann, T. Prohaska and J. Irrgeher, Development of a versatile analytical protocol for the comprehensive determination of the elemental composition of smartphone compartments on the example of printed circuit boards, Anal. Methods, 2018, 10, 3864–3871, 10.1039/c8ay01192c.
- D. F. Andrade, R. C. Machado, M. A. Bacchi and E. R. Pereira, Proposition of electronic waste as a reference material – part 1: sample preparation, characterization and chemometric evaluation, J. Anal. At. Spectrom., 2019, 34, 2394–2401, 10.1039/c9ja00283a.
- D. F. Andrade, R. C. Machado and E. R. Pereira, Proposition of electronic waste as a reference material – part 2: homogeneity, stability, characterization, and uncertainties, J. Anal. At. Spectrom., 2019, 34, 2402–2410, 10.1039/c9ja00284g.
- A. L. Hauswaldt, O. Rienitz, R. Jaehrling, N. Fischer, D. Schiel, G. Labarraque and B. Magnusson, Uncertainty of standard addition experiments: a novel approach to include the uncertainty associated with the standard in the model equation, Accredit. Qual. Assur., 2012, 17, 129–138, DOI:10.1007/s00769-011-0827-5.
- O. Rienitz, A. L. Hauswaldt and R. Jaehrling, Solution to standard addition challenge, Anal. Bioanal. Chem., 2012, 404, 2117–2118, DOI:10.1007/s00216-012-6377-9.
- F. De Corte, The standardization of standardless NAA, J. Radioanal. Nucl. Chem., 2001, 248, 13–20, DOI:10.1023/a:1010601403010.
- R. R. Greenberg, P. Bode and E. N. Fernandes, Neutron activation analysis: a primary method of measurement, Spectrochim. Acta, Part B, 2011, 66, 193–241, DOI:10.1016/j.sab.2010.12.011.
- A. Hubau, A. Chagnes, M. Minier, S. Touzé, S. Chapron and A. G. Guezennec, Recycling-oriented methodology to sample and characterize the metal composition of waste printed circuit boards, Waste Manage., 2019, 91, 62–71, DOI:10.1016/j.wasman.2019.04.041.
- S. Touze, S. Guignot, A. Hubau, N. Devau and S. Chapron, Sampling waste printed circuit boards: achieving the right combination between particle size and sample mass to measure metal content, Waste Manage., 2020, 118, 380–390, DOI:10.1016/j.wasman.2020.08.054.
- T. Zimmermann, M. von der Au, A. Reese, O. Klein, L. Hildebrandt and D. Pröfrock, Substituting HF by HBF4 – an optimized digestion method for multi-elemental sediment analysis via ICP-MS/MS, Anal. Methods, 2020, 12, 3778–3787, 10.1039/D0AY01049A.
- D. Profrock and A. Prange, Inductively coupled plasma-mass spectrometry (ICP-MS) for quantitative analysis in environmental and life sciences: a review of challenges, solutions, and trends, Appl. Spectrosc., 2012, 66(8), 843–868, DOI:10.1366/12-06681.
- J. R. de Laeter, J. K. Bohlke, P. De Bievre, H. Hidaka, H. S. Peiser, K. J. R. Rosman and P. D. P. Taylor, Atomic weights of the elements. Review 2000 (IUPAC technical report), Pure Appl. Chem., 2003, 75, 683–800, DOI:10.1351/pac200375060683.
- J. Kragten, Tutorial review. Calculating standard deviations and confidence intervals with a universally applicable spreadsheet technique, Analyst, 1994, 119, 2161–2165, 10.1039/an9941902161.
- EURACHEM/CITAC, EURACHEM/CITAC Guide: Quantifying Uncertainty in Analytical Measurements, EURACHEM, 3rd edn, 2012 Search PubMed.
- S. T. Lancaster, T. Prohaska and J. Irrgeher, Characterisation of gas cell reactions for 70+ elements using N2O for ICP tandem mass spectrometry measurements, J. Anal. At. Spectrom., 2023, 38, 1135–1145, 10.1039/D3JA00025G.
- S. Trimmel, T. C. Meisel, S. T. Lancaster, T. Prohaska and J. Irrgeher, Determination of 48 elements in 7 plant CRMs by ICP-MS/MS with a focus on technology-critical elements, Anal. Bioanal. Chem., 2023, 415, 1159–1172, DOI:10.1007/s00216-022-04497-3.
- BIPM, IEC, IFCC, ILAC, ISO, IUPAC, IUPAP, and OIML, Joint Committee for Guides in Metrology – Evaluation of Measurement Data, Guide to the Expression of Uncertainty in Measurement JCGM 100:2008. http://www.bipm.org/utils/common/documents/jcgm/JCGM_100_2008_E.pdf, accessed 8 March , 2024 Search PubMed.
- M. Di Luzio, M. Oddone, M. Prata, D. Alloni and G. D'Agostino, Measurement of the neutron flux parameters f and α at the pavia TRIGA mark II reactor, J. Radioanal. Nucl. Chem., 2017, 312, 75–80, DOI:10.1007/s10967-017-5191-4.
- HyperLab, Quick Start Guide for Main Module, HyperLabs Software, Budapest, Hungary, 2014 Search PubMed.
- M. Di Luzio and G. D'Agostino, The k0-INRIM software version 2.0: presentation and an analysis vademecum, J. Radioanal. Nucl. Chem., 2023, 332, 3411–3420, DOI:10.1007/s10967-022-08622-5.
- Kayzero for Windows (KayWin®), User's Manual for Reactor Neutron Activation Analysis (NAA) Using the K0 Standardization Method, Version 3.30, 2017 Search PubMed.
- A. Pramann and O. Rienitz, The molar mass of a new enriched silicon crystal: maintaining the realization and dissemination of the kilogram and mole in the new SI, Eur. Phys. J. Appl. Phys., 2019, 88, 20904, DOI:10.1051/epjap/2019190284.
- J. Vogl, et al. , Intercalibration of Mg Isotope delta scales and realisation of SI traceability for Mg isotope amount ratios and isotope delta values, Geostand. Geoanal. Res., 2020, 44, 439–457, DOI:10.1111/ggr.12327.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ja00235k |
| This journal is © The Royal Society of Chemistry 2024 |