
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper ions-intercalated manganese dioxide self-supporting mesoporous carbon electrode for aqueous zinc-ion batteries†
Richeng Jin,
Yuan Fang,
Beibei Gao,
Ying Wan,
Yi Zhou,
Guofeng Rui,
Wei Sun*,
Pengpeng Qiu* and
Wei Luo *
*
State Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science and Engineering, Institute of Functional Materials, Donghua University, Shanghai 201620, China. E-mail: weisun@dhu.edu.cn; qiupengpeng@dhu.edu.cn; wluo@dhu.edu.cn
First published on 31st May 2024
Abstract
In aqueous zinc-ion batteries (AZIB), layered manganese dioxide (δ-MnO2) is considered to be a suitable cathode material due to its high theoretical capacity, suitable operating voltage and Zn2+/H+ co-intercalation mechanism. However, the strong coulomb interaction between Zn2+ and δ-MnO2 results in the slow diffusion dynamics of Zn2+ in the electrochemical process, which affects the structural stability of the cathode. Herein, we report a structural design that stabilizes the δ-MnO2-layered structure by pre-intercalation of Cu2+ to expand the layer spacing, and thus improve H+-transfer kinetics. Compared with the bulk δ-MnO2, the modified cathode showed excellent electrochemical performances, including a highly reversible capacity of 280 mA h g−1 at 1 A g−1 and 62.5% capacity retention after 1500 cycles at 5 A g−1. The results shown above confirmed the possibility of increasing the capacity contribution of H+ through structural design, and provides a novel idea for the development of high-performance cathode materials.
Keywords: Aqueous zinc-ion batteries; Layered manganese dioxide; Pre-intercalation; Self-supporting electrode.
1 Introduction
The excessive consumption of traditional fossil fuels has led to the development of reliable and low-cost electrochemical energy storage systems (EESs). The latter are used to attain a wide application of clean energies such as nuclear and geothermal energy.1–7 Aqueous Zn-ion batteries (AZIB) with a suitable redox potential (−0.76 V vs. standard hydrogen electrode) and large specific capacity (820 mA h g−1) are considered to be promising EESs due to their advantages of low cost, safety, and non-toxicity.8–10 However, the structural phase transition of the cathode and dissolution of the active substance limit the whole performance of ZIB.11,12 Therefore, the exploration of a suitable cathode material is particularly important for the commercialization of AZIB.Manganese-based oxides, vanadium-based oxides, Prussian blue analogues, and organic compounds have been extensively studied.13–17 Among them, manganese oxide has attracted tremendous interest because of its high specific capacity of ∼308 mA h g−1, large operating voltage of ∼1.35 V, and low toxicity. Compared with the other phases of MnO2 (α-, β-, γ-, ε-, λ-MnO2), the layered structure of δ-MnO2 has a large layer spacing (∼0.7 nm), which makes it theoretically more suitable for the intercalation of Zn ions.18–20 However, δ-MnO2 still has the problems of poor kinetics of ion diffusion and unstable structure, which lead to poor magnification performance and rapid capacity decay in practical applications. To address these problems, pre-intercalation,21 oxygen defects,22 structural design,23 and construction of composite materials24 are usually employed to boost the electrochemical properties of the cathode. In particular, the pre-intercalation strategy is of great importance because it can increase the conductivity of MnO2 and expand the lattice spacing to improve the ion-transport dynamics. For example, Zhang et al. prepared a PVP-intercalated layered manganese dioxide by a hydrothermal method. It showed a high performance for Zn-ion storage by accelerating electron and interlayer ion transport.25 Jing et al. reported a dual-ion intercalation strategy to incorporate Mg2+ and K+ into δ-MnO2, which showed high stability during an electrochemical process.21 The above studies show that the pre-intercalation of different ions affects the contribution ratio of H+ and Zn2+. However, Zn2+ has a strong electrostatic interaction with the base material, resulting in slow kinetics, while H+ has faster reaction kinetics and weak interaction with the main structure. Hence, it is a practicable scheme to increase the capacity contribution of H+.
Herein, we prepared a Cu2+-intercalated MnO2 self-supporting electrode as the cathode of AZIB. In order to improve the intrinsic low wettability of the original carbon cloth, mesoporous structures were constructed in situ on the carbon cloth to provide a ‘cradle’ for the growth of δ-MnO2. Then, Cu2+-intercalated δ-MnO2 was grown on the substrate by a hydrothermal method. The Cu2+ intercalation greatly expanded the lattice spacing and strengthened the diffusion kinetics of H+ while maintaining the original layered structure. Benefiting from the superior structural design, the Cu–MnO2 cathode delivered a high specific capacity and excellent cycling stability.
2 Results and discussion
Fig. 1 shows the synthetic scheme of the Cu–MnO2 cathode. The polyethylene oxide-b-styrene (PEO-b-PS) copolymer template was first prepared by an atom-transfer radical-polymerization method.26 Gel permeation chromatography of PEO-b-PS (Fig. S1†) showed that the molecular weight was 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 343.0 g mol−1 and the polydispersity index was ∼1.0, indicating the high quality of PEO-b-PS. Then, mesoporous carbon layers were grown on both sides of the carbon cloths (CCs) through assembly of interfacial monomicelles, followed by a carbonization process in N2. Finally, Cu ions-intercalated δ-MnO2 was deposited on the surface of mesoporous carbon through a hydrothermal reaction.27,28 As a comparison, δ-MnO2 was prepared by a similar method without addition of the Cu precursor.
343.0 g mol−1 and the polydispersity index was ∼1.0, indicating the high quality of PEO-b-PS. Then, mesoporous carbon layers were grown on both sides of the carbon cloths (CCs) through assembly of interfacial monomicelles, followed by a carbonization process in N2. Finally, Cu ions-intercalated δ-MnO2 was deposited on the surface of mesoporous carbon through a hydrothermal reaction.27,28 As a comparison, δ-MnO2 was prepared by a similar method without addition of the Cu precursor.
