
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient electrosynthesis of oximes from nitrates and carbonyl compounds in acidic media†
Cheng
Xue
ab,
Shuaiqiang
Jia
*ab,
Jiapeng
Jiao
ab,
Xiao
Chen
ab,
Zhanghui
Xia
ab,
Mengke
Dong
ab,
Ting
Deng
ab,
Hailian
Cheng
ab,
Chunjun
Chen
 ab,
Haihong
Wu
*ab,
Mingyuan
He
ab and
Buxing
Han
*abc
ab,
Haihong
Wu
*ab,
Mingyuan
He
ab and
Buxing
Han
*abc
aShanghai Key Laboratory of Green Chemistry and Chemical Processes, State Key Laboratory of Petroleum Molecular & Process Engineering, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China. E-mail: sqjia@chem.ecnu.edu.cn; hhwu@chem.ecnu.edu.cn; hanbx@iccas.ac.cn
bInstitute of Eco-Chongming, 20 Cuiniao Road, ChenjiaTown, Chongming District, Shanghai 202162, China
cBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid and Interface and Thermodynamics, Center for Carbon Neutral Chemistry, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
First published on 26th August 2024
Abstract
Electrocatalytic nitrate reduction reaction (NO3RR) coupled with organic compounds to synthesize oximes for value-added conversion of waste pollutants has very promising prospects. However, due to the susceptibility of the feedstock to hydrogenation, the relatively low reaction efficiency, and the poor stability of oximes, the reaction in acidic electrolytes remains a formidable challenge. Herein, we report a novel strategy for the one-step synthesis of oximes from nitrite and aldehydes/ketones in acidic electrolytes using a Zn-based catalyst (multilayered Zn nanosheet catalyst, M-ZnNSs). 99% yield and 99% selectivity of phenylacetaldehyde oxime were achieved at a constant current of −12 mA cm−2. Moreover, various aldehydes/ketones were efficiently converted to oximes with yield and selectivity up to >90%, demonstrating the high versatility of this method. Furthermore, the catalyst exhibited remarkable long-term stability (>100 h). This work proposes a green strategy to promote the recycling of nitrogen resources, enhance the value of NO3− conversion products, and develop new ideas for electrochemical C–N coupling.
Introduction
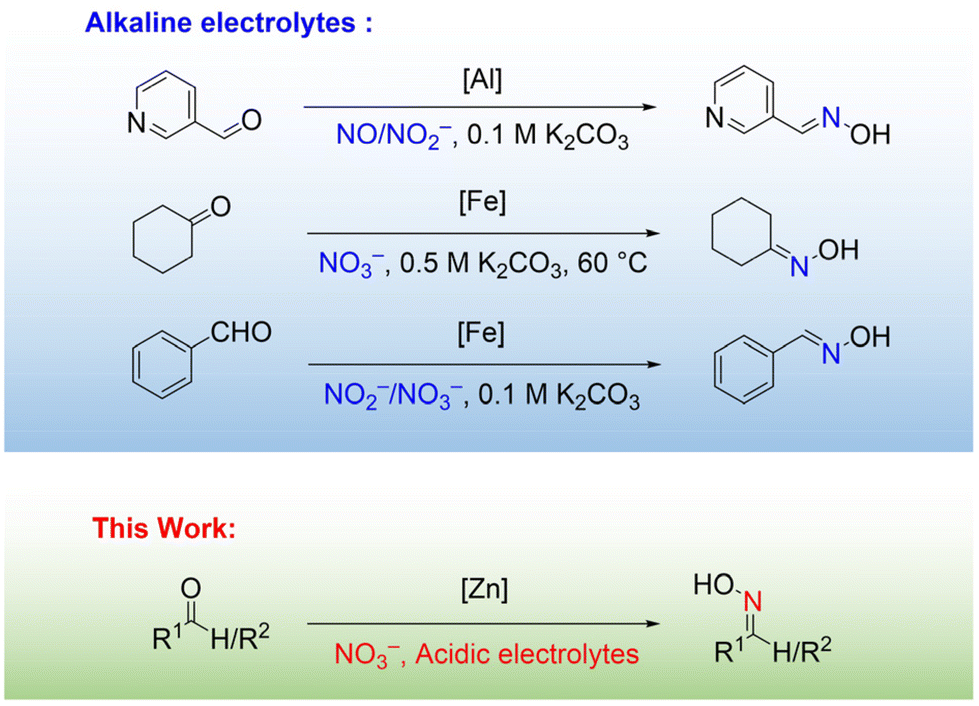
Anthropogenic emissions of nitrogen oxides (NOx) have increased rapidly over the past century, posing a threat to human health and ecosystems.1,2 Various methods have been employed to remove NOx.3–5 Among them, electrocatalysis has received much attention from researchers due to its environmental friendliness, energy saving, simple operation and controllable products.6,7 Most of the studies on electrocatalytic nitrate reduction reactions are based on the in-situ generation of NH2OH for the preparation of C–N coupling products in tandem with other suitable reactions to transform waste pollutants into high value-added chemicals.8–16 Oximes are important intermediates in organic synthesis and have proved to be industrially useful as fine chemicals.17–19 For instance, cyclohexanone oxime serves as a key precursor in the production of caprolactam,20 while perillartine finds extensive application in the flavor industry.21Recently, researchers explored the electrocatalytic NOx reduction coupling reaction and demonstrated the preliminary feasibility of electrocatalytic synthesis of oximes using NOx as a nitrogen source (Fig. 1). For instance, existing literature reports that oximes such as pyridine aldoxime,22 cyclohexanone oxime,23 and benzaldoxime24 have been synthesized using Al-based and Fe-based catalysts with NO, NO2− or NO3− as nitrogen sources. These electrochemical coupling strategies are typically conducted under alkaline conditions because the alkaline environment restricts the electrochemical hydrogenation of substrates, thereby driving the coupling step.25,26 However, in neutral or alkaline environments, the proton concentration is low, with most of the hydrogen in NH2OH originating from water.27
 | ||
| Fig. 1 Electrochemical C–N coupling of aldehydes and ketones with NOx. | ||
Furthermore, currently available electrocatalysts are still limited, and the balance between stability and activity under alkaline conditions needs further optimization.28–31 In view of this, there is an urgent need to develop catalytic systems for the electrocatalytic synthesis of oximes under acidic conditions. Acidic conditions offer advantages such as increased proton concentration, enhanced system stability, and potential yield improvement, aiding in overcoming limitations associated with alkaline conditions and providing new avenues and higher yields for the reaction.32–35 Although Wu et al. proposed a reaction route for the synthesis of cyclohexanone by NO2− electrochemical reduction coupling of cyclo-hexanone under acidic conditions.36 It is still a challenge to achieve efficient C–N coupling under acidic conditions with NO3− as the nitrogen source.
Herein, we report a novel electrochemical strategy for the synthesis of high value-added oximes from aldehyde/ketone and NO3− in an acidic electrolyte using electrodeposited modified Zn catalyst (multilayered Zn nanosheets catalyst, M-ZnNSs) as an electrode. 99% yield and 99% selectivity of phenylacetaldehyde oxime were achieved at a constant current of −12 mA cm−2. The durability of the catalyst was verified by maintaining high performance after 10 cycle tests (>100 h). A series of controlled experiments, combined with in-situ attenuated total reflectance infrared spectroscopy (ATR-FTIR) and online differential electrochemical mass spectrometry (DEMS) tests, were conducted to elucidate the reaction pathway: NO3− → NH2OH → oxime. In addition, isotope labeling experiments demonstrated that the N source was derived from NO3−. Importantly, this strategy exhibits broad applicability, not only for aldehydes but also for ketones. This work introduces a novel catalytic system for the value-added conversion of NOx, with significant implications for energy and environmental sustainability.
Results and discussion
The synthesis of oximes from aldehydes and NO3− involves two main steps: NO3− reduction to *NH2OH at the electrode surface, followed by nucleophilic addition–elimination with aldehydes to form oximes and eliminate water. This process includes electrochemical (e.g., hydrogenation and hydrodeoxygenation) and non-electrochemical pathways.24 Therefore, controlling the electrochemical hydrogenation process in acidic media without affecting intermediate production rates is crucial for target product formation. It is worth mentioning that oxime is unstable in acidic conditions and may convert to other organic nitrogen compounds. This places extreme demands on both the electrode and electrolyte.Based on the understanding of the reaction mechanism described above, we first utilized metallic materials commonly employed in electrocatalytic reactions as electrodes to directly electrosynthesize oximes, aiming to identify suitable catalysts (Fig. 2a). In the initial screening experiments, a weakly acidic buffer solution (1 M PBS, pH = 5.8) was used as the electrolyte to limit the electrochemical hydrogenation process of the organics and to ensure that the products could exist stably. Phenylacetaldehyde (PAH) and KNO3 were used as organic compounds and NOx model compounds, respectively, and electrolyzed at −12 mA cm−2 for 10 h. Although different electrode materials have different electrosynthesis oxime yields and selectivity in the co-reduction process of KNO3 and PAH, all electrode generated products can stably exist in the electrolyte. Pt and Fe catalysts primarily drive hydrogenation reactions due to their strong reducing abilities (ESI Fig. S1†). Despite improved selectivity for oximes, catalysts like Pd, Ni, Cu, etc., still struggle to effectively limit hydrogenation. Encouragingly, Zn catalysts show a different selectivity, drastically reducing aldehyde hydrogenation and favoring C–N coupling to form oximes as the main reaction pathway.
 | ||
| Fig. 2 Electrosynthesis of phenylacetaldehyde oxime performance from KNO3. (a) Screening of catalysts for the electrosynthesis of phenylacetaldehyde oxime using phenylacetaldehyde and KNO3 as feedstock. Reaction conditions: substrate (0.4 mmol), metal electrode (working area: 1.1 × 1.5 cm2), 1 mol L−1 PBS containing 0.4 mol L−1 KNO3 (5 mL), −12 mA cm−2, 10 h. (b) Effect of different pH on the electrosynthesis of phenylacetaldehyde oxime over Zn foil. (c) Effect of different current densities for phenylacetaldehyde oxime yield and selectivity with a total 720 C of charge using M-ZnNSs electrode. (d) The effect of molar ratio of KNO3 to PAH over different electrodes. (e) Time-dependent PAH conversion, phenylacetaldehyde oxime yield and FE using M-ZnNSs electrode. (f) The long-term stability test for phenylacetaldehyde oxime yield and FE on M-ZnNSs. | ||
We further investigated the effect of acidic electrolyte at different pH on the electrosynthesis of oximes when Zn foil was used as catalyst. The pH of the electrolyte played a decisive role in maintaining the stability of the oximes. In addition, although pH was not directly related to the non-electrochemical coupling process, it will inhibit the non-electrochemical coupling by affecting the electrochemical hydrogenation of key intermediates and the hydrodeoxygenation of aldehydes (hydride yield at different pH in ESI Fig. S2†). Acidic electrolytes with different pH were realized by preparing different buffer solutions (for detailed steps see ESI†). As shown in Fig. 2b, when the electrolyte was at medium acidity (pH = 3.2), the feedstock was dominated by the hydrogenation reaction (32% yield of alcohols), which inhibited the coupling reaction. Under strong acid conditions, there were some hydrolysis products of oxime, which also led to the decrease of the yield and selectivity of the target product. The stability of product improved as the pH was raised gradually. At pH = 5, hydrolysis of the oxime hardly happened, but some alcohol by-product was produced. Optimal selectivity and yield of oxime were achieved at pH = 5.8. However, increasing the pH further led to some side reactions in the feedstock, slightly reducing oxime selectivity.
After identifying the metal and optimal electrolyte, we proceeded to electrochemically modify commercial Zn foil, thus obtaining different modified Zn electrodes (for detailed steps see ESI†). The modified-Zn electrodes were prepared via a two-electrode constant-current deposition method, controlling deposition parameters such as current and time, while utilizing different Zn salt precursors and KOH concentrations to fabricate a series of Zn-based electrodes (ESI Fig. S3†). The performance of modified Zn electrodes with different Zn salts were examined by electrolysis at −12 mA cm−2 for 10 h, the results were shown in ESI Fig. S4.† The multilayered Zn nanosheets catalysts prepared with ZnCl2 (M-ZnNSs) showed the best catalytic performance, with 99% selectivity for oxime (ESI Fig. S5 and S6†). The desired nanosheets were obtained when ZnCl2 was used as the deposition solution at −90 mA cm−2 for 2 min. Scanning electron microscopy (SEM) revealed densely arranged layered Zn flakes on the electrode, contrasting with the smooth surface of unmodified Zn foil (ESI Fig. S7 and S8†), with an overall thickness of the nanosheets about 3 μm (ESI Fig. S9†). X-ray diffraction (XRD) analysis confirmed their highly crystalline nature, with diffraction peaks matching those of bulk Zn foil (ESI Fig. S10†). X-ray photoelectron spectroscopy (XPS) indicated Zn valence states close to 0 (ESI Fig. S11†), consistent with the zero-valence state of Zn atoms observed in X-ray absorption near-edge structure (XANES) spectra (ESI Fig. S12†).
To further improve the yield of oxime, different current densities in the range of −3 to −30 mA cm−2 was used for electrolysis while ensuring the passage of the same amount of charge (720 C). As shown in Fig. 2c, at lower current densities, the yield and selectivity of oxime were lower, the key intermediate *NH2OH was less and the feedstock failed to combine with it in time and other side reactions occurred. Increasing the current initially boosted oxime production, reaching a peak selectivity of 99% and yield of 82% at −12 mA cm−2. Yet, further current increase led to declining yield and selectivity due to raw material instability. This triggered more self-hydrogenation, raising phenylethanol by-product levels and impeding the desired reaction (ESI Fig. S13†).
Subsequently, the performance of electrocatalytic synthesis of oximes was evaluated at different KNO3/PAH molar ratios. As illustrated in Fig. 2d, when the molar ratio was at 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, although the selectivity of oxime was high, PAH was not completely converted, indicating that the *NH2OH produced during the KNO3 reduction process was not sufficient to completely convert the PAH into oxime. Increasing the molar ratio to 10:1, all PAH in the system was basically converted to oxime. To show the superiority of the electrochemically modified Zn foil strategy, we used unmodified Zn foil for the electrosynthesis of oximes at the optimum molar ratio, and its performance was not as good as that of M-ZnNSs. To further enhance the yield and selectivity of electrochemically synthesizing oxime, we also investigated the effect of reaction time. As shown in Fig. 2e, the optimal reaction time was 10 h, PAH was completely converted into oxime. Moreover, the durability of the electrocatalyst was also evaluated (Fig. 2f). After 10 consecutive electrical cycles (about 100 h), there was no obvious decrease in the yield and selectivity of oximes, which demonstrated the excellent stability of the M-ZnNSs catalysts and proved their excellent recyclability, which is important for practical applications. Moreover, the crystallographic structure, and compositions of M-ZnNSs electrode remained stable even after 100 h of reaction (ESI Fig. S14†).
:1, although the selectivity of oxime was high, PAH was not completely converted, indicating that the *NH2OH produced during the KNO3 reduction process was not sufficient to completely convert the PAH into oxime. Increasing the molar ratio to 10:1, all PAH in the system was basically converted to oxime. To show the superiority of the electrochemically modified Zn foil strategy, we used unmodified Zn foil for the electrosynthesis of oximes at the optimum molar ratio, and its performance was not as good as that of M-ZnNSs. To further enhance the yield and selectivity of electrochemically synthesizing oxime, we also investigated the effect of reaction time. As shown in Fig. 2e, the optimal reaction time was 10 h, PAH was completely converted into oxime. Moreover, the durability of the electrocatalyst was also evaluated (Fig. 2f). After 10 consecutive electrical cycles (about 100 h), there was no obvious decrease in the yield and selectivity of oximes, which demonstrated the excellent stability of the M-ZnNSs catalysts and proved their excellent recyclability, which is important for practical applications. Moreover, the crystallographic structure, and compositions of M-ZnNSs electrode remained stable even after 100 h of reaction (ESI Fig. S14†).
To gain a deeper understanding of the reaction pathways and excellent performance of M-ZnNSs electrocatalysts, a series of mechanistic experiments were conducted. The C–N coupling reactions were carried out using K14NO3 and K15NO3 as nitrogen sources to reveal the source of nitrogen in the resulting oximes (Fig. 3a and b). A molecular ion peak of 136 appears in the K15NO3 solution, while the K14NO3 system shows a molecular ion peak of 135, demonstrating that the nitrogen in the coupling product is derived from nitrate. In addition, in-situ ATR-FTIR spectroscopy was used to gain insight into the reaction pathway of C–N coupling on M-ZnNSs electrocatalysts. As shown in Fig. 3c, with the increase of electrolysis time, the M-ZnNSs showed IR absorption peaks at 1632 cm−1, which may be the characteristic peak of NH2OH.37 The persistence of this absorption peak indicates that NH2OH is stable during the electrolysis process. To further verify that NH2OH intermediates were produced during the reaction process, an online DEMS was used to analyze the reaction with optimal reaction conditions. As shown in Fig. 3d, applying the current, the mass spectral signal of NH2OH could be detected, which proved that NH2OH was produced during the reduction of KNO3 (other nitrogen-containing products were shown in ESI Fig. S15†).
 | ||
| Fig. 3 Reaction mechanism investigation. (a) The mass-spectrometry spectrum of the electrocatalytic synthesis of phenylacetaldehyde oxime from K14NO3 and PAH. (b) The mass-spectrometry spectrum of the electrocatalytic synthesis of phenylacetaldehyde oxime from K15NO3 and PAH. (c) Time-gradient electrolysis test using in-situ ATR-FTIR on M-ZnNSs under working condition. (d) Online DEMS measurements for the NO3RR over the M-ZnNSs electrode. | ||
To demonstrate the versatility of this catalytic system, we carried out electrosynthesis of various oximes bearing substituent groups over the M-ZnNSs electrode, such as aldehyde oxime and ketone oxime (as shown in Fig. 4 and ESI Fig. S16–27†). It is shown that this method was applicable to synthesizing different types of aldehyde oximes in high yields (yield and selectivity >90%), demonstrating the high versatility of this method. The above results indicate that the electrochemical in-situ production of hydroxylamine offers high feasibility for converting nitrogen-containing waste gases and wastewater into high-value organic nitrogen compounds.
 | ||
| Fig. 4 Substrate expansion and universality studies. (a) Reaction scope of the direct electrosynthesis of aldehyde oxime from KNO3 and R-CHO. Reaction conditions: RCHO (40 mM), M-ZnNSs (working area: 1.1 × 1.5 cm2), 1 mol L−1 PBS containing 0.4 mol L−1 KNO3 (5 mL), −12 mA cm−2, 10 h, isolated yield. (b) Reaction scope of the direct electrosynthesis of ketone oxime from KNO3 and ketone. Reaction conditions: R1COR2 (40 mM), M-ZnNSs (working area: 1.1 × 1.5 cm2), 1 mol L−1 PBS containing 0.4 mol L−1 KNO3 (5 mL), −12 mA cm−2, 10 h, isolated yield. | ||
Conclusions
In conclusion, we present a novel catalytic system for the synthesis of oximes using NH2OH generated by KNO3 electrocatalytic reduction coupled with aldehyde/ketone compounds under acidic conditions over Zn-based catalysts. The catalytic system achieved outstanding results in oxime electrosynthesis, offering high yield, selectivity, broad substrate compatibility, functional group tolerance, and long-term stability. It also excelled in synthesizing cinnamaldehyde oximes for food flavors and butyl ketones oximes for coating antioxidants. This study not only lays the groundwork for designing efficient catalytic systems for C–N coupling, enabling the conversion of nitrates into valuable oximes, but also expands efficient NH2OH production methods with promising applications.Author contributions
C. X., S. Q. J., H. H. W. and B. X. H. proposed the project, designed the experiments, and wrote the manuscript. C. X. performed the whole experiments. J. P. J., X. C., Z. H. X., M. K. D., T. D., H. L. C. and C. J. C. performed the analysis of experimental data. S. Q. J., H. H. W., M. Y. H. and B. X. H. co-supervised the whole project. All authors discussed the results and commented on the manuscript.Data availability
All data generated or analyzed in this study are included in this manuscript and ESI.† The datasets used and/or analyzed in this study are available upon request from the corresponding authors. The data are available in the publicly available in a public repository.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The work was supported by the National Key R&D Program of China (2023YFA1507901, 2020YFA0710201), the National Natural Science Foundation of China (22293015, 22121002), the China Postdoctoral Science Foundation (2023M731096), and the Fundamental Research Funds for the Central Universities.References
- C. J. Stevens, Science, 2019, 363, 578–580 CrossRef CAS PubMed.
- O. S. G. P. Soares, J. J. M. Órfão, J. Ruiz-Martínez, J. Silvestre-Albero, A. Sepúlveda-Escribano and M. F. R. Pereir, Chem. Eng. J., 2010, 165, 78–88 CrossRef CAS.
- J. Li, X. Han, X. Zhang, A. M. Sheveleva, Y. Cheng, F. Tuna, E. J. L. McInnes, L. J. M. McPherson, S. J. Teat, L. L. Daemen, A. J. Ramirez-Cuesta, M. Schröder and S. Yang, Nat. Chem., 2019, 11, 1085–1090 CrossRef CAS PubMed.
- C. Mao, J. Wang, Y. Zou, Y. Shi, C. J. Viasus, J. Y. Y. Loh, M. Xia, S. Ji, M. Li, H. Shang, M. Ghoussoub, Y.-F. Xu, J. Ye, Z. Li, N. P. Kherani, L. Zheng, Y. Liu, L. Zhang and G. A. Ozin, J. Am. Chem. Soc., 2023, 145, 13134–13146 CrossRef CAS PubMed.
- M. Wu, J. Li, A. O. Leu, D. V. Erler, Z. Yuan, S. J. McIlroy, T. Stark, G. W. Tyson and J. Guo, Nat. Commun., 2022, 13, 6115 CrossRef CAS PubMed.
- K. Liu, H. Li, M. Xie, P. Wang, Z. Jin, Y. Liu, M. Zhou, P. Li and G. Yu, J. Am. Chem. Soc., 2024, 146, 7779–7790 CrossRef CAS PubMed.
- D. Xu, Y. Li, L. Yin, Y. Ji, J. Niu and Y. Yu, Front. Environ. Sci. Eng., 2018, 12, 1–14 CAS.
- Y. Cheng, S. Liu, J. Jiao, M. Zhou, Y. Wang, X. Xing, Z. Chen, X. Sun, Q. Zhu, Q. Qian, C. Wang, H. Liu, Z. Liu, X. Kang and B. Han, J. Am. Chem. Soc., 2024, 46, 10084–10092 CrossRef PubMed.
- M. He, Y. Wu, R. Li and B. Zhang, Nat. Commun., 2023, 14, 5088 CrossRef CAS PubMed.
- S. Jia, L. Wu, H. Liu, R. Wang, X. Sun and B. Han, Angew. Chem., Int. Ed., 2024, 63, e202400033 CrossRef CAS PubMed.
- M. Li, Y. Wu, B.-H. Zhao, C. Cheng, J. Zhao, C. Liu and B. Zhang, Nat. Catal., 2023, 6, 906–915 CrossRef CAS.
- J. Wu, L. Xu, Z. Kong, K. Gu, Y. Lu, X. Wu, Y. Zou and S. Wang, Angew. Chem., Int. Ed., 2023, 62, e202311196 CrossRef CAS PubMed.
- Y. Wu, Z. Jiang, Z. Lin, Y. Liang and H. Wang, Nat. Sustainability, 2021, 4, 725–730 CrossRef.
- Y. Wu, M. Li, T. Li, J. Zhao, Z. Song and B. Zhang, Sci. China: Chem., 2023, 66, 1854–1859 CrossRef CAS.
- J. Xian, S. Li, H. Su, P. Liao, S. Wang, R. Xiang, Y. Zhang, Q. Liu and G. Li, Angew. Chem., Int. Ed., 2023, 62, e202306726 CrossRef CAS PubMed.
- J. Xian, S. Li, H. Su, P. Liao, S. Wang, Y. Zhang, W. Yang, J. Yang, Y. Sun, Y. Jia, Q. Liu, Q. Liu and G. Li, Angew. Chem., Int. Ed., 2023, 62, e202304007 CrossRef CAS PubMed.
- D. S. Bolotin, N. A. Bokach, M. Y. Demakova and V. Y. Kukushkin, Chem. Rev., 2017, 117, 13039–13122 CrossRef CAS PubMed.
- S. Ji, L. Zhao, B. Miao, M. Xue, T. Pan, Z. Shao, X. Zhou, A. Fu and Y. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202304434 CrossRef CAS PubMed.
- W. Xue, Y. Jiang, H. Lu, B. You, X. Wang and C. Tang, Angew. Chem., Int. Ed., 2023, 62, e202314364 CrossRef CAS PubMed.
- R. J. Lewis, K. Ueura, X. Liu, Y. Fukuta, T. E. Davies, D. J. Morgan, L. Chen, J. Qi, J. Singleton, J. K. Edwards, S. J. Freakley, C. J. Kiely, Y. Yamamoto and G. J. Hutchings, Science, 2022, 376, 615–620 CrossRef CAS PubMed.
- M. S.-N. Gabriela Juárez, J. L. Alonso, E. R. Alonso and I. León, Molecules, 2022, 27, 1924 CrossRef PubMed.
- R. Xiang, S. Wang, P. Liao, F. Xie, J. Kang, S. Li, J. Xian, L. Guo and G. Li, Angew. Chem., Int. Ed., 2023, 62, e202312239 CrossRef CAS PubMed.
- Y. Wu, W. Chen, Y. Jiang, Y. Xu, B. Zhou, L. Xu, C. Xie, M. Yang, M. Qiu, D. Wang, Q. Liu, Q. Liu, S. Wang and Y. Zou, Angew. Chem., Int. Ed., 2023, 62, e202305491 CrossRef CAS PubMed.
- W. Chen, Y. Wu, Y. Jiang, G. Yang, Y. Li, L. Xu, M. Yang, B. Wu, Y. Pan, Y. Xu, Q. Liu, C. Chen, F. Peng, S. Wang and Y. Zou, J. Am. Chem. Soc., 2024, 146, 6294–6306 CrossRef CAS PubMed.
- D. C. Cantu, A. B. Padmaperuma, M.-T. Nguyen, S. A. Akhade, Y. Yoon, Y.-G. Wang, M.-S. Lee, V.-A. Glezakou, R. Rousseau and M. A. Lilga, ACS Catal., 2018, 8, 7645–7658 CrossRef CAS.
- X. H. Chadderdon, D. J. Chadderdon, J. E. Matthiesen, Y. Qiu, J. M. Carraher, J.-P. Tessonnier and W. Li, J. Am. Chem. Soc., 2017, 139, 14120–14128 CrossRef CAS PubMed.
- G. Zhang, X. Li, K. Chen, Y. Guo, D. Ma and K. Chu, Angew. Chem., Int. Ed., 2023, 62, e202300054 CrossRef CAS PubMed.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- E. Fabbri, M. Nachtegaal, T. Binninger, X. Cheng, B.-J. Kim, J. Durst, F. Bozza, T. Graule, R. Schäublin, L. Wiles, M. Pertoso, N. Danilovic, K. E. Ayers and T. J. Schmidt, Nat. Mater., 2017, 16, 925–931 CrossRef CAS PubMed.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS PubMed.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148 RSC.
- Y. Guo, R. Zhang, S. Zhang, Y. Zhao, Q. Yang, Z. Huang, B. Dong and C. Zhi, Energy Environ. Sci., 2021, 14, 3938–3944 RSC.
- J. Yu, Y. Qin, X. Wang, H. Zheng, K. Gao, H. Yang, L. Xie, Q. Hu and C. He, Nano Energy, 2022, 103, 107705 CrossRef CAS.
- R. Zhang, C. Li, H. Cui, Y. Wang, S. Zhang, P. Li, Y. Hou, Y. Guo and C. Zhi, Nat. Commun., 2023, 14, 8036 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, A. Vasileff and S.-Z. Qiao, Angew. Chem., Int. Ed., 2018, 57, 7568–7579 CrossRef CAS PubMed.
- Y. Wu, B.-H. Zhao, Z. Song, C. Liu, J. Zhao, C. Wang, T. Li and B. Zhang, Nat. Commun., 2023, 14, 3057 CrossRef CAS PubMed.
- R. Hao, S. Fang, L. Tian, R. Xia, Q. Guan, L. Jiao, Y. Liu and W. Li, Chem. Eng. J., 2023, 467, 143371 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc03429e |
| This journal is © The Royal Society of Chemistry 2024 |