
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Auto-relay catalysis for the oxidative carboxylation of alkenes into cyclic carbonates by a MOF catalyst†
Ha Phan a,
Pol de la Cruz-Sáncheza,
María Jesús Cabrera-Afonsoab and
Belén Martín-Matute*a
a,
Pol de la Cruz-Sáncheza,
María Jesús Cabrera-Afonsoab and
Belén Martín-Matute*a
aDepartment of Organic Chemistry, Stockholm University, 106 91 Stockholm, Sweden. E-mail: belen.martin.matute@su.se
bOrganic Chemistry Department, Universidad Autónoma de Madrid (UAM), Avda. Francisco Tomás y Valiente 7, Cantoblanco 28049, Madrid, Spain
First published on 30th January 2025
Abstract
In this study, we present the preparation and application of a new manganoporphyrin Hf-MOF catalyst, Hf-PCN-222(Mn) for the direct oxidative carboxylation of alkenes with CO2, leading to the effective formation of cyclic organic carbonates (COCs). In contrast to the conventional two-step process, this one-step methodology eliminates the need for the preparation, purification, and handling of epoxides. Hf-PCN-222(Mn) operates under very mild conditions, enabling the synthesis of a wide variety of COCs from alkenes (23 examples, up to 75% yield), as well as the chemoselective and size-selective carboxylation of dienes (7 examples, up to 61% yield). Additionally, we observed that Hf-PCN-222(Mn) could be recycled multiple times without significant loss of activity, providing insight into the sustainability of this approach.
Green foundation1. In this study, we introduce Hf-PCN-222(Mn), a new MOF-based catalyst for the direct oxidative carboxylation of alkenes with CO2. This methodology advances in the application of CO2 as a C1 synthon for the preparation of high-value compounds, contributing to the circularity CO2 and to the reduction of the carbon footprint. For the first time we show that the methodology can be applied to a broad range of substrates. The catalyst shows size and chemoselectivity.2. Preparing Hf-PCN-222(H2) with microwave-assisted techniques lowers preparation time significantly (from days to hours) and minimizes “non-green” or hazardous solvent/reagents use. The MOF-catalyst efficiently yields cyclic organic carbonates from alkenes using CO2 as feedstock, eliminating toxic epoxide preparation and handling. The reaction runs solvent-free at low temperatures and atmospheric CO2. That allows the exploration of a wide range of alkenes (30 examples) with diverse functional groups. Additionally, this heterogeneous catalyst is reusable up to five times without yield loss. 3. Future efforts should focus on (1) replacing hazardous solvents in MOF synthesis and (2) substituting PhIO with a more environmentally friendly oxidant. |
Introduction
Carbon dioxide is an abundant, inexpensive, non-flammable, and non-toxic carbon source that can be used as a C1-synthon for the preparation of organic compounds.1–12 Recycling carbon from CO2 as a precursor contributes to the circularity of CO2, and to the reduction of the carbon footprint.One of the ways to use CO2 as a carbon feedstock for the synthesis of organic molecules is the preparation of cyclic organic carbonates (COCs). These species are rather stable compounds that present low toxicity, low flammability, and high boiling point. Due to these properties, COCs are broadly used as polar aprotic solvents, lithium battery electrolytes, and as monomers for the synthesis of cyclic carbonate functional polymers.4,13–19 They also serve as synthons or intermediates for the preparation of a variety of functionalized organic compounds, such as 1,2-diols,20 β-hydroxycarbamates,21 and other functionalized intermediates22 by replacing other toxic and difficult to handle reagents, such as phosgene or cyanates.
Currently, one of the most widely used process for the synthesis of COCs is the cycloaddition reaction of CO2 with epoxides (Fig. 1A, right).23,24 In this well-known process, a Lewis acid species activates the epoxide through the oxygen atom while, at the same time, a nucleophile facilitates the ring opening of the epoxide enabling the insertion of CO2 (Fig. 1A, left).17,25 However, the major drawbacks for this methodology, especially in the metal-free examples, have been the limited selectivity, and the need to use high temperatures and very high pressures.26–33 However, recent advances in catalyst design have enabled the cycloaddition reaction under milder conditions, and using atmospheric CO2 pressure.6,34–44 Furthermore, the sustainability of metal-based catalysts for the synthesis of COCs has been reviewed by North and co-workers.45
 | ||
| Fig. 1 Strategies to synthesize COCs from CO2 and this work. | ||
Although epoxides are readily available substrates, accessible via oxidation of alkenes,46–48 they are highly toxic (potentially mutagenic)49,50 and unstable, requiring special conditions for transportation, handling, and storage. To address the challenges associated with the inherent properties of epoxides, recent research has focused on the in situ generation of epoxides from alkenes and their subsequent conversion into cyclic carbonates. Therefore, the direct conversion of alkenes to COCs via oxidative carboxylation using readily available alkenes avoids the direct handling of epoxides, being a less toxic, safer and more sustainable alternative (Fig. 1B).51–62 Ideally, a single catalyst is used for both steps under the same reaction conditions, a process referred to as auto-relay catalysis.63,64 However, this approach remains challenging since it requires a catalyst that would mediate two catalytic reactions of rather different nature, namely, alkene epoxidation using an oxidant, and a cycloaddition reaction of CO2, a gas reagent, to the epoxide.51–62 In this context, metal–organic frameworks (MOFs) with their high affinities towards CO2,65–70 and their structural and chemical tunability, have the potential to catalyze tandem or auto-relay reactions that involve the use of CO2 as a C1 feedstock.51–53 The use of MOFs as catalysts in CO2 cycloadditions to epoxides has been previously demonstrated.71–75 For example, we reported that Zr-PCN-222(Co) catalyzes the CO2 cycloaddition to epoxides under very mild conditions (atmospheric pressure and at room temperature).76 These advances have prompted several research groups to develop MOF-based systems as sole catalysts for the tandem oxidative carboxylation of alkenes.54,58,77–87 However, the application of MOFs for the oxidative cycloaddition of alkenes with CO2 suffer limitations due to the need of high temperatures58,79–83,86,87 and high pressures.54,77,79,80,84,85 They often require the use of chlorinated solvents (CH2Cl2),84,86 or other solvents such as decane, classified as a volatile organic compound (VOC).57,78–83,85,87 A major common drawback that is also encountered is the lack of selectivity88–91 when the reaction is applied to styrene structures, due to formation of styrene diols, phenyl acetaldehydes (e.g. 4a, vide infra), benzaldehydes and polystyrene during the epoxidation step, as well as due to unconsumed styrene oxide intermediate,84 Further, the scope has been limited to primarily styrene as the substrate. Therefore, a catalyst for the selective and high-yielding oxidative carboxylation of a large scope alkenes remains elusive.
Towards achieving this goal, an alkene oxidation method reported by Nam and co-workers caught our attention.92 The authors reported the stochiometric use of Mn(III)-iodosylarene porphyrin adducts to mediate the epoxidation of alkenes at low temperatures. However, catalytic loading of Mn was only tolerated when 20 equiv. of alkene were used with respect to the oxidant, PhIO. This opened the possibility for the development of a manganese-based catalytic system able to conduct simultaneously the oxidation of the alkene and the cycloaddition step in a two-step one-pot manner without the use of solvent.
Therefore, with these antecedents, herein we present a new and straightforward method to rapidly synthesize, for the first time, manganoporphyrinic Hf-based MOF catalyst Hf-PCN-222(Mn), as well as its application in the auto-relay catalytic oxidative carboxylation of alkenes, affording a wide range of cyclic organic carbonates (COCs, Fig. 1C). This catalyst overcomes limitations of previous systems: the reaction is solventless, runs at mild temperature (40 °C), and under 1 atm of CO2. It does not suffer from lack of selectivity, and it mediates the chemoselective oxidative carboxylation of dienes. Its catalytic efficiency is demonstrated in the oxidative carboxylation of a large variety of alkenes, giving access to COCs, important intermediates in synthetic organic chemistry. Taking advantage of its porous nature, size-selectivity is also demonstrated. Furthermore, recyclability test showed that Hf-PCN-222(Mn) could be reused multiple times for the oxidative carboxylation of alkenes without loss of activity or crystalline structure, giving insights on the sustainability of the process.
Results and discussion
Regarding the preparation of our MOF catalytic system, in 2015, Farha's group reported the first synthesis of Hf-PCN-222(Fe),93 a three-dimensional Hf6-oxo cluster nodes connected by Fe-porphyrin linkers with alternate hexagonal and triangular 1D channels. In their approach, the MOF-catalyst was prepared by using the pre-metalated linker under solvothermal conditions. More recently, the group of Su reported a similar protocol for the synthesis of Hf-PCN-222(Pd), also under solvothermal conditions.94 However, the pre-metalation approach is only applicable when the metal (M) in the porphyrin does not interfere with the carboxylate functionalities of the linker, and when metal does not leach from the porphyrin to the MOF structure under synthetic conditions resulting in lower metal (M) incorporation. Preparing these PCN-222(M) MOFs requires complex and long procedures, involving preparation of the metalated porphyrin linkers prior to the MOF synthesis. Commonly, carboxylic group protection – deprotection steps to prevent metal (M)-carboxylate coordination (M = Fe and Mn), are needed. Therefore, we first focused on developing a method to prepare Hf-PCN-222(M), (M = Mn or Co). For that, we used a modified strategy for the preparation of the related Zr-PCN-222(M) MOFs (Fig. 2A).76 | ||
| Fig. 2 Synthesis (A) and PXRD patterns (B) of Hf-PCN-222 (H2) and metalated Hf-PCN-222(M). (C) CO2 adsorption – desorption isotherms of Hf-PCN-222 (H2) and metalated Hf-PCN-222(M). | ||
First, the non-metalated Hf-PCN-222(H2) was obtained using a microwave-assisted strategy. The Hf6-oxo clusters were prepared by reacting commercially available bis(cyclopentadienyl)hafnium(IV) dichloride (HfCp2Cl2) with 2-fluorobenzoic acid (2-FBA), the modulator, for 5 min at 140 °C in DMF under microwave irradiation. This mixture was then treated with tetrakis(4-carboxyphenyl)porphyrin (H2TCPP) and trifluoroacetic acid (TFA) for 3 h at 150 °C under microwave irradiation. An acidic treatment (2 M HCl at 100 °C, see the ESI† for a full optimization of this step) of as-synthesized Hf-PCN-222(H2) is essential to remove the unreacted linkers trapped within the pores, as well as to remove 2-FBA modulators on Hf6-oxo clusters. The activated non-metalated Hf-PCN-222(H2), obtained as bright purple crystals in 81% yield from H2TCPP, has increased surface area compared to that of the as-synthesized MOF, and high crystallinity (Fig. S1†). The use of DMF to prepare PCN-222-MOFs is essential, and unsuccessful efforts to replace it by a greener alternative95 have been reported.96 Its role is to modulate the structure of the MOF, ensure mixture homogeneity and regular crystal growth, among others.96 Replacement by DMSO resulted in a MOF structure with low crystallinity,96 that influences the MOF catalytic and CO2 capture capabilities. Although the DMF solvent could not be replaced during catalyst synthesis, the microwave method reported here requires reduced reaction time and lower amounts of DMF and of modulators than those reported for the preparation of Hf-PCN-222(M) structures (see ESI, Table S1†).
With the optimized preparation of Hf-PCN-222(H2) in hand, we could implement a divergent metalation protocol, opening up the possibility of introducing many metals in a facile manner without the need to consider possible interferences or incompatibilities with the MOF structure. Thus, by reaction in aqueous solutions with either MnCl2 or CoCl2 under hydrothermal conditions, Hf-PCN-222(Mn) and Hf-PCN-222(Co) were obtained in 80% overall yield and >95% metalation efficiency in both instances (see ESI 2.3†).
The crystallinity and phase purity of activated Hf-PCN-222(H2) was confirmed by powder X-ray diffraction (PXRD) analysis (Fig. 2B). Additionally, the diffraction patterns of Hf-PCN-222(Mn) and Hf-PCN-222(Co) closely matched with those of Hf-PCN-222(H2) as well as with the simulated pattern, proving evidence of the preservation of the framework structure and crystallinity after the post-synthetic metalation. N2 gas adsorption–desorption analysis showed that metalated Hf-PCN-222(M) has only a slightly lower surface area compared to non-metalated Hf-PCN-222(H2) (Fig. S4†), confirming that the porous structure of the MOFs is preserved.
Carbon dioxide adsorption isotherms of Hf-PCN-222(H2), Hf-PCN-222(Mn) and Hf-PCN-222(Co) (Fig. 2C) showed excellent adsorption values: Hf-PCN-222(H2) = 3.0 mmol g−1 (273 K, 1 bar) and 1.8 mmol g−1 (298 K, 1 bar); Hf-PCN-222(Mn) = 2.5 mmol g−1 (273 K, 1 bar) and 1.6 mmol g−1 (298 K, 1 bar); Hf-PCN-222(Co) = 2.7 mmol g−1 (273 K, 1 bar) and 1.6 mmol g−1 (298 K, 1 bar). The oxophilicity of Hf on the clusters97,98 and the confined structure of the MOF can be involved in the CO2 adsorption via coordination, which may explain the good adsorption values observed. Additional information regarding the characterization of the synthesized materials is collected in the ESI,† including thermogravimetric analysis (Fig. S2†), scanning electron microscopy (Fig. S8†), elemental analysis (Table S3†), UV-Vis (Fig. S7†), and FT-IR spectroscopies (Fig. S6†).