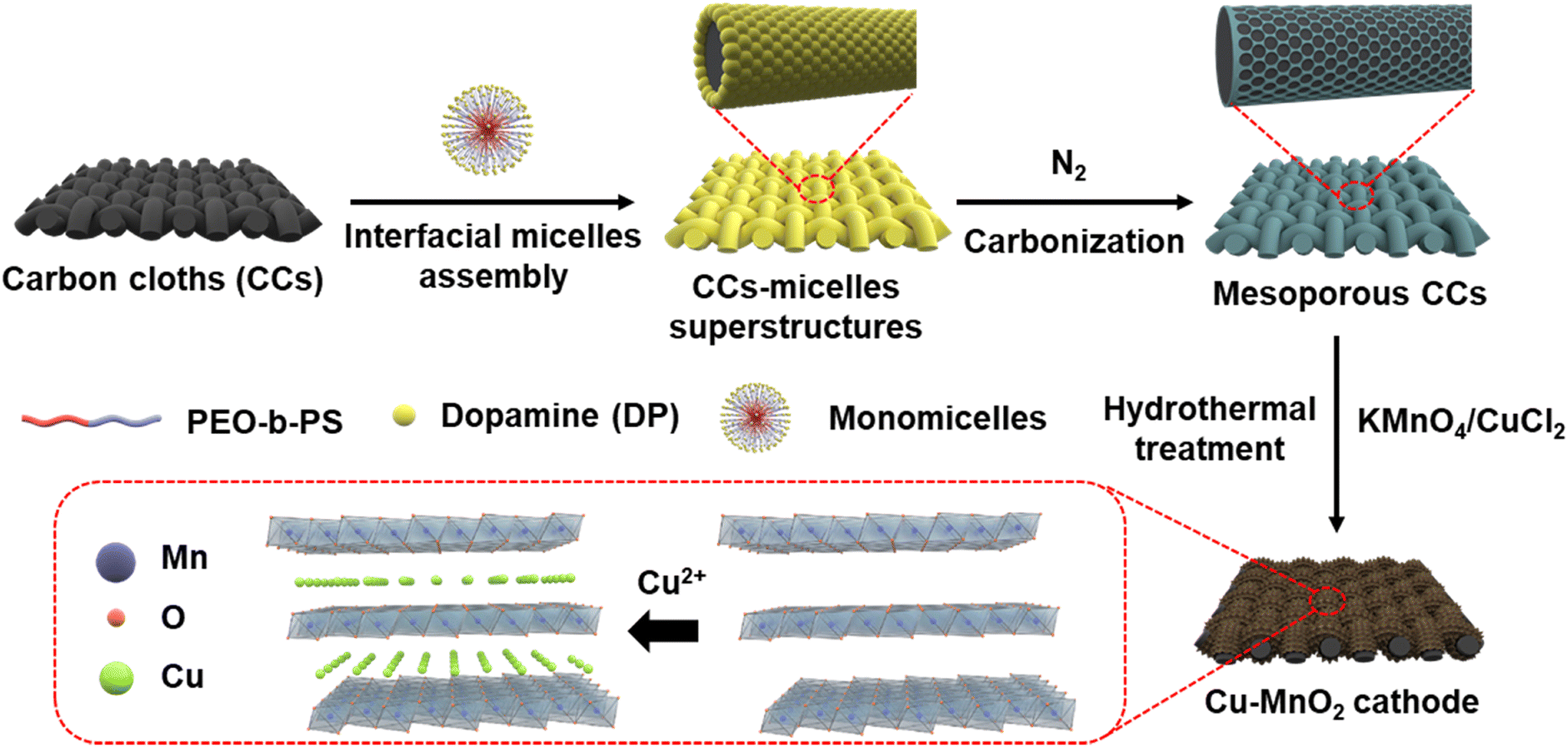 | ||
| Fig. 1 Synthesis of Cu–MnO2 (schematic). | ||
Field-emission scanning electron microscopy (FESEM) and high-resolution transmission electron microscopy (HRTEM) were used to observe the morphology and microstructure of samples. Compared with the original carbon cloth, mesoporous carbon structures were clearly deposited on the surface of the carbon cloth (Fig. S2a–d†). After hydrothermal treatment, Cu–MnO2 nanosheets grew uniformly on the surface of the mesoporous carbon cloth with an ordered nanoarray morphology (Fig. 2a–c and S3a†). TEM images (Fig. 2d and e and S3b†) also show similar nanoarray structures with a width of 75 nm. Fig. 2e displays the high-resolution HRTEM of Cu–MnO2, which clearly revealed a lamellar structure with a d-spacing of 0.75 nm which belongs to the (001) crystal plane of δ-MnO2 (JCPDS no. 80-1098). The distinct diffraction rings in the SAED pattern showed that Cu2+ intercalation did not change the phase type of δ-MnO2. However, compared with δ-MnO2 (Fig. S3c†), the d-spacing of Cu–MnO2 increased, which was caused by Cu2+ intercalation. Energy dispersive X-ray spectroscopy (EDX) (Fig. 2g) exhibited a uniform distribution of elements in Cu–MnO2, indicating the average distribution of Cu2+ into δ-MnO2.
 | ||
| Fig. 2 (a–c) SEM images, (d) TEM image, (e) HRTEM image, (f) SAED pattern, and (g) HAADF-STEM image and corresponding elemental mapping images of Cu–MnO2. | ||
The crystal structures of cathodes were examined by XRD. In the XRD patterns of δ-MnO2, the diffraction peaks at 12°, 26.2°, and 37.5° correspond well to the (001), (002), and (−111) planes of δ-MnO2 (JCPDS no. 80-1098), indicating the synthesis of layered manganese dioxide. Cu–MnO2 showed a similar XRD pattern to that of δ-MnO2, further indicating that the intercalation of Cu2+ did not break the phase structure of δ-MnO2. Raman spectra of the cathodes (Fig. 3b) showed characteristic peaks at 500, 575 and 649 cm−1 belonging to the Mn–O–Mn stretching vibration, Mn–O vibration along the skeleton chain, and the symmetric stretching vibration Mn–O bond, respectively.29,30 These data suggested that these two samples had a typical crystal structure of birnessite-type. In addition, the difference in relative intensity of the Raman band between the two samples reflected the species variation between layered δ-MnO2 layers.31 The N2 sorption isotherms of Cu–MnO2 exhibited a typical type-IV characteristic curve and a H3-type hysteresis loop, suggesting that Cu–MnO2 had a mesoporous structure.32,33 The Brunauer–Emmett–Teller (BET) surface area and pore size of Cu–MnO2 were calculated to be 3.7 nm and 23.0 m2 g−1, respectively. In order to determine the source of the mesoporous structure of the electrode, nitrogen absorption and desorption tests were carried out on the treated carbon cloth. As shown in Fig. S2c,† the treated carbon cloth had a mesoporous structure, and its specific surface area and pore size were 3.096 m2 g−1 and 7.4 nm, respectively. This result showed that the mesoporous structure of the electrode was mainly derived from the accumulation of Cu–MnO2 nanosheets rather than mesoporous carbon cloth. The elemental composition of Cu–MnO2 was investigated by X-ray photoelectron spectroscopy (XPS). In the Mn 2p XPS spectrum of MnO2 (Fig. 3d), dominant peaks of Mn 2p3/2 and Mn 2p1/2 were located at 642.4 and 654.3 eV, respectively. In the Mn 2p XPS spectrum of Cu–MnO2, both Mn4+ and Mn3+ were present.34 Among them, the amount of Mn4+ was 71.98% (Fig. S4b†) and the average chemical valence of Mn was +3.72. The valence state of Mn can be explained by the representative multiplet splitting caused by the exchange of electrons at its 3s–3d level.35–37 As shown in Fig. 3e, the splitting energy (ΔE) of the two 3s peaks increased from 4.73 eV of δ-MnO2 to 5.03 eV of Cu–MnO2, indicating an increase in the amount of Mn3+ in order to balance the valence imbalance caused by Cu2+ intercalation. XPS of O 1s (Fig. 3f) showed lattice oxygen (Olat) and O (OMn–OH) which belonged to Mn–OH.34 We found that after pre-intercalation of Cu2+, Olat was transferred in a more negative direction so that Olat had more electrons, which was due to electron transfer between the embedded Cu2+ and lattice oxygen. The increase in the charge around the O atom greatly strengthened the adsorption capacity of Cu–MnO2 for H+, improved the diffusion capacity of H+, and more H+ participated in the electrode reaction, thereby showing higher electrochemical performance. In Fig. S4,† characteristic peaks of Cu2+ were found at 934.3 eV and 954 eV, and belonged to Cu 2p 3/2 and Cu 2p 1/2, respectively. There were oscillating satellite peaks of Cu2+ at 942.7 eV and 962.4 eV, suggesting that Cu2+ had embedded within δ-MnO2.
