
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn overview of the applications of total reflection X-ray fluorescence spectrometry in food, cosmetics, and pharmaceutical research
Eva
Marguí
 *a,
Diane
Eichert
b,
Jasna
Jablan
c,
Fabjola
Bilo
de,
Laura E.
Depero
de,
Ana
Pejović-Milić
f,
Armin
Gross
g,
Haegen
Stosnach
g,
Aldona
Kubala-Kukuś
h,
Dariusz
Banaś
hi and
Laura
Borgese
de
*a,
Diane
Eichert
b,
Jasna
Jablan
c,
Fabjola
Bilo
de,
Laura E.
Depero
de,
Ana
Pejović-Milić
f,
Armin
Gross
g,
Haegen
Stosnach
g,
Aldona
Kubala-Kukuś
h,
Dariusz
Banaś
hi and
Laura
Borgese
de
aDepartment of Chemistry, Faculty of Sciences, Girona, Spain. E-mail: eva.margui@udg.edu
bElettra Sincrotrone Trieste, Trieste, Italy
cDepartment of Analytical Chemistry, University of Zagreb Faculty of Pharmacy and Biochemistry, Zagreb, Croatia
dINSTM & Chemistry for Technologies Laboratory, Department of Mechanical and Industrial Engineering, University of Brescia, Brescia, Italy
eSmart Solutions s.r.l, Brescia, Italy
fDepartment of Physics, Toronto Metropolitan University, Toronto, Canada
gBruker Nano Analytics, Berlin, Germany
hInstitute of Physics, Jan Kochanowski University, Kielce, Poland
iHoly Cross Cancer Center, Kielce, Poland
First published on 6th June 2024
Abstract
The need for elemental analysis in quality control of food, pharmaceuticals and cosmetics, taking into account human safety, has become an essential requirement for any product destinated for commercialization and targets in particular potentially toxic elements. The main objective is to obtain reliable data on elemental content that enables end-users to make informed decisions and to address problems effectively. To align with the principles of green and sustainable analytical chemistry, recent efforts have focused on incorporating new, low-cost screening and quantitative analytical tools such as total reflection X-ray fluorescence spectrometry (TXRF). This paper provides an overview of recent TXRF applications in the elemental analysis of food, pharmaceuticals, and cosmetic samples. The state-of-the-art procedures for sample preparation with details on the sample type and amount, dilution steps, mixing agents, analysed elements, and addition of internal calibration standards are presented together with analytical parameters such as limits of detection and quantification. The current challenges for applying TXRF to each of these research fields are discussed.
1 Introduction
The determination of elemental composition in consumer products such as foodstuffs, pharmaceuticals, drugs, supplements, and cosmetics is a necessary step in industry and quality control laboratories to ensure product quality and safety.The analytical practices involve the examination of many samples and, in most cases, the consumption and generation of high amounts of reagents and wastes, especially when atomic spectrometry techniques such as atomic absorption spectrometry (AAS) and inductively coupled plasma (ICP) based spectrometry are used. Consequently, the running costs are high, and these methods may involve time-consuming1 sampling and sample procedures steps, when the sample cannot be directly measured. To mitigate these issues, new low-cost analytical tools which incorporate green and sustainable analytical chemistry principles are desirable.2 These tools should be able to assess the elemental composition while presenting the following characteristics: minimum sample treatment, high throughput, and adequate specificity and sensitivity for monitoring the elements of interest at the expected concentration levels.
Energy dispersive X-ray fluorescence (EDXRF) and wavelength dispersive X-ray fluorescence (WDXRF) spectrometry have been widely used in many industrial applications as screening tools and for quantitative analysis of major, minor, and trace elements in solid samples. However, the lack of matrix-matched reference materials for calibration limits their widespread use.3 Total reflection X-ray fluorescence (TXRF) is an alternative approach. TXRF takes advantage of the very low background and the higher excitation level that are achieved when a suitable sample is placed onto a reflective carrier, on which is shined an X-ray beam at an angle below the critical angle of the sample reflector material.4 Consequently, in comparison to other X-ray fluorescence (XRF) techniques, TXRF presents lower limits of detection (ranging from 10−7 g to 10−12 g) that may be applied to solid and liquid samples for simultaneous multi-elemental information. Reliable quantitative analysis may be obtained incorporating an internal calibration standard (ICS) to the sample, adequately prepared and deposited on a reflective carrier to comply with the thin film quantification approach. A benefit of TXRF is the possibility of analysing samples using simple and straightforward sample treatments, compared to acid digestion steps required by other atomic spectrometry techniques, offering thus unique screening possibility. Screening is undoubtedly a strategic way of investigation which may answer to some specific characterization demands, especially within industrial or regulatory contexts, where elemental occurrence and concentration thresholds are answers highly sought, but do not necessarily require accurate quantifications. TXRF has, however, the potential to be applied for both screening and qualitative or quantitative purposes. Qualitative and quantitative analysis aim at identifying the elements present into a sample, and at determining the concentration or mass, together with the related uncertainty, of one or more elements present into the sample, respectively. This, especially in the case of quantitative analysis, may require the development of specific sample preparation and handling procedures. In contrast, more expeditious procedures solely intending to identify, and select from a starting sample' set, a group of samples that will contain one or more elements, eventually above a pre-set concentration level, is the goal of screening procedures. There, some shortcuts in the sample handling may be considered, as a yes/no answer is usually sought, without the need of thorough analysis. As TXRF unravel multi-elemental content, simultaneously, it is a promising analytical technique for consumer products. These advantages open interesting new applications for TXRF, especially in food, pharmaceuticals, and cosmetics.
In the last decade, the development and commercialization of benchtop TXRF spectrometers, offering extreme simplicity of operation in a low-cost compact design, have promoted the application of these systems in many research fields, including environment,5,6 geological,7,8 biological,9,10 medical,11 and nuclear materials.12 The use of TXRF in pharmaceuticals and cosmetics is very seldom, while in the field of beverages and foodstuffs it has been more widely employed.13 However, TXRF analysis of consumer products is still limited to research purposes and is rarely conducted as a daily routine analysis within an industrial framework (with the exception of Si wafers). Some of the limitations impairing the widespread use of TXRF, rely on the fact that TXRF is not considered as a technique of reference, due to a poor normative framework and lack of matrix-matched reference materials for calibration purposes.3 It, furthermore, lacks associated standard operating procedures, and as such is not included by regulatory bodies in the instructions or guidelines for compulsory testing.
The introduction of new techniques and methods in regulations and standards can be challenging due to the difficulty of accreditation procedures, the need to achieve a consensus between the elemental analysis experts, and consequently of worldwide recognition. Such ventures are based on a thorough assessment of the analytical performance, embracing the available guidelines of each application field and on proper validation of the developed alternative methods,14via for instance proficiency tests. This review presents an overview of the published methods, protocols, sample preparation and handling procedures for the TXRF analysis, according to the needs and regulations in the various application fields for quality and safety.
2 Elemental analysis for quality and safety of consumer products
2.1 Foodstuff
The definition of food quality is global and comprises both positive attributes, such as appearance, origin, texture, flavor, and nutritional content and value, as well as negative ones, such as contamination, discoloration, off-odors, and spoilage. Quality standards are essential in the European market to meet consumers' and regulatory bodies' requirements, and specifically target products' attributes, consequently affecting the value of the product for both producers and consumers. For industry, those attributes are evaluated via three key factors: conformity with the product's intended purpose and regulatory requirements, safety, and satisfaction of consumer expectations and perceptions.Food safety refers to all hazards, whether chronic or acute, that may negatively impact consumer health according to the Food and Agriculture Organization (FAO) and the World Health Organization (WHO) (2003).15 The EU Food Safety Authorities (EFSA),16 regulates food content, ingredients, product description, chemical safety, sanitary conditions, hygiene, and other product specifications. The legislation on contaminants is based on scientific evidence and on the principle of keeping contaminants' levels as low as reasonably achievable by using good working practices. Maximum concentration levels acceptable in products have been established for specific contaminants, including heavy metals, to protect public health.
Nonetheless, the contamination of food with heavy metals remains a relevant issue for public health. Therefore, it is essential to ensure food safety by using various analytical techniques for metals identification and quantification. In particular, the EU Regulation (EC) No. 1881/2006 sets the maximum levels for potentially toxic metals in food. For example, the allowed Pb levels range from 0.02 to 2 mg kg−1 wet weight for raw milk and food supplements, respectively. Cadmium limits are set between 0.005 mg kg−1 wet weight for liquid infant formulae from cow milk and 3 mg kg−1 for food supplements from seaweed. The permitted mercury levels are 0.1 mg kg−1 wet weight in food supplements and 1 mg kg−1 in muscle fish. The European Commission also established maximum levels for inorganic tin in canned baby foods, infant formulae and other canned foods, excluding beverages.17
Besides the interest in toxic elements, the determination of other elements presents in food samples, such as major essential elements, trace essential elements, ultra-trace essential elements, and non-essential trace elements, is relevant.18 Major elements are usually found in the percent range, while trace and ultra-trace elements span over a wide concentration range from ppm to ppb.19 The level of these elements varies widely in food samples, even within similar matrices. For example, the content of Ca within a group of 35 different alcoholic beverages, like beers, wines, and liqueurs, may vary from 3 to 50 mg g−1; in cookies the content of Fe may fluctuate from 0.2 to 12 mg g−1; and in raw apples the content of Mg may be from 0 to 4%.20 Some of these elements, such as Na, Ca, Mg, K, and Cl, are macronutrients required in larger quantities for healthy nutrition, while others, such as F, V, Cr, Mn, Fe, Co, Cu, Zn, Se, Mo, and I, are micronutrients essential for plants, animals, and human nutrition. Excessive amounts of these elements can have phytotoxic and/or zootoxic effects.21 Fe, Zn and Mn, for instance, are toxic to humans if their daily intake level is exceeded.22 Thus, the determination of the elemental composition and concentration in food is crucial for making informed recommendations for consumption, especially for children, pregnant women, and people with specific elemental allergies or medical conditions. The interest in elemental analysis is increasing in agriculture and the food industry, with many papers focusing on the elemental composition of seasoning products,19 salts, and spices.23,24 Contamination due to the environment22,25 and food processing must be avoided26 or reduced27 whenever possible, but in any case controlled. Consequently, there is a strong need for fast screening tools and techniques with high sensitivity and applicable over a wide dynamic range of concentrations.
The Association of Official Agricultural Chemists (AOAC) developed the standard method performance requirements (SMPRs) for heavy metals in foods and beverages to ensure reliable monitoring through routine surveillance methods, defining the required values for concentration range, limit of quantification, repeatability, reproducibility, and recovery irrespective of the analytical techniques.28 Atomic and inorganic mass spectrometries are widely used to obtain accurate information about trace levels of metals in food. Furnace atomic absorption spectrometry (FAAS), graphite furnace atomic absorption spectrometry (GFAAS), inductively coupled plasma optical emission spectrometry (ICP-OES) and inductively coupled plasma mass spectrometry (ICP-MS) are the most used analytical techniques due to the availability of good laboratory practices and standard operating procedures, which, when followed, aim to ensure analysis quality.29 For a solid sample analysis, the X-ray fluorescence spectrometry is still the method most widely employed.30,31
2.2 Pharmaceuticals, drugs, and supplements
The interest in the determination of elemental impurities in pharmaceuticals has increased in recent years due to greater awareness of their potential toxicity, changes in regulatory requirements, and the need for updating limit tests in pharmacopeia.32 These impurities can arise from various sources, including catalyst residues, raw materials, solvents, and container/closure systems, at different stages of the manufacturing process. Thus, their identification is required during chemistry route scouting and optimization, process development phase, or catalyst screening and loading.33 While some elements may be intentionally added as active pharmaceutical ingredients (i.e., platinum-based chemotherapeutics agents, antimicrobials containing Fe, Ag, and Au, among others),34 others pose no therapeutic benefit to the patient and must be maintained within acceptable limits. The relevant side-effects of metal impurities on the safety of drugs (adverse pharmacological and toxicological effects), or on their stability and efficacy (causes of intolerance and degradation), led to an increasing interest in their contents in drugs.35 Regulatory bodies such as the European Medicines Agency (EMEA) in the International Council for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) from the Q3D working group on elemental impurities, known as ICH Q3D guidelines,36 and the United States Pharmacopoeia (USP) in the USP<232> chapter32 have categorized elemental impurities into three classes and established concentration limits according to a risk-based strategy and their toxicological properties, daily dosage budget fraction, and route of administration. The elements included in each class are: Class 1 (As, Cd, Hg, Pb), Class 2 (Co, V, Ni, Tl, Au, Pd, Ir, Os, Rh, Ru, Se, Ag and Pt), and Class 3 (Li, Sb, Ba, Mo, Cu, Sn, and Cr). Class 2 elements are divided into subclasses 2A and 2B based on their relatively probability of occurrence in the drug. In Table 1, concentration limits in (μg g−1) for components used in oral drug products with a 10 g maximum daily dose are reported.| 10 | Class | Drug products, drug substances and excipients | Parenteral drug products | Inhalation drug products | Dietary supplementsa | |||
|---|---|---|---|---|---|---|---|---|
| ICH Q3D | USP<232> | ICH Q3D | USP<232> | ICH Q3D | USP<232> | USP<2232> | ||
| a Finished dosage forms. b Inorganic form: speciation is not necessary when the total mercury or arsenic is less than the limit. c Mercury (total). d Methylmercury (as Hg): speciation is not necessary when the total mercury is less than the limit. | ||||||||
| Cd | 1 | 0.5 | 0.5 | 0.2 | 0.2 | 0.3 | 0.2 | 0.5 |
| Pb | 1 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| As | 1 | 1.5 | 1.5b | 1.5 | 1.5 | 0.2 | 0.2 | 1.5b |
| Hg | 1 | 3 | 3b | 0.3 | 0.3 | 0.1 | 0.1 | 1.5c |
| Methyl-Hg | 1 | — | — | — | — | — | — | 0.2d |
| Co | 2A | 5 | — | 0.5 | — | 0.3 | — | — |
| V | 2A | 10 | 10 | 1 | 1 | 0.1 | 0.1 | — |
| Ni | 2A | 20 | 20 | 2 | 2 | 0.5 | 0.5 | — |
| Tl | 2B | 0.8 | — | 0.8 | — | 0.8 | — | — |
| Au | 2B | 30 | — | 10 | — | 0.1 | — | — |
| Pd | 2B | 10 | 10 | 1 | 1 | 0.1 | 0.1 | — |
| Ir | 2B | 10 | 10 | 1 | 1 | 0.1 | 0.1 | — |
| Os | 2B | 10 | 10 | 1 | 1 | 0.1 | 0.1 | — |
| Rh | 2B | 10 | 10 | 1 | 1 | 0.1 | 0.1 | — |
| Ru | 2B | 10 | 10 | 1 | 1 | 0.1 | 0.1 | — |
| Se | 2B | 15 | — | 8 | — | 13 | — | — |
| Ag | 2B | 15 | — | 1 | — | 0.7 | — | — |
| Pt | 2B | 10 | 10 | 1 | 1 | 0.1 | 0.1 | — |
| Li | 3 | 55 | — | 25 | — | 2.5 | — | — |
| Sb | 3 | 120 | — | 9 | — | 2 | — | — |
| Ba | 3 | 140 | — | 70 | — | 30 | — | — |
| Mo | 3 | 300 | 300 | 150 | 150 | 1 | 1 | — |
| Cu | 3 | 300 | 300 | 30 | 30 | 3 | 3 | — |
| Sn | 3 | 600 | — | 60 | — | 6 | 0.1 | — |
| Cr | 3 | 1100 | 1100 | 110 | 110 | 0.3 | 0.3 | — |
Recently, the USP also proposed a general chapter <2232> about elemental contaminants in dietary supplements (DS)37 to be monitored in finished dosage forms (e.g., powders, tablets, capsules, liquids) and not in dietary ingredients.38 The permitted daily exposure (PDE) endorsed by the FAO/WHO has been used to set the limits of the four significant elements of toxicological concern (As, Cd, Pb, Hg).
