
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Uranium isotopic analysis in unpurified solutions by ICP-MS†
Sean R.
Scott
 *,
Kirby P.
Hobbs
,
Amanda D.
French
,
Isaac J.
Arnquist
,
Sonia
Alcantar Anguiano
,
Daniel L.
Sullivan
and
Staci M.
Herman
*,
Kirby P.
Hobbs
,
Amanda D.
French
,
Isaac J.
Arnquist
,
Sonia
Alcantar Anguiano
,
Daniel L.
Sullivan
and
Staci M.
Herman
Pacific Northwest National Laboratory, USA. E-mail: sean.scott@pnnl.gov
First published on 24th June 2024
Abstract
Mass spectrometry is a widely used tool for analysis of uranium isotopic composition. For solution based inductively coupled plasma mass spectrometry, uranium isotopes are typically analyzed after purification from complex matrices. In this work, we tested the ability of three mass spectrometers (ThermoScientific iCAP TQ, ThermoScientific Neoma, and Agilent 8900) to analyze uranium isotopes in an unpurified NIST reference material (SRM2780a, Hard Rock Mine Waste) digest solution. Results indicate that 235U/238U can be analyzed within 1% of the true value. 234U/238U is a more challenging analysis due to low count rates and potential isobar interferences, but strategies to mitigate these effects, such as the use of reaction gases in a collision cell and desolvating nebulizer introduction system, are effective for the triple quadrupole instruments. However, the use of the Neoma MS/MS in reaction mode using O2 gas was problematic. Nevertheless, analysis of unpurified solutions for quick assessment of uranium isotope compositions is practical, especially when high precision is not required.
1. Introduction
Mass spectrometry analysis of uranium isotopic composition is a widespread practice with applications in nuclear,1–9 geological,10–18 biological,19–23 environmental,24–29 and materials30,31 science. Improvements in mass spectrometry technology have led to enhanced precision and lower detection limits that have strengthened understanding of a variety of natural and man-made processes from the micro to the macro scale.32–37 For bulk sample analysis, two types of mass spectrometers are typically used. Thermal ionization mass spectrometry (TIMS) has been considered the conventional method with decades of supporting method development and high precision analytical data using either partial or total evaporation methods.38–41 Inductively coupled plasma mass spectrometry (ICP-MS), including quadrupole based single collector and magnetic sector multi collector, is also used for isotopic analysis and improves sample throughput.42–45 Multi collector instruments have primarily followed the path of TIMS instruments, focusing on the analysis of purified samples.46,47 Most method development studies regardless of the analytical technique have focused on pure uranium standards and/or sample solutions that have undergone a purification process.48–53Purifications provide two benefits for mass spectrometry analysis by either TIMS or ICP-MS. For samples with high matrix and relatively low uranium contents, the process of purification allows for preconcentration of a relatively small total amount of uranium analyte.45,46,54–58 The sample load size requirements for TIMS generally requires a high-purity uranium aliquot with techniques used to enhance ionization and detection.59,60 The presence of additional matrix elements during ICP-MS analysis can create isobaric interferences on uranium masses,61 as well as impart matrix effects during sample introduction that can affect the accuracy of high precision isotopic data.46 The drawback of sample purification is the amount of time required, including performing chromatographic separations and concentrating the final uranium aliquot eluted from ion exchange columns. Given technological advances in ICP-MS, especially the use of collision-reaction cells, the potential for reducing or even eliminating the need for extensive purification procedures is becoming more viable.
Here we present uranium isotopic data in purified and unpurified aliquots of a standard reference material from both triple quadrupole and multi-collector ICP-MS instruments. The intention of these analyses is to determine the accuracy of uranium isotopic composition data in unpurified samples and compare performance of the triple quadrupole and multi collector instruments. Purified samples were measured using typical methods associated with high precision analysis to establish a baseline value for uranium isotope ratios in the SRM2780a material, and unpurified analyses were compared to this baseline value to determine accuracy. In doing so, we show that uranium isotopes (235U/238U, 234U/238U) can be analyzed accurately (within 1% and 10% of the true values, respectively) without prior purification. This uncertainty is sufficient to determine whether uranium isotopes in a sample are nominally natural or have been altered from the natural isotopic composition, as well as a determination of whether the 234U/238U is near secular equilibrium. Thus, analysis of uranium isotopes without purification can be useful when high precision is not required.
2. Analysis of uranium isotopes in unpurified matrices
All sample preparation and analytical methods were carried out in either the Ultratrace Laboratory or Radiation Detection Laboratory at Pacific Northwest National Laboratory. All acid reagents were diluted from Optima Grade, and all H2O used was 18.2 MΩ cm. Analytical experiments were conducted to test the accuracy and precision of uranium isotopic ratios in an unpurified digest compared to high-precision analyses of purified uranium fractions. Samples were prepared from a primary solution containing of NIST SRM2780a (Hard Rock Mine Waste). Uranium isotopes (234U, 235U, and 238U) were analyzed in purified and unpurified aliquots on the ThermoScientific NeptunePlus MC-ICPMS, ThermoScientific Neoma MC-ICPMS (and later upgraded to the Neoma MS/MS), Agilent 8900 QQQ-ICPMS, and ThermoScientific iCAP TQ ICPMS. Accurate isotope ratios were measured as metal ions on the MC-ICP-MS instruments, and metals, oxides, or double oxides on the QQQ-ICPMS. Analysis of double oxides on the Neoma MS/MS was also attempted. Unpurified NIST2780a solutions were diluted to match the uranium concentration of standards.2.1 Preparation of the NIST SRM2780a solution
NIST SRM2780a contains a certified uranium concentration of 4 mg kg−1. The material also contains relative high abundances of Al, Si, S, and Fe (mass fractions 8.43, 24.1, 8.85, and 8.75%, respectively).62 Approximately 10 grams of material were weighed into a clean glass beaker, and the sample was placed into an oven at 110 °C for two hours to dry. After cooling the dry weight was recorded. The aliquot was then quantitatively transferred to a Teflon beaker using 33 mL of concentrated (16 M) nitric acid (HNO3). This solution was dried, then 20 mL of aqua regia (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio of concentrated hydrochloric acid (HCl):HNO3) was added and the sample was refluxed for one hour, then dried. The sample was then transposed to nitrate using three sequential additions and dry downs of 10 mL of concentrated HNO3. This was followed by three sequential additions and dry downs of 10 mL, 20 mL, and 10 mL of concentrated (29 M) hydrofluoric acid (HF). The sample was again transposed to nitrate using three sequential additions and dry downs of 10 mL of concentrated HNO3. Then a 1:1 mixture of concentrated HNO3 and concentrated (9.5 M) perchloric acid (HClO4) was added and the sample was dried, followed by two more additions and dry downs of 10 mL of concentrated HClO4. The HF step was repeated, then the HClO4 step was repeated. The sample was transposed to chloride using sequential additions and dry downs of 10 mL of concentrated (11 M) HCl, followed by two sequential additions and dry downs of 30 mL of 2 M HCl. The sample was then quantitatively transferred to a pre-weighed 2 liter PFA bottle containing 150 mL of H2O using concentrated HCl, and the sample was diluted to provide approximately 2 L of 2 M HCl containing a final concentration of 4.84 mg of sample per g of solution.