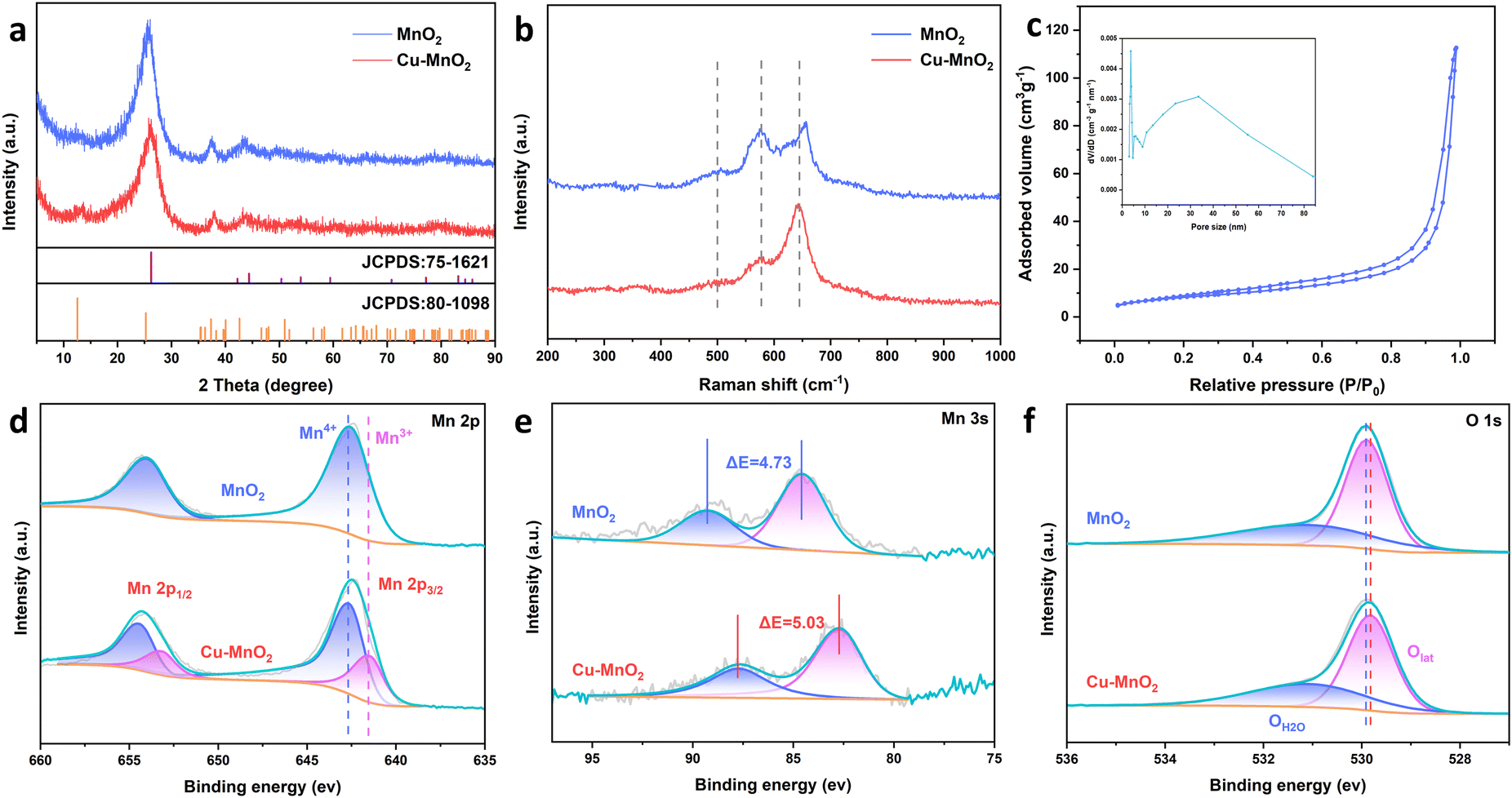 | ||
| Fig. 3 (a) XRD curves, (b) Raman spectra, (c) N2 sorption isothermals and pore-size distribution of Cu–MnO2. (d) XPS spectra of Mn 2p. (e) XPS spectra of Mn 3s. (f) O 1s XPS spectra of Cu–MnO2. | ||
For research into the electrochemical properties of Cu–MnO2, a coin cells configuration with a zinc anode, glass fiber film (Whatman, GF/D), and 2 M ZnSO4 and 0.2 M MnSO4 as the electrolyte were employed. The addition of MnSO4 can inhibit the dissolution of Mn2+ during discharge processes.38,39 The cyclic voltammetry (CV) curves of Cu–MnO2 and δ-MnO2 at 0.1 mV s−1 are shown in Fig. 4a and S5a,† respectively. The two pairs of redox peaks of 1.56/1.38 V and 1.6/1.26 V corresponded to the deintercalation/intercalation of H+ and Zn2+, respectively. The almost overlapping CV curve after the first two cycles (Fig. 4a) illustrated the stable and reversible charge and discharge processes of the Cu–MnO2 cathode material. At the same time, the larger CV area of Cu–MnO2 compared with that of δ-MnO2 also indicated that Cu–MnO2 provided more capacity. The galvanostatic charge–discharge (GCD) voltage profiles of Cu–MnO2 are shown in Fig. 4b, and display two charge–discharge platforms, which is consistent with CV curve. The reduction in capacity after the first cycle may have been due to the phase transition of Cu–MnO2 caused by incomplete deintercalation/intercalation of Zn2+ in Cu–MnO2. No obvious change of charge/discharge curves in subsequent cycles indicated that the Cu–MnO2 electrode had excellent cyclic stability. The cycling performance of Cu–MnO2 and δ-MnO2 cathode under the rate of 1 A g−1 is shown in Fig. 4c. The Cu–MnO2 cathode stabilized at 300 mA h g−1 after the initial capacity reduction, and was maintained at 280 mA h g−1 after 400 cycles, with a capacity attenuation rate of 7%. By comparison, the δ-MnO2 cathode exhibited poor cyclic performance, with 70% capacity fading after 400 cycles. It is worth noting that an increase in capacity was found in Fig. 4c. This was because as the cycle progresses, H+ is consumed in the electrolyte, and the pH gradually increases, triggering the deposition of additional Mn2+, resulting in an increase in capacity.39 Fig. 4d shows the rate capability of Cu–MnO2 and δ-MnO2 cathodes measured at a rate of 0.3–5 A g−1. As the current density increased, the specific capacity of electrodes decreased due to the limitation of electrochemical reaction kinetics. The Cu–MnO2 cathode exhibited a discharge capacity of 617.85, 487.36, 315.14, 130.64, and 87.33 mA h g−1 at a rate of 0.3, 0.5, 1, 3, and 5 A g−1, respectively. After restoration to 0.3 A g−1, the capacity of the Cu–MnO2 cathode reached up to 506 mA h g−1. In contrast, δ-MnO2 exhibited much lower capacity than Cu–MnO2 at the same rate. This finding suggested that the insertion of copper ions could modulate the electronic configuration and efficient ion/charge storage sites of δ-MnO2 and, thus, improve the electron-transport and ion-diffusion capacities of the δ-MnO2 cathode. The greater-than-theoretical-capacity of Cu–MnO2 was due to the additional capacity provided by the 2 e− deposition reaction of Mn2+ added in the electrolytic liquid.39,40 To deeply research long-term cycling stability at high rates, Cu–MnO2 and δ-MnO2 cathodes were tested at a rate of 5 A g−1. As shown in Fig. 4e, Cu–MnO2 maintained a capacity of 114.23 mA h g−1 after 1500 cycles of 5 A g−1, which was much higher than that of δ-MnO2 (80.4 mA h g−1). At the same time, Cu–MnO2 could maintain a stable coulombic efficiency of nearly 100% under long cycles while δ-MnO2 showed an unstable coulombic efficiency. These results showed that Cu2+ could be used as the pillars in the interlayer of Cu–MnO2 to obtain excellent electrochemical properties compared with δ-MnO2.21,41 For a more comprehensive study of the electrochemical properties of Cu–MnO2, we tested its performance in different electrolytes at a current density of 1 A g−1. It can be seen from Fig. S6† that Cu–MnO2 exhibited a relatively stable long cycle performance in different electrolytes, but its specific capacity was different in different electrolytes. In an aqueous electrolyte without Mn2+, Cu–MnO2 showed the highest electrochemical performance in 2 M ZnSO4, but poor specific capacity in 3 M and 2 M Zn(CF3SO3)2. This was because SO4− easily binds to H2O and forms a water-rich layer around the cathode, which provides kinetic support for H+ embedding, while CF3SO3− is hydrophobic and preferentially adsorbed at the cathode to create a low-water environment, which hinders H+ embedding and results in reduced capacity.42 In a non-aqueous point solution, DMSO electrolyte with 5% H2O showed better electrochemical performance than that of pure DMSO. This was because the high electrostatic interaction within the ion pair (Zn2+/CF3SO3−) in DMSO and the high desolvation penalty at the electrode–electrolyte interface will reduce ion transport and increase the energy loss of the charge-storage reaction.43 At the same time, the lack of H+ in DMSO stops it providing capacity contribution, so it shows poor specific capacity. The addition of H2O causes MnO2 to adsorb water during the discharge process and form superhydrated zinc pyroxene, which greatly promotes the diffusion of Zn2+ and improves the reactivity of the Zn-insertion reaction. In addition, we compared Cu–MnO2 with other reported advanced ZIBs cathodes (Table S1†). Cu–MnO2 showed better electrochemical properties than that of most cathodes.
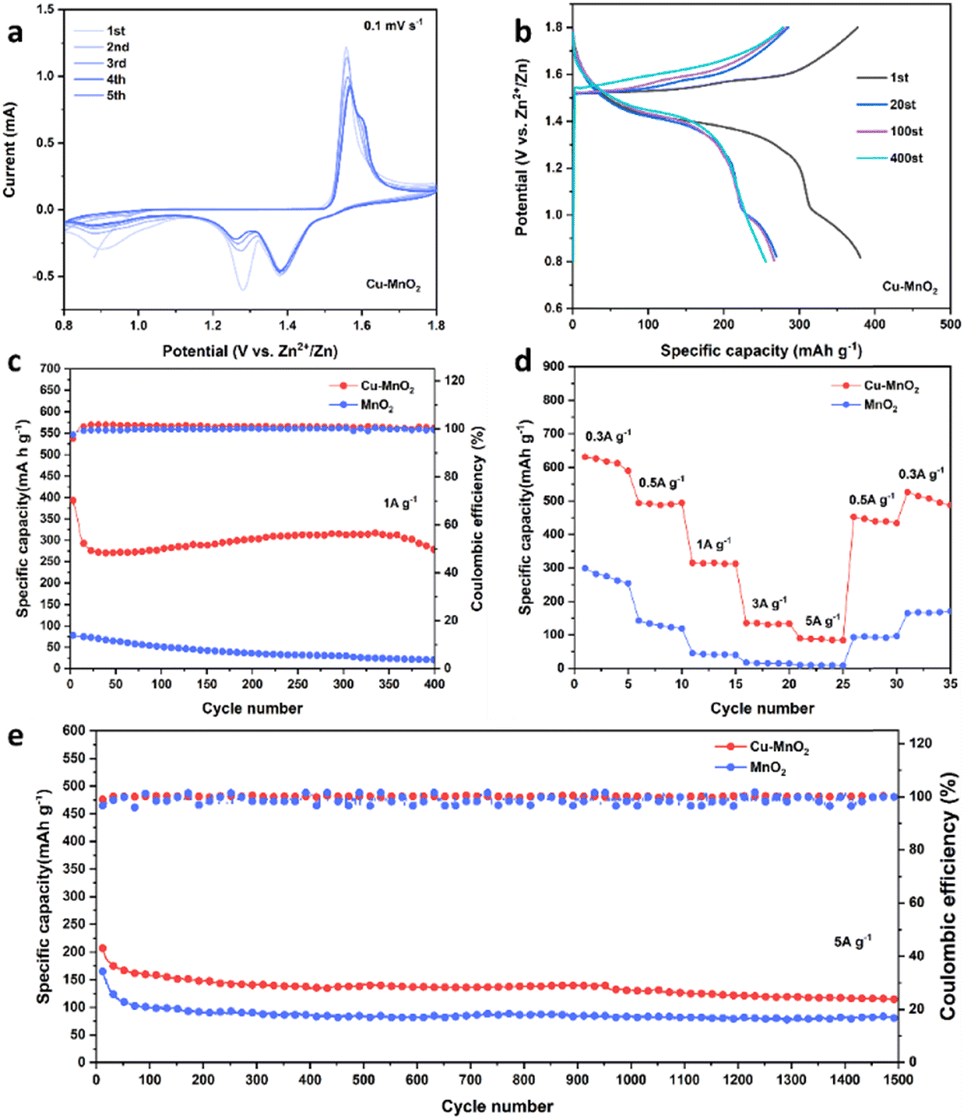 | ||
| Fig. 4 (a) CV curves at 0.1 mV s−1. (b) GCD curves at 1.0 A g−1 of Cu–MnO2. (c) Cycling performance of Cu–MnO2 and δ-MnO2 at a current density of 1.0 A g−1. (d) Rate performance for Cu–MnO2 and δ-MnO2. (e) Long-cycling performance at 5.0 A g−1. | ||
In order to study how Cu2+ improves the electrochemical performance of δ-MnO2, the ion diffusion kinetics and energy storage mechanism of the cathode were investigated by CV, galvanostatic intermittent titration technique (GITT) and electrochemical impedance spectroscopy (EIS). As shown in Fig. 5a, the CV curves of Cu–MnO2 at different scanning rates were measured from 0.1 to 0.5 mV s−1. Two pairs of redox peaks were detected during the scan, which corresponded to the reversible insertion–extraction of Zn2+/H+. Although polarization during the intercalation/deintercalation process led to a slight peak shift, the well-maintained shape of the CV curves indicated a reversible redox reaction of the Cu–MnO2 cathode. The variation of peak current (i) with scan rate (ν) follows an empirical power-law, which can be expressed by the following formula:44
| i = aνb | (1) |
| i(ν) = k1ν + k2ν1/2 | (2) |
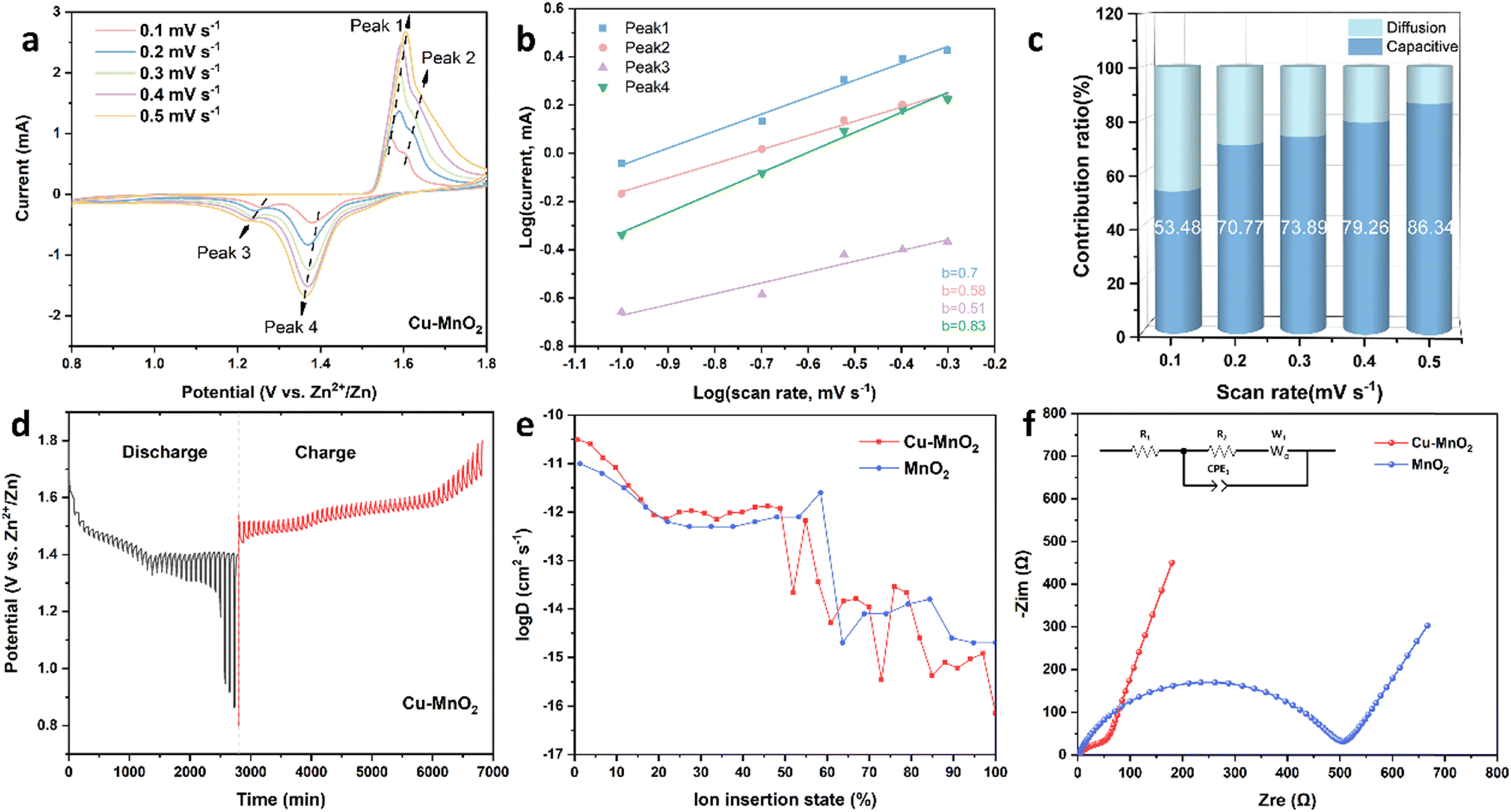 | ||
| Fig. 5 (a) CV curves of 0.1–0.5 mV s−1. (b) Corresponding dependence of logi versus logv at selected oxidation/reduction states. (c) Capacitive-controlled ratio at various scan rates. (d) GITT curves of Cu–MnO2. (e) Diffusion coefficient of Cu–MnO2 and δ-MnO2. (f) EIS spectra. | ||
The response currents generated by capacitive and diffusion-controlled behaviour were calculated using k1ν and k2ν1/2. As shown in Fig. 5c, at the scanning rates of 0.1, 0.2, 0.3, 0.4 and 0.5 mV s−1, the capacitance contribution was 53.48%, 70.77%, 73.89%, 79.26% and 86.34%, respectively, indicating that capacitor control was dominant in Cu–MnO2. However, the δ-MnO2 cathode exhibited a lower capacitance ratio, as shown in Fig. S6,† indicating that Cu2+ could introduce more capacitance to promote the electrochemical performance of δ-MnO2.
The ion diffusion coefficient was measured by constant current intermittent titration (GITT) and calculated according to the following formula:45
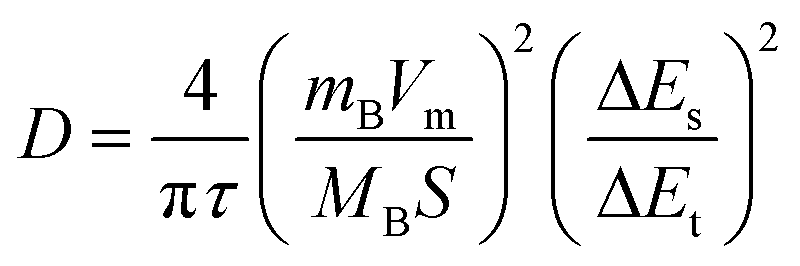 | (3) |
τ: constant current pulse duration
mB: mass loading of the reactant
MB: molecular volume
Vm: molar volume
S: electrode–electrolyte interface area
ΔEs: change in steady-state voltage
ΔEt: change in discharge/charge voltage
Fig. 5d and S7† are the GITT curves of Cu–MnO2 and δ-MnO2 cathodes. The appearance of the two platforms was caused by the sequential intercalation of H+ and Zn2+.46,47 The ion diffusion coefficient (D) is shown in Fig. 5e. The diffusion coefficient before 50% was greater than the diffusion coefficient after 50%, which corresponded to the intercalation process of H+ and Zn2+, respectively. This observation indicated that the diffusion kinetics of H+ and Zn2+ were different in the electrochemical process. Compared with δ-MnO2, the H+ diffusion coefficient of the Cu–MnO2 cathode was one order of magnitude higher, indicating that the pre-intercalation of Cu2+ accelerated the diffusion rate of H+, improved the electrochemical performance and enhanced the capacity of Cu–MnO2. Fig. 5f shows the Nyquist plot of Cu–MnO2 and δ-MnO2 cathodes. The Rct (charge transfer impedance) of Cu–MnO2 and δ-MnO2 obtained by circuit fitting were 57.92 (Ω) and 505.04 (Ω), respectively, which further confirmed the improvement of the kinetics performance of δ-MnO2 by Cu2+.
To further investigate the ion-transfer behaviour and energy-storage mechanism of the Cu–MnO2 cathode in AZIB, the crystal configuration, valence state and micromorphology of the cathode were studied by ex situ XRD, XPS and SEM. Fig. 6a and b show the XRD patterns of Cu–MnO2 in different states during one cycle. When fully discharged (0.8 V), there were characteristic peaks at 34.6° and 58.69° which were attributed to Zn2MnO4 (JCPDS no. 19-1459). Diffraction peaks were observed at 28.14° and 32.8°, which corresponded to Zn4SO4(OH)6·H2O (ZSH) (JCPDS no. 39-0690), while the characteristic peak of MnOOH (JCPDS no. 24-0713) was observed at 21.13°. The formation of MnOOH and ZSH during discharge indicated that H+ intercalated into δ-MnO2. With more H+ being consumed, a large amount of OH− was produced in the electrolyte, and excess OH−, Zn2+ and SO42− reacted to obtain ZSH. The formation of Zn2MnO4 was due to the insertion of Zn2+ into the interlayer of δ-MnO2. The shift of the characteristic peaks of δ-MnO2 from 37.39° and 12.52° to 35.86° and 10.93° confirmed the behaviour of ion intercalation. In the process of charging, the characteristic peaks of MnOOH and ZSH gradually disappeared, which suggested the good cyclic reversibility of H+. The characteristic peaks of Zn2MnO4 could still be observed when the charge was up to 1.8 V, indicating the poor reaction kinetics of Zn2+. The process stated above suggested the reaction mechanism of H+ and Zn2+ intercalation. The ex situ SEM results (Fig. 6c and f) showed that ZSH nanosheets were formed when discharged to 0.8 V, and disappeared when charged to 1.8 V, revealing the good reversibility of the microstructure. Fig. 6d and e show the ex situ XPS spectra of Mn 2p and O 1s at full charge (1.8 V) and complete discharge (0.8 V). When fully discharged (0.8 V), the peak area of Mn3+ increased significantly, further demonstrating the embedding of Zn2+ and H+, and leading to the reduction of Mn4+ to Mn3+. When fully charged (1.8 V), the content of Mn4+ increased, indicating that the deintercalation of ions re-oxidized Mn3+ to Mn4+. The XPS spectrum of Zn 2p is shown in Fig. S8.† The peak of Zn 2p decreased when charging from 0.8 to 1.8 V, which indicated the deintercalation behaviour of Zn2+. The observation of the peak of Zn at 1.8 V suggested that the Zn2+ mentioned above was not completely deintercalated, indicating the low diffusion kinetics of Zn2+. In addition, Fig. 6d reveals the change of O valence state during charge and discharge. When discharging to 0.8 V, the peak area of OH2O was increased significantly due to the formation of ZSH, while the peak area of Olat decreased due to the intercalation of ions. The opposite process during charging indicated that H+ and Zn2+ left the lattice of Cu–MnO2 and the large chunks of ZSH disappeared, which showed that Cu–MnO2 had good reversibility. The above results showed that the Cu–MnO2 cathode exhibited good cyclic reversibility, and it was a feasible strategy to improve the embedding and removal ability of H+ by intercalation of Cu2+. In addition, we studied the structural changes during the long-term cycling of the battery. As shown in Fig. S10a,† an obvious characteristic peak of ZSH appeared in the XRD curve of the electrode after 1500 cycles, indicating that irreversible phase transition occurred during the long cycle. As mentioned above, H+ in the electrolyte was consumed during the electrochemical cycle, which led to an increase in pH and triggered ZSH deposition. The deposition of a large amount of ZSH would hinder the contact between Cu–MnO2 and the electrolyte, resulting in a decrease in capacity. Fig. S10b† shows the SEM image of the electrode after 1500 C, and many ZSH patches in the figure confirm this interpretation.
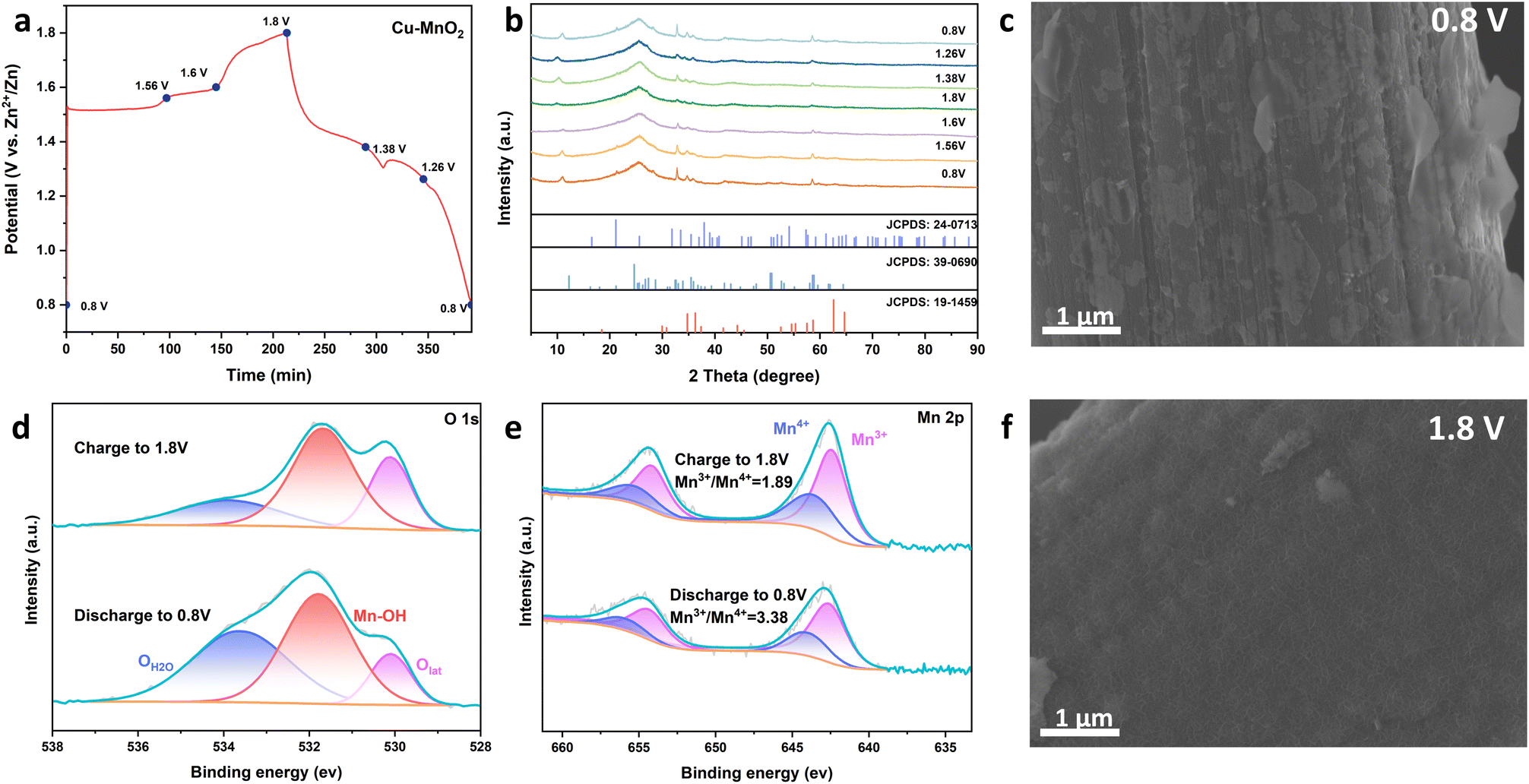 | ||
| Fig. 6 (a) Typical cycle curves. (b) Ex situ XRD patterns at selected potentials. SEM images of (c) 0.8 V and (f) 1.8 V. XPS spectra of (d) O1s and (e) Mn2p. | ||
3 Conclusions
Cu2+-intercalated δ-MnO2 (Cu–MnO2) was grown in situ on a mesoporous CCs substrate by a hydrothermal method as the cathode of AZIB, and its electrochemical properties were investigated. The designed Cu–MnO2 cathodes showed excellent specific capacity, rate performance and cycle stability in AZIB. Cu–MnO2 exhibited a reversible capacity of 280 mA h g−1 at the rate of 1 A g−1, maintained 93% capacity after 400 cycles, and had a high specific capacity of 114.23 mA h g−1 at 5 A g−1 after 1500 cycles. The combined use of CV, GITT, EIS, ex situ XRD and XPS showed that the intercalated Cu2+ formed a strong ionic bond with the oxygen atom, thus improving the stability of the layered structure and enhancing the H+ transfer kinetics.4 Experimental section
4.1 Chemicals and materials
PEO-5000, 2-bromoisobutyryl bromide, and N,N,N′,N′,N′-pentamethyldiethylenetriamine (PMDETA) were purchased from Aladdin. Tetrahydrofuran (THF) (>99%), pyridine (>99%), cuprous bromide (CuBr), ammonium hydroxide (NH4OH, 28–30 wt%), dopamine hydrochloride, and copper(II) chloride dihydrate (CuCl2·2H2O) were obtained from Greagent. Potassium permanganate (KMnO4) and ethanol were sourced from Sinopharm Group Chemical Reagents. All reagents were of analytical grade and did not require further purification. Deionized water was used for all experiments.4.2 Synthesis of Cu–MnO2 and δ-MnO2
Cu–MnO2 and δ-MnO2 was prepared by a one-step hydrothermal method. KMnO4 and CuCl2·2H2O at a molar ratio of 10:1 were dissolved in 10 ml of water under vigorous stirring for 10 min. One piece of carbon cloth (2 × 2) was placed into the above solution, transferred into a Teflon™-lined stainless-steel autoclave and heated at 180 °C for 2 h. After cooling down to room temperature, the obtained sample was washed with DI water thrice, and dried using a vacuum oven at 60 °C for 12 h. δ-MnO2 was synthesized using a similar procedure without CuCl2·2H2O.
4.3 Materials characterization
The crystal structure of the sample was characterized by X-ray diffraction (XRD) using a D/Max-2550 PC diffractometer (Rigaku). The microstructure of electrodes was characterized by scanning electron microscopy (SEM) using the MAIA3 system (TESCAN) and transmission electron microscopy (TEM) employing a 2100F system (JEM). HAADF-STEM and EDS elemental mapping were done using a Talos F200S setup operating at 200 kV. Raman spectra were acquired using a reflex Raman microscope with laser excitation at 532 nm. N2 sorption was carried out with an Autosorb-iQ system (Quantachrome) to measure the Brunauer–Emmett–Teller (BET) surface area and pore-size distribution. The BET method was employed for the determination of the specific surface area of catalysts. The Barrett–Joyner–Halenda (BJH) model was employed to analyze the pore-size distribution and pore volume. Electrode surface valence and chemical environment were investigated using an 250Xi setup (Escalab) equipped with Al Kα radiation.4.4 Electrochemical measurements
Cu–MnO2 was prepared on carbon cloth by in situ growth and used as the cathode (area: 0.25 cm2) without conductive additives or adhesives. The mass loading of electrodes was 1.5 mg cm−2. CR2032 coin cells were assembled in air. The configuration was with a zinc anode (diameter: 14 mm), glass fiber film (GF/D; Whatman), and 2 M ZnSO4 and 0.2 M MnSO4 as the electrolyte to estimate the properties of cathodic active materials. The galvanostatic discharge/charge (GCD) curve and GITT curve were created on the NEWARE battery test system, and the test voltage was 0.8–1.8 V. The specific capacity of the battery was calculated using this formula:where Qs represents the specific capacity (mA h g−1), I represents the current (mA), m denotes the mass of the active substance (g) and Δt represents the discharge time.
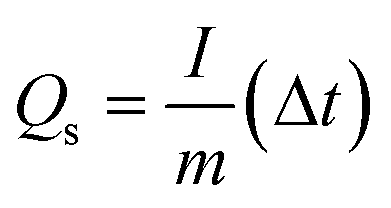
The discharge GITT profiles were obtained at 0.1 A g−1 for 1200 s followed with a rest for 1 h. The CV curve and EIS curve were measured by an electrochemical workstation (CHI600E). The frequency range of EIS was 100 kHz to 0.01 Hz. All tests were performed at room temperature.