Previous pharmacopoeia tests for heavy metals in the European Pharmacopoeia (Ph. Eur.), USP, British Pharmacopoeia and Japanese Pharmacopoeia were based on the reaction of metals with the thioacetamide reagent, resulting in the formation of coloured sulphides and their precipitation from aqueous solution.39 This visual colorimetric test suffers from serious limitations such as the lack of sensitivity, specificity, and recovery to monitor properly the actual levels for some metals. For instance, elements of toxicological concern (i.e., Cr, V, Ni) cannot be determined by this classical limit test, and biased results were reported for elements such as As, Cd, Mo, Pd, Pt and In with poor recovery values between 30–50%.40
The USP chapter <233> includes two analytical procedures involving ICP-OES and ICP-MS.41 One of the major limitations of such procedures is the treatment of solid pharmaceutical samples that must be solubilized to be compatible with the sample introduction systems of ICP-OES and ICP-MS instrumentation. Furthermore, pharmaceutical products are not sufficiently soluble in deionized water or dilute acids,42 and solubilization using organic chemicals or concentrated acids is required.43 When the sample is difficult to dissolve, a digestion method based on different acid mixtures (i.e., mixtures of HNO3 with HCl, HF, HClO4, H3PO4, and H2SO4) is employed.34 Notwithstanding, some pharmaceutical products are extremely difficult to digest even with the use of high-pressure systems and concentrated acids44 and consequently, a universal method of sample decomposition, which applies to all dosage forms of pharmaceutical samples, does not exist.
An additional inconvenience of the use of ICP-based techniques for the determination of elemental impurities in pharmaceutical products is the presence of spectral and non-spectral interferences and thus, a previous evaluation of the matrix composition should be carried out before even starting the analysis of the sample, to identify those. For instance, it is interesting to mention that almost 20% of the active pharmaceutical ingredients are hydrochlorides and spectral interferences of chlorine in ICP-MS analysis are expected.45 For this reason, although ICP platforms are predominant for most applications dealing with inorganic impurity analysis in pharmaceutical laboratories, the use of other analytical approaches which involve a simpler sample treatment is worth of investigation. In fact, the USP chapter <233> describes the criteria for the acceptability of alternative procedures equivalent to ICP-OES and ICP-MS ones.41Table 2 summarize those analytical characteristics and acceptance criteria, which are required in the validation of alternate quantitative approaches and test limit procedures.
| Analytical parameter | Acceptance criteria | Quantitative method | Test limit | |
|---|---|---|---|---|
| a Limits are reported in Table 1 (USP<232>). b Spiked sample 1: 1.0 × Limit. c Standard solution: 1.0 × Limit. d Spiked sample 2: 0.8 × Limit. | ||||
| Accuracy | Recovery: 70–150% (n = 3) | Yes | No | |
| Levels: 50, 100, 150% limita | ||||
| Precision | Repeatability | RSD ≤ 20% (n = 6) | Yes | Yes |
| Level: 100% limita | ||||
| Intermediate precision | RSD ≤ 25% | Yes | No | |
| Repeatability study on different days, instruments and analysts | ||||
| Specificity | Unequivocally assessment of the analyte in the presence of other target elements and matrix components | Yes | Yes | |
| Detectability | Signal spiked sample 1b ± 15% of the average signal value for standard solutionc | No | Yes | |
| Signal spiked sample 2d lower than the average signal value for standard solution | ||||
| Quantification limit (LOQ) | Demonstrated by meeting the accuracy requirement | Yes | No | |
| Linearity | Demonstrated by meeting the accuracy requirement | Yes | No | |
| Range | Demonstrated by meeting the accuracy requirement | Yes | No | |
In this view, the direct analysis of solid pharmaceutical samples by means of XRF technique demonstrated promising results, for instance, for the determination of Pd, Zn, Fe and Ni residues in active pharmaceutical ingredients (API).46,47 The major drawback reported in these studies was the lack of suitable reference materials with a matrix similar to the real samples for validation and calibration purposes. To overcome this problem, the use of synthetic standards made of cellulose simulating the API matrix appeared to be an effective means to obtain reliable calibration curves when no suitable pharmaceutical reference materials were available. More recently, XRF potential for screening oral solid dosage drug products for elemental impurities was demonstrated.48 The proposed method can be used as a limit test for class 1 and class 2A elements, and is fully compliant with the method validation requirements according to the ICH Q3D guidelines. It is worth to mention that in 2015 the USP included XRF as an instrumental method that can be used for both qualitative and quantitative analysis of liquids, powders, and solid materials (USP chapter <735> X-ray fluorescence Spectrometry),49 but recommends only the EDXRF and WDXRF techniques. The same applies to the last edition of Ph. Eur.50
2.3 Cosmetics
Cosmetics have drawn attention because of the potential negative effects of the chemicals they might contain. For cosmetic companies, proposing safe and high-quality cosmetic products become crucial, not only for regulatory purposes, but also because customers have been increasingly looking for documentation of efficacy, compliance, and quality on raw materials to finished products.1 Potentially toxic metals may originate from various sources such as raw materials, metal-coated equipment during manufacturing, and intentional addition as pigments and ingredients.51 To mitigate against the potential toxic effects of some heavy metals, the European Union (EU) Directive prohibits their intentional use, for instance of Sb, As, Cd, Cr, Co, Hg, Ni, and Pb.52 The amount of metals permissible in cosmetic products is assessed based on a broad concept approach of risk evaluation because the EU regulations have not set so far specific limits. Using estimated margins of safety values that take into account both potential negative effects and systematic exposure dose, the Scientific Committee Consumer Safety (SCCS) of the European Commission has determined recommended maximum thresholds for heavy metals in cosmetic items. As visible in Table 3, other international agencies and governments from all over the world have also established metal concentration limits for inorganic contaminants in cosmetic products. While the amount of Hg, Pb, As, Cd, and Sb are regulated, other metals such as Cr, Ni, and Co are not always considered due to their lower health risk. The strictest limitations are those enforced in Germany,53 which are based mostly on levels that can be practically avoided rather than on risk. There isn't much agreement, though, on which metals or inorganic species, and in which amount, require control in cosmetics. Currently, cosmetics manufacturers are not required to disclose the metal contaminants found in their products, and consequently customers have no way to know the risks involved in using cosmetics on themselves. Several studies have demonstrated the presence of metals causing eye allergy in coloured eyeshadows like Co, Cr, and Ni, and Pb above 10 μg g−1 in some lip products.54,55 Hence, it is obvious that there is a critical need for harmonized international regulations to control the amount of metals in cosmetic products. In addition to monitoring hazardous metals, the cosmetic industries must also keep an eye on other substances used as ingredients in the manufacturing of cosmetics, such as Fe, Zn, Ti, and Ba, which may have a dual effect.56| Element | FDAa (US) | BVLb (Germany) | FDA-CRc (Canada) | SCCS ECd (Europe) | ||||
|---|---|---|---|---|---|---|---|---|
| Limit (μg g−1) | Cosmetic type | Limit | Cosmetic type | Limit (μg g−1) | Cosmetic type | Limit | Cosmetic type | |
| a Food and drug administration (FDA). b Country's federal office of consumer protection and food safety (BVL). c Food and drug act (FDA) and Cosmetic regulations (CR). d Scientific committee consumer safety of the European Commission (SCCS EC). e For make-up powder, rouge, eye shadow, eyeliner, kajal as well as theatre, fan or carnival make-up: 5 μg g−1. f For theatre, fan or carnival make-up: 2.5 μg g−1. g Soluble Ni according to the DIN EN 71. | ||||||||
| Hg | 65 | Eye products | 0.1 | All | 1 | All | 1 | All |
| 1 | Other cosmetics and colour additives | 0.1 | Toothpaste | |||||
| Pb | 10 | Externally applied cosmetics | 2e | All | 10 | All | 20 | All |
| 20 | Colour additives | 0.5 | Toothpaste | |||||
| As | 3 | Colour additives | 0.5f | All | 3 | All | 5 | All |
| 0.5 | Toothpaste | |||||||
| Cr | 50 | Colour additive (FD&CBlue No.1) | — | — | 100 (Cr III) | All | ||
| 25 (Cr VI) | All | |||||||
| Cd | — | 0.1 | All | 3 | All | 5 | All | |
| 0.1 | Toothpaste | |||||||
| Sb | — | 0.5 | All | 5 | All | 100 | All | |
| 0.5 | Toothpaste | |||||||
| Ni | — | 10g | All | — | 200 | All | ||
| Co | — | — | — | 70 | All | |||
Elemental analysis of cosmetics is not trivial because of the wide range of elements and their respective concentrations from μg g−1 to mg g−1; of a wide variety of presentations, i.e., lipsticks, eyeshadows, blushes, lotions, mascaras, foundations, body powders, creams, face paints; and of the complex matrices present such as oils, creams, and powders. For instance, lipsticks are composed of many ingredients, including waxes, oils, dyes and pigments, including refractory minerals, such as alumina, silica, titanium dioxide and mica,57 which may hamper chemical analyses. Over the years, various analytical methods have been developed and proposed for cosmetic analysis. Most of them are based on the use of flame atomic absorption spectrometry (FAAS),58 electrothermal atomic absorption spectrometry (ETAAS),58 inductively coupled plasma optical emission spectroscopy (ICP-OES)59 and inductively coupled plasma mass spectrometry (ICP-MS).58 The need of sample solubilization for these techniques remains problematic for cosmetics. Usually, a single digestion step using a mixture of concentrated inorganic acids (i.e., HNO3, HCl, H2SO4 and HClO4) and an oxidant agent, such as H2O2 is required.60 In 2009, a study performed by the Food and Drug Administration (FDA)-US demonstrated the necessity of using a microwave-assisted digestion procedure for lipstick samples based on a combination of HNO3 and HF followed by an addition of boric acid to ensure the solubility of any formed fluorides. Even with aggressive sample treatments, clear digests were not obtained, and portion size effects were observed.61 This is unfortunately almost systematically observed in the case of cosmetics. Their digestion is described as a critical step in the International Standard ISO 21392, which focuses on the measurement of heavy metals traces in cosmetics finished products using ICP-MS technique.
XRF spectrometry has been evaluated as a multi-elemental screening tool for cosmetic product analysis, but revealed poor sensitivity in determining some relevant toxic metals, and suffered from the presence of matrix effects which vary with the type of cosmetic products considered.62 Some recent applications using hand-held XRF instrumentation were published on the analysis of kohl products63 and skin creams.8 The lack of international guidelines for validating analytical procedures in elemental determination in cosmetics, and the lack of dedicated reference materials to verify the quality of results present a crucial analytical problem for cosmetic manufacturers, legislators, and policymakers. Indeed, the external calibration procedure is hampered by the lack of reference materials with a matrix similar to the one of the samples considered. When using not-matching reference materials for building external calibration curves, some influence coefficients are determined in a purely empirical way. Unfortunately, those are hardly verifiable and consistent with the physic processes undergoing into the sample system they represent, in particular with respect to the fundamental parameters. Alternatively, purely theoretical calibrations are determined, based on the fundamental parameters of pure elemental solutions, which consequently are not considering the sample characteristics under study. In both cases, the uncertainty budget linked to these external calibrations may be extremely important, and the results disputable.
A few commercial reference materials are available but mostly only certify the concentration of a single element in a specific cosmetic product, e.g., CBW09302 cream cosmetic with a Pb concentration of 14.6 ± 0.9 mg kg−1. To increase confidence in the quality of results, participating in proficiency tests organized by global producers of reference materials, such as the Laboratory of the Government Chemist (LGC Ltd, UK), may be useful to at least get a feedback on the methodology employed.
3 Total reflection X-ray fluorescence spectroscopy: experimental background
3.1 Elemental quantification and figures of merit
Quantification in TXRF analysis relies on the so-called “thin film” approximation, which requires that the sample, which is deposited onto a reflective carrier behaves as an “infinitely thin layer”, so that any matrix effect may be considered as negligeable. Then, a direct relationship between the XRF emission peak associated to a specific element visible in the spectrum, and its concentration, may be established. To obtain the best signal to noise ratio, and thus a proper enhancement of the elemental XRF emissions, two specific components must be optimised from the sample side: the reflector or sample carrier, and the sample itself.Reflectors are typically 30 mm diameter and 3 mm thick disks made of materials like quartz glass, glassy carbon, sapphire, or acrylic glass, and which are chemically pure, smooth, and flat, and free of impurities. The carriers have to reflect X-rays efficiently, present low background levels, and be chemically inert and stable during analysis. The carrier surface needs to be cleaned prior to use to avoid memory effects, and cleaning procedures vary based on the carrier material and previously analysed samples. Cleaning can take up to 3–4 hours and involves precleaning with acetone, followed by several steps with cleaning solutions and diluted acid nitric. Usually a set of reflectors is cleaned and preserved for future use to save time. Improper handling, storage, or contamination may alter carrier properties, affecting analysis results.
The quantification approach making use of the internal calibration standard (ICS) is, and by far, the most used one as it is the simplest one. It relies on a mono-element solution of known concentration, added during the sample preparation stage to the sample to be analyzed to act as ICS. It also requires that the sample under analysis complies with TXRF requirements, i.e., a controlled mass deposition, no matrix effect, homogeneous sample, and a homogeneous distribution of the ICS and of the various elements onto the reflector' surface. The choice of the ICS is crucial, as it should be absent from the sample analytes, its concentration should lay in the average concentration range of all analytes present in the sample, its XRF emission lines should be free of any type of spectral interference, and its atomic number (Z) should frame the other elements present. Rare elements like Ga and Y are commonly used as ICS when Mo- and W-anode X-ray tubes are employed as source of X-ray excitation energy, for instance. However, their choice as ICS should not be systematic, but in line with the required properties as listed above.
Knowing the instrumental sensitivity Si for the analyte i and SICS for the ICS element, the concentration CICS of the ICS, the unknown concentration Ci of the respective analyte in the sample can be determined from the retrieved net analyte intensities Ni and NICS based on eqn (1), with an associated uncertainty αCi calculated as eqn (2).
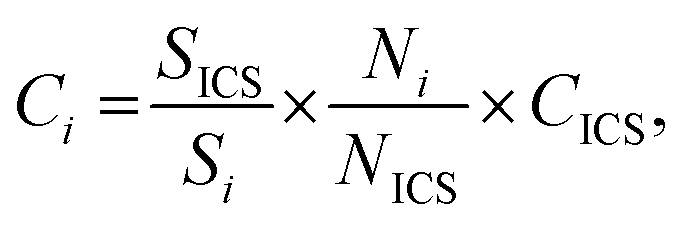 | (1) |
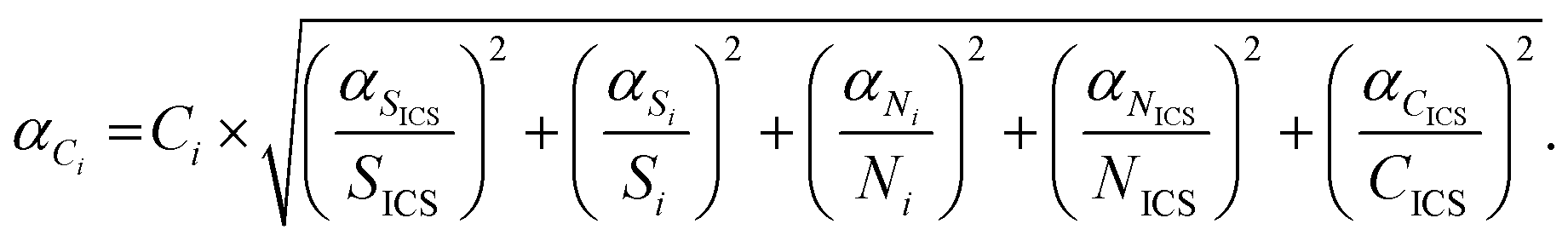 | (2) |
On a first approximation basis: αSICS and αSi are the uncertainties related to the determination of the slope value; 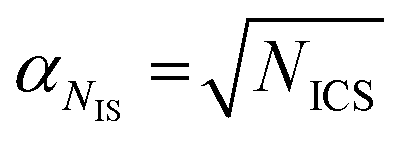 and
and 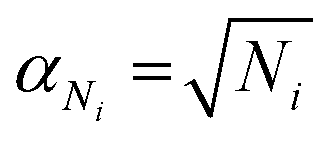 , are the uncertainties linked to the net intensity for the ICS and the analyte, respectively, and αCICS is the uncertainty of the ICS concentration which can be roughly considered as a systematic uncertainty resulting from the ICS preparation procedure. The uncertainty αNi is a significant part of the total uncertainty (eqn (2)) for elements with a low content in the sample.
, are the uncertainties linked to the net intensity for the ICS and the analyte, respectively, and αCICS is the uncertainty of the ICS concentration which can be roughly considered as a systematic uncertainty resulting from the ICS preparation procedure. The uncertainty αNi is a significant part of the total uncertainty (eqn (2)) for elements with a low content in the sample.
The most employed figures of merit in TXRF analysis are the Limit of Detection (LOD) and the Limit of Quantification (LOQ), which aim at describing the performance characteristics of the technique in a specific analytical process. Although their meaning is clear, a wide variety of definitions may be found across analytical techniques and application fields, and consequently various calculation procedures are applied to their determination. This lack of clarity has created and still creates confusion and lack of consistency for the scientific community, as well as for the regulators and industries. For these reasons, the LODs and LOQs values reported in the following sections cannot be compared easily, and the readers are encouraged to check in the cited literature or previous related works how the authors have considered and determined those parameters, as these procedures are unfortunately seldomly reported.
3.2 Sample preparation and handling procedures
TXRF analysis requires a small amount of sample (1–10 μL of liquids or 1–200 μg of solids) to be dropped onto a reflector and dried, while ensuring that the entire sample is contained within the illuminated area by the X-ray beam. Sample mass deposition must be adjusted based on the sample matrix, the analytes present and the X-ray excitation energy employed. Sample homogeneity must also be ensured, both in terms of surface and thickness, and in terms of elemental distributions, especially for the ICS.Many procedures proved to be suitable for the analysis of solid and liquid samples by TXRF.64 Various strategies have been used for screening and quantitative analysis of foodstuffs, pharmaceuticals and cosmetics samples, and the selection depends on the purpose of the analysis, the sample availability, and the matrix complexity. An overview is presented in Fig. 1. An internal calibration standard solution is added for quantification purposes, and a few microliters of the mixed solution are deposited on a suitable carrier for analysis. Blanks, duplicates, and replicates should be prepared and analysed at each step of the sample handling and preparation procedure.
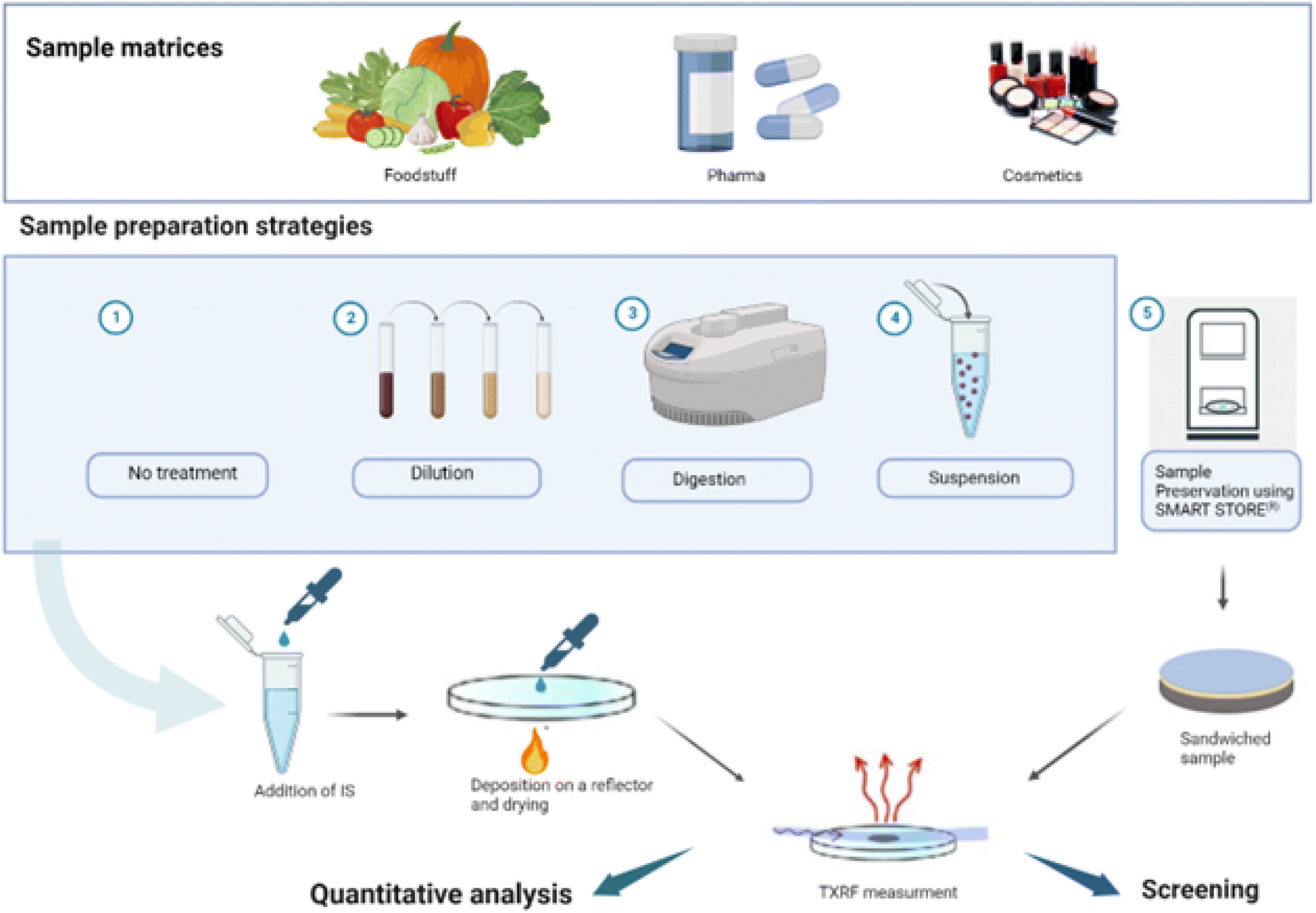 | ||
| Fig. 1 Overview of sample preparation methods for TXRF analysis of foodstuff, pharmaceuticals, and cosmetics samples (created with http://BioRender.com). | ||
Direct sample analysis is simple, efficient, and mostly preferred for liquid samples, either as-is or following dilution. Ultrapure water is usually employed as diluting agent to avoid contamination, and the dilution factor is kept in the range of 1–30, depending on the complexity of the sample and the analyte concentration. However, it should be avoided with samples containing a complex organic matrix, where sample solubilisation is recommended to improve accuracy and detection limits. Solid samples are typically decomposed by dry or wet digestion before analysis, as digestion produces in general a homogeneous liquid mixture. Indeed, solid particles are not present in the solution and in the deposited sample, which ensures a more homogeneous residue deposition of the sample on the reflector. Most of the digestion procedures are similar to those used in elemental analysis by other atomic spectrometric methods (mainly AAS- and ICP-based). The choice of the best decomposition procedure for a specific solid sample should be determined based on the matrix and analyte under study, and the result verified. For instance, biological samples are relatively easy to decompose via a mixture of nitric acid and hydrogen peroxide, whereas for some plant samples, the addition of hydrofluoric acid is mandatory to obtain a complete digestion due to their high silicon content (up to 10%).65 The use of H2SO4 is not recommended due to the high temperature needed to dry the digested sample deposited on the reflector. Similarly, the use of HF should be avoided as it might damage the quartz reflector surface. To digest samples, open systems (working at atmospheric pressure) have been employed using conventional sources of heating such as sand baths, heating plates, etc. Microwave ovens are often used for sample digestion, as they can shorten the dissolution time and the related procedure may minimize analyte losses and cross-contamination. Block heaters for digestion of small amounts of vegetation samples (several mg) have also been successfully employed.66
Another approach for analysing solid samples is to prepare suspensions mixing milligrams of powdered sample (10–50 mg) in an adequate disperser and depositing few μL (5–10 μL) of the resulting suspension on a reflective carrier. The amount of the analysis sample onto the carrier is highly dependent on the sample itself and its matrix, as well as of the spectrometer employed (beam size, excitation energy, flux density, etc.), thus there is not one only volume or mass range which may be appropriate, but several of them, and as mentioned already depending on the sample type. Therefore, it is important to work in a volume/mass range adequate to obtain a thin layer on the reflector. If the deposited sample it is too thick, we will suffer from matrix effects, commonly present in EDXRF and WDXRF spectrometry.4
In the case of preparing the solid sample as a suspension, another interesting point is to ensure the homogeneity of the original sample and to select the appropriate amount and type of solvent and dispersing agents. The particle size of the powder material is another parameter relevant to achieve a homogeneous suspension, and reference materials usually present a particle size below 75 μm which is still too big for a TXRF analysis. Cryogenic grinders and ball mixer mills are used to obtain a good suspension homogeneity in the case of real food, pharmaceutical and cosmetic samples. While suspension offers the advantage of reduced cost and the elimination of hazardous chemicals, it may be challenging to optimise and to ensure sample homogeneity, and it may also limit the determination of analytes present in low concentration levels.
Despite digestion and suspension being the most used sample preparation strategies for TXRF analysis of solid samples, additional research is being carried out to develop faster, simpler, and more environmentally friendly sample treatment approaches, especially if elemental screening is the analytical goal. Among those, a novel device called Smart Store® has been developed, which encloses the sample between two thin plastic films, then cuts out a 30 mm diameter disc to be stuck to a reflector and inserted into a TXRF system.67 Promising results have been obtained using this approach for the rapid screening of heavy metals content in cosmetics68 revealing a high potential of this device for fast and easy preparation of many types of samples to be analysed with a TXRF spectrometer.