We first tested the newly prepared PCN-catalysts in the cycloaddition of CO2. Reaction optimization was performed on styrene oxide (2a, Table 1), which was used as reagent and solvent simultaneously. We could observe that, when using Hf-PCN-222(Mn) (1 mol% based on porphyrin linker), tetrabutylammonium bromide (TBAB) (6 mol%), at 25 °C for 8 h (Fig. 3), 45% yield of styrene carbonate (3a) was obtained (Table 1, entry 1). Replacing Hf-PCN-222(Mn) by non-metalated Hf-PCN-222(H2) or by the cobalt analogue Hf-PCN-222(Co) resulted in lower yields under otherwise identical conditions (Table 1, entries 2 and 3). Reaction with using Zr-PCN-222(Mn) analogue yielded only 17% of the desired COC (Table 1, entries 2 vs. 4). This behaviour can be attributed to the fact that Hf is more oxophilic than Zr.97,98 Indeed, negligible to no yields were observed in the absence of either MOF catalyst, TBAB co-catalyst, or CO2 (Table 1, entries 5–7). Moreover, the yield could be increased to a remarkable 93% when the reaction time was prolonged from 8 to 24 h (Table 1, entry 8).
 | ||
| Fig. 3 Scope of CO2 cycloaddition to epoxides 2. Conditions: 2 (0.2 mmol), TBAB (6 mol%), Hf-PCN-222(Mn) (1 mol%), CO2 (1 bar, balloon), neat, 25 °C, 24 h. 1H NMR yields are reported. Isolated yields are in parentheses. aReaction carried out at 50 °C. bReaction carried out at 60 °C, 3n and 3n′ were obtained from 2n in the same reaction. | ||
|
||
|---|---|---|
| Entry | Deviations from standard conditionsa | Yield 3a (%) |
| a Conditions: 2a (0.2 mmol), Hf-PCN-222(Mn) (1 mol% based on porphyrin linker), TBAB (6 mol%), 25 °C, 8 h. 1H NMR yields were obtained using 1,3,5-trimethoxybenzene as an internal standard. | ||
| 1 | None | 45 |
| 2 | Hf-PCN-222(H2) instead of Hf-PCN-222(Mn) | 27 |
| 3 | Hf-PCN-222(Co) instead of Hf-PCN-222(Mn) | 29 |
| 4 | Zr-PCN-222(Mn) instead of Hf-PCN-222(Mn) | 17 |
| 5 | Absence of Hf-PCN-222(Mn) | 1 |
| 6 | Absence of TBAB | 6 |
| 7 | Absence of CO2 | n.d. |
| 8 | 24 h instead of 8 h | 93 |

With the optimal conditions in hand, we proceeded to study the scope and limitations of the CO2 cycloaddition to epoxides (Fig. 3). Styrene oxide (2a), epichlorohydrin (2b), and epibromohydrin (2c) yielded the corresponding cyclic carbonates in near quantitative yields. Furthermore, glycerol carbonate derivatives were obtained in 83% (MeO–, 3d), 95% (nBuO–, 3e), 89% (PhO–, 3f) under identical conditions. Notably, alkyl-epoxides 2g–2j underwent CO2 cycloaddition to give cyclic carbonates 3g–3j in excellent yield up to >99%. Unsaturated epoxides 2k and 2l yielded 3k and 3l in 92% and 50% yield, respectively, without interference of the double bond moiety present in their structure. Interestingly, when bis-epoxide 2m bearing terminal and internal epoxide moieties was used, only the terminal epoxide underwent the cycloaddition reaction with CO2, giving 3m in 70% yield. Terminal bis-epoxide 2n formed the bis(cyclic carbonate) 3n in 48% yield, together with 52% yield of the mono carbonate by-product 3n′. Formation of 3n′ indicates that the cycloaddition rate of alkyl epoxides is higher than that of aryl epoxides.
Obtaining chiral styrene carbonate 3a in high enantiomeric purity from easily accessible chiral (R/S)-styrene oxide 2a is challenging. That is due to the fact that the carbocation formed under Lewis acid conditions accounts for the racemization of chiral styrene oxide derivatives.99 The reported procedures for the synthesis of chiral styrene carbonate (63% to 99% ee) usually require high temperature (100 to 150 °C) and/or high CO2 pressure (10 to 80 bar).100–108 Despite that, we obtained (R)-3a and (S)-3a in quantitative yields and >99% ee at room temperature under atmospheric pressure of CO2. Likewise, (R)-3o and (S)-3o with >99% ee were synthesized in quantitative yield under identical conditions.
Unfortunately, internal epoxides 2p and 2q did not yield the desired products. Low yield (16%) of internal carbonate 3r was obtained when the CO2 cycloaddition reaction was carried out at a higher temperature of 50 °C.
Catalyst recyclability experiments were conducted. We were happy to see that Hf-PCN-222(Mn) can be reused for 10 consecutive runs without significant loss in reactivity and at a comparable rate (see ESI, Fig. S17†).
Encouraged by the results in the cycloaddition of CO2 into epoxides, we then turned into developing an efficient auto-relay catalytic oxidative carboxylation of alkenes. We started evaluating 4-chlorostyrene (1a) as model substrate to form COC 3s, by performing the epoxidation and the cycloaddition catalytic reactions using Hf-PCN-222(Mn) as a sole catalyst and with all the reagents in one-pot fashion (Table 2). We were pleased to observe that 2 mol% of Hf-PCN-222(Mn) efficiently catalyzed the oxidation when using 1.5 equiv. of iodosobenzene (PhIO) as the oxidant. The reaction runs in neat conditions without the use of organic solvent using 1.5 equiv. of PhIO. Furthermore, COC 3s was isolated in 75% at 40 °C, using a CO2 atmosphere (1 bar) and in the presence of TBAB (12 mol%) (for full optimization, see the ESI, Table S6†). Notably, the reaction outcome was also highly selective, providing COC 3s in 75% yield, with only traces of the corresponding epoxide intermediate 2v (Table 2, entry 1). No other by-products were detected after 24 h of reaction time. This is quite remarkable, as side reactions, forming styrene diol, phenylacetaldehyde, benzaldehyde and polystyrene, during styrene epoxidation often limits high yield and selectivity of epoxidation reactions.88–91,109–112
|
|||||
|---|---|---|---|---|---|
| Entry | Deviations from standard conditionsa | Conv. (%) | Yield (%) | ||
| 2v | 3s | 4a | |||
| n.d. = not detected.a Conditions: 1a (0.2 mmol), Hf-PCN-222(Mn) (2 mol% based on porphyrin linker), TBAB (12 mol%), 40 °C, 24 h. 1H NMR yields and conversions were obtained using 1,3,5-trimethoxybenzene as an internal standard.b PhI(OAc)2 (1.5 equiv.) and H2O (1.5 equiv.) was used instead of PhIO (1.5 equiv.).c Hf-PCN-222(Co) (2 mol%) was used instead of Hf-PCN-222(Mn) (2 mol%). | |||||
| 1 | None | 96 | 2 | 75 | n.d. |
| 2 | No TABAB | 90 | 3 | n.d. | 15 |
| 3 | No PhIO | 91 | n.d. | n.d. | n.d. |
| 4b | PhI(OAc)2 + H2O instead of PhIO | 92 | n.d. | n.d. | n.d. |
| 5 | No Hf-PCN-222(Mn) | 97 | 7 | 2 | 4 |
| 6c | Hf-PCN-222(Co) instead of Hf-PCN-222(Mn) | 67 | n.d. | 8 | 1 |
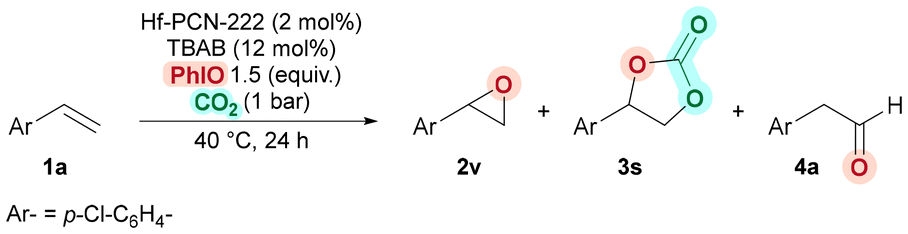
Control experiments conducted without PhIO, TBAB, or Hf-PCN-222(Mn) confirmed that each component is essential to achieve the auto-relay oxidative carboxylation (Table 2, entries 2–6). In these instances, although high conversions were obtained, unidentified by-products (likely polymers) are produced, along with small amounts (<15%) of epoxide 2v, carbonate 3s, or aldehyde 4a. Additionally, these experiments proved that Hf-PCN-222(Mn) was involved not only in the cycloaddition step, but also in the epoxidation92 as only traces of COC 3s and epoxide 2v were detected without this catalyst (Table 2, entry 5). The use of the analogous cobalt catalyst, Hf-PCN-222(Co), resulted in negligible amount of product 3s (Table 2, entry 6). Further, since Hf-PCN-222(Co) is active in the cycloaddition reaction (Table 1, entry 3) we attributed this lack of activity to the so-called “oxo-wall effect”, which prevented the formation of the epoxide intermediate.113
Then, the scope and limitations of the tandem oxidative carboxylation of a range of alkenes was explored (Fig. 4). Halide handles are important functional groups as they enable further diversification via well-known procedures.114,115 Thus, para-Cl, -Br and -F-substituted styrene derivatives were tested, affording COCs 3s–3u in good, isolated yields. Electron-poor (R = CF3, CN, NO2) and electron-rich (R = Me, tBu, OMe) para-substituted styrenes gave COCs 3a, 3v–3aa in moderate to good yields. meta-Me 3ab and ortho-Me 3ae COCs were synthesized in 47% and 54% yield, respectively. meta-OMe styrene carbonate 3ac was formed in 39%. Likewise, meta-NO2 3ad and ortho-NO2 3af COCs were obtained in 51% and 38% yield, respectively. Pentafluorobenzene COC 3ag was obtained in 40% yield under the optimized conditions. Heterocycles such as pyridine (3ah) and benzofuran (3ai) were obtained moderate to low yields. Interestingly, (E)-1,3-Butadien-1-ylbenzene underwent oxidative carboxylation yielding unsaturated cyclic carbonate 3aj in 42% yield. Although the yields range from moderate to good, with most being moderate, it is important to highlight that we could achieve synthetically useful yields for a wide scope of olefins in a two-steps one-pot process. This kind of methodology eliminates the need for isolation, purification, and handling of epoxides which is a significant advantage from an atom-economy perspective.
 | ||
| Fig. 4 Scope of oxidative carboxylation of alkenes 1. Conditions: 1 (0.2 mmol), TBAB (12 mol%), Hf-PCN-222(Mn) (2 mol%), CO2 (1 bar, balloon), neat, 40 °C, 24 h. 1H NMR yields are reported. Isolated yields are in parentheses. aReaction carried out for 48 h. bReaction carried out for 72 h. | ||
Several attempts were made to convert non-conjugated alkenes to the corresponding COCs (3ak–3ap); however, they were unproductive. The exception was pentafluorobenzyl-substituted ethylene carbonate 3ak, obtained in 26% yield. In any case, we decided to take advantage of this apparent limitation and develop a chemoselective carboxylation of dienes with both conjugated and non-conjugated alkenes present in the same molecule.
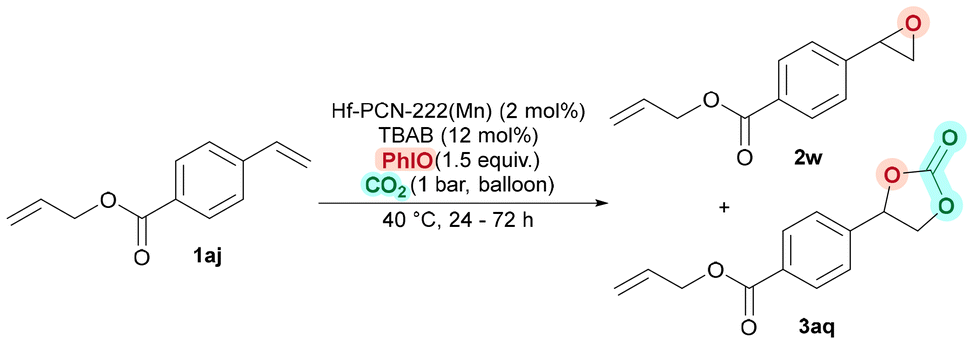
Therefore, diene 1aj, was used as the model substrate (Table 3). First catalytic tests showed that, the en-cyclic carbonate 3aq was formed in 26% yield, together with 41% yield of monoepoxide 2w after 24 h at 40 °C (Table 3, entry 1). No oxidized products related to the non-conjugated alkene moiety of 1aj were observed. Hence, to get a higher yield of 3aq, the reaction time was extended. We were pleased to see, 3aq was formed in 46% and 68% yields after 48 h and 72 h, respectively (Table 3, entries 2 and 3). Then, with optimized reaction conditions, several dienes with different length of the unsaturated alkyl chain were tested (Fig. 5). In all cases, only the conjugated alkene underwent oxidative carboxylation. Notably, as the size of the diene substrates increased, a steady decrease in yield was observed, likely due to the confined space effects within the MOF's channels (see the ESI, Table S8 and Fig. S22, S23†); en-cyclic carbonates 3aq–3at were formed in 68%, 53%, 40%, and 15% yield for n = 1, 2, 3, and 8, respectively. Ether 3aw was obtained in 40% yield under identical conditions. The reaction showed high sensitivity to steric effects in the aryl ring, as no yield was obtained for ortho-3aw, in comparison to 44% yield for meta-3av and 68% yield for para-3aq. Unsaturated mono-cyclic carbonate derived from mushroom alcohol 3ax was also obtained in full selectivity, albeit in low yields likely due to their large size.
 | ||
| Fig. 5 Scope of chemoselective oxidative carboxylation of dienes. | ||
|
||||
|---|---|---|---|---|
| Entry | Timea (h) | Conv. (%) | Yield 2w (%) | Yield 3aq (%) |
| a Conditions: 1aj (0.2 mmol), Hf-PCN-222(Mn) (2 mol% based on porphyrin linker), TBAB (12 mol%), 40 °C, 24–72 h. 1H NMR yields and conversions were obtained using 1,3,5-trimethoxybenzene as an internal standard. | ||||
| 1 | 24 | 83 | 41 | 26 |
| 2 | 48 | 83 | 21 | 46 |
| 3 | 72 | 83 | 4 | 68 |
Finally, the recyclability of the catalyst Hf-PCN-222(Mn) used for the oxidative carboxylation of 4-chlorostyrene (1a) was investigated (Fig. 6). After 5 consecutive runs, Hf-PCN-22(Mn) keeps a stable activity, affording 3s in a remarkable 61% yield and epoxide 2v in 8% yield after the fifth run. This is comparable to the results after the first run, namely 68% of 3s and 1% yield of 2v. PXRD pattern and SEM analysis showed that the recycled Hf-PCN-222(Mn) samples after 1 run and 5 runs have similar crystallinity and morphology compared to the pristine MOF. SEM-EDS showed that the leaching of Mn from the MOF structure is negligible (see ESI, Table S9†).
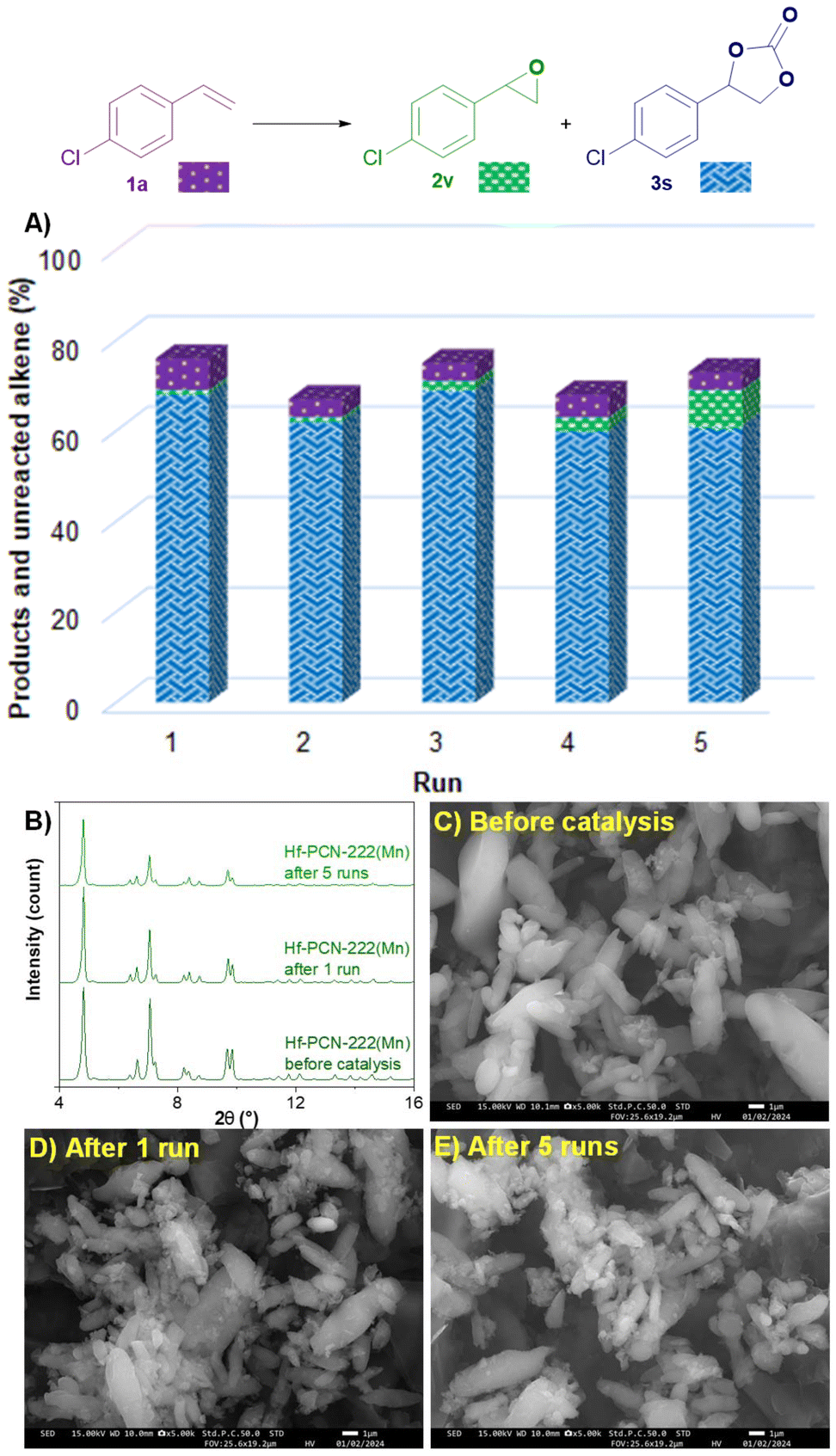 | ||
| Fig. 6 Recyclability experiment. (A) Reaction outcome. (B) PXRD patterns of Hf-PCN-222(Mn) before and after catalyst. SEM images of Hf-PCN-222(Mn) before catalysis (C), after 1 run (D), and after 5 runs (E). | ||
Conclusions
The porous Hf-based MOF with manganoporphyrin carboxylate linkers, namely Hf-PCN-222(Mn), has been prepared for the first time using a post-metalation synthetic approach, enabled by microwave radiation. This methodology reduces the synthesis time and overall quantity of reagents used. Furthermore, the development of a post-metalation approach of the Hf-PCN-222(H2) makes possible the introduction of a wide metal diversity avoiding interferences between the MOF structure and the metal of choice. Through this method, elusive porphyrin-MOFs metallated with Mn can be prepared with high metalation yield.Hf-PCN-222(Mn) proved to be active in the cycloaddition of CO2 to over 20 epoxides, achieving excellent yields (up to >99% yield) using mild pressure and temperature conditions. Furthermore, when applied to the tandem oxidative carboxylation of alkenes with CO2, Hf-PCN-222(Mn) provided organic cyclic carbonates from 23 alkenes, as well as the chemoselective and size-selective carboxylation of 7 dienes using the same mild conditions.
Furthermore, a comparison of the reaction conditions, catalytic activity and scope with other relevant MOF-based systems reveals that our system is able to perform the oxidative carboxylation of alkenes under mild temperatures and at ambient pressure without sacrificing efficiency. Thus, enabling the exploration of a broader scope for the one-step synthesis of cyclic carbonates from different readily accessible olefins with a variety of functional groups. We also eliminated completely the use of solvent in the catalysis, contributing in the overall sustainability of the process.
Furthermore, Hf-PCN-222(Mn) could be reused 5 times without loss of activity in the tandem oxidative carboxylation of alkenes with CO2. This is quite remarkable, as the MOF is not degraded by the oxidative reaction conditions, and keeps its catalytic activity for several runs, increasing the sustainability of the process.
Author contributions
All authors have given approval to the final version of the manuscript.Data availability
The data supporting the submitted work can be found in the ESI† of the article.The ESI† also contains further details regarding safety, as well as characterization information of the prepared compounds (1H NMR, 13C NMR, 19F NMR, and HRMS).
The raw NMR data files for all compounds reported in the article are deposited at Zenodo and will be made publicly available after acceptance (DOI: 10.5281/zenodo.13365877). The numbering of the files will then be matched to those in the article.
Each parent folder in Zenodo will contain subfolders with different files. In order to process the data, the full parent folder must be dragged into either Mestrenova or Topspin and then the data is automatically processed. If the name of the raw data files are renamed, the software (Mestrenova or Topspin) will not be able to process the files.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr V. García-Vázquez is gratefully acknowledged for scientific discussions of the oxidative carboxylation reaction. We thank Dr S. Carrasco for discussions regarding Co-MOF synthesis via pre-metalation protocols. The authors are grateful for financial supports from the Swedish Foundation for Strategic Environmental Research (Mistra SafeChem, project number 2018/11), Nordic Consortium for CO2 conversion (NordCO2, project number 85378), the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement (No. 859910, CO2PERATE ITN), Knut and Alice Wallenberg Foundation (CATSS, Catalytic Composites for Sustainable Synthesis), and The Swedish Research Council (VR).References
- S.-L. Hou, J. Dong, X.-Y. Zhao, X.-S. Li, F.-Y. Ren, J. Zhao and B. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202305213 CrossRef CAS PubMed.
- E. Loccufier, G. Watson, Y. Zhao, M. Meledina, R. Denis, P. G. Derakhshandeh, P. Van Der Voort, K. Leus, D. P. Debecker, K. De Buysser and K. De Clerck, Appl. Catal., B, 2023, 320, 121972 CrossRef CAS.
- S. Das, R. C. Turnell-Ritson, P. J. Dyson and C. Corminboeuf, Angew. Chem., Int. Ed., 2022, 61, e202208987 CrossRef CAS PubMed.
- P. P. Pescarmona, Curr. Opin. Green Sustainable Chem., 2021, 29, 100457 CrossRef CAS.
- J. Hu, L. Yu, J. Deng, Y. Wang, K. Cheng, C. Ma, Q. Zhang, W. Wen, S. Yu, Y. Pan, J. Yang, H. Ma, F. Qi, Y. Wang, Y. Zheng, M. Chen, R. Huang, S. Zhang, Z. Zhao, J. Mao, X. Meng, Q. Ji, G. Hou, X. Han, X. Bao, Y. Wang and D. Deng, Nat. Catal., 2021, 4, 242–250 CrossRef CAS.
- L. Guo, K. J. Lamb and M. North, Green Chem., 2021, 23, 77–118 RSC.
- B. Grignard, S. Gennen, C. Jérôme, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC.
- T. Niemi and T. Repo, Eur. J. Org. Chem., 2019, 1180–1188 CrossRef CAS.
- N. A. Tappe, R. M. Reich, V. D'Elia and F. E. Kühn, Dalton Trans., 2018, 47, 13281–13313 RSC.
- J. Song, Q. Liu, H. Liu and X. Jiang, Eur. J. Org. Chem., 2018, 696–713 CrossRef CAS.
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804–9838 CrossRef.
- Z. Zhang, T. Ju, J.-H. Ye and D.-G. Yu, Synlett, 2017, 741–750 Search PubMed.
- J. Sun, S.-I. Fujita and M. Arai, J. Organomet. Chem., 2005, 690, 3490–3497 CrossRef CAS.
- M. O. Sonnati, S. Amigoni, E. P. Taffin de Givenchy, T. Darmanin, O. Choulet and F. Guittard, Green Chem., 2013, 15, 283–306 RSC.
- Y. Hu, J. Steinbauer, V. Stefanow, A. Spannenberg and T. Werner, ACS Sustainable Chem. Eng., 2019, 7, 13257–13269 CrossRef CAS.
- C.-C. Su, M. He, R. Amine, Z. Chen, R. Sahore, N. D. Rago and K. Amine, Energy Storage Mater., 2019, 17, 284–292 CrossRef.
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC.
- C. N. Tounzoua, B. Grignard and C. Detrembleur, Angew. Chem., Int. Ed., 2022, 61, e202116066 CrossRef PubMed.
- G. Fiorani, A. Perosa and M. Selva, Green Chem., 2023, 25, 4878–4911 RSC.
- V. Zubar, Y. Lebedev, L. M. Azofra, L. Cavallo, O. El-Sepelgy and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 13439–13443 Search PubMed.
- S. Sopeña, V. Laserna, W. Guo, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Adv. Synth. Catal., 2016, 358, 2172–2178 CrossRef.
- J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663–674 CrossRef CAS.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC.
- M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 48, 2946–2948 CrossRef CAS PubMed.
- T.-D. Hu, Y.-W. Sun and Y.-H. Ding, J. CO2 Util., 2018, 28, 200–206 CrossRef CAS.
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed.
- T. Ema, Y. Miyazaki, J. Shimonishi, C. Maeda and J.-Y. Hasegawa, J. Am. Chem. Soc., 2014, 136, 15270–15279 CrossRef CAS PubMed.
- L. Álvarez-Miguel, J. D. Burgoa, M. E. G. Mosquera, A. Hamilton and C. J. Whiteoak, ChemCatChem, 2021, 13, 4099–4110 CrossRef.
- J. D. Burgoa, L. Álvarez-Miguel, M. E. G. Mosquera, A. Hamilton and C. J. Whiteoak, Inorg. Chem., 2024, 63, 15376–15387 CrossRef PubMed.
- J. Sun, S.-I. Fujita, B. M. Bhanage and M. Arai, Catal. Today, 2004, 83–95, 383–388 CrossRef.
- W. Natongchai, D. Crespy and V. D'Elia, Chem. Commun., 2025, 61, 419–440 RSC.
- T. Yan, H. Liu, Z. X. Zeng and W. G. Pan, J. CO2 Util., 2023, 68, 102355 CrossRef CAS.
- M. Usman, A. Rehman, F. Saleem, A. Abbas, V. C. Eze and A. Harvey, RSC Adv., 2023, 13, 22717–22743 RSC.
- F. Mundo, S. Caillol, V. Ladmiral and M. A. R. Meier, ACS Sustainable Chem. Eng., 2024, 12, 6452–6466 CrossRef CAS.
- K. Liu, S. Jiao, H. Zhao, F. Cao and D. Ma, Green Chem., 2021, 23, 1766–1771 RSC.
- Y.-H. Zou, Y.-B. Huang, D.-H. Si, Q. Yin, Q.-J. Wu, Z. Weng and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 20915–20920 CrossRef CAS PubMed.
- X.-J. Bai, X.-Y. Lu, R. Ju, H. Chen, L. Shao, X. Zhai, Y.-N. Li, F.-Q. Fan, Y. Fu and W. Qi, Angew. Chem., Int. Ed., 2021, 60, 701–705 CrossRef CAS PubMed.
- G. Li, S. Dong, P. Fu, Q. Yue, Y. Zhou and J. Wang, Green Chem., 2022, 24, 3433–3460 RSC.
- Y. Li, F. Gao, J. Xue, G.-P. Yang and Y.-Y. Wang, Cryst. Growth Des., 2023, 23, 3702–3710 CrossRef CAS.
- K. Cai, P. Liu, Z. Chen, P. Chen, F. Liu, T. Zhao and D.-J. Tao, Chem. Eng. J., 2023, 451, 138946 CrossRef CAS.
- C. Lu, Y.-Y. Zhang, X.-F. Zhu, G.-W. Yang and G.-P. Wu, ChemCatChem, 2023, 15, e202300360 CrossRef CAS.
- S. Sarkar, S. Ghosh, R. Sani, J. Seth, A. Khan and S. M. Islam, ACS Sustainable Chem. Eng., 2023, 11, 14422–14434 CrossRef CAS.
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- X. Chen, Q. Duez, G. L. Tripodi, P. J. Gilissen, D. Piperoudis, P. Tinnemans, J. A. A. W. Elemans, J. Roithová and R. J. M. Nolte, Eur. J. Org. Chem., 2022, e202200280 CrossRef CAS PubMed.
- G. Grigoropoulou, J. H. Clark and J. A. Elings, Green Chem., 2003, 5, 1–7 RSC.
- A. A. Ryan, S. D. Dempsey, M. Smyth, K. Fahey, T. S. Moody, S. Wharry, P. Dingwall, D. W. Rooney, J. M. Thompson, P. C. Knipe and M. J. Muldoon, Org. Process Res. Dev., 2023, 27, 262–268 CrossRef CAS PubMed.
- C. E. Voogd, J. J. van der Stel and J. J. J. A. A. Jacobs, Mutat. Res./Genet. Toxicol., 1981, 89, 269–282 CrossRef CAS PubMed.
- D. R. Wade, S. C. Airy and J. E. Sinsheimer, Mutat. Res./Genet. Toxicol., 1978, 58, 217–223 CrossRef CAS PubMed.
- L. Wang, S. Que, Z. Ding and E. Vessally, RSC Adv., 2020, 10, 9103–9115 RSC.
- R. Calmanti, M. Selva and A. Perosa, Green Chem., 2021, 23, 1921–1941 RSC.
- F. Han, H. Li, H. Zhuang, Q. Hou, Q. Yang, B. Zhang and C. Miao, J. CO2 Util., 2021, 53, 101742 CrossRef CAS.
- K. Yu, P. Puthiaraj and W.-S. Ahn, Appl. Catal., B, 2020, 273, 119059 CrossRef CAS.
- F. Ono, K. Qiao, D. Tomida and C. Yokoyama, Appl. Catal., A, 2007, 333, 107–113 CrossRef CAS.
- D. Bai and H. Jing, Green Chem., 2010, 12, 39–41 RSC.
- K. Jasiak, T. Krawczyk, M. Pawlyta, A. Jakóbik-Kolon and S. Baj, Catal. Lett., 2016, 146, 893–901 CrossRef CAS.
- H. T. D. Nguyen, Y. B. N. Tran, H. N. Nguyen, T. C. Nguyen, F. Gándara and P. T. K. Nguyen, Inorg. Chem., 2018, 57, 13772–13782 CrossRef CAS PubMed.
- Z. Shi, G. Niu, Q. Han, X. Shi and M. Li, Mol. Catal., 2018, 461, 10–18 CrossRef CAS.
- S.-C. Ke, T.-T. Luo, G.-G. Chang, K.-X. Huang, J.-X. Li, X.-C. Ma, J. Wu, J. Chen and X.-Y. Yang, Inorg. Chem., 2020, 59, 1736–1745 CrossRef CAS PubMed.
- H. Pallathadka, H. K. Mohammed, Z. H. Mahmoud, A. A. Ramírez-Coronel, F. M. A. Altalbawy, M. A. Gatea and M. Kazemnejadi, Inorg. Chem. Commun., 2023, 154, 110944 CrossRef CAS.
- T. Zhao, G. Long, H. Liang, W. Xiong and X. Hu, Microporous Mesoporous Mater., 2023, 356, 112576 CrossRef CAS.
- J. E. Camp, Eur. J. Org. Chem., 2017, 425–433 CrossRef CAS.
- S. Martínez, L. Veth, B. Lainer and P. Dydio, ACS Catal., 2021, 11, 3891–3915 CrossRef.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- J. Yu, L.-H. Xie, J.-R. Li, Y. Ma, J. M. Seminario and P. B. Balbuena, Chem. Rev., 2017, 117, 9674–9754 CrossRef CAS PubMed.
- Z. I. Zulkifli, K. L. Lim and L. P. Teh, ChemistrySelect, 2022, 7, e202200572 CrossRef CAS.
- S. Mahajan and M. Lahtinen, J. Environ. Chem. Eng., 2022, 10, 108930 CrossRef CAS.
- R. A. Maia, B. Louis, W. Gao and Q. Wang, React. Chem. Eng., 2021, 6, 1118–1133 RSC.
- M. Pander, M. Janeta and W. Bury, ACS Appl. Mater. Interfaces, 2021, 13, 8344–8352 CrossRef CAS PubMed.
- F. N. Al-Rowaili, U. Zahid, S. Onaizi, M. Khaled, A. Jamal and E. M. Al-Mutairi, J. CO2 Util., 2021, 53, 101715 CrossRef CAS.
- I. Campello, A. Sepúlveda-Escribano and E. V. Ramos-Fernández, in Engineering Solutions for CO2 Conversion, 2021, pp. 407–427 Search PubMed.
- T. K. Pal, D. De and P. K. Bharadwaj, Coord. Chem. Rev., 2020, 408, 213173 CrossRef CAS.
- M. H. Beyzavi, C. J. Stephenson, Y. Liu, O. Karagiaridi, J. T. Hupp and O. K. Farha, Front. Energy Res., 2015, 2, 63 Search PubMed.
- S. Carrasco, A. Sanz-Marco and B. Martín-Matute, Organometallics, 2019, 38, 3429–3435 CrossRef CAS.
- O. V. Zalomaeva, N. V. Maksimchuk, A. M. Chibiryaev, K. A. Kovalenko, V. P. Fedin and B. S. Balzhinimaev, J. Energy Chem., 2013, 22, 130–135 CrossRef CAS.
