
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid and accurate determination of chlorine isotopic ratios with ICP-MS/MS using O2 reaction gas†
Tyler D.
Schlieder
 *,
Nicole D.
Rocco
,
Maria Laura
di Vacri
,
Isaac J.
Arnquist
,
Danny
Bottenus
,
Zachary
Huber
and
Bruce
McNamara
*,
Nicole D.
Rocco
,
Maria Laura
di Vacri
,
Isaac J.
Arnquist
,
Danny
Bottenus
,
Zachary
Huber
and
Bruce
McNamara
Pacific Northwest National Laboratory, Richland, WA 99352, USA. E-mail: tyler.schlieder@pnnl.gov
First published on 13th September 2024
Abstract
Chlorine isotopic ratio measurements are useful for stable isotope tracing, isotopic abundance measurements in nuclear chemistry, and accurate determination of concentrations using isotope dilution methods. Accurate and precise determination of Cl isotopic ratios using inductively coupled plasma mass spectrometry (ICP-MS) methods is challenging due to major polyatomic interferences of 16O18O1H+ and 36Ar1H+ on 35Cl+ and 37Cl+, respectively. Previous work has demonstrated that using tandem mass spectrometry (ICP-MS/MS) with either H2 or O2 gas in the collision/reaction cell can significantly improve the precision, but not necessarily the accuracy, of chlorine isotopic measurements over single-quadrupole techniques. In this work, we further investigate ICP-MS/MS, using O2 as a reaction gas, as a technique for accurate determination of Cl isotopic ratios. Using the methodology developed herein we measure both natural and enriched chlorine isotopic ratios in diverse samples matrices, targeting 37Cl isotope enrichment efforts, without the need for complex front-end chemistry (i.e., ion exchange chromatography), while maintaining a typical accuracy and precision better than ∼1%. The reduced need for time-consuming sample processing afforded by this method results in higher sample throughput (>80 measurements/day) relative to other analytical techniques (e.g., thermal ionization mass spectrometry, accelerator mass spectrometry, etc.). This work demonstrates that ICP-MS/MS with O2 as a reaction gas can be a useful tool for making rapid and accurate chlorine isotopic ratio measurements.
1 Introduction
Chlorine isotopic ratio measurements are a powerful tool used within a number of disciplines. Applications include tracking the production and migration of organic environmental contaminants,1–5 tracking the efficiency of bioremediation efforts,6 probing the origin/evolution of the earth and solar system,7–12 and the quantification of chlorine concentrations with isotope dilution methods.13 Furthermore, chlorine isotopic ratio measurements are relevant in nuclear science, for example, as a means of validating 37Cl enrichment efforts for use in molten salt reactors.14–16Chlorine isotopic ratio measurements can be made using a variety of analytical techniques but are most commonly performed using thermal ionization mass spectrometry (TIMS),17–20 accelerator mass spectrometry (AMS),21,22 and isotope ratio mass spectrometry (IRMS).23–26 While these techniques are capable of accurate and precise chlorine isotopic ratio measurements (typically ≤0.2–0.6 per mil18–20,23,24,27), they generally involve costly instrumentation and complicated/time-consuming front-end chemistry (e.g., Nb-assisted AgCl precipitations for AMS,27 AgCl precipitation followed by CH3Cl production/separation via chromatography and subsequent cryogenic purification for IRMS,26 cation exchange chromatography and conversion to CsCl for TIMS26).