4 Research applications of TXRF
4.1 Foodstuffs
The elemental concentration range and sensitivity of TXRF make it suitable for the analysis of food, where it can simultaneously determine trace levels of contaminants, macro, and micronutrients. Indeed, TXRF has been applied in the analysis of a wide range of liquids, like drinks and beverages (see Table 4), and solids like vegetables, fruits, dietary supplements, animal tissues and cereals (see Table 5).13| (a) Dilution | ||||||||
|---|---|---|---|---|---|---|---|---|
| Sample type | Sample amount (g) | Solvent | Dilution factor (g![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :mL) :mL) |
Elements | ICS | LOD (mg L−1) | Validationb | Ref. |
| a n. a – not available, MW: microwave. b Validation parameters: linearity (L), precision (P), accuracy (A), recovery (R). | ||||||||
| Honey | 0.3–5 | Ultrapure water | 3:100–5:2 |
Multielemental: P, S, Cl, K, Ca, Cr, Mn, Fe, Ni, Cu, Zn, As, Pb, Br, Rb | Ga, Y | 0.31–20.3 | — | 120–123 |
| Soy sauce | 1 | Ultrapure water | 1:5 |
Multielemental: P, S, Cl, K, Ca, Ti, Cr, Mn, Fe, Co, Ni, Cu, As, Br, Pb, Ba, Rb, Sr | Zn | 0.1–10 | R, P | 105 |
| Pineapple juice | n.a.a | Ultrapure water | 1:4 |
Mn | Se | n.a.a | — | 124 |
| Wine | 1 | Ultrapure water | 1:1 |
Multielemental: K, Ca, Mn, Fe, Ni, Cu, Zn, Rb, Sr, Pb | Ga | 0.01–0.2 (Mo-TXRF) 0.2-5 (W-TXRF) | L, P, R | 79 |
| (b) Digestion | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Sample type | Sample amount | Digestion | Elements | ICS | LOD (μg L−1) | Validationb | Ref. | ||
| Acid mixture | Energy source | Time | |||||||
| Bovine milk | 2 mL | HNO3 | Muffle furnace | 20 min | Multielemental: P, S, K, Ca, Ti, Cr, Mn, Fe, Ni, Cu, Zn, Se, Rb, Sr | Ga | 0.08–34 | A, P | 98 |
| Honey | 1 g | HNO3 + H2O2 | MW furnace | 15 min | Multielemental: Ti, Cr, Mn, Cu, Zn, As, Se, Br, Rb, Sr, Pb | Ga | n.a.a | — | 99 |
| 5 mL | Ashing + HCl | Muffle furnace | 20 min | Multielemental: K, Ca, Ti, Cr, Mn, Fe, Ni, Cu, Zn, Se, Br, S | Ga | n.a.a | A | 100 | |
| 6 mL | HCl | Muffle furnace | 20 min | Multielemental: Ti, K, Ca, Fe, Mn, Zn, Cr, Ni, Cu, Se, Br, Sr | Ga | n.a.a | A | 101 | |
| Soft drinks | 1 mL | HNO3 | MW furnace | 15 min | Multielemental: P, S, K, Ca, Fe, Cu, Zn, As, Pb, Br, Sr | Ti | n.a.a | — | 102 |
| 0.8–1 mL | HNO3 + H2O2 | MW furnace | 15 min | Multielemental: P, S, K, Ca, Fe, Cu, Zn, As, Pb, Br, Sr | Ti | 2–350 | — | 103 | |
| Block heater | 60 min | 8–1100 | |||||||
| Wine | 10 mL | HNO3 | MW furnace | 50 min | Multielemental: Al, Cd, Mn, Cu, Fe, Cr, Ni, Pb, Zn | Mo | 10 | — | 104 |
| Wine | 0.7 mL | HNO3 + H2O2 | Hot surface | Multielemental: P, S, K, Ca | Ga | 60–780 | P | 77 | |
| Soy sauce | 1 g | HNO3 + H2O2 | MW furnace | 60 min | Multielemental: P, S, Cl, K, Ca, Ti, Cr, Mn, Fe, Co, Ni, Cu, As, Br, Pb, Ba, Rb, Sr | Ga | 0.1–10 | R, P | 105 |
| Sugar-cane spirits | 10 mL | Ashing HNO3 | MW furnace | n.a.a | Fe, Cu, Zn | Ga | 8–10.9 | — | 106 |
| (a) Digestion | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Sample type | Sample amount (g) | Digestion | Elements | Internal standard | LOD (mg kg−1) | Validationb | Ref. | ||
| Acid mixture | Energy source | Time | |||||||
| a n.a. – not available, PVA: polyvinyl alcohol, MW: microwave, TM: thermo block. b Validation parameters: linearity (L), precision (P), accuracy (A), recovery (R), limit of quantification (LOQ). | |||||||||
| Eggs | 0.5–3 | HNO3 + H2O2 | TM heater | n.a.a | Multielemental: P, S, K, Ca, Cr, Cu, Fe, Ni, Mn, Pb, Se, Zn | Ga | n.a.a | — | 108 |
| 0.5–1.5 | HNO3 + HClO4 + H2O2 | TM heater | 90 min | Multielemental: Ba, Cu, Fe, Mn, Ni, Pb, Rb, Se, Sr, V, Zn | Ga | n.a.a | A, P | 109 | |
| 1.46–1.61 | HNO3 + H2O2 | TM heater | 90 min | Multielemental: Ba, Cu, Fe, Mn, Ni, Pb, Rb, Se, Sr, V, Zn | Ga | n.a.a | A, P | 109 | |
| Rice grain | 0.5 | HNO3 | MW furnace | 10 min | Se | Ga | 0.05 | R | 125 |
| 0.3 | HNO3 | MW furnace | 15 min | Multielemental: K, Ca, Ti, Cr, Mn, Fe, Ni, Cu, Zn, Ba, Rb, Pb, Br, Sr, Ba | Ga | n.a.a | — | 126 | |
| 1 | HCl:HNO3 (1:3) |
TM heater | 2 h | Multielemental: As, Ca, K, Mn, P, Rb, S, Se, Zn | Co | n.a.a | R | 127 | |
| 0.5 | HNO3 + H2O2 | MW furnace | 15 min | Multielemental: K, Ca, Mn, Fe, Cu, Zn, Br, Rb, Sr | Ga | 0.2–99 | L, P, A | 66 | |
| 0.02 | HCl/HNO3, (3:1) |
TM heater | 2 h | ||||||
| Beans | 0.15 | HNO3 + H2O2 | MW furnace | 25 min | Multielemental: K, Ca, Mn, Fe, Ni, Cu, Zn, Rb, Sr, Pb | Ga | n.a.a | A, P | 87 |
| Onions | 0.2 | HNO3 + H2O2 | Hot plate | n.a.a | As | Co | n.a.a | A, P | 128 |
| Porridge flour | 1 | HNO3 | TM heater | n.a.a | Multielemental: Mn, Fe, Cu, Zn | Ga | n.a.a | A, P | 129 |
| Maize flour | 0.35 | HNO3 | TM heater | n.a.a | Multielemental: Mn, Fe, Cu, Zn | Ga | n.a.a | — | 130 |
| Candies | 0.5 | HNO3 + HCl + H2O2 | MW furnace | n.a.a | Multielemental: Ca, Ti, Cr, Mn, Fe, Cu, Zn, Br, Rb, Sr, Pb | Ga | 0.02–0.1 | A, P | 107 |
| Chocolate | 0.4 | HNO3 + H2O2 | MW furnace | 25 min | Multielemental: P, S, K, Ca, Ti, Cr, Mn, Fe, Ni, Cu, Zn, Br, Rb, Sr, Ba | Ga | n.a.a | — | 112 |
| Pork meat | 0.3 | HNO3 | MW furnace | n.a.a | Multielemental: P, S, Cl, K, Ca, Ti, Mn, Fe, Ni, Cu, Zn, Se, Br, Rb, Sr, Ba, Pb | Ga | 0.01–1 | A, P | 131 |
| Edible Clams | 0.5 | HNO3 + H2O2 | MW furnace | 15 min | Multielemental: Ti, Mn, Cu, Zn, As, Br, Sr | Y | n.a.a | A | 92 |
| (b) Suspension | ||||||||
|---|---|---|---|---|---|---|---|---|
| Sample type | Sample amount (mg) | Disperser agent | Disperser (mL) | Elements | Internal standard | LOD (mg kg−1) | Validationb | Ref. |
| Beans root | 10 | Triton X 100 (1% wt) | 0.99 | Multielemental: K, Ca, Mn, Fe, Ni, Cu, Zn, Rb, Sr, Pb | Ga | n.a.a | A, P | 87 |
| Microgreens | 100 | Triton X-100 | 5 | Multielemental: P, S, K, Ca, Fe, Zn, Mn, Cu, Ni | Ga | n.a.a | A | 88 |
| Multielemental: P, S, K, Ca, Cl, Mn, Fe, Ni, Cu, Zn, Br, Rb, Sr | 0.2–30 | A, P, LOQ | 91 | |||||
| Vegetables | 20 | Ultrapure water | 1 | Multielemental: K, Ca, Mn, Fe, Cu, Zn, Rb, Sr | Ga | 2–100 | L, A(R), P | 66,89 |
| Edible Clams | 100 | Ultrapure water | 1 | Multielemental: Ti, Mn, Cu, Zn, As, Br, Sr | Y | n.a.a | A | 92 |
| Wheat flour | 50 | Triton X-100 (1% wt) | 2.5 | Multielemental: P, S, Cl, K, Ca, Mn, Fe, Ni, Cu, Zn, Se, Br, Rb, Sr | Y | 0.13–28 | A(R) | 132 |
| 1 | Multielemental: Cu, Fe, Mn, Zn | Ga | 0.27–0.44 | A(R) | 133 | |||
| Meat | 100 | Triton X-100, (0.1% wt) + PVA | 5 | Multielemental: K, Ca, Fe, Cu, Zn | Ga | 0.08–1.67 | — | 93 |
| 200 | Ultrapure water + Triton X-100 (0.1% wt) + PVA (0.2%) | 5 | Multielemental: P, Ca, Fe | Ga | n.a.a | — | 90 | |
| Cheese | 200 | Ultrapure water + Triton X-100 (0.1% wt) + PVA (0.2%) | 5 | Multielemental: P, S, Cl, K, Ca, Fe, Zn | Ga | n.a.a | — | 90 |
| Raw meat, salami, sausages, fish, mussels | 50 | Triton X-100 (0.5% wt) | 4 | Multielemental: P, S, Cl, K, Ca, Cr, Mn, Fe, Ni, Cu, Zn, Br, Rb, Sr | Ga, V | n.a.a | A(R) | 94 |
A direct analysis is frequently performed for the multi-element analysis of beers,69 distilled alcoholic beverages,70 food contact simulant,71 infusions,72–75 milk,76 wine77–84 and whisky.85 The recommended volume of liquids deposited on a sample carrier ranges from 5 μL to 20 μL, depending on the sample type, in order to avoid matrix effects.86 In some cases, to reduce the mass deposition and overcome matrix effect phenomena, a minimal treatment as a sample dilution step is necessary. For example, direct quantitative analysis of honey is quite problematic due to its very high concentration of sugars and high viscosity, making it also very difficult to dry and obtain a flat solid residue. In the case of wine analysis, it was demonstrated that the analytical quality of TXRF results can be improved if measurements are performed after sample dilution, especially for light Z elements like K and Ca.79 In Table 4(a) summary of relevant publications about liquid foodstuff samples measured by TXRF after dilution is provided.
TXRF analysis of suspended solid residues (see Table 5(b)) offers a fast and straightforward way to perform screening and quantitative analysis for food safety, traceability, and quality control purposes. Non-ionic surfactants such as Triton X-100 (ref. 87 and 88) and ultrapure water66,89 are commonly used as dispersing agents. Polyvinyl alcohol can also be added to improve sample deposition on the reflective carrier.90 Before deposition, the suspension mixture should be homogenized using ultrasonic baths or vortex shakers.66,87,91,92 The use of polyvinyl alcohol was demonstrated to be effective in improving sample deposition for TXRF analysis of meat and cheese.90 A novel method aiming to distinguish between mechanically separated meat (MSM) and fresh meat by monitoring K, Ca and Fe and using suspension for sample preparation and TXRF analysis is reported.93,94 The proposed procedure represents a powerful tool in food routine controls for consumer protection. Likewise, suspension proved to be a suitable analytical approach to check easily the presence of additives as polyphosphates (P), verify the content of salt (Cl) and detect some accidental metal contaminations (Cr, Co, Ni). In addition to the advantage of being a minimum sample treatment, suspension preparation can be of particular interest for the analysis of mass-limited samples (e.g., soybean roots) since the amount of material needed to prepare the suspension (10–200 mg) is much lower than the one required for digestion.95 Despite the number of benefits that there is in preparing suspension for the TXRF analysis of solid materials, only 15% of the sample treatment methods use it64 due to calibration issues, weighing errors, and sample/suspension inhomogeneity that need to be properly addressed.96,97 In addition, the results for lighter elements, mainly K and Ca, are not as good as for mid-high Z elements in some food matrices because of their complicated matrix and of their effects, among other limiting parameters. In these situations, external calibration is performed to ameliorate the results utilizing a set of reference materials with a matrix resembling that of the actual samples.66
Decomposition of the liquid to be deposited onto the reflector is also a common sample preparation method to improve the limits of detection. This pre-treatment is reported for the TXRF analysis of milk,98 honey,99–101 soft drinks,102,103 wine,104 soy sauce105 and sugar-cane spirits.106 As shown in Table 4(b), sample decomposition is usually performed by wet ashing procedures involving the use of concentrated acids (HNO3, HCl) or some mixtures of acids in combination with oxidizing agents (HNO3 + H2O2). To overcome problems of contamination or volatilization during the digestion process, microwave ovens are preferred as heating sources. Dry ashing by incineration of the liquid samples at 500 °C and dissolution of the resulting ashes in HNO3 or HCl has been used as a digestion procedure for the determination of Fe, Cu and Zn in sugar cane spirits106 or multi-elemental analysis of honey.100 For the complete dissolution of food solid samples, a mass of about 0.1 g to 1 g is mixed with acids and other oxidating compounds. Often, concentrated HNO3 or a mixture of HNO3 + H2O2 are employed but in some cases the additional use of HCl or HClO4 has been reported to get the total solubilisation of vegetables,66 candies107 or eggs.108,109 Microwave ovens and thermo block heaters are two types of heating sources employed during digestion. The main advantage of those systems is the possibility to digest a low quantity of sample, therefore using reduced acid volumes, as it was done for a very small amount of vegetables (20 mg) and only 1 mL of aqua regia (HCl:HNO3, 3:1).66 Compared to liquid samples, digestion of solid foodstuffs requires a longer time due to the complexity of sample matrices.
Microextraction and selective adsorption procedures can also be considered to enhance TXRF microanalytical capability. For instance in the case of Se and Hg determination of low level (0.6 g L−1) in seafood samples110 and in the determination of Sb in milk after nanoparticles immobilization reflectors,111 respectively.
Promising results have been obtained using the Smart Store® system for the rapid screening of chocolate samples. In this case, chocolate was first dissolved in 3 mL of Ga solution in bain-marie conditions. Then, the total dissolved sample was placed on a polymeric foil and dried at room temperature. The plasticized sample was put onto a quartz sample carrier with a diameter of 30 mm to be measured with the TXRF spectrometer.112 Though underestimation of the low Z elements occurred, the procedure demonstrated to be a fast and easy approach for screening.
4.2 Pharmaceuticals, drugs, and supplements
At present, there is a lack of general procedures in the pharmacopoeias for a reliable application of TXRF method in pharmaceutical analysis. Over the past few years, many studies have investigated the potential of TXRF for determining elemental impurities in pharmaceutical products and dietary supplements (DS). Table 6 summarizes the research published on this topic between 2000–2020, in which TXRF has been applied to analyse various dosage forms of pharmaceutical products, including APIs, drug substances, and metal-containing drugs. However, only finished dosage forms of DS have been considered. Solid samples can be dissolved by organic solvent-based diluents, such as DMF, DMSO, and methanol33,113–115 or suspended using an aqueous non-ionic surfactant solution.116 More intricate sample treatments, such as microwave digestion, have also been used, primarily for the analysis of more complex matrices.38 One critical factor for determining elemental impurities in pharmaceutical samples with a commercial TXRF spectrometer is the instrumental setup and, in particular, the X-ray anode source determining the excitation energy. This aspect is particularly important for platinum group metals (PGMs), which are commonly used in medicinal chemistry. The determination of Pd and other PGMs has been the focus of many studies. Better results were obtained using X-ray sources equipped with W and Ag anodes that allow elemental determination through the Pd–K emission lines, free of interferences,114 while with Mo X-ray sources spectral interferences overlapping with Pd–L emission lines occur.113 However, Mo X-ray sources provides better sensitivity for mid-Z elements (i.e., Fe, Ni, Zn, Cr, Cu, etc.).33 Therefore, a trade-off must be made between PGMs analysis versus sensitivity for most other elements of interest, and a thorough analysis may require the use of TXRF systems equipped with both Mo and W X-ray tubes. The quantification limits reported for TXRF applications in the field of pharmaceutical products and dietary supplement analysis are suitable for controlling class 2 and class 3 impurities (see Table 1). Yet, the sensitivity of most of the published methods is not adequate to monitor the concentration limits for elements included in class 1 (limit range: 0.5–3 μg g−1).| (A) Pharmaceutical products | ||||||||
|---|---|---|---|---|---|---|---|---|
| Samples | Analytes | Sample preparation details | Internal standard | TXRF measurement | LOQ (μg g−1) | Purpose of the method | Validationb | Ref. |
| a API: active pharmaceutical ingredient, MW: microwave, THF: tetrahydrofurane, DMSO: dimethylsulfoxide, DMF: dimethylformamide, PVA: polyvinyl alcohol. b Validation parameters: specificity (S), linearity (L), precision (P), accuracy (A), limit of detection (D). c Cisplatin, auranofin. d Liquid form (0.2 ppm). e Ca-folinate, Levodopa, Enalapril maleate. f Triprolidine HCl, diphenhydramine HCl, Chlorpheniramine maleate, pseudoephedrine HCl, ephedrine sulfate, scopolamine HBr. g Komplivit (DS with capsule), Multifort (DS without capsule). h Fat burner, laxative, tejocote root, omega oil 6. i In the case of Hg (Ga solution contain EDTA to avoid Hg loses during drying). | ||||||||
| API (solid) | K, Ca, Fe, Ni, Sr, Pd | 10 mg dissolution (THF, DMSO), 10–20 μL deposition | Ga | Mo-TXRF (1000 s) | 10–20 | Quantitative | S, L, P, A, D | 49 |
| W-TXRF (2000s) | ||||||||
| API (solid) | PGMs metals (Rh, Pd, Ir, Pt) | 150–500 mg dissolution (water, methanol), 10 μL deposition | Y | W-TXRF (1000 s) | 1.9–4.1 | Screening | S, P, A | 113 |
| 500 mg MW digestion (HNO3 +HCl), 10 μL deposition | Y | W-TXRF (1000 s) | 1.7–8.7 | Quantitative | ||||
| Metal containing drugsc | Au, Pt | Dissolution (DMF + water + PVA), 4–10 μL deposition | Mn | Mo-TXRF (250 s) | 0.2d | Quantitative | P, A | 114 |
| Drug Substancese | Ca, Fe, Zn, Sr | Dissolution (0.2–0.3 M HNO3) 1% sample solution, 10 μL deposition | Se | Mo-TXRF (600 s) | — | Screening | — | 115 |
| Drug Substancesf | Pd, Rh, Pt, Cr, Fe, Cu | Dissolution (50% methanol + 2% HNO3 + 0.5% HCL) 5 mg/1 mL, 10 μL deposition | Sr | Mo-TXRF (1000 s) | <2–10 | Screening platform | L, P, A, D | 134 |
| W-TXRF (2000s) | ||||||||
| Pd, Rh, Pt, Cr, Fe, Cu | Dissolution (50% methanol + 2% HNO3 + 0.5% HCL) 10 mg mL−1, 5 μL deposition | V | WL-TXRF (500 s) | 2–15 | ||||
| AgK-TXRF (500 s) | ||||||||
| (B) Dietary supplements, DS (finished dosage forms) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Samples | Analytes | Sample preparation details | Internal standard | TXRF measurement | LOQ (μg g−1) | Purpose of the method | Validationb | Ref. |
| Commercial DS (tablets)g | P, Ca, Mn, Fe, Cu, Zn, K, Cr, Se, I | DS without capsule: dissolution (water), 2 μL deposition | Ni, Ga, Y | Mo-TXRF (250 s) | — | Screening | A | 33 |
| DS with capsule: dissolution (HNO3), 2 μL deposition | Quantitative | |||||||
| Commercial DS (different dosage forms)h | Pb, As, Cr, Hg | 20–50 mg digestion (HNO3+HCl + H2O2) filtration | Gai | Mo-TXRF | 0.2–3.1 | Quantitative | L, P, A, D | 38 |
| Commercial DS and seaweed products | I | 500 mg digestion (NH3), 25 μL deposition | Cd, Ag, Y | W-TXRF (1000 s) | — | Screening | None | 135 |
| Commercial DS (tablets) | Se | 50 mg powder supension (2.5 mL 1% Triton X100) 10 μL deposition | Ga | Mo-TXRF (1000 s) | 0.3–0.6 | Quantitative | P, A | 136 |
As shown in Table 6, rare elements like Ga and Y have been widely used as ICS for determination of elemental impurities by TXRF. Nonetheless, for specific applications, other elements such as Mn, Se, Sr, V and Ni have also been reported.
4.3 Cosmetics
TXRF is a technique that has been explored less for the analysis of cosmetics, and the number of published articles on this topic is still limited. As demonstrated in Table 7, TXRF has shown great potential in analysing lipsticks, eyeshadows, foundations, liners, and creams, as well as plant-based cosmetic ingredients.117 In addition to heavy metal quantification, such as Pb,118 TXRF has also revealed its potential as a multi-elemental screening tool.60,68 It offers the opportunity to determine major (>1%), minor (0.01–1%), and trace elements (<0.01%), sometimes even without having to dilute the sample. This is particularly advantageous for cosmetic producers who need to control the quantity of elements introduced as ingredients and pigments in their final products.| Cosmetic type | Analytes | Sample preparation details | Internal standard | TXRF measurement | LOD (μg g−1) | Purpose of the method | Validationa | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Validation parameters: accuracy (A), precision (P), limit of detection (D). b LOD for low atomic number elements (K, Ca). c Lotus corniculatus, Medicago lupulina, Knautia arvensis and Plantago major. | ||||||||
| Lipstick | Pb (screening other elements) | 100–400 mg suspension (1% Triton X-100), liquefaction/solidification steps | Y | Mo-TXRF (900 s) | 0.04 | Quantitative | A, P, D | 137 |
| Lipstick | K, Ca, Ti, Mn, Fe, Cu, Zn, Rb, Sr, Ba, Sn | 10 mg suspension (Chloroform) | Mo | W-TXRF (2000s) | 2 (100)b | Screening | A, P, D | 118 |
| Eye shadow | 20 mg suspension (1% Triton X-100) | Pd | 5 (150)b | |||||
| Cream | Pb, Cd, Cr, Ni, Sb, Se | 20 mg suspension (1% Triton X-100) | Pd | 1–8 | ||||
| Eyeshadows, lipsticks, foundation, liners | K, Ca, Ti, Cr, Mn, Fe, Ni, Cu, Zn, Pb, Br, Rb, Sr | Digestion (300 mg, USEPA 305260) |
Ga | Mo-TXRF (600 s) | Pb digestion: 0.9 | Screening | — | 67 |
| Suspension (10 mg, 1% Triton X-100) | Suspension: 1.2 | |||||||
| SMART STORE®27 | SMART STORE®: 5.7 | |||||||
| Plants (cosmetic ingredients)c | Ca, Zn, Se, Fe, Cr, Pb, Ni, Cd | 20 mg powder suspension (ultrapure water)68 | Ga | Mo-TXRF (600 s) | — | Screening | — | 66 |
The most common sample preparation procedure involves suspending several mg of sample (10–400 mg) in an appropriate disperser agent followed by sonication (5–15 minutes). Since only a small amount of sample is analyzed, it is crucial to guarantee the homogeneity of the original cosmetic sample to ensure good precision and representativeness of the results. It is also necessary to obtain homogeneous suspensions to analyze, which is possible using an aqueous solution of 1% Triton X-100 in water for most cosmetic products (eye shadows, liners, foundations, and creams) or ultrapure water for plant-based cosmetic ingredients. Yet, lipstick samples require additional steps of liquefaction/solidification118 or the use of an organic solvent60 to obtain a homogeneous suspension. Unfortunately, suspension is not a suitable approach to quantify elements present at ultra-trace concentrations (<1–2 μg g−1). However, based on the limits of detection shown in Table 7, it could be useful for screening some major and minor toxic elements present in targetted cosmetic samples and checking their compliance with the recommended values (see Table 3). It is also interesting to note that, for most commercial TXRF spectrometers operating with a Mo or W excitation X-ray source, the limits of detection for light elements (e.g., K, Ca) are higher in comparison to the ones for mid-Z elements due to their low fluorescence yields and lower energies of analytical lines, which are also partly absorbed by air. Nevertheless, this is not a critical point for the analysis of cosmetics because light elements are usually present at high concentrations (1000–10000 μg g−1)60 and do not present a high relevance for the quality control. Of notice is that the sample treatment approach selected also impacts the limit of detection. A comparison among results obtained using TXRF to analyze the same lipsticks and eyeshadows samples prepared by different procedures revealed an effect on the limit of detection of metals of interest. In particular, the limit of detection for Pb found was 5.7 μg g−1, 1.2 μg g−1, and Pb: 0.9 μg g−1 when digestion, suspension, and Smart Store® sample treatment approaches were respectively used.68
As highlighted in Table 7, in all publications reporting on the analysis of cosmetics by TXRF, quantification was carried out by internal calibration standardization. Typically, Ga and Y are used as internal calibration standards in systems equipped with Mo X-ray sources. Still, other higher atomic number elements, such as Pd and Mo, are used as internal calibration standards when using W X-ray sources to avoid spectral overlaps with many elements present in cosmetic samples within the energy range of 2–18 keV. For the specific case of lipstick samples suspended in an organic solvent, a metalorganic internal calibration standard has been employed instead of the more widely used aqueous standards to ensure a proper mixing of the internal calibration standard and the suspended sample.60
5 Concluding remarks
Though the scientific community is developing and publishing valuable procedures, protocols, and methods to obtain reliable analytical results based on TXRF, and addressing both needs for quantitative analysis and screening of consumer products, no references to TXRF applications to quality control and safety in industry and quality control or regulatory bodies' laboratories are available.As outlined in this paper, TXRF is successfully used for both elemental identification and quantification, especially of impurities or unwanted contaminations. While promising results have been obtained to quantify most mid-Z and high-Z elements, additional research is required to improve the quality of the results obtained for quantifying the low-Z and volatile elements (such as Hg). Furthermore, spectral overlaps between various elemental fluorescent lines, such as Ba L lines with Ti K lines, or As K lines with Pb L lines, deserve more attention. The determination of the figures of merit of analytical methods based on TXRF are limited by the lack of fit-for-purpose and certified reference materials, though frequently some parameters, such as linearity range, accuracy, precision, reproducibility, sensitivity, LOD and LOQ, are determined using calibration samples developed in-house or reference materials designed for AAS and ICP. Unfortunately, the different approaches to calculating the detection limits prevent an objective assessment of the TXRF performance. To assess the limits of validity of the TXRF results, to claim a certain level of confidence, or to allow for comparison with techniques of reference such as ICP-OES or ICP-AES, it is fundamental to define properly a total uncertainty budget, with all its potential contributors under control. Again, this factual situation is partially due to the lack of fit-for-purpose reference materials, which would allow to probe deeper the impact of instrumental and experimental parameters on the results, especially the sample handling procedures, the spectral analysis, and to control, and potentially validate, more easily a measurement procedure for a given type of samples. The quantification of elements present in consumer products should be done along with the corresponding validation steps using suitable reference materials. In this context, the biggest challenge comes with the biggest opportunity, particularly in cosmetics applications, where regulations are still under development and product screening is an important analytical feature. However, without the development of specific reference materials to cosmetics, the efforts to create robust TXRF-based methods have limited use for quality control and applications in the cosmetic industry.
The wide acceptance of TXRF by the analytical community is thus hindered by the lack of proper standard operating procedures and fit-for-purpose reference materials, which are both needed to claim the validity of results at a certain level of confidence. Establishing the uncertainty budget is also a priority to ensure the physical and chemical traceability of results and reliability in quantification. Such awareness has initiated already pre-normative research activities tackling the above-mentioned issues.119 Those ventures are essential to establish all the physical and chemical interdependencies linked to the use of TXRF, and prior to developing any normative documentation (e.g., standard) under the ISO/TC201/SC10 framework. Only then, TXRF may be proposed as a standardized chemical method and labelled as the technique of reference, similar to AAS or ICPs techniques, for the elemental analysis of consumer products such as food, pharmaceutical, and cosmetic samples.
Author contributions
Eva Marguí: conceptualization, visualization, writing – original draft, writing – review & editing, Fabjola Bilo: visualization, writing – original draft, writing – review & editing, Laura E. Depero: writing – review & editing, Diane Eichert: visualization, writing – original draft, writing – review & editing; Ana Pejović-Milić: writing – review & editing, Jasna Jablan: visualization, writing – original draft, writing – review & editing, Armin Gross: writing – review & editing, Haegen Stosnach: writing – review & editing, Aldona Kubala-Kukuś: writing – review & editing, Dariusz Banaś: writing – review & editing, Laura Borgese: funding acquisition, visualization, writing – original draft, writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This article/publication is based upon work from COST Action CA18130 ENFORCE TXRF European network for chemical elemental analysis by total reflection X-ray fluorescence (https://www.cost.eu/actions/CA18130/), supported by COST (European Cooperation in Science and Technology, http://www.cost.eu/). Open access fees are covered by CSUC-UdG.References
- M. Maithani, R. Raturi, P. Sharma, V. Gupta and P. Bansal, Elemental impurities in pharmaceutical products adding fuel to the fire, Regul. Toxicol. Pharmacol., 2019, 108, 104435, DOI:10.1016/j.yrtph.2019.104435.
- S. Garrigues and M. de la Guardia, Challenges in Green Analytical Chemistry, Royal Society of Chemistry, Cambridge, 2nd edn, 2020, 10.1039/9781788016148.
- V. Balaram, Recent advances in the determination of elemental impurities in pharmaceuticals – Status, challenges and moving frontiers, Trends Anal. Chem., 2016, 80, 83–95, DOI:10.1016/j.trac.2016.02.001.
- R. Klockenkämper and A. von Bohlen, Total-Reflection X-Ray Fluorescence Analysis and Related Methods, John Wiley & Sons, Inc., Hoboken, New Jersey, USA, 2nd edn, 2015, DOI:10.1002/9781118985953.
- L. Borgese, F. Bilo, A. Zacco, S. Federici, A. W. Mutahi, E. Bontempi, K. Trzepla, N. Hyslop, S. Yatkin, P. Wobrauschek, J. Prost, D. Ingerle and L. E. Depero, The assessment of a method for measurements and lead quantification in air particulate matter using total reflection X-ray fluorescence spectrometers, Spectrochim. Acta, Part B, 2020, 167, 105840, DOI:10.1016/j.sab.2020.105840.
- E. Marguí, B. Zawisza and R. Sitko, Trace and ultratrace analysis of liquid samples by X-ray fluorescence spectrometry, TrAC, Trends Anal. Chem., 2014, 53, 73–83 CrossRef.
- H. Gallardo, I. Queralt, J. Tapias, L. Candela and E. Margui, Bromine and bromide content in soils: Analytical approach from total reflection X-ray fluorescence spectrometry, Chemosphere, 2016, 156, 294–301, DOI:10.1016/j.chemosphere.2016.04.136.
- G. V. Pashkova, T. S. Aisueva, A. L. Finkelshtein, T. Yu. Cherkashina and A. A. Shchetnikov, Quantitative approaches to the determination of elements in lake sediments by total reflection X-ray fluorescence, Microchem. J., 2018, 143, 264–271, DOI:10.1016/j.microc.2018.08.020.
- N. Szoboszlai, Z. Polgári, V. G. Mihucz and G. Záray, Recent trends in total reflection X-ray fluorescence spectrometry for biological applications, Anal. Chim. Acta, 2009, 633, 1–18, DOI:10.1016/j.aca.2008.11.009.
- E. Marguí, J. Jablan, I. Queralt, F. Bilo and L. Borgese, Potential of total-reflection X-ray spectrometry for multielement analysis of biological samples using dilution or suspension sample preparation techniques, X-Ray Spectrom., 2022, 51, 230–240, DOI:10.1002/xrs.3230.
- U. Majewska, P. Łyżwa, A. Kubala-Kukuś, D. Banaś, J. Wudarczyk-Moćko, I. Stabrawa and S. Góźdź, Total reflection X-ray fluorescence medical applications: Elemental analysis of human urine, Spectrochim. Acta, Part B, 2018, 147, 121–131, DOI:10.1016/j.sab.2018.05.014.
- S. Dhara and N. L. Misra, Elemental characterization of nuclear materials using total reflection X-ray fluorescence spectrometry, TrAC, Trends Anal. Chem., 2019, 116, 31–43, DOI:10.1016/j.trac.2019.04.017.
- L. Borgese, F. Bilo, R. Dalipi, E. Bontempi and L. E. Depero, Total reflection X-ray fluorescence as a tool for food screening, Spectrochim. Acta, Part B, 2015, 113, 1–15, DOI:10.1016/j.sab.2015.08.001.
- A. Teasdale, D. Elder and R. W. Nims, ICH Quality Guidelines: an Implementation Guide, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2017, DOI:10.1002/9781118971147.
- Nutrition Division, Assuring Food Safety and Quality, Guidelines for Strengthening National Food Control Systems: Food and Nutrition Paper 76, FAO, Rome, Italy, 2003, https://www.fao.org/publications/card/en/c/92f82d38-5557-4ca1-b361-be14cd129db6/, accessed September 19, 2022 Search PubMed.
- European Food Safety Authority–EFSA, European Union, https://european-union.europa.eu/institutions-law-budget/institutions-and-bodies/search-all-eu-institutions-and-bodies/european-food-safety-authority-efsa_en, accessed February 11, 2024.
- Commission Regulation (EC) No 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs (Text with EEA relevance),https://Webarchive.Nationalarchives.Gov.Uk/Eu-Exit/Https://Eur-Lex.Europa.Eu/Legal-Content/EN/TXT/?Uri=CELEX:02006R1881-20201014. https://www.legislation.gov.uk/eur/2006/1881, accessed September 19, 2022.
- M. de la Guardia and S. Garrigues, Handbook of Mineral Elements in Food: de la Guardia/Handbook of Mineral Elements in Food, John Wiley & Sons, Ltd, Chichester, UK, 2015, DOI:10.1002/9781118654316.
- A. Howe, L. H. Fung, G. Lalor, R. Rattray and M. Vutchkov, Elemental composition of Jamaican foods 1: a survey of five food crop categories, Environ. Geochem. Health, 2005, 27, 19–30, DOI:10.1007/s10653-004-5671-7.
- Canadian Nutrient File (CNF), 2015, https://www.canada.ca/en/health-canada/services/food-nutrition/healthy-eating/nutrient-data/canadian-nutrient-file-2015-download-files.html, accessed September 22, 2022.
- B. R. Singh, S. K. Gupta, H. Azaizeh, S. Shilev, D. Sudre, W. Y. Song, E. Martinoia and M. Mench, Safety of food crops on land contaminated with trace elements, J. Sci. Food Agric., 2011, 91, 1349–1366, DOI:10.1002/jsfa.4355.
- T. Tongesayi, P. Fedick, L. Lechner, C. Brock, A. Le Beau and C. Bray, Daily bioaccessible levels of selected essential but toxic heavy metals from the consumption of non-dietary food sources, Food Chem. Toxicol., 2013, 62, 142–147, DOI:10.1016/j.fct.2013.08.052.
- I. De La Calle, M. Costas, N. Cabaleiro, I. Lavilla and C. Bendicho, Fast method for multielemental analysis of plants and discrimination according to the anatomical part by total reflection X-ray fluorescence spectrometry, Food Chem., 2013, 138, 234–241, DOI:10.1016/j.foodchem.2012.09.105.
- A. Gonzálvez, S. Armenta, M. L. Cervera and M. de la Guardia, Elemental composition of seasoning products, Talanta, 2008, 74, 1085–1095, DOI:10.1016/j.talanta.2007.09.039.
- M. Puschenreiter, O. Horak, W. Friesl and W. Hartl, Low-cost agricultural measures to reduce heavy metal transfer into the food chain – a review, Plant, Soil Environ., 2011, 51, 1–11, DOI:10.17221/3549-PSE.
- Z. Dong, L. Lu, Z. Liu, Y. Tang and J. Wang, Migration of Toxic Metals from Ceramic Food Packaging Materials into Acid Food Simulants, Math. Probl. Eng., 2014, 2014, 1–7, DOI:10.1155/2014/759018.
- P. Hajeb, J. J. Sloth, Sh. Shakibazadeh, N. A. Mahyudin and L. Afsah-Hejri, Toxic Elements in Food: Occurrence, Binding, and Reduction Approaches, Compr. Rev. Food Sci. Food Saf., 2014, 13, 457–472, DOI:10.1111/1541-4337.12068.
- AOAC SMPR 2012.007, Standard method performance requirements for determination of heavy metals in a variety of foods and beverages, J. AOAC Int., 2013, 96, 704, DOI:10.5740/jaoac.int.2012.007.
- AOAC-Method-2015.01.pdf, https://brooksapplied.com/wp-content/uploads/download/AOAC-Method-2015.01.pdf, accessed September 19, 2022.
- L. Perring and J. Blanc, Validation of quick measurement of mineral nutrients in milk powders: comparison of energy dispersive X-ray fluorescence with inductively coupled plasma-optical emission spectroscopy and potentiometry reference methods, Sens. Instrum. Food Qual. Saf., 2008, 2, 254–261 CrossRef.
- L. Perring and J. Blanc, Faster Measurement of Minerals in Milk Powders: Comparison of a High Power Wavelength Dispersive XRF System with ICP-AES and Potentiometry Reference Methods, Food Anal. Methods, 2008, 1, 205–213, DOI:10.1007/s12161-008-9030-7.
- c232-usp-39.pdf, https://www.usp.org/sites/default/files/usp/document/our-work/chemical-medicines/key-issues/c232-usp-39.pdf, accessed September 16, 2022.
- B. J. Shaw, D. J. Semin, M. E. Rider and M. R. Beebe, Applicability of total reflection X-ray fluorescence (TXRF) as a screening platform for pharmaceutical inorganic impurity analysis, J. Pharm. Biomed. Anal., 2012, 63, 151–159 CrossRef CAS PubMed.
- J. S. Barin, P. A. Mello, M. F. Mesko, F. A. Duarte and E. M. M. Flores, Determination of elemental impurities in pharmaceutical products and related matrices by ICP-based methods: a review, Anal. Bioanal. Chem., 2016, 408, 4547–4566, DOI:10.1007/s00216-016-9471-6.
- R. Nageswara Rao and M. V. N. K. Talluri, An overview of recent applications of inductively coupled plasma-mass spectrometry (ICP-MS) in determination of inorganic impurities in drugs and pharmaceuticals, J. Pharm. Biomed. Anal., 2007, 43, 1–13, DOI:10.1016/j.jpba.2006.07.004.
- EMA, ICH Q3D Elemental impurities, European Medicines Agency, 2018, https://www.ema.europa.eu/en/ich-q3d-elemental-impurities, accessed September 18, 2022 Search PubMed.
- General Chapter Elemental Contaminants in Dietary Supplements, USP-NF, 2017, https://www.uspnf.com/notices/general-chapter-elemental-contaminants-dietary-supplements, accessed September 18, 2022.
- B. G. Beltrán, V. Ramos-Sanchez, D. Chávez-Flores, R. Rodríguez-Maese and E. Palacio, Total Reflection X-Ray Fluorescence Spectroscopy (TXRF) Method Validation: Determination of Heavy Metals in Dietary Supplements, J. Chem., 2020, 1–9, DOI:10.1155/2020/8817393.
- T. Wang, J. Wu, R. Hartman, X. Jia and R. S. Egan, A multi-element ICP-MS survey method as an alternative to the heavy metals limit test for pharmaceutical materials, J. Pharm. Biomed. Anal., 2000, 23, 867–890, DOI:10.1016/s0731-7085(00)00361-7.
- P. Raghuram, I. V. Soma Raju and J. Sriramulu, Heavy metals testing in active pharmaceutical ingredients: an alternate approach, Pharmazie, 2010, 65, 15–18 CAS.
- 233_ElementalImpuritiesProcedures.pdf, https://www.usp.org/sites/default/files/usp/document/our-work/chemical-medicines/key-issues/233_ElementalImpuritiesProcedures.pdf, accessed September 18, 2022.
- J. S. Barin, B. Tischer, R. S. Picoloto, F. G. Antes, F. E. B. da Silva, F. R. Paula and E. M. M. Flores, Determination of toxic elements in tricyclic active pharmaceutical ingredients by ICP-MS: a critical study of digestion methods, J. Anal. At. Spectrom., 2014, 29, 352–358, 10.1039/C3JA50334H.
- Q. Tu, T. Wang and V. Antonucci, High-efficiency sample preparation with dimethylformamide for multi-element determination in pharmaceutical materials by ICP-AES, J. Pharm. Biomed. Anal., 2010, 52, 311–315 CrossRef CAS PubMed.
- C. Støving, H. Jensen, B. Gammelgaard and S. Stürup, Development and validation of an ICP-OES method for quantitation of elemental impurities in tablets according to coming US pharmacopeia chapters, J. Pharm. Biomed. Anal., 2013, 84, 209–214 CrossRef PubMed.
- A. L. Muller, J. S. Oliveira, P. A. Mello, E. I. Muller and E. M. Flores, Study and determination of elemental impurities by ICP-MS in active pharmaceutical ingredients using single reaction chamber digestion in compliance with USP requirements, Talanta, 2015, 136, 161–169 CrossRef CAS PubMed.
- E. Marguí, C. Fontàs, A. Buendía, M. Hidalgo and I. Queralt, Determination of metal residues in active pharmaceutical ingredients according to European current legislation by using X-ray fluorescence spectrometry, J. Anal. At. Spectrom., 2009, 24, 1253–1257, 10.1039/B904064A.
- E. Marguí, K. Van Meel, R. Van Grieken, A. Buendía, C. Fontàs, M. Hidalgo and I. Queralt, Method for the determination of Pd-catalyst residues in active pharmaceutical ingredients by means of high-energy polarized-beam energy dispersive X-ray fluorescence, Anal. Chem., 2009, 81, 1404–1410, DOI:10.1021/ac8021373.
- B. Sauer, Y. Xiao, M. Zoontjes and C. Kroll, Application of X-ray fluorescence spectrometry for screening pharmaceutical products for Elemental Impurities according to ICH guideline Q3D, J. Pharm. Biomed. Anal., 2020, 179, 113005 CrossRef CAS PubMed.
- X-Ray Fluorescence Spectrometry, DOI:10.31003/USPNF_M7469_03_01.
- European Pharmacopoeia(Ph. Eur.), European Directorate for the Quality of Medicines & HealthCare – Liferay DXP, European Directorate for the Quality of Medicines & HealthCare, 10th edn, https://www.edqm.eu/en/, accessed September 18, 2022 Search PubMed.
- S. Borowska and M. M. Brzóska, Metals in cosmetics: implications for human health, J. Appl. Toxicol., 2015, 35, 551–572 CrossRef CAS PubMed.
- P. UNION, Regulation (EC) No 1223/2009 of the european parliament and of the council, OJEU, 2009, vol. 342, pp. 59.
- The Federal Office for Consumer Protection and Food Safety (BVL), https://www.bvl.bund.de/EN/Home/home_node.html.
- E. L. Sainio, R. Jolanki, E. Hakala and L. Kanerva, Metals and arsenic in eye shadows, Contact Dermatitis, 2000, 42, 5–10 CrossRef CAS PubMed.
- A. Zakaria and Y. B. Ho, Heavy metals contamination in lipsticks and their associated health risks to lipsticks consumers, Regul. Toxicol. Pharmacol., 2015, 73, 191–195 CrossRef CAS PubMed.
- V. Coger, N. Million, C. Rehbock, B. Sures, M. Nachev, S. Barcikowski, N. Wistuba, S. Strauß and P. M. Vogt, Tissue concentrations of zinc, iron, copper, and magnesium during the phases of full thickness wound healing in a rodent model, Biol, Trace Elem. Res., 2019, 191, 167–176 CrossRef CAS PubMed.
- E. Pinto, K. Paiva, A. Carvalhido and A. Almeida, Elemental impurities in lipsticks: results from a survey of the Portuguese and Brazilian markets, Regul. Toxicol. Pharmacol., 2018, 95, 307–313 CrossRef CAS PubMed.
- M. G. Volpe, M. Nazzaro, R. Coppola, F. Rapuano and R. P. Aquino, Determination and assessments of selected heavy metals in eye shadow cosmetics from China, Italy, and USA, Microchem. J., 2012, 101, 65–69 CrossRef CAS.
- A. Zakaria and Y. B. Ho, Heavy metals contamination in lipsticks and their associated health risks to lipstick consumers, Regul. Toxicol. Pharmacol., 2015, 73, 191–195 CrossRef CAS PubMed.
- E. Marguí, R. Dalipi, L. Borgese, L. E. Depero and I. Queralt, Possibilities and drawbacks of total reflection X-ray fluorescence spectrometry as a fast, simple and cost-effective technique for multielement analyses of cosmetics, Anal. Chim. Acta, 2019, 1075, 27–37 CrossRef PubMed.
- N. M. Hepp, W. R. Mindak and J. Cheng, Determination of total lead in lipstick: development and validation of a microwave-assisted digestion, inductively coupled plasma-mass spectrometric method, J. Cosmet. Sci., 2009, 60, 405 CAS.
- SPECTRO XEPOS Case Study: L'Oreal, https://www.spectro.com/landingpages/www.spectro.com/landingpages/xrf-xepos-case-study-achieving-beautiful-results-for-cosmetics-analysis-at-loreal, accessed September 18, 2022.
- M. Filella, A. Martignier and A. Turner, Kohl containing lead (and other toxic elements) is widely available in Europe, Environ. Res., 2020, 187, 109658 CrossRef CAS PubMed.
- I. De La Calle, N. Cabaleiro, V. Romero, I. Lavilla and C. Bendicho, Sample pretreatment strategies for total reflection X-ray fluorescence analysis: A tutorial review, Spectrochim. Acta, Part B, 2013, 90, 23–54, DOI:10.1016/j.sab.2013.10.001.
- E. Marguí, I. Queralt, M. L. Carvalho and M. Hidalgo, Comparison of EDXRF and ICP-OES after microwave digestion for element determination in plant specimens from an abandoned mining area, Anal. Chim. Acta, 2005, 549, 197–204 CrossRef.
- R. Dalipi, E. Marguí, L. Borgese and L. E. Depero, Multi-element analysis of vegetal foodstuff by means of low power total reflection X-ray fluorescence (TXRF) spectrometry, Food Chem., 2017, 218, 348–355 CrossRef CAS PubMed.
- Smart Store – Keep your sample and data safe forever, Smart Store, https://www.smartstore.systems/, accessed September 17, 2022.
- MT2_2020_Online_OK.pdf, https://www.ceceditore.com/wpcontent/uploads/2021/01/MT2_2020_Online_OK.pdf, accessed September 18, 2022.
- E. M. Gama, C. C. Nascentes, R. P. Matos, G. de C. Rodrigues and G. D. Rodrigues, A simple method for the multi-elemental analysis of beer using total reflection X-ray fluorescence, Talanta, 2017, 174, 274–278, DOI:10.1016/j.talanta.2017.05.059.
- Z. Ajtony, N. Laczai, G. Dravecz, N. Szoboszlai, Á. Marosi, B. Marlok, C. Streli and L. Bencs, Fast and direct screening of copper in micro-volumes of distilled alcoholic beverages by high-resolution continuum source graphite furnace atomic absorption spectrometry, Food Chem., 2016, 213, 799–805, DOI:10.1016/j.foodchem.2016.06.090.
- R. Dalipi, L. Borgese, A. Casaroli, M. Boniardi, U. Fittschen, K. Tsuji and L. E. Depero, Study of metal release from stainless steels in simulated food contact by means of total reflection X-ray fluorescence, J. Food Eng., 2016, 173, 85–91, DOI:10.1016/j.jfoodeng.2015.10.045.
- R. Dalipi, L. Borgese, K. Tsuji, E. Bontempi and L. E. Depero, Elemental analysis of teas, herbs and their infusions by means of total reflection X-ray fluorescence, J. Food Compos. Anal., 2018, 67, 128–134, DOI:10.1016/j.jfca.2018.01.010.
- E. Marguí and M. Voutchkov, Multielement Analysis of Tea and Mint Infusions by Total Reflection X-ray Fluorescence Spectrometry, Food Anal. Methods, 2018, 11, 282–291, DOI:10.1007/s12161-017-0998-8.
- A. Winkler, M. Rauwolf, J. H. Sterba, P. Wobrauschek, C. Streli and A. Turyanskaya, Total reflection X-ray fluorescence analysis of elemental composition of herbal infusions and teas, J. Sci. Food Agric., 2020, 100, 4226–4236, DOI:10.1002/jsfa.10463.
- A. S. Maltsev, E. V. Chuparina, G. V. Pashkova, J. V. Sokol'nikova, O. V. Zarubina and A. N. Shuliumova, Features of sample preparation techniques in the total-reflection X-ray fluorescence analysis of tea leaves, Food Chem., 2021, 343, 128502, DOI:10.1016/j.foodchem.2020.128502.
- A. N. Smagunova and G. V. Pashkova, Choice of optimal conditions for X-ray fluorescence analysis of milk products with varying fat content: X-ray fluorescence analysis of dairy products, X-Ray Spectrom., 2013, 42, 546–551, DOI:10.1002/xrs.2519.
- A. S. Maltsev, R. A. Yusupov and S. A. Bakhteev, Overcoming absorption effects in the determination of light elements in beverages by total-reflection X-ray spectrometry, X-Ray Spectrom., 2022, 3283, DOI:10.1002/xrs.3283.
- D. Vitali Čepo, M. Karoglan, L. Borgese, L. E. Depero, E. Marguí and J. Jablan, Application of benchtop total-reflection X-ray fluorescence spectrometry and chemometrics in classification of origin and type of Croatian wines, Food Chem.: X, 2022, 13, 100209, DOI:10.1016/j.fochx.2022.100209.
- R. Dalipi, E. Marguí, L. Borgese, F. Bilo and L. E. Depero, Analytical performance of benchtop total reflection X-ray fluorescence instrumentation for multielemental analysis of wine samples, Spectrochim. Acta, Part B, 2016, 120, 37–43, DOI:10.1016/j.sab.2016.04.001.
- M. J. Anjos, R. T. Lopes, E. F. O. de Jesus, S. Moreira, R. C. Barroso and C. R. F. Castro, Trace elements determination in red and white wines using total-reflection X-ray fluorescence, Spectrochim. Acta, Part B, 2003, 58, 2227–2232, DOI:10.1016/j.sab.2003.07.004.
- X. Gruber, P. Kregsamer, P. Wobrauschek and C. Streli, Total-reflection X-ray fluorescence analysis of Austrian wine, Spectrochim. Acta, Part B, 2006, 61, 1214–1218, DOI:10.1016/j.sab.2006.08.006.
- S. Pessanha, M. L. Carvalho, M. Becker and A. von Bohlen, Quantitative determination on heavy metals in different stages of wine production by Total Reflection X-ray Fluorescence and Energy Dispersive X-ray Fluorescence: Comparison on two vineyards, Spectrochim. Acta, Part B, 2010, 65, 504–507, DOI:10.1016/j.sab.2010.04.003.
- M. M. Castiñeira, R. Brandt, A. von Bohlen and N. Jakubowski, Development of a procedure for the multi-element determination of trace elements in wine by ICP-MS, Fresenius. J. Anal. Chem., 2001, 370, 553–558, DOI:10.1007/s002160100862.
- R. Dalipi, L. Borgese, A. Zacco, K. Tsuji, E. Sangiorgi, R. Piro, E. Bontempi and L. E. Depero, Determination of trace elements in Italian wines by means of total reflection X-ray fluorescence spectroscopy, Int. J. Environ. Anal. Chem., 2015, 95, 1208–1218, DOI:10.1080/03067319.2015.1036861.
- C. A. Shand, R. Wendler, L. Dawson, K. Yates and H. Stephenson, Multivariate analysis of Scotch whisky by total reflection x-ray fluorescence and chemometric methods: A potential tool in the identification of counterfeits, Anal. Chim. Acta, 2017, 976, 14–24, DOI:10.1016/j.aca.2017.04.041.
- L. Borgese, R. Dalipi, A. Riboldi, F. Bilo, A. Zacco, S. Federici, M. Bettinelli, E. Bontempi and L. E. Depero, Comprehensive approach to the validation of the standard method for total reflection X-ray fluorescence analysis of water, Talanta, 2018, 181, 165–171, DOI:10.1016/j.talanta.2017.12.087.
- F. Bilo, L. Borgese, A. Zacco, P. Lazo, C. Zoani, G. Zappa, E. Bontempi and L. E. Depero, Total Reflection X-Ray Fluorescence Spectroscopy to Evaluate Heavy Metals Accumulation in Legumes, J. Anal. Bioanal. Tech., 2015, 7, 1–7 Search PubMed.
- V. M. Paradiso, M. Castellino, M. Renna, C. E. Gattullo, M. Calasso, R. Terzano, I. Allegretta, B. Leoni, F. Caponio and P. Santamaria, Nutritional characterization and shelf-life of packaged microgreens, Food Funct., 2018, 9, 5629–5640, 10.1039/C8FO01182F.
- R. Dalipi, L. Borgese, E. Marguí, E. Sangiorgi and L. E. Depero, Total Reflection X-Ray Fluorescence Technique for Multi-Elemental Analysis of Food, Spectrosc. Europe/World, 2017, vol. 29, p. 4 Search PubMed.
- IMEKO-TC23-2016-030.pdf, https://imeko.net/publications/tc23-2016/IMEKO-TC23-2016-030.pdf, accessed September 21, 2022.
- I. Allegretta, C. E. Gattullo, M. Renna, V. M. Paradiso and R. Terzano, Rapid multi-element characterization of microgreens via total-reflection X-ray fluorescence (TXRF) spectrometry, Food Chem., 2019, 296, 86–93, DOI:10.1016/j.foodchem.2019.05.187.
- E. Marguí, A. de Fátima Marques, M. de Lurdes Prisal, M. Hidalgo, I. Queralt and M. L. Carvalho, Total Reflection X-ray Spectrometry (TXRF) for Trace Elements Assessment in Edible Clams, Appl. Spectrosc., 2014, 68, 1241–1246, DOI:10.1366/13-07364.
