
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the study of paintings' stratigraphy by fs-LIBS and MA-XRF techniques
E.
Kechaoglou
 a,
K. A.
Agrafioti
b,
G. P.
Mastrotheodoros
bc,
D. F.
Anagnostopoulos
b and
C.
Kosmidis
*a
a,
K. A.
Agrafioti
b,
G. P.
Mastrotheodoros
bc,
D. F.
Anagnostopoulos
b and
C.
Kosmidis
*a
aDepartment of Physics, University of Ioannina, Ioannina, Greece. E-mail: kkosmid@uoi.gr
bDepartment of Materials Science and Engineering, University of Ioannina, Ioannina, Greece
cConservation of Antiquities & Works of Art Department, West Attica University, Athens, Greece
First published on 10th January 2024
Abstract
Works of art, especially paintings, often involve successive layers of materials of diverse compositions and properties. The current study explores the potentialities of the combination of femtosecond Laser Induced Breakdown Spectroscopy (fs-LIBS) and Macro X-ray Fluorescence (MA-XRF) for the study of paintings. In this framework, two mock-up samples that were manufactured by using traditional post-byzantine painting methods and numerous inorganic and organic pigments was thoroughly investigated. The selected pigments were either applied as single paint layers on top of a calcium carbonate substrate or as a combination of two and/or three successive paint layers over the substrate. The authors attempt to disclose the layered structure of the paints by inspecting some carefully chosen LIBS spectral features (markers) as a function of the laser shot number. The various paint layers are identified by applying less than ten laser shots while the diameter of the induced craters is limited to under 100 μm. Additionally, it is shown that through the proper treatment and evaluation of complementary MA-XRF data, one may overcome some of the fs-LIBS limitations and thoroughly investigate a painting's stratigraphy. Given the micro-invasive character of fs-LIBS and the non-invasive character of MA-XRF, combining the selected analytical methods is a promising tool for painting investigation.
Introduction
Understanding the constituent materials and structure of a work of art (and especially paintings) is an essential prerequisite for its proper preservation. It contributes considerably towards a better assessment of its artistic and other (e.g., historical) inherent values.1 With this knowledge, it is easier to ensure whether an artwork is preserved in its original state and, therefore, the potential restoration or conservation interventions may be carried out correctly.Conventionally, micro-samples are mainly collected from artworks by means of surgical equipment (e.g., scalpels), and through their proper manipulation (e.g., epoxy resin embedment and creation of polished cross-sections), it is often possible to achieve a rather thorough cross-sectional characterization of a painting.2,3 Quite recently, innovative and far less invasive methods for sample extraction have evolved, such as, e.g., laser cutting using focused fs pulses, which minimizes the induced collateral damage on the studies item.4 Nevertheless, being precious and rare objects, there is a tendency to investigate paintings and other pertinent cultural heritage objects by non-invasive analytical methods5–8 yet, quite often, these very techniques may not be able to differentiate between successive layers on an object, due to inherent limitations of the experimental process.9,10
The non-destructive and non-invasive X-Ray Fluorescence (XRF) allows rapid and thorough multi-elemental analysis, with a probing depth in the order of few to hundreds of micrometers.11 The measured photon intensity of characteristic transition is a complicated function of the pigment's complex structure, which may consist of successive layers differing in thickness, elemental composition, and density. The exact correlation between the measured intensity and the specimen's physical properties is described by the XRF fundamental parameters theory.12 The probing depth depends not only on the specimen's physical properties (structure, atomic elements properties, atomic weight concentration, density), but also: (i) on the spectral distribution of the incoming ionization radiation and the energy of the recorded characteristic fluorescence radiation, and (ii) from the radiation beam path relative to the specimen's surface (incoming angle of the ionization radiation and the outgoing characteristic transition). The dependence of probing depth on the energy of characteristic radiation has allowed the development of techniques to extract information for the in-depth distribution by evaluating the intensity ratio of different characteristics transitions of the same element, like Kβ/Kα, Lβ/Lα, Mα/Lα transitions rates.13 Moreover, the dependence of probing depth on the radiation beam path allowed the implementation of XRF measurements using spectrometers with variable geometry to extract in-depth information.14,15 The inherent problem of conventional XRF spectroscopy to extract differential in-depth information has been resolved by the recently developed confocal micro-X-ray fluorescence spectroscopy.16,17 Due to its capability to provide depth-resolved information on the elemental distribution, confocal micro-XRF has become a state-of-the-art technique for the study of cultural heritage, like paintings.
On the other hand, MA-XRF imaging allows the fast extraction of the elemental intensity distribution maps across the surface, with limited capability regarding the in-depth elemental distribution, relative to the confocal μ-XRF. In the present work, we applied MA-XRF to take advantage of its imaging abilities combined with the fs-Laser Induced Breakdown Spectroscopy (LIBS), in order to overcome its drawback for detailed in-depth information.
LIBS is a micro-destructive experimental technique that is employed for the determination of the composition of multi-layered analytes, as well as for the elemental analysis of various materials,10,18–34 and its main advantage over other techniques is that it does not require removal and displacement of part of the object for these purposes. In brief, this technique involves the collection and analysis of the radiation emitted after the ablation of the surface material of an object, induced by a pulsed laser beam at intensities higher than 109 W cm−2. Additionally, the use of a tightly focused, low-energy, ultra-fast (fs) laser system ensures that the surface damage is kept to a minimum, due to limited thermalization of the area around the beam spot.4,31,32,34–38 Undoubtedly there are also other laser-based analytical techniques that can provide information on the layering of materials. Micro-SORS is an analytical technique that can be used to non-destructively provide information on the composition of thin layered materials.39,40 Although its many benefits, a key advantage of the micro-destructive LIBS technique, is the ability to simultaneously provide the atomic and molecular composition of the sample, while micro-SORS is mainly focused on the later.
Moreover, LIBS is substantially effective in the detection and classification of lighter elements, such as carbon and nitrogen, which may not be detected by conventional techniques that use X-rays, and it can also directly detect molecular compounds and fragments, such as C2 and CN, with high efficacy.41–44 The complementarity of LIBS and XRF is evident when considering that the distinction between (or even the mere identification of) some elements can be challenging using one of the techniques, while it can be a straightforward process using the other, and vice versa.
Overall, LIBS offers a very powerful tool for revealing the stratigraphy of multi-layered items such as paintings. At the same time, it provides a micro-invasive and highly effective identification of the elemental composition of the studied materials. In particular, its ability to detect and classify light elements and molecular compounds, combined with its complementarity with XRF, makes LIBS a valuable technique in the field of conservation science, which currently experiences a boost in applications.
In the present work authors thoroughly investigate the composition and layered structure of several mock-up paint layers by combining state-of-the-art fs-LIBS and MA-XRF.6,45,46 The samples were produced following the typical byzantine/post-byzantine painting technique, i.e. by applying mixtures of pigments plus egg yolk over an inert substrate/gesso that covers a wooden substrate.47 The selected pigments include silicon, aluminum, and titanium oxides (SiO2, Al2O3, TiO2), hematite (iron oxides, Fe3O4, Fe2O3, FeO3), synthetic ultramarine/sodium-aluminum-sulfo-silicate (Al6Na8O24S3Si6), lead, mercury, and arsenic compounds, as well as synthetic organic compounds, while the base/preparation layer is composed of calcium carbonate (CaCO3) plus hide glue.
There is an extended literature on the complementarity of XRF and LIBS techniques in cultural heritage,9,10,18,29,48 yet it has been proved that the commonly employed ns laser LIBS systems are prone to inducing thermal effects in the vicinity of the irradiated area. These phenomena can be greatly reduced using an ultra-fast (fs) laser system while maintaining LIBS's analytical advantages. The selection of the pigments employed in the current work was due to their content in specific light elements such as Si, Na, Al, C, and N, and molecular compounds that are very difficult to detect using XRF methods; conversely, some of the selected pigments contain atoms that are challenging to detect using the LIBS method. The pigments were mixed with egg yolk (binder) and deposited as a single layer and as combinations of two successive layers over a calcium carbonate plus hide glue substrate (see next for details).
Materials and methods
The mock-up
A 23 × 13 cm2 piece of plywood was covered by a thin coating of hide glue and then received numerous successive thin layers of a CaCO3 plus hide glue priming; upon the setting of the priming, its surface was treated using various grades of silicon carbide sandpapers. Subsequently, small quantities of selected pigments were mixed with a tempera medium (egg yolk![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water – 1:1 v/v) and were applied in the form of thin paint layers on small squares marked on the surface of the primed panel. In particular, the selected pigments include contemporary commercial products (Kremer Pigmente), namely mars black (corresponding paint layer code: “A”, Kremer Pigmente code: 48400), synthetic ultramarine (B/45010), hematite (C/48600), alizarin dark (D/23610), red mine ochre (E/40351), gold French ochre (F/40070), caput mortum (G/48750), rutile Ti-white (H/46200), terra Ercolano (I/41600) and raw sienna (J/40392); for reference purposes, a small square on the bare preparation was covered by the pure medium (egg yolk + water) (Fig. 1). The paints were applied by using thin synthetic brushes in pairs and in a standard sequence; for example, in the case of the pigments' pair A & B, the corresponding paints were applied in the following manner: from left to right, pure A (A), B over A (B/A), A over B (A/B), pure B (B) (Fig. 1). Note that every single paint layer was applied by successive thin brushstrokes on its substrate until the latter was fully covered. A second mock-up was also manufactured by using the same procedures and by employing three additional pigments, namely vermilion (paint L/42000), lead white (M/46000) and orpiment (N/10700).
:water – 1:1 v/v) and were applied in the form of thin paint layers on small squares marked on the surface of the primed panel. In particular, the selected pigments include contemporary commercial products (Kremer Pigmente), namely mars black (corresponding paint layer code: “A”, Kremer Pigmente code: 48400), synthetic ultramarine (B/45010), hematite (C/48600), alizarin dark (D/23610), red mine ochre (E/40351), gold French ochre (F/40070), caput mortum (G/48750), rutile Ti-white (H/46200), terra Ercolano (I/41600) and raw sienna (J/40392); for reference purposes, a small square on the bare preparation was covered by the pure medium (egg yolk + water) (Fig. 1). The paints were applied by using thin synthetic brushes in pairs and in a standard sequence; for example, in the case of the pigments' pair A & B, the corresponding paints were applied in the following manner: from left to right, pure A (A), B over A (B/A), A over B (A/B), pure B (B) (Fig. 1). Note that every single paint layer was applied by successive thin brushstrokes on its substrate until the latter was fully covered. A second mock-up was also manufactured by using the same procedures and by employing three additional pigments, namely vermilion (paint L/42000), lead white (M/46000) and orpiment (N/10700).
 | ||
| Fig. 1 A photo of the mock-up with single and double-layered samples is presented. | ||
In order to accurately measure the mock up paint layers' thickness, micro-samples were extracted (using surgical scalpel) from all the painted squares and were embedded into epoxy resin. Upon the curing of the resin, samples were subjected to grinding and polishing and transformed into polished cross-sections; the latter were subsequently examined and photographed under an optical microscope (OM, Leica, DMRXP) at magnifications up to 200×.