More recently, inductively coupled plasma mass spectrometry (ICP-MS) techniques have garnered increased attention for making chlorine isotopic ratio measurements. Advantages of ICP-MS over aforementioned analytical techniques include relatively low instrumentation costs, faster analysis, and increased sample throughput. Furthermore, ICP-MS has a high ionization efficiency which enables the partial ionization of analytes with high ionization potential such as chlorine (12.97 eV). However, determination of chlorine isotopic ratios by ICP-MS is hindered primarily by spectral isobaric interferences from polyatomic ions generated in the plasma, primarily 16O18O1H+ and 36Ar1H+, which interfere with 35Cl+ and 37Cl+, respectively. These interferences produce high backgrounds (>1 × 106 cps) at m/z = 35 and 37, complicating the accurate measurement of Cl isotopes on mass. One way to overcome this hindrance is with the use of the high mass resolving power (R). A resolution of about 1100 and 4000 are required to resolve 16O18O1H+ from 35Cl+ and 36Ar1H+ from 37Cl+, respectively. The resolving power achievable with a sector field mass analyzer coupled with a multi-collector detector (MC-ICP-MS; R ≈ 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) is sufficient to resolve these interferences, as reported in previous work.28 While this approach has been demonstrated to be effective, it still requires costly instrumentation and significant sample preparation to isolate the target analyte from the sample matrix (i.e., ion exchange chromatography).28 Additionally, given the high mass resolving power necessary for determining chlorine isotopic ratios by MC-ICP-MS, Cl concentrations >∼70 ppm are required.28 While Cl contents of many industrial, biological, and environmental samples are high (i.e., >0.1%29–31), Cl concentrations often extend to significantly lower values (i.e., <1–100 ppm12,21,22,30,31), particularly in environmental samples (e.g., ground/surface waters, soils, etc.22,30,31). Furthermore, samples with complex and/or high concentration matrices may require significant dilution prior to analysis, thus a methodology for measuring Cl isotope ratios at concentrations <70 ppm is valuable.
000) is sufficient to resolve these interferences, as reported in previous work.28 While this approach has been demonstrated to be effective, it still requires costly instrumentation and significant sample preparation to isolate the target analyte from the sample matrix (i.e., ion exchange chromatography).28 Additionally, given the high mass resolving power necessary for determining chlorine isotopic ratios by MC-ICP-MS, Cl concentrations >∼70 ppm are required.28 While Cl contents of many industrial, biological, and environmental samples are high (i.e., >0.1%29–31), Cl concentrations often extend to significantly lower values (i.e., <1–100 ppm12,21,22,30,31), particularly in environmental samples (e.g., ground/surface waters, soils, etc.22,30,31). Furthermore, samples with complex and/or high concentration matrices may require significant dilution prior to analysis, thus a methodology for measuring Cl isotope ratios at concentrations <70 ppm is valuable.
An alternative approach to eliminating problematic polyatomic interferences is through the use of tandem mass spectrometry (ICP-MS/MS) using a reaction gas.32,33 This approach uses a gas, such as O2 or H2, within a reaction cell to circumvent polyatomic interferences, either via a mass shift or charge transfer reaction.32,33 Much of the work using this method has primarily focused on quantifying chlorine concentrations13,33,34 as single-collector detectors in quadrupole-based ICP-MS generally limit precision and accuracy of isotope ratio measurements relative to their multi-collector counterparts. Nevertheless, recent work has begun investigating the utility of this approach for accurate determination of chlorine isotopic ratios. For example, it has been demonstrated that using ICP-MS/MS with O2 or H2 can significantly improve the analytical precision (from ∼14% to <1%), but not necessarily accuracy (±3–5.5%), of chorine isotopic ratio measurements over single quadrupole ICP-MS.32
This study builds upon previous work and provides a methodology for chorine isotopic ratio measurements using triple quadrupole (QQQ)-ICP-MS/MS with an O2 reaction gas, with a typical accuracy and precision better than ∼1%. The method was validated for samples with natural and enriched chorine isotopic ratios (>95% 37Cl) and for a diverse suite of sample matrices that were associated with 37Cl enrichment efforts.35 Additionally, for the measured sample matrices, no chemical separation was required prior to analysis, which greatly increases sample throughput relative to alternative methods for measuring chlorine isotopic ratios.