:1 volume ratio of concentrated hydrochloric acid (HCl):HNO3) was added and the sample was refluxed for one hour, then dried. The sample was then transposed to nitrate using three sequential additions and dry downs of 10 mL of concentrated HNO3. This was followed by three sequential additions and dry downs of 10 mL, 20 mL, and 10 mL of concentrated (29 M) hydrofluoric acid (HF). The sample was again transposed to nitrate using three sequential additions and dry downs of 10 mL of concentrated HNO3. Then a 1:1 mixture of concentrated HNO3 and concentrated (9.5 M) perchloric acid (HClO4) was added and the sample was dried, followed by two more additions and dry downs of 10 mL of concentrated HClO4. The HF step was repeated, then the HClO4 step was repeated. The sample was transposed to chloride using sequential additions and dry downs of 10 mL of concentrated (11 M) HCl, followed by two sequential additions and dry downs of 30 mL of 2 M HCl. The sample was then quantitatively transferred to a pre-weighed 2 liter PFA bottle containing 150 mL of H2O using concentrated HCl, and the sample was diluted to provide approximately 2 L of 2 M HCl containing a final concentration of 4.84 mg of sample per g of solution.
2.2 Purification of uranium
Two purification schemes were testing for purification of uranium from a digest of SRM2780a. The first purification test involved AG1-X8, 100–200 mesh anion exchange resin using methods similar to those previously published.18,63,64 Two mL of resin were loaded into a column made from a disposable pipette tip. The resin was cleaned with washes of H2O, 9 M HCl, and 1 M HCl, and H2O, then conditioned with 7.5 M HNO3. The sample was loaded in 7.5 M HNO3, then the bulk sample matrix was eluted with additional washes of 7.5 M HNO3. Thorium (Th) was eluted with 9 M HCl, then U was eluted with 1.2 M HCl. The uranium fraction was transposed to 2% HNO3 in preparation for analyses on the NeptunePlus and Neoma MC-ICPMS instruments. The uranium chemical yields of this procedure from literature studies is near quantitative. Procedural blanks were not explicitly processed during the separation of the SRM2780a material in this study, however blanks processed using the same or similar chemistries during the time period of this study were < 12 pg.The second purification scheme used a 2 mL Eichrom UTEVA, 50–100 mesh resin cartridge and a vacuum box. UTEVA resin extraction methods improve sample processing times and blank contributions.49,65,66 The resin was cleaned using 0.02 M HNO3 and conditioned with 3 M HNO3. The sample was loaded in 3 M HNO3, then the resin was washed with 3 M HNO3 and 4 M HCl to remove the bulk sample matrix. Uranium was eluted with 0.02 M HNO3. The uranium fraction was transposed to 2% HNO3 in preparation for analysis. The uranium chemical yields of this procedure from literature studies is near quantitative. Procedural blanks were not explicitly processed during the separation of the SRM2780a material in this study, however blanks processed using the same or similar chemistries during the time period of this study were typically lower than instrumental background. The estimated procedural blank is <2 pg.
2.3 Isotopic analyses
Instrument setup and acquisition parameters for the unpurified sample analyses are provided in Table 1. In general, the Agilent 8900 was configured to provide the most rapid isotopic analysis, whereas the iCAP was configured to optimize performance of isotopic composition analysis resulting in longer analysis times. The running configurations for each instrument are described in more detail below.| Instrument | Agilent 8900 | iCAP | iCAP | Neoma | Neoma MS/MS |
|---|---|---|---|---|---|
| Sensitivity spec (kcps per ppb 238U) | 1000 | 330 | 330 | 3100 | 3100 |
| Introduction | Spray chamber | Spray chamber | Aridus II | Spray chamber | Spray chamber |
| Acquisition time (s) | 90 | 300 | 1010 (long) | 160 | 160 |
| 101 (rapid) |
Method one used an Apex Ω desolvating system to introduce purified uranium solutions into the plasma (dry plasma). Uranium isotopes were analyzed in static mode on Faraday cups, with 238U and 235U measured on the L1 and H1 cups with 1011 ohm resistors, and 234U measured on the L2 cup with a 1013 ohm resistor. The SRM2780a uranium fraction was diluted to provide >1 × 109 counts per second 238U (∼10 ppb). Beam intensities were measured for 40 cycles of 4 second integrations. Mass bias was externally corrected using solutions of either U030a or NIST CRM-129a.
Methods using traditional nebulization included analyses of both purified and unpurified fractions of SRM2780a. Samples were introduced into the plasma using a dual cyclonic spray chamber (wet plasma). Uranium isotopes were analyzed in static mode using a multiple ion counting array (nuclear package). 234U and 235U were measured on SEMs and 238U was measured on a compact discrete dynode (CDD) ion counter. In addition, isotopes were measured in dynamic mode using the center SEM/RPQ. Purified fractions were diluted to provide ∼ 4 × 105 counts per second 238U (∼100 ppt). Beam intensities for 234U were ∼22 cps at this concentration. Unpurified fractions were tested at multiple dilutions to evaluate matrix effects on sensitivity. Beam intensities were measured for 40 cycles of 4 second integrations. Mass and detector bias was externally corrected using 100 ppt solutions of either U030a or NIST CRM-129a. In dynamic mode, no detector bias correction is required, and no mass bias correction was applied.
Uranium isotopes were also measured as double oxides using O2 (0.05 mL min−1) as a reaction gas in the collision/reaction cell. This method was tested to compare the mass shifting capabilities of the Neoma MS/MS to the triple quadrupole instruments. Uranium double oxide ions were measured in static mode using the multiple ion counting array. 234U and 235U were measured on SEMs and 238U was measured on a CDD ion counter. Mass and detector bias was externally corrected using 100 ppt solutions of NIST CRM-129a. Discrepancies in isotopic compositions (reported below) were investigated using mass scans from 228 amu to 272 amu covering the range of masses from thorium metal to uranium double oxide.