The LIBS setup
The radiation source for the LIBS setup is a 30 fs, 5 mJ per pulse laser system (DUO Legend Elite) centered at 800 nm, operating at 10 Hz. The laser beam has an initial diameter of 1 cm and is focused using a convex lens with an effective focal distance of 23 cm (diameter of spot size ∼26 μm). A constant flow of argon (6 l min−1) prevents chemical reactions between the atoms and ions of the plume with atmospheric air;38,49 thus the resulting excited moieties consist only of elements of the irradiated material. Control over the peak intensity is achieved by employing a couple of stepwise neutral density filters, which attenuate the beam to 150 μJ per pulse. These filters are also used as samplers, reflecting part of the pulse to two photodiodes that trigger a shutter and the spectrograph, which are described later.The radiation emitted by the laser-induced plasma, is collected by utilizing a collimator/collector (Andor ME-OPT-0007) and then analyzed using a gated spectrograph (Andor iStar). In order to be able to simultaneously study a wide part of the emitted spectrum (200–600 nm), a 150 l mm−1 grating is utilized. This also guarantees better light collection efficiency for the gated iCCD camera, although it admittedly lowers the spectral resolution to around 0.6 nm. The width of the temporal gate is set to 600 ns, opening approximately 30 ns after the white light produced by the inverse bremsstrahlung has faded. Furthermore, the setup incorporates an electronically controlled bespoke and a 2-axis stage that enables accurate selection of the ablation target over the mock-up.
A homemade, electronically controlled shutter is incorporated to ensure that the subject only interacts with a preset number of pulses. This feature guarantees repeatability over the experimental process. Finally, the diameter of the created craters has been determined by utilizing a Leica DM-4000 optical microscope.
The MA-XRF set-up
The MA-XRF analysis of the mock-up samples was conducted by using the M6-Jetstream scanner (Bruker), which is equipped with a 30 W rhodium anode X-ray tube (maximum high voltage: 50 kV) with polycapillary optics and a silicon drift detector (active area: 30 mm2, energy resolution: 139 eV at Mn Kα). The polycapillary full lens focuses the X-ray radiation from the tube on the target. For the mock-up study, the X-ray tube was operated in a voltage and current setting of 50 kV and 600 μA, respectively. The beam spot size was set to a diameter of 100 μm and the scanning step/pixel size to 300 μm (no filter), while the dwell time was set to 20 ms per pixel. These parameters resulted in an overall scanning time of 90 minutes, a measuring time of 30 minutes, and in collecting 208310 individual spectra from the selected mock-up area. In all the cases, the collected spectra were processed and visualized (i.e., transformed into elemental distribution maps) using the built-in M6-Jetstream software.
Results and discussion
Study of the single paint layers
Table 1 includes the detailed list of the pigments used in the mock-up under investigation. The latter consists of square regions where either single paint layers or successive layers of two distinct paints (each paint composed of a different, pure pigment) have been applied (Fig. 1); note that no mixing of the individual pigments has been performed. The first objective is to investigate the elemental composition of the pure pigments, to create an identification tool based on the LIBS fingerprinting. This is achieved by inspecting the LIBS spectra collected from the single-layered regions. Acquisition of the spectra from only the top layer is accomplished by irradiating the desired target with a very limited number of pulses: to record the emission from plasma originating solely from the surface material, only four shots are used, ensuring that the paint layer is not penetrated. However, for improved statistics, a total of 100 shots is recorded and averaged, resulting from the irradiation of 25 different points on each of the single-paint layer squares.| Codename | Pigment | Chemical formulas of components | Selected spectral peaks and bands |
|---|---|---|---|
| A | Mars black | Fe3O4 | Fe II 274.7 nm |
| B | Ultramarine blue | Na6–8Al6Si6O24S2–4 | Si I 251.6 nm |
| C | Hematite | Fe2O3 | Fe II 274.7 nm |
| D | Alizarine dark | Organic, C14H8O4 | CN band 386 nm |
| E | English red ochre | Unspecified composition inc. Fe2O3 | Fe II 274.7 nm |
| F | Gold French ochre | SiO2 + AlO3 + FeO3 | Fe II 274.7 nm, Al II 559.3 nm |
| G | Caput mortum | Fe2O3 | Fe II 274.7 nm |
| H | Titanium white rutile | TiO2 | Ti II 323.5, 335.0 & 453.4 nm |
| I | Terra ercolano | Fe2O3 | Fe II 274.7 nm |
| J | French raw sienna | Fe2O3 + SiO2 + Al2O3 | Fe II 274.7 nm |
| K | Egg white + water | Organic inc. Ca, Mg, S etc. | CN band 386 nm |
| Substrate | Calcium carbonate | CaCO3 | Ca II 317.9 nm |
The first target to be investigated is the binding medium (egg yolk and water) used to prepare the paints, codenamed “K”. The relevant LIBS spectrum is seen in Fig. 2(a) and includes some standard features observed in the spectra of the other paint layers. These spectral lines are attributed to transitions of carbon, calcium, and magnesium atoms, as well as some molecular bands assigned to radiative transitions from excited electronic states of molecular fragments like C2 and CN. Carbon and magnesium atoms are expected to exist as trace elements within the egg yolk.51 Nevertheless, due to the high cross-section of these transitions, their contribution to the emission spectra is stronger than that of elements like carbon, oxygen, nitrogen, etc., which comprise a considerable percentage of the composition of the irradiated materials.
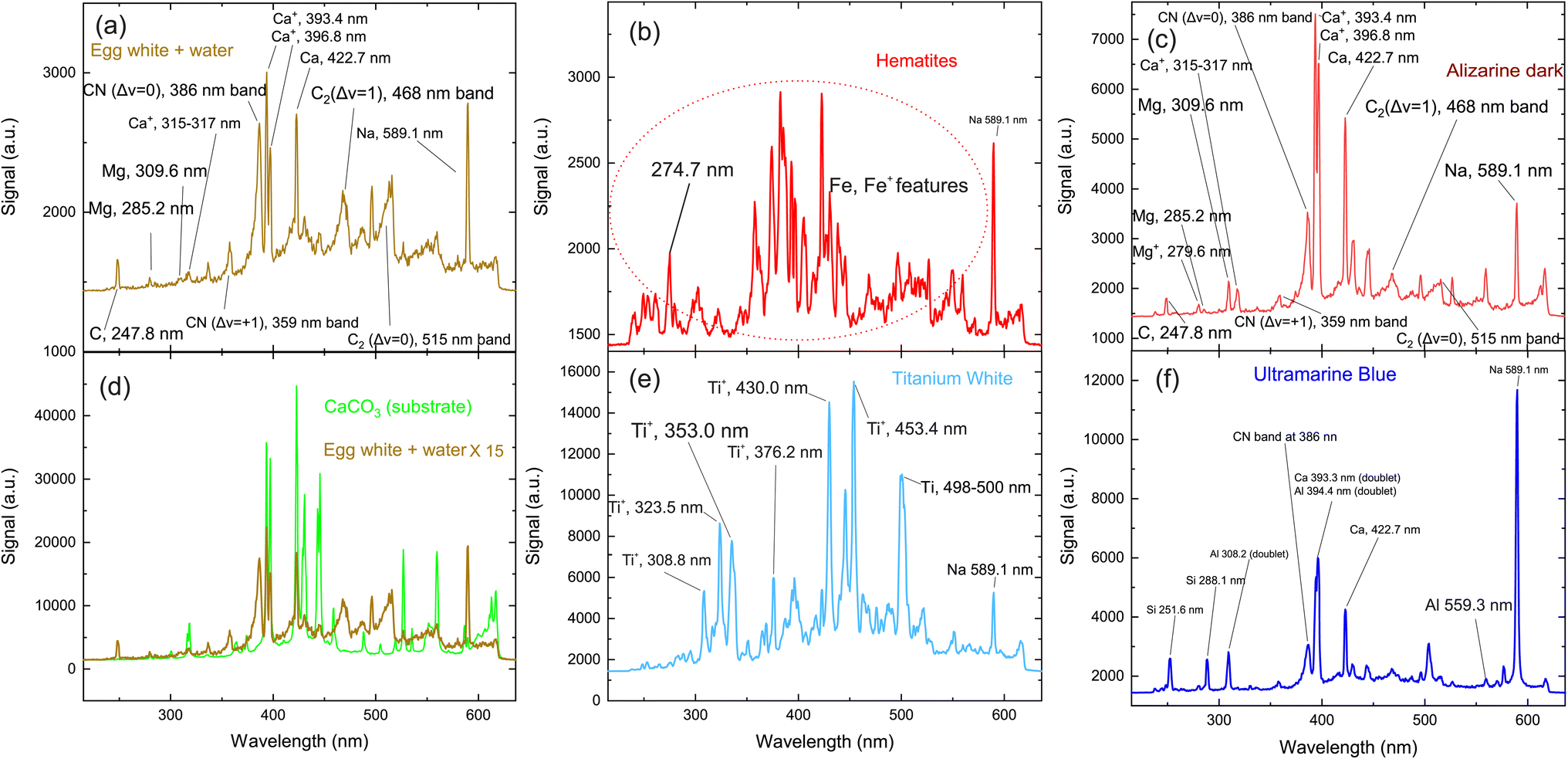 | ||
| Fig. 2 Typical LIBS spectra of the pigments. The item irradiated on each graph is: (a) the binder used for the production of the paints (“K”) (b) pigment containing iron oxides (the graph presents common spectral features of the pigments “A”, “C”, “E”, “F”, “G”, “I” and “J”), (c) organic pigment “D” (d) calcium carbonate and gelatin (substrate), (e) pigment “H” (f) pigment “B”. | ||
The structures mentioned above are more or less observed within all of the spectra and even in the substrate, which consists of hide glue (proteinaceous organic matter) and calcium carbonate. Due to its composition, the spectrum obtained from the substrate includes molecular bands, but overall, it is dominated by calcium atomic and ionic lines, as seen in Fig. 2(d). This is somewhat problematic, as Ca and Ca+ emission spectra are exceptionally intense and may mask nearby atomic spectral lines and molecular bands. It is, therefore, crucial to find spectral areas which, on the one hand, contain atomic lines from the elements found in the pigments, but on the other, are not overwhelmed by calcium spectral lines. In this respect, the presence of Ca in the material is validated by inspecting the 317.9 nm and 318.1 nm double ionic lines (the lines are seen as one, due to limitations of the grating used for these series of spectra) which correspond to the 3p64p 2P° (j = 3/2) ← 3p64d 2D (j = 5/2) and 3p64p 2P° (j = 3/2) ← 3p64d 2D (j = 3/2) transitions.