2 Experimental
2.1 Reagents and materials
All samples were prepared using ultrapure deionized water (>18.2 MΩ cm) from a MilliQ system (Merk Millipore GmbH, Burlington, MA, USA). Hydrochloric (HCl) acid solutions were prepared using Optima grade HCl (Fisher Scientific, Pittsburg, PA, USA). Dilute (1%) Optima grade ammonium hydroxide (NH4OH; Fisher Scientific, Pittsburg, PA, USA) was used as a rinsing solution between samples during ICP-MS analysis. A natural chlorine isotopic ratio standard (∼10 ppm Cl) was produced gravimetrically by dissolving certified reference material NIST SRM 975a (NaCl; 37Cl/35Cl = 0.31970) in ultrapure water. Enriched 37Cl isotopic ratio standards were produced using an enriched 37Cl reference material (ERM®-AE642, 37Cl/35Cl = 52.247; Cl concentration = ∼164.63 ppm in water). All samples were prepared in cleaned perfluoroalkoxy alkane (PFA) vials from Savillex (Eden Prairie, MN). Prior to use, PFA vials were cleaned by leaching in Optima grade 2.8 M HCl followed by 6 M HNO3 for at least 24 hours at 80 °C and triply rinsed with ultrapure water. To validate the cleaning procedure, 10 vials were randomly sampled after cleaning and filled with ultrapure water. After at least 24 hours contact, the water was analyzed by ICP-MS for chlorine, to ensure vial backgrounds were at or below instrumental background. In no case did the cleaned PFA vials yield chlorine signals above instrument background, indicating negligible chlorine is contributed to solutions from PFA vials after cleaning.2.2 Instrumentation and analytical method
Analyses were performed using an Agilent 8900 triple-quadrupole (QQQ)-ICP-MS/MS (Agilent Technologies Inc., Santa Clara, CA, USA). Experimental operating conditions are listed in Table 1. All determinations were performed in mass shift mode (MS/MS), setting Q1 on m/z 35 and 37, and Q2 on m/z 35 + 16 = 51 and 37 + 16 = 53. Instrument parameters (Table 1) were optimized using a solution of Optima grade HCl, diluted to ∼10 ppm Cl, to maximize sensitivity and instrument stability (low RSD) for ClO+ at m/z = 51 and 53. The optimal O2 flow rate for ClO+ production was found be 0.225 mL min−1, consistent with previous research.32 Other critical parameters for maximizing ClO+ sensitivity include axial acceleration, energy discrimination, OctP bias, and deflect (see Table 1). Sensitivities of ∼96 k and ∼31 k CPS/ppm Cl were achieved for m/z 51 and 53, respectively. The concentration of Cl in the analyzed solutions was chosen to ensure the detector was in pulse counting mode (<1.3 × 106 cps) for both the major and minor isotopes to maximize the accuracy of isotopic ratio measurements. Following optimization for sensitivity, Cl isotopic ratios was fine-tuned using the Q1 bias (Table 1).| RF power | 1600 W |