3. Results
3.1 Purified fractions
Results of analyses of purified fractions of NIST2780a are provided in Table 2 and shown in Fig. 1. No systematic differences were observed between the two purification methods within uncertainty, and the values reported in Table 2 are averages of values measured from both purification processes as available. The high-precision analyses of SRM2780a (a combination of 15 analyses from the Neptune and Neoma) gave an average 235U/238U of 0.007255 ± 0.000003 and 234U/238U of 0.0000630 ± 0.0000001 (uncertainties are 2 standard error). The 235U/238U is in good agreement with the assumed value for natural uranium as measured in CRM960 (0.0072549 ± 0.0000008 (ref. 67)). The 234U/238U indicates secular disequilibrium in this reference material giving a (234U)/(238U) activity ratio of 1.146 assuming the secular equilibrium 234U/238U of Cheng et al., 2013 (54.970 × 10−6). Analyses of uranium isotopes using all other methods gave 235U/238U from 0.00723 to 0.00728, and 234U/238U from 0.000063 to 0.000066.| Instrument | Introduction | Method | 235U/238U | 1σa | 234U/238U | 1σa | n |
|---|---|---|---|---|---|---|---|
| a For n = 1, the standard deviation from the single measurement is reported. | |||||||
| Agilent 8900 (rapid) | Spray chamber | SQ | 0.00723 | 0.00001 | 0.000065 | 0.0000004 | 2 |
| Agilent 8900 (rapid) | Spray chamber | TQ-NO | 0.00727 | 0.00001 | 0.000066 | 0.000002 | 2 |
| iCAP | Aridus II | SQ | 0.00728 | 0.00003 | 0.000064 | 0.000001 | 6 |
| iCAP | Aridus II | TQ-O2 | 0.00724 | 0.00002 | 0.000063 | 0.000001 | 6 |
| Neptune | Spray chamber | Faraday-IC | 0.007248 | 0.000001 | 0.0000631 | 0.0000005 | 1 |
| Neoma | ApexΩ | Faraday | 0.007259 | 0.000012 | 0.0000632 | 0.0000001 | 2 |
| Neoma | Spray chamber | MIC | 0.00727 | 0.00002 | 0.000064 | 0.000003 | 5 |
| Neoma MS/MS | ApexΩ | Faraday | 0.007255 | 0.000004 | 0.0000629 | 0.0000002 | 12 |
| Neoma MS/MS | Spray chamber | MIC | 0.00724 | 0.00023 | 0.000065 | 0.000007 | 1 |
| Neoma MS/MS | Spray chamber | SC | 0.007242 | 0.000016 | 0.000063 | 0.000002 | 2 |
| Average and 2StdErr of faraday measurements | — | — | 0.007255 | 0.000003 | 0.0000630 | 0.0000001 | 15 |
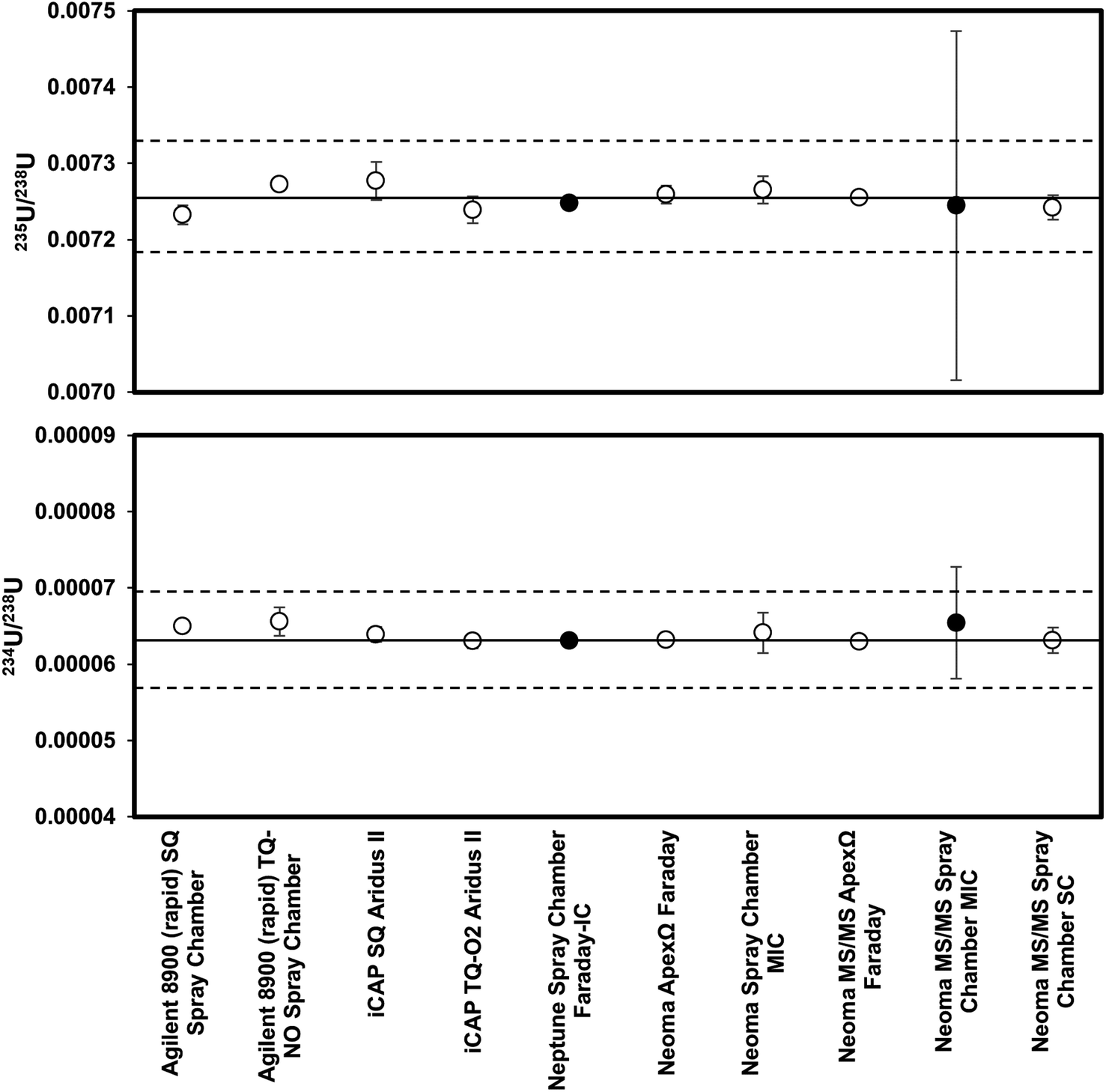 | ||
| Fig. 1 235U/238U (top) and 234U/238U (bottom) in purified aliquots of NIST SRM2780a compared to the high precision value (black solid lines) and 1% and 10% deviations (dashed lines), respectively. Filled circles indicate n = 1, and open circles indicate n > 1. SQ = single quad; TQ = triple quad; IC = ion counter; MIC = multiple ion counting; SC = single collector. | ||
3.2 Unpurified solutions
Results of analyses of unpurified solutions of NIST2780a are provided in Table 3 and shown in Fig. 2. Analyses of the unpurified solutions using the multiple ion counting (MIC) method on the Neoma gave 235U/238U = 0.00731 ± 0.00008 and 234U/238U = 0.000075 ± 0.000009 (1σ, n = 5). After installation of the MS/MS module, analyses of the unpurified solutions using the MIC method gave 235U/238U = 0.00727 ± 0.00003 and 234U/238U = 0.000069 ± 0.000002 (1σ, n = 7). Single collector mode gave 235U/238U = 0.00722 ± 0.00012 and 234U/238U = 0.000065 ± 0.000011 (1σ, n = 2). The analysis of the double oxides gave values significantly different than any other method, 235U/238U = 0.0147 ± 0.0008 and 234U/238U = 0.107 ± 0.011 (1σ, n = 9). The origins of these discrepancies are discussed below, and mass scans across the 228 amu to 272 amu range are provided in the ESI file.† Analyses of uranium isotopes on the triple quadrupole instruments gave 235U/238U from 0.00718 to 0.00732, and 234U/238U from 0.000055 to 0.000075.| Instrument | Introduction | Method | 235U/238U | 1σa | 234U/238U | 1σa | n |
|---|---|---|---|---|---|---|---|
| a For n = 1, the standard deviation from the single measurement is reported. | |||||||
| Agilent 8900 (rapid) | Spray chamber | SQ | 0.00718 | 0.00017 | 0.000068 | 0.000007 | 1 |
| Agilent 8900 (rapid) | Spray chamber | TQ-NO | 0.00727 | 0.00015 | 0.000065 | 0.000012 | 1 |
| iCAP TQ | Spray chamber | SQ | 0.00728 | 0.00006 | 0.000075 | 0.000004 | 3 |
| iCAP TQ | Spray chamber | TQ-O2 | 0.00721 | 0.00003 | 0.000055 | 0.000005 | 3 |
| iCAP TQ | Aridus II | SQ | 0.00726 | 0.00001 | 0.000065 | 0.000001 | 3 |
| iCAP TQ | Aridus II | TQ-O2 | 0.00732 | 0.00001 | 0.000065 | 0.000002 | 3 |
| iCAP TQ (rapid) | Aridus II | SQ | 0.00728 | 0.00002 | 0.000063 | 0.000004 | 3 |
| iCAP TQ (rapid) | Aridus II | TQ-O2 | 0.00722 | 0.00004 | 0.000061 | 0.000005 | 3 |
| Neoma | Spray chamber | MIC | 0.00731 | 0.00008 | 0.000075 | 0.000009 | 5 |
| Neoma MS/MS | Spray chamber | MIC | 0.00727 | 0.00003 | 0.000069 | 0.000002 | 7 |
