New natural garnet reference materials for determining the oxidation state of iron in garnet using the electron microprobe flank method
Received
10th April 2024
, Accepted 18th November 2024
First published on 19th November 2024
Abstract
The oxidation state of iron (e.g., Fe3+/ΣFe) in minerals is a direct proxy for the oxygen fugacity of magma and fluid, which plays a key role in the formation of various types of ore deposits. Although many techniques have been developed to determine the Fe3+/ΣFe ratio in minerals, the electron microprobe flank method is particularly notable for its easy accessibility and high efficiency. However, the application of this method is limited by a shortage of suitable calibration standards. In this study, we collected a series of natural, euhedral garnet grains and gem-quality garnet fragments, which were carefully crushed and separated under a binocular microscope. Following a detailed examination of their major element compositions and Mössbauer spectroscopy measurements for their Fe3+/ΣFe ratios, we report ten new garnet samples (three belonging to the andradite–grossular series and seven to the almandine–pyrope–grossular series) that can be used as reference materials to calibrate the Fe3+/ΣFe ratio of garnet using the flank method. The andradite–grossular samples are highly enriched in Fe3+, exhibiting Fe3+/ΣFe ratios ranging from 0.89 ± 0.03 to 1.00 ± 0.03, while the almandine–pyrope–grossular samples contain minimal Fe3+ with Fe3+/ΣFe ratios ranging from 0.01 ± 0.02 to 0.03 ± 0.01. One andradite sample (And1902) and one almandine sample (Ald1906) were identified as ideal for determining the flank positions for Fe Lα and Fe Lβ. These two end-members, along with the other eight samples, can be employed to quantify the relationship between Fe Lβ/Lα at flank positions and the Fe2+ or ΣFe content. The results indicate that the Fe2+ contents and Fe3+/ΣFe ratios of the ten garnet samples align with those obtained through Mössbauer spectroscopy, with an uncertainty of ±1 wt% for Fe2+ and ±0.05 for Fe3+/ΣFe, respectively. Consequently, these well-characterized natural garnet samples can serve as reliable reference materials when synthetic garnet standards are unavailable.
1. Introduction
Iron (Fe) is generally a major element in many mafic silicate minerals (e.g., olivine, pyroxene, garnet, amphibole, and biotite), oxides (e.g., spinel, chromite, and magnetite), and sulfides (e.g., pyrrhotite and pyrite). The oxidation state of Fe (expressed as Fe3+/ΣFe) in the minerals is commonly used to constrain the oxygen fugacity of magma/fluid evolution.1–5 However, obtaining the accurate Fe3+/ΣFe ratio at the scale of individual mineral grains remains challenging, even though their Fe content can be easily measured.
Many techniques have been developed to determine the Fe3+/ΣFe ratio of minerals, including wet chemistry, Mössbauer spectroscopy, X-ray absorption near-edge structure (XANES) spectroscopy, electron energy loss spectroscopy (EELS), X-ray photoelectron spectroscopy (XPS), and electron probe microanalysis (EPMA). However, each method has its advantages and disadvantages.6 Wet chemistry and conventional Mössbauer spectroscopy are commonly used to determine the Fe3+/ΣFe ratio of powdered samples,7 they are inadequate for analyzing small areas (micron-scale) of individual minerals. Over the past twenty years, Mössbauer spectroscopy has significantly improved in spatial resolution with the development of “milliprobe” capabilities, achieving resolutions as small as 50 μm; however, the process of acquiring spectra remains time-consuming.8,9In situ micro-Mössbauer and XANES techniques facilitate precise measurements of the Fe3+/ΣFe ratio at spatial resolutions down to a few microns.10–13 However, these methods necessitate a synchrotron source of X-rays, which is only accessible at limited synchrotron facilities. The EELS method offers exceptional spatial resolution on the nanometer scale; however, the standards used for calibration may be inhomogeneous at this scale, and beam damage is often significant.14 XPS can provide information about the elemental and chemical states of the atoms, but a major drawback is its strong surface sensitivity, as it typically samples only the top 5 nm.6 The EPMA is suitable for in situ routine analysis of the Fe3+/ΣFe ratio in natural minerals of millimeter size; however, it necessitates multiple mineral standards for data calibration.
Höfer et al.15 conducted the pioneering study on the EPMA “flank method”, which was improved later for accurate determination of the Fe3+/ΣFe ratio of garnet.16–18 This method has been successfully applied to measure the Fe3+/ΣFe ratio of natural garnet from metamorphic rocks,19–22 arclogite, and deep-seated garnet pyroxenite,23 and has been extended to amphibole and biotite,24 silicate glass,25,26 and synthetic majoritic garnet from high-pressure experiments.27 The principle of this method is based on the difference in the self-absorption of Fe2+ and Fe3+ from the systematic correlation of Fe Lβ/Lα at flank positions and Fe2+ content.17 In these studies, two synthetic pure garnet reference materials, one Fe2+ end-member (almandine) and one Fe3+ end-member (andradite), were used to identify the flank positions of Fe Lα and Fe Lβ. Using multiple well-characterized garnet reference materials, a linear correlation of Fe Lβ/Lα at flank positions and Fe2+ content of garnet can be obtained and used for determining the Fe3+/ΣFe ratio of unknown garnets. It is suggested that the EPMA flank method can yield an accuracy of Fe3+/ΣFe at ±0.04 for garnets with 10 wt% total Fe.17
The advantages of the EPMA such as easy accessibility, small spot size (micrometer) and high speed of analysis, make the EPMA flank method an ideal choice for determining Fe3+/ΣFe ratios of natural garnet. Garnet is widespread in metamorphic rocks, a primary constituent of the upper mantle, and is also found in some igneous rocks and sediments.28 However, the application of the flank method to natural garnet is hindered by the limited availability of garnet calibration standards, which are present in only a few EPMA laboratories. In this study, we collected a series of natural, euhedral garnet grains and gem-quality garnet fragments to identify potential reference materials for the EPMA flank method. A total of ten garnet samples were ultimately selected, which can be used to determine the flank positions of Fe Lα and Fe Lβ, as well as to calibrate the Fe3+/ΣFe ratio of unknown garnets.
2. Samples and analytical methods
2.1. Sample preparation
A total of twenty natural garnet samples were collected during the preparation. Three of these samples belong to the andradite–grossular series, while the remaining seventeen belong to the almandine–pyrope–grossular series (Fig. 1). These samples consist of euhedral garnet grains and gem-quality garnet fragments collected from China, Brazil, and Switzerland, all of which were purchased from online mineral retailers. The garnet samples were crushed to a particle size of 30–60 mesh. Subsequently, the garnet fragments were meticulously handpicked under a binocular microscope. Approximately 20 garnet fragments from each sample were mounted on epoxy resin and polished with diamond paste. The mounted garnet fragments were carefully examined using Raman spectroscopy to identify the phases of the garnets, while backscattered electron (BSE) images were employed to evaluate the potential zoning of the garnet grains. Finally, three samples from the andradite–grossular series (And1902, Grs1928, and MelS01) and seven samples from the almandine–pyrope–grossular series (Ald1906, Ald1915, Ald1917, Ald1905, Ald1907, Ald1925, and Ald1926) were selected (Fig. 1).
 |
| | Fig. 1 Photographs of garnet collections. Abbreviations: Adr-andradite. The scale bar in all photographs is 1 cm in length. | |
2.2. Raman analysis
Raman spectra of natural garnet fragments were obtained using a Renishaw inVia™ Reflex (UK) confocal Raman system at the Electron Microscopy Center, Guangzhou Institute of Geochemistry, Chinese Academy of Sciences (GIGCAS). The excitation source was a 532 nm laser (220 V) paired with a 50× objective lens to focus the excitation laser onto a spot approximately 1 μm in diameter. The laser power was set to 10 mW, and spectra were collected in the range of 50–1799 cm−1 with a 3-second exposure time using a grating of 1800 grooves per mm (vis).