| Sample depth | 4.0 mm |
| Nebulizer gas (Ar) | 0.75 L min−1 |
| Makeup gas (Ar) | 0.25 L min−1 |
| Plasma gas (Ar) | 15.0 L min−1 |
| Auxiliary gas (Ar) | 0.90 L min−1 |
| Spray chamber | Quartz double pass |
| Nebulizer | MircoFlow PFA-100 |
| Cones | Pt skimmer and sampler cone |
| Extract 1 | 4.0 V |
| Extract 2 | −250 V |
| Omega bias | −150 V |
| Omega lens | 12.5 V |
| Q1 entrance | −5 V |
| Q1 exit | 0.0 V |
| Cell focus | 1.0 V |
| Cell entrance | −60 V |
| Cell exit | −85 V |
| Deflect | 3.0 V |
| Plate bias | −60 V |
| Q1 bias | −1.0 V |
| Q1 prefilter bias | −20.0 V |
| Q1 postfilter bias | −10.0 V |
| 4th gas flow | 15% O2 (0.225 mL min−1) |
| OctP bias | −4.0 V |
| Axial acceleration | 2 V |
| OctP RF | 180 V |
| Energy discrimination | −10 V |
| Measured mass (m/z) | 35 (Q1) → 51 (Q2) |
| 37 (Q1) → 53 (Q2) | |
| Points per peak | 1 |
| Number of sweeps | 1000 |
| Acquisition time | 3.5 s |
| Wait time offset | 3 ms |
Signal to noise (S/N) ratios were studied as a function of the Cl content in the analyzed solution, using different concentrations of HCl and ultrapure deionized water as the background measurement. At ∼10 ppm Cl, S/N ratios were determined as ∼155 and ∼154 for m/z 51 and 53, respectively. Improved S/N ratios were obtained through rinsing between samples, which decreased carryover effects. Several rinsing solutions were tested to minimize carryover: 2% HNO3, 1% TMAH (tetramethylammonium hydroxide), and 1% NH4OH. Best results were obtained with a 30 s rinse in NH4OH, as was observed in previous work.28
3 Results and discussion
The accuracy and precision of this method was evaluated using a 10 ppm solution of natural chlorine isotope certified reference material NIST SRM 975a (37Cl/35Cl = 0.31970 ± 0.00048; Fig. 1). The average ratio from 16 analyses of NIST SRM 975a was measured at 37Cl/35Cl = 0.31993 ± 0.00069 (1σ), which is within the certified uncertainty of the NIST SRM 975a standard. These data demonstrate that this method allows for accurate measurement of natural Cl isotopic ratios to within ∼0.1%. While the standard deviation (2σ) of the 16 replicate measurements is small (∼0.45%) the instrumental uncertainty on individual measurements is larger (typically <1%), therefore we conservatively estimate the typical precision of this method to be <1% when measuring natural Cl isotopic ratios. Accuracy and precision plus additional figures of merit are summarized in Table 2.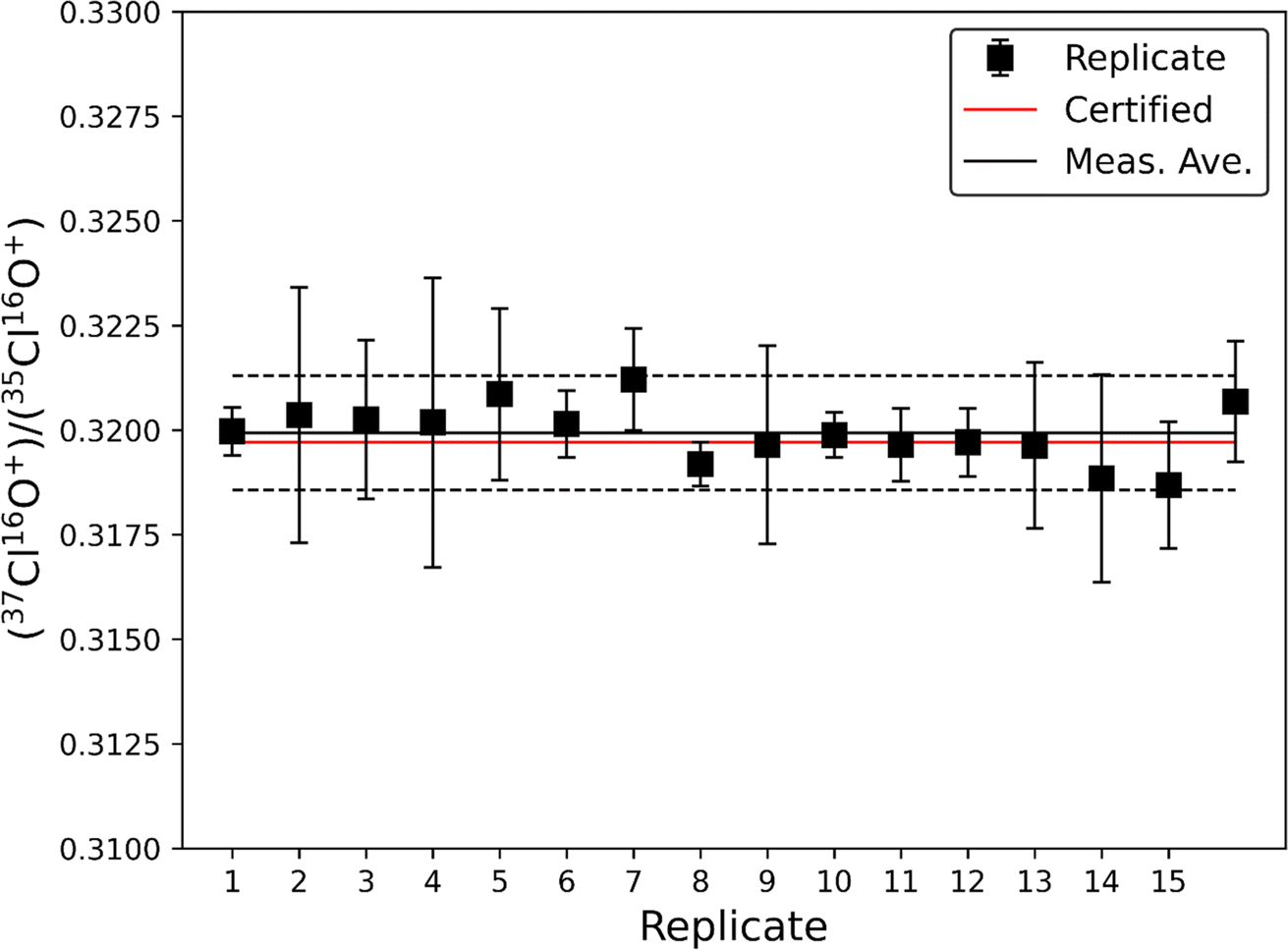 | ||
| Fig. 1 Replicate analyses (n = 16) of NIST SRM 975a NaCl standard (∼10 ppm Cl) using QQQ-ICP-MS/MS and O2 as a reaction gas. The solid red line represents the certified 37Cl/35Cl of NIST SRM 975a, the solid and dashed black lines are the average and 2σ standard deviation of our 16 measurements, respectively. Uncertainties on individual replicate measurements are 1σ. | ||
| Accuracy | ±0.1% |
| Precision (2σ) | ±1% |
| LOD [ng g−1] | 3.4 |
| LOQ [ng g−1] | 11.5 |
| Throughput | ∼80 measurements/8 h |
The method was also tested with isotopically enriched Cl solutions. Enriched isotopic solutions were produced gravimetrically by spiking natural 37Cl/35Cl ratio solutions (NIST SRM 975a Cl isotopic ratio standard and Optima grade HCl; Fig. 2) with a known amount of an enriched Cl isotopic reference material (ERM®-AE642, 37Cl/35Cl = 52.247; 164.63 ppm Cl). In all cases measured Cl isotopic ratios are within 1–2% of the calculated value (ESI 1†). The largest measured deviation from the calculated 37Cl/35Cl ratio (2.2%) was determined for a direct measurement of ERM®-AE642 and is within the stated uncertainty of the certified value (2.51%). In both solutions, measured 37Cl/35Cl ratios were in very good agreement with calculated 37Cl/35Cl ratios (r2 > 0.999). The highest measured 37Cl/35Cl ratio (18.25), which equates to ∼95% 37Cl, was determined within an HCl matrix (Fig. 2B). These data demonstrate that this method is valid for measuring natural and enriched 37Cl/35Cl isotopic ratios.
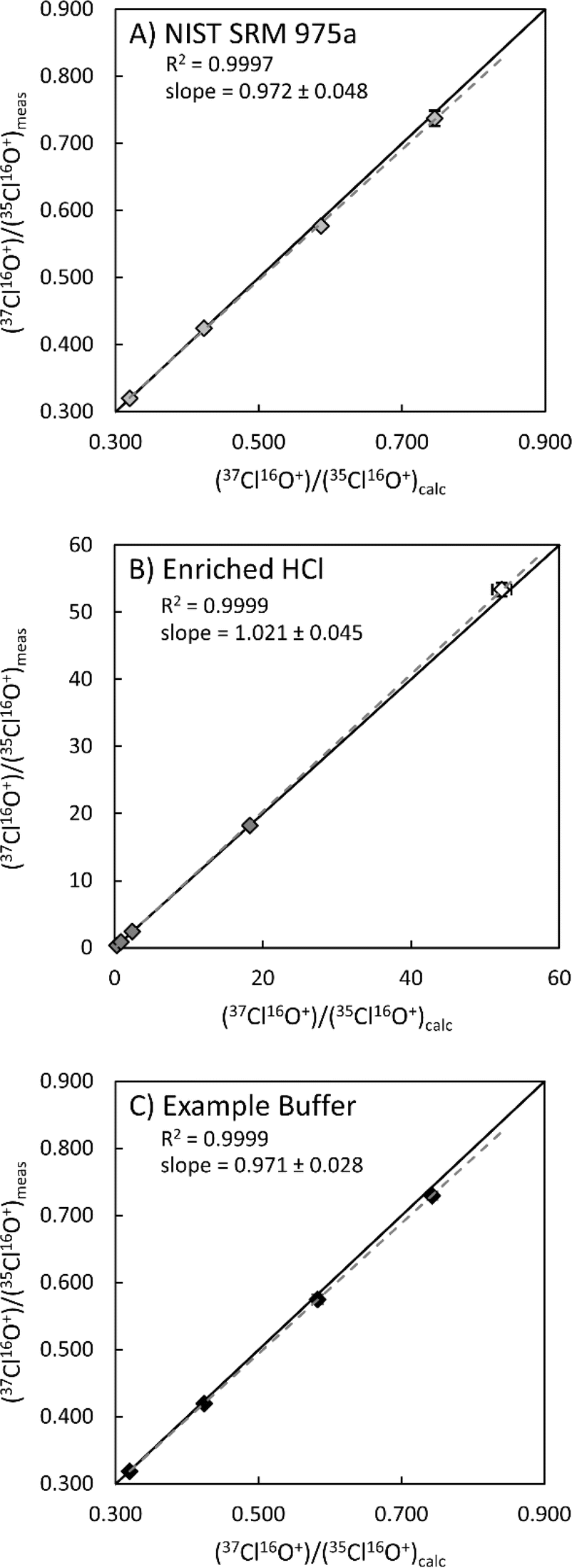 | ||
| Fig. 2 Measured 37Cl/35Cl ratios vs. calculated 37Cl/35Cl ratios for solutions of NIST SRM 975a certified reference material (A), HCl (B), and example buffer solution (C). The open symbol in B is a direct measurement of the enriched 37Cl standard used to make enriched Cl solutions. Uncertainties (2σ) are shown when larger than symbols. Solid lines represent a 1:1 ratio of calculated to measured 37Cl/35Cl ratios, where dashed lines represent best fit lines for each dataset. Uncertainties on slope were estimated by calculating minimum and maximum slopes using the 2σ uncertainties on measured and calculated 37Cl/35Cl ratios. All solutions contain ∼10 ppm chlorine. See ESI 1† for details. | ||
Many applications require determinations of chlorine isotopic ratios in complex sample matrices such as organic solvents,1,2 hydrocarbons,3 terrestrial and chondritic materials,9–11 and 37Cl isotope enrichment efforts for molten salt reactor (MSR) research.35 Therefore, we evaluated the robustness of this methodology for measuring Cl isotopic ratios within different sample matrices. Targeted sample matrices (Table 3) were selected for their relevance to Cl isotope enrichment efforts via isotachophoresis ongoing at Pacific Northwest National Laboratory (PNNL).35 For these experiments, matrix matched buffer solutions were produced, using starting solutions in Table 3, to mimic the conditions of 37Cl isotope enrichment experiments. These solutions were then spiked gravimetrically with a known amount of NIST SRM975a and ERM®-AE642 to produce natural and enriched Cl solutions with concentrations of ∼10 ppm Cl, which were then measured using the method develop here (Fig. 2C and ESI 1†). As with the NIST SRM975a and HCl experiments, measured 37Cl/35Cl values within mixed buffer solutions match with calculated ratios to within 1–2% (r2 > 0.999; Fig. 2C). These results indicate the method presented herein is robust for measuring natural and enriched Cl isotopic ratios within more complex matrices.
| Matrix | Composition | Approx. starting concentration* |
|---|---|---|
| Hydrobromic acid | HBr | 10 mM |
| Tris(hydroxymethyl)aminomethane [tris] | C4H11NO3 | Variable (used for titration) |
| ε-aminocaproic acid [EACA] | C6H13NO2 | 60 mM |
| Orange G | C16H10N2Na2O7S2 | 0.4 mM |
| Ammonium hydroxide | NH4OH | Variable (used for titration) |
| Ammonium thiosulfate | (NH4)2S2O3 | 10 mM |
| Barium hydroxide | Ba(OH)2 | 5 mM |
| HEPES | C8H18N2O4S | 10 mM |
Major challenges with ICP-MS analysis of samples in complex matrices are: (1) the matrix can modify the ionization efficiency and ion transmission of the analyte of interest, and/or (2) the matrix can produce additional polyatomic interferences that lead to inaccurate results. Considering most chlorine samples contain relatively high Cl concentrations (>100–1000 ppm Cl)9,12,28–31 we can mitigate many issues associated with high matrix concentrations by diluting samples with ultra-pure deionized water. We find that diluting samples to ∼10 ppm Cl overcomes many issues associated with high matrix concentrations, in addition to maintaining signals from both Cl isotopes within the detector pulse mode, allowing for robust Cl isotopic ratio measurements even in complex sample matrices. While the list of matrix compositions tested here is by no means exhaustive, we observed no matrix effects for the examined solutions, suggesting simple dilution is sufficient to overcome issues associated with high matrix concentrations, at least for the selected matrices.
The reduced need for time-consuming front-end chemistry (i.e., ion exchange chromatography to remove matrix) greatly simplifies the sample processing required prior to analysis. This simplified preparation procedure coupled with the relatively short analytical time (<5–6 min per sample) required for ICP-MS/MS analysis significantly increases sample throughput (Table 2) relative to alternative methods for performing chlorine isotopic ratio measurement. We estimate a throughput of ∼80 samples per day (8 hours) can be routinely achieved using this approach. Thus, this method provides a valuable tool for any application in which rapid validation of chorine enrichment is necessary (e.g., chlorine isotopic enrichment efforts for molten salt reactor research14–16,35).
4 Conclusions
In this work, we present a method for accurately determining Cl isotopic ratios using ICP-MS/MS with O2 as a reaction gas. This approach offers rapid analysis with minimal sample preparation, increased accuracy compared to similar approaches, high sample throughput, with reduced cost of instrumentation. The method was successfully validated for determinations of Cl isotopic ratios in diverse sample matrices. The method developed in this work and the capability of ICP-MS/MS technology for minimizing (or removing) polyatomic interferences lays the groundwork for novel methods using up-and-coming instrumentation, such as collision cell MC-ICP-MS,36,37 that could provide unparalleled precision. While the method defined here does not provide the same accuracy and precision as a MC-ICP-MS, it provides a faster, less expensive, high throughput method for those applications for which this level of accuracy is sufficient. Additionally, compared to previously proposed ICP-MS/MS methods for Cl isotope measurements using a collision/reaction gas, this method can attain improved accuracy/precision and is reliable for samples with lower Cl concentrations compared to the current state of the art with MC-ICP-MS.Data availability
They data supporting this manuscript are available in the main text, figures, and tables, as well as the supplementary dataset provided.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We would like to acknowledge the U.S. Department of Energy (DOE) Office of Nuclear Energy and in particular Patricia Paviet. We would also like to thank the reviewers for providing insightful comments and suggestions, which helped to improve this manuscript.References
- S. Cretnik, A. Bernstein, O. Shouakar-Stash, F. Löffler and M. Elsner, Molecules, 2014, 19, 6450–6473 CrossRef PubMed.
- O. Shouakar-Stash, S. K. Frape and R. J. Drimmie, J. Contam. Hydrol., 2003, 60, 211–228 CrossRef CAS PubMed.
- J. Zimmermann, L. J. Halloran and D. Hunkeler, Chemosphere, 2020, 244, 125476 CrossRef CAS PubMed.
- E. Van Warmerdam, S. Frape, R. Aravena, R. Drimmie, H. Flatt and J. Cherry, Appl. Geochem., 1995, 10, 547–552 CrossRef CAS.
- D. Hunkeler, B. M. Van Breukelen and M. Elsner, Environ. Sci. Technol., 2009, 43, 6750–6756 CrossRef CAS PubMed.
- N. C. Sturchio, P. B. Hatzinger, M. D. Arkins, C. Suh and L. J. Heraty, Environ. Sci. Technol., 2003, 37, 3859–3863 CrossRef CAS PubMed.
- F. Dhooghe, J. De Keyser, N. Hänni, K. Altwegg, G. Cessateur, E. Jehin, R. Maggiolo, M. Rubin and P. Wurz, Mon. Not. R. Astron. Soc., 2021, 508, 1020–1032 CrossRef CAS.
- J. W. Boyce, A. H. Treiman, Y. Guan, C. Ma, J. M. Eiler, J. Gross, J. P. Greenwood and E. M. Stolper, Sci. Adv., 2015, 1, e1500380 CrossRef PubMed.
- Z. Sharp, J. Barnes, A. Brearley, M. Chaussidon, T. Fischer and V. Kamenetsky, Nature, 2007, 446, 1062–1065 CrossRef CAS PubMed.
- Z. Sharp, C. Shearer, K. McKeegan, J. Barnes and Y. Wang, Science, 2010, 329, 1050–1053 CrossRef CAS PubMed.
- Z. Sharp, J. Mercer, R. Jones, A. Brearley, J. Selverstone, A. Bekker and T. Stachel, Geochim. Cosmochim. Acta, 2013, 107, 189–204 CrossRef CAS.
- J. D. Barnes, Z. D. Sharp and T. P. Fischer, Geology, 2008, 36, 883–886 CrossRef CAS.
- M. Ohata and A. Wada, Anal. Methods, 2017, 9, 4004–4010 RSC.
- B. Hombourger, J. Krepel and A. Pautz, EPJ Nuclear Sciences & Technologies, 2019, 5, 15 Search PubMed.
- B. Merk, A. Detkina, D. Litskevich, S. Atkinson and G. Cartland-Glover, Energies, 2020, 13, 1609 CrossRef CAS.
- A. Mourogov and P. Bokov, Energy Convers. Manage., 2006, 47, 2761–2771 CrossRef CAS.
- T. Fujitani and N. Nakamura, Geostand. Geoanal. Res., 2006, 30, 113–120 CrossRef CAS.
- A. Vengosh, A. R. Chivas and M. T. McCulloch, Chem. Geol., 1989, 79, 333–343 CAS.
- J. Gui, X. Chen, X. Han, Z. Wang and Y. Ma, Anal. Lett., 2022, 55, 2564–2573 CrossRef CAS.
- D. Banks, R. Green, R. Cliff and B. Yardley, Geochim. Cosmochim. Acta, 2000, 64, 1785–1789 CrossRef CAS.
- L. Di Nicola, C. Schnabel, K. Wilcken and K. Gméling, Quat. Geochronol., 2009, 4, 501–507 CrossRef.
- C. Bouchez, J. Pupier, L. Benedetti, P. Deschamps, V. Guillou, K. Keddadouche, G. Aumaître, M. Arnold and D. Bourlès, Chem. Geol., 2015, 404, 62–70 CrossRef CAS.
- O. Shouakar-Stash, R. J. Drimmie, M. Zhang and S. K. Frape, Appl. Geochem., 2006, 21, 766–781 CrossRef CAS.
- J. Renpenning, K. L. Hitzfeld, T. Gilevska, I. Nijenhuis, M. Gehre and H.-H. Richnow, Anal. Chem., 2015, 87, 832–2839 Search PubMed.
- M. Bonifacie, N. Jendrzejewski, P. Agrinier, M. Coleman, F. Pineau and M. Javoy, Chem. Geol., 2007, 242, 187–201 CrossRef CAS.
- A. Godon, N. Jendrzejewski, H. G. Eggenkamp, D. A. Banks, M. Ader, M. L. Coleman and F. Pineau, Chem. Geol., 2004, 207, 1–12 CrossRef CAS.
- T. Anderson, A. Hidy, J. Boyce, F. McCubbin, S. Tumey, J.-M. Dudley, N. Haney, G. Bardoux and M. Bonifacie, Int. J. Mass Spectrom., 2022, 477, 116849 CrossRef CAS.
- J. S. de Gois, M. Costas-Rodríguez, P. Vallelonga, D. L. Borges and F. Vanhaecke, J. Anal. At. Spectrom., 2016, 31, 537–542 RSC.
- S. Frais, Y.-L. Ng and K. Gulabivala, Int. Endod. J., 2001, 34, 206–2015 CrossRef CAS PubMed.
- T. Svensson, H. Kylin, M. Montelius, P. Sandén and D. Bastviken, Environ. Sci. Pollut. Res., 2021, 28, 7691–7709 CrossRef CAS PubMed.
- T. P. Barnum and J. D. Coates, Geobiology, 2022, 20, 634–649 CrossRef CAS PubMed.
- M. Ohata, Y. Zhu and N. Nonose, Anal. Sci., 2017, 33, 375–380 CrossRef CAS PubMed.
- B. Russell, S. Goddard, H. Mohamud, O. Pearson, Y. Zhang, H. Thompkins and R. Brown, J. Anal. At. Spectrom., 2021, 36, 2704–2714 RSC.
- J. Nelson, L. Poirier and F. Lopez-Linares, J. Anal. At. Spectrom., 2019, 34, 1433–1438 RSC.
- D. Bottenus, N. Rocco, T. Schlieder, M. Salalila, S. Branch, C. Ivory, and H. Hallikainen, 2023 Internal Pacific Northwest National Lab Report PNNL-34997, 2023, [Unpublished] Search PubMed.
- J. Wang, D. Tang, B. Su, Q. Yuan, W. Li, B. Gao, K. Chen, Z. Bau and Y. Zhao, J. Anal. At. Spectrom., 2022, 37, 1869 RSC.
- N. Dauphas, T. Hopp, G. Craig, Z. Zhang, M. Valdes, P. Heck, B. Charlier, E. Bell, T. Harrison, A. Davis, L. Dussubieux, P. Willians, M. Krawczynski, C. Bouman, N. Lloyd, D. Tollstrup and J. Schwieters, J. Anal. At. Spectrom., 2022, 37, 2420–2441 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ja00191e |
| This journal is © The Royal Society of Chemistry 2024 |