Some of the chemical elements in the paint layers emit brightly in the optical part of the spectrum (Na, Si, Al), while others in the UV (Fe, Ca, Ti, CN). Typical LIBS spectra from the paint layers in consideration are presented in Fig. 2(a)–(f). Each one of the spectra is characteristic of the elemental composition of the corresponding pigment. However, to study the layered structure, one should pinpoint at least one part of the spectrum which can be utilized for accurate identification of each paint layer. The accurate assessment of the spectra recorded from each painted square, lead us to use the following spectral features as the identification criteria for the employed pigments:52
• iron oxides are found in pigments A, C, E, F, G, I and J with a particularly well-defined ionic line at 274.7 nm (3d6(5D)4s 4D (j = 3/2) ← 3d6(5D)4p 4F° (j = 5/2)) (Fig. 2b).
• For the organic pigment E and the binder K, the CN band around 386 nm is particularly pronounced and therefore used as a marker (Fig. 2(a)).
• Titanium dioxide (pigment H) is a quite easily distinguishable pigment, due to the strong spectral features of Ti and Ti+. Multiple lines are well separated from the rest of the elements and can be seen in Fig. 2(e). Thus, as a Ti marker used the spectral lines at 323.5 nm and 335.0 nm originated from the 3d2(3F)4s 4F ← 3d2(3F)4p 4F°, 3d2(3F)4s 4F ← 3d2(3F)4p 4G° and transitions of Ti+ ion respectively.
• Aluminum is found in pigments B, F and J, but is quite a difficult element to distinguish, particularly in the presence of calcium and iron. Only the feature at 559.3 nm can be safely used as an Al marker because is attributed to the 3s4p 1P° ← 3s4d 1D transition of Al+ ion. Two sets of double lines, the first at 308.2 nm and 309.2 nm, and another at 394.3 nm and 396.1 nm respectively appeared in the LIBS spectra, however, they could not be considered as handy markers, because they reside very close to strong atomic lines of Mg and Ca; the latter elements exist within the egg yolk and were not distinguishable under the present experimental conditions.
• Pigments B, F and J contain SiO2. Similarly, to the case of Al, a strong atomic line is expected at 386.2 nm, however, is not easily distinguishable from the Ca lines that are emitted from the ablation of the substrate. The Si 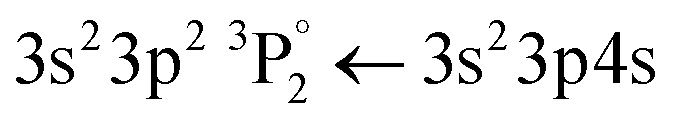 transition
transition 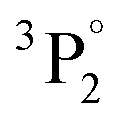 is expected at 251,6 nm while the 3s23p 2P° ← 3s24d 2D Si+ transition in the atomic ion is expected at 505.6 nm. However, they are clearly distinguishable only in the spectrum of pigment B.
is expected at 251,6 nm while the 3s23p 2P° ← 3s24d 2D Si+ transition in the atomic ion is expected at 505.6 nm. However, they are clearly distinguishable only in the spectrum of pigment B.
• Finally, sodium is easily recognizable due to the (double) line at 589.1 nm (2p63s 2S (j = 1/2) ← 2p63p 2P° j = 3/2, 1/2). It seems that the binder (sample K) contained a detectable amount of sodium and thus was observed in all the spectra. Thus, for the case of pigment B, where Na, Al and Si coexist, the study is based on the silicon emission lines mentioned above.
In Table 1 the spectral features used as markers for the identification of each pigment/paint are presented.
Once the individual “markers” (i.e., fingerprinting peaks) have been defined, the response (durability) of each paint was studied. A general goal of studying the layers of paints, is to remove as little material per shot as possible, while collecting the maximum amount of information form the emitted radiation. By doing so, even very fine/thin paint layers can be individually studied and at the same time, their relative thickness can be deduced, given that the durability of each paint is known, under certain experimental condition (pulse energy, duration, focal length of the focusing lens, position after the lens). In the current study, the number of laser pulses needed to completely remove a single paint layer and thus reach the substrate of the mock-up ranges from 8–14 pulses while, in the case of the ablation of two successive paint layers, 10–26 pulses. The spectra induced by two laser pulses was recorded and the area under the appropriate peak is calculated. This quantity is then used as an indicator of each element present in the paint layer.
In Fig. 3, the peak intensities of iron and calcium, which are used as markers for pigment A is presented as a function of the laser shot number. We aim to study the composition of the materials qualitatively, and therefore the signals are normalized from zero to unity for eye-guiding purposes. In this case (pigment A) it is evident that the upper layer has a high concentration of iron, which starts to faint as the laser ablates it after approximately eight shots. Once the laser starts interacting with the substrate, the Ca emission lines become more pronounced and then slowly disappear because the intensity of the laser has been reduced due to defocusing. It should be noted that this graph arises from spectra acquired by the same point on the paint, while on the contrary, Fig. 3b depicts the average of the spectra recorded from 10 different points of the same pigment (A). The resemblance between the two graphs is highly promising since a potential real-life application of LIBS to works of art should be as less destructive as possible, meaning that the number of irradiated spots should be as limited as possible. Note however that in the following graphs, only the ten-spot average will be presented for clarity reasons.
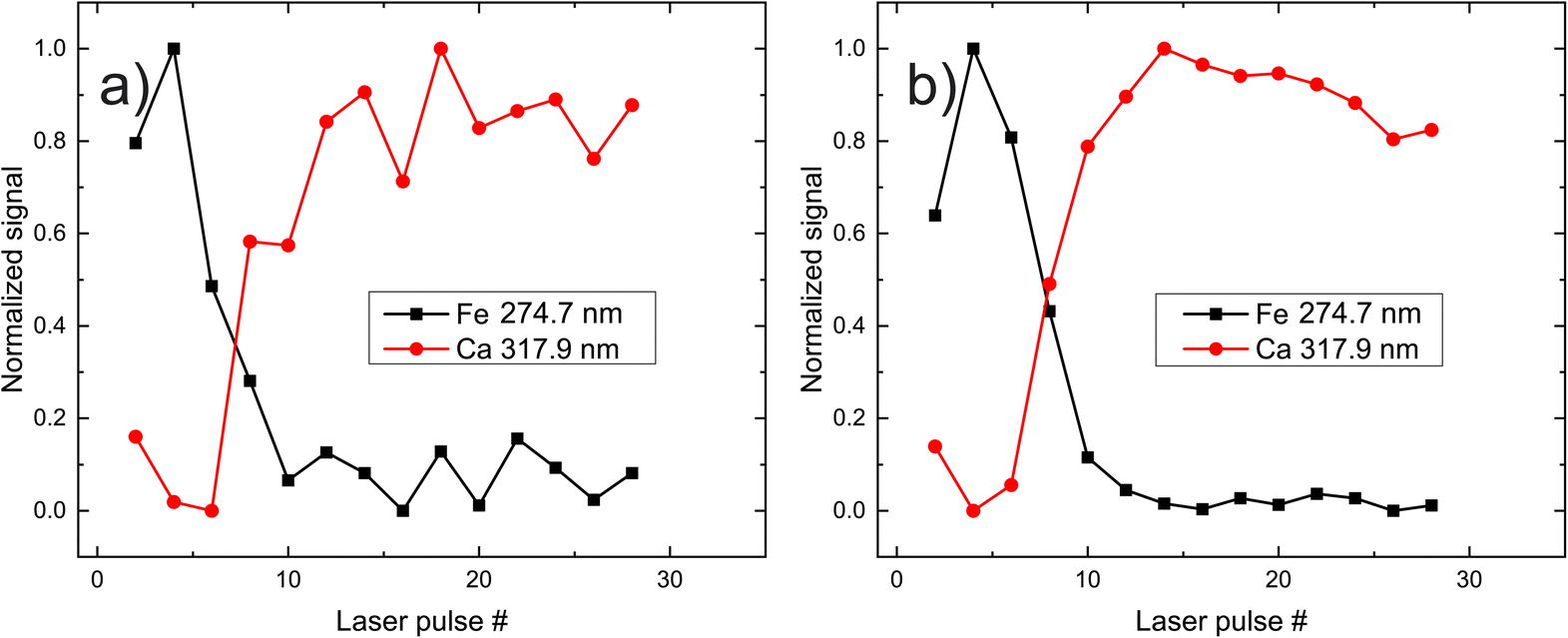 | ||
| Fig. 3 Dependence of Fe and Ca signal following paint layer “A” irradiation, as a function of the laser shot number. (a) Data accumulated from a single position, (b) data averaged after accumulation from ten different positions. | ||
In Fig. 4 the signal variation of the peaks characterized as pigment markers (Fig. 1) is presented as a function of the laser-shot number. Most of the pigments exhibit similar tendencies, lasting approximately eight pulses before the signal from the substrate becomes dominant, apart from samples E, J and K, where this transition is observed after approximately ten shots. The latter is understandable assuming a small difference of the layer thickness, which is expected given the free-hand manufacturing of the mock-up.
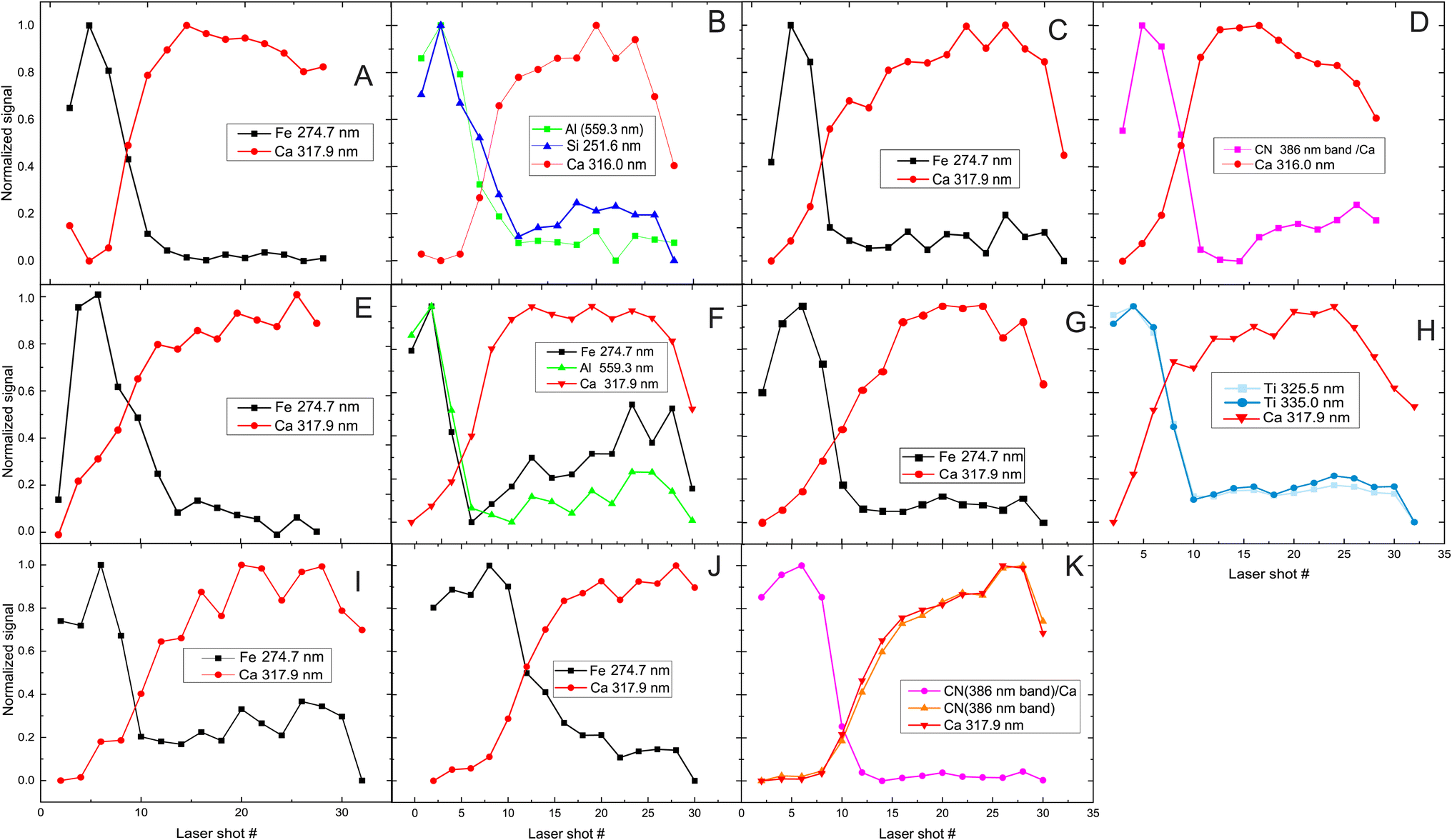 | ||
| Fig. 4 Signal recorded after laser irradiation of a single layer of each paint as a function of the laser pulse number. The insert in each plot (letters A–K) refers to the investigated paint and the markers used for its identification. | ||
In the case of the binder K and paint D, the signal assigned to the CN fragment is affected by the substrate contribution as the etching of the paint proceeds. This is because this band resides closely with some strong lines of Ca, which distort the characteristic shape of the band when the substrate is irradiated. This is evident in Fig. 4K, where CN and Ca signals increase in parallel. Nevertheless, the difference between the paints and the substrate is revealed by plotting their signal ratio. This approach can also be applied to distinguish the contribution from two layers of materials that exhibit similar compositions (the pigment, binder, and substrate incorporate Ca atoms and CN compounds) and can be particularly useful for studying the layered structure of multiple paint layers, even when organic pigments (like D) are employed. In some cases, the LIBS signal originating from the substrate decreases after 30 pulses. Initially it was inferred that this could imply laser penetration and irradiation of the lower parts of the mock-up (hide glue and plywood). To clarify that, we recorded the LIBS signal of the mock-up for 60 pulses in various spots on the mock-up, and then we analyzed the CN/Ca signal ratio, which was expected to show a considerable variation between the mock-up's priming substrate (hide glue plus CaCO3) and the first layer of pure hide glue that was applied on the bare wood. However, we observed that even though the overall signal decreases, the CN/Ca ratio remains constant throughout the 60 pulses, indicating thus that the substrate has not been pierced even after 60 pulses.
It is clear that the identification of various pigments is plausible by taking advantage of the proper markers in the LIBS spectra as discussed in detail above. In the case of paints/materials of similar composition, the identification is safer by utilizing the signal ratio of these markers.
Study of the superimposed paint layers
The next step is to investigate the potentialities of the employed fs-LIBS set-up for the investigation of the superimposed paint layers, i.e., the twelve square areas of the mock-up that were painted with two consecutive (yet different in terms of composition) paint layers (paints A–J). In Fig. 5 the recorded peak intensities corresponding to the defined markers (see above) are depicted as a function of the laser shot number.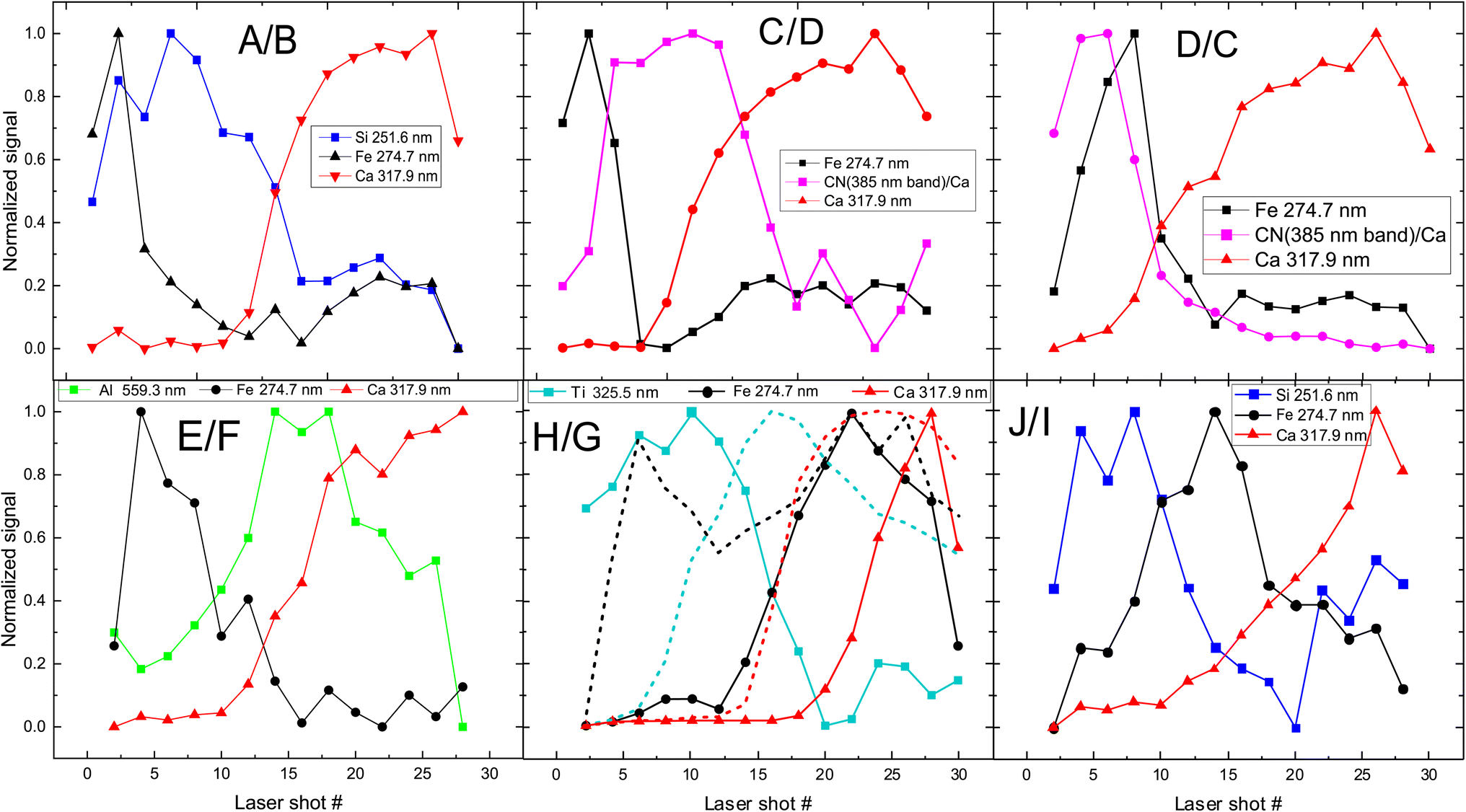 | ||
| Fig. 5 Depth profile of layered samples. Two paints (composed of discrete pigments) are applied in each sample/square. The inserts in graphs are referred to markers used to investigate pigments (see Fig. 1 for indexing purposes). The combinations of paints are presented in each graph too, with the first letter indicating the upper layer. In the case of H/G (i.e., H over G), the dotted lines represent the inverse layering case, i.e., G/H. | ||
The etching rate of the paint layers is expected to be the same regardless of the order of deposition/application, given that the layers are sufficiently thin, i.e., comparable to the Rayleigh length of the focused laser beam. Therefore, variation in the pulse number required for removing a layer is a clear indication of thickness difference among the layers. Such differences are observed in the cases where C & D and H & G paints are applied on the mock-up and therefore, as illustrated in Fig. 5 for both layer arrangements.
Furthermore, we found that the shots needed to penetrate the ensemble of paints is about double than the single-layer case. Of course, there are some exceptions, and this may be attributed to the paint layers being inhomogeneous (i.e., of varying thickness). For example, A/B shows that the markers of first layer (paint A) are only seen in the six first pulses, and then the emission lines corresponding to the marker of the lower layer (B) begin to appear in the spectra. On the other hand, in the case of H/G, titanium oxide (H) is evident in the spectra even after 18 pulses, while in G/H (dotted line in Fig. 5 (H/G)) the TiO2 layer lasts 12 to 14 pulses. This incongruity could be related to physical process associated with the width of each layer. Similar conclusions are drawn from Fig. 5 (C/D) and (D/C), where the organic pigment D is significantly thicker in the first case, resisting for almost double the number of pulses compared to the later. The latter case might well be linked to the need to apply a significantly thicker layer of alizarin lake to fully cover a white substrate (i.e., the preparation ground, case C/D) than a dark red substrate (i.e., hematite, case D/C).