2.3. EPMA analysis of major elements
Major element compositions of selected garnets were determined in conjunction with flank-method measurements using a JEOL JXA-8230 EPMA equipped with five wavelength-dispersive spectrometers at the Key Laboratory of Mineralogy and Metallogeny at GIGCAS (see the Discussion section). The operating conditions of 15 kV, 60 nA, and a 5 μm beam were employed for all elements. For separate quantitative elemental analysis, we utilized 15 kV, 20 nA, and a 1 μm beam. Counting times on the peak and upper/lower background positions of elements were 20 and 10 seconds for Si, Al, Fe, Mg, and Ca; 40 and 20 seconds for Mn, Cr, and Ti; and 10 and 5 seconds for Na and K. Analytical results were processed using ZAF correction routines. Primary standards used for data calibration included chrome-diopside (Si and Ca), olivine (Mg), magnetite (Fe), almandine (Al), albite (Na), Cr2O3 (Cr), rutile (Ti), rhodonite (Mn), and orthoclase (K). An in-house almandine was employed as the secondary standard to monitor data accuracy and precision. Major element contents (e.g., SiO2, MgO, FeO, Al2O3, and CaO) were within ±2% of preferred values, while minor element contents (<1 wt%) were within 10% of preferred values. Relative precisions were within 2% for major elements and 7% for minor elements. High-resolution elemental mapping of individual garnet fragment was conducted using the same EPMA under operating conditions of 20 kV, 100 nA, and a beam diameter of 1–1.5 μm. The dwell time for each pixel was set to 60 ms.
2.4. Mössbauer analysis of the Fe3+/ΣFe ratio
The Fe3+/ΣFe ratios of selected garnet samples were determined using Mössbauer spectroscopy. Garnet fragments of the selected samples were cleaned in an ultrasonic bath, first with ethanol for 5 minutes and then with Milli-Q water for an additional 5 minutes to remove impurities. The cleaned garnet grains were then powdered to a 200-mesh size using an agate mortar prior to Mössbauer analyses. Mössbauer spectra of the garnet powder were collected using two WSS-10 Mössbauer spectrometers at the GIGCAS (And1902, Ald1905, and Ald1906) and China University of Geosciences (Wuhan) (CUG) (Grs1928, MelS01, Ald1907, Ald1915, Ald1917, Ald1925, and Ald1926), respectively. A 57Co source in a rhodium matrix was used at room temperature in both laboratories. Measurements were carried out in transmission geometry with a moving absorber. A standard absorber, α-Fe foil, with a thickness of 7 μm (GIGCAS) or 25 μm (CUG) was used to calibrate the velocity scale.29 The Mössbauer spectra were fitted using the least squares method with the MossWinn 4.0 program, employing a Lorentzian line shape. The quality of the fits to the Mössbauer spectra indicates that the 1σ uncertainty of Fe3+/∑Fe is 0.01–0.04 for the different garnet samples (Table 1).
Table 1 Chemical compositions and Fe3+/∑Fe ratios of selected natural garnetsb
| Series |
Andradite–grossular |
Almandine–pyrope–grossular |
| Sample no. |
And1902-Band1 |
And1902 |
Grs1928 |
MelS01 |
Ald1906 |
Ald1915 |
Ald1917 |
Ald1905 |
Ald1907 |
Ald1925 |
Ald1926 |
| n |
20 |
σ
|
20 |
σ
|
12 |
σ
|
20 |
σ
|
20 |
σ
|
16 |
σ
|
20 |
σ
|
20 |
σ
|
20 |
σ
|
16 |
σ
|
12 |
σ
|
|
The FeO and Fe2O3 contents were calculated using the total FeO and Mössbauer Fe3+/∑Fe ratios.
bdl-below detection limit.
|
| SiO2 |
34.68 |
0.33 |
36.41 |
0.54 |
38.07 |
0.23 |
34.24 |
0.23 |
37.43 |
0.24 |
37.29 |
0.23 |
37.16 |
0.29 |
40.19 |
0.23 |
39.65 |
0.21 |
38.71 |
0.16 |
39.33 |
0.15 |
| TiO2 |
bdl |
0.00 |
0.06 |
0.05 |
0.05 |
0.03 |
2.80 |
0.26 |
bdl |
0.00 |
bdl |
0.00 |
0.02 |
0.01 |
0.03 |
0.01 |
0.01 |
0.01 |
0.06 |
0.01 |
0.02 |
0.01 |
| Al2O3 |
0.71 |
0.05 |
5.85 |
2.78 |
17.93 |
0.33 |
6.55 |
0.09 |
21.10 |
0.11 |
21.05 |
0.10 |
20.97 |
0.13 |
22.79 |
0.12 |
22.48 |
0.14 |
21.71 |
0.11 |
22.05 |
0.09 |
| Cr2O3 |
bdl |
0.00 |
bdl |
0.00 |
bdl |
0.00 |
0.01 |
0.00 |
0.01 |
0.01 |
0.01 |
0.01 |
0.05 |
0.01 |
0.02 |
0.01 |
0.01 |
0.00 |
0.07 |
0.01 |
bdl |
0.00 |
| FeOa |
bdl |
0.00 |
bdl |
0.00 |
0.72 |
0.03 |
0.62 |
0.00 |
31.55 |
0.26 |
30.97 |
0.36 |
30.30 |
0.32 |
17.90 |
0.09 |
19.38 |
0.08 |
19.69 |
0.06 |
18.56 |
0.07 |
| Fe2O3a |
30.10 |
0.19 |
23.71 |
3.38 |
6.26 |
0.25 |
19.86 |
0.14 |
0.30 |
0.00 |
1.83 |
0.02 |
0.98 |
0.01 |
0.34 |
0.00 |
0.68 |
0.00 |
1.40 |
0.00 |
1.64 |
0.01 |
| MnO |
0.50 |
0.03 |
0.48 |
0.20 |
0.32 |
0.04 |
0.22 |
0.02 |
2.84 |
0.06 |
0.90 |
0.11 |
0.65 |
0.16 |
0.75 |
0.06 |
0.72 |
0.06 |
0.76 |
0.06 |
0.86 |
0.06 |
| MgO |
0.02 |
0.01 |
0.11 |
0.04 |
0.09 |
0.05 |
0.56 |
0.04 |
5.19 |
0.08 |
5.27 |
0.19 |
4.92 |
0.23 |
14.51 |
0.09 |
13.07 |
0.12 |
9.61 |
0.07 |
12.44 |
0.04 |
| CaO |
32.73 |
0.16 |
33.68 |
0.24 |
35.56 |
0.38 |
33.41 |
0.11 |
1.58 |
0.10 |
2.15 |
0.18 |
3.97 |
0.12 |
3.90 |
0.06 |
3.74 |
0.03 |
6.97 |
0.06 |
4.43 |
0.04 |
| Na2O |
bdl |
0.01 |
bdl |
0.01 |
bdl |
0.00 |
0.01 |
0.01 |
0.01 |
0.01 |
0.01 |
0.01 |
0.01 |
0.01 |
bdl |
0.01 |
bdl |
0.00 |
0.01 |
0.01 |
0.01 |
0.01 |
| Total |
98.75 |
100.30 |
99.02 |
98.28 |
100.01 |
99.50 |
99.01 |
100.44 |
99.73 |
98.99 |
99.34 |
| Fe3+/∑Fe Mössbauer |
1.00 |
0.03 |
1.00 |
0.03 |
0.89 |
0.03 |
0.97 |
0.04 |
0.01 |
0.03 |
0.05 |
0.02 |
0.03 |
0.01 |
0.02 |
0.03 |
0.03 |
0.01 |
0.06 |
0.03 |
0.07 |
0.02 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|
Endmembers
|
| Andradite |
96.68% |
|
71.73% |
|
18.84% |
|
72.06% |
|
1.64% |
|
1.39% |
|
1.66% |
|
1.90% |
|
1.48% |
|
2.31% |
|
2.61% |
|
| Almandine |
— |
|
— |
|
— |
|
— |
|
67.63% |
|
69.97% |
|
66.64% |
|
33.68% |
|
38.76% |
|
41.63% |
|
38.47% |
|
| Pyrope |
0.09% |
|
0.44% |
|
0.34% |
|
0.90% |
|
20.47% |
|
20.86% |
|
19.52% |
|
53.03% |
|
48.56% |
|
36.59% |
|
46.60% |