| Neoma MS/MS | Spray chamber | SC | 0.00722 | 0.00012 | 0.000065 | 0.000011 | 1 |
| Neoma MS/MS | Spray chamber | MS/MS O2 | 0.0147 | 0.0008 | 0.107 | 0.011 | 9 |
 | ||
| Fig. 2 235U/238U (top) and 234U/238U (bottom) in the unpurified NIST SRM2780a digest compared to the high precision value (black solid lines) and 1% and 10% deviations (dashed lines), respectively. Filled circles indicate n = 1, and open circles indicate n > 1. All ICP-MS analyses produced 235U/238U within 1% of the high precision value. In general, 234U/238U were within 10% of the high precision value. In some cases, results were biased high, likely due to molecular interferences that have an outsized impact on the 234U due to relatively low count rates. However, the iCAP TQ analyses using the spray chamber with O2 in the collision cell produced low-based 234U/238U. Other than generally low count rates on the 234U, the reasons for the low bias were not immediately clear. SQ = single quad; TQ = triple quad; IC = ion counter; MIC = multiple ion counting; SC = single collector. | ||
4. Discussion
4.1 Comparison of purified and unpurified results
One benefit of the purification process is the ability to concentrate an analyte that is typically found in μg g−1 quantities or less in sample materials. Analyses using Faraday cups benefit from this process, whereby uranium that is purified from a solution containing a dissolved reference material is purified and concentrated. The NIST2780a solution used in this study contained ∼4.84 mg of soil per gram of solution. NIST2780a contains a certified uranium concentration of 4.0 mg kg−1, giving a uranium concentration in the digest solution of 0.019 μg g−1 (19 ppb). This concentration is above that used for high precision uranium isotopic analysis on the Neoma (10 ppb) but is accompanied by the soil matrix that can include more than 1000 ppm of additional elements (primarily major elements such as Al, Fe, K) in solution. Although not tested in this study, the impacts of matrix effects and interferences would likely prevent any ability to measure uranium isotopes with high precision in unpurified samples.Analyses of the 235U/238U in unpurified NIST2780a solution produced accurate results (within 1%) compared to the high precision value measured in the purified fractions using Faraday cups. No systematic biases exist between the different mass spectrometers, indicating that accurate results can be achieved using either the single collector ICP-MS (iCAP, 8900) or multiple ion counting methods on the MC-ICP-MS (Neoma). However, compared to the results of purified samples, results tend to be biased towards higher 234U/238U (10–20%), except for the 234U/238U measured in TQ mode on the iCAP with the spray chamber that is biased low. This is likely a result of a combination of interferences and relatively low count rates on the minor 234U isotope. Potential improvements could be achieved by increasing total integration times on the low abundance 234U isotope.
4.2 Origins of discrepancies in unpurified digests
Analyses of uranium isotopic ratios in unpurified samples are problematic due to the presence of matrix elements that can form molecular species in the argon plasma, leading to interferences at uranium masses. This is supported by the high-biased 234U/238U ratios in the unpurified digests. Mass shifting the uranium isotopes using gases in the collision cell for measurement as oxides or double oxides mitigates the bulk of the effects of these interferences. However, especially for the minor 234U isotope, we found no significant difference between single quadrupole and triple quadrupole measurements on the iCAP using the Aridus II. This can likely be attributed to the enhanced sensitivity afforded by using the desolvating nebulizer. In addition, the use of a desolvating nebulizer reduces oxide and hydride formation rates, which would decrease the impacts of some molecular interferences that form in the plasma (e.g., lead nitrides or oxides).61Analyses of uranium isotopes as double oxides in “MS/MS” mode on the Neoma gave spurious results. The origins of these discrepancies are at least two-fold. The band-pass window of the double Wien filter of the Neoma MS/MS is trapezoidal68,69 with much lower mass resolution compared to the quadrupole-based mass filters on the triple-quadrupole instruments. It is therefore unavoidable to pass thorium through the pre-filter along with the uranium isotopes. We hypothesize that the significant deviations from the high precision isotope ratios, especially in 234U/238U, are partially the result of the formation of thorium oxide species that also contain additional hydrogen atoms (Fig. 3; additional mass scans are provided in the ESI file†).