The above interpretation of the variation laser etching rate is in agreement with paint layers thickness measured by inspecting with an optical microscope the micro-samples extracted from the mock-up. From Fig. 6 it is evident that the thickness of the layers varies across the painted surface. In Table 2 the thickness variation range of each pigment is presented.
 | ||
| Fig. 6 (Up) Paint layer E (red ochre) over paint layer F (gold French ochre); OM, 200×. (Bottom) Paint layer H (Ti white) over the calcium carbonate substrate; OM, 200×. | ||
| Pigment | Codename | Paint layer thickness range (μm) | |
|---|---|---|---|
| Minimum | Maximum | ||
| Vermilion | A | 18 | 35 |
| Ultramarine blue | B | 17 | 32 |
| Hematite | C | 13 | 47 |
| Alizarin dark | D | 17 | 38 |
| Red ochre | E | 17 | 72 |
| Gold French ochre | F | 10 | 42 |
| Caput mortum | G | 20 | 37 |
| Titanium white | H | 12 | 65 |
| Terra ercolano | I | 27 | 73 |
| French raw sienna | J | 24 | 66 |
| Egg yolk + water | K | 64 | 82 |
Therefore, it is clear that for the vast majority of the two-layered samples it is possible to safely identify the individual, successive paint layers. Thus, the potential of LIBS for the distinction of the layers containing some common elements is revealed, as demonstrated in the case of the organic pigment D and the inorganic pigment C. Even for layers that incorporate similar elements like E/F and J/I combinations, in which cases both pigments contain primarily iron oxides, LIBS spectra offer the ability for discrimination by considering the emission signal of accompanying elements, like silicon and aluminum.
XRF imaging measurements were performed to extract the two-dimensional elemental maps of the under-investigation mock-up. The Fe Kα intensities map shown in Fig. 7 (left) contains about 200k pixels/spectra, while the measuring time per pixel/spectrum was 20 ms. Fe Kα intensity variations are observed between the different pigment “squares”. These intensity variations are related, through the X-ray fundamental parameters theory, to the in-depth Fe weight concentration distribution and matrix composition. The X-ray characteristic intensities variations are observed not only between the different pigment “squares” but also within the same “square”. On the square “A/B”, in Fig. 7 (left), two marked areas (“1” and “2”) differ in the Fe Kα brightness. Each area consists of 238 pixels, with a total measuring time of 4.8 s per area. The average MA-XRF spectra (sum spectrum divided by the number of pixels) of areas “1” and “2” are depicted in Fig. 7 (right). Inspecting the XRF spectra, differences are observed concerning the intensity of the characteristic transitions and scattered radiation. The observed intensities variances are due to the elemental distribution and the specimen's structure across the probing depth, without allowing the extraction of information as a function of depth. The Fe and Mn K transitions are higher in the spectrum of area “1” (spectrum “1”) relative to the spectrum of area “2” (spectrum “2”). In the rest of spectrum “1” the intensity is lower than the spectrum “2”. The lower scattered radiation intensity in spectrum “1” reveals a higher mean atomic number in area “1” relative to area “2”.12,53 This should be attributed to higher “effective” Fe concentration across the probing depth in area “1”. The acquired LIBS spectrum in the square “A/B”, shown in Fig. 5 A, reveals the presence of a Fe-rich top layer, followed in sequence by Si- and Ca-containing layers (the LIBS measurement agrees with the structure of the mock-up: mars black over ultramarine over the CaCO3 preparation ground). The LIBS point-extracted structure allows us to assume that it is representative of the pigment's structure over all the square “A/B”. The LIBS provided information that Fe is only on the top layer, allowing us to conclude from the XRF spectra that the top layer is thicker in area “1” relative to area “2”. The observed top layer (mars black) thickness variations correspond to the difference in brush strokes, which is expected given that the paints were applied free-hand. The XRF spectrum “2” shows higher intensities of the Si, S, K, and Ca K X-rays relative to the spectrum “1”. The Si K X-rays originate from the second top layer, as has been deduced from the LIBS measurement. The presence of Si, Al, and Na in the LIBS spectra (Fig. 2f) (S is hardly detected by LIBS), and the intensity correlation between S Kα and the weak Si Kα transition in the XRF spectrum supports the presence of ultramarine. Possible thickness variations of the ultramarine layer are not expected to contribute to the intensity variations S and Si Kα transitions, as a few μm thick ultramarine layer is infinite thick from these transition energies11 (the ultramarine thickness as extracted from the sample's cross-section is in the range of about 20–30 μm). Consequently, the higher intensity of S and Si Kα transitions in the XRF spectrum “2” confirms that the top layer is thinner in area “2”. Finally, the Ca K transitions originate from the bulk CaCO3 substrate/preparation ground. The intensity variations as a function of the position are attributed to the above-lying layers, while the Ca K transition intensities are in anticorrelation with the Fe K transition intensities.
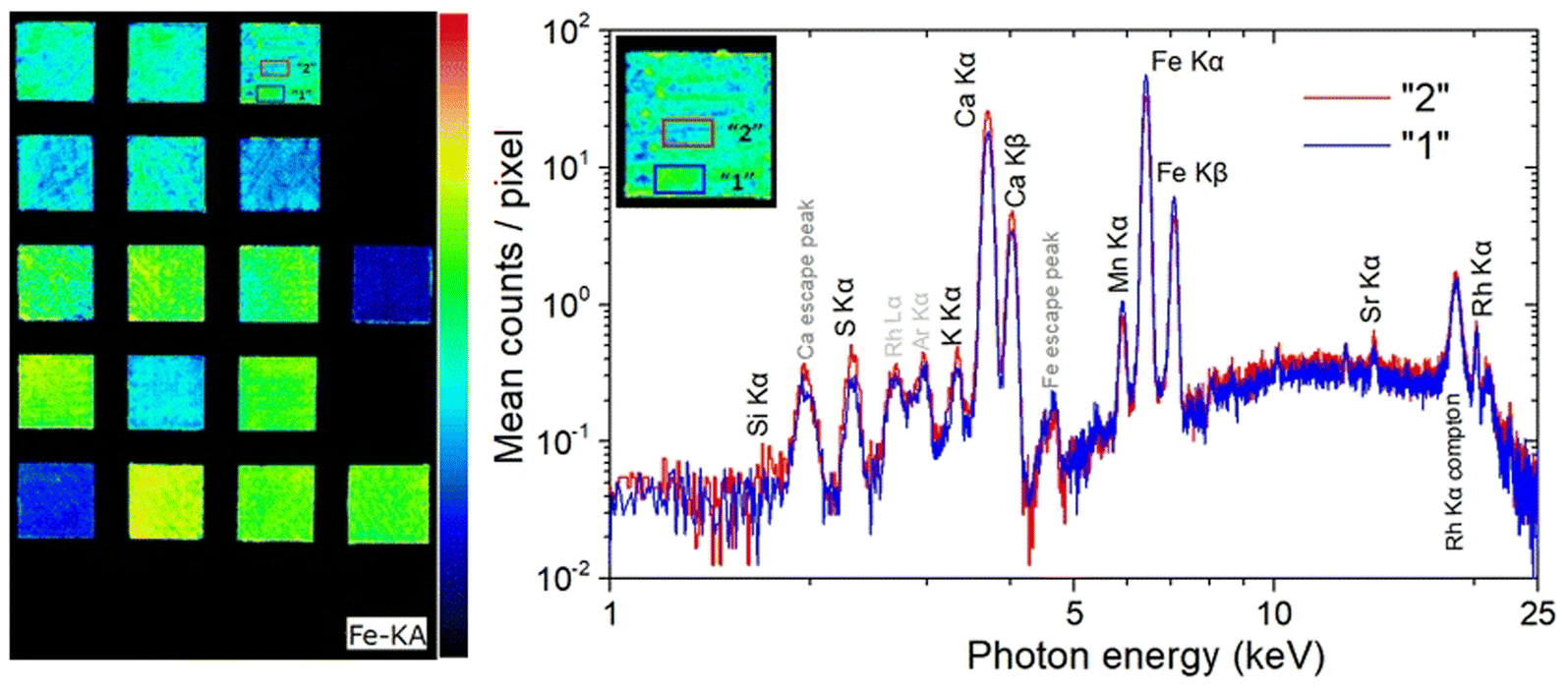 | ||
| Fig. 7 (Left) Fe Kα intensity map obtained in the framework of MA-XRF analysis. In specimen “A/B” (mars black over ultramarine) two different in Fe intensity areas are marked with blue (“1”) and red (“2”). Each area consists of 238 pixels. (Right) MA-XRF mean spectra of areas “1” and “2”. | ||
Evaluation of the fs-laser interaction
Having shown that the irradiation of a single location on the mock-up paints with only two pulses provides a solid ground for the extraction of safe conclusions on their elemental composition and layering, the next step is to examine the size of the induced damage on the subject. Therefore, photomicrographs of the produced craters on paints H and C are presented in Fig. 8. Note that these two paints showed the smallest (∼40 μm) and largest crater (∼110 μm) respectively, induced by the employed laser beam (λ = 800 nm, 30 fs pulse duration, 150 μJ per pulse, ∼1014 W cm−2). Additionally, in the case of paint H, slight discoloration of the layer is observed around the crater area. Moreover, in case of the paints A, D and E, authors were unable to spot the induced craters through the employed microscope. As for the paints B, F, G, I, J and K, the diameters of the corresponding craters were found to be equal to 60, 80, 90, 90, 80 and 60 μm respectively (estimated error ± 10 μm) with clear edges (no discoloration).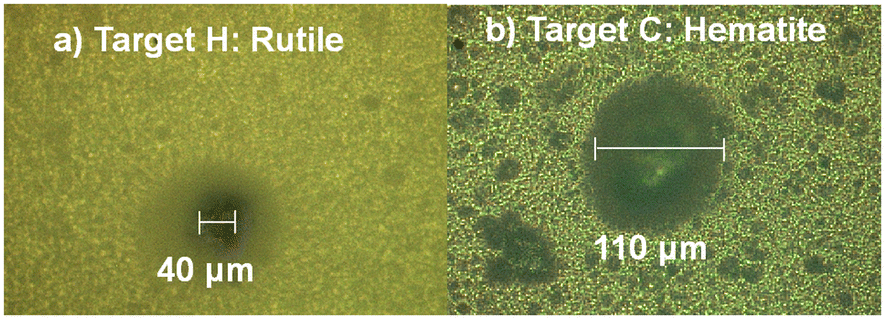 | ||
| Fig. 8 Photos from the irradiated areas of paints “H” (a) and C (b), captured using an optical microscope. | ||
Compared to the values reported in the pertinent literature, the diameters of the induced craters are considered acceptable. Indeed, most of the previous reports have used ns laser systems which despite having the advantage of being semi-portable, cause thermal effects which create extensive damage on the object.32–34 Moreover, Bruder et al., studied ultramarine-based paint layers using a macro-LIBS setup, creating holes that where measured to range between 340 and 465 μm.54 In comparison, our measurement on the same paint layer is approximately 60 μm. Other studies,30,33,55,56 reported diameters in various paint layers that where near or above 100 μm. At this point we can make a rough estimation of the amount of pigment that is removed by each laser pulse. By combining the data in Table 2 and those of the Fig. 5 we can estimate that the average thickness removed per laser pulse is roughly ∼3 μm.