|
| Grossular |
1.54% |
|
26.59% |
|
79.42% |
|
23.28% |
|
2.80% |
|
4.71% |
|
9.52% |
|
8.29% |
|
8.48% |
|
16.57% |
|
9.31% |
|
| Spessartine |
— |
|
— |
|
— |
|
— |
|
6.37% |
|
2.03% |
|
1.46% |
|
1.57% |
|
1.52% |
|
1.65% |
|
1.83% |
|
| Uvarovite |
0.01% |
|
0.01% |
|
0.00% |
|
0.02% |
|
0.03% |
|
0.03% |
|
0.15% |
|
0.06% |
|
0.03% |
|
0.20% |
|
0.01% |
|
3. Results
3.1. Major element compositions of garnet
The sample And1902 is identified as andradite (Fig. 2), exhibiting zoning in the BSE image, which consists of alternating dark and bright bands that correspond to variations in Fe and Al content (Fig. 3a). The MnO and MgO contents of And1902 are very low, at 0.5 wt% and 0.1 wt%, respectively (Table 1). A compositional profile across one fragment reveals opposing trends in the total FeO and Al2O3 contents (Fig. 4a and b). The brightest band of the fragment (Band 1) in the BSE image contains the highest total FeO content of 26.7 wt% and the lowest Al2O3 content of 0.62 wt% (Fig. 4b and c). Utilizing charge balance principles, the molar proportions of garnet end-members were calculated from EPMA results using an Excel spreadsheet developed by Locock.30 The results show that the andradite fragment is primarily composed of 64–94% andradite (Ca3(Fe3+, Ti)2Si3O12) and 2–31% grossular (Ca3Al2Si3O12) end-members throughout the alternating bands (Fig. 4c and Table 2). A clear negative correlation is observed between the octahedral Fe3+ and Al atoms per formula unit (Fig. 4d). A random analysis of twenty points of the And1902 sample reveals an average total FeO of 21.33 wt% and Al2O3 of 5.85 wt%, corresponding to 72% andradite and 27% grossular end-members (Fig. 5a). The MelS01 sample is a melanite, a black Ti-bearing variety of andradite (Fig. 2). It is homogeneous and contains 17.87 wt% total FeO, 2.80 wt% TiO2, and 6.55 wt% Al2O3, corresponding to 72% andradite and 23% grossular end-members (Table 1 and Fig. 5a). The Grs1928 sample exhibits a uniform composition with 5.64 wt% total FeO and 17.93 wt% Al2O3, comprising 79% grossular and 19% andradite end-members (Table 1 and Fig. 5a). For the andradite–grossular series samples (And1902, MelS01, and Grs1928), CaO and MgO exhibit limit variation (Fig. 5b and d), while Al2O3 and total FeO show large variation and a clear negative correlation (Fig. 5c).
 |
| | Fig. 2 Representative Raman spectra of garnet fragments, with background correction applied. Reference spectra for andradite, grossular, and almandine are included for comparison. | |
 |
| | Fig. 3 Backscattered electron images and high-resolution X-ray maps of Fe, Al, and Ca of selected garnet standards. (a) The andradite (And1902) fragments show clear chemical zoning of Fe and Al. (b and c) The almandine (Ald1906 and Ald1905) grains are homogeneous in both BSE images and elemental maps. | |
 |
| | Fig. 4 Compositional profile across the representative andradite (And1902) fragment. (a) BSE image showing the analysis locations (white circles) across the bright and dark bands. (b) Variations of total FeO and Al2O3 contents, and (c) calculated mole percentage (%) of andradite and grossular end-members across the profile. (d) Covariation of Fe3+ and AlVI atoms per formula unit (apfu) calculated on the basis of 8 cations per 12 oxygens. | |
Table 2 Compositional profile across the andradite grain
| Sample no. |
Distance (μm) |
SiO2 |
TiO2 |
Al2O3 |
Cr2O3 |
FeOtot |
MnO |
MgO |
CaO |
Na2O |
K2O |
Total |
Fe3+ |
AlVI |
End-members |
| Oxides (wt%) |
apfu |
apfu |
Andradite |
Grossular |
| And1902-1 |
313 |
35.18 |
0.00 |
0.62 |
0.01 |
26.65 |
0.59 |
0.04 |
33.50 |
0.03 |
0.00 |
96.60 |
1.883 |
0.033 |
94.15% |
1.65% |
| And1902-2 |
304 |
35.34 |
0.00 |
0.76 |
0.00 |
26.56 |
0.56 |
0.03 |
33.54 |
0.03 |
0.00 |
96.81 |
1.871 |
0.053 |
93.57% |
2.65% |
| And1902-3 |
271 |
35.68 |
0.02 |
3.04 |
0.01 |
23.91 |
0.57 |
0.01 |
33.84 |
0.01 |
0.00 |
97.09 |
1.666 |
0.270 |
83.28% |
13.49% |
| And1902-4 |
249 |
36.55 |
0.20 |
5.54 |
0.00 |
20.70 |
0.72 |
0.01 |
34.07 |
0.01 |
0.00 |
97.79 |
1.418 |
0.527 |
70.88% |
26.37% |
| And1902-5 |
233 |
36.27 |
0.05 |
3.96 |
0.00 |
22.90 |
0.69 |
0.02 |
34.36 |
0.00 |
0.01 |
98.26 |
1.570 |
0.357 |
78.51% |
17.85% |
| And1902-6 |
211 |
36.13 |
0.25 |
4.87 |
0.00 |
21.67 |
0.61 |
0.05 |
34.26 |
0.00 |
0.01 |
97.85 |
1.487 |
0.436 |
74.37% |
21.80% |
| And1902-7 |
191 |
36.20 |
0.71 |
6.37 |
0.00 |
19.20 |
0.67 |
0.06 |
34.14 |
0.00 |
0.01 |
97.34 |
1.317 |
0.586 |
65.85% |
29.28% |
| And1902-8 |
172 |
35.92 |
0.54 |
6.68 |
0.00 |
19.21 |
0.72 |
0.05 |
34.29 |
0.00 |
0.02 |
97.43 |
1.316 |
0.586 |
65.79% |
29.29% |
| And1902-9 |
153 |
36.29 |
0.57 |
6.99 |
0.01 |
18.92 |
0.69 |
0.09 |
34.22 |
0.01 |
0.00 |
97.79 |
1.289 |
0.628 |
64.47% |
31.05% |
| And1902-10 |
134 |
36.31 |
0.52 |
6.60 |
0.03 |
19.16 |
0.67 |
0.08 |
34.70 |
0.04 |
0.00 |
98.11 |
1.302 |
0.582 |
65.10% |
29.12% |
| And1902-11 |
115 |
35.84 |
0.04 |
4.08 |
0.03 |
22.15 |
0.41 |
0.05 |
34.39 |
0.00 |
0.01 |
97.01 |
1.536 |
0.370 |
76.78% |
18.50% |
| And1902-12 |
94 |
35.97 |
0.03 |
3.41 |
0.01 |
23.62 |
0.31 |
0.05 |
34.93 |
0.00 |
0.01 |
98.34 |
1.620 |
0.279 |
81.02% |
13.97% |
| And1902-13 |
77 |
36.04 |
0.05 |
3.78 |
0.01 |
22.85 |
0.35 |
0.03 |
34.55 |
0.01 |
0.00 |
97.66 |
1.576 |
0.339 |
78.79% |
16.95% |
| And1902-14 |
58 |
36.08 |
0.04 |
3.91 |
0.00 |
22.48 |
0.33 |
0.04 |
34.50 |
0.00 |
0.00 |
97.39 |
1.553 |
0.362 |
77.67% |
18.10% |
| And1902-15 |
39 |
36.29 |
0.19 |
4.81 |
0.00 |
21.47 |
0.41 |
0.10 |
34.52 |
0.00 |
0.01 |
97.79 |
1.473 |
0.442 |
73.64% |
22.08% |
| And1902-16 |
20 |
36.54 |
0.10 |
5.52 |
0.01 |
20.59 |
0.43 |
0.07 |
34.91 |
0.00 |
0.00 |
98.15 |
1.403 |
0.506 |
70.14% |