- R. Dalipi, R. Berneri, M. Curatolo, L. Borgese, L. E. Depero and E. Sangiorgi, Total reflection X-ray fluorescence used to distinguish mechanically separated from non-mechanically separated meat, Spectrochim. Acta, Part B, 2018, 148, 16–22, DOI:10.1016/j.sab.2018.06.002.
- R. Piro and E. Sangiorgi, Nutritional Mineral in Foods by Txrf: an Easy, Fast and Reliable Approach for Food Characterization, 2nd IMEKOFOODS Promoting Objective and Measurable Food Quality & Safety Benevento (Italy), 2016, vol. 4 Search PubMed.
- E. de Almeida, G. S. Montanha, H. W. Pereira de Carvalho and E. Marguí, Evaluation of energy dispersive X-ray fluorescence and total reflection X-ray fluorescence spectrometry for vegetal mass-limited sample analysis: Application to soybean root and shoots, Spectrochim. Acta, Part B, 2020, 170, 105915, DOI:10.1016/j.sab.2020.105915.
- M. J. Cal-Prieto, M. Felipe-Sotelo, A. Carlosena, J. M. Andrade, P. López-Mahía, S. Muniategui and D. Prada, Slurry sampling for direct analysis of solid materials by electrothermal atomic absorption spectrometry (ETAAS). A literature review from 1990 to 2000, Talanta, 2002, 56, 1–51, DOI:10.1016/s0039-9140(01)00543-4.
- S. L. C. Ferreira, M. Miró, E. G. P. da Silva, G. D. Matos, P. S. dos Reis, G. C. Brandao, W. N. L. dos Santos, A. T. Duarte, M. G. R. Vale and R. G. O. Araujo, Slurry Sampling—An Analytical Strategy for the Determination of Metals and Metalloids by Spectroanalytical Techniques, Appl. Spectrosc. Rev., 2010, 45, 44–62, DOI:10.1080/05704920903435474.
- U. B. de Araújo, A. C. M. da Costa, D. F. de Oliveira, E. F. O. de Jesus, M. J. Anjos, E. T. Mársico, C. da Silva Carneiro, R. de Oliveira Resende Ribeiro and R. T. Lopes, Analysis of milk trace elements with a home-made portable automated total reflection x-ray fluorescence system, Radiat. Phys. Chem., 2019, 156, 216–221, DOI:10.1016/j.radphyschem.2018.10.026.
- C. Enrich, S. Boeykens, N. Caracciolo, G. Custo and C. Vázquez, Honey characterization by total reflection x-ray fluorescence: evaluation of environmental quality and risk for the human health, X-Ray Spectrom., 2007, 36, 215–220, DOI:10.1002/xrs.944.
- R. de O. R. Ribeiro, E. T. Mársico, E. F. O. de Jesus, C. da Silva Carneiro, C. A. C. Júnior, E. de Almeida and V. F. do N. Filho, Determination of trace elements in honey from different regions in Rio de Janeiro State (Brazil) by total reflection X-ray fluorescence, J. Food Sci., 2014, 79, T738–T742, DOI:10.1111/1750-3841.12363.
- R. de Oliveira Resende Ribeiro, E. T. Mársico, C. da Silva Carneiro, J. S. Simoes, M. da Silva Ferreira, E. F. O. de Jesus, E. Almeida and C. A. C. Junior, Seasonal variation in trace and minor elements in Brazilian honey by total reflection X-ray fluorescence, Environ. Monit. Assess., 2015, 187, 96, DOI:10.1007/s10661-015-4284-1.
- A. von Bohlen and R. Fernández-Ruiz, Experimental evidence of matrix effects in total-reflection X-ray fluorescence analysis: Coke case, Talanta, 2020, 209, 120562, DOI:10.1016/j.talanta.2019.120562.
- R. Fernández-Ruiz, A. von Bohlen, E. J. Friedrich K and M. J. Redrejo, Analysis of coke beverages by total-reflection X-ray fluorescence, Spectrochim. Acta, Part B, 2018, 145, 99–106, DOI:10.1016/j.sab.2018.04.013.
- S. Galani-Nikolakaki, N. Kallithrakas-Kontos and A. A. Katsanos, Trace element analysis of Cretan wines and wine products, Sci. Total Environ., 2002, 285, 155–163, DOI:10.1016/s0048-9697(01)00912-3.
- D. Monticelli, A. Cinosi, G. Siviero and L. Seralessandri, Trace element determination in soy sauce: A novel total reflection X-ray fluorescence procedure and comparison with inductively coupled plasma–mass spectrometry, Spectrochim. Acta, Part B, 2018, 146, 16–20, DOI:10.1016/j.sab.2018.04.022.
- R. M. Cunha e Silva, E. Almeida, E. P. E. Valencia and V. F. N. Filho, Determination of Fe, Cu and Zn in sugar-cane spirits commercialized in Southeastern Brazil by TXRF, J. Radioanal. Nucl. Chem., 2004, 260, 3–7, DOI:10.1023/B:JRNC.0000027053.80091.7c.
- T. Martinez, J. Lartigue, G. Zarazua, P. Avila-Perez, M. Navarrete and S. Tejeda, Total reflection X-ray fluorescence analysis of trace-elements in candies marketed in Mexico, Spectrochim. Acta, Part B, 2010, 65, 499–503, DOI:10.1016/j.sab.2010.04.002.
- Z. Vincevica-Gaile and M. Klavins, Concentration of Elements in Food: How Can It Reflect Impact of Environmental and Other Influencing Factors, Scientific Journal of Riga Technical University, Environ. Clim. Technol., 2013, 12, 15–19, DOI:10.2478/rtuect-2013-0011.
- Z. Vincevica-Gaile, M. Klavins, V. Rudovica and A. Viksna, Research review trends of food analysis in Latvia: major and trace element content, Environ. Geochem. Health, 2013, 35, 693–703, DOI:10.1007/s10653-013-9549-4.
- V. Romero, I. Costas-Mora, I. Lavilla and C. Bendicho, Silver nanoparticle-assisted preconcentration of selenium and mercury on quartz reflectors for total reflection X-ray fluorescence analysis, J. Anal. At. Spectrom., 2014, 29, 696, 10.1039/c3ja50361e.
- V. Romero, I. Costas-Mora, I. Lavilla and C. Bendicho, Room temperature trapping of stibine and bismuthine onto quartz substrates coated with nanostructured palladium for total reflection X-ray fluorescence analysis, Spectrochim. Acta, Part B, 2015, 107, 125–131, DOI:10.1016/j.sab.2015.03.006.
- F. Bilo, L. Borgese, R. Dalipi, A. Zacco, E. Bontempi and L. E. Depero, A Smart Sample Preparation Method for Chocolate Analysis, 2nd IMEKOFOODS Promoting Objective and Measurable Food Quality & Safety Benevento (Italy), 2016, vol. 5 Search PubMed.
- F. J. Antosz, Y. Xiang, A. R. Diaz and A. J. Jensen, The use of total reflectance X-ray fluorescence (TXRF) for the determination of metals in the pharmaceutical industry, J. Pharm. Biomed. Anal., 2012, 62, 17–22 CrossRef CAS PubMed.
- E. Marguí, I. Queralt and M. Hidalgo, Determination of platinum group metal catalyst residues in active pharmaceutical ingredients by means of total reflection X-ray spectrometry, Spectrochim. Acta, Part B, 2013, 86, 50–54 CrossRef.
- A. Meyer, S. Grotefend, A. Gross, H. Wätzig and I. Ott, Total reflection X-ray fluorescence spectrometry as a tool for the quantification of gold and platinum metallodrugs: determination of recovery rates and precision in the ppb concentration range, J. Pharm. Biomed. Anal., 2012, 70, 713–717 CrossRef CAS PubMed.
- G. Cristoforetti, A. De Giacomo, M. Dell'Aglio, S. Legnaioli, E. Tognoni, V. Palleschi and N. Omenetto, Local thermodynamic equilibrium in laser-induced breakdown spectroscopy: beyond the McWhirter criterion, Spectrochim. Acta, Part B, 2010, 65, 86–95 CrossRef.
- M. Marijan, J. Jablan, L. Jakupović, M. Jug, E. Marguí, R. Dalipi, E. Sangiorgi and M. Zovko Končić, Plants from Urban Parks as Valuable Cosmetic Ingredients: Green Extraction, Chemical Composition and Activity, Agronomy, 2022, 12, 204 CrossRef CAS.
- E. DaSilva, A. M. David and A. Pejović-Milić, The quantification of total lead in lipstick specimens by total reflection X-ray fluorescence spectrometry, X-Ray Spectrom., 2015, 44, 451–457, DOI:10.1002/xrs.2629.
- Versailles Project on Advanced Materials and Standards (VAMAS), http://www.vamas.org/, accessed March 4, 2024.
- T. Golob, U. Doberšek, P. Kump and M. Nečemer, Determination of trace and minor elements in Slovenian honey by total reflection X-ray fluorescence spectroscopy, Food Chem., 2005, 91, 593–600, DOI:10.1016/j.foodchem.2004.04.043.
- U. Kropf, M. Korošec, J. Bertoncelj, N. Ogrinc, M. Nečemer, P. Kump and T. Golob, Determination of the geographical origin of Slovenian black locust, lime and chestnut honey, Food Chem., 2010, 121, 839–846, DOI:10.1016/j.foodchem.2009.12.094.
- M. Nečemer, I. J. Košir, P. Kump, U. Kropf, M. Jamnik, J. Bertoncelj, N. Ogrinc and T. Golob, Application of total reflection X-ray spectrometry in combination with chemometric methods for determination of the botanical origin of slovenian honey, J. Agric. Food Chem., 2009, 57, 4409–4414, DOI:10.1021/jf900930b.
- J. Braziewicz, I. Fijal, T. Czyewski, M. Jaskóla, A. Korman, D. Banaś, A. Kubala-Kuku, U. Majewska and L. Zemlo, PIXE and XRF analysis of honey samples, Nucl. Instrum. Methods Phys. Res., Sect. B, 2002, 187, 231–237, DOI:10.1016/S0168-583X(01)00942-9.
- A. W. C. Patza, C. Cescutti and H. Dietrich, Manganese screening of pineapple by total-reflection X-ray fluorescence (TXRF) spectroscopy, Deutsche Lebensmittel-Rundschau, Z. Lebensm. Unters. Forsch., 2013, 109, 315–319 Search PubMed.
- P. F. Boldrin, V. Faquin, S. J. Ramos, K. V. F. Boldrin, F. W. Ávila and L. R. G. Guilherme, Soil and foliar application of selenium in rice biofortification, J. Food Compos. Anal., 2013, 31, 238 CrossRef.
- F. Bilo, M. Lodolo, L. Borgese, A. Bosio, L. Benassi, L. E. Depero and E. Bontempi, Evaluation of Heavy Metals Contamination from Environment to Food Matrix by TXRF: The Case of Rice and Rice Husk, J. Chem., 2015, 2015 DOI:10.1155/2015/274340.
- C. N. Grant, H. T. Dennis, J. M. R. Antoine, L. A. Hoo-Fung and G. C. Lalor, Agglomerative hierarchical clustering analysis of twenty-six rice samples analysed by instrumental neutron activation analysis and other techniques, J. Radioanal. Nucl. Chem., 2013, 297, 233–239 CrossRef CAS.
- M. Lué-Merú, Determination of total As in onion plants growing in contaminated substrates by total reflection X-ray fluorescence, J. Radioanal. Nucl. Chem., 2011, 287, 479–484, DOI:10.1007/s10967-010-0834-8.
- K. P. Kageliza, Determination of Mn, Fe, Cu and Zn in Indigenous Complementary Infant Flour from Kenya by Total-Reflection X-Ray Fluorescence, Journal of Food and Nutrition Sciences, 2014, 2, 110–116, DOI:10.11648/j.jfns.20140204.13.
- D. M. Maina, M. M. Gatari, S. K. Bartilol and M. K. Nguli, Essential trace metals in selected maize flour brands consumed in Nairobi, Kenya: application of txrf, 2nd IMEKOFOODS Promoting Objective and Measurable Food Quality & Safety Benevento (Italy), 2016, vol. 5 Search PubMed.
- K. Bielecka, W. Kurtek, D. Banaś, A. Kubala-Kukuś, J. Braziewicz, U. Majewska, M. Pajek, J. Wudarczyk-Moćko and I. Stabrawa, X-ray Diffraction and Elemental Analysis of Medical and Environmental Samples, Acta Phys. Pol., A, 2014, 125, 911–918, DOI:10.12693/APhysPolA.125.911.
- H. Stosnach, Analytical determination of selenium in medical samples, staple food and dietary supplements by means of total reflection X-ray fluorescence spectroscopy, Spectrochim. Acta, Part B, 2010, 65, 859–863, DOI:10.1016/j.sab.2010.07.001.
- I. Machado, S. Mondutey, N. Pastorino, V. Arce and M. Pistón, A green analytical method for the determination of Cu, Fe, Mn, and Zn in wheat flour using total reflection X-ray fluorescence, J. Anal. At. Spectrom., 2018, 33, 1264–1268, 10.1039/C8JA00144H.
- A. Lasztity, A. Kelko-Levai, I. Varga, K. Zih-Perenyi and E. Bertalan, Development of atomic spectrometric methods for trace metal analysis of pharmaceuticals, Microchem. J., 2002, 73, 59–63 CrossRef CAS.
- D. V. Danilov, P. Y. Sharanov and N. V. Alov, Determination of the Elemental Composition of Dietary Supplements by Total Reflection X-Ray Fluorescence Spectrometry, J. Anal. Chem., 2020, 75, 764–768 CrossRef CAS.
- I. Varga, Iodine determination in dietary supplement products by TXRF and ICP-AES spectrometry, Microchem. J., 2007, 85, 127–131 CrossRef CAS.
- LGC Standards: Reference Materials, Standards & Testing, https://www.lgcstandards.com/HR/en/, accessed September 18, 2022.
| This journal is © The Royal Society of Chemistry 2024 |