Of course, even such a micro-destruction is often considered problematic in case of an art/archaeological object. In some instances, it can be avoided by applying XRF techniques. In the current study, MA-XRF measurements were performed alongside LIBS on the investigated mock-up, and the relevant analytical results were comparatively assessed. Regarding the presence of heavier elements, it is resolved absolute agreement between LIBS and XRF data. Note, however, that even though LIBS induces craters on the analyzed object, the technique cannot be deemed as obsolete, mainly because it offers (a) the ability to detect some lighter elements like Na and Al (whose detection is rather problematic via conventional XRF set-ups) and molecular fragments like C2 and CN which are of importance in many cases, and (b) the possibility to study the elemental profiling of the analyzed object. On the other hand, elements like chlorine and sulfur are difficult to safely assign using LIBS (especially in the spectral region examined in the current study), yet they can be easily detected using XRF. For instance, the Kα X-ray characteristics transitions of S (2.31 keV) and Cl (2.62 keV) are readily observable on XRF spectra. The S and Cl MA-XRF elemental map for the scanned mock-up is presented arr Fig. 9. The S distribution is profoundly seen in target I (terra ercolano) and in target I/J (terra ercolano over raw sienna), and it pertains to the presence of sulfur in gypsum which is a constituent of the terra ercolano pigment. The intensity difference between targets I and I/J should be attributed to the different thicknesses of the terra ercolano layer. Moreover, the S vanishing in the target J/I (raw sienna over terra ercolano) is attributed to the absorbance of the substrate-emitting sulfur K X-ray characteristics from the raw sienna overlayer. The S distribution is also profoundly seen in target B (ultramarine) and target B/A (ultramarine over mars black). The MA-XRF sum spectrum of target B is shown in Fig. 9b.
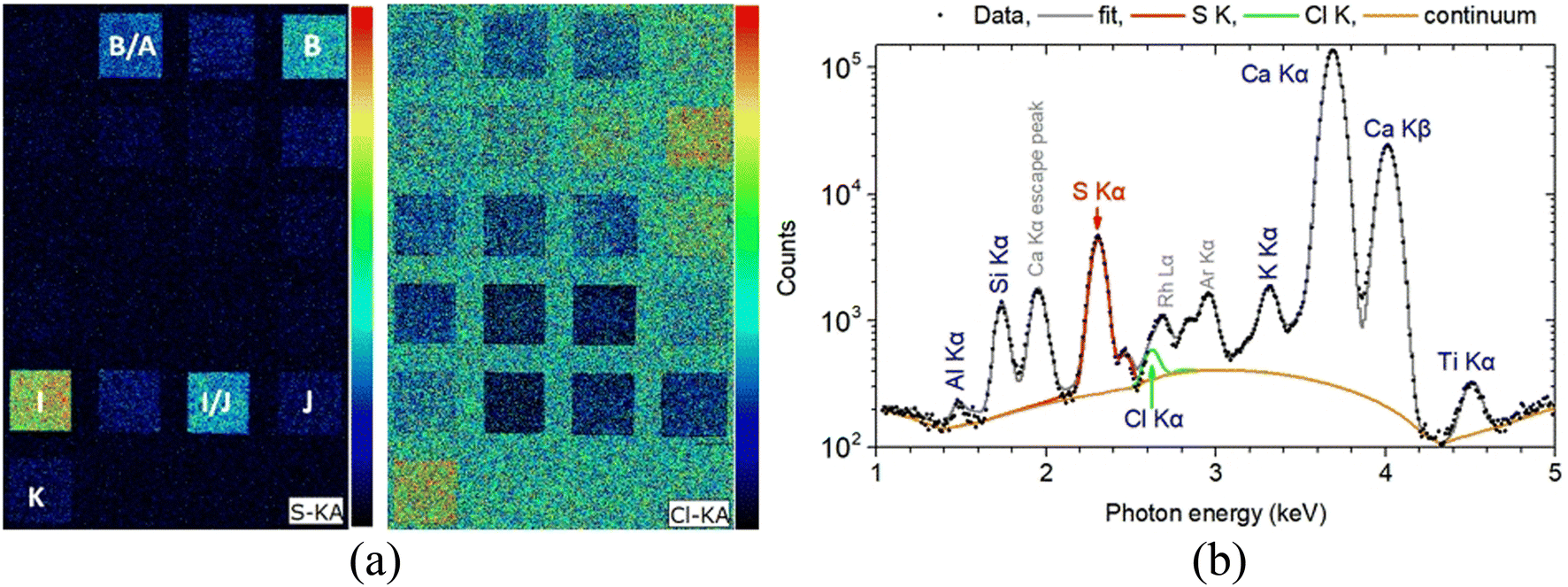 | ||
| Fig. 9 (a) Left: S and Cl Kα intensity map obtained in the framework of MA-XRF analysis. (b) Right: MA-XRF sum spectrum of 3190 pixels from target B (ultramarine) in the 1–5 keV energy range. The fitted by PyMca56 characteristic X-ray transitions and the scattered spectral lines are shown. | ||
The complementarity of LIBS and XRF techniques in the present set of experiments, is also revealed in the case of ultramarine blue where aluminum exists in the sample, yet as seen in Fig. 9b, its detection is difficult in the XRF spectra. Conversely, as seen in Fig. 2(f), tracing this element is a straightforward process in LIBS.
Finally, an observation also indicative of the complementarity of the employed techniques is related to sodium detection. As mentioned earlier, sodium is found in all the paint layers studied, although based on their presumable elemental composition, it should only be detected in ultramarine (paint B). Both techniques are important to validate the origin of the detected sodium, as LIBS allows for the detection of sodium and XRF for chlorine. Besides, the combination of the results from both techniques indicates that the detected sodium might reflect the presence of a sodium-chloride (NaCl) component.
A step further: three-layered samples
From the above data analysis of two-layered samples, it is evident that the use of the fs-LIBS technique offers very good results in terms of pigment characterization, while the created crater on the surface is rather significantly limited compared to that reported by applying ns pulses.This fact reinforces the belief that the technique could be exploited for stratigraphy studies. As a first step in this direction, a new mockup was made by using the above-mentioned construction technique. The difference is that, in this case, the samples under investigation consist of three paint layers made of different pigments. In order to enlarge the number of paint layers studied, three new pigments were used. In particular, single-layered samples of vermilion/cinnabar (HgS), lead white (2PbCO3·Pb(OH)2), and orpiment (As2S3) and their combinations were applied on a panel primed with CaCO3 pus hide glue. A photograph of the mockup is depicted in Fig. 10, while in Table 3, the codenames of the corresponding paint layers and six-layer combinations of the three distinct paints in different overlap sequences are noted.
 | ||
| Fig. 10 A photo of the mock-up with the three-layered samples is presented. | ||
| Codename | Pigment | Chemical formula | Selected spectral features |
|---|---|---|---|
| L | Cinnabar | HgS | Hg 312.5 nm |
| M | Lead white | 2PbCO3·Pb(OH)2 | Pb 261.4 nm |
| N | Orpiment | As2S3 | As 278.0 nm |
| Substrate | Calcium carbonate | CaCO3 | Ca 317.9 nm |
Furthermore, in order to better interpret the LIBS and MA-XRF data, the corresponding paint thickness of the layers was determined on cross-sections of microsamples extracted from the second mock-up (Fig. 11). Thus, the thickness of the layers found to vary in a value range of 8 to 33 μm for cinnabar, 10 to 64 μm for lead white and 10 to 56 μm for orpiment.
 | ||
| Fig. 11 (Left) Paint layer M (lead white) over paint layers L (cinnabar) and N (orpiment); OM, 200×. (Right) Paint layer N (orpiment) over paint layers L (cinnabar) and M (lead white); OM, 200×. | ||
XRF imaging measurements were performed to extract the two-dimensional elemental maps of the under-investigation mock-up. The pixel size was 300 μm, while the dwell time was 20 ms per pixel. The elemental transitions maps of the Ca Kα, Hg Lα, As Kα, and Pb Lα are shown in Fig. 12. The Ca Kα originates from the CaCO3 preparation ground layer, while Hg Lα, As Kα, and Pb Lα originate from the cinnabar, orpiment and lead white, respectively. The XRF analysis in the case of the co-existence of As, Hg, and Pb is not trivial due to the energy overlap of the As Kα transition (10.54 keV) with the Pb Lα transition (10.55 keV) and the As Kβ transition (11.73 keV) with the Hg Lβ transition (11.82 keV). The data analysis was completed by applying spectral deconvolution using the M6-Jetstream software. The intensity anti-correlation between the Ca Kα and the rest of the transitions is apparent in the case of single-layer samples. In the case of the multilayers, the Ca Kα is not observed due to its attenuation. Intensity variations across the samples allow information to be obtained about the pigments' thickness homogeneity but not about the stratigraphy.