25.31% |
| And1902-17 |
0 |
36.13 |
0.00 |
3.44 |
0.01 |
22.26 |
0.27 |
0.06 |
34.10 |
0.01 |
0.01 |
96.29 |
1.556 |
0.339 |
77.80% |
16.74% |
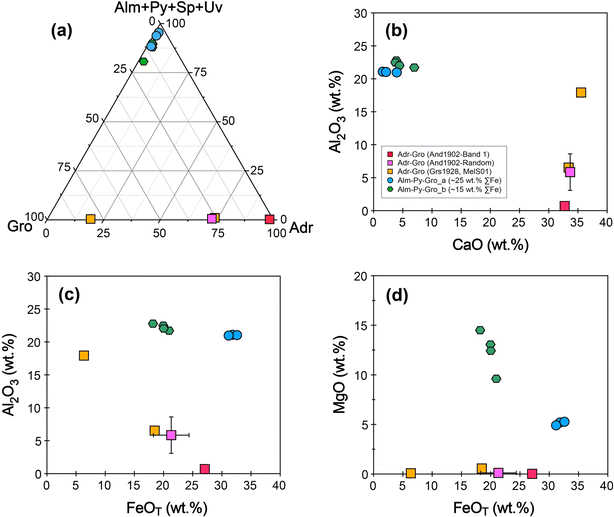 |
| | Fig. 5 Garnet end-members and chemical compositions of the selected garnet samples. (a) A triangular diagram of [almandine (Alm, Fe32+Al2Si3O12) + pyrope (Py, Mg3Al2Si3O12) + spessartine (Sp, Mn3Al2Si3O12) + uvarovite (Uv, Ca3Cr2Si3O12)], [grossular (Gro, Ca3Al2Si3O12)], and [andradite (Adr, Ca3(Fe3+, Ti)2Si3O12)] end-member. (b) Binary plot of CaO versus Al2O3 content. (c and d) Binary plots of Al2O3 and MgO versus total FeO contents. | |
The almandine–pyrope–grossular series samples are homogeneous in the BSE images and EPMA X-ray intensity maps (Fig. 3b and c). They exhibit a large variation in FeO and MgO content, with a restricted range of Al2O3 (Fig. 5b–d). These samples can be divided into two groups based on their Fe content: Alm-Py-Gro_a and Alm-Py-Gro_b. Alm-Py-Gro_a (Ald1906, Ald1915, and Ald1917) contains 30.19–32.62 wt% total FeO and 4.92–5.27 wt% MgO, corresponding to 67–70% almandine (Fe2+3Al2Si3O12), 20–21% pyrope (Mg2+3Al2Si3O12), 3–10% grossular, 1–6% spessartine (Mn3Al2Si3O12), and 1–2% andradite. In contrast, Alm-Py-Gro_b contains lower total FeO (18.21–20.96 wt%) and higher MgO (9.61–14.51 wt%), corresponding to 34–42% almandine (Fe2+3Al2Si3O12), 37–53% pyrope (Mg2+3Al2Si3O12), 8–17% grossular, 1–2% spessartine (Mn3Al2Si3O12), and 1–3% andradite.
3.2. Fe3+/ΣFe ratio of garnet obtained by Mössbauer spectroscopy
The Mössbauer results of the Fe3+/ΣFe ratio of the selected garnet samples are presented in Table 1 and illustrated in Fig. 6. The And1902 sample contains nearly pure ferric iron, exhibiting a Fe3+/ΣFe ratio of 1.00 ± 0.03. The Grs1928 and MelS01 samples are enriched in ferric iron, with Fe3+/ΣFe ratios of 0.89 ± 0.03 and 0.97 ± 0.04, respectively. In contrast, the almandine–pyrope–grossular garnets contain trace amounts of ferric iron, with Fe3+/ΣFe ratios ranging from 0.01 to 0.07.
 |
| | Fig. 6 Mössbauer spectra of the selected garnet samples and their Fe3+/∑Fe ratios. | |
4. Discussion
A comprehensive measurement procedure for the EPMA flank method was summarized by Höfer and Brey,17 encompassing two primary parts: (a) spectrometer calibration (a daily procedure) and (b) flank method measurement. To assess the reliability of the garnet samples in this study, we followed the procedures outlined by Höfer and Brey17 using the JEOL JXA-8230 EPMA at GIGCAS.
4.1. Spectrometer calibration
The reproducibility and stability of the wavelength-dispersive spectrometer in the EPMA can be affected by various factors, such as electromagnetic interference and laboratory temperature change during long-term analysis. These factors may lead to a shift in the spectrum position. Therefore, it is essential to correct the offset of the spectrometer position before conducting flank method measurements.17,31
Höfer and Brey17 provided a daily calibration procedure for the spectrometer to correct the spectral shifts. In contrast to the calibration procedure, we performed spectrometer initialization on a thallium acid phthalate (TAP) spectrometer (slit width = 300 μm) on Day 1, prior to conducting a search for the FeKα1 9th order peak on the in-house Fe metal standard (Fe = 99.99 wt%). The operational conditions of 25 kV, 80 nA, and a 1 μm beam were applied to the qualitative scan using the TAP crystal in integral pulse height analysis mode. The scan range on the TAP crystal extends from 189.3 mm to 189.5 mm, with a movement step of 10 μm and a dwell time of 20000 ms. The FeKα1 9th order (fe0) peak on Day 1 was recorded at 189.40 mm. Subsequently, we performed a 50 μm interval shift on both sides (fe1 and fe2) of the fe0 peak to measure the counts at these two positions (Fig. 7). The fe1 and fe2 positions were then shifted by ±5 μm, and counts were measured at the corresponding offset positions (Fig. 7).31 For each of the six positions (fe1, fe1+5, fe1−5, fe2, fe2+5, and fe2−5), a duration of 180 seconds was allocated to obtain sufficient counts, and three repeated measurements were conducted. The averaged intensity ratio of cps fe1/cps fe2 was then calculated (Table 3). The resulting three ratios are plotted against the spectrometer shift of their center (−5, 0, +5) and fitted with a regression line to determine the “true” position of the peak maximum. We arbitrarily defined the cps fe1/cps fe2 ratio of 0.957 measured on Day 1 as the “true” spectrometer peak position. On Days 2 and 3, we conducted two runs of spectrometer calibration each day. The first run followed the same procedures as Day 1 without spectrometer initialization. In the second run, we performed spectrometer initialization followed by the routine calibration procedure. The cps fe1/cps fe2 ratios before spectrometer initialization measured on Days 2 and 3 was 0.898 and 0.933, respectively (Table 3), indicating a relatively large shift from the “true” position established on Day 1 (Fig. 7). In contrast, the cps fe1/cps fe2 ratio on Days 2 and 3 after spectrometer initialization were 0.952 and 0.945, respectively (Table 3), which are very close to the “true” position (Fig. 7). Thus, the spectrometer initialization effectively reduces the spectrometer shift. The complete spectrometer calibration procedure suggested by Höfer and Brey17 takes approximately 40 minutes in our laboratory. In contrast, the spectrometer initialization operation requires 5 minutes. Therefore, we can perform wave scans for the flank position once on the first day of each session, and the spectrometer shift can be corrected through spectrometer initialization.
 |
| | Fig. 7 Comparison of the spectrometer position before and after the operation of spectrometer initialization (SPI) for 3 working days. The peak shift was accurately defined by the peak maximum of Fe FeKα1 9th order peak on Fe metal (see the text for details). | |
Table 3 Results of the spectrometer shift for 3 working daysa
| Measurements |
Spectrometer shift (μm) |
Day 1_after SPI |
Day 2_before SPI |
Day 2_after SPI |
Day 3_before SPI |
Day 3_after SPI |
| 3 |
σ
|
3 |
σ
|
3 |
σ
|
3 |
σ
|
3 |
σ
|
|
SPI-spectrometer initialization.
|
| cps fe1/cps fe2 |
−5 |
0.883 |
0.003 |
0.837 |
0.013 |
0.887 |
0.009 |
0.864 |
0.005 |
0.879 |
0.009 |
| cps fe1/cps fe2 |
0 |
0.957 |
0.005 |
0.898 |
0.009 |
0.952 |
0.006 |
0.933 |
0.008 |
0.945 |
0.024 |
| cps fe1/cps fe2 |
5 |
1.041 |
0.009 |
0.985 |
0.014 |
1.055 |
0.008 |
1.025 |
0.004 |
1.047 |
0.013 |
4.2. Flank method measurement
4.2.1. Identifying the flank positions of Fe Lα and Fe Lβ.
The EPMA flank method is based on the Fe L-line X-ray emission spectra, which exhibit a concomitant change in both the intensity and wavelength of the soft Fe Lα and Fe Lβ emission lines.17 The prerequisite for flank method measurements is the determination of the flank positions of Fe Lα and Fe Lβ, which can be derived from the difference spectrum between pure garnet end-member standards (Fe2+ and Fe3+).
To evaluate the potential influence of chemical zoning in andradite on the determination of the Fe Lα and Fe Lβ flank positions, we conducted wave scans on Band 1 and Band 4 of the And1902 sample (Fe3+/ΣFe = 1.00 ± 0.03). These bands exhibit different total FeO and Al2O3 contents, as well as varying mole percentages of calculated end-members (Fig. 4). Additionally, we scanned another andradite sample, MelS01, which has a comparable FeO content to And1902-Band 4 but a slightly lower Fe3+/ΣFe ratio (0.97 ± 0.04). For the Fe2+ garnet end-member, the Ald1906 sample (Fe3+/ΣFe = 0.01 ± 0.03) was selected for the wave scan.
We conducted the wave scan using an accelerating voltage of 15 kV, a probe current of 80 nA, and a beam diameter of 10 μm, employing a differential pulse height analysis mode. To achieve high intensities, we set an accumulation of twenty measurements for each garnet sample. The scanning range of the L value for the TAP spectrometer extends from 186 to 193 mm. The raw spectra of And1902-Band 1, And1902-Band 4, MelS01, and Ald1906 are presented in Fig. 8a. It shows that the Fe Lα and Fe Lβ lines of Ald1906 (Fe2+ end-member) are shifted to a higher wavelength compared to those of the andradite samples due to self-absorption.17 And1902-Band 4 and MelS01 exhibit nearly identical peak intensities. In contrast, And1902-Band 1 displays higher peak intensities for Fe Lα and Fe Lβ. Nevertheless, the peak positions of Fe Lα and Fe Lβ for Band 1, Band 4 and MelS01 are nearly identical (Fig. 8a). The flank positions of Fe Lα and Fe Lβ were defined in the smoothed difference spectrum, with the minimum value representing the Fe Lβ flank position and the maximum value representing the Fe Lα flank position (Fig. 8b). Despite the differences in chemical compositions among And1902-Band 1, Band 4, and MelS01 their smoothed difference spectra show almost identical Fe Lα and Fe Lβ flank positions (Fig. 8b). The raw spectra of these garnets, along with their difference spectrum, are comparable to those determined from synthetic garnets, as shown in Li et al.24 Given that And1902-Band 1 contains a higher FeO content, which can yield greater intensity during the wave scan, the combination of And1902-Band 1 and Ald1906 is used for identifying the flank positions.
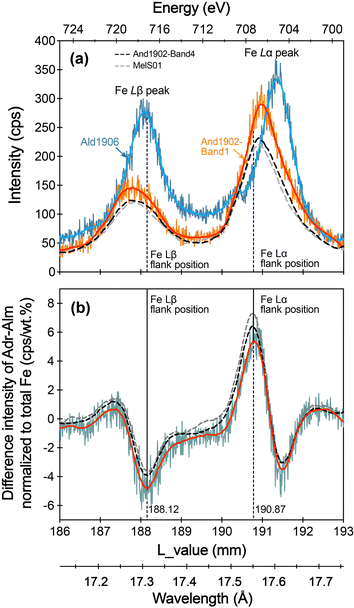 |
| | Fig. 8 The flank positions of Fe Lα and Fe Lβ based on andradite and almandine samples. (a) Raw spectra of the Fe L-line of And1902-Band 1, And1902-Band 4, MelS01, and Ald1906, which were collected at 15 kV, 80 nA, and a probe diameter of 10 μm in the differential analysis mode. (b) Difference spectra between And1902-Band 1, And1902-Band4, MelS01 and Ald1906 were normalized by using their total Fe content, respectively. The flank positions of Fe Lα and Fe Lβ were located by using the maximum and minimum intensities on the smoothed difference spectrum (red lines). | |
We also evaluated the robustness of the samples under prolonged beam bombardment, using an accelerating voltage of 15 kV, a probe current of 200 nA, and a beam size of 20 μm.22 The results show that the mean intensities of Fe Lβ and Fe Lα at flank positions, as well as the Fe Lβ/Lα ratios for And1902 and Ald1906, remain consistent for up to 1200 s (Fig. 9). This stability suggests that these garnets can withstand extended exposure to high electron beam current.
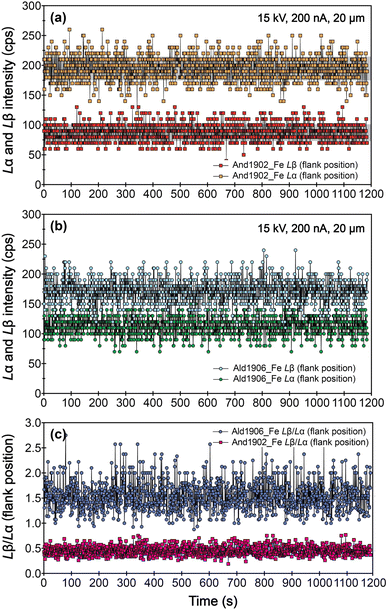 |
| | Fig. 9 Variation of intensities of Fe Lα (a) and Fe Lβ (b) at flank positions, and the Lβ/Lα ratios of And1902 and Ald1906 (c) as a function of time. | |
4.2.2. Analyses of Fe Lβ and Fe Lα at the flank position.
Using the flank positions of Fe Lα and Fe Lβ determined from And1902-Band 1 and Ald1906, we can obtain the intensities of Fe Lβ and Fe Lα at these flank positions. In this study, we added two “fake” elements at the TAP spectrometer for the flank method measurements: arsenic (As) for the Fe Lβ flank position and bromine (Br) for the Fe Lα flank position. Major elements were arranged on the remaining four spectrometers. The operational conditions included an accelerating voltage of 15 kV, a probe current of 60 nA, and a beam size of 5 μm. Hezel et al.18 suggested that the mean of 9 to 16 flank method analyses per sample yields the same result as the mean of 25 analyses per sample. In this case, for each garnet sample, at least twelve points were analyzed, with a counting time of 180 s for the flank positions of Fe Lβ and Fe Lα, respectively. To verify the reliability of the flank method on our EPMA, we also analyzed the Fe Lβ and Fe Lα at the flank positions of nine garnet reference standards with known Fe3+/∑Fe ratios (0.03 to 1.01) as reported by Tao et al.27 as monitor standards (provided by Dr H. Höfer), including AND1, FRA1, UA5, UA10, Mir1, Mir2, Mir13, Mir23, and Damknolle. The results of the Fe Lβ/Lα ratios at the flank positions of the nine garnet reference standards are listed in Table 4.
Table 4 Chemical compositions and Fe3+/ΣFe ratios of garnet reference standards
| Sample |
AND1 |
FRA1 |
Damknolle |
Mir1 |
Mir2 |
Mir13 |
Mir23 |
UA5 |
UA10 |
| Measurements |
10 |
σ
|
9 |
σ
|
10 |
σ
|
10 |
σ
|
10 |
σ
|
10 |
σ
|
10 |
σ
|
10 |
σ
|
14 |
σ
|
|
The Fe3+/ΣFe ratios of reference garnet standards are sourced from Tao et al.27
|
| SiO2 |
35.22 |
0.18 |
36.17 |
0.14 |
39.41 |
0.18 |
41.22 |
0.15 |
41.02 |
0.17 |
40.63 |
0.15 |
41.37 |
0.13 |
41.73 |
0.28 |
41.84 |
0.31 |
| TiO2 |
0.01 |
0.01 |
0.00 |
0.00 |
0.34 |
0.01 |
0.40 |
0.01 |
0.34 |
0.01 |
0.45 |
0.01 |
0.41 |
0.02 |
0.69 |
0.03 |
0.06 |
0.01 |
| Al2O3 |
0.69 |
0.07 |
19.74 |
0.12 |
21.67 |
0.10 |
22.99 |
0.06 |
22.78 |
0.08 |
22.56 |
0.15 |
23.03 |
0.09 |
19.02 |
0.12 |
19.96 |
0.22 |
| Cr2O3 |
0.00 |
0.01 |
0.00 |
0.00 |
0.01 |
0.01 |
0.01 |
0.01 |
0.01 |
0.01 |
0.00 |
0.01 |
0.01 |
0.01 |
4.31 |
0.06 |
4.90 |
0.13 |
| FeO |
26.61 |
0.18 |
42.58 |
0.15 |
19.31 |
0.16 |
11.02 |
0.15 |
13.00 |
0.14 |
13.52 |
0.10 |
10.50 |
0.14 |
7.96 |
0.06 |
7.46 |
0.11 |
| MnO |
1.13 |
0.03 |
0.02 |
0.02 |
0.56 |
0.03 |
0.34 |
0.03 |
0.36 |
0.02 |
0.38 |
0.02 |
0.32 |
0.03 |
0.31 |
0.02 |
0.39 |
0.03 |
| MgO |
0.12 |
0.01 |
0.44 |
0.01 |
11.29 |
0.04 |
18.82 |
0.12 |
17.16 |
0.07 |
16.65 |
0.13 |
19.35 |
0.07 |
21.48 |
0.12 |
21.36 |
0.22 |
| CaO |
33.75 |
0.12 |
0.55 |
0.01 |
7.60 |
0.06 |
5.77 |
0.05 |
5.98 |
0.09 |
6.22 |
0.04 |
5.54 |
0.05 |
5.36 |
0.06 |
5.11 |
0.04 |
| Na2O |
0.01 |
0.01 |
0.02 |
0.02 |
0.03 |
0.01 |
0.02 |
0.02 |
0.01 |
0.01 |
0.02 |
0.02 |
0.02 |
0.01 |
0.05 |
0.01 |
0.02 |
0.01 |
| Total |
97.53 |
0.34 |
99.52 |
0.24 |
100.23 |
0.29 |
100.61 |
0.34 |
100.67 |
0.27 |
100.42 |
0.32 |
100.55 |
0.20 |
100.90 |
0.46 |
101.11 |
0.39 |
| Fe3+/ΣFea Mössbauer |
1.01 |
— |
0.03 |
— |
0.04 |
— |
0.05 |
— |
0.04 |
— |
0.04 |
— |
0.06 |
— |
0.13 |
— |
0.07 |
— |
|
L
β/Lα (net) flank method |
0.49 |
0.01 |
1.87 |
0.07 |
1.21 |
0.03 |
0.97 |
0.03 |
1.01 |
0.01 |
1.03 |
0.03 |
0.95 |
0.02 |
0.82 |
0.02 |
0.84 |
0.00 |
The Fe Lβ/Lα ratios of the nine garnet reference standards exhibit a strong positive correlation with their Fe2+ contents (Fig. 10). A simple linear regression analysis of these standards yields an equation of Lβ/Lα = 0.0410 × Fe2+ + 0.5965 (R2 = 0.9835), which is comparable to the equation obtained by Tao et al.27i.e., Lβ/Lα = 0.0423 × Fe2+ + 0.5714 (R2 = 0.9893). Therefore, And1902 and Ald1906 serve as reliable garnet reference materials for determining the flank positions of Fe Lα and Fe Lβ.
 |
| | Fig. 10 Simple linear regression of Fe Lβ/Lα intensity ratios (flank ratios) against Fe2+ contents for nine garnet reference standards. Note that the simple linear regression for the nine garnet reference standards refer to Tao et al.27 | |
4.3. Calculation of garnet Fe2+ content and the Fe3+/∑Fe ratio
Höfer and Brey17 demonstrated that the Fe Lβ/Lα ratios exhibit a linear correlation with Fe2+ content for garnet, with a secondary dependence on total Fe. This correlation leads to the development of a linear equation that enables the quantification of Fe2+ content and Fe3+/∑Fe in unknown garnet samples.
Ten garnet samples, with Fe2+ contents (calculated from the total FeO and Mössbauer Fe3+/∑Fe ratios) ranging from 0 to 24.5 wt%, were used as calibration standards to establish the linear equation for garnet. The Fe Lβ/Lα ratios of the ten garnet samples are positively correlated with Fe2+ contents (Fig. 11a). A simple linear regression analysis yields an equation of
| | | Lβ/Lα = 0.0392 × Fe2+ + 0.5328 (R2 = 0.9864) | (1) |
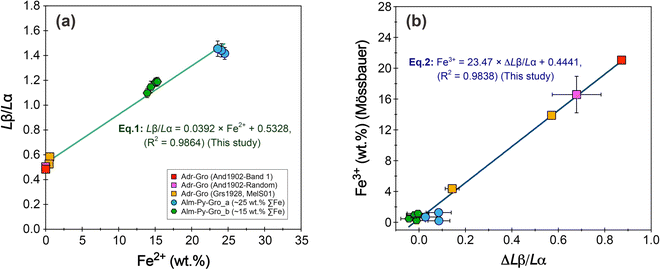 |
| | Fig. 11 (a) Simple linear regression of Fe Lβ/Lα intensity ratios (flank ratios) against Fe2+ contents for the ten garnet samples. (b) Plot of the ΔLβ/Lα (defined as the difference of the corresponding point on the Fe3+/∑Fe = 0 equi-line from the measured Fe Lβ/Lα ratio at a given total Fe, cf., Höfer and Brey.17) versus Fe3+ contents (as calculated from the EPMA analyses of total FeO and Mössbauer analyses of Fe3+/ΣFe ratios). | |
Based on eqn (1), we recalculated the Fe2+ content and Fe3+/∑Fe of each garnet from the measured Fe Lβ/Lα ratios and total FeO (Table 5). The results indicate that the deviation of the calculated Fe2+ content and the Fe3+/∑Fe ratios exceeded ±1 wt% and ±0.05, respectively, for most of the samples when compared to the data obtained through Mössbauer spectroscopy (Fig. 12a and b). This discrepancy arises because the simple linear regression solely examines the relationship between Fe2+ contents and the Fe Lβ/Lα ratios. It fails to consider the secondary self-absorption effect that results from the total Fe content, which includes a small contribution from Fe3+.17
Table 5 Chemical compositions and Fe3+/ΣFe ratios of garnet reference standards
| Classification |
Andradite–grossular series |
Almandine–pyrope–grossular |
| Sample |
And1902-Band1 |
And1902 |
Grs1928 |
MelS01 |
Ald1906 |
Ald1915 |
Ald1917 |
Ald1905 |
Ald1907 |
Ald1925 |
Ald1926 |
| Measurements |
20 |
20 |
12 |
20 |
20 |
16 |
20 |
20 |
20 |
16 |
12 |
| Fe Lβ/Lα |
0.485 |
0.504 |
0.584 |
0.526 |
1.417 |
1.442 |
1.455 |
1.098 |
1.185 |
1.190 |
1.147 |
| SD |
0.010 |
0.021 |
0.025 |
0.011 |
0.048 |
0.054 |
0.062 |
0.037 |
0.034 |
0.035 |
0.047 |
| ∑Fe (wt%) |
21.06 |
16.59 |
4.95 |
14.38 |
24.75 |
25.37 |
24.26 |
14.16 |
15.54 |
16.30 |
15.58 |
|
|
Recalculated based on the Mössbauer Fe
3+
/∑Fe ratios
|
| Fe2+_Moss |
0.00 |
0.00 |
0.56 |
0.48 |
24.54 |
24.09 |
23.57 |
13.92 |
15.07 |
15.32 |
14.43 |
| SD |
0.00 |
0.00 |
0.02 |
0.00 |
0.20 |
0.28 |
0.25 |
0.07 |
0.06 |
0.05 |
0.06 |
| Fe3+_Moss |
21.06 |
16.59 |
4.38 |
13.90 |
0.21 |
1.28 |
0.69 |
0.24 |
0.47 |
0.98 |
1.15 |
| SD |
0.13 |
2.37 |
0.17 |
0.09 |
0.00 |
0.02 |
0.01 |
0.00 |
0.00 |
0.00 |
0.00 |
|
|
Recalculated using the simple linear regression equation (eqn (1)): L
β
/L
α
= 0.0392 × Fe
2+
+ 0.5328
|
| Fe2+ |
−1.21 |
−0.73 |
1.31 |
−0.18 |
22.57 |
23.21 |
23.53 |
14.42 |
16.63 |
16.78 |
15.68 |
| SD |
0.27 |
0.54 |
0.64 |
0.27 |
1.21 |
1.37 |
1.58 |
0.95 |
0.86 |
0.89 |
1.21 |
| Fe3+/∑Fe |
1.06 |
1.04 |
0.74 |
1.01 |
0.09 |
0.09 |
0.03 |
−0.02 |
−0.07 |
−0.03 |
−0.01 |
| SD |
0.01 |
0.03 |
0.14 |
0.02 |
0.05 |
0.05 |
0.07 |
0.07 |
0.06 |
0.05 |
0.08 |
|
|
Recalculated using the delta equation (eqn (2)): Fe
3+
= 23.47 × ΔL
β
/L
α
+ 0.4441
|
| ΔLβ/Lα |
0.87 |
0.68 |
0.14 |
0.57 |
0.09 |
0.08 |
0.03 |
−0.01 |
−0.04 |
−0.02 |
0.00 |
| SD |
0.01 |
0.10 |
0.03 |
0.01 |
0.05 |
0.05 |
0.06 |
0.04 |
0.03 |
0.03 |
0.05 |
| Fe2+ |
0.13 |
0.22 |
1.16 |
0.54 |
22.30 |
22.94 |
23.15 |
13.95 |
16.10 |
16.29 |
15.22 |
| SD |
0.24 |
0.43 |
0.58 |
0.25 |
1.12 |
1.27 |
1.45 |
0.88 |
0.79 |
0.82 |
1.11 |
| Fe3+/∑Fe |
0.99 |
0.99 |
0.77 |
0.96 |
0.10 |
0.10 |
0.05 |
0.01 |
−0.04 |
0.00 |
0.02 |
| SD |
0.01 |
0.03 |
0.12 |
0.02 |
0.04 |
0.05 |
0.06 |
0.06 |
0.05 |
0.05 |
0.07 |
|
|
Recalculated using the multiple linear regression equation (eqn (3)): Fe
2+
= −11.18 + 19.05 × L
β
/L
α
−0.001 × ∑Fe + 0.22 × Lβ/Lα× ∑Fe |
| Fe2+ |
0.26 |
0.22 |
0.57 |
0.46 |
23.41 |
24.22 |
24.18 |
13.09 |
15.37 |
15.69 |
14.54 |
| SD |
0.24 |
0.41 |
0.49 |
0.23 |
1.17 |
1.33 |
1.50 |
0.83 |
0.76 |
0.79 |
1.06 |
| Fe3+/∑Fe |
0.99 |
0.99 |
0.89 |
0.97 |
0.05 |
0.05 |
0.00 |
0.08 |
0.01 |
0.04 |
0.07 |
| SD |
0.01 |
0.02 |
0.10 |
0.02 |
0.05 |
0.05 |
0.06 |
0.06 |
0.05 |
0.05 |
0.07 |
 |
| | Fig. 12 Comparison of Fe2+ content and Fe3+/∑Fe ratios of the garnet samples determined by the flank method and by Mössbauer spectroscopy. The Fe2+ content and Fe3+/∑Fe ratios obtained by the flank method in (a and b) using simple linear regression of the flank-method data and Fe2+ content (eqn (1)), in (c and d) using the ΔLβ/Lα-Fe3+ equation (eqn (2)), and in (e and f) multiple regression of the flank-method data and total Fe content (eqn (3)). The error bar is 1σ. The solid line is a 1:1 line. The dotted lines denote an uncertainty of ±1 wt% for Fe2+ and ±0.05 for the Fe3+/∑Fe ratio. | |
Höfer and Brey17 found that for a garnet with an unknown Fe3+ but a known total Fe content, the Fe Lβ/Lα ratio of the garnet will plot below the reference line of Fe3+/∑Fe = 0. The magnitude of the deviation from the reference line (Δ ratio) is positively correlated with the Fe3+ content of the garnet. If a series of garnets with different known Fe3+ are available, the linear equation relating ΔLβ/Lα to the Fe3+ content of these garnets can be used to determine the Fe3+ content of unknown garnet samples.17 Based on eqn (1), we calculated the ΔLβ/Lα for ten garnet samples (Table 5). The results show that the ΔLβ/Lα is positively correlated with the Fe3+ contents (calculated from the total FeO by EPMA analyses and Mössbauer analyses of Fe3+/ΣFe ratios) (Fig. 11b), yielding a linear regression of
| | | Fe3+ = 23.47 × ΔLβ/Lα + 0.4441 (R2 = 0.9838) | (2) |
The Fe2+ contents and Fe3+/ΣFe ratios calculated using eqn (2) were plotted on a 1:1 line against the Mössbauer values (Fig. 12c and d). While most samples exhibit a deviation in Fe2+ contents of less than ±1 wt%, half of these samples show a deviation in Fe3+/ΣFe ratios greater than ±0.05. The almandine–pyrope–grossular samples show a larger discrepancy compared to the andradite–grossular samples, as the former contain very low Fe3+ content but with dispersed Fe Lβ/Lα ratios, which increases the error during the calculation.
Finally, we adopted the flank-method data and total Fe content to calculate the Fe2+ content of these samples.17 Specifically, the equation is given as Fe2+ = A + B(Lβ/Lα) + C∑Fe + D∑Fe × (Lβ/Lα), where the coefficients A, B, C, and D can be determined through multiple regression analysis of the flank-method data, Fe2+ content, and the total Fe contents of the ten garnet samples. The multiple regression equation, derived from a total of 196 data points, is expressed as
| | | Fe2+ = −11.18(0.737) + 19.05(0.799) × (Lβ/Lα) − 0.001(0.040) × ∑Fe + 0.22(0.039) × ∑Fe × (Lβ/Lα) | (3) |
with the numbers in parentheses representing the standard error. This equation was subsequently used to calculate the Fe
2+ and Fe
3+ contents of garnet samples. When compared to the results obtained from Mössbauer spectroscopy, the Fe
2+ contents of the garnet samples exhibit a discrepancy within ±1 wt% (
Fig. 12e), while the Fe
3+/∑Fe ratios of these standards show a discrepancy within ±0.05 (
Fig. 12f). Therefore, in contrast to the simple linear equation and the derived Δ
Lβ/
Lα-Fe
3+ equation, the multiple regression analysis of the flank-method data, Fe
2+ and total FeO contents of these natural garnet samples provides a higher accuracy for Fe
3+/∑Fe, particularly for garnet with low Fe
3+ or total Fe contents.