Author contributions
Conceptualization: P. Q., R. J. and Y. F. Methodology: P. Q., R. J., W. S. and Y. F. Validation: P. Q. and R. J. Data analysis, R. J. Investigation: R. J. Resources, P. Q. and W. L. Data curation: R. J., Y. Z., G. R., Y. W. and B. G. Writing (preparation of original draft): R. J., W. S. and P. Q. Writing (review and editing): P. Q., W. S. and R. J. Visualization: R. J. Supervision: P. Q. and W. L. Project administration: P. Q. and W. L. Funding acquisition: P. Q. and W. L.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52225204, 52173233, 52002059 and 52202085), Innovation Program of Shanghai Municipal Education Commission (2021-01-07-00-03-E00109), Natural Science Foundation of Shanghai (23ZR1479200), “Shuguang Program” supported by the Shanghai Education Development Foundation and Shanghai Municipal Education Commission (20SG33), DHU Distinguished Young Professor Program (LZA2022001 and LZB2023002) and the Fundamental Research Funds for Central Universities (2232023G-07, 2232024Y-01).References
- J. H. Lim, J. H. Won, M. K. Kim, D. S. Jung, M. Kim, C. Park, S.-M. Koo, J.-M. Oh, H. M. Jeong, H. Sohn and W. H. Shin, Synthesis of flower-like manganese oxide for accelerated surface redox reactions on nitrogen-rich graphene of fast charge transport for sustainable aqueous energy storage, J. Mater. Chem. A, 2022, 10, 7668–7676 RSC.
- S.-J. Tan, J. Yue, Z. Chen, X.-X. Feng, J. Zhang, Y.-X. Yin, L. Zhang, J.-C. Zheng, Y. Luo, S. Xin and Y.-G. Guo, Asymmetric fire-retardant quasi-solid electrolytes for safe and stable high-voltage lithium metal battery, Energy Mater. Adv., 2024, 5, 0076 CrossRef CAS.
- Z. Zheng, X. Zhong, Q. Zhang, M. Zhang, L. Dai, X. Xiao, J. Xu, M. Jiao, B. Wang, H. Li, Y. Jia, R. Mao and G. Zhou, An extended substrate screening strategy enabling a low lattice mismatch for highly reversible zinc anodes, Nat. Commun., 2024, 15, 753 CrossRef CAS PubMed.
- M. Fu, H. Yu, S. Huang, Q. Li, B. Qu, L. Zhou, G.-C. Kuang, Y. Chen and L. Chen, Building sustainable saturated fatty acid-zinc interfacial layer toward ultra-stable zinc metal anodes, Nano Lett., 2023, 23, 3573–3581 CrossRef CAS PubMed.
- H. Xia, W. Zhang, S. Cao and X. Chen, A figure of merit for fast-charging Li-ion battery materials, ACS Nano, 2022, 16, 8525–8530 CrossRef CAS PubMed.
- L. Miao, R. Wang, W. Xin, L. Zhang, Y. Geng, H. Peng, Z. Yan, D. Jiang, Z. Qian and Z. Zhu, Three-functional ether-based co-solvents for suppressing water-induced parasitic reactions in aqueous Zn-ion batteries, Energy Storage Mater., 2022, 49, 445–453 CrossRef.
- Z. Zhu, S. Cao, X. Ge, S. Xi, H. Xia, W. Zhang, Z. Lv, J. Wei and X. Chen, Enabling the high-voltage operation of layered ternary oxide cathodes via thermally tailored interphase, Small Methods, 2022, 6, 2100920 CrossRef CAS PubMed.
- L. Ma, S. Chen, C. Long, X. Li, Y. Zhao, Z. Liu, Z. Huang, B. Dong, J. A. Zapien and C. Zhi, Achieving high-voltage and high-capacity aqueous rechargeable zinc ion battery by incorporating two-species redox reaction, Adv. Energy Mater., 2019, 9, 1902446 CrossRef CAS.
- F. Ming, Y. Zhu, G. Huang, A. H. Emwas, H. Liang, Y. Cui and H. N. Alshareef, Co-solvent electrolyte engineering for stable anode-free zinc metal batteries, J. Am. Chem. Soc., 2022, 144, 7160–7170 CrossRef CAS PubMed.
- D. Selvakumaran, A. Pan, S. Liang and G. Cao, A review on recent developments and challenges of cathode materials for rechargeable aqueous Zn-ion batteries, J. Mater. Chem. A, 2019, 7, 18209–18236 RSC.
- X. Jia, C. Liu, Z. G. Neale, J. Yang and G. Cao, Active materials for aqueous zinc ion batteries: Synthesis, crystal structure, morphology, and electrochemistry, Chem. Rev., 2020, 120, 7795–7866 CrossRef CAS PubMed.
- X. Ji and L. F. Nazar, Best practices for zinc metal batteries, Nat. Sustain., 2024, 7, 98–99 CrossRef.
- S. Chang, J. F. F. Gomez, S. Katiyar, G. Morell and X. Wu, Trivalent indium metal as a high-capacity, high-efficiency, low-polarization, and long-cycling anode for aqueous batteries, J. Am. Chem. Soc., 2023, 145, 24746–24754 CAS.
- Y. Zhu, G. Liang, X. Cui, X. Liu, H. Zhong, C. Zhi and Y. Yang, Engineering hosts for Zn anodes in aqueous Zn-ion batteries, Energy Environ. Sci., 2024, 17, 369–385 RSC.
- X. Hu, G. Wang, J. Li, J. Huang, Y. Liu, G. Zhong, J. Yuan, H. Zhan and Z. Wen, Significant contribution of single atomic Mn implanted in carbon nanosheets to high-performance sodium-ion hybrid capacitors, Energy Environ. Sci., 2021, 14, 4564–4573 RSC.
- S. Liu, L. Kang, J. M. Kim, Y. T. Chun, J. Zhang and S. C. Jun, Recent advances in vanadium-based aqueous rechargeable zinc-ion batteries, Adv. Energy Mater., 2020, 10, 2001769 CrossRef.
- J. Zhao, Z. Xu, Z. Zhou, S. Xi, Y. Xia, Q. Zhang, L. Huang, L. Mei, Y. Jiang, J. Gao, Z. Zeng and C. Tan, A safe flexible self-powered wristband system by integrating defective MnO2-x nanosheet-based zinc-ion batteries with perovskite solar cells, ACS Nano, 2021, 15, 10597–10608 CrossRef CAS PubMed.
- J. Wang, J. G. Wang, H. Liu, C. Wei and F. Kang, Zinc ion stabilized MnO2 nanospheres for high capacity and long lifespan aqueous zinc-ion batteries, J. Mater. Chem. A, 2019, 7, 13727–13735 RSC.
- T. Xiong, Y. Zhang, W. S. V. Lee and J. Xue, Defect engineering in manganese-based oxides for aqueous rechargeable zinc-ion batteries: A review, Adv. Energy Mater., 2020, 10, 2001769 CrossRef CAS.
- X. Zhu, Z. Cao, W. Wang, H. Li, J. Dong, S. Gao, D. Xu, L. Li, J. Shen and M. Ye, Superior-performance aqueous zinc-ion batteries based on the In Situ growth of MnO2 nanosheets on V2CTX MXene, ACS Nano, 2021, 15, 2971–2983 CrossRef CAS PubMed.
- F. Jing, Y. Liu, Y. Shang, C. Lv, L. Xu, J. Pei, J. Liu, G. Chen and C. Yan, Dual ions intercalation drives high-performance aqueous Zn-ion storage on birnessite-type manganese oxides cathode, Energy Storage Mater., 2022, 49, 164–171 CrossRef.
- Y. Zhao, S. Zhang, Y. Zhang, J. Liang, L. Ren, H. J. Fan, W. Liu and X. Sun, Vacancy-rich Al-doped MnO2 cathodes break the trade-off between kinetics and stability for high-performance aqueous Zn-ion batteries, Energy Environ. Sci., 2024, 17, 1279–1290 RSC.
- H. Tang, W. Chen, N. Li, Z. Hu, L. Xiao, Y. Xie, L. Xi, L. Ni and Y. Zhu, Layered MnO2 nanodots as high-rate and stable cathode materials for aqueous zinc-ion storage, Energy Storage Mater., 2022, 48, 335–343 CrossRef.