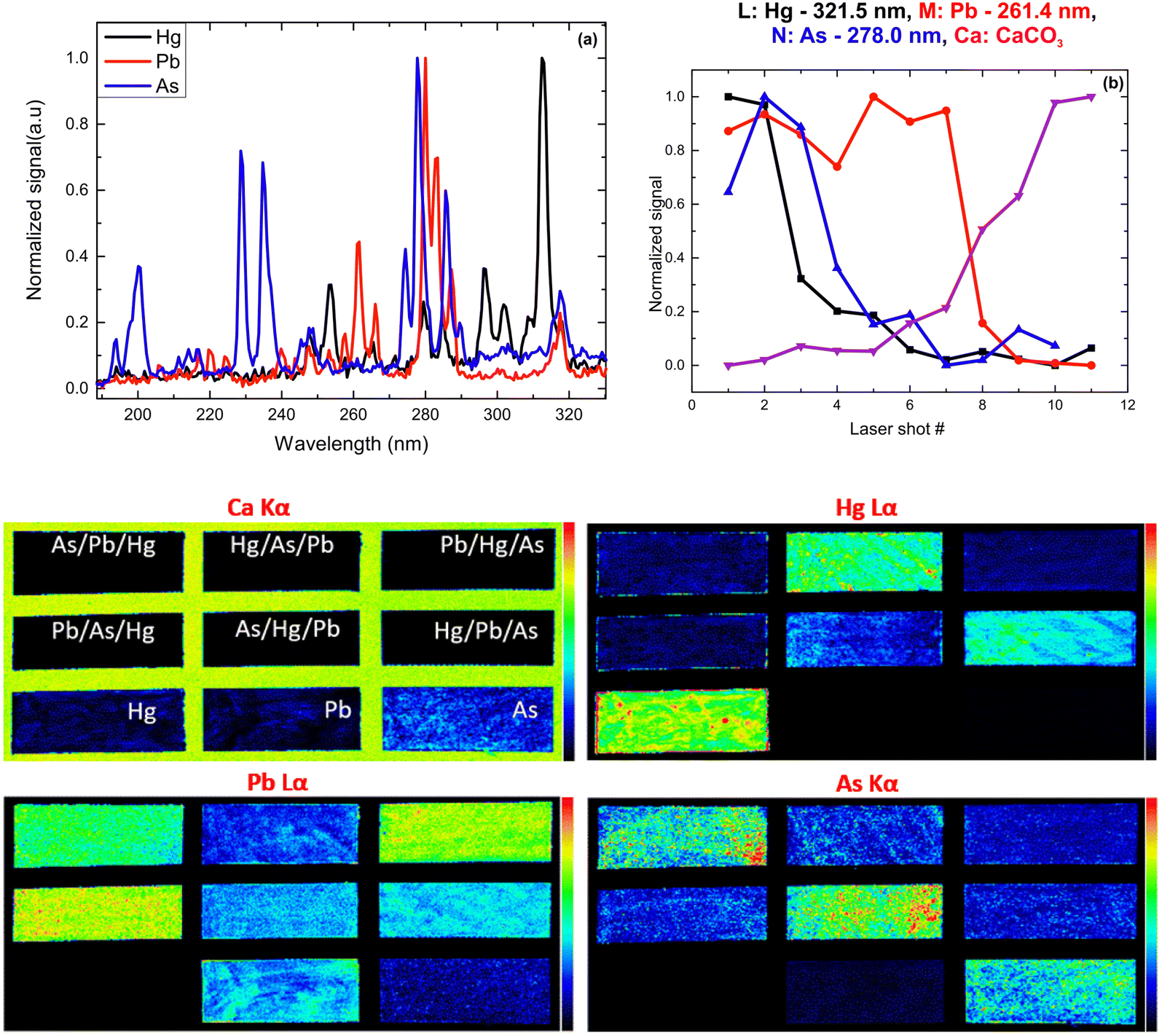 | ||
| Fig. 12 (Up) Typical LIBS spectra of the pigments in the UV spectral region (a) and single-layer signal dependence on the laser shot number (b). (Bottom) XRF-intensity distribution maps Ca Kα, Hg Lα, Pb Lα, and As Kα. | ||
Following the same methodology as before, we initially determined applying LIBS measurements the characteristic spectral feature of the elemental composition of each corresponding pigment by investigating the single layered samples (Fig. 12). Thus, the existence of cinnabar in the irradiated sample is associated with the observation of a mercury (Hg) transition 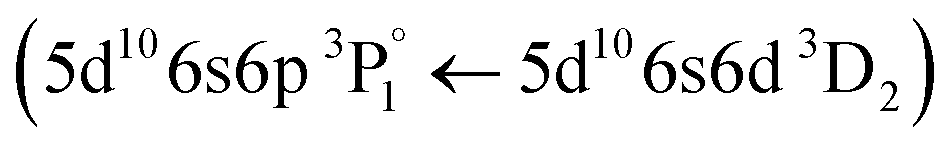 at 312.5 nm while for lead white and orpiment the spectral features at 261.4 nm
at 312.5 nm while for lead white and orpiment the spectral features at 261.4 nm 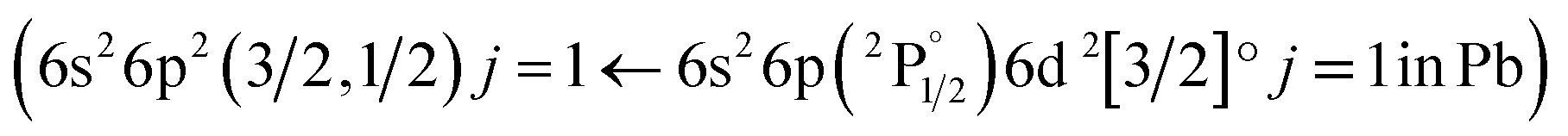 and 278.0 nm
and 278.0 nm 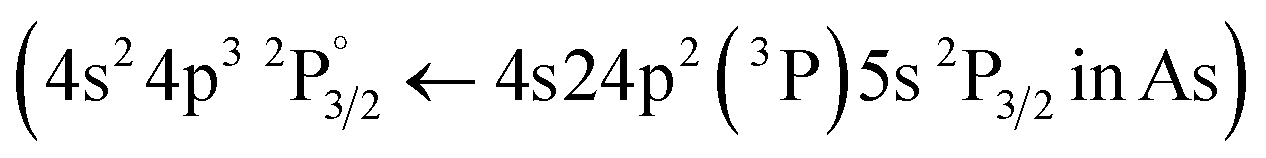 are respectively employed.
are respectively employed.
From the fs -LIBS spectra of the three-layered samples and by utilizing these spectral features as markers it is possible to identify the different paint layers that make up each sample. In Fig. 13 the intensity of the characteristic spectral markers as a function of the number of laser pulses incident to the surface is presented. Obviously, the number of pulses required to penetrate each layer is a function of the laser beam intensity and in this case, it is noted that the pulse energy of the 30 fs pulse was 160 μJ focused on an area of ∼550 μm2.
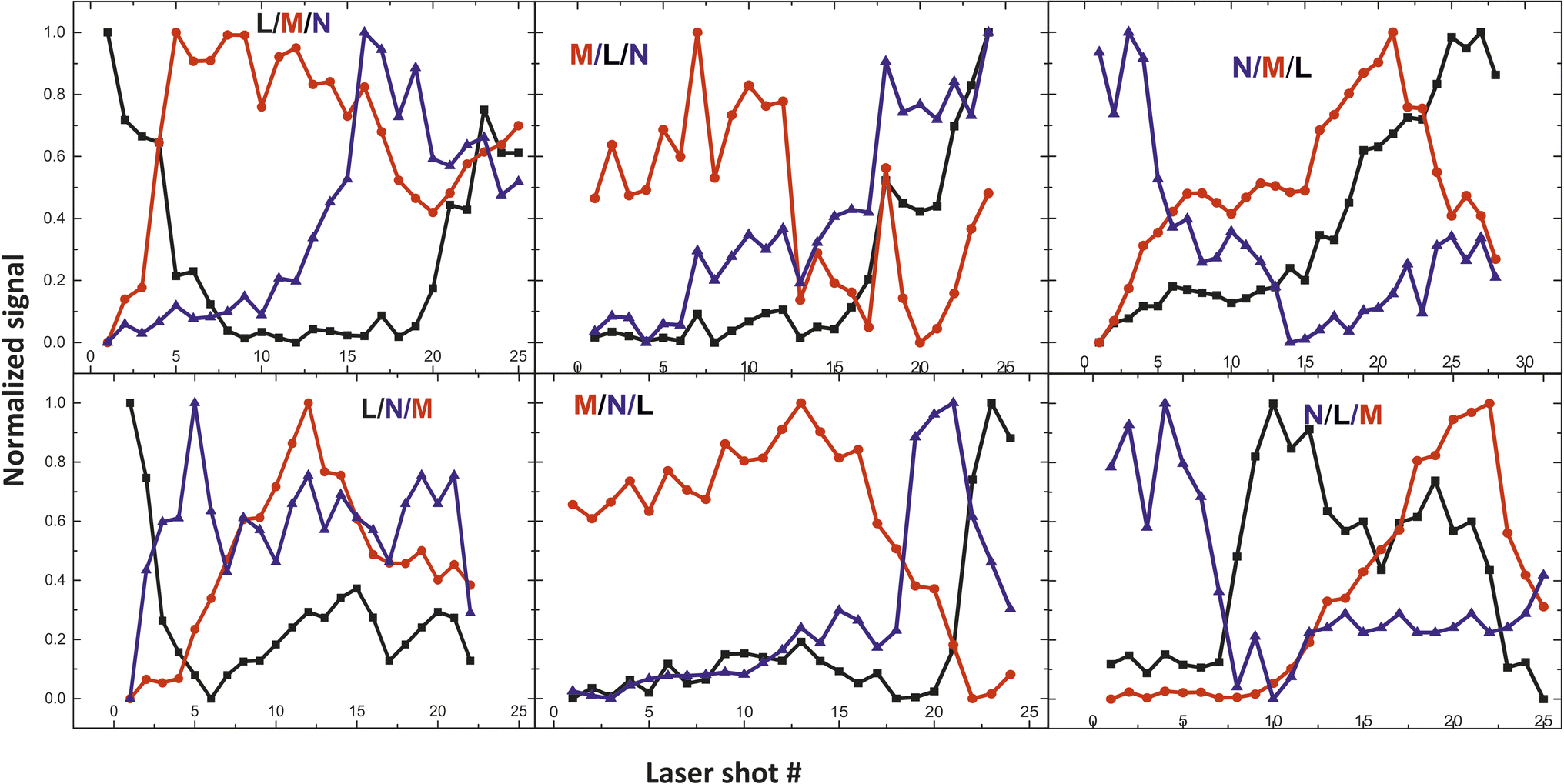 | ||
| Fig. 13 Depth profile of the three-layered samples. Three paints (composed of discrete pigments) are applied in each area. The inserts in graphs are referred to the layer order (see Table 2). | ||
Conclusions
In summary, the elemental composition of a mock-up painting sample comprising ten pigments/paints, a painting medium, and a painting substrate was investigated using Laser Induced Breakdown Spectroscopy. Analyzing the recorded spectra of the single-layered paints leads to tagging some spectral features as characteristic markers of the pigments. A remarkable fact is that sample irradiation by only a few low-energy fs laser pulses results in accurate identification of the paints.Taking it a step further, two- and three-layered samples were investigated. Thus, accurate depth profile analysis can be safely performed through LIBS based on the data collected from mono-layer samples. It is important to note that the depth profile analysis is independent of the paint placement order in the multilayered sample. These results can be considered as indicative of the potential of the fs-LIBS technique for stratigraphy studies. Obviously, there is a need for further investigation by performing experiments at different wavelength and laser intensities.
The assessment of the possibilities of the fs-LIBS technique is also strengthened by the fact that the well-known disadvantage of LIBS i.e., the creation of craters on the surface of the sample under investigation, is minimized. In the present experiments, it is found that the destruction of the paint surface was kept to a minimum, with the diameter of the craters being approximately 80 μm for most of the paints.
XRF measurements were performed too. The results show a good agreement between LIBS & XRF regarding the presence of heavier elements; however, it has been shown that these techniques are, in fact, complementary as some components were only clearly detected using LIBS (Na, Al, CN, etc.). In contrast, others were identified solely by XRF spectroscopy (Cl, S). Thus, the complementarity of LIBS and XRF spectroscopy is revealed when comprehensive elemental and depth analysis is concerned.