- Q. Han, B. Qi, W. Ren, C. He, J. Niu and C. Duan, Nat. Commun., 2015, 6, 10007 CrossRef PubMed.
- P. T. K. Nguyen, H. T. D. Nguyen, H. N. Nguyen, C. A. Trickett, Q. T. Ton, E. Gutiérrez-Puebla, M. A. Monge, K. E. Cordova and F. Gándara, ACS Appl. Mater. Interfaces, 2018, 10, 733–744 CrossRef CAS PubMed.
- A. Valverde-González, M. C. Borrallo-Aniceto, U. Díaz, E. M. Maya, F. Gándara, F. Sánchez and M. Iglesias, J. CO2 Util., 2023, 67, 102298 CrossRef.
- Y. Yu, X. Chen, X. Wang, X. Feng, S. Liu, C. Duan, Y. Wu and H. Xi, Chem. Eng. Sci., 2023, 278, 118898 CrossRef CAS.
- Y. Yu, X. Feng, Z. Xie, Y. Wu and H. Xi, AIChE J., 2024, 70, e18290 CrossRef CAS.
- X. Zhao, G. Chang, H. Xu, Y. Yao, D. Dong, S. Yang, G. Tian and X. Yang, ACS Appl. Mater. Interfaces, 2024, 16, 7364–7373 CrossRef CAS PubMed.
- N. Sharma, S. S. Dhankhar, S. Kumar, T. J. D. Kumar and C. M. Nagaraja, Chem. – Eur. J., 2018, 24, 16662–16669 CrossRef CAS PubMed.
- N. V. Maksimchuk, I. D. Ivanchikova, A. B. Ayupov and O. A. Kholdeeva, Appl. Catal., B, 2016, 181, 363–370 CrossRef CAS.
- R. Das, S. Kamra and C. M. Nagaraja, Inorg. Chem. Front., 2023, 10, 2088–2099 RSC.
- Y. B. N. Tran, P. T. K. Nguyen, V.-A. Dao and V.-D. Le, New J. Chem., 2024, 48, 5300–5310 RSC.
- M. Aresta, A. Dibenedetto and I. Tommasi, Appl. Organomet. Chem., 2000, 14, 799–802 CrossRef CAS.
- M. Aresta and A. Dibenedetto, J. Mol. Catal. A: Chem., 2002, 182–183, 399–409 CrossRef CAS.
- J. Sun, S.-I. Fujita, F. Zhao, M. Hasegawa and M. Arai, J. Catal., 2005, 230, 398–405 CrossRef CAS.
- J. Chen, X. Liu, P. Zhang, H. Zhou, L. Li, H. Luo, H. Wang and Y. Sun, ChemSusChem, 2024, 17, e202301567 CrossRef CAS PubMed.
- M. Guo, Y.-M. Lee, M. S. Seo, Y.-J. Kwon, X.-X. Li, T. Ohta, W.-S. Kim, R. Sarangi, S. Fukuzumi and W. Nam, Inorg. Chem., 2018, 57, 10232–10240 CrossRef CAS PubMed.
- H. Beyzavi, N. A. Vermeulen, A. J. Howarth, S. Tussupbayev, A. B. League, N. M. Schweitzer, J. R. Gallagher, A. E. Platero-Prats, N. Hafezi, A. A. Sarjeant, J. T. Miller, K. W. Chapman, J. F. Stoddart, C. J. Cramer, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 13624–13631 CrossRef CAS PubMed.
- S. Li, H.-M. Mei, S.-L. Yao, Z.-Y. Chen, Y.-L. Lu, L. Zhang and C.-Y. Su, Chem. Sci., 2019, 10, 10577–10585 RSC.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehadad and P. J. Dunne, Green Chem., 2016, 18, 288–296 RSC.
- A. Zuliani, M. C. Castillejos and N. Khiar, Green Chem., 2023, 25, 10596–10610 RSC.
- E. W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC Press/Taylor And Francis, Boca Raton, FL, 2016 Search PubMed.
- M. Khononov, N. Fridman, M. Tamm and M. S. Eisen, Eur. J. Org. Chem., 2020, 3153–3160 CrossRef CAS.
- A. Boukhari, R. Blida and F. Ismail, C. R. Chim., 2010, 13, 1440–1442 CrossRef CAS.
- Y. Du, J.-Q. Wang, J.-Y. Chen, F. Cai, J.-S. Tian, D.-L. Kong and L.-N. He, Tetrahedron Lett., 2006, 47, 1271–1275 CrossRef CAS.
- W.-M. Ren, Y. Liu and X.-B. Lu, J. Org. Chem., 2014, 79, 9771–9777 CrossRef CAS PubMed.
- A. Buonerba, A. De Nisi, A. Grassi, S. Milione, C. Capacchione, S. Vagin and B. Rieger, Catal. Sci. Technol., 2015, 5, 118–123 RSC.
- T. Ema, K. Fukuhara, T. Sakai, M. Ohbo, F.-Q. Bai and J.-Y. Hasegawa, Catal. Sci. Technol., 2015, 5, 2314–2321 RSC.
- Z. Zhou, C. He, J. Xiu, L. Yang and C. Duan, J. Am. Chem. Soc., 2015, 137, 15066–15069 CrossRef CAS PubMed.
- T. Yano, H. Matsui, T. Koike, H. Ishiguro, H. Fujihara, M. Yoshihara and T. Maeshima, Chem. Commun., 1997, 1129–1130 RSC.
- M. Aresta, A. Dibenedetto, L. Gianfrate and C. Pastore, Appl. Catal., A, 2003, 255, 5–11 CrossRef CAS.
- L. Wang, L. Lin, G. Zhang, K. Kodama, M. Yasutake and T. Hirose, Chem. Commun., 2014, 50, 14813–14816 RSC.
- Q.-W. Song, L.-N. He, J.-Q. Wang, H. Yasuda and T. Sakakura, Green Chem., 2013, 15, 110–115 RSC.
- P. A. A. Ignacio-de Leon, C. A. Contreras, N. E. Thornburg, A. B. Thompson and J. M. Notestein, Appl. Catal., A, 2016, 511, 78–86 CrossRef CAS.
- F. M. Rabagliati and J. M. Contreras, Eur. Polym. J., 1987, 23, 63–67 CrossRef CAS.
- R. Zhang, S. Liu, C. Zhuo, H. Cao and X. Wang, Macromolecules, 2024, 57, 132–141 CrossRef.
- S. Aoshima and S. Kanaoka, Chem. Rev., 2009, 109, 5245–5287 CrossRef CAS PubMed.
- V. A. Larson, B. Battistella, K. Ray, N. Lehnert and W. Nam, Nat. Rev. Chem., 2020, 4, 404–419 CrossRef CAS PubMed.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- P. Kaur, V. Kumar and R. Kumar, Catal. Rev., 2020, 62, 118–161 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc06360k |
| This journal is © The Royal Society of Chemistry 2025 |