- X. Xiao, L. Zhang, W. Xin, M. Yang, Y. Geng, M. Niu, H. Zhang and Z. Zhu, Self-assembled layer of organic phosphonic acid enables highly stable MnO2 cathode for aqueous znic batteries, Small, 2024, 2309271 CrossRef CAS PubMed.
- A. Zhang, R. Zhao, Y. Wang, J. Yue, J. Yang, X. Wang, C. Wu and Y. Bai, Hybrid superlattice-triggered selective proton Grotthuss intercalation in δ-MnO2 for high-performance zinc-ion battery, Angew. Chem., Int. Ed., 2023, 62, e202313163 CrossRef CAS PubMed.
- P. Qiu, Y. Yao, W. Li, Y. Sun, Z. Jiang, B. Mei, L. Gu, Q. Zhang, T. Shang, X. Yu, J. Yang, Y. Fang, G. Zhu, Z. Zhang, X. Zhu, T. Zhao, W. Jiang, Y. Fan, L. Wang, B. Ma, L. Liu, Y. Yu and W. Luo, Sub-nanometric manganous oxide clusters in nitrogen doped mesoporous carbon nanosheets for high-performance lithium–sulfur batteries, Nano Lett., 2021, 21, 700–708 CrossRef CAS PubMed.
- H. Yao, H. Yu, Y. Zheng, N. W. Li, S. Li, D. Luan, X. W. Lou and L. Yu, Pre-intercalation of ammonium ions in layered δ-MnO2 nanosheets for high-performance aqueous zinc-ion batteries, Angew. Chem., Int. Ed., 2023, 62, e202315257 CrossRef CAS PubMed.
- J. Zhang, W. Li, J. Wang, X. Pu, G. Zhang, S. Wang, N. Wang and X. Li, Engineering p-band center of oxygen boosting H+ intercalation in δ-MnO2 for aqueous zinc ion batteries, Angew. Chem., Int. Ed., 2023, 62, e202215654 CrossRef CAS PubMed.
- G. Wang, Y. Wang, B. Guan, J. Liu, Y. Zhang, X. Shi, C. Tang, G. Li, Y. Li, X. Wang and L. Li, Hierarchical K-birnessite-MnO2 carbon framework for high-energy-density and durable aqueous zinc-ion battery, Small, 2021, 17, 2104557 CrossRef CAS PubMed.
- R. Cao, P. Zhang, Y. Liu and X. Zheng, Ammonium-treated birnessite-type MnO2 to increase oxygen vacancies and surface acidity for stably decomposing ozone in humid condition, Appl. Surf. Sci., 2019, 495, 143607 CrossRef CAS.
- M. Ju, Z. Chen, H. Zhu, R. Cai, Z. Lin, Y. Chen, Y. Wang, J. Gao, X. Long and S. Yang, Fe(III) docking-activated sites in layered birnessite for efficient water oxidation, J. Am. Chem. Soc., 2023, 145, 11215–11226 CrossRef CAS PubMed.
- F. Wei, T. Zhang, R. Dong, Y. Wu, W. Li, J. Fu, C. Jing, J. Cheng, X. Feng and S. Liu, Solution-based self-assembly synthesis of two-dimensional-ordered mesoporous conducting polymer nanosheets with versatile properties, Nat. Protoc., 2023, 18, 2459–2484 CrossRef CAS PubMed.
- J. Li, R. Li, W. Wang, K. Lan and D. Zhao, Ordered mesoporous crystalline frameworks toward promising energy applications, Adv. Mater., 2024, 36, 2311460 CrossRef CAS PubMed.
- T. Sun, Q. Nian, S. Zheng, J. Shi and Z. Tao, Layered Ca0.28MnO2·0.5H2O as a high performance cathode for aqueous zinc-ion battery, Small, 2020, 16, 2000597 CrossRef CAS PubMed.
- G. Zhu, W. Zhu, Y. Lou, J. Ma, W. Yao, R. Zong and Y. Zhu, Encapsulate α-MnO2 nanofiber within graphene layer to tune surface electronic structure for efficient ozone decomposition, Nat. Commun., 2021, 12, 4152 CrossRef CAS PubMed.
- H. Wang, R. Guo, H. Li, J. Wang, C. Du, X. Wang and Z. Zheng, 2D metal patterns transformed from 3D printed stamps for flexible Zn//MnO2 in-plane micro-batteries, Chem. Eng. J., 2022, 429, 132196 CrossRef CAS.
- W. Ma, K. W. Kwan, R. Wu and A. H. W. Ngan, High-performing, linearly controllable electrochemical actuation of c-disordered δ-MnO2/Ni actuators, J. Mater. Chem. A, 2021, 9, 6261–6273 RSC.
- C. Li, R. Kingsbury, A. S. Thind, A. Shyamsunder, T. T. Fister, R. F. Klie, K. A. Persson and L. F. Nazar, Enabling selective zinc-ion intercalation by a eutectic electrolyte for practical anodeless zinc batteries, Nat. Commun., 2023, 14, 3067 CrossRef CAS PubMed.
- H. Yang, T. Zhang, D. Chen, Y. Tan, W. Zhou, L. Li, W. Li, G. Li, W. Han, H. J. Fan and D. Chao, Protocol in evaluating capacity of Zn–Mn aqueous batteries: A clue of pH, Adv. Mater., 2023, 35, 2300053 CrossRef CAS PubMed.
- J. Huang, J. Zeng, K. Zhu, R. Zhang and J. Liu, High-performance aqueous zinc–manganese battery with reversible Mn2+/Mn4+ double redox achieved by carbon coated MnOx nanoparticles, Nano-Micro Lett., 2020, 12, 110 CrossRef CAS PubMed.
- K. Ma, Q. Li, C. Hong, G. Yang and C. Wang, Bi doping-enhanced reversible-phase transition of α-MnO2 raising the cycle capability of Aqueous Zn–Mn batteries, ACS Appl. Mater. Interfaces, 2021, 13, 55208–55217 CrossRef CAS PubMed.
- O. Zhanadilov, H. J. Kim, A. Konarov, J. Jeong, J.-H. Park, K. Y. Chung, Z. Bakenov, H. Yashiro and S.-T. Myung, Layered manganese oxide cathode boosting high-capacity and long-term cyclability in aqueous Zinc-Ion batteries, Energy Storage Mater., 2024, 67, 103283 CrossRef.
- W. Kao-ian, J. Sangsawang, P. Kidkhunthod, S. Wannapaiboon, M. Suttipong, A. Khampunbut, P. Pattananuwat, M. T. Nguyen, T. Yonezawa and S. Kheawhom, Unveiling the role of water in enhancing the performance of zinc-ion batteries using dimethyl sulfoxide electrolyte and the manganese dioxide cathode, J. Mater. Chem. A, 2023, 11, 10584–10595 RSC.
- X.-Z. Zhai, J. Qu, J. Wang, W. Chang, H.-J. Liu, Y.-H. Liu, H. Yuan, X. Li and Z.-Z. Yu, Diffusion-driven fabrication of yolk-shell structured K-birnessite@mesoporous carbon nanospheres with rich oxygen vacancies for high-energy and high-power zinc-ion batteries, Energy Storage Mater., 2021, 42, 753–763 CrossRef.
- N. Zhang, M. Jia, Y. Dong, Y. Wang, J. Xu, Y. Liu, L. Jiao and F. Cheng, Hydrated layered vanadium oxide as a highly reversible cathode for rechargeable aqueous zinc batteries, Adv. Funct. Mater., 2019, 29, 1807331 CrossRef.
- J. Wang, J.-G. Wang, H. Liu, Z. You, Z. Li, F. Kang and B. Wei, A highly flexible and lightweight MnO2/graphene membrane for superior zinc-ion batteries, Adv. Funct. Mater., 2021, 31, 2007397 CrossRef CAS.
- D. Wang, L. Wang, G. Liang, H. Li, Z. Liu, Z. Tang, J. Liang and C. Zhi, A superior δ-MnO2 cathode and a self-healing Zn-δ-MnO2 battery, ACS Nano, 2019, 13, 10643–10652 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4im00042k |
| This journal is © Institute of Process Engineering of CAS 2024 |