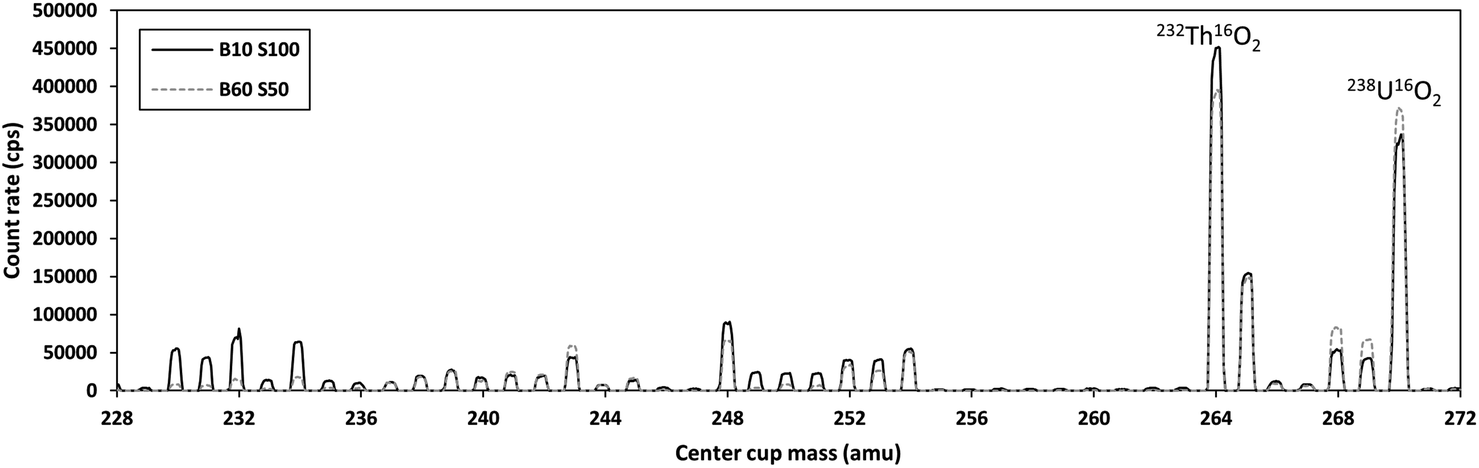 | ||
| Fig. 3 Mass scan in the NIST SRM2780a digest (diluted to provide ∼100 ppt U) from 228 amu to 272 amu with O2 in the collision/reaction cell at a flow rate of 0.05 mL min−1. Both the Th and U are converted to dominantly double oxide species. The solid black line labeled B10 S100 indicates a pre-filter magnetic field of 10% with the pre-filter slit 100% open. The dashed grey line labeled B60 S50 indicates a pre-filter magnetic field of 60% with the pre-filter slit 50% open. When the B field was set to 10%, the E field was approximately 40 volts. When the B field was set to 60%, the E field was approximately 240 volts. A reduction in beam intensities of the lower mass range (<236 amu) is achieved by changing the pre-filter settings. However, only slight changes are observed in the double oxide mass range of Th and U (>263 amu). Large interfering peaks on the minor uranium isotopes (234U and 235U) persist regardless of the pre-filter setting. | ||
Despite using the purest available oxygen gas (99.995% purity), introduction of oxygen into the collision cell raised background levels significantly in blank solutions. For example, background count rates on 234U were in some cases above 1000 cps, and over 100 cps on 235U (see ESI file†). Typical background count rates are <10 cps for 235U and <1 cps for 234U when measured as metal ions without any gas in the collision cell. Furthermore, a sample with a 238U count rate of 4 × 105 cps would give 234U count rate of ∼22 cps (assuming secular equilibrium), making the background issue with collision/reaction gases untenable. This suggests that even the high purity gases introduce additional impurities that make measurements of small ion beams on the ion counters problematic. These impurities (e.g., water) could also be causing the formation of the additional thorium species that cause large deviations in the measured isotope ratios.
4.3 Benefits of triple quadrupole ICPMS for uranium isotope analysis in unpurified digests
The primary benefit of the MC-ICP-MS is the ability to measure relatively high beam intensities simultaneously, allowing for high precision isotopic analysis comparable to TIMS. Given the uncertainties, there is minimal benefit to measuring purified solutions at low concentrations using multiple ion counting on the MC-ICP-MS compared to analysis of unpurified solutions on any of the instruments used in this study. However, analyses of unpurified digests remains unsuitable when high precision is required.Regardless of the mass spectrometer, precision generally improves when acquisition times are longer and sample consumption is higher. For example, the best precision achieved was using the iCAP equipped with the Aridus II in single quadrupole mode. However, precision of individual analyses across mass spectrometers was similar for ion-counter only measurements (data provided in the ESI file†). Nevertheless, data presented here indicate that triple quadrupole ICP-MS can provide accurate uranium isotopic results that can be acquired without purification, significantly reducing sample preparation time. This “early time” data can be used to enhance sample processing (e.g., provide information for optimized sample/spike ratios) when higher precision data are required. In addition, this type of analysis can be especially useful when rapid analysis is crucial, such as in emergency response situations. When rapid analyses are required, the analysis times can be minimized (in this case, analysis time per sample was as low as 90 seconds) while maintaining accuracy.
5. Conclusions
Here we presented uranium isotopic composition data measured in an unpurified digest solution of NIST SRM2780a by multi-ion counting ICP-MS (Neoma) and triple quadrupole ICP-MS (8900, iCAP). These data were compared to high precision uranium isotopic data measured in purified fractions by multi-collector ICP-MS (Neptune and Neoma). Results show that triple quadrupole ICP-MS can produce uranium isotopic compositions within 1% of the high precision value for the critical 235U/238U isotope ratio and 234U/238U generally within 10% of the high precision value. These results were generated using several different conditions (desolvator vs. spray chamber, long vs. short acquisition times). There is no clear benefit to analyzing unpurified solutions using the multi-ion counting array of the Neoma compared to the iCAP or 8900. Analyses of uranium isotopes as double oxides on the Neoma MS/MS was problematic and requires additional development, potentially in the use of higher purity gases to reduce formation of oxy-hydroxide species. Analysis of uranium isotopes in unpurified solutions can provide a rapid assessment of isotopic composition and can be especially useful when samples are expected to contain an isotopic composition outside of the range of naturally occurring uranium.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Norbert Gajos is acknowledged for providing comments and discussion on analysis of uranium isotopes. This work was sponsored by the Office of the Deputy Assistant Secretary of Defense for Nuclear Matters. Pacific Northwest National Laboratory is a multi-program national laboratory operated for the U.S. Department of Energy (DOE) by Battelle Memorial Institute under contract number DE-AC05-76RL01830.References
- T. L. Spano, A. Simonetti, E. Balbonia, C. Dorais and P. C. Burns, Trace element and U isotope analysis of uraninite and ore concentrate: Applications for nuclear forensics applications, Appl. Geochem., 2017, 84, 277–285 CrossRef CAS.
