
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInitial demonstration of microplasma ionization/Orbitrap mass spectrometry for molecular screening of perfluorinated compounds†
Joseph V.
Goodwin
 a,
Claudia
Masucci
bc,
Davide
Bleiner
bc and
R. Kenneth
Marcus
*a
a,
Claudia
Masucci
bc,
Davide
Bleiner
bc and
R. Kenneth
Marcus
*a
aDepartment of Chemistry Clemson University, Clemson, South Carolina, USA. E-mail: marcusr@clemson.edu
bSwiss Federal Laboratories for Materials Science and Technology (Empa), Überlandstrasse 129, Dübendorf, CH-8600, Switzerland
cUniversity of Zurich, Winterthurerstrasse 90, Zurich CH 8057, Switzerland
First published on 27th August 2024
Abstract
The rapid determination of per and polyfluoroalkyl (PFAS) persistent organic pollutants is of growing interest but remains instrumentally challenging. Traditional techniques require some preliminary knowledge of the target species and often time-consuming multistep procedures. Often, the concentration and compositional range of sample contamination is unknown. This is a limitation in investigating the level and fate of a new material's environmental footprint. The liquid sampling – atmospheric pressure glow discharge (LS-APGD) is a microplasma ionization source which provides combined atomic and molecular (CAM) information about analytes. To extend upon the demonstrated applications of the ionization source, the LS-APGD was coupled to an Orbitrap Fourier transform mass spectrometer (FT-MS) to characterize its capabilities towards the analysis of PFAS compounds, including perfluorooctanoic acid (PFOA) and perfluorooctyl sulfonic (PFOS) acid, extending to the perfluoro sulfonamides, acrylates, and telomer alcohols (FTOH). Across the board, these compounds pose incredible analytical challenges regarding the diverse matrices where they are found, their ubiquitous nature (including the laboratory), the lack of a universal ionization method, and the necessity for complex preconcentration/separation prior to MS analysis. The efforts here set the basic characteristics for such analyses, with the caveat that this laboratory is not outfitted for high-sensitivity PFAS analysis, setting up opportunities for more in-depth developments in the future. The mass spectral features for the respective compound types are very uniform, with those of PFOA, PFOS, sulfonamides, and acrylates dominated by their respective M–H (deprotonated) pseudomolecular ions. FTOH compounds were determined by identifying a common characteristic fragmentation pathway. The simplicity of the spectra and high mass resolution/accuracy suggest that determinations might be made without chemical separations. Linear response curves are realized for all species, with limits of detection of 20 pg mL−1 (PFOA) and 310 pg mL−1 (PFOS) obtained, without pre-concentration, for 60 μL infusions. In contrast to the established electrospray ionization (ESI-MS) methods, the CAM/Orbitrap coupling provides species selectivity across the entire breadth of the PFAS compounds and the potential for mixture discrimination without prior chromatographic separation or preconcentration.
1 Introduction
Perfluorooctanoic acid (C7F15COOH, PFOA) and perfluorooctyl sulfonic acid (C8HF17SO3, PFOS) are persistent organic pollutants (POP) and are among the so-called “forever chemicals”, banned under the Stockholm Convention.1,2 Due to their persistence and bioaccumulation, the EPA regards these chemicals as contaminants of emerging concern. PFOS and PFOA are part of the larger group of compounds called per- and polyfluoroalkyl (PFAS) substances; these are compounds in which some or all of the terminal hydrogens on the carbon backbone have been substituted with fluorine (except functional groups). Perfluoroalkyl substances have all hydrogens on the carbon backbone replaced, while polyfluoroalkyl substances have at least two hydrogens, but not all on the chain, have been replaced by fluorine. The perfluoroalkyl moiety is chemically inert and exhibits very low attractive intermolecular forces due to the strength of the C–F bond (485 kJ mol−1) and the very low van-der-Waals forces exhibited by the CF3–(CF2)x>2 moieties. Several commercially produced polyfluorinated precursor compounds, including PFOA and PFOS, yield fluorinated metabolites when subjected to biological or non-biological degradation processes, i.e., fluorotelomer acids via atmospheric oxidation reactions or microbial degradation processes.3,4 PFAS compounds have been widely used in industrial and consumer products due to their unique properties, such as heat resistance and repellency to water and oil. These properties make them more functional than their hydrocarbon analogs. Within the last 50 years, they have been employed in the production of diverse polymer systems, including use in metal plating, aqueous fire-fighting foams, cleaning products, paints, cookware, and electronic components.5–7In 2014, the US EPA recognized PFOA and PFOS as threatening to human health.8 Based on new science and considering lifetime exposure, in 2022, the US EPA updated interim advisories for PFOA and PFOS in drinking water.9 The updated advisory levels indicate that adverse health effects occur even with drinking water concentrations of these two POPs in the sub-ppt range. The US EPA put forth a “roadmap to action”,10 noting that PFAS are ubiquitous in the environment due to their widespread use and emissions. A broad range of these substances have been detected in the soils, streams, water, air, wildlife, and humans,11–13 most probably a minimal representation of such chemicals' actual extent and load. Novel materials in private homes or workplaces, drinking water pipelines, food packaging, waste containers, fire extinguishers, personal care products, and more continually show appreciable fractions and strong emissivity. Because of their proven and potential toxicity, the rapid, selective, and full screening of PFOS, PFOA, and other PFAS compounds in diverse matrices is of high urgency.14
Given the characteristics mentioned above for these compounds and their unknown health effects, selective and sensitive methods are needed to provide researchers and regulators with the necessary information to make informed decisions. While standard procedures, such as US EPA methods 533 (ref. 15) and 537.1,16 are available for analyzing PFAS substances in drinking water, advanced analytical methodologies for determining PFAS, e.g., within complex matrices, are needed. The rapid determination and complete characterization across the spectrum of PFAS compounds remains an unmet analytical challenge due to their chemical complexity, diverse sources, and broad range of compositional variation.17 A recent survey found more than 200 uses for more than 1400 different PFAS across 64 different use categories as diverse as watchmaking and musical instruments.18 In 2018, the Organization for Economic Co-operation and Development (OECD) updated its PFAS database to now include 4730 PFAS and PFAS-related CAS numbers.19 Indeed, it is likely that most PFAS compounds have not been identified in environmental settings, and consequently, their potential adverse impact is not fully understood due to incomplete data coverage.20 Among the broad group of PFAS compounds, the vast structural diversity leads to different chemical and physical characteristics, thus requiring a diversity of analytical techniques. Established techniques involve complex multistep methods.21 Indeed, for target PFAS analysis, high-performance liquid chromatography-electrospray ionization tandem MS (HPLC-ESI-MS/MS) has been routinely employed in specific methods.22,23 Even so, the methods are not amenable to determinations across the full diversity of PFAS compounds; particularly challenging are the fluorotelomer alcohols. As a complement, researchers have recently developed alternative methods for targeted and non-targeted analysis (NTA) of PFAS, for instance, using high-resolution continuum source graphite furnace molecular absorption spectrometry24 to determine the total extractable organic fluorine in place of combustion ion chromatography25 or ICP-MS as an elemental (fluorine) detector (following gas phase modification of the fluoride ion to form a molecular ion).26 To accentuate the complexity and breadth of the subject, a topical collection was recently edited by Baker and Knappe.6
Still, the high diversity and wide mass range of unidentified PFAS and the complexity of the analytical matrices punctuate the importance of developing new, effective instrumental methods. The diversity of structures and chemical–physical properties of PFAS require a versatile technique, while their toxicity requires selectivity and sensitivity. The liquid sampling – atmospheric pressure glow discharge (LS-APGD) is a microplasma ionization source with combined atomic and molecular (CAM) spectral capabilities from a single analysis.27–29 While initially envisioned for elemental analysis,30–33 the LS-APGD has been proven capable of ionizing small polar compounds, polyaromatic hydrocarbons (PAHs), and proteins,34–36 offering CAM ionization across a wide diversity of analytes. In addition, the LS-APGD has proved able to form representative ions from species in complex mixtures not readily amenable to ESI ionization, such as the work of Prather et al. in which a mixture of triglycerides was analyzed in 10 mM solution of sodium chloride, while ESI spectra were composed totally of salt-related species.37 This wide breadth of application is complemented by an equally wide range of mass spectrometer platforms to which the ion source has been coupled, including single- and triple-quadrupoles,35,38 quadrupole ion traps,34,39 and Orbitraps.30,31,40 As such, the mass analyzer can be chosen based on the challenges at hand. Given these characteristics, the LS-APGD may be uniquely positioned to become a viable and versatile ionization source for environmental monitoring.
The omics market has driven the impressive progress of ultrahigh-resolution (UHR) mass spectrometry (e.g., Fourier Transform-Mass Spectrometry (FT-MS)),41–44 both in terms of performance and flexibility. The biological, pharmaceutical, and medical domains have dominated the academic and industrial context in which manufacturers orient their R&D tool development. It is often a point of open discussion as to whether or not chemical separations or tandem mass spectrometry approaches can be alleviated when mass-resolving powers above 40k are available. If chemical separations could be eliminated, dramatic savings in time and cost could be realized for environmentally relevant samples, including the analysis of PFAS compounds. Beyond biomolecule analysis, there is an extensive range of applications in materials science, leading to the release of complex inorganic/organic chemical mixtures. Here, the study of toxic compounds with a growing environmental footprint is of particular importance. These mixtures can only partially be investigated through inorganic mass spectrometry, e.g., ICP-MS, which only provides information about the compound's metal constituents. Information about metal speciation can be inferred only by retention time matching from HPLC-ICP-MS and comparison to known standards, but no direct measurement of the ligated complex is possible with traditional inorganic mass spectrometry. Such approaches virtually eliminate any possibility for NTA of mixture components. Such analyses demand state-of-the-art mass spectrometry, e.g., FT-MS, coupled with flexible ionization sources for the parallel investigation of inorganic and organic components. The LS-APGD, a unique CAM ionization source, has the potential to impact NTA for mixtures containing both organic and inorganic constituents while retaining information about the ligated inorganic complex.
The aim of this work was to investigate the capabilities of LS-APGD coupled with an ultra-high-resolution (UHR) Orbitrap45 Fourier transform mass spectrometer for the determination of PFAS compounds of high environmental (and potentially toxicological) relevance. The structurally-simple mass spectra generated by the solution-based microplasma ionization source,27,34 coupled with high mass accuracy and resolution (m/Δm = 70k at m/z = 200) afforded by the Orbitrap MS,29 provides high confidence in peak assignments without the use of either chromatographic or tandem mass spectrometry approaches. We demonstrate the basic spectral characteristics and preliminary sensitivity figures of merit for this coupling. The product mass spectra are easily interpreted, resulting from class-specific modes of ionization, proton extraction for the acidic sulfonamide and acrylate PFAS compounds, and a straightforward cleavage reaction for the fluorotelomer alcohols. These traits are observed simultaneously under identical plasma operation conditions, illustrating versatility that is not obtained from any other ionization source. While all environmental-level PFAS determinations must be performed under ultraclean room conditions, the sensitivity demonstrated in a standard, open laboratory is very encouraging. The basic characteristics demonstrated here are believed to provide new opportunities in the continuously evolving field of PFAS analyses, extending perhaps to the realm of NTA of PFAS compounds.
2 Materials and methods
2.1 Instrumentation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 methanol:water).
:30 methanol:water).
 | ||
| Fig. 1 Graphical representation of coupling the LS-APGD to the Exactive Q Focus Orbitrap. | ||
000 (m/z = 200) was employed for all work, determined by the time-length of the FT-processed ion transients; 256 ms in this case. While the resolution of the Orbitrap does decrease with increasing mass, the resolution found for 1H,1H,2H,2H-perfluoro-n-dodecyl acrylate (m/z = 618), the highest-mass analyte utilized during this study, was over 40000, still well within the ultrahigh mass resolution regime. The ion injection time maximum was set to 50 ms with automatic gain control of 1 million charges. The ion transfer capillary temperature was maintained at 150 °C. The Q Exactive Focus Orbitrap is equipped with two modes of collisional dissociation. In-source collisional dissociation occurs after the mass spectrometer entrance but before the quadrupole mass filter, while higher energy collisional dissociation (HCD) occurs after initial ion accumulation in the C-trap. However, for the sake of simplicity and direct comparisons, no collisional dissociation was employed in any of the reported measurements. The negative ion mode was used for all measurements. Different digitization m/z ranges were used for PFOA (403–423 Da), PFOS (484–514 Da), and the PFAS mixture (100–800 Da). Spectral measurements were performed for the PFOA and PFOA samples based on 60 μL infusions (2 min acquisitions), while for the volume-limited PFAS mixture, 20 μL injections were employed, resulting in 0.67 min injection transients. All measurements were performed in triplicate.
2.2 Samples
An aqueous solution 70:30 (v/v) of methanol (Sigma-Aldrich, Burlington, MA, USA) and deionized water (PURELAB flex 18.2 MΩ cm−1 water purification system, Velia Water Technologies, High Wycombe, England) was used as the carrier solvent/electrolyte and spectroscopic blank throughout these studies. Perfluorooctanesulfonic acid (PFOS) was obtained from Matrix Scientific (Columbia, SC, USA), and perfluorooctanoic acid (PFOA) was obtained from Tokyo Chemical Industry (TCI America, Portland, OR, USA). 6:2 PFOH was purchased from Sigma-Aldrich (Burlington, MA. USA) and analyzed as a fluorotelomer alcohol standard. 6:2 FTAC was purchased from Oakwood Products (Estill, SC, USA) and was analyzed as a standard for the fluorotelomer acrylates. As the purchase of high-purity, single-component samples of PFAS compounds is difficult and perhaps cost prohibitive, a mixture (50 mg mL−1, each) of 9 polyfluorinated compounds (PFAS Mix 09, Chiron, Trondheim, Norway) was obtained, consisting of N-ethyl-N-(2-hydroxyethyl)perfluorooctyl sulfonamide (N-EtFOSE), N-(2-hydroxyethyl)-N-methylperfluorooctyl sulfonamide (N-MeFOSE), 1H,1H,2H,2H-perfluorohexan-1-ol (4:2 FTOH), 1H,1H,2H,2H-perfluorooctan-1-ol (6:2 FTOH), 1H,1H,2H,2H-perfluorodecan-1-ol (8:2 FTOH), 1H,1H,2H,2H-perfluorododecan-1-ol (10:2-FTOH), 1H,1H,2H,2H-perfluoro-n-octyl acrylate (6:2-FTAC), 1H,1H,2H,2H-perfluoro-n-decyl acrylate (8:2-FTAC), and 1H,1H,2H,2H-perfluoro-n-dodecyl acrylate (10:2-FTAC) was employed. To be clear, this is, in all respects, a complex mixture of PFAS compounds, which would pose a challenge in many mass spectrometric analyses in the absence of prior chromatographic separation. In fact, the use of such a mixture to set analytical benchmarks is akin to operation in an NTA situation.
All solution media were stored in polypropylene vials which were rinsed with solvent and allowed to dry before use. Other than pre-rinsing, no other methods were employed to reduce background.
3 Results and discussion
3.1 Spectral characteristics of PFOA and PFOS
The tested PFAS compounds can be grouped into three classes based on their chemical properties/functionalities. The first class of compounds has low pKa values (class A), the second has high pKa values with sites susceptible to deprotonation (class B), and the third class of neutral compounds (class C), with examples in each class shown in Fig. 2. Specific pKa values for the various PFAS compounds are generally not available. The available model-predicted and experimental pKa values range from −0.5 to 3.8, with PFOA and PFOS having pKa <1.0.46 PFOA and PFOS are strong acids due to the electron-withdrawing effects of fluorine extending to their acidic functional groups. These compounds readily dissociate in water and other environmental matrices, so PFOA and PFOS are present in the dissociated anionic form rather than the protonated, acid form. As such, it is not surprising that the base peaks in the LS-APGD spectra for PFOA and PFOS (class A) represent the deprotonated, psuedomolecular ions [M–H]− at m/z 412.97 and 498.93 Da, respectively, as shown in Fig. 3. Perhaps surprising, given the 2100–3000 K kinetic temperature of microplasma,47 is the minimal amount of fragmentation seen in the mass spectra for these compounds. The losses observed for PFOA, –COOH, and CF2OH make reasonable chemical sense, given the structure of the molecule. Additionally, no other analyte-related spectral signatures appear above the 5% relative abundance level for PFOS, reflecting a lack of appreciable dissociation. As discussed previously, the simplicity of the product spectra bodes well for multispecies determinations without the need for extensive separations. | ||
| Fig. 2 Complete list and chemical structures of per and polyfluoroalkyl substances tested. Class A includes compounds with low pKa. Class B represents compounds with high pKa but with ionizable functional groups like perfluoro sulfonamides and polyfluoro acrylates. Class C represents high pKa, neutral PFAS such as fluorotelomer alcohols. | ||
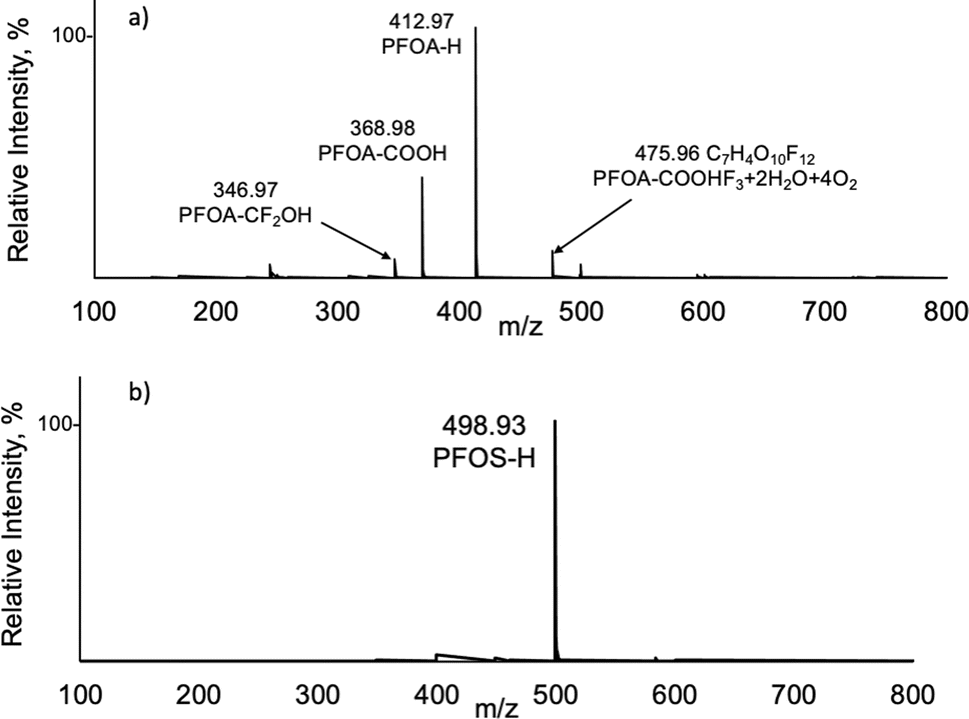 | ||
| Fig. 3 Orbitrap mass spectra of (a) perfluorooctanoic acid (PFOA) and (b) perfluorooctanesulfonic acid (PFOS) from 20 μL injections of a 10 μg mL−1 solution. | ||
After understanding the spectral characteristics of the class A PFAS compounds, response curves were generated for PFOA and PFOS across the concentration ranges 0.5–50 ng mL−1 and 0.05–25 ng mL−1, respectively. Spectral data were acquired for a direct infusion into the LS-APGD source over 2 minutes (equivalent to 60 μL), monitoring the most abundant ion [M–H]−. No preconcentration (i.e., solid phase extraction) was performed before this analysis. The respective response curves are displayed in Fig. 4 for (a) PFOA and (b) PFOS, with the respective linear plots exhibiting a goodness-of-fit R2 > 0.99 in both cases. Precision is indicated in Fig. 4 by the error bars, which represent the standard deviation for n = 3 injections at each level. The %RSD for PFOS ranged between 0.1% and 10%, while the %RSD for PFOA ranged from 1.9% to 6.0%. Higher %RSD values were typically seen from lower concentration solutions. Extended log–log linearity plots are shown in the inserts, highlighting the linearity of the method covering four orders of magnitude in concentration. Sensitivities for PFOA and PFOS are taken as the slope of the calibration curve and are ∼400 and 7500 counts per pg for PFOA and PFOS, respectively, clearly indicating higher sensitivity for PFOS. Limits of detection (LOD) determinations are complicated with the standard Orbitrap mass spectrometer data system due to the automatic baseline subtraction that occurs with the default software; i.e., all values below a certain value are “0”. Given this subtraction, the standard deviation of the lowest point on the calibration curve is used instead of the standard deviation of a blank measurement. Limits of detection of 0.02 and 0.31 ng mL−1 were calculated, respectively, for PFOA and PFOS based on three times the standard deviation of the lowest point of the curve divided by the slope of the linearity curve (LOD = 3σlow/m). These values correspond to solute masses of 1.2 and 19 picograms, respectively, for the two analytes. The worse (higher) LOD found for PFOS, even though the sensitivity for PFOS is higher, is a result of having a higher standard deviation (and average value) at the lowest injection concentration for PFOS in comparison to the standard deviation of the lowest point in the PFOA curve. Given the ubiquitous nature of these compounds in the environments and instrument system components, the sensitivity observed here is quite promising for these preliminary results.
 | ||
| Fig. 4 Response curves for (a) perfluorooctanoic acid (PFOA) and (b) perfluorooctanesulfonic acid (PFOS) showing a good linearity (R2 > 0.99) across >3 orders of magnitude and a calculated LOD (3 s lowest point) of 0.02 and 0.31 ng mL−1 respectively for PFOA and PFOS. Insertions display the log–log response curves to highlight the dynamic range of the Orbitrap capabilities. | ||
3.2 Spectral characteristics of PFAS sulfonamides, acrylates, and telomers
As described above, the primary source of examples of the sulfonamide, acrylate, and fluorotelomer alcohol compounds for this characterization exercise was a mixture containing nine of those species. Knowing the identities of the constituents of the mixture, the respective signature ions could be mined, so long as their LS-APGD mass spectra are of fairly simple structure and the mass spectrometer has sufficient mass resolution/accuracy. In this regard, the process is akin to the methodology necessary for the development of capabilities needed for NTA of PFAS compounds. To that end, an understanding of the ionization characteristics of the chemical classes that are expected to be present is required. As a first step towards this goal, the mixture containing fluorinated sulfonamide, acrylate, and alcohol was analyzed, focussing on what might be expected to be the most simplistic product ions for each compound type.The most acidic compounds were considered first, with the expected products being related to what was observed for the sulfonic and carboxylic acids. As with PFOA and PFOS, fluorinated sulfonamides and acrylates (class B) were detected as [M–H]− ions as well, except for 1H,1H,2H,2H-perfluoro-n-octyl acrylate (6:2 FTAC) that was detected primarily with the addition of an oxygen atom [M–H + O]−. Perfluoro sulfonamides N-MeFOSE and N-EtFOSE were detected primarily as the [M–H]− species from the mixture. A representative spectrum of perfluoro sulfonamides (N-MeFOSE) is shown in Fig. 5a, with the complementary spectrum of N-EtFOSE included in the ESI (Fig. S1).† Polyfluoro acrylates displayed a more complex ionization pattern, not only appearing as the [M–H]− species but also appearing at the [M–2H]˙− radical ion and as the [M–H + O]− species with the exception of 6:2 FTAC which only appeared as the [M–H + O]− species as mentioned above. Representative spectra of the polyfluoro acrylates (6:2 FTAC and 8:2 FTAC) are shown in Fig. 5b and c, with additional spectra in the ESI (Fig. S2).† Given that 6:2 FTAC did not result in a [M–H] peak as seen for 8:2 FTAC and 10:2 FTAC, an individual solution of 6:2 FTAC was injected to confirm the ionization and is shown in red in Fig. 5b. In fact, that compound showed no other related fragment/adduct species with relative abundances of >10% relative to the base peak at m/z = 430.0 Da. The relatively “soft” nature within the micro-plasma source is demonstrated here, again boding well for complex mixture analysis.
 | ||
| Fig. 5 Mass spectrum obtained from the mixture solution of (a) N-(2-hydroxyethyl)-N-methylperfluorooctyl sulfonamide (N-MeFOSE), (b) 1H,1H,2H,2H-perfluoro-n-octyl acrylate (6:2 FTAC) (c) 1H,1H,2H,2H-perfluoro-n-decyl acrylate (8:2 FTAC), and (d) 1H,1H,2H,2H-perfluorooctan-1-ol (6:2 FTOH). The spectra shown in red in (b) and (d) were obtained from injecting individual solutions of 6:2 FTAC and 6:2 FTOH, respectively, to confirm the ionization since the [M–H]− peak was not found. | ||
While a potential acidic moiety, the hydrogen atom associated with the alcohol group of the fluorotelomers, none were detected as [M–H]− ions. However, the formation of a characteristic fragment ion was consistently observed as a result of an intramolecular nucleophilic or radical attack of the hydroxy group that leads to the loss of neutral ethylene oxide (C2H4O). In fact, a signature ion composed solely of the remaining fluorocarbon backbone is the product in each case. A representative spectrum of the fluorotelomer alcohols (6:2 FTOH) is shown in Fig. 5d, with additional spectra of the three other fluorotelomers presented in the ESI (Fig. S3).†Fig. 6 summarizes the proposed fragmentation processes that are consistent across all the tested fluorotelomer alcohols, resulting in the common loss and mass spectra, which directly represent the parent fluoro-alkyl chains. As both a nucleophilic or a radical attack may occur in the microplasma environment, both mechanisms are presented. For all of the fluorotelomer alcohols, the [M-C2H4O–H]− fragments enable their detection in the same mixture with the above-discussed class B compounds. The fragment was confirmed to be related to fluorotelomer alcohols by injecting an individual standard of 6:2 FTOH, shown in red in Fig. 5d. These spectra demonstrated the ability to identify fluorotelomer alcohols as representative ions and not in the form of adducts, i.e., no prior derivatization steps are required for the LS-APGD analysis as they are required for high-sensitivity ESI/APCI-MS. In the absence of derivatization, FTOH complexation with acetate in the LC mobile phase occurs to allow their ionization, but indeed, the respective compounds do not produce unique spectra.48 The identified fragment might also occur for other PFAS compound classes; however, it is not a prominent fragment in any of those spectra. If it were common to other PFAS classes, correction factors could be established to compensate for potential overlaps.
 | ||
| Fig. 6 A proposed mechanism for forming a characteristic fragment ion [M-C2H4O–H]− for the fluorotelomer alcohols. The proposed mechanisms consist of either a nucleophilic attack (1) or in radical attack (2) that could lead to a neutral loss of ethylene oxide (C2H4O). | ||
The results from this simulated non-target (full-mixture) analysis are quite encouraging in terms of unambiguous compound identification; however, further studies are needed to understand better the formation of the (albeit minor) adducts that may be occurring. Importantly, no chromatography was needed to distinguish the different compounds in the mixture in this initial screening experiment. After identifying the relevant species, calibration curves were generated to get a preliminary assessment of measurement sensitivity. The standard mixture of perfluoro sulfonamides, polyfluoro acrylate, and fluorotelomer alcohols was analyzed, and response curves were generated for each of the compounds across the concentration range of 0.1–10 μg mL−1. To be clear, these concentrations are not in the realm of practicality, but as will be discussed subsequently, the nature of the commercial mixture hampers efforts in lower-concentration determinations. The response curves for the fluorotelomer alcohols are presented in Fig. 7, with those of the sulfonamides and acrylates presented in the ESI (Fig. S4 and S5).† The key here as well is that each of these PFAS species was determined simultaneously, without any form of chromatography or sophisticated MS/MS approaches such as selected reaction monitoring (SRM). Precision is indicated in Fig. 7, S4, and S5† by the error bars, which represent the standard deviation found for n = 3 injections. For the fluorotelomer alcohols, the %RSD ranged from 2–25% across all different chain lengths. For the polyfluoro acrylates, the %RSD ranged between 7–26% for both (8:2) and (10:2) FTAC. finally, for the perfluoro sulfonamides, the %RSD ranged between 7–27% for both N-MeFOSE and N-EtFOSE.
 | ||
| Fig. 7 Response curves for all fluorotelomer alcohols 20 μL injections of a mixture across a concentration range of 0.1 to 10 μg mL−1. | ||
A cursory comparison of the response curves for the acids and the fluorotelomers points to a limiting feature of the Orbitrap detection strategy. Specifically, the sensitivities for the fluorotelomer alcohols are between ∼0.6 and ∼6 counts per pg in comparison to a sensitivity of ∼400 counts per pg for PFOA. The low recoveries are due in large part to the nature of ion accumulation in the Orbitrap across large m/z ranges (100 to 800 Da). Since ions are collected until a preset maximum number of charges are accumulated in the C-trap, the trap is filled with background species and other more abundant ions, as well as the target ions. Effectively, this limits the sensitivity as well as the signal-to-background characteristics of trace analytes. These issues can be mitigated by using chromatography to separate the components of the mixture before ionization, the use of collisional dissociation modalities to reduce background species while leaving the molecular ion intact, or by using narrow quadrupole ranges only allowing for the transmission of the analyte of interest. It should also be reiterated that no optimization of plasma discharge conditions or mass spectrometric measurement conditions was performed for this initial proof-of-concept screening. Given these factors and based on the linearity observed at the lower concentrations for the class A acidic compounds, much lower levels of detectability are likely achievable with further optimization. Finally, it must be reiterated that these efforts were undertaken in a standard academic laboratory, replete with background PFAS species, and not sub-class 100 cleanrooms common to commercial or governmental laboratories.
4 Conclusions
Perfluoroalkyl substances in the environment pose some of the greatest challenges encountered by analytical chemists. Many of the challenges are imposed by the simple, ubiquitous nature of the compounds, the diversity of homologs, and the incredibly complex matrices from which they must be determined. Advantages might be projected for methods that can be more uniformly applied across the different compound classes or might reduce the reliance on complex chromatography/pre-concentration protocols. This initial study illustrates the great promise of the combination of high resolution and mass accuracy provided by the Orbitrap mass spectrometer and the CAM ionization provided by the LS-APGD. Uniquely, the LS-APGD simultaneously ionizes PFAS compounds of divergent characteristics (classes A, B, C) in parallel, without varying any measurement parameters and without the benefit of prior chromatographic separations or derivatization.Deprotonated, pseudomolecular ions (M–H)− of PFOA and PFOS were readily produced in near-exclusion of any types of fragmentation or adduct formation. Those species could be used to generate response curves with linearity over two-orders of magnitude and LODs of 0.02 and 0.3 ng mL−1, respectively, for PFOA and PFOS, for 60 μL infusions of the test compounds. It is worth reiterating that these results were generated without pre-concentration steps, such as solid phase extraction, before analysis. In addition, this was a preliminary evaluation study with no optimization of the microplasma conditions on an instrument which is admittedly housed in a multipurpose academic laboratory.
As a far more difficult challenge, the mass spectrometric characteristics of perfluoro sulfonamides, polyfluoro acrylates, and fluorotelomers were evaluated on a mixture of 9 compounds; very much akin to a non-targeted analysis (NTA) situation. Deprotonated ions (M–H)− were also observed for perfluoro sulfonamides and polyfluoro acrylates, with the exception of 6:2 FTAC, which only appeared as the [M–H + O]−. Uniquely, the fluorotelomer compounds were all observed to fragment in a common modality, with the loss of an ethanol unit, to yield ions representing the base fluorocarbon structures, indicative of fluorocarbon chain length. This promising result suggests an opening toward other PFAS compounds which exhibit poor ESI ionization. For instance, it had been held that FTOH compounds could not form deprotonated molecular species with ESI;49 instead, FTOHs formed acetate adducts,50 which did not form unique fragments in MS/MS experiments.51 It was not until Berger et al. removed all traces of acetate from the HPLC system that M–H peaks were observed for FTOH's.48 As would be expected, the responsivity of the compounds in the mixture was suppressed by virtue of the limited ion processing capacities of the Orbitrap. That said, the simultaneous detection across the different classes of PFAS within a mixture illustrates the promise of this instrumental platform for conducting non-targeted analysis of PFAS compounds within a complex mixture. Future studies should be focused on the optimization of plasma and instrumentation conditions for maximum sensitivity, determining figures of merit from the optimized conditions, and exploring in detail the possible formation of adduct species in more complex sample matrices. True samples should be tested using the fully developed method conditions. In addition, the use of HPLC to perform species separations prior to analysis should be investigated for the potential to further simplify spectra and perhaps mitigate any matrix effects. Ultimately, though, such analyses must be performed in class 100 or better laboratories, on systems specifically designed to minimize the ubiquitous presence of PFAS compounds.
Declarations
Empa purchased the LS-APGD ionization source from Clemson University.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Joseph V. Goodwin: methodology, data curation, visualization, writing – original draft preparation; Claudia Masucci: methodology, data curation, visualization, writing – original draft preparation; Davide Bleiner: conceptualization, supervision, writing – reviewing and editing; R. Kenneth Marcus: conceptualization, supervision, writing – reviewing and editing.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
CM is supported by the awarded Empa Internal Research Call (IRC) Funding ref. 2760-FOKO. Continuing studies at Clemson University (JVG) have been supported by the Oak Ridge National Laboratory, managed by UT-Battelle for the Department of Energy under Contract DE-AC05-000R22725.References
- W. Xu, X. Wang and Z. Cai, Anal. Chim. Acta, 2013, 790, 1–13 CrossRef.
- P. E. Hagen and M. P. Walls, Nat. Resour. Environ., 2005, 19, 49–52 Search PubMed.
- M. J. A. Dinglasan, Y. Ye, E. A. Edwards and S. A. Mabury, Environ. Sci. Technol., 2004, 38, 2857–2864 CrossRef.
- A. A. Jensen and M. Warming, Short-chain Polyfluoroalkyl Substances (PFAS), Danish Environmental Protection Agency, 2015 Search PubMed.
- C. E. Müller, A. C. Gerecke, A. C. Alder, M. Scheringer and K. Hungerbühler, Environ. Pollut., 2011, 159, 1419–1426 CrossRef PubMed.
- E. S. Baker and D. R. Knappe, Anal. Bioanal. Chem., 2022, 414, 1187–1188 CrossRef PubMed.
- E. Kissa, Fluorinated Surfactants and Repellents, CRC Press, 2001 Search PubMed.
- EPA, Proposed PFAS Drinking Water Regulation - Public Meeting on Environmental Justice Considerations, https://www.epa.gov/sdwa/and-polyfluoroalkyl-substances-pfas, accessed 06/24/2024.
- EPA, Addressing PFAS Discharges in National Pollutant Discharge Elimination System (NPDES) Permits and Through the Pretreatment Program and Monitoring Programs, https://www.epa.gov/newsreleases/epa-issues-guidance-states-reduce-harmful-pfas-pollution, accessed 06/24/2024.
- PFAS Strategic Roadmap: EPA's Commitments to Action 2021-2024, https://www.epa.gov/pfas/pfas-strategic-roadmap-epas-commitments-action-2021-2024, accessed 06/24/2024.
- R. Buck, J. Franklin, U. Berger, J. Conder, I. Cousins and P. Voogt, Integr. Environ. Assess. Manage., 2011, 7, 513–541 CrossRef CAS PubMed.
- A. M. Calafat, Z. Kuklenyik, J. A. Reidy, S. P. Caudill, J. S. Tully and L. L. Needham, Environ. Sci. Technol., 2007, 41, 2237–2242 CrossRef CAS PubMed.
- J. P. Giesy and K. Kannan, Environ. Sci. Technol., 2001, 35, 1339–1342 CrossRef CAS.
- S. Taniyasu, K. Kannan, M. K. So, A. Gulkowska, E. Sinclair, T. Okazawa and N. Yamashita, J. Chromatogr. A, 2005, 1093, 89–97 CrossRef CAS.
- EPA, Method 533: Determination of Per- and Polyfluoroalkyl Substances in Drinking Water by Isotope Dilution Anion Exchange Solid Phase Extraction and Liquid Chromatography/Tandem Mass Spectrometry, https://www.epa.gov/dwanalyticalmethods/method-533-determination-and-polyfluoroalkyl-substances-drinking-water-isotope, accessed 06/24/2024.
- EPA, Method 537.1: Determination of Selected Per- And Polyfluorinated Alkyl Substances in Drinking Water By Solid Phase Extraction And Liquid Chromatography/Tandem Mass Spectrometry (LC/MS/MS), https://cfpub.epa.gov/si/si_public_record_Report.cfm?Lab=NERL%26dirEntryId=343042, accessed 06/24/2024.
- H. Mahoney, Y. Xie, M. Brinkmann and J. P. Giesy, Eco Environ. Health, 2022, 117–131 CrossRef PubMed.
- J. Glüge, M. Scheringer, I. T. Cousins, J. C. DeWitt, G. Goldenman, D. Herzke, R. Lohmann, C. A. Ng, X. Trier and Z. Wang, Environ. Sci.: Processes Impacts, 2020, 22, 2345–2373 RSC.
- OECD, Toward A New Comprehensive Global Database of Per- And Polyfluoroalkyl Substances (PFASS): Summary Report On Updating The OECD 2007 List fo Per- And Polyfluoroalkyl Substances (PFASS), https://one.oecd.org/document/ENV/JM/MONO(2018)7/en/pdf, accessed 06/24/2024.
- S. Kurwadkar, J. Dane, S. R. Kanel, M. N. Nadagouda, R. W. Cawdrey, B. Ambade, G. C. Struckhoff and R. Wilkin, Sci. Total Environ., 2022, 809, 151003 CrossRef CAS PubMed.
- C. E. Müller, A. C. Gerecke, C. Bogdal, Z. Wang, M. Scheringer and K. Hungerbühler, Environ. Pollut., 2012, 169, 196–203 CrossRef.
- C. Tang, Y. Liang, K. Wang, J. Liao, Y. Zeng, X. Luo, X. Peng, B. Mai, Q. Huang and H. Lin, npj Clean Water, 2023, 6, 6 CrossRef CAS.
- D. J. Beale, S. Nilsson, U. Bose, N. Bourne, S. Stockwell, J. A. Broadbent, V. Gonzalez-Astudillo, C. Braun, B. Baddiley, D. Limpus, T. Walsh and S. Vardy, Sci. Total Environ., 2022, 817, 153019 CrossRef CAS PubMed.
- T. Nxumalo, A. Akhdhar, V. Mueller, F. Simon, M. von der Au, A. Cossmer, J. Pfeifer, E. M. Krupp, B. Meermann and A. Kindness, Anal. Bioanal. Chem., 2023, 415, 1195–1204 CrossRef CAS PubMed.
- A. Akhdhar, M. Schneider, A. Orme, L. Schultes, A. Raab, E. M. Krupp, J. P. Benskin, B. Welz and J. Feldmann, Talanta, 2020, 209, 120466 CrossRef CAS PubMed.
- S. Heuckeroth, T. N. Nxumalo, A. Raab and J. Feldmann, Anal. Chem., 2021, 93, 6335–6341 CrossRef CAS PubMed.
- R. K. Marcus, B. T. Manard and C. D. Quarles, J. Anal. At. Spectrom., 2017, 32, 704–716 RSC.
- R. K. Marcus, E. D. Hoegg, K. A. Hall, T. J. Williams and D. W. Koppenaal, Mass Spectrom. Rev., 2023, 44, 652–673 CrossRef PubMed.
- D. W. Koppenaal and R. K. Marcus, Appl. Spectrosc., 2023, 77, 885–906 CrossRef CAS PubMed.
- R. K. Marcus, C. D. Quarles Jr, C. J. Barinaga, A. J. Carado and D. W. Koppenaal, Anal. Chem., 2011, 83, 2425–2429 CrossRef CAS PubMed.
- E. D. Hoegg, C. J. Barinaga, G. J. Hager, G. L. Hart, D. W. Koppenaal and R. K. Marcus, J. Am. Soc. Mass Spectrom., 2016, 27, 1393–1403 CrossRef.
- T. J. Williams, E. D. Hoegg, J. R. Bills and R. K. Marcus, Int. J. Mass Spectrom., 2021, 464, 116572 CrossRef.
- J. V. Goodwin, B. T. Manard, B. W. Ticknor, P. Cable-Dunlap and R. K. Marcus, J. Anal. At. Spectrom., 2022, 37, 814–822 RSC.
- L. X. Zhang and R. K. Marcus, J. Anal. At. Spectrom., 2016, 31, 145–151 RSC.
- T. J. Williams and R. K. Marcus, J. Anal. At. Spectrom., 2020, 35, 1910–1921 RSC.
- T. J. Williams, J. R. Bills and R. K. Marcus, J. Anal. At. Spectrom., 2020, 35, 2475–2478 RSC.
- M. R. Alves, J. S. Sauer, K. A. Prather, V. H. Grassian and C. L. Wilkins, Anal. Chem., 2020, 92, 8845–8851 CrossRef CAS PubMed.
- T. J. Williams and R. K. Marcus, J. Anal. At. Spectrom., 2019, 34, 1468–1477 RSC.
- L. X. Zhang, B. T. Manard, S. Konegger-Kappel and R. K. Marcus, Anal. Bioanal. Chem., 2014, 406, 7497–7509 CrossRef CAS PubMed.
- E. D. Hoegg, S. Godin, J. Szpunar, R. Lobinski, D. W. Koppenaal and R. K. Marcus, J. Am. Soc. Mass Spectrom., 2019, 30, 1163–1168 CrossRef CAS.
- A. G. Marshall, T. C. L. Wang and T. L. Ricca, J. Am. Chem. Soc., 1985, 107, 7893–7897 CrossRef.
- J. R. Yates, J. Mass Spectrom., 1998, 33, 1–19 CrossRef.
- Q. Z. Hu, R. J. Noll, H. Y. Li, A. Makarov, M. Hardman and R. G. Cooks, J. Mass Spectrom., 2005, 40, 430–443 CrossRef PubMed.
- A. C. Peterson, J. D. Russell, D. J. Bailey, M. S. Westphall and J. J. Coon, Mol. Cell. Proteomics, 2012, 11, 1475–1488 CrossRef.
- S. Kim, D. Kim, M. J. Jung and S. Kim, Mass Spectrom. Rev., 2022, 41, 352–369 CrossRef CAS PubMed.
- J. Cheng, E. Psillakis, M. Hoffmann and A. Colussi, J. Phys. Chem. A, 2009, 113, 8152–8156 CrossRef CAS PubMed.
- W. C. Davis and R. K. Marcus, Spectrochim. Acta, Part B, 2002, 57, 1473–1486 CrossRef.
- U. Berger, I. Langlois, M. Oehme and R. Kallenborn, Eur. J. Mass Spectrom., 2004, 10, 579–588 CrossRef CAS PubMed.
- B. Szostek, K. B. Prickett and R. C. Buck, Rapid Commun. Mass Spectrom., 2006, 20, 2837–2844 CrossRef CAS PubMed.
- C. Gremmel, T. Frömel and T. P. Knepper, Chemosphere, 2016, 160, 173–180 CrossRef CAS PubMed.
- J. F. Ayala-Cabrera, F. Javier Santos and E. Moyano, Anal. Bioanal. Chem., 2018, 410, 4913–4924 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ja00157e |
| This journal is © The Royal Society of Chemistry 2024 |