It should be noted that the calculated Fe3+/∑Fe ratio of the Grs1928 sample exhibits a larger standard deviation than that of the other samples, which may be attributed to its low FeO content.22 Recent studies have demonstrated that garnet flank method measurements conducted at 80 nA and 120 nA yield improved analytical precision compared to those performed at 60 nA.18,21,22 Therefore, a higher probe current could be utilized for garnet during the flank method measurement in the future analysis.
5. Conclusions
We report ten new natural garnets for determining the Fe oxidation state of garnet using the EPMA flank method. This collection includes three samples from the andradite–grossular series and seven samples from the almandine–pyrope–grossular series. The samples And1902 and Ald1906 can serve as primary standards for identifying the flank positions of Fe Lα and Fe Lβ. All ten garnet samples can be utilized as reference materials during flank method measurements to calibrate the Fe3+/∑Fe ratio of unknown garnet. To determine the Fe2+ and Fe3+ with the flank method, we recommend employing multiple regression analysis of the Fe Lβ/Lα at flank positions, along with Fe2+ content and the total Fe content of garnet. This approach yields a low uncertainty in the Fe3+/∑Fe ratio. Prior to conducting flank method measurement, it is advisable to initialize the spectrometer for daily calibration, as this can reduce the spectrometer shift and save time.
Data availability
Data for this article are presented in the tables.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Renbiao Tao is thanked for sharing the nine garnet reference standards and for providing valuable suggestions. Puqiu Wu and Qiuxia Wang are recognized for their assistance with the Mössbauer analysis. This study was financially supported by the National Key R&D Program of China (2018YFA0702600) and the Science and Technology Planning Project of Guangdong Province, China (2023B1212060048).
References
- B. J. Wood and D. Virgo, Geochim. Cosmochim. Acta, 1989, 53, 1277–1291 CrossRef CAS.
- T. C. Feeley and Z. D. Sharp, Geol, 1996, 24, 1021 CrossRef CAS.
- M. Enders, D. Speer, W. V. Maresch and C. A. McCammon, Contrib. Mineral. Petrol., 2000, 140, 135–147 CrossRef CAS.
- M. C. McCanta, M. D. Dyar, M. J. Rutherford and J. S. Delaney, Am. Mineral., 2004, 89, 1685–1693 CrossRef CAS.
- A. J. Berry, G. M. Yaxley, B. J. Hanger, A. B. Woodland, M. D. De Jonge, D. L. Howard, D. Paterson and V. S. Kamenetsky, Geology, 2013, 41, 683–686 CrossRef CAS.
-
C. A. McCammon, International Kimberlite Conference: Extended Abstracts, 1998, vol. 7, pp. 555–557 Search PubMed.
- M. D. Dyar, D. G. Agresti, M. W. Schaefer, C. A. Grant and E. C. Sklute, Annu. Rev. Earth Planet. Sci., 2006, 34, 83–125 CrossRef CAS.
- C. A. McCammon, W. L. Griffin, S. R. Shee and H. S. C. O'Neill, Contrib. Mineral. Petrol., 2001, 141, 287–296 CrossRef CAS.
-
C. McCammon, in Modern Mössbauer Spectroscopy: New Challenges Based on Cutting-Edge Techniques, ed. Y. Yoshida and G. Langouche, Springer, Singapore, 2021, pp. 221–266 Search PubMed.
- M. Wilke, F. Farges, P.-E. Petit, G. E. Brown Jr and F. Martin, Am. Mineral., 2001, 86, 714–730 CrossRef CAS.
- A. J. Berry, H. S. C. O'Neill, K. D. Jayasuriya, S. J. Campbell and G. J. Foran, Am. Mineral., 2003, 88, 967–977 CrossRef CAS.
- E. Borfecchia, L. Mino, D. Gianolio, C. Groppo, N. Malaspina, G. Martinez-Criado, J. A. Sans, S. Poli, D. Castelli and C. Lamberti, J. Anal. At. Spectrom., 2012, 27, 1725–1733 RSC.
- G. Marras, G. Carnevale, A. Caracausi, S. G. Rotolo and V. Stagno, Eur. J. Mineral., 2023, 35, 665–678 CrossRef CAS.
- K. D. Burgess, R. M. Stroud, M. D. Dyar and M. C. McCanta, Am. Mineral., 2016, 101, 2677–2688 CrossRef.
- H. E. Höfer, G. P. Brey, B. Schulz-Dobrick and R. Oberhänsli, East. J. Med., 1994, 6, 407–418 Search PubMed.
- H. E. Höfer, Hyperfine Interact., 2002, 144, 239–248 CrossRef.
- H. E. Höfer and G. P. Brey, Am. Mineral., 2007, 92, 873–885 CrossRef.
- D. C. Hezel, H. E. Höfer and A. Fichtner, Am. Mineral., 2024, 109, 1387–1393 CrossRef.
- N. Malaspina, S. Poli and P. Fumagalli, J. Petrol., 2009, 50, 1533–1552 CrossRef CAS.
- X. Li, S. Song, L. Zhang and E. H. Höfer, Sci. Bull., 2018, 63, 300–305 CrossRef CAS PubMed.
- M. Holycross, E. Cottrell, J. Ague, A. Lanzirotti and M. Newville, Chem. Geol., 2024, 647, 121937 CrossRef CAS.
- C. Wang, R. Tao, J. B. Walters, H. E. Höfer and L. Zhang, Geochim. Cosmochim. Acta, 2022, 336, 269–290 CrossRef CAS.
- M. Tang, C.-T. A. Lee, G. Costin and H. E. Höfer, Earth Planet. Sci. Lett., 2019, 528, 115827 CrossRef CAS.
- X. Li, C. Zhang, R. R. Almeev, X.-C. Zhang, X.-F. Zhao, L.-X. Wang, J. Koepke and F. Holtz, Chem. Geol., 2019, 509, 152–162 CrossRef CAS.
- C. Zhang, R. R. Almeev, E. C. Hughes, A. A. Borisov, E. P. Wolff, H. E. Höfer, R. E. Botcharnikov and J. Koepke, Am. Mineral., 2018, 103, 1445–1454 CrossRef.
- E. C. Hughes, B. Buse, S. L. Kearns, J. D. Blundy, G. Kilgour, H. M. Mader, R. A. Brooker, R. Balzer, R. E. Botcharnikov, D. Di Genova, R. R. Almeev and J. M. Riker, Am. Mineral., 2018, 103, 1473–1486 CrossRef.
- R. Tao, Y. Fei, E. S. Bullock, C. Xu and L. Zhang, Geochim. Cosmochim. Acta, 2018, 225, 1–16 CrossRef CAS.
- E. F. Baxter, M. J. Caddick and J. J. Ague, Elements, 2013, 9, 415–419 CrossRef CAS.
- Q. Wang, X. Ping, J. Zheng and H. Dai, Earth Sci., 2023, 48, 1217–1231 Search PubMed.
- A. J. Locock, Comput. Geosci., 2008, 34, 1769–1780 CrossRef CAS.
- H. E. Höfer, S. Weinbruch, C. A. Mccammon and G. P. Brey, East. J. Med., 2000, 12, 63–71 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *abc,
Christina Yan
Wang
abc,
Xianquan
Ping
d and
Xiaoju
Lin
abc
*abc,
Christina Yan
Wang
abc,
Xianquan
Ping
d and
Xiaoju
Lin
abc