- S. F. Boulyga and J. S. Becker, Isotopic analysis of uranium and plutonium using ICP-MS and estimation of burn-up of spent uranium in contaminated environmental samples, J. Anal. At. Spectrom., 2002, 17, 1143–1147 RSC.
- R. C. Marin, J. E. S. Sarkis and M. R. L. Nascimento, The use of LA-SF-ICP-MS for nuclear forensics purposes: uranium isotope ratio analysis, J. Radioanal. Nucl. Chem., 2012, 295, 99–104 CrossRef.
- S. F. Boulyga, C. Testa, D. Desideri and J. S. Becker, Optimisation and application of ICP-MS and alpha-spectrometry for determination of isotopic ratios of depleted uranium and plutonium in samples collected in Kosovo, J. Anal. At. Spectrom., 2001, 16, 1283–1289 RSC.
- M. Krachler, Z. Varga, A. Nicholl, M. Wallenius and K. Mayer, Spatial distribution of uranium isotopes in solid nuclear materials using laser ablation multi-collector ICP-MS, Microchem. J., 2018, 140, 24–30 CrossRef CAS.
- S. F. Boulyga, A. Koepf, S. Konegger-Kappel, Z. Macsik and G. Stadelmann, Uranium isotope analysis by MC-ICP-MS in sub-ng sized samples, J. Anal. At. Spectrom., 2016, 31, 2272–2284 RSC.
- Y. Shi, R. Collins and C. Broome, Determination of uranium, thorium, and plutonium isotopes by ICP-MS, J. Radioanal. Nucl. Chem., 2012, 296, 509–515 CrossRef.
- M. J. Kristo, A. M. Gaffney, N. E. Marks, K. Knight, W. S. Cassata and I. D. Hutcheon, Nuclear forensic science: analysis of nuclear material out of regulatory control, Annu. Rev. Earth Planet. Sci., 2016, 44, 555–579 CrossRef CAS.
- Z. Varga, J. Krajkó, M. Peńkin, M. Novák, Z. Eke, M. Wallenius and K. Mayer, Identification of uranium signatures relevant for nuclear safeguards and forensics, J. Radioanal. Nucl. Chem., 2017, 312, 639–654 CrossRef CAS PubMed.
- S. Turner, P. van Calsteren, N. Vigier and L. Thomas, Determination of thorium and uranium isotope ratios in low-concentration geological materials using a fixed multi-collector-ICP-MS, J. Anal. At. Spectrom., 2001, 16, 612–615 RSC.
- P. Deschamps, R. Doucelance, B. Ghaleb and J.-L. Michelot, Further investigations on optimized tail correction and high-precision measurement of uranium isotopic ratios using multi-collector ICP-MS, Chem. Geol., 2003, 201, 141–160 CrossRef CAS.
- J. Fietzke, V. Liebetrau, A. Eisenhauer and Ch. Dullo, Determination of uranium isotope ratios by multi-static MIC-ICP-MS: method and implementation for precise U- and Th-series isotope measurements, J. Anal. At. Spectrom., 2005, 20, 395–401 RSC.
- R.-M. Wang and C.-F. You, Precise determination of U isotopic compositions in low concentration carbonate samples by MC-ICP-MS, Talanta, 2013, 107, 67–73 CrossRef CAS PubMed.
- H.-W. Chiang, Y. Lu, W. Wang, K. Lin and X. Liu, Optimizing MC-ICP-MS with SEM protocols for determination of U and Th isotope ratios and 230Th ages in carbonates, Quat. Geochronol., 2019, 50, 75–90 CrossRef.
- B. Seth, M. F. Thirlwall, S. L. Houghton and C.-A. Craig, Accurate measurements of Th-U isotope ratios for carbonate geochronology using MC-ICP-MS, J. Anal. At. Spectrom., 2003, 18, 1323–1330 RSC.
- D. L. Hoffmann, J. Prytulak, D. A. Richards, T. Elliott, C. D. Coath, P. L. Smart and D. Scholz, Procedures for accurate U and Th isotope measurements by high precision MC-ICPMS, Int. J. Mass Spectrom., 2007, 264, 97–109 CrossRef CAS.
- K. A. Matthews, M. T. Murrell, S. J. Goldstein, A. J. Nunn and D. E. Norman, Uranium and thorium concentration and isotopic composition in five glasses (BHVO-2G, BCR-2G, NKT-1G, T1-G, ATHO-G) and two powder (BHVO-2, BCR-2) reference materials, Geostand. Geoanal. Res., 2010, 35, 227–234 CrossRef.
- K. W. W. Sims, S. R. Hart, M. K. Reagan, J. Blusztajn, H. Staudigel, R. A. Sohn, G. D. Layne, L. A. Ball and J. Andrews, 238U-230Th-226Ra-210Pb-210Po, 232Th-228Ra, and 235U-231Pa constraints on the ages and petrogenesis of Vailulu’u and Malumalu Lavas, Samoa, Geochem., Geophys., Geosyst., 2008, 9, Q04003 CrossRef.
- J. S. Becker, H. Sela, J. Dobrowolska, M. Zoriy and J. S. Becker, Recent applications on isotope ratio measurements by ICP-MS and LA-ICP-MS on biological samples and single particles, Int. J. Mass Spectrom., 2008, 270, 1–7 CrossRef CAS.
- J. G. Arnason, C. N. Pellegri and P. J. Parsons, Determination of total uranium and uranium isotope ratios in human urine by ICP-MS: results of an interlaboratory study, J. Anal. At. Spectrom., 2015, 30, 126–138 RSC.
- M. V. Zoriy, M. Kayser, A. Izmer, C. Pickhardt and J. S. Becker, Determination of uranium isotopic ratios in biological samples using laser ablation inductively coupled plasma double focusing sector field mass spectrometry with cooled ablation chamber, Int. J. Mass Spectrom., 2005, 242, 297–302 CrossRef CAS.
- J. W. Ejnik, T. I. Todorov, F. G. Mullick, K. Squibb, M. A. McDiarmid and J. A. Centeno, Uranium analysis in urine by inductively coupled plasma dynamic reaction cell mass spectrometry, Anal. Bioanal. Chem., 2005, 382, 73–79 CrossRef CAS PubMed.
- P. Krystek and R. Ritsema, Determination of uranium in urine – measurement of isotope ratios and quantification by use of inductively coupled plasma mass spectrometry, Anal. Bioanal. Chem., 2002, 374, 226–229 CrossRef CAS PubMed.
- S. F. Boulyga, J. L. Matusevich, V. P. Mironov, V. P. Kudrjashov, L. Halicz, I. Segal, J. A. McLean, A. Montaser and J. S. Becker, Determination of 236U/238U isotope ratio in contaminated environmental samples using different ICP-MS instruments, J. Anal. At. Spectrom., 2002, 17, 958–964 RSC.
- F. Pointurier, A. Hubert, N. Baglan and P. Hemet, Evaluation of a new generation quadrupole-based ICP-MS for uranium isotopic measurements in environmental samples, J. Radioanal. Nucl. Chem., 2008, 276, 505–511 CrossRef CAS.
- S. Uchida, R. Garcia-Tenorio, K. Tagami and M. Garcia-Leon, Determination of U isotopic ratios in environmental samples by ICP-MS, J. Anal. At. Spectrom., 2000, 15, 889–892 RSC.
- D. G. Kelly, L. G. I. Bennett, M. M. Fill, K. M. Mattson, K. S. Nielsen, S. D. White and S. F. Allen, Analytical methods for the environmental quantification of uranium isotopes: Method of validation and environmental application, J. Radioanal. Nucl. Chem., 2008, 278, 807–811 CrossRef CAS.
- M. Forte, R. Rusconi, C. Margini, G. Abbate, S. Maltese, P. badalamenti and S. Bellinzona, Determination of uranium isotopes in food and environmental samples by different techniques: A comparison, Radiat. Prot. Dosim., 2001, 97, 325–328 CrossRef CAS PubMed.
- T. F. Kraemer, M. W. Doughten and T. D. Bullen, Use of ICP/MS with ultrasonic nebulizer for routine determination of uranium activity ratios in natural water, Environ. Sci. Technol., 2002, 36, 4899–4904 CrossRef CAS PubMed.
- N. S. Lloyd, R. R. Parrish, M. S. A. Horstwood and S. R. N. Chenery, Precise and accurate isotopic analysis of microscopic uranium-oxide grains using LA-MC-ICP-MS, J. Anal. At. Spectrom., 2009, 24, 752–758 RSC.
- M. Krachler, Z. Varga, A. Nicholl and K. Mayer, Analytical considerations in the determination of uranium isotope ratios in solid uranium materials using laser ablation multi-collector ICP-MS, Anal. Chim. Acta: X, 2019, 2, 100018 CAS.
- B. T. Manard, K. T. Rogers, B. W. Ticknor, S. C. Metzger, N. A. Zirakparvar, B. D. Roach, D. A. Bostick and C. R. Hexel, Direct uranium isotopic analysis of swipe surface by microextraction-ICP-MS, Anal. Chem., 2021, 93, 11133–11139 CrossRef CAS PubMed.
- R. Park, C.-G. Lee, K. H. Chung and J. Park, Quantitative and isotopic analysis of single micrometer-sized uranium particles using multiple mass spectrometric techniques, J. Radioanal. Chem., 2023, 332, 2833–2840 CrossRef CAS.
- H. Jaegler, A. Gourgiotis, P. Steier, R. Golser, O. Diez and C. Cazala, Pushing limits of ICP-MS/MS for the determination of ultralow 236U/238U isotope ratios, Anal. Chem., 2020, 92, 7869–7876 CrossRef CAS PubMed.
- S. Kappel, S. F. Boulyga and T. Prohaska, Direct uranium isotope ratio analysis of single micrometer-sized glass particles, J. Environ. Radioact., 2012, 113, 8–15 CrossRef CAS PubMed.
- A. M. Gothmann, J. A. Higgins, J. F. Adkins, W. Broecker, K. A. Farley, R. McKeon, J. Stolarski, J. Planavsky, N. Planavsky, X. Wang and M. L. Bender, A Cenozoic record of seawater uranium in fossil corals, Geochim. Cosmochim. Acta, 2019, 250, 173–190 CrossRef CAS.
- M. A. Kipp, H. Li, M. J. Ellwood, S. G. John, R. Middag, J. F. Adkins and F. L. H. Tissot, 238U, 235U, and 234U in seawater and deep-sea corals: A high-precision reappraisal, Geochim. Cosmochim. Acta, 2022, 336, 231–248 CrossRef CAS.
- J. H. Chen and G. J. Wasserburg, Isotopic determination of uranium in picomole and .ubpicomole quantities, Anal. Chem., 1981, 53, 2060–2067 CrossRef CAS.
- J. D. Fassett and W. R. Kelly, Interlaboratory isotopic ratio measurement of nanogram quantities of uranium and plutonium on resin beads by thermal ionization mass spectrometry, Anal. Chem., 1984, 56, 550–556 CrossRef CAS.
- E. L. Callis and R. M. Abernathey, High-precision isotopic analyses of uranium and plutonium by total sample volatilization and signal integration, Int. J. Mass Spectrom. Ion Processes, 1991, 103, 93–105 CrossRef CAS.
- S. Richter and A. S. Goldberg, Improved techniques for high accuracy isotope ratio measurements of nuclear materials using thermal ionization mass spectrometry, Int. J. Mass Spectrom., 2003, 229, 181–197 CrossRef CAS.
- P. Lindahl, G. Olszewski and M. Eriksson, Performance and optimization of triple quadrupole ICP-MS for accurate measurement of uranium isotopic ratios, J. Anal. At. Spectrom., 2021, 36, 2164–2172 RSC.
- J. Wimpenny, K. M. Samperton, P. Sotorrio, M. S. Wellons, S. M. Scott, D. Willingham and K. Knight, Rapid isotopic analysis of uranium particles by laser ablation MC-ICP-MS, J. Anal. At. Spectrom., 2023, 38, 827–840 RSC.
- S. Xing, M. Luo, Y. Wu, D. Liu and X. Dai, Rapid determination of uranium isotopes in calcium fluoride sludge by tandem quadrupole ICP-MS/MS, J. Anal. At. Spectrom., 2019, 34, 2027–2034 RSC.
- K. Tagami and S. Uchida, Rapid uranium preconcentration and separation method from fresh water samples for total U and 235U/238U isotope ratio measurements by ICP-MS, Anal. Chim. Acta, 2007, 592, 101–105 CrossRef CAS PubMed.
- L. Rovan and M. Štrok, Optimization of the sample preparation and measurement protocol for the analysis of uranium isotopes by MC-ICP-MS without spike addition, J. Anal. At. Spectrom., 2019, 34, 1882–1891 RSC.
- S. C. Metzger, K. T. Rogers, D. A. Bostick, E. H. McBay, B. W. Ticknor, B. T. Manard and C. R. Hexel, Optimization of uranium and plutonium separations using TEVA and UTEVA cartridges for MC-ICP-MS analysis of environmental swipe samples, Talanta, 2019, 198, 257–262 CrossRef CAS PubMed.
- J. Zheng and M. Yamada, Determination of U isotope ratios in sediments using ICP-QMS after sample cleanup with anion-exchange and extraction chromatography, Talanta, 2006, 68, 932–939 CrossRef CAS PubMed.
- S. C. Metzger, B. W. Ticknor, K. T. Rogers, D. A. Bostick, E. H. McBay and C. R. Hexel, Automated separation of uranium and plutonium from environmental swipe samples for multiple collector inductively coupled plasma mass spectrometry, Anal. Chem., 2018, 90, 9441–9448 CrossRef CAS PubMed.
- R. Martinelli, T. Hamilton, R. Williams and S. Kehl, Separation of uranium and plutonium isotopes for measurement by multi-collector inductively coupled plasm mass spectroscopy, J. Radioanal. Nucl. Chem., 2009, 282, 343–347 CrossRef CAS.
- M. H. Lee, J. H. Park, S. Y. Oh, H. J. Ahn, C. H. Lee, K. Song and M. S. Lee, Determination of Pu and U isotopes in safeguard swipe sample with extraction chromatographic techniques, Talanta, 2011, 86, 99–102 CrossRef CAS PubMed.
- N. Veerasamy, A. Takamasa, R. Murugan, S. Kasar, T. Aono, K. Inoue, M. Fukushi and S. K. Sahoo, Chemical separation of uranium and precise measurement of 234U/238U and 235U/238U ratios in soil samples using multi collector inductively coupled plasma mass spectrometry, Molecules, 2021, 25, 2138 CrossRef PubMed.
- R. Murugan, N. Veerasamy, Y. Zhao, T. Aono and S. K. Sahoo, Precise measurement of uranium isotope ratios in Fukushima soils using multi-collector inductively coupled plasma mass spectrometry, Int. J. Mass Spectrom., 2021, 467, 116623 CrossRef CAS.
- X. Luo, M. Rehkämper, D.-C. Lee and A. N. Halliday, High precision 230Th/232Th and 234U/238U measurements using energy-filtered ICP magnetic sector multiple collector mass spectrometry, Int. J. Mass Spectrom. Ion Processes, 1997, 171, 105–117 CrossRef CAS.
- M. Abe, N. Seko, H. Hoshina, S. Wada, S. Yamasaki, K. Sueki and A. Sakaguchi, Simple and convenient preconcentration procedure for the isotopic analysis of uranium in seawater, Anal. Methods, 2024, 16, 2478–2488 RSC.
- W. Wang, R. D. Evans and H. E. Evans, A rapid, automated system for the separation, preconcentration and measurement of 90Sr, and U, Am, and Pu isotopes, Talanta, 2021, 233, 122507 CrossRef CAS PubMed.
- E. P. Horwitz, M. L. Dietz, R. Chiarizia, H. Diamond, A. M. Essling and D. Graczyk, Separation and preconcentration of uranium from acidic media by extraction chromatography, Anal. Chim. Acta, 1992, 266, 25–37 CrossRef CAS.
- V. G. Torgov, M. G. Demidova, A. I. Saprykin, I. V. Nikolaeva, T. V. Us and E. P. Chebykin, Extraction preconcentration of uranium and thorium traces in the analysis of bottom sediments by inductively coupled plasma mass spectrometry, J. Anal. Chem., 2002, 57, 303–310 CrossRef CAS.
- M. Kraiem, K. Mayer, T. Gouder, A. Seibert, T. Wiss, H. Thiele and J.-P. Hiernaut, Experimental investigation of the ionization mechanisms of uranium in thermal ionization mass spectrometry in the presence of carbon, Int. J. Mass Spectrom., 2010, 289, 108–118 CrossRef CAS.
- J. D. Fassett and W. R. Kelly, Interlaboratory isotopic ratio measurement of nanogram quantities of uranium and plutonium on resin beads by thermal ionization mass spectrometry, Anal. Chem., 1984, 56, 550–556 CrossRef CAS.
- A. D. Pollington, W. S. Kinman, S. K. Hanson and R. E. Steiner, Polyatomic interferences on high precision uranium isotope ratio measurements by MC-ICP-MS: applications to environmental sampling for nuclear safeguards, J. Radioanal. Nucl. Chem., 2016, 307, 2109–2115 CrossRef CAS.
- C. A. Gonzalez and S. J. Choquette, Certificate of Analysis: Standard Reference Materials 2780a Hard Rock Mine Waste, National Institute of Standards and Technology, Gaithersburg, MD, USA. 2018 Search PubMed.
- S. R. Scott, K. W. W. Sims, M. K. Reagan, L. Ball, J. B. Schwieters, C. Bouman, N. S. Lloyd, C. L. Waters, J. J. Standish and D. L. Tollstrup, The application of abundance sensitivity filters to the precise and accurate measurement of uranium series nuclides by plasma mass spectrometry, Int. J. Mass Spectrom., 2019, 435, 321–332 CrossRef CAS.
- S. R. Scott, F. C. Ramos and J. B. Gill, Spreading dynamics of an intermediate Ridge: Insights from U-series disequilibria, Endeavour Segment, Juan de Fuca Ridge, J. Petrol., 2018, 59, 1847–1868 CrossRef CAS.
- S. C. Metzger, K. T. Rogers, D. A. Bostick, E. H. McBay, B. W. Ticknor, B. T. Manard and C. R. Hexel, Optimization of uranium and plutonium separations using TEVA and UTEVA cartridges for MC-ICP-MS analysis of environmental swipe samples, Talanta, 2019, 198, 257–262 CrossRef CAS PubMed.
- A. Morgenstern, C. Apostolidis, R. Carlos-Marquez, K. Mayer and R. Molinet, Single-column extraction chromatographic separation of U, Pu, Np and Am, Radiochim. Acta, 2002, 90, 81–85 CrossRef CAS.
- S. Richter, R. Eykens, H. Kuhn, Y. Aregbe, A. Verbruggen and S. Weyer, New average values for the n(238U)/n(235U) isotope ratios of natural uranium standards, Int. J. Mass Spectrom., 2010, 295, 94–97 CrossRef CAS.
- G. Craig, H. Wehrs, D. G. Bevan, M. Pfeifer, J. Lewis, C. D. Coath, T. Elliott, C. Huang, N. S. Lloyd and J. B. Schwieters, Project Vienna: A novel precell mass filter for collision/reaction cell MC-ICPMS/MS, Anal. Chem., 2021, 93, 10519–10527 CrossRef CAS PubMed.
- P. Télouk, E. Albalat, B. Bourdon, F. Albarède and V. Balter, Performance of the double-Wien filter of the Neoma MC-ICPMS/MS with an application to copper stable isotope compositions, J. Anal. At. Spectrom., 2023, 38, 1973–1983 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ja00130c |
| This journal is © The Royal Society of Chemistry 2024 |