Author contributions
E. Kechaoglou: data curation, formal analysis investigation, methodology, formal analysis, writing – original draft, K.A. Agrafioti: data curation, formal analysis, investigation, methodology, visualization, G. P. Mastrotheodoros: resources, methodology, writing – review & editing, D. F. Anagnostopoulos: conceptualization, data curation, investigation, methodology, resources, supervision, writing – review & editing, C. Kosmidis: conceptualization, data curation, investigation, methodology, resources, supervision, project administration, writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The LIBS experiments were performed at the Central Laser Facility of University of Ioannina. We acknowledge support of this work by the project “Center for research quality analysis of cultural heritage materials and communication of science (MIS 5047233) which is implemented under the Action ‘‘Reinforcement of the Research and Innovation Infrastructure”, funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation”, Greece (NSRF 2014–2020) and co-financed by Greece and the European Union (European Regional Development Fund).References
- K. Janssens and R. Van Grieken, Compr. Anal. Chem., 2004, 42, 1–11 CrossRef.
- G. P. Mastrotheodoros, K. G. Beltsios and Y. Bassiakos, Archaeometry, 2021, 63, 753–778 CrossRef.
- I. Karapanagiotis, D. Lampakis, A. Konstanta and H. Farmakalidis, J. Archaeol. Sci., 2013, 40, 1471–1478 CrossRef.
- T. Harada, S. Spence, A. Margiolakis, S. Deckoff-Jones, R. Ploeger, A. N. Shugar, J. F. Hamm, K. M. Dani and A. R. Dani, Materials, 2017, 10, 107 CrossRef PubMed.
- B. Brunetti, C. Miliani, F. Rosi, B. Doherty, L. Monico, A. Romani and A. Sgamellotti, Analytical Chemistry for Cultural Heritage, ed. R. Mazzeo, Springer International Publishing, Cham, 2017, pp. 41–75 Search PubMed.
- K. Janssens, G. Van der Snickt, F. Vanmeert, S. Legrand, G. Nuyts, M. Alfeld, L. Monico, W. Anaf, W. De Nolf, M. Vermeulen, J. Verbeeck and K. De Wael, Top. Curr. Chem., 2016, 374, 1–52 CrossRef PubMed.
- C. Cucci, J. K. Delaney and M. Picollo, Acc. Chem. Res., 2016, 49, 2070–2079 CrossRef.
- M. Albini, S. Ridolfi, C. Giuliani, M. Pascucci, M. P. Staccioli and C. Riccucci, Front. Chem., 2020, 8, 515326 Search PubMed.
- M. F. Alberghina, R. Barraco, M. Brai, D. Fontana and L. Tranchina, Period. Mineral., 2015, 84, 569–589 Search PubMed.
- E. Pospíšilová, K. Novotný, P. Pořízka, D. Hradil, J. Hradilová, J. Kaiser and V. Kanický, Spectrochim. Acta, Part B, 2018, 147, 100–108 CrossRef.
- M. Mantler and M. Schreiner, X-Ray Spectrom., 2000, 29, 3–17 CrossRef.
- B. Beckhoff, Handbook of Practical X-Ray Fluorescence Analysis, Springer, 2006 Search PubMed.
- T. Trojek, T. Čechák and L. Musílek, Appl. Radiat. Isot., 2010, 68, 871–874 CrossRef.
- L. Musílek, T. Trojek and R. Prokeš, Radiat. Phys. Chem., 2020, 167, 108344 CrossRef.
- Y. Kataoka, H. Kohno, E. Furusawa and M. Mantler, X-Ray Spectrom., 2007, 36, 221–225 CrossRef.
- B. Kanngießer, W. Malzer and I. Reiche, Nucl. Instrum. Methods Phys. Res., Sect. B, 2003, 211, 259–264 CrossRef.
- R. Prokeš, V. Antušková, R. Šefců, T. Trojek, Š. Chlumská and T. Čechák, Radiat. Phys. Chem., 2018, 151, 59–64 CrossRef.
- A. Botto, B. Campanella, S. Legnaioli, M. Lezzerini, G. Lorenzetti, S. Pagnotta, F. Poggialini and V. Palleschi, J. Anal. At. Spectrom., 2019, 34, 81–103 RSC.
- D. M. Wong, A. A. Bol’shakov and R. E. Russo, Encycl. Spectrosc. Spectrom., 2016, 533–538 Search PubMed.
- I. Osticioli, M. Wolf and D. Anglos, Appl. Spectrosc., 2008, 62, 1242–1249 CrossRef CAS PubMed.
- S. Choi, J. Choi and C. Park, Appl. Phys. B, 2023, 129, 1–11 CrossRef.
- S. Mahmood, S. A. Abbasi, S. Jabeen and M. A. Baig, J. Quant. Spectrosc. Radiat. Transfer, 2010, 111, 689–695 CrossRef CAS.
- E. N. Rao, S. Sunku and S. V. Rao, Acta Radiol., 2015, 56, 1342–1354 CrossRef.
- F. Ruan, T. Zhang and H. Li, Appl. Spectrosc. Rev., 2019, 54, 573–601 CrossRef CAS.
- K. Melessanaki, M. Mateo, S. C. Ferrence, P. P. Betancourt and D. Anglos, Appl. Surf. Sci., 2002, 197–198, 156–163 CrossRef CAS.
- S. Tzortzakis, D. Anglos and D. Gray, Opt. Lett., 2006, 31, 1139–1141 CrossRef.
- A. Giakoumaki, K. Melessanaki and D. Anglos, Anal. Bioanal. Chem., 2007, 387, 749–760 CrossRef CAS.
- A. Giakoumaki, I. Osticioli and D. Anglos, Appl. Phys. A, 2006, 83, 537–541 CrossRef CAS.
- A. Arafat, M. Na’es, V. Kantarelou, N. Haddad, A. Giakoumaki, V. Argyropoulos, D. Anglos and A.-G. Karydas, J. Cult. Herit., 2013, 14, 261–269 CrossRef.
- E. Pospíšilová, K. Novotný, P. Pořízka, J. Hradilová, J. Kaiser and V. Kanický, Chem. Pap., 2019, 73, 2937–2943 CrossRef.
- G. Galbács, Anal. Bioanal. Chem., 2015, 407, 7537–7562 CrossRef.
- A. Elhassan, A. Giakoumaki, D. Anglos, G. M. Ingo, L. Robbiola and M. A. Harith, Spectrochim. Acta, Part B, 2008, 63, 504–511 CrossRef.
- A. Brysbaert, K. Melessanaki and D. Anglos, J. Archaeol. Sci., 2006, 33, 1095–1104 CrossRef.
- V. Piñon, C. Fotakis, G. Nicolas and D. Anglos, Spectrochim. Acta, Part B, 2008, 63, 1006–1010 CrossRef.
- T. A. Labutin, V. N. Lednev, A. A. Ilyin and A. M. Popov, J. Anal. At. Spectrom., 2016, 31, 90–118 RSC.
- S. Sheta, Z. Hou, Y. Wang and Z. Wang, Sci. Rep., 2021, 11, 1–23 CrossRef.
- B. Stuart, M. Feit, S. Herman, A. Rubenchik, B. Shore and M. Perry, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 1749–1761 CrossRef.
- B. Zhang, M. He, W. Hang and B. Huang, Anal. Chem., 2013, 85, 4507–4511 CrossRef.
- C. Conti, C. Colombo, M. Realini and P. Matousek, J. Raman Spectrosc., 2015, 46, 476–482 CrossRef.
- C. Conti, A. Botteon, C. Colombo, D. Pinna, M. Realini and P. Matousek, J. Cult. Herit., 2020, 43, 319–328 CrossRef.
- P. Veis, A. Marín-Roldán and J. Krištof, Plasma Sources Sci. Technol., 2018, 27, 95001 CrossRef.
- M. A. Khater, Opt. Spectrosc., 2013, 115, 574–590 CrossRef.
- S. J. Mousavi, M. Hemati Farsani, S. M. R. Darbani, A. Mousaviazar, M. Soltanolkotabi and A. Eslami Majd, Appl. Phys. B, 2016, 122, 106 CrossRef.
- L. J. Fernández-Menéndez, C. Méndez-López, C. Abad, J. Fandiño, C. González-Gago, J. Pisonero and N. Bordel, Spectrochim. Acta, Part B, 2022, 190, 106390 CrossRef.
- M. Alfeld and L. de Viguerie, Spectrochim. Acta, Part B, 2017, 136, 81–105 CrossRef.
- G. P. Mastrotheodoros, A. Asvestas, T. Gerodimos and D. F. Anagnostopoulos, Heritage, 2023, 6, 1903–1920 CrossRef.
- Dionysios of Fourna, The ‘Painter's Manual’ of Dionysius of Fourna: an English Translation [from the Greek] with Commentary of cod. gr. 708 in the Saltykov-Shchedrin State Public Library, Leningrad, Oakwood, London, 1996 Search PubMed.
- S. Raneri, A. Botto, B. Campanella, M. Momčilović, V. Palleschi, F. Poggialini, C. Sciuto, G. Gattiglia, F. Volpintesta, T. Selvaraj, S. Živković, G. Lorenzetti and S. Legnaioli, Spectrochim. Acta, Part B, 2022, 194, 106482 CrossRef.
- D. Sun, X. Li, Y. Yin, Y. Zhang, W. Han, Y. Wang, M. Su, C. Dong, Z. Yu and B. Su, J. Cult. Herit., 2022, 55, 399–408 CrossRef.
- Kremer Pigments, https://www.kremer-pigmente.com/en/, accessed November 2023.
- A. Staicu, I. Apostol, A. Pascu, I. Urzica, M. L. Pascu and V. Damian, Opt. Laser Technol., 2016, 77, 187–192 CrossRef.
- A. Kramida, Yu. Ralchenko, J. Reader and NIST ASD Team, NIST Atomic Spectra Database (ver. 5.2), 2014, available: http://physics.nist.gov/asd Search PubMed.
- G. P. Mastrotheodoros, A. Asvestas, T. Gerodimos and D. F. Anagnostopoulos, Heritage, 2023, 6, 1903–1920 CrossRef.
- R. Bruder, D. Menut and V. Detalle, Lasers Conserv. Artworks, 2007, 367–375 Search PubMed.
- V. Lazic, M. Romani, L. Pronti, M. Angelucci, M. Cestelli-Guidi, M. Mangano and R. Fantoni, Spectrochim. Acta, Part B, 2023, 201, 106601 CrossRef CAS.
- Y. Yin, D. Sun, M. Su, Z. Yu, B. Su, B. Shui, C. Wu, W. Han, Z. Shan and C. Dong, Opt. Laser Technol., 2019, 120, 105689 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |