
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A sequential flow process of CO2 capture and conversion using cost-effective porous organic polymers†
Zhongqi
Wu‡
a,
Zhong
Li‡
a,
Lei
Hu
a,
Samson
Afewerki
 b,
Maria
Strømme
b,
Qian-Feng
Zhang
a and
Chao
Xu
*ab
b,
Maria
Strømme
b,
Qian-Feng
Zhang
a and
Chao
Xu
*ab
aInstitute of Molecular Engineering and Applied Chemistry, Anhui University of Technology, Ma'anshan, 243002, P. R. China
bDivision of Nanotechnology and Functional Materials, Department of Materials Science and Engineering, Ångström Laboratory, Uppsala University, 75121 Uppsala, Sweden. E-mail: chao.xu@angstrom.uu.se
First published on 24th September 2024
Abstract
Porous organic polymers (POPs) have shown significant potential for CO2 capture and utilization due to their high surface areas, tunable porosity, high stability, and ease of modification. Developing POPs for CO2 capture and catalytic conversion offers a viable solution to rising CO2 emissions. This study presents POPs composed of pyridine units, serving as dual functional materials that act as sorbents for CO2 capture and as substrates supporting silver chalcogenolate clusters (SCCs) for catalytic CO2 conversion. The scalable and cost-effective synthesis of these POPs enabled the design of pilot-scale breakthrough apparatus with two parallel POP sorbent beds for continuous CO2 capture from simulated flue gas, achieving a high working capacity of 20 Lflue gas kgPOP−1 h−1 for flue gas separation. Given the practical feasibility of using POPs for CO2 capture and the high catalytic activity of POPs loaded with SCCs in CO2 cycloaddition, a sequential process that integrates capturing CO2 from simulated flue gas and directly converting the captured CO2 into oxazolidinone achieves a high space–time yield of up to 9.6 g LPOP−1 day−1 in continuous operation. This study provides a viable strategy for CO2 capture and utilization using cost-effective, dual-functional porous materials.
Introduction
The increasing emissions of CO2 and the accumulation of this greenhouse gas in the atmosphere have significantly impacted climate change, posing a threat to the human living environment.1 Post-combustion capture of CO2 from point sources is considered a feasible strategy to reduce CO2 emission.2–4 In this process, CO2 is selectively captured from emissions such as flue gas generated by burning fossil fuels in various industries, employing technologies like absorption, adsorption, or membrane separation. The captured CO2 can be either permanently stored underground or converted into valuable products such as fine chemicals, polymers, and fuels via catalytic processes. Compared to the costly CO2 storage method involving compression, transportation, injection, and the risk of leakage, direct utilization of CO2 presents a more economically friendly approach. Therefore, carbon capture and utilization emerge as a promising strategy for mitigating CO2 emissions.5,6Porous organic polymers (POPs) represent a novel class of functional porous materials constructed by linking purely organic linkers via covalent bonds, exhibiting significant potential in both CO2 capture and CO2 conversion.7–12 Compared to traditional inorganic porous materials, POPs offer distinct advantages such as high surface area, tunable pore size, and rich surface chemistry. Moreover, the synthetic diversity and ease of modification of POPs facilitate the design of materials with tailored properties and functionalities for specific applications.13,14 One common strategy for functionalizing POPs involves introducing heteroatoms into the framework.15–17 For instance, incorporating nitrogen-containing pyridine units into POPs can create strong interactions with CO2 molecules via Lewis acid–base interactions and electrostatic forces.18 Additionally, the nitrogen atoms in pyridine units, with their lone pair electrons, can act as coordination sites for bonding with transition metal ions, thereby introducing catalytically active centers into POPs.19–21 Consequently, designing POPs with specific functional groups represents a viable approach for developing dual-functional materials capable of both CO2 capture and conversion. Although POPs have been extensively studied as sorbents for CO2 capture and as catalysts or catalyst supports for CO2 conversion separately, the coupling of CO2 capture and CO2 conversion in a flow process utilizing POPs as dual-functional materials has not yet been developed. Furthermore, practical applications of POPs in CO2 capture and conversion remain limited primarily due to their high synthesis cost, harsh synthesis conditions, and the resulting challenges associated with scale-up synthesis.
Remarkably, recent studies have demonstrated the feasibility of synthesizing POPs through cost-effective and environmentally friendly approaches, facilitated by the wealth of available organic linkers and the versatility of polymer chemistry. For instance, Xu et al. reported the kilogram-scale synthesis of crystalline covalent triazine frameworks—a subclass of POPs—using the low-cost linker 1,4-dicyanobenzene catalyzed by polyphosphoric acid.22 Similarly, Zhang et al. reported the synthesis of olefin-linked covalent organic frameworks (COFs)—a type of crystalline POP—via a melt polymerization method employing inexpensive monomers tetramethylpyrazine and terephthalaldehyde.23 Recently, we also developed the green and low-cost synthesis of imine and aminal-linked POPs using diamine and dialdehyde linkers in green solvents such as water and ethanol at room temperature.24 The advantages of low cost and scalability in the synthesis process are highly beneficial for their practical applications, such as in adsorption and catalysis. Additionally, we achieved the aqueous synthesis of a series of imine-linked COFs with high surface areas under ambient conditions.25 These results have inspired us to integrate low-cost and scalable POP-based materials into chemical engineering processes such as separation, purification, and catalysis, to advance their real-world applications. Herein, we developed cost-effective pyridine-based porous organic polymers (POPs) as sorbents for CO2 capture and as substrates to immobilize catalysts composed of silver chalcogenolate clusters (SCCs) for catalytic CO2 conversion. By coupling the CO2 capture and catalytic CO2 conversion processes, we enabled continuous capture of CO2 from flue gas and its direct conversion into fine chemicals (Fig. 1).
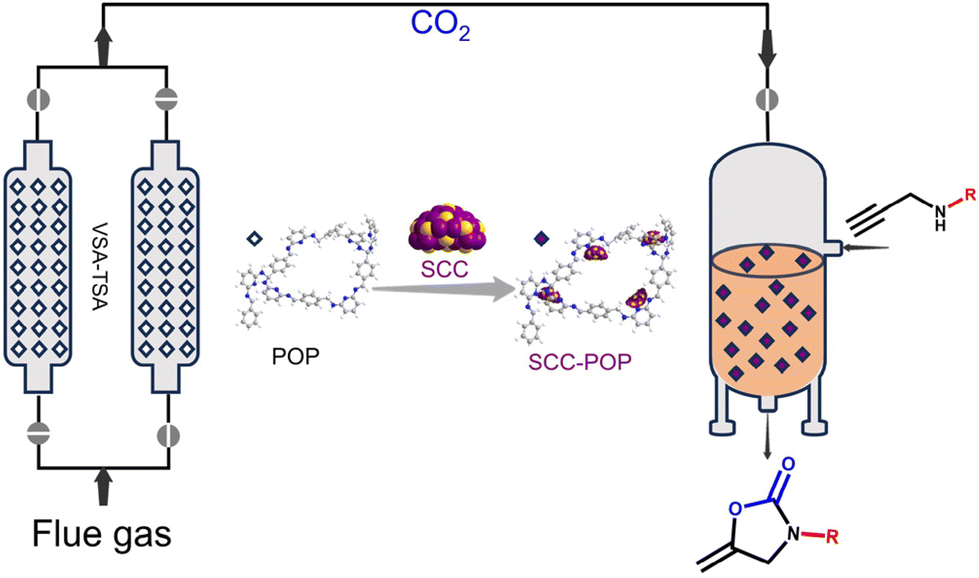 | ||
| Fig. 1 Illustration of the sequential flow CO2 capture and conversion process. | ||
Results and discussion
Synthesis and characterization of POPs
The POPs incorporating pyridine units were synthesized via Schiff-base condensation between organic monomers, specifically diaminopyridine and aromatic dialdehyde.24 By employing cross-condensation reactions of distinct diamines, namely 3,5-diaminopyridine (3,5-DAP) and 2,6-diaminopyridine (2,6-DAP), with dialdehydes such as terephthalaldehyde (TPA), isophthalaldehyde (IPA), and 4,4′-biphenyldicarboxaldehyde (BDA) in ethanol with aqueous acetic acid as the catalyst, six unique POPs were synthesized (Fig. 2a). Key synthesis parameters, including monomer and acetic acid concentration, reaction temperature, and reaction time were optimized, aiming to achieve high specific surface areas for the POPs (Tables S1–3†). The synthesis conducted on a sub-gram scale resulted in POPs with surface areas ranging between 340 and 590 m2 g−1, under solvothermal conditions at 120 °C. All of the POPs exhibited a notable N2 uptake at low partial pressures (P/P0 < 0.05) at 77 K, indicating the formation of microporous structures (Fig. 2b and S1†). Pore size distribution analysis revealed micropores within the range of 0.5–1.6 nm in the POPs. The POPs showed mainly an amorphous structure with high thermal stability (Fig. S2 and 3†). The molecular structure of the POPs was characterized using solid-state 13C nuclear magnetic resonance (NMR) spectroscopy and infrared (IR) spectroscopy. The 13C NMR spectra showed characteristic bands corresponding to carbon atoms in aminal and imine bonds at chemical shifts of 52 and 162 ppm, respectively. The intense bands observed between 110 and 150 ppm were attributed to aromatic carbons, while a weak band at 193 ppm indicated the presence of carbon atoms in residual aldehyde end groups (Fig. 2c).26,27 Consistently, IR spectra displayed stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds at 1634 cm−1 and those of the N–H bonds of aminal groups at 3390 cm−1, respectively. These results confirmed the formation of imine and aminal linkages in the POPs (Fig. 2d). Notably, the formation of aminal linkages could result in a branch network, which is crucial for achieving high porosity in the polymer synthesized via [2 + 2] Schiff-base condensation.
N bonds at 1634 cm−1 and those of the N–H bonds of aminal groups at 3390 cm−1, respectively. These results confirmed the formation of imine and aminal linkages in the POPs (Fig. 2d). Notably, the formation of aminal linkages could result in a branch network, which is crucial for achieving high porosity in the polymer synthesized via [2 + 2] Schiff-base condensation.
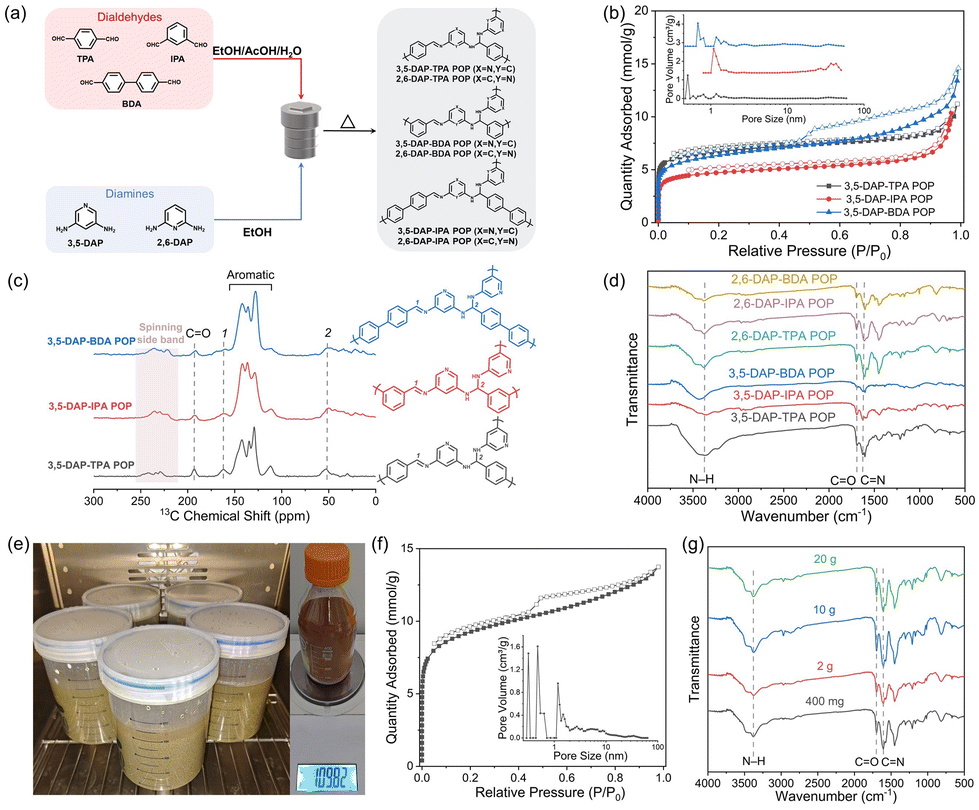 | ||
| Fig. 2 (a) Synthesis route of POPs; (b) N2 sorption isotherms of the POPs recorded at 77 K. The inset shows the pore size distribution calculated from the adsorption branches; (c) solid-state 13C NMR spectra, and (d) infrared spectra of the POPs; (e) optical images of sub-kilogram synthesis of 2,6-DAP-TPA POP and the product; (f) N2 sorption isotherm and pore size distribution of 2,6-DAP-TPA POP synthesized at a sub-kilogram scale; and (g) infrared spectra of 2,6-DAP-TPA POP synthesized at different scales. | ||
To enable large-scale production of POPs and to further their practical applications, we scale up the synthesis while reducing the reaction temperature. In this context, cost-effective monomers 2,6-DAP and TPA were selected for the scale-up synthesis of the POP. Through careful optimization of the synthesis conditions, we successfully synthesized 2,6-DAP-TPA POP at a lower temperature of 80 °C, achieving scales of ∼2 g, 10 g, and 20 g and a high yield of ∼98% (Table S4†). Notably, the moderate synthesis conditions enabled the production of POPs in polypropylene plastic buckets instead of autoclaves, significantly facilitating large-scale production. As shown in Fig. 2e, five parallel synthesis batches yielded a total of ∼110 g product, denoted as LSS-2,6-DAP-TPA POP (LSS: large scale synthesized). The scaled-up reactions resulted in products with high specific surface areas reaching up to 745 m2 g−1 (Fig. 2f). Importantly, scaling up the synthesis did not result in significant changes to the molecular structure of 2,6-DAP-TPA POP (Fig. 2g). The synthesis costs of the POP were calculated to be ∼63 USD per kg and ∼2420 USD per ton at different production scales (Table S5†), which are comparable to that of commercial porous materials such as activated carbon and zeolite, and much cheaper than the production costs of most reported POPs, covalent organic frameworks, and metal–organic frameworks.
CO2 adsorption and breakthrough experiments
The performance of the POPs in CO2 capture was initially evaluated through the study of their CO2 and N2 sorption isotherms (Fig. 3a and S4–6†). The POPs demonstrated curved CO2 adsorption isotherms with significant adsorption capacities reaching up to 2.95 mmol g−1 at 1 bar and 1.08 mmol g−1 at 0.15 bar, both measured at 273 K. Based on the single-component CO2 and N2 adsorption isotherms recorded under identical conditions, the CO2/N2 selectivities of the POPs were found to range from 48 to 94 using Ideal Adsorbed Solution Theory (IAST)28 for a simulated flue gas (15 v% CO2/85 v% N2) at 273 K (Fig. 3b and Tables S6, 7†), which are comparable with those of reported POPs (Table S8†). The notable CO2 uptake and the high CO2/N2 selectivity can be attributed to the presence of (ultra)micropores and the N species in the POPs, which promote strong interactions with CO2 molecules. Based on the adsorption isotherms recorded at different temperatures (273, 293, and 298 K), the heat of adsorption of CO2 (Qst) for POPs was calculated to be 29.4–32.7 kJ mol−1 (Fig. 3c). The moderate Qst values enabled efficient temperature swing adsorption (TSA) and regeneration of the adsorbent at lower energy costs. As expected, up to 45 wt%, 60 wt%, and 90 wt% of captured CO2 can be released from LSS-2,6-DAP-TPA POP when the temperature is increased from 25 °C to 50 °C, 70 °C, and 100 °C, respectively, under TSA conditions, and the sorbent can be fully regenerated at 150 °C (Fig. 3d). Moreover, the sorbents demonstrate excellent stability during cyclic adsorption, with no significant changes in CO2 uptake observed across several TSA and vacuum swing adsorption (VSA) cycles (Fig. S7†). | ||
Fig. 3 (a) CO2 adsorption isotherms of POPs recorded at 273 K; (b) CO2-over-N2 selectivity of POPs at 273 K; (c) heat of adsorption of CO2 for POPs; (d) gravimetric CO2 uptake of LSS-2,6-DAP-TPA POP at 298 K and elevated temperatures. (e–l) Column breakthrough experiments using LSS-2,6-DAP-TPA POP for CO2 capture from simulated flue gases at 293 K: (e) breakthrough curves of simulated flue gases at 293 K under dry and humid conditions, and in the presence of SO2 and NO2, with a volumetric ratio of CO2 to N2 in the gas mixtures of 15%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85%; (f) adsorption and desorption processes of the breakthrough test; (g) breakthrough curves of gas mixtures with different compositions (CO2/N2 = 15 v%/85 v%, 66 v%/34 v%, 87 v%/13 v%, from left to right) for mimicking a multi-bed separation process, and (h) the resulting increased CO2 concentration in the purified gas mixture; (i) optical images of the column and the sample (15 g of LSS-2,6-DAP-TPA POP) used in the pilot-scale breakthrough test; (j) results of pilot-scale breakthrough experiments at different flow rates; and (k) schematic view and (l) optical image of the continuous CO2 capture apparatus coupled with 2 POP beds (MFC = Mass Flow Controller). :85%; (f) adsorption and desorption processes of the breakthrough test; (g) breakthrough curves of gas mixtures with different compositions (CO2/N2 = 15 v%/85 v%, 66 v%/34 v%, 87 v%/13 v%, from left to right) for mimicking a multi-bed separation process, and (h) the resulting increased CO2 concentration in the purified gas mixture; (i) optical images of the column and the sample (15 g of LSS-2,6-DAP-TPA POP) used in the pilot-scale breakthrough test; (j) results of pilot-scale breakthrough experiments at different flow rates; and (k) schematic view and (l) optical image of the continuous CO2 capture apparatus coupled with 2 POP beds (MFC = Mass Flow Controller). | ||
To demonstrate the effectiveness of POPs in post-combustion CO2 capture, the POP powders were packed into a column as a fixed bed sorbent to separate a simulated flue gas (15 v% CO2/85 v% N2) in breakthrough experiments.29,30 Specifically, the breakthrough experiments were carried out at 293 K, using 1.0 g of POP powder. The flow rate of the gas mixture was set at 2 mL min−1, and the pressure was maintained at 1 bar throughout the tests. The POPs exhibit distinct selectivity for CO2 over N2. Pure N2 elutes rapidly at 2.4 min, whereas CO2 breaks through the column at 18–26.4 min (Fig. 3e and S8†). This finding suggests that the sorbent bed selectively captures CO2 molecules from the simulated flue gas and, hence, a purified gas with increased CO2 concentration can be obtained by applying elevated temperature and/or vacuum on the bed. Moreover, neither the presence of moisture (at a relative humidity of 85%) nor the presence of NO2 and SO2 (totaling 100 ppm) in the simulated flue gas significantly affected the separation efficiency, demonstrating the potential of using the POPs for CO2 capture from real flue gas. Calculations based on breakthrough curves indicate that the CO2 concentration in the purified gas could reach up to 66 v%, significantly higher than the initial 15% in the simulated flue gas. When the saturated column was flushed with helium (He) at a flow rate of 2 mL min−1 at room temperature for 20 min, most of the N2 was eluted, while a significant amount of the captured CO2 remained in the column (Fig. 3f). This is due to the relatively higher binding affinity between CO2 and the POP bed compared to that between N2 and the POP. On increasing the temperature to 150 °C after the He flush, the desorption curves showed only CO2 signals, indicating that pure CO2 gas with a high purity of >99 v% could be collected. To simulate the multi-bed separation process, a gas mixture (comprising 66 v% CO2 and 34 v% N2), mimicking the gas component obtained from the first separation process, was subjected to breakthrough experiments (Fig. 3g). The results demonstrate that the CO2 concentration in the purified gas could increase up to 87 v% and 93 v% after the second and third upgrading processes, respectively (Fig. 3h). Furthermore, the POP bed showed significant separation efficiency at an elevated temperature of 313 K, which is close to the operational temperature used in practical post-combustion CO2 capture processes (Fig. S9†).
To further assess the working capacity and efficiency of POPs for practical CO2 capture from flue gas, we conducted a pilot-scale breakthrough experiment using a larger column filled with 15 g of 2,6-DAP-TPA POP (Fig. 3i). At a flow rate of 2 mL min−1, CO2 elutes from the sorbent bed after a prolonged period of 249.6 min, with CO2 adsorption reaching equilibrium at 729.6 min. In contrast, N2 breaks through the column in a much shorter time of 9.6 min (Fig. 3j). It was calculated that 120.17 mL of CO2 and 50.89 mL of N2 were adsorbed on the sorbent bed. Subsequently, a pre-vacuumed flask with a volume of 250 mL was connected to the column, which was then heated to 150 °C to release the captured gas. Monitoring the pressure change in the flask over time revealed the desorption kinetics, indicating that 120 and 150 mL of gas could be captured in 10 and 30 min, respectively (Fig. S10†). Gas chromatographic analysis of the collected gas revealed a CO2/N2 ratio of 66.5 v%/33.5 v%, which is close to the value calculated from the breakthrough curves. When the flow rate is increased to 10 mL min−1, CO2 starts to elute from the sorbent bed at 48.0 min, while N2 elutes at 4.8 min. The results suggested that the CO2 concentration in purified gas could reach up to 60 v%. This result indicated that increasing the gas flow rate from 2 to 10 mL min−1 did not significantly affect the separation efficiency, probably because of the rapid kinetics of CO2 adsorption on the POPs (Fig. S11†). A higher flow rate could significantly increase the overall CO2 separation working capacity. By integrating two POP sorbent beds for simultaneous operation—one absorbing and the other desorbing and releasing purified CO2—continuous gas capture was enabled (Fig. 3k and l). The alternating roles of the beds enabled continuous separation under combined TSA–VSA conditions, achieving a CO2 capture working capacity of ∼20 L of flue gas per kg of POP sorbent per hour.
Catalytic CO2 conversion
Given the high porosity and abundant pyridine units serving as coordination sites in POPs, we anchored SCCs onto the polymer to impart catalytic activity and facilitate CO2 transformation (Fig. 4a). The selection of SCCs is due to their ability to act as π-activators of the carbon–carbon triple bond, enabling efficient cycloaddition of alkynes.31–33 Specifically, a molecular SCC, [Ag33S3(tBuS)16(CF3COO)11(CH3CN)4],34 was immobilized onto the POPs via coordination between the Ag atoms in the cluster and the N atoms in the pyridine unit of the POPs, resulting in six hybrid composites Ag33-3,5-DAP-TPA POP, Ag33-3,5-DAP-IPA POP, Ag33-3,5-DAP-BDA POP, Ag33-2,6-DAP-TPA POP, Ag33-2,6-DAP-IPA POP, and Ag33-2,6-DAP-BDA POP. The Ag loadings on the composites were determined to be 2.47, 1.05, 1.41, 1.76, 2.75, and 1.52 wt%, respectively, by Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES). All the composites showed an amorphous structure based on X-ray diffraction (XRD) analyses, similar to their POP counterparts (Fig. 4b and S13†). The absence of diffraction peaks for the silver cluster indicates that no isolated cluster molecules were present in the composites. Additionally, Ag33-POPs exhibited comparable high thermal stability to POPs, with an onset decomposition temperature of ∼400 °C in air, which is crucial for their utilization at elevated temperatures. The residual materials obtained after the thermal decomposition of Ag33-POPs at 800 °C under air were identified as elemental Ag through XRD analysis, confirming the successful immobilization of the Ag33 cluster (Fig. S14†). More significantly, the rich microporosity, high surface area, and high CO2 adsorption capacity were retained in the POPs after the immobilization of the cluster, which is greatly beneficial for their application in catalytic CO2 conversion (Fig. 4c and S15†). The surface area and CO2 adsorption capacity of the POPs slightly decreased upon loading Ag33 due to the increased bulk density. The composites showed a similar nanosphere morphology to their POP counterparts, with a diameter of 1–2 μm (Fig. 4d, e and S16, 17†). Elemental mapping using energy-dispersive X-ray spectroscopy indicated the uniform distribution of Ag and S elements on the surface of the substrate (Fig. 4f). The composites and the molecular Ag33 cluster showed similar high-resolution Ag 3d and S 2p X-ray photoelectron spectra (XPS), indicating that the molecular structure of the Ag33 cluster remained intact upon coordination bonding to the POPs (Fig. 4g and h). Additionally, analysis of N 1s and C 1s XPS spectra and infrared spectra indicated that the immobilization of Ag33 did not influence the molecular structure of the POPs (Fig. 4i and S18, 19†).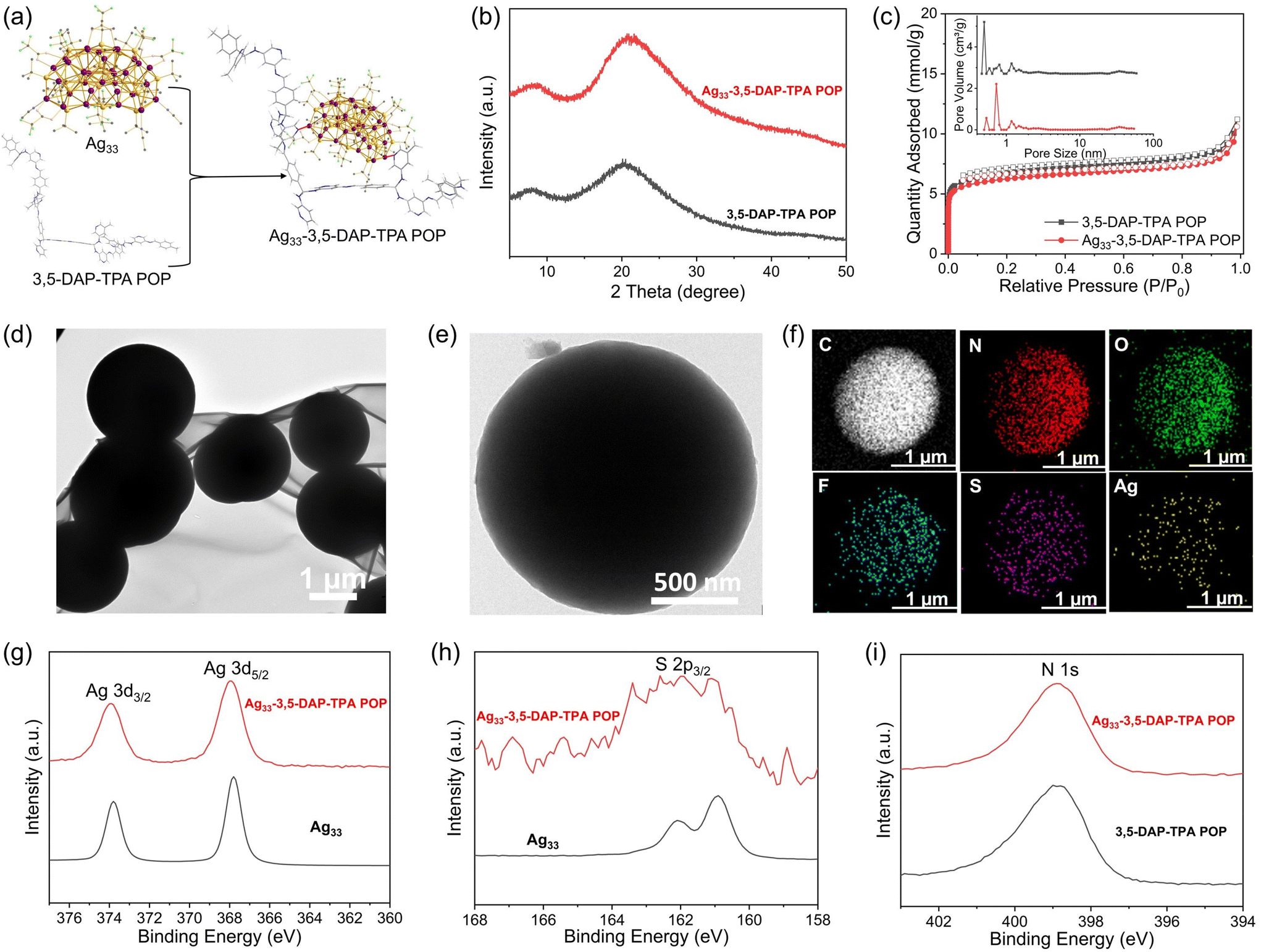 | ||
| Fig. 4 (a) Synthesis route of Ag33-3,5-DAP-TPA POP; (b) XRD patterns of 3,5-DAP-TPA POP and its counterparts loaded with Ag33 clusters; (c) N2 sorption isotherms and pore size distribution of 3,5-DAP-TPA POP and Ag33-3,5-DAP-TPA POP; (d and e) TEM images and (f) EDS elemental mapping images of Ag33-3,5-DAP-TPA POP; and Ag 3d (g) and S 2p (h) XPS spectra of Ag33 and Ag33-3,5-DAP-TPA POP. (i) High resolution N 1s XPS spectra of 3,5-DAP-TPA POP and Ag33-3,5-DAP-TPA POP. Ag33 = [Ag33S3(tBuS)16(CF3COO)11(CH3CN)4]. | ||
The catalytic activities of Ag33-POPs were evaluated through CO2 cycloaddition with N-methylpropargylamine (1a) in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as the co-catalyst under 1 bar pressure of CO2. Key parameters, including the solvent, reaction time, and reaction temperature, were optimized through screening experiments using Ag33-3,5-DAP-TPA POP as the catalyst (Table S9†). The use of CH3CN as the solvent resulted in higher yields than with other solvents such as CH3OH, THF, and DMF (Table S9, entries 1–4†). Notably, catalysis at room temperature achieved yields of up to 80%, which increased to 95% when the temperature was elevated to 60 °C (Table S9, entries 4 and 5†). The catalytic reactions exhibited fast kinetics, achieving yields of 50%, 85%, and 90% after 1, 3, and 6 h, respectively, with a maximum yield of 95% after 24 h (Fig. 5a). More significantly, the catalyst showed high activity for CO2 cycloaddition even at low CO2 concentrations. A relatively high yield of 66% was achieved using a simulated flue gas (15 v% CO2/85 v% N2) under ambient pressure. When the CO2 concentration in the gas mixture reached 60% or higher, the catalytic efficiency was comparable to that when using pure CO2 (Fig. 5b and Table S10†). The high catalytic activity of Ag33-POPs can be attributed to the homogeneous distribution of high-density silver centers as π-Lewis acid catalytic sites within the porous polymer networks, enabling efficient activation of alkyne substrates and facilitating rapid mass transport for catalytic conversion (Fig. 5c). In addition, the high CO2 adsorption capacity of Ag33-POPs enabled increased local concentration of CO2 on the porous surface, which accelerated reaction kinetics and improved the overall conversion efficiency.35,36 Control experiments using Ag33 and its precursors tBuSAg and CF3COOAg resulted in significantly lower yields of 53%, 32%, and 29%, respectively, while using pure POP resulted in a negligible yield of 9%. These results demonstrated the importance of integrating POPs and Ag33, highlighting the synergistic effect of the high catalytic activity centers and the porous structure of Ag33-POP for highly efficient CO2 conversion.
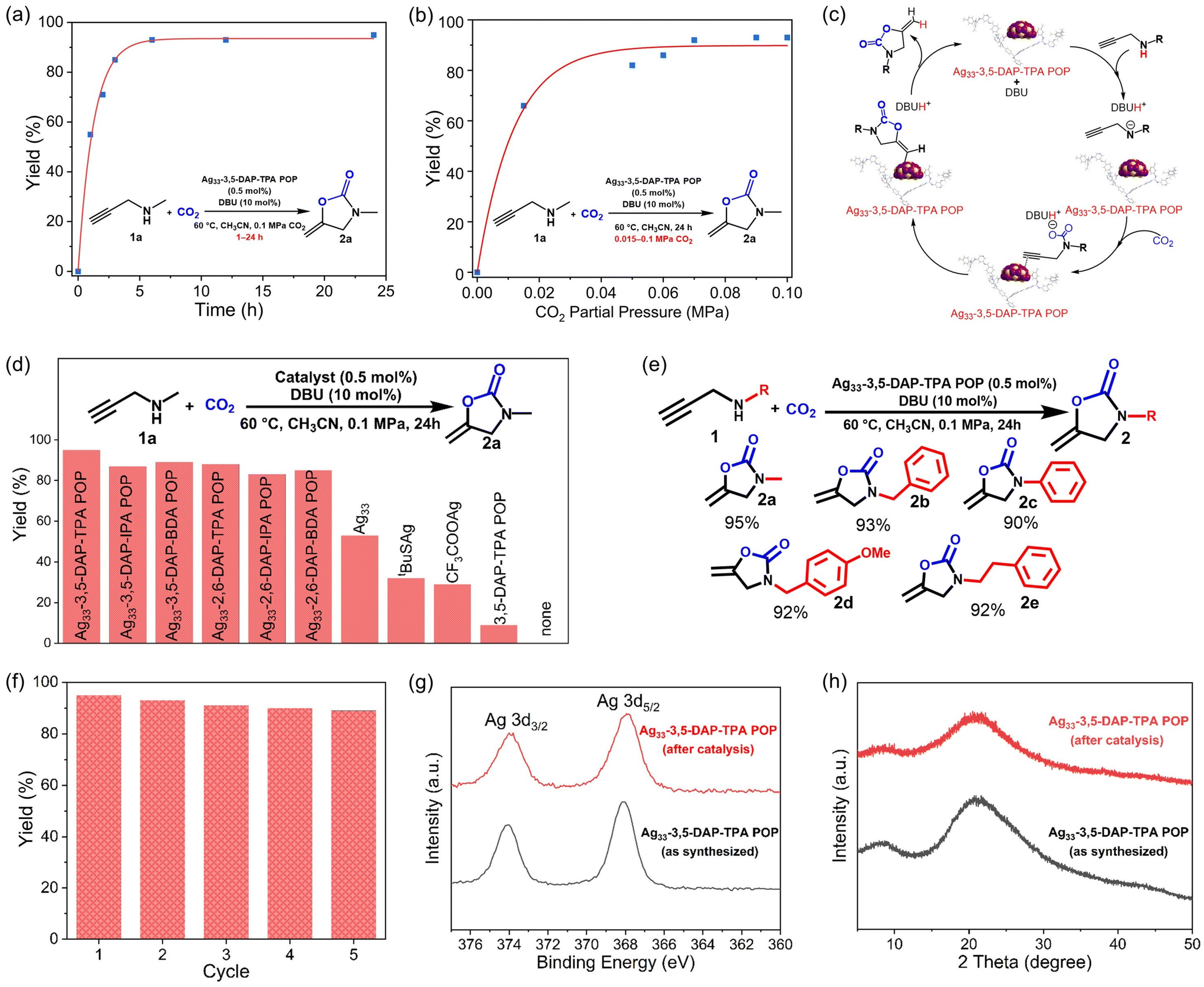 | ||
| Fig. 5 (a) Kinetics of CO2 cycloaddition with 1a using Ag33-3,5-DAP-TPA POP and the results of the hot filtration experiment; (b) influence of CO2 partial pressure on CO2 cycloaddition with 1a using Ag33-3,5-DAP-TPA POP; (c) proposed catalytic mechanism using Ag33-3,5-DAP-TPA POP for CO2 cycloaddition with 1; (d) yields of different catalysts for CO2 cycloaddition with N-methylpropargylamine (1a); (e) catalytic results using Ag33-3,5-DAP-TPA POP for CO2 cycloaddition with propargylamines with different N-substituents; (f) cyclic catalytic performance using Ag33-3,5-DAP-TPA POP for CO2 cycloaddition with 1a; and comparison of (g) Ag 3d XPS spectra and (h) XRD patterns of the as-synthesized Ag33-3,5-DAP-TPA POP and recycled catalyst after the reaction. | ||
Under the same conditions, other Ag33-POPs gave slightly lower yields of 80–89% after 24 h (Fig. 5d). It was observed that loading Ag33 onto 2,6-DAP-derived POPs resulted in slightly lower catalytic activity compared to those synthesized from 3,5-DAP. This difference in catalytic efficiency can be attributed to the steric effects introduced by the different positions of Ag33 coordination on the POPs. In the 2,6-DAP-derived POPs, the side groups at the 2 and 6 positions of the pyridine ring, where the Ag33 cluster is coordinated, create a crowded environment around Ag33 clusters. This steric hindrance restricts the access of reactant molecules, reducing the catalytic efficiency. Conversely, in the 3,5-DAP-derived POPs, the side groups at the 3 and 5 positions are further apart, creating a less crowded environment around the active sites. This reduced steric hindrance allows easier access for reactant molecules to the silver clusters, enhancing the catalytic efficiency. Using various propargylamine substrates for the catalytic conversions also resulted in high yields of 90–93% for different oxazolidinones (2b–e) (Fig. 5e and Table S11†). Remarkably, a gram-scale catalytic reaction was conducted, and the catalytic efficiency did not significantly decrease with scaling up of the reaction, yielding approximately 1.47 g of the product 3-methyl-5-methylene-2-oxazolidinone (2a) (Scheme S2†). The catalytic efficiency of Ag33-POPs is comparable to that of previously reported silver cluster and POP-based catalysts in similar CO2 cycloaddition reactions (Table S12†).
Moreover, the catalysts demonstrated good reusability, maintaining high catalytic activity for at least five consecutive cycles (Fig. 5f). Hot filtration experiments confirmed that no further catalytic reaction occurred once the catalyst was removed from the mixture, indicating negligible leaching of the silver ions or clusters into the solution during the reaction (Fig. S19†). Consistently, XPS and XRD studies revealed that the composition and structure of the catalyst remained intact during the catalytic reactions (Fig. 4g, h and S20†). The high stability of the catalyst can be attributed to the strong coordination between the POP substrate and the Ag33 clusters.
Sequential flow CO2 capture and conversion
Given the high catalytic activity and good recyclability of the Ag33-POPs in CO2 cycloaddition under mild conditions, the high CO2 capture performance of the POPs, and the cost efficiency and scalability of both the POP sorbents and catalysts, we envisioned the feasibility of using the purified flue gas from the POP sorbent beds as feedstock for highly efficient CO2 conversion (Fig. 6a and b). Consequently, we subjected the collected gas, with a volume of 150 mL and a CO2 concentration of 66% obtained from the pilot-scale breakthrough experiment at a flow rate of 2 mL min−1, to catalytic conversion using Ag33-3,5-DAP-TPA POP as the catalyst. This resulted in a high yield of 90% for the product 2a. Remarkably, the presence of 50 ppm of NO2 and 50 ppm of SO2 in the simulated flue gas had no significant influence on the catalytic efficiency (yield: 88%). Furthermore, we coupled the developed 2-bed TSA–VSA process with the catalysis process, enabling continuous CO2 capture from flue gas and subsequent CO2 conversion. To achieve a high space–time yield (STY) for practical production, we increased the flow rate of simulated flue gas from 2 to 10 mL min−1. As observed in the breakthrough studies, increasing the flow rate reduced the separation time in a single POP bed from 10 h to 2 h, resulting in a significant improvement in the working capacity for flue gas separation from 4 to 20 Lflue gas kgPOP−1 h−1 and a slight decrease in the CO2 concentration in the purified gas from 66% to 60%. The use of the collected gas with slightly lower CO2 concentration gave a high yield of 86%. Under the optimized conditions, the sequential CO2 capture and conversion process resulted in a high STY of 9.6 g LPOP−1 day−1 for the production of 2a (Fig. 6c, d and Table S13†).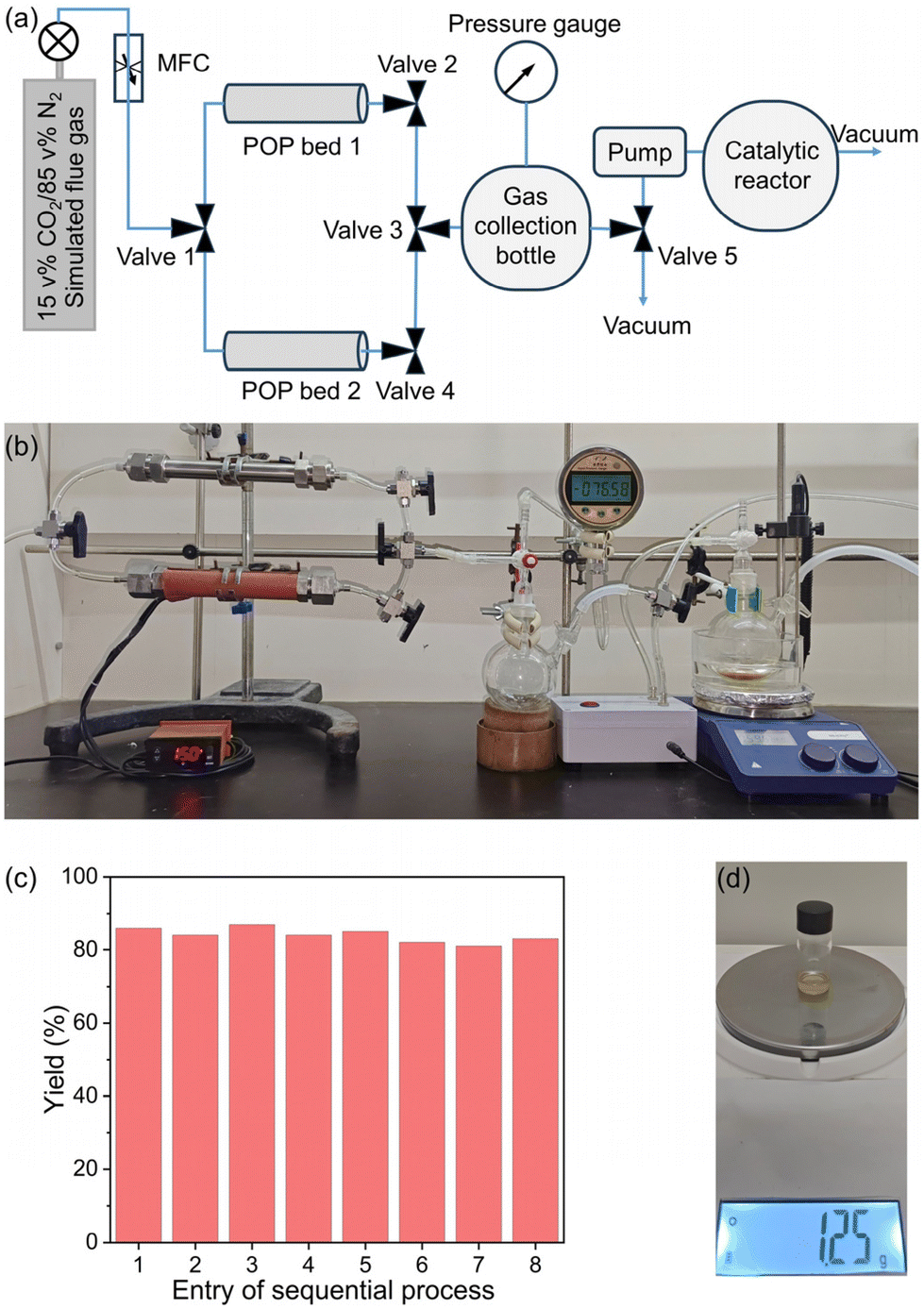 | ||
| Fig. 6 (a) Schematic view and (b) optical image of the apparatus for sequential CO2 capture and conversion from simulated flue gas (MFC = Mass Flow Controller); (c) catalytic results of 8 continuous sequential CO2 capture and conversion processes; and (d) image of the purified product obtained from several entries of sequential processes. | ||
Conclusions
To conclude, a series of POPs serving as dual functional materials have been successfully synthesized in a cost-effective and scalable manner. The rich microporosity of the POPs enabled the capture of CO2 from simulated flue gas with high adsorption capacity and selectivity. The pyridine units in the POPs provide anchoring sites for immobilizing silver chalcogenolate clusters, facilitating the design of efficient heterogeneous catalysts for CO2 cycloaddition into oxazolidinones. The POPs were used as sorbents in a pilot-scale breakthrough unit, enabling continuous CO2 capture from simulated flue gas with high working capacity by employing a combined TSA and VSA technique. Moreover, by integrating CO2 capture with CO2 conversion units, a sequential flow process was developed that captures CO2 from flue gas and directly converts the captured CO2 into fine chemicals. Future studies may focus on modifying or hybridizing the scalable POPs to optimize CO2 capture performance further, aiming for higher working capacity, selectivity, and lower energy costs. Additionally, the design of metal-free POP-based catalysts for highly efficient CO2 conversion will significantly enhance the sustainability of the catalytic process. Furthermore, investigating how to retrofit the developed materials and techniques with existing industrial processes for CO2 capture and utilization is crucial for their practical applications.Author contributions
C. X. conceived and designed the study. Z. W., Z. L., and L. H. performed the synthesis, characterization and catalysis studies. C. X. wrote the manuscript together with all the authors.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from the Swedish Research Council, Formas, Vinnova, ÅForsk, and the Swedish Energy Agency is gratefully acknowledged.References
- S. Solomon, G.-K. Plattner, R. Knutti and P. Friedlingstein, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1704–1709 CrossRef CAS PubMed.
- C. Chao, Y. Deng, R. Dewil, J. Baeyens and X. Fan, Renewable Sustainable Energy Rev., 2021, 138, 110490 CrossRef CAS.
- S. Chu, Science, 2009, 325, 1599–1599 CrossRef CAS PubMed.
- R. L. Siegelman, E. J. Kim and J. R. Long, Nat. Mater., 2021, 20, 1060–1072 CrossRef CAS PubMed.
- P. Gabrielli, M. Gazzani and M. Mazzotti, Ind. Eng. Chem. Res., 2020, 59, 7033–7045 CrossRef CAS.
- K. M. G. Langie, K. Tak, C. Kim, H. W. Lee, K. Park, D. Kim, W. Jung, C. W. Lee, H.-S. Oh, D. K. Lee, J. H. Koh, B. K. Min, D. H. Won and U. Lee, Nat. Commun., 2022, 13, 7482 CrossRef CAS PubMed.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS.
- N. Chaoui, M. Trunk, R. Dawson, J. Schmidt and A. Thomas, Chem. Soc. Rev., 2017, 46, 3302–3321 RSC.
- K. S. Song, P. W. Fritz and A. Coskun, Chem. Soc. Rev., 2022, 51, 9831–9852 RSC.
- N. Huang, X. Chen, R. Krishna and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 2986–2990 CrossRef CAS PubMed.
- X.-Y. Dong, Y.-N. Si, Q.-Y. Wang, S. Wang and S.-Q. Zang, Adv. Mater., 2021, 33, 2101568 CrossRef CAS PubMed.
- W. Zhou, Q.-W. Deng, G.-Q. Ren, L. Sun, L. Yang, Y.-M. Li, D. Zhai, Y.-H. Zhou and W.-Q. Deng, Nat. Commun., 2020, 11, 4481 CrossRef CAS PubMed.
- W. Chen, P. Chen, G. Zhang, G. Xing, Y. Feng, Y.-W. Yang and L. Chen, Chem. Soc. Rev., 2021, 50, 11684–11714 RSC.
- K. S. Song, T. Ashirov, S. N. Talapaneni, A. H. Clark, A. V. Yakimov, M. Nachtegaal, C. Copéret and A. Coskun, Chem, 2022, 8, 2043–2059 CAS.
- W. Lu, J. P. Sculley, D. Yuan, R. Krishna, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 7480–7484 CrossRef CAS PubMed.
- J. F. Van Humbeck, T. M. McDonald, X. Jing, B. M. Wiers, G. Zhu and J. R. Long, J. Am. Chem. Soc., 2014, 136, 2432–2440 CrossRef CAS PubMed.
- Q. Sun, B. Aguila, Y. Song and S. Ma, Acc. Chem. Res., 2020, 53, 812–821 CrossRef CAS PubMed.
- C. Liu, L. Shi, J. Zhang and J. Sun, Chem. Eng. J., 2022, 427, 131633 CrossRef CAS.
- X. Zhao, P. Pachfule, S. Li, T. Langenhahn, M. Ye, C. Schlesiger, S. Praetz, J. Schmidt and A. Thomas, J. Am. Chem. Soc., 2019, 141, 6623–6630 CrossRef CAS PubMed.
- X. Wang, Z. Fu, L. Zheng, C. Zhao, X. Wang, S. Y. Chong, F. McBride, R. Raval, M. Bilton, L. Liu, X. Wu, L. Chen, R. S. Sprick and A. I. Cooper, Chem. Mater., 2020, 32, 9107–9114 CrossRef CAS.
- H.-P. Liang, Q. Chen and B.-H. Han, ACS Catal., 2018, 8, 5313–5322 CrossRef CAS.
- T. Sun, Y. Liang, W. Luo, L. Zhang, X. Cao and Y. Xu, Angew. Chem., Int. Ed., 2022, 61, e202203327 CrossRef CAS PubMed.
- Z. Wang, Y. Zhang, E. Lin, S. Geng, M. Wang, J. Liu, Y. Chen, P. Cheng and Z. Zhang, J. Am. Chem. Soc., 2023, 145, 21483–21490 CrossRef CAS PubMed.
- D. Luo, T. Shi, Q.-H. Li, Q. Xu, M. Strømme, Q.-F. Zhang and C. Xu, Angew. Chem., Int. Ed., 2023, 62, e202305225 CrossRef CAS PubMed.
- X. Kong, Z. Wu, M. Strømme and C. Xu, J. Am. Chem. Soc., 2024, 146, 742–751 CrossRef CAS PubMed.
- A. Laybourn, R. Dawson, R. Clowes, J. A. Iggo, A. I. Cooper, Y. Z. Khimyak and D. J. Adams, Polym. Chem., 2012, 3, 533–537 RSC.
- M. G. Schwab, B. Fassbender, H. W. Spiess, A. Thomas, X. Feng and K. Müllen, J. Am. Chem. Soc., 2009, 131, 7216–7217 CrossRef CAS PubMed.
- S. Hug, L. Stegbauer, H. Oh, M. Hirscher and B. V. Lotsch, Chem. Mater., 2015, 27, 8001–8010 CrossRef CAS.
- Q. Dong, Y. Huang, J. Wan, Z. Lu, Z. Wang, C. Gu, J. Duan and J. Bai, J. Am. Chem. Soc., 2023, 145, 8043–8051 CrossRef CAS PubMed.
- H. Zeng, X.-J. Xie, T. Wang, M. Xie, Y. Wang, R.-J. Wei, W. Lu and D. Li, Nat. Chem. Eng., 2024, 1, 108–115 CrossRef.
- Z. Zhou, C. He, L. Yang, Y. Wang, T. Liu and C. Duan, ACS Catal., 2017, 7, 2248–2256 CrossRef CAS.
- M. Zhao, S. Huang, Q. Fu, W. Li, R. Guo, Q. Yao, F. Wang, P. Cui, C.-H. Tung and D. Sun, Angew. Chem., Int. Ed., 2020, 59, 20031–20036 CrossRef CAS PubMed.
- L. Li, Y. Lv, H. Sheng, Y. Du, H. Li, Y. Yun, Z. Zhang, H. Yu and M. Zhu, Nat. Commun., 2023, 14, 6989 CrossRef CAS PubMed.
- Y.-L. Shen, J.-L. Jin, G.-X. Duan, P.-Y. Yu, Y.-P. Xie and X. Lu, Chem. – Eur. J., 2021, 27, 1122–1126 CrossRef CAS PubMed.
- L. Liang, C. Liu, F. Jiang, Q. Chen, L. Zhang, H. Xue, H.-L. Jiang, J. Qian, D. Yuan and M. Hong, Nat. Commun., 2017, 8, 1233 CrossRef PubMed.
- L. Zhang, X.-X. Li, Z.-L. Lang, Y. Liu, J. Liu, L. Yuan, W.-Y. Lu, Y.-S. Xia, L.-Z. Dong, D.-Q. Yuan and Y.-Q. Lan, J. Am. Chem. Soc., 2021, 143, 3808–3816 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc03494e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |