
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
anti-Dihydroxylation of olefins enabled by in situ generated peroxyacetic acid†
Michael
Tapera‡
,
Mohit
Chotia‡
,
Jan Lukas
Mayer-Figge
,
Adrián
Gómez-Suárez
 * and
Stefan F.
Kirsch
*
* and
Stefan F.
Kirsch
*
Organic Chemistry, Bergische Universität Wuppertal, Gaußstr. 20, 42119 Wuppertal, Germany. E-mail: gomezsuarez@uni-wuppertal.de; sfkirsch@uni-wuppertal.de
First published on 5th September 2024
Abstract
Herein, we report a general and green protocol for the anti-dihydroxylation of unactivated alkenes. Combining H2O2 and acetic acid at 50 °C results in the formation of peroxyacetic acid, which enables the efficient synthesis of a wide range of anti 1,2-diols in moderate to good yields without the need for hazardous solvents or expensive transition metals as catalysts.
Since the establishment of the 12 principles of Green Chemistry over 25 years ago,1 chemists have devoted considerable efforts and resources towards the development of more efficient, environmentally friendly, and sustainable chemical processes.2,3 Good examples of this trend are the emergence of catalytic methods employing organometallic complexes based on earth-abundant metals (e.g., Cu, Fe, Co, etc.);4–6 the move towards the use of continuous-flow technologies to increase safety standards and improve process scalability;7–9 and the conception of several metrics to quantify the efficiency or the environmental impact of a chemical process.10–13
The dihydroxylation of alkenes is a fundamental reaction in synthetic organic chemistry, which enables the preparation of either syn or anti 1,2-diols, key structural motifs found in a wide range of natural products and biologically active molecules.14–17 While the syn-dihydroxylation of alkenes has been extensively studied, resulting in the development of a myriad of protocols,18–20 the corresponding anti-dihydroxylation has received comparatively less attention.21,22 This reaction typically occurs through epoxidation of the targeted alkene, which can be promoted by transition metal catalysis,23–27 organocatalysis,28–33 photocatalysis,34,35 electrocatalysis36,37 or biocatalysis,38–40 followed by hydrolysis to generate the targeted anti 1,2-diols (Scheme 1A). Most of these processes require using (toxic) organic solvents, leading to increased waste generation,1 or strong and non-sustainable oxidants to promote the key epoxidation step. Therefore, there is a need to develop greener protocols for the anti-dihydroxylation of alkenes based on the use of sustainable resources.
 | ||
| Scheme 1 anti-Dihydroxylation of alkenes using in situ generated peroxy acids – state of the art vs. this work. | ||
In 1926, Hilditch reported the anti-dihydroxylation of oleic and elaidic acids, as well as their corresponding methyl esters, using a combination of H2O2 and acetic acid to generate peroxyacetic acid, a strong oxidant capable of promoting the key epoxidation step (Scheme 1B).41 While it took nine days at room temperature to produce the targeted products, the process could be significantly accelerated by increasing the reaction temperature. It was postulated that the existence of a dynamic equilibrium during the formation of the peroxy acid species was the main reason behind the slow reactivity at room temperature and the need for a high excess of H2O2.42 Later, Swern reported that using catalytic amounts of sulfuric acid or replacing acetic acid by formic acid, shifts the equilibrium towards the formation of the key peroxy acid species and drives the reaction forward.22,43 However, the use of these strong acids results in highly exothermic processes, thus increasing the overall risks associated with these reactions. To overcome this issue, researchers have established several strategies. For example: (a) the use of alternative acids (e.g., p-toluenesulfonic acid) to form the key peroxy acid species catalytically,44,45 (b) pre-forming the peroxy acid species ex situ by premixing the corresponding acid and H2O2,46,47 or (c) the use of flow technology to increase the overall safety and scalability of the process.48 Nevertheless, these approaches still rely on the use of strong and corrosive acids to form the reactive peroxy acid species, or convoluted approaches to avoid highly exothermic processes. Therefore, developing more sustainable and environmentally friendly processes for the anti-dihydroxylation of alkenes is highly desirable.
From a sustainability point of view, Hilditch's seminal contribution is almost ideal. It employs an eco-friendly and renewable solvent, such as acetic acid,49,50 a relatively mild oxidant, such as H2O2,51–53 and the sole by-product of the reaction is the formation of H2O. More importantly, since the equilibrium constant for the formation of the peroxyacetic acid is low when mixing H2O2 and acetic acid at room temperature,42,54 it does not result in a highly exothermic reaction, as it is the case when using formic acid,43,48 thus reducing the hazards associated with the transformation. However, the process was never fully optimised, it was sluggish, and the reaction scope was restricted to oleic and elaidic acid derivatives. Overall, these factors limited its synthetic applicability and uptake by the wider chemical community. Therefore, with the aim of developing a more sustainable process, we decided to revisit Hilditch's original work. Our goals were: (1) to optimise the reaction conditions, leading to efficient in situ formation of peroxyacetic acid and reduced reaction times, (2) to evaluate the reaction scope, and (3) to demonstrate the suitability of this sustainable strategy for the large-scale production of anti 1,2-diols. Herein, we report the results of our efforts: a general and environmentally friendly approach for the anti-dihydroxylation of alkenes, using acetic acid and a slight excess of H2O2, for the synthesis of a wide range of anti 1,2-diols (Scheme 1C).
We began the optimisation of the reaction conditions using cyclohexene 1a as the model substrate (Table 1). The reactions were carried out in deuterated acetic acid, and the yield of the product(s) was calculated by 1H NMR spectroscopy using trichloroethylene as the internal standard. Initially, a large excess of H2O2 (8.0 equiv.) was tested at 50 °C (Table 1, entry 1). To our delight, we observed complete consumption of 1 and formation of trans-cyclohexane-1,2-diol (2) and acetylated species 2′ in 40% and 25%, respectively, after 3 h. Decreasing the amount of H2O2 to 2.0 equiv. resulted in incomplete conversion of 1 after 6 h; however, increasing the reaction time to 16 h resulted in complete conversion and formation of 25% of 2 and 66% of 2′ (Table 1, entries 2 & 3). Further decreasing the equivalents of H2O2 to either 1.5 or 1.25 resulted in longer reaction times (up to 24 h) or incomplete conversion of 1 (Table 1, entries 4–6). Therefore, we selected to move forward using 2.0 equiv. of H2O2 at 50 °C for 16 h as our standard reaction conditions.§ Finally, the mixture of 2 and 2′ was readily converted to pure 2via saponification with a 5 M aqueous NaOH solution.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | H2O2 (equiv.) | T (°C) | t (h) | 1 (%) | 2 (%) | 2′ (%) |
| a Reactions were carried out on a 0.1 mmol scale. Yields determined by 1H NMR using trichloroethylene as the internal standard. | ||||||
| 1 | 8.0 | 50 | 3 | 0 | 40 | 25 |
| 2 | 2.0 | 50 | 6 | 38 | 12 | 29 |
| 3 | 2.0 | 50 | 16 | 0 | 25 | 66 |
| 4 | 1.5 | 50 | 16 | 20 | 16 | 49 |
| 5 | 1.5 | 50 | 24 | 0 | 23 | 67 |
| 6 | 1.25 | 50 | 24 | 17 | 60 | 25 |

With the optimised conditions in hand, we explored the scope of the methodology (Scheme 2). For selected reactions where incomplete conversion of the starting olefin was observed, increasing the amount of H2O2 to 8.0 equiv. usually afforded increased yields of the targeted anti 1,2-diols in shorter reaction times (4 h). Cyclic alkenes can be smoothly converted to the corresponding diols (2–6) in moderate to excellent yields (36–84%). Interestingly, compound 6 bearing a nucleophilic tosyl-protected amine motif could be isolated in 36% yield. Meanwhile, non-cyclic internal olefins often afforded diminished yields of the vicinal diols (7–10) using 2.0 equiv. of H2O2, excellent yields (up to 86%) were obtained using 8.0 equiv. of H2O2. The anti-dihydroxylation of oleic acid proceeded smoothly under this new reaction conditions, affording the targeted diol 10 in 78% yield. Terminal olefins were also well tolerated, and the corresponding diols (11–15) were isolated in excellent yields (74–85%) using either 2.0 or 8.0 equiv. of H2O2.
 | ||
Scheme 2 Scope of the methodology. Reaction conditions: a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1. alkene (0.5 mmol), H2O2 (2.0 equiv.), HOAc (0.5 M), 50 °C, 16 h; 2. aqueous Na2S2O3 (1.0 M), aqueous NaOH (5.0 M), RT, 1 h. bH2O2 (8.0 equiv.), 4 h. 1. alkene (0.5 mmol), H2O2 (2.0 equiv.), HOAc (0.5 M), 50 °C, 16 h; 2. aqueous Na2S2O3 (1.0 M), aqueous NaOH (5.0 M), RT, 1 h. bH2O2 (8.0 equiv.), 4 h. | ||
Next, we tested our protocol for the dihydroxylation of styrene derivatives (16–25). While styrene produced the targeted diol 16 in 68% yield, decorating the aromatic ring with electron-withdrawing groups, such as CF3, afforded diminished yields even when 8.0 equiv. of H2O2 were used (17, 40%). The addition of electron-donating/withdrawing substituents at either the ortho or meta positions was also tolerated, and the corresponding 1,2-diols (20–21) were isolated in moderate yields (42–45%). The introduction of α-substituents on the styrene core was also investigated. Both α-phenylstyrene and α-methylstyrene afforded the respective diols 23 and 24 in 65% and 71% yield. Finally, dihydroxylation of 1-phenyl-1-cyclohexene yielded the anti 1,2-diol 25 in 83% yield.
To test the functional group compatibility of our method, we decided to conduct an additive-based screening (Scheme 3).55–58 Cyclohexene (1) and equimolar amounts of a given additive (A–P) were subjected to the standard reaction conditions using 2.0 equiv. of H2O2 at 50 °C in acetic acid (0.5 M), and the outcome analysed by GC-FID using methyl laurate as the internal standard. This screening revealed that free alcohol (A), alkyl bromides (B), acrylates (C), ketones (D), nitriles (E), alkynes (H), phenols (I), benzofurans (J), benzoyl-protected anilines (K), pyridines (M) and thiophenes (N) are tolerated (remaining additive 78–100%) and have minimal impact on the yield of the reaction (82–90% vs. 94% yield without additive). While aldehydes (F), acetals (G), and Boc-protected pyrroles (O) are affected by our reaction conditions (remaining additive 0–58%), the targeted products 2 and 2′ could still be obtained in good yields (73–89%). However, unprotected amines (L and P) completely hamper the dihydroxylation reaction. Overall, the additive screening revealed that our method displays an excellent functional group compatibility, affording excellent yields of the targeted product, while preserving the additive in most cases.
 | ||
| Scheme 3 Additive screening to assess the functional group tolerance of the reaction. | ||
To evaluate how our method compares to the current state-of-the-art for the anti-dihydroxylation of olefins using in situ generated peroxy acids, we evaluated the dihydroxylation of three distinct types of olefins – internal (26), cyclic (1), and terminal (27) – using comparable methodologies (Scheme 4). For the anti-dihydroxylation of 26, our conditions using 8.0 equiv. of H2O2 in HOAc (0.5 M) and 50 °C afforded 10 in 78% yield after 4 h, while Hilditch's original conditions yielded 45% of 10 after 9 days at room temperature.41 For the anti-dihydroxylation of cyclohexene (1), there is more available data for comparison. Using the highly exothermic combination of formic acid and H2O2, Gregory obtained 2 in 70% yield after 2 h at 65–70 °C,59 while Kraft obtained 88% of 2 after just 1 min at 50 °C using flow chemistry.48 The combination of sulfonic acids and H2O2 is another popular combination for the anti-dihydroxylation of cyclohexene. Using a resin-bound catalyst and 2.0 equiv. of H2O2, Sato obtained 98% of 2 after 20 h at 70 °C;44 while Afonso obtained 98% of 2 after 21 h combining 20 mol% of para-toluenesulfonic acid (PTSA) and 2.0 equiv. of H2O2 at 70 °C.45 In comparison, our method afforded 90% of 2 after 16 h using 2.0 equiv. of H2O2 in HOAc (0.5 M) at 50 °C. Finally, for the dihydroxylation of 1-hexene (27), Sato's conditions afforded only 40% yield of the targeted product (12) after 20 h,44 while Afonso managed to obtain 87% yield after 21 h.45 In contrast, we were able to obtain 12 in 81% yield after 4 h using 8.0 equiv. of H2O2 in HOAc (0.5 M) at 50 °C. Overall, our method affords similar yields of the targeted 1,2-diols compared to reported methods using similar conditions, albeit in shorter reaction times (in batch), and bypassing the need for formic acid – and the hazards associated with it, i.e., high exotherm – to generate the key peroxy acid species.
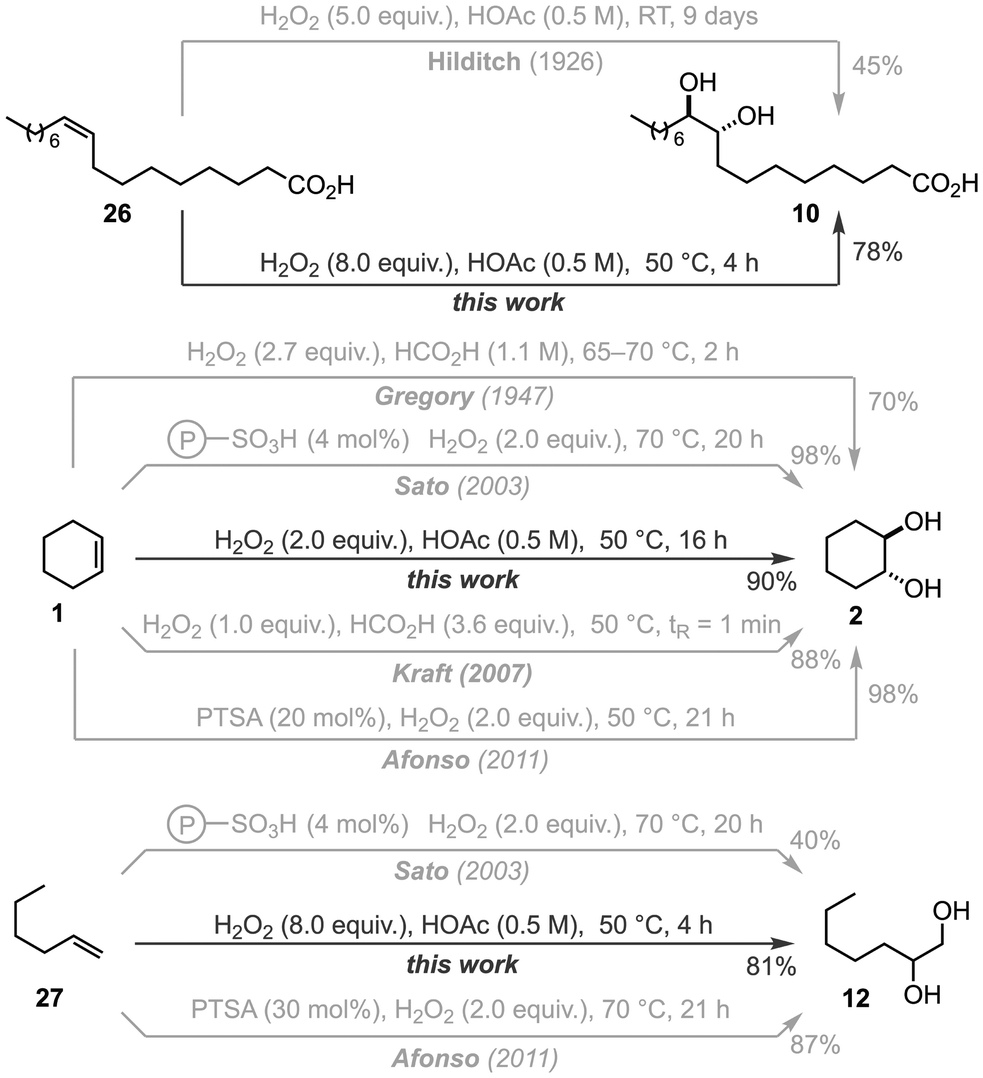 | ||
| Scheme 4 Comparison between this work and previously reported methods using in situ generated peroxy acids. | ||
To fully exploit the applicability of this sustainable and environmentally friendly protocol, it is crucial to test its scalability (Scheme 5). First, the anti-dihydroxylation of cyclohexene (1) was carried out in 10 mmol scale. Gratifyingly, this enabled the synthesis of diol 2 in 86% yield (0.99 g) without modification of the standard reaction conditions. With this encouraging result, the reaction was further scaled up to 100 mmol scale, affording diol 2 in 78% isolated yield (9.1 g).
 | ||
| Scheme 5 Multigram-scale reactions in batch. | ||
While these are excellent results for laboratory scale reactions, further scaling up of our process required addressing some potential safety risks. The key species in our protocol, peroxyacetic acid, is a strong oxidant and a highly reactive and corrosive species; therefore, it is desirable to avoid its use in high concentrations.60,61 Our standard protocol enables the generation of peroxyacetic acid in situ, therefore circumventing hazards associated with its transport and manipulation.61 However, for large-scale synthesis, this would still require the generation of large quantitates of peroxyacetic acid, increasing potential safety risks. We hypothesised that we might be able to address these safety concerns by employing a continuous-flow process. The use of flow technology has proven beneficial in reducing reaction times while increasing safety and scalability, which is key for bulk applications in the chemical industry.62–65 One of the main advantages offered by flow chemistry is that it enables the safe and effective use of small concentrations of highly reactive species.12,66,67 With this in mind, we envisioned that we might be able to exploit the low equilibrium constant for the formation of peroxyacetic acid from H2O2 and HOAc at room temperature to prepare a stable solution of both reagents, which would not be possible with formic acid due to the high exothermic reaction occurring when mixing it with H2O2.68 This H2O2 solution in HOAc would mix with a solution of the alkene in HOAc at room temperature using a T-mixer. The reaction mixture would then flow through a heated coiled reactor, generating peroxyacetic acid in situ and enabling the targeted alkene anti-dihydroxylation (Scheme 6A). With this setup, only small concentrations of peroxyacetic acid would be generated at any given time, therefore decreasing the safety risks associated with the process. Using this flow protocol, we were able to prepare 2 and 8 in 5.0 mmol scale in good yields (65–85%) using a flow rate (f) of 31.5 μL min−1 (residence time (tR) = 39 min) at 70 °C, and 8.0 equiv. of H2O2 (Scheme 6B).¶
 | ||
| Scheme 6 Continuous-flow setup and application. | ||
Conclusions
In conclusion, this work demonstrates an enhanced and sustainable protocol for the anti-dihydroxylation of unactivated alkenes, achieved through the in situ formation of peroxyacetic acid. This method employs an eco-friendly and renewable solvent, acetic acid, and a relatively mild oxidant, hydrogen peroxide. This catalyst-free process exhibits a broad substrate scope with good functional group tolerance, and it is amenable to large-scale synthesis in batch (e.g. 100 mmol scale). Moreover, to address safety hazards associated with the generation of large quantities of highly reactive peroxyacetic acid in large-scale synthesis, a continuous flow setup for the anti-dihydroxylation of olefins was also developed. Overall, our protocol offers a simple, efficient, and sustainable method for the synthesis of anti 1,2-diols on a multigram scale.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- P. T. Anastas and J. C. Warner, Green Chemistry, Oxford University Press, 2000 Search PubMed.
- C. J. Clarke, W.-C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed.
- I. T. Horváth, Chem. Rev., 2018, 118, 369–371 CrossRef PubMed.
- R. M. Bullock, J. G. Chen, L. Gagliardi, P. J. Chirik, O. K. Farha, C. H. Hendon, C. W. Jones, J. A. Keith, J. Klosin, S. D. Minteer, R. H. Morris, A. T. Radosevich, T. B. Rauchfuss, N. A. Strotman, A. Vojvodic, T. R. Ward, J. Y. Yang and Y. Surendranath, Science, 2020, 369, eabc3183 CrossRef CAS PubMed.
- B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS PubMed.
- U. Schneider and S. Thomas, Catalysis with Earth-abundant Elements, The Royal Society of Chemistry, 2020 Search PubMed.
- F. Darvas, G. Dormán, V. Hessel and S. V. Ley, Flow Chemistry – Applications, De Gruyter, 2021 Search PubMed.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed.
- H. P. L. Gemoets, Y. Su, M. Shang, V. Hessel, R. Luque and T. Noël, Chem. Soc. Rev., 2016, 45, 83–117 RSC.
- D. Dallinger and C. O. Kappe, Curr. Opin. Green Sustainable Chem., 2017, 7, 6–12 CrossRef.
- L. Rogers and K. F. Jensen, Green Chem., 2019, 21, 3481–3498 RSC.
- M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC.
- J. Britton and C. L. Raston, Chem. Soc. Rev., 2017, 46, 1250–1271 RSC.
- G. Franz and R. A. Sheldon, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2003 Search PubMed.
- M. D. Reddy, H. Kobori, T. Mori, J. Wu, H. Kawagishi and E. B. Watkins, J. Nat. Prod., 2017, 80, 2561–2565 CrossRef CAS PubMed.
- N. V. S. Mudiganti, S. Claessens, P. Habonimana and N. de Kimpe, J. Org. Chem., 2008, 73, 3867–3874 CrossRef CAS PubMed.
- N. K. Adlington, P. S. Siedlecki, I. Derrick, S. D. Yates, A. D. Campbell, P. Tomlin and T. Langer, Org. Process Res. Dev., 2022, 26, 2337–2350 CrossRef CAS.
- H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS.
- M. J. Rawling and N. C. O. Tomkinson, Org. Biomol. Chem., 2013, 11, 1434–1440 RSC.
- C. J. R. Bataille and T. J. Donohoe, Chem. Soc. Rev., 2011, 40, 114–128 RSC.
- C. Wang, Asian J. Org. Chem., 2018, 7, 509–521 CrossRef CAS.
- D. Swern, Chem. Rev., 1949, 45, 1–68 CrossRef CAS.
- M. Mugdan and D. P. Young, J. Chem. Soc., 1949, 2988–3000 RSC.
- I. Cristea, E. Kozma and C. Batiu, Tetrahedron: Asymmetry, 2002, 13, 915–918 CrossRef CAS.
- W. A. Herrmann, R. W. Fischer and D. W. Marz, Angew. Chem., Int. Ed. Engl., 1991, 30, 1638–1641 CrossRef.
- P. Gogoi, S. D. Sharma and D. Konwar, Lett. Org. Chem., 2007, 4, 249–252 CrossRef CAS.
- P. Fan, S. Su and C. Wang, ACS Catal., 2018, 8, 6820–6826 CrossRef CAS.
- Ł. Albrecht, H. Jiang, G. Dickmeiss, B. Gschwend, S. G. Hansen and K. A. Jørgensen, J. Am. Chem. Soc., 2010, 132, 9188–9196 CrossRef PubMed.
- S. Rani and Y. D. Vankar, Tetrahedron Lett., 2003, 44, 907–909 CrossRef CAS.
- X. Wang and Y. R. Lee, Tetrahedron Lett., 2007, 48, 6275–6280 CrossRef CAS.
- T. Li and C. Li, J. Agric. Food Chem., 2013, 61, 12522–12530 CrossRef CAS PubMed.
- B. J. Deadman, S. Gian, V. E. Y. Lee, L. A. Adrio, K. Hellgardt and K. K. Hii, Green Chem., 2022, 24, 5570–5578 RSC.
- M.-Z. Zhang, P. Wang, H.-Y. Liu, D. Wang, Y. Deng, Y.-H. Bai, F. Luo, W.-Y. Wu and T. Chen, ChemSusChem, 2023, 16, e202300583 CrossRef CAS PubMed.
- Y. Masuda, D. Ikeshita and M. Murakami, Helv. Chim. Acta, 2021, 104, e2000228 CrossRef CAS.
- C. Hampton, M. Simonetti and D. Leonori, Angew. Chem., Int. Ed., 2023, 62, e202214508 CrossRef CAS PubMed.
- M. Liu, T. Feng, Y. Wang, G. Kou, Q. Wang, Q. Wang and Y. Qiu, Nat. Commun., 2023, 14, 6467 CrossRef CAS PubMed.
- W. Jud, C. O. Kappe and D. Cantillo, Electrochem. Sci. Adv., 2021, 1, e2100002 CrossRef CAS.
- X. M. Zhang, A. Archelas and R. Furstoss, J. Org. Chem., 1991, 56, 3814–3817 CrossRef CAS.
- Y. Xu, A. Li, X. Jia and Z. Li, Green Chem., 2011, 13, 2452 RSC.
- S. Wu, Y. Chen, Y. Xu, A. Li, Q. Xu, A. Glieder and Z. Li, ACS Catal., 2014, 4, 409–420 CrossRef CAS.
- T. P. Hilditch, J. Chem. Soc., 1926, 129, 1828–1836 RSC.
- W. C. Smit and J. Böeseken, Recl. Trav. Chim. Pays-Bas, 1930, 49, 675–685 CrossRef CAS.
- D. Swern, G. N. Billen, T. W. Findley and J. T. Scanlan, J. Am. Chem. Soc., 1945, 67, 1786–1789 CrossRef CAS.
- Y. Usui, K. Sato and M. Tanaka, Angew. Chem., Int. Ed., 2003, 42, 5623–5625 CrossRef CAS PubMed.
- A. A. Rosatella and C. A. M. Afonso, Adv. Synth. Catal., 2011, 353, 2920–2926 CAS.
- J. T. Scanlan and D. Swern, J. Am. Chem. Soc., 1940, 62, 2305–2309 CrossRef CAS.
- J. T. Scanlan and D. Swern, J. Am. Chem. Soc., 1940, 62, 2309–2311 CrossRef CAS.
- A. Hartung, M. A. Keane and A. Kraft, J. Org. Chem., 2007, 72, 10235–10238 CrossRef CAS PubMed.
- M. Tobiszewski, J. Namieśnik and F. Pena-Pereira, Green Chem., 2017, 19, 1034–1042 RSC.
- T. Freese, R. Kat, S. D. Lanooij, T. C. Böllersen, C. M. de Roo, N. Elzinga, M. Beatty, B. Setz, R. R. Weber, I. Malta, T. B. Gandek, P. Fodran, R. Pollice and M. M. Lerch, A guidebook for sustainability in laboratories, 2024 Search PubMed.
- C. W. Jones, J. H. Clark and M. J. Braithwaite, Applications of Hydrogen Peroxide and Derivatives, The Royal Society of Chemistry, 1999 Search PubMed.
- R. Ciriminna, L. Albanese, F. Meneguzzo and M. Pagliaro, ChemSusChem, 2016, 9, 3374–3381 CrossRef CAS PubMed.
- A. Agosti, G. Bertolini, G. Bruno, C. Lautz, T. Glarner and W. Deichtmann, Org. Process Res. Dev., 2017, 21, 451–459 CrossRef CAS.
- M. Janković and S. Sinadinović-Fišer, J. Am. Oil Chem. Soc., 2005, 82, 301–303 CrossRef.
- K. D. Collins and F. Glorius, Nat. Chem., 2013, 5, 597–601 CrossRef CAS PubMed.
- K. D. Collins, A. Rühling and F. Glorius, Nat. Protoc., 2014, 9, 1348–1353 CrossRef CAS PubMed.
- K. D. Collins, A. Rühling, F. Lied and F. Glorius, Chem. – Eur. J., 2014, 20, 3800–3805 CrossRef CAS PubMed.
- T. Gensch, M. Teders and F. Glorius, J. Org. Chem., 2017, 82, 9154–9159 CrossRef CAS PubMed.
- J. English and J. D. Gregory, J. Am. Chem. Soc., 1947, 69, 2120–2122 CrossRef CAS.
- Safety Data Sheet, available at: https://www.sigmaaldrich.com/DE/en/sds/SIGALD/269336?userType=undefined, accessed July 2024.
- W.-C. Chen, J.-R. Lin, M.-S. Liao, Y.-W. Wang and C.-M. Shu, J. Therm. Anal. Calorim., 2017, 127, 1019–1026 CrossRef CAS.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- B. Gutmann and C. O. Kappe, J. Flow Chem., 2017, 7, 65–71 CrossRef CAS.
- M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Org. Process Res. Dev., 2020, 24, 1802–1813 CrossRef CAS.
- L. Capaldo, Z. Wen and T. Noël, Chem. Sci., 2023, 14, 4230–4247 RSC.
- D. Mazzarella, J. Stanić, M. Bernús, A. S. Mehdi, C. J. Henderson, O. Boutureira and T. Noël, JACS Au, 2024, 4, 2989–2994 CrossRef CAS PubMed.
- A. Bonner, P. Naik, R. Crawford and M. Baumann, Curr. Opin. Green Sustainable Chem., 2024, 47, 100907 CrossRef.
- A. Reinold, S. Leininger and A. Hellwig, US Pat, US9573893B2, 2017 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc03540b |
| ‡ These authors contributed equally. |
| § See Table S1 in the ESI† for further optimisation studies. |
| ¶ Flow process was run under unoptimised reaction conditions. |
| This journal is © The Royal Society of Chemistry 2024 |