
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproving both activity and stability for direct conversion of cellulose to ethanol by decorating Pt/WOx with mononuclear NbOx†
Weixiang
Guan
a,
Chen
Cao
ab,
Fei
Liu
a,
Aiqin
Wang
 *a and
Tao
Zhang
a
*a and
Tao
Zhang
a
aCAS Key Laboratory of Science and Technology on Applied Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: aqwang@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 11th September 2024
Abstract
Chemocatalytic conversion of cellulose to ethanol provides an alternative route for biofuel production with a theoretical carbon yield of 100%; however, it faces significant challenges of high catalyst cost and poor catalyst stability. In this work, we report a new strategy to decrease the use of expensive noble metals, by decorating mononuclear NbOx on a low-Pt Pt/WOx catalyst surface. The resulting 0.1Nb/0.5Pt/WOx catalyst gave rise to an ethanol yield of 33.7% together with an ethylene glycol yield of 21.8%, and the noble metal efficiency reached 25.90 gethanol gPt−1 h−1, an increase by a factor of 2–10 compared to those in the literature. Moreover, the catalyst stability was significantly enhanced by the decoration of mononuclear NbOx, allowing for recycling at least 7 times without obvious activity decay. Characterization revealed that Pt was highly dispersed at subnanometer and single atom scales, and modification with mononuclear NbOx facilitated hydrogen spillover and created more oxygen vacancies on the WOx surface upon hydrogen reduction, thus generating a higher density of Brønsted acid sites. This effect not only favored cellulose conversion to ethylene glycol but also promoted the hydrogenolysis of ethylene glycol to ethanol.
Introduction
With increasing concerns about climate change caused by CO2 emissions, carbon-neutral biomass is being considered as an environmentally benign feedstock for producing liquid fuels and chemicals.1 Among various platform and end-use biomass-derived chemicals, ethanol is one of the most important under consideration because it has been widely used as a drop-in biofuel in gasoline.2,3 Although ethanol can be directly produced from cellulose via the bio-catalysis of cellulase, the recalcitrance of lignocellulose as well as the vulnerability of cellulase make the bioconversion process less efficient.4,5 As an alternative to the enzymatic process, chemocatalytic conversion of cellulose into ethanol has recently been developed.6–10 For this method, both cellulose and hemicellulose could be converted in one pot to ethanol without the need for pre-separation, and the theoretical carbon atom utilization could reach 100% without producing CO2. These inherent advantages make the chemocatalytic process appealing.Based on the reaction pathway shown in Scheme 1, cellulose is first transformed into ethylene glycol (EG) which is subsequently converted to ethanol via hydrogenolysis. Previous works have shown that tungsten-based catalysts, such as Ni-promoted tungsten carbide,11 dual catalysts composed of tungsten oxide (or tungstic acid) and transition metals (typically Ni, Ru, Pd and Pt),12,13 are highly efficient for cellulose conversion to EG. Modification of these catalysts to promote EG hydrogenolysis enabled the one-pot conversion of cellulose to ethanol. For instance, Song et al. reported that the binary catalyst, H2WO4 + Pt/ZrO2, could afford an ethanol yield of 32% for the one-pot conversion of cellulose at 250 °C and 4 MPa H2.3 Li et al. also employed a binary catalyst, 5%Ru–WOx/HZSM-5 + 5%Ru/WOx, for this reaction and obtained a 87.5% yield of ethanol at 235 °C and 3.0 MPa H2.7 Similarly, Wu et al. used a physical mixture of 5%Pt/WOx and 5%Pt@HZSM-5 (hollow) to catalyse the reaction and achieved an ethanol yield of 54.4%.10 Although high yields of ethanol could be obtained by these dual catalyst systems, they are facing two grand challenges. One is the low efficiency of expensive noble metals (ranging from 0.3 to 8.1 gethanol gmetal−1 h−1), and the other is the poor catalyst stability. Both of these are big barriers that need to be overcome for practical applications. As an alternative to the binary catalysts, multifunctional W-based catalysts capable of breaking both C–C and C–O bonds are advantageous due to improved synergy among different sites.6 Previously, we developed a multifunctional Mo/Pt/WOx catalyst which afforded an ethanol yield of 43.2% and the noble metal efficiency reached 12.4 gethanol gPt−1 h−1, demonstrating the great potential of this strategy for improving the synergy between different functional constituents. The success of this strategy lies in that the mononuclear MoOx could modulate the interaction between Pt and WOx while not covering the exposed Pt surface. Nevertheless, Mo was not stable under hydrothermal reaction conditions and the leaching of Mo led to a remarkable decrease in ethanol yield in the second run. Therefore, it is highly desirable to develop an efficient multifunctional heterogeneous catalyst with both high metal efficiency and good hydrothermal stability.
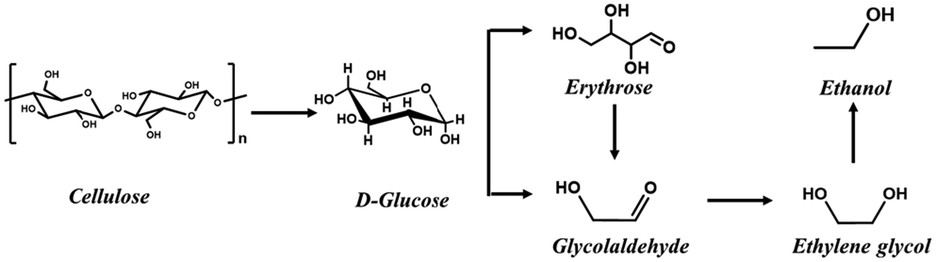 | ||
| Scheme 1 Reaction network for the conversion of cellulose into ethanol. | ||
Niobium is adjacent to molybdenum in the periodic table of the elements. The oxophilic character of Nb and the excellent stability of Nb2O5 in acidic hot water have made Nb-based catalysts widely used in biomass upgrading.14,15 Therefore, we envision that by decorating a low-Pt Pt/WOx catalyst with mononuclear NbOx, the activity and hydrothermal stability might be simultaneously enhanced for the one-pot conversion of cellulose to ethanol.
Experiment
Catalyst preparation
The WOx support was synthesized following the procedure described in a previous study.16 Typically, 10.0 g of WCl6 was fully dissolved in 200 mL of ethanol with continuous stirring, and the resulting clear solution was then transferred to a Teflon-lined autoclave followed by heating at 160 °C in an oven for 36 h. After cooling to room temperature, the dark blue precipitate was recovered through filtration and washed with ethanol and water several times. Finally, the precipitate was freeze-dried for 7 h to obtain WOx.1.0 g of the as-prepared WOx was then impregnated with 0.0663 g of an aqueous solution of H2PtCl6 with a Pt concentration of 7.54 wt%. The impregnated material was dried at 120 °C for 4 h and then reduced under a H2 atmosphere at 300 °C for 1 h to produce 0.5Pt/WOx with a Pt mass content of 0.5 wt%. Subsequently, Nb was introduced to 0.5Pt/WOx by the wet impregnation method using an ethanol solution of NbCl5 (0.5 g NbCl5/10.0 g ethanol). After that, the sample was dried at 120 °C for 4 h, calcined at 400 °C for 1 h in air and finally reduced at 300 °C for 1 h under a H2 atmosphere to obtain xNb/0.5Pt/WOx with various Nb contents (where x represents the weight percent of Nb). For comparison, other transition metals including Mo, W, and Ta were introduced to the 0.5Pt/WOx in a similar manner to Nb, except that NbCl5 was replaced by other metal precursors.
Materials
Ethanol (99%) was supplied by Tianjin Fuyu Fine Chemical Co., Ltd. H2PtCl6·6H2O (99%) was purchased from Tianjin Fengchuan Chemical Reagent Co., Ltd. WCl6, NbCl5 and other transition metal precursors (99%) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd.Characterization
The nitrogen adsorption/desorption measurements were conducted on an Autosorb iQ instrument. The specific surface areas of catalysts were determined by the Brunauer–Emmett–Teller (BET) method and the average pore size diameter was determined by the Barrett–Joyner–Halenda (BJH) method.The X-ray diffraction (XRD) patterns were recorded on a PAN-alytical X'pert diffractometer in the range of 5–90° at 40 kV per 40 mA with Cu Kα1 radiation (λ = 1.5418 Å).
The Raman spectra were collected on a Jobin Yvon HR 800 Dispersive Raman Spectrometer with a resolution of 2 cm−1 in the range of 200–1200 cm−1 at room temperature, a wavelength of 532 nm and a power of 0.1 mW was used.
The UV-vis diffuse reflectance spectra of solid samples were recorded on a Shimadzu UV2600 spectrometer in the range of 200–800 nm with BaSO4 as the background.
Scanning electron microscopy (SEM) images were acquired on a JSM-7800F electron microscope operating at 20 kV. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images were obtained on JEM-2100F and JEM-ARM200 transmission electron microscopes operating at 200 kV and equipped with an energy dispersive spectroscopy (EDS) microanalysis system.
X-ray photoelectron spectroscopy (XPS) was performed on the ESCALAB 250 X-ray photoelectron spectrometer with an Al Kα X-ray radiation source. All the binding energies were calibrated using the C 1s peak at 284.6 eV as a reference.
The chemisorption experiments using H2 probe molecules were conducted on a Micromeritics AutoChem II 2920 chemisorption instrument. Before the measurements, all the samples were pre-reduced at 300 °C under pure H2 for 1 h, and then purged at 310 °C with an inert gas flow (Ar) for 30 min. After cooling down to 50 °C, a pulse of 10% H2/Ar was introduced for H2 chemisorption until saturation was reached. The consumed H2 was detected with TCD and the uptakes were calculated by calibration with the standard sample.
The hydrogen temperature-programmed reduction (H2-TPR) and the temperature-programmed desorption of NH3 (NH3-TPD) experiments were conducted using the same chemisorption instrument. For the H2-TPR experiments, 100 mg of the sample was loaded into a U-shaped quartz tube reactor and pretreated at 150 °C for 1 h under an Ar atmosphere. After cooling down to −80 °C under an Ar flow with a cold trap, the Ar gas was switched to 10 vol% H2/Ar and the sample was heated to 800 °C at a ramp rate of 10 °C min−1 with a gas flow rate of 20 mL min−1. The consumption of H2 was monitored using a TCD detector and calibrated with the standard sample. For the NH3-TPD experiments, 100 mg of sample was pretreated in H2 gas at 300 °C for 1 h and heated to 310 °C under a He flow for 30 min, then cooled to 100 °C. After that, pulses of NH3 were introduced to the sample for adsorption until saturation was reached. Subsequently, the sample was heated to 800 °C at a ramp rate of 10 °C min−1 and the desorbed NH3 was quantified with a TCD detector and calibrated with the standard sample.
The 2-butanol dehydration reaction was used as a probe to detect the dynamic Brønsted acid sites. The reaction was conducted using a fixed-bed reaction system (Fig. S1†). For a typical reaction, 50 mg of the catalyst was placed at the bottom of a U-tube reactor, with quartz wool packed at both ends of the catalyst. The catalyst was first reduced in situ with H2 (20 mL min−1) at 300 °C for 1 h, and then cooled to 140 °C for the reaction. 2-Butanol was introduced into the reactor by passing through a bubbler in a water bath at 60 °C, with the carrier gas being either H2 or N2 (with a gas flow rate of 20 mL min−1). The gaseous products were analyzed online using an Agilent 7890B GC with a HayeSep Q packed column with a TCD detector and a DB FFAP capillary column with a FID detector.
Reaction tests
The catalytic conversion of cellulose was performed in a 50 mL autoclave reactor lined with quartz. In detail, 0.1 g of microcrystalline cellulose, 0.1 g of catalyst and 9.9 g of water were placed in the reactor. After the reactor was sealed and purged with H2 three times, 6.0 MPa H2 was charged at room temperature. The autoclave reactor was then heated to 245 °C and kept for 2 h under vigorous stirring. After the reaction had finished, the reactor was quickly cooled using ice water. The liquid products were separated from the solid catalyst by centrifugation and analyzed with HPLC (Shimadzu equipped with a Phenomenex Rezex RQA-Organic Acid H+ (8%) column and a differential refractive index detector) and GC (Agilent 7890B with an HP-INNO WAX capillary column (30 m × 0.32 mm × 0.5 μm, FID detector)) using an external standard method. The conversion of cellulose and the yield of liquid product i were calculated as follows:| Conv. (%) = (m(cellulose input) − m(cellulose output))/m(cellulose input) × 100 | (1) |
| Yi (%) = ni/n(cellulose input) × 100 | (2) |
The total organic carbon in the liquid products after the reaction was determined with a TOC analyzer (Shimadzu). The mass of metal leached in the liquid after the reaction was determined by ICP-OES on a PerkinElmer 7300DV.
Results and discussion
Physicochemical properties of catalysts
In this work, a series of oxophilic metals were introduced to the freshly reduced 0.5Pt/WOx catalyst by the impregnation method in order to create direct interaction between the additive metal and Pt.17 The specific surface area of 0.5Pt/WOx was hardly altered by the addition of oxophilic metals, being maintained at 60–65 m2 g−1, while the pore volumes were slightly increased during the calcination process (Table S1†). Fig. 1 displays the XRD patterns and Raman spectra of the series of 0.1M/0.5Pt/WOx samples (where M represents NbOx, TaOx and MoOx, abbreviated as Nb, Ta and Mo). Compared to the parent WOx and 0.5Pt/WOx, the three 0.1M/0.5Pt/WOx showed sharper and better-resolved XRD peaks which can be indexed as WO2.83 (JCPDS no. 36-0103). The improvement in crystallinity of WOx by the introduction of M can be attributed to the calcination treatment. However, neither Pt nor promoter M crystalline reflections can be found, which suggests that both elements are highly dispersed in the WOx support. This is not surprising considering that the oxygen-vacancy-rich WOx and the strong interaction with Pt facilitate the dispersion of Pt, as we previously reported that Pt could be atomically dispersed in the WOx support at a loading of up to 2.5 wt%.18 In particular, in the case of an extremely low concentration of the additive metal (0.1 wt%), the oxophilic metal oxide additives tend to be atomically dispersed and cannot be detected by XRD, which is similar to our previously reported 0.1Mo/2Pt/WOx.6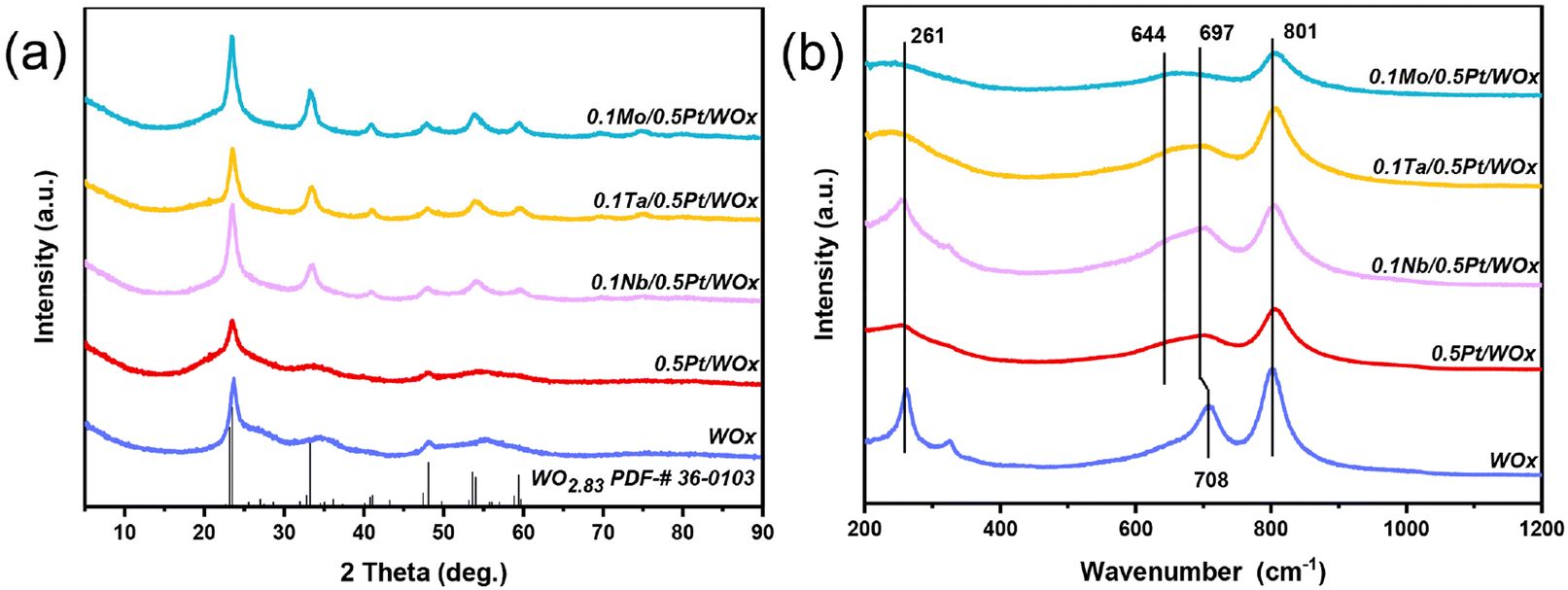 | ||
| Fig. 1 XRD patterns (a) and Raman spectra (b) of various WOx-based catalysts. | ||
Raman spectroscopy was further employed to detect the changes associated with W–O bonding before and after the introduction of additive M. The bare WOx support exhibited three prominent bands at 261, 708 and 801 cm−1, which could be ascribed to the W–O–W deformation, W–O bending and W–O stretching modes of WOx, respectively.8,19,20 Compared with this, the loading of Pt caused the band at 708 cm−1 to shift to a lower wavenumber of 697 cm−1, together with the emergence of a new shoulder band at around 644 cm−1. According to the literature, this phenomenon should be associated with the creation of surface oxygen vacancies in the WOx support.21–23 With the further introduction of Nb, Ta and Mo species, the band at 697 cm−1 became intensified and the highest intensity was observed on 0.1Nb/0.5Pt/WOx, which suggests that it has the highest number of surface oxygen vacancies.
Consistent with the Raman spectroscopy results, the UV-vis spectra (Fig. S2 and Table S1†) showed that the electronic edge (Eg) values decreased in the order of WOx > 0.5Pt/WOx > 0.1Ta/0.5Pt/WOx > 0.1Mo/0.5Pt/WOx > 0.1Nb/0.5Pt/WOx. It was reported that the smaller the Eg, the greater the number of oxygen vacancies present in the materials.24,25 Thus, the sequential introduction of Pt followed by the incorporation of the second metal oxides (NbOx, MoOx, and TaOx) resulted in the generation of an increased number of oxygen vacancies within the WOx structure. In particular, the lowest Eg value of 0.1Nb/0.5Pt/WOx means that it contains the largest number of surface oxygen vacancies, which will be conducive to delocalizing the negative charge and therefore increasing the number of Brønsted acid sites.26
Fig. 2 shows the SEM and HAADF-STEM images of the 0.5Pt/WOx and 0.1Nb/0.5Pt/WOx catalysts. Both samples present a morphology of aggregates of small flakes with sizes of several hundred nanometers, and the introduction of Nb did not cause an appreciable change in the morphology. The HAADF-STEM images showed the presence of mesopores in the WOx support; however, no Pt nanoparticles could be observed in either sample, which suggests that Pt should be dispersed as subnanometer clusters or even single atoms. Unfortunately, due to the very poor contrast between Pt and W (the atomic number difference between the two elements is only 4), the Pt species at subnanometer and single-atom scale could not be resolved even with the sub-angstrom resolution aberration-corrected HAADF-STEM imaging. Lattice fringes with an interplanar distance of 0.379 nm could be clearly observed, which were indexed to the (010) plane of WO2.83 (Fig. 2g), in line with the XRD results. The uniform dispersion of Pt and Nb could be indicated by elemental mapping which showed the superimposition of W, Pt and Nb signals in the entire particle.
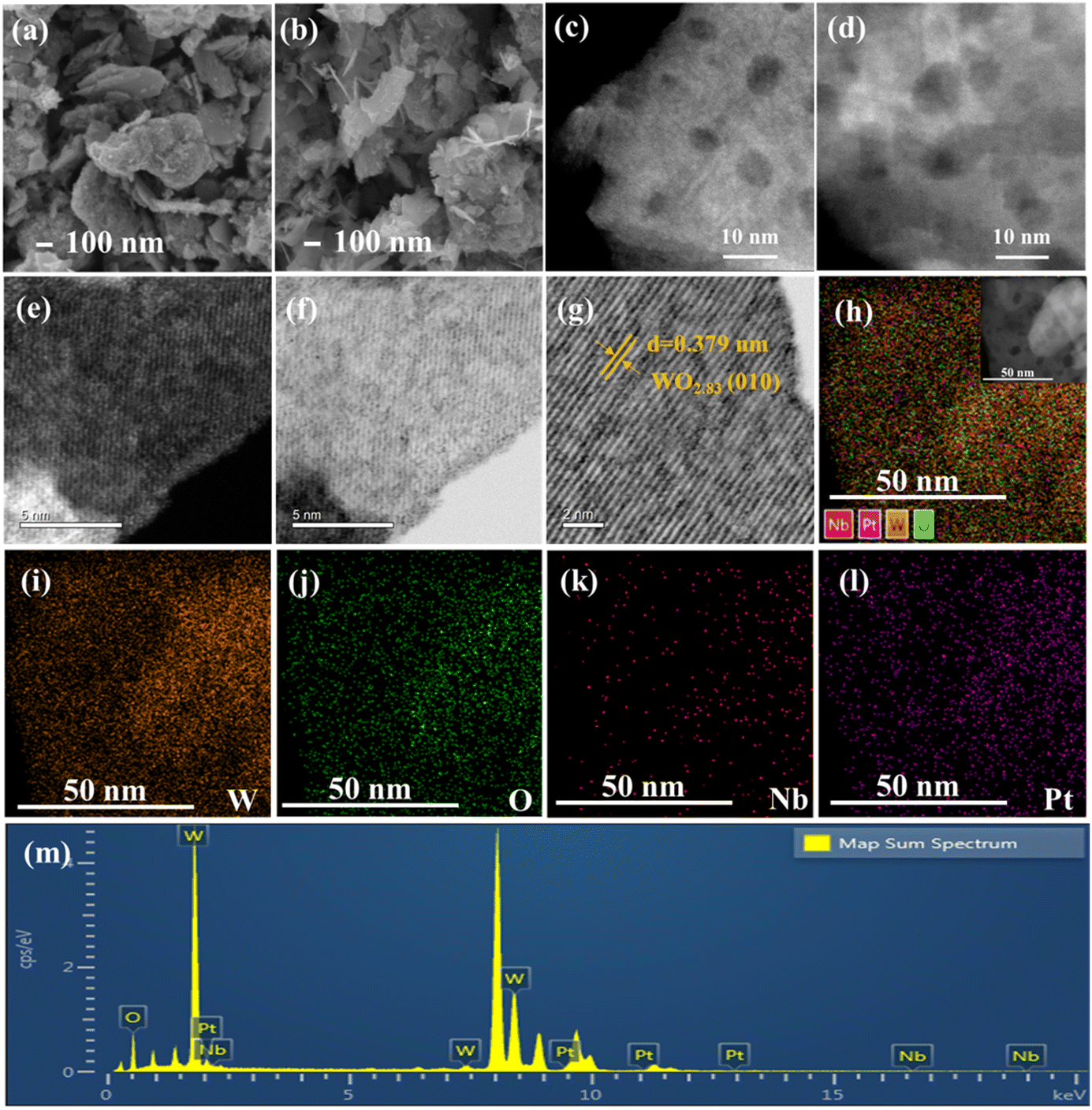 | ||
| Fig. 2 SEM and HAADF-STEM images of 0.5Pt/WOx (a and c) and 0.1Nb/0.5Pt/WOx (b and d); aberration-corrected HAADF-STEM images (e–g) and the corresponding elemental mappings (h–l) of 0.1Nb/0.5Pt/WOx, and the EDX spectrum (m). | ||
Fig. 3 displays the H2-TPR profiles of the series of catalysts. Compared to the parent 0.5Pt/WOx, the three 0.1M/0.5Pt/WOx catalysts present distinctive H2-TPR features, with the low-temperature reduction peak shifting to a higher temperature in the order of 0.5Pt/WOx (8.4 °C) < 0.1Nb/0.5Pt/WOx (17.9 °C) < 0.1Ta/0.5Pt/WOx (45.1 °C) < 0.1Mo/0.5Pt/WOx (54.6 °C). This result suggests that the decoration of oxophilic metal oxide on Pt has made Pt more difficult to reduce. Moreover, accompanied by the first major peak (peak 2), a minor reduction peak appeared below 0 °C for each of the 0.1M/0.5Pt/WOx catalysts (peak 1), which could be due to the reduction of Pt4+ in direct contact with the additive M since the reduction of the 0.5Pt/WOx catalyst did not produce any peak at such a low temperature. Integration of the peak area followed by calibration indicated that the H2 consumption for peak 1 was 0.103, 0.086, and 0.022 mmol gcat−1 for 0.1Nb/0.5Pt/WOx, 0.1Ta/0.5Pt/WOx, and 0.1Mo/0.5Pt/WOx (Table 1), respectively. It was noted that the theoretical H2 consumption for reducing Pt4+ to Pt0 of the 0.5Pt/WOx catalyst was only 0.05 mmol gcat−1. Obviously, the H2 consumptions of all the investigated catalysts far exceeded the theoretical value, which strongly indicates the occurrence of hydrogen spillover. Based on the H2 consumption, it was found that the hydrogen spillover took place to the most pronounced extent over the 0.1Nb/0.5Pt/WOx catalyst, which was in line with the highest number of oxygen vacancies in this catalyst.
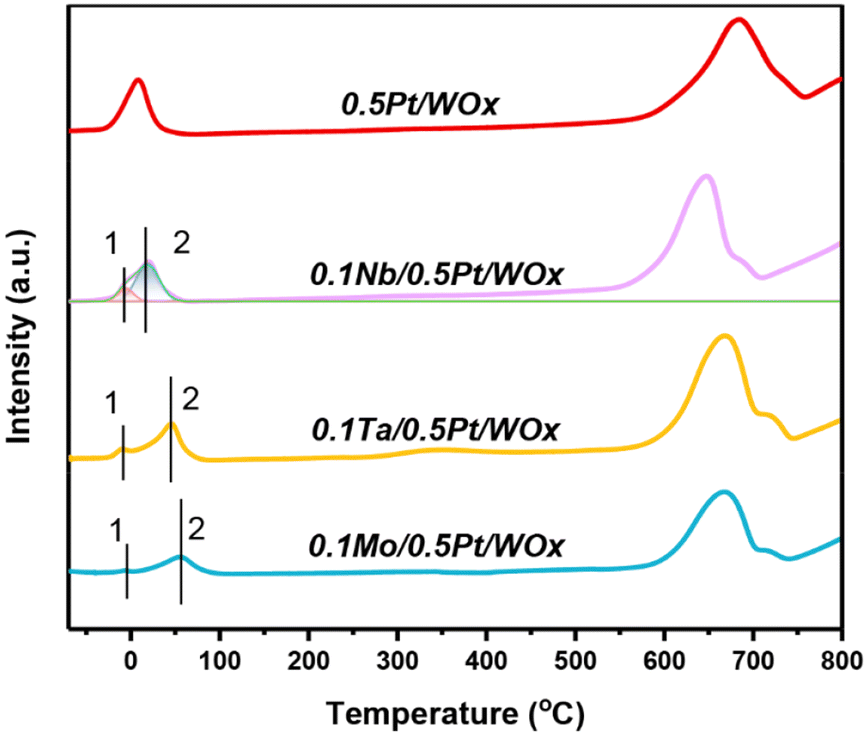 | ||
| Fig. 3 H2-TPR profiles of 0.1M/0.5Pt/WOx catalysts. | ||
| Sample | Reduction temperature (°C) | H2 consumption (mmol gcat−1) | H2 uptake (μmol g−1) | Dispersiona (%) | ||
|---|---|---|---|---|---|---|
| Peak 1 | Peak 2 | Peak 1 | Peak 2 | |||
| a Determined by assuming H/Pt = 1/1. | ||||||
| 0.5Pt/WOx | — | 8.4 | — | 0.826 | 12.9 | 103.4 |
| 0.1Nb/0.5Pt/WOx | −8.9 | 17.9 | 0.103 | 0.774 | 14.6 | 114.5 |
| 0.1Ta/0.5Pt/WOx | −8.7 | 45.1 | 0.086 | 0.683 | 14.1 | 110.6 |
| 0.1Mo/0.5Pt/WOx | −6.0 | 54.6 | 0.022 | 0.514 | 13.4 | 105.1 |
The enhanced hydrogen spillover due to the introduction of Nb can also be seen from the hydrogen chemisorption data. As shown in Table 1, the H2 uptake increased from 12.9 μmol g−1 to 14.6 μmol g−1 after adding 0.1 wt% Nb to 0.5Pt/WOx. The Pt dispersion, estimated based on the stoichiometric ratio of H/Pt = 1/1, reached the highest value of 114% over 0.1Nb/0.5Pt/WOx, highlighting a significant hydrogen spillover effect, which is consistent with the H2-TPR results.
The electronic interaction between Nb, Pt and W could be characterized by XPS. Due to the interference of W 5s, the Pt 4f doublet peaks were unfortunately attenuated such that they could not be accurately quantified. However, it could still be seen that, compared to the parent 0.5Pt/WOx, the three 0.1M/0.5Pt/WOx catalysts showed stronger Pt 4f peaks (Fig. 4a), indicating that the M additive weakened the SMSI effect between Pt and W such that more Pt sites could be exposed on the surface.27 Moreover, compared to 0.5Pt/WOx, the Pt 4f peaks of all three 0.1M/0.5Pt/WOx samples shifted to lower binding energies, suggesting that there is electron transfer from M to Pt due to the larger electronegativity of Pt relative to M. On the other hand, except for 0.1Nb/0.5Pt/WOx, the W 4f XPS spectra (Fig. 4b) of the other three samples show the spin–orbit splitting doublet peaks of 4f7/2 and 4f5/2 located at 35.8 eV and 37.9 eV, respectively, in between those for W6+ (36.2 and 38.3 eV) and W5+ (35.0 and 37.3 eV).28 Compared to them, the W 4f doublet peaks in 0.1Nb/0.5Pt/WOx shifted to lower binding energies at 35.5 eV and 37.6 eV, respectively, which indicated that the valence state of W in 0.1Nb/0.5Pt/WOx is the lowest, in agreement with it having the most oxygen vacancies in this sample.
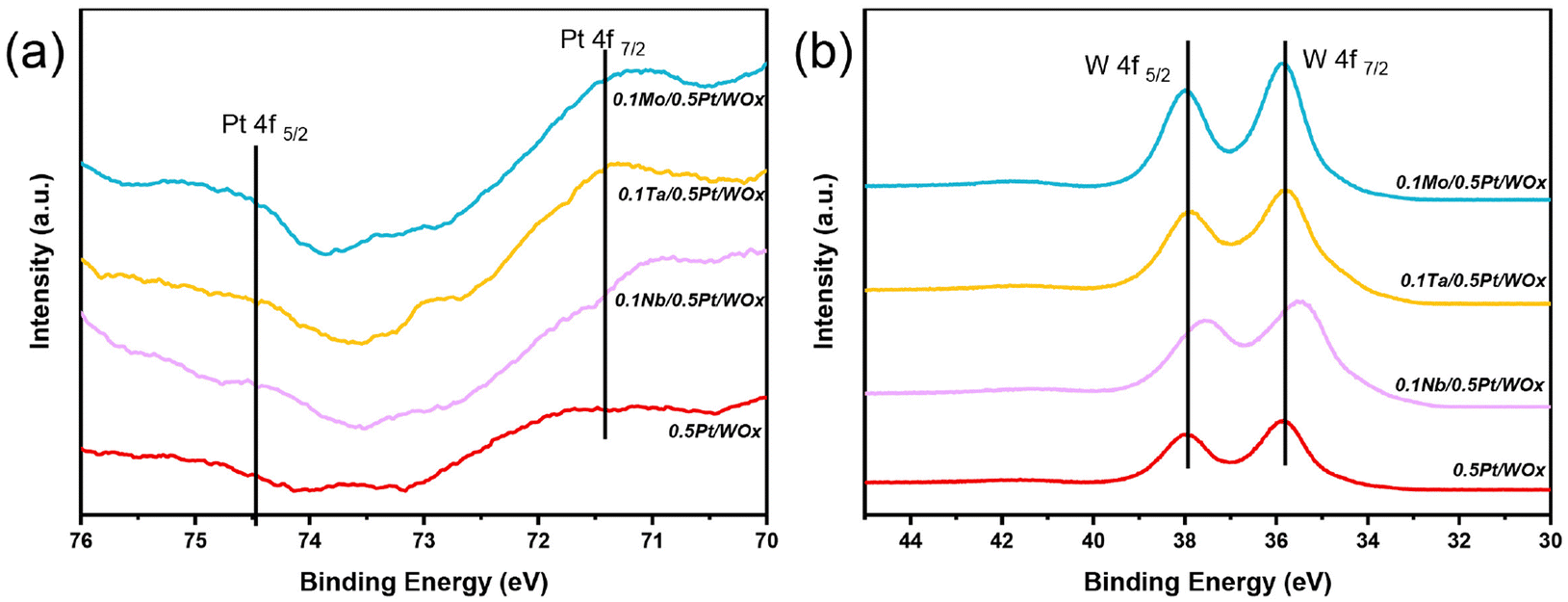 | ||
| Fig. 4 XPS spectra of Pt 4f (a) and W 4f (b) for 0.1M/0.5Pt/WOx catalysts. | ||
Catalytic performance
Promotional effect of different oxophilic metal additives
The one-pot conversion of cellulose to ethanol involves two sequential reactions, cellulose conversion to EG followed by EG hydrogenolysis to ethanol, and the latter is regarded as the rate-determining step.6 Given that oxophilic metals can effectively promote C–O cleavage by lowering the energy barrier,29 several oxophilic metals including Nb, Ta, Mo, Fe and Zn were introduced to the surface of 0.5Pt/WOx by a sequential impregnation method and evaluated in the one-pot conversion of cellulose. The results are summarized in Table 2. Compared to the parent 0.5Pt/WOx catalyst which gave rise to EG and ethanol yields of 15.2% and 17.7% (entry 1), respectively, the addition of Nb resulted in remarkable enhancement of the ethanol yield (entries 2–4). Moreover, the promotional effect was highly sensitive to the Nb amount following a volcano-type relationship. The best result was achieved with 0.1Nb/0.5Pt/WOx which afforded EG and ethanol with yields of 21.8% and 33.7%, increases of 6.6% and 16.0% compared with 0.5Pt/WOx, respectively. An Nb amount lower or higher than 0.1 wt% led to a decrease in the EG yield but the enhancement of the ethanol yield was still remarkable. This result strongly suggests that the addition of a small amount of Nb contributes mainly to the hydrogenolysis of EG to ethanol, and this point was proved by the EG hydrogenolysis test (Table S2†).| Entrya | Catalyst | Carbon yield (%) | |||||
|---|---|---|---|---|---|---|---|
| EG | Ethanol | 1,2-PG | Propanol | Butanol | TOC | ||
| a Reaction conditions: 0.1 g catalyst, 0.1 g cellulose, 9.9 g H2O, 245 °C, 6.0 MPa H2, 2 h, 700 rpm. Under the reaction conditions, cellulose was completely converted with all of the investigated catalysts. 1,2-PG stands for 1,2-propanediol. b 0.1 g 0.5Pt/WOx + 0.1 g Nb2O5; 0.1 g 0.5Pt/Nb2O5 + 0.1 g WOx. | |||||||
| 1 | 0.5Pt/WOx | 15.2 | 17.7 | 1.6 | 5.7 | 3.3 | 67.0 |
| 2 | 0.05Nb/0.5Pt/WOx | 11.7 | 27.7 | 1.3 | 6.3 | 3.6 | 58.0 |
| 3 | 0.1Nb/0.5Pt/WOx | 21.8 | 33.7 | 3.2 | 5.5 | 4.9 | 73.1 |
| 4 | 0.2Nb/0.5Pt/WOx | 9.6 | 26.6 | 0.8 | 6.2 | 4.0 | 71.0 |
| 5 | 0.1Nb–0.5Pt/WOx | 7.7 | 19.9 | 2.2 | 5.5 | 3.1 | 54.1 |
| 6 | 0.1Ta/0.5Pt/WOx | 24.8 | 22.0 | 4.3 | 3.2 | 3.4 | 79.1 |
| 7 | 0.1Mo/0.5Pt/WOx | 26.1 | 20.5 | 4.0 | 4.7 | 2.5 | 78.8 |
| 8 | 0.1Fe/0.5Pt/WOx | 12.4 | 22.4 | 1.5 | 7.0 | 3.6 | 78.1 |
| 9 | 0.1Zn/0.5Pt/WOx | 14.8 | 19.9 | 3.3 | 5.5 | 4.4 | 65.2 |
| 10 | 0.1 W/0.5Pt/WOx | 23.0 | 15.7 | 4.6 | 3.4 | 3.5 | 75.1 |
| 11b | 0.5Pt/WOx + Nb2O5 | 1.3 | 6.5 | 3.3 | 2.7 | 1.8 | 46.5 |
| 12b | 0.5Pt/Nb2O5 + WOx | 2.4 | 6.3 | 1.2 | 5.7 | 3.8 | 46.0 |
In addition to Nb, other oxophilic metal additives including Ta, Mo and Fe also exhibited a promotional effect on ethanol formation (entries 6–8, for more data see Table S3†) although to a lesser extent than Nb. In particular for Ta and Mo, both the EG and ethanol yields were enhanced compared to the additive-free Pt/WOx sample, suggesting that they, like Nb, promote both the C–C and the C–O bond cleavage of cellulose. Unlike these metals, the W additive did not show a promotional effect on ethanol formation although the EG yield was increased to some extent (entry 10), and the Zn additive had a negligible effect on both EG and ethanol formation (entry 9) possibly due to the lack of redox properties. Obviously, Nb was the best additive among various oxophilic metals for the one-pot conversion of cellulose to ethanol over the 0.5Pt/WOx catalyst. The ethanol formation rate reached 25.90 gethanol gPt−1 h−1 over 0.1Nb/0.5Pt/WOx, which was 2–10 times higher than those over other catalysts reported earlier (Table S4†). It was noted that many previous efforts focused on increasing the cellulosic ethanol yield by increasing the noble metal contents,7,9,10,30 which would bring about a large increase in the catalyst cost. In fact, it was found that increasing the Pt content of Pt/WOx from 0.5 wt% to 4.0 wt% could only increase the ethanol yield from 16.2% to 20.8% (Table S5†), demonstrating the inefficient use of expensive Pt. In contrast to earlier reports, we revealed a cost-effective method by modifying the existing low-Pt catalyst with a very small amount of Nb.
It was noted that the loading sequence of Nb relative to Pt had a significant effect on the catalytic performance. When Nb was introduced together with Pt, i.e., co-impregnation of WOx with NbCl5 and H2PtCl6, the resulting 0.1Nb–0.5Pt/WOx catalyst gave EG and ethanol in yields of only 7.7% and 19.9% (Table 2, entry 5), respectively, much lower than those for the 0.1Nb/0.5Pt/WOx catalyst. Similar phenomena were also reported for glycerol hydrogenolysis over the Au/Pt/WOx system,27 implying that polyol hydrogenolysis was highly structure-sensitive. Only when Nb was deposited on the freshly reduced Pt/WOx surface was direct interaction between Nb and Pt enabled. Evidently, the Nb–Pt–W interface played a key role in the one-pot conversion of cellulose to ethanol. To further demonstrate the indispensable role of the Nb–Pt–W interface, another two control samples, 0.5Pt/Nb2O5 + WOx and 0.5Pt/WOx + Nb2O5, were also evaluated in the catalytic transformation of cellulose (Table 2, entries 11 and 12). As expected, both of them gave very low yields of EG and ethanol with poor carbon balance. Meanwhile, large amounts of humins were formed, which might be caused by the strong acidity of Nb2O5.31 When Pt was directly loaded on Nb2O5, Ta2O5 and MoO3, the total yield of ethanol and EG was only around 10% (Table S6†), much lower than that for Pt/WOx, which indicates that the Pt–W interface was the major active site for the C–C and C–O cleavage of cellulose, while the decoration of Nb, Ta, and Mo, most likely as mononuclear oxide species, on Pt/WOx modulates the electronic properties of the Pt–W interface, thus giving rise to a remarkably improved activity for the cleavage of C–C and C–O bonds of cellulose.
Since the 0.1Nb/0.5Pt/WOx catalyst offered the highest activity for cellulose conversion to ethanol, we then investigated its catalytic performance at an increased cellulose concentration (Table S7†). In contrast to the dramatic drop in the EG yield upon increasing the cellulose concentration, the ethanol yield decreased at a much slower rate, indicating that EG hydrogenolysis to ethanol should not be affected by a higher feeding concentration. Considering that a glucose feeding concentration of up to 30% could be tolerated in cellulose conversion to EG by optimizing the reactor engineering and kinetics,32,33 it could be expected that a high concentration of cellulose/glucose would be allowed in the future for cellulose conversion to ethanol.
To understand the effect of the NbOx additive on the acidity of the catalyst, we first conducted the NH3-TPD experiment. Nevertheless, the two catalysts, 0.5Pt/WOx and 0.1Nb/0.5Pt/WOx, showed quite similar NH3-TPD profiles and total acid amounts (Fig. S3†), which suggests that the addition of 0.1 wt% Nb has little effect on the “static” acidity of the catalyst. Based on our previous work on the hydrogenolysis of glycerol over the Pt–WOx system,18,34 “dynamic” rather than “static” acid sites were found to be responsible for the hydrogenolysis activity. To probe the “dynamic” Brønsted acid sites, we here used 2-butanol dehydration in the presence or absence of hydrogen as the probe reaction.18,34–36 As shown in Fig. 5, for either 0.5Pt/WOx or 0.1Nb/0.5Pt/WOx, the yields of acid-catalyzed products under a hydrogen atmosphere were much higher than those under a nitrogen atmosphere, indicating that “dynamic” Brønsted acid sites are indeed created under a hydrogen atmosphere. More interestingly, the yield of acid-catalyzed products over the 0.1Nb/0.5Pt/WOx catalyst was almost twice that over the 0.5Pt/WOx catalyst, which provides strong evidence that the decoration of mononuclear NbOx on the Pt significantly enhanced the number of “dynamic” acid sites by improving the hydrogen spillover from Pt to WOx. The “dynamic” acid sites can act as active sites for EG hydrogenolysis to ethanol.
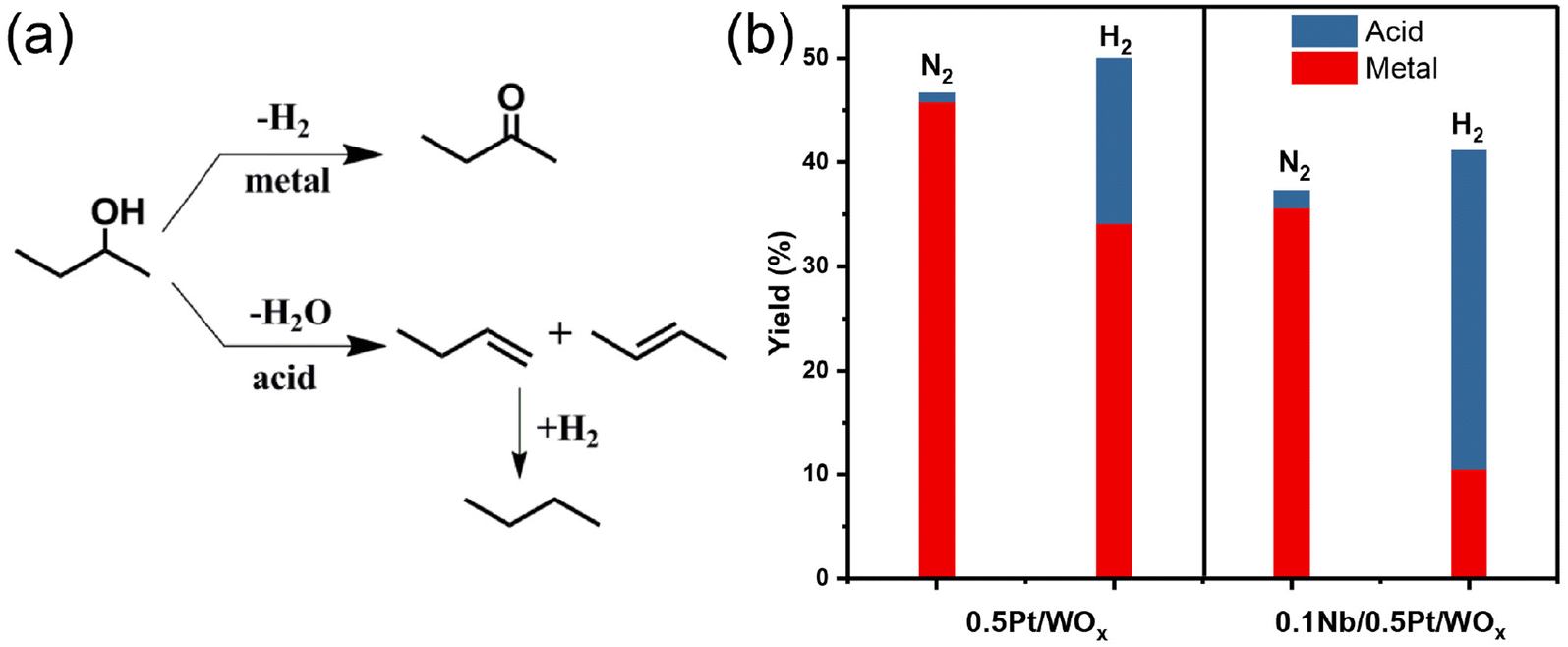 | ||
| Fig. 5 (a) Reaction pathways for the dehydration/dehydrogenation of 2-butanol and (b) the performance of 2-butanol dehydration/dehydrogenation in the presence of N2 or H2 over 0.5Pt/WOx and 0.1Nb/0.5Pt/WOx samples. | ||
Catalyst stability
For the one-pot conversion of cellulose to ethanol, the reaction proceeded in hot water and under pressurized hydrogen, posing a significant challenge to the catalyst stability under such harsh reaction conditions. Since 0.1Nb/0.5Pt/WOx offered the best yield of ethanol, we then investigated its reaction stability. Fig. 6 illustrates the stability of the 0.1Nb/0.5Pt/WOx catalyst in recycling tests. To our delight, the 0.1Nb/0.5Pt/WOx catalyst could be reused 7 times without obvious decay in the EG and ethanol yields. In sharp contrast, both 0.5Pt/WOx and 0.1Mo/0.5Pt/WOx began to decay after only three or four runs (Fig. S4†). The ICP-OES analysis of the liquids after each reaction revealed that Nb was rather resistant to leaching in hot water whereas Mo was continuously leached in each run (Fig. S4e†). The HAADF-STEM imaging of the used catalyst after the 7th cycle showed no sign of Pt aggregation (Fig. S5†). Evidently, the introduction of Nb to Pt/WOx not only greatly enhanced the activity for the cleavage of both C–C and C–O bonds, but also significantly improved the stability of the catalyst.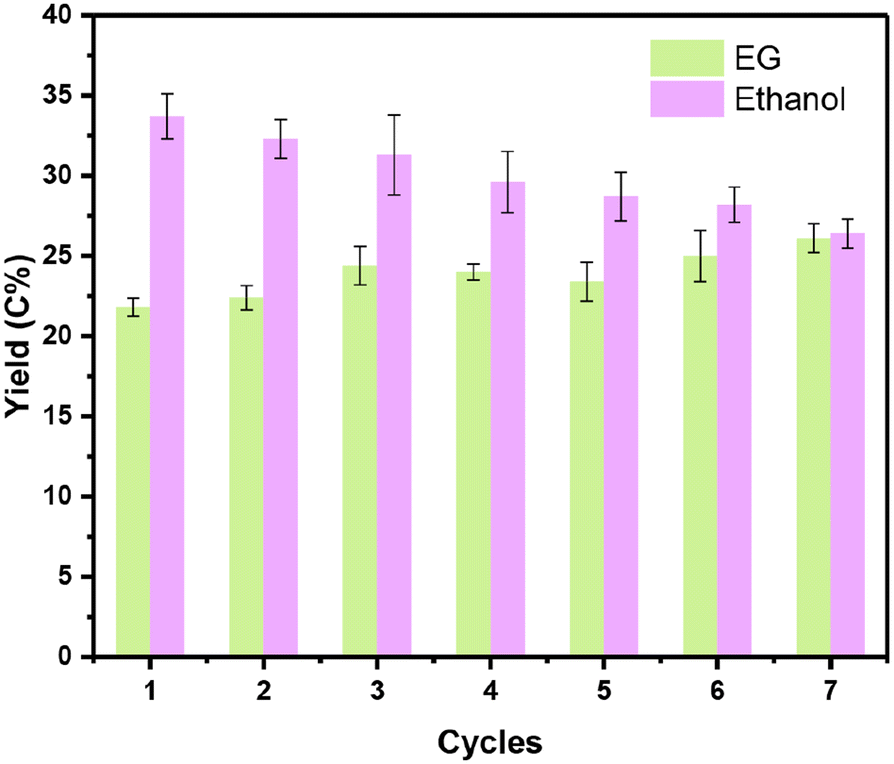 | ||
| Fig. 6 The stability tests for the catalytic conversion of cellulose over 0.1Nb/0.5Pt/WOx. Reaction conditions: 0.1 g cellulose, 0.1 g catalyst, 9.9 g H2O, 245 °C, 6.0 MPa, 2 h, 700 rpm. | ||
Conclusion
We have developed a highly active and robust catalyst 0.1Nb/0.5Pt/WOx for the one-pot conversion of cellulose to ethanol. By decorating mononuclear oxophilic NbOx on the low-Pt Pt/WOx surface, more surface oxygen vacancies were created upon hydrogen reduction, thus creating a higher density of Brønsted acid sites, and therefore leading to an enhancement in activity for C–O hydrogenolysis. As a result, the formation rate of ethanol reached 25.90 gethanol gPt−1 h−1, increased by 2–10 fold compared to literature reports. Moreover, the decoration of NbOx significantly enhanced the catalyst stability, allowing for recycling up to 7 times without obvious activity decay. The strategy of mononuclear oxide decoration on the precious metal surface will provide a new avenue towards enhancing the noble metal efficiency and stability in various biomass upgrading reactions.Author contributions
Weixiang Guan performed the catalyst preparation, characterization studies and reaction tests and also wrote the draft. Chen Cao and Fei Liu performed some of the catalyst preparations and characterization studies. Aiqin Wang and Tao Zhang conceived the idea and revised the paper.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the support from the National Key R&D Program of China (2023YFA1506803), the National Natural Science Foundation of China (22209171 and 22132006), and the NSFC Center for Single-Atom Catalysis (Grant No. 22388102).References
- P. Sudarsanam, R. Zhong, S. Van den Bosch, S. M. Coman, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2018, 47, 8349–8402 RSC.
- Q. Liu, H. Wang, H. Xin, C. Wang, L. Yan, Y. Wang, Q. Zhang, X. Zhang, Y. Xu, G. W. Huber and L. Ma, ChemSusChem, 2019, 12, 3977–3987 CrossRef CAS PubMed.
- H. Song, P. Wang, S. Li, W. Deng, Y. Li, Q. Zhang and Y. Wang, Chem. Commun., 2019, 55, 4303–4306 RSC.
- D. Kennes, H. N. Abubackar, M. Diaz, M. C. Veiga and C. Kennes, J. Chem. Technol. Biotechnol., 2015, 91, 304–317 CrossRef.
- N. Yan and S. Ding, Trends Chem., 2019, 1, 457–458 CrossRef CAS.
- M. Yang, H. Qi, F. Liu, Y. Ren, X. Pan, L. Zhang, X. Liu, H. Wang, J. Pang, M. Zheng, A. Wang and T. Zhang, Joule, 2019, 3, 1937–1948 CrossRef CAS.
- C. Li, G. Xu, C. Wang, L. Ma, Y. Qiao, Y. Zhang and Y. Fu, Green Chem., 2019, 21, 2234–2239 RSC.
- Y. Liu, Y. Liu and Y. Zhang, Appl. Catal., B, 2019, 242, 100–108 CrossRef CAS.
- D. Chu, Z. Luo, Y. Xin, C. Jiang, S. Gao, Z. Wang and C. Zhao, Fuel, 2021, 292, 120311–120318 CrossRef CAS.
- Y. Wu, C. Dong, H. Wang, J. Peng, Y. Li, C. Samart and M. Ding, ACS Sustainable Chem. Eng., 2022, 10, 2802–2810 CrossRef CAS.
- N. Ji, M. Zheng, A. Wang, T. Zhang and J. G. Chen, ChemSusChem, 2012, 5, 939–944 CrossRef CAS PubMed.
- N. Li, Y. Zheng, L. Wei, H. Teng and J. Zhou, Green Chem., 2017, 19, 682–691 RSC.
- A. Wang and T. Zhang, Acc. Chem. Res., 2013, 46, 1377–1386 CrossRef CAS PubMed.
- Y. Shao, Q. Xia, L. Dong, X. Liu, X. Han, S. F. Parker, Y. Cheng, L. L. Daemen, A. J. Ramirez-Cuesta, S. Yang and Y. Wang, Nat. Commun., 2017, 8, 16104–16112 CrossRef PubMed.
- W. Guan, X. Chen, H. Hu, C.-W. Tsang, J. Zhang, C. S. K. Lin and C. Liang, Fuel Process. Technol., 2020, 203, 106392–106401 CrossRef CAS.
- G. Xi, J. Ye, Q. Ma, N. Su, H. Bai and C. Wang, J. Am. Chem. Soc., 2012, 134, 6508–6511 CrossRef CAS PubMed.
- L. Liu, T. Asano, Y. Nakagawa, M. Tamura, K. Okumura and K. Tomishige, ACS Catal., 2019, 9, 10913–10930 CrossRef CAS.
- J. Wang, X. Zhao, N. Lei, L. Li, L. Zhang, S. Xu, S. Miao, X. Pan, A. Wang and T. Zhang, ChemSusChem, 2016, 9, 784–790 CrossRef CAS PubMed.
- B. Zhao, Y. Liang, L. Liu, Q. He and J. Dong, Green Chem., 2020, 22, 8254–8259 RSC.
- D. Yun, E. Z. Ayla, D. T. Bregante and D. W. Flaherty, ACS Catal., 2021, 11, 3137–3152 CrossRef CAS.
- J. Ma, Y. Ren, X. Zhou, L. Liu, Y. Zhu, X. Cheng, P. Xu, X. Li, Y. Deng and D. Zhao, Adv. Funct. Mater., 2018, 28, 1705268–1705279 CrossRef.
- R. Jolly Bose, N. Illyaskutty, K. S. Tan, R. S. Rawat, M. V. Matham, H. Kohler and V. P. Mahadevan Pillai, Europhys. Lett., 2016, 114, 66002–66008 CrossRef.
- J. Yan, T. Wang, G. Wu, W. Dai, N. Guan, L. Li and J. Gong, Adv. Mater., 2015, 27, 1580–1586 CrossRef CAS PubMed.
- Y. Sun, C. J. Murphy, K. R. Reyes-Gil, E. A. Reyes-Garcia, J. M. Thornton, N. A. Morris and D. Raftery, Int. J. Hydrogen Energy, 2009, 34, 8476–8484 CrossRef CAS.
- I. M. Szilágyi, B. Fórizs, O. Rosseler, Á. Szegedi, P. Németh, P. Király, G. Tárkányi, B. Vajna, K. Varga-Josepovits, K. László, A. L. Tóth, P. Baranyai and M. Leskelä, J. Catal., 2012, 294, 119–127 CrossRef.
- W. Zhou, N. Soultanidis, H. Xu, M. S. Wong, M. Neurock, C. J. Kiely and I. E. Wachs, ACS Catal., 2017, 7, 2181–2198 CrossRef CAS.
- B. Wang, F. Liu, W. Guan, A. Wang and T. Zhang, ACS Sustainable Chem. Eng., 2021, 9, 5705–5715 CrossRef CAS.
- X. Yu, H. Tian, Z. Fu, F. Pei, L. Peng, G. Meng, F. Kong, Y. Chen, C. Chen, Z. Chang, X. Cui and J. Shi, ACS Catal., 2023, 13, 2834–2846 CrossRef CAS.
- X. Wang, M. Arai, Q. Wu, C. Zhang and F. Zhao, Green Chem., 2020, 22, 8140–8168 RSC.
- Y. Weng, Y. Wang, M. Zhang, X. Wang, Q. Sun, S. Mu, H. Wang, M. Fan and Y. Zhang, Catal. Today, 2023, 407, 89–95 CrossRef CAS.
- J. J. Wiesfeld, P. Peršolja, F. A. Rollier, A. M. Elemans-Mehring and E. J. M. Hensen, Mol. Catal., 2019, 473, 110400–110409 CrossRef CAS.
- G. Zhao, M. Zheng, R. Sun, Z. Tai, J. Pang, A. Wang, X. Wang and T. Zhang, AIChE J., 2016, 63, 2072–2080 CrossRef.
- G. Zhao, M. Zheng, J. Zhang, A. Wang and T. Zhang, Ind. Eng. Chem. Res., 2013, 52, 9566–9572 CrossRef CAS.
- N. Lei, X. Zhao, B. Hou, M. Yang, M. Zhou, F. Liu, A. Wang and T. Zhang, ChemCatChem, 2019, 11, 3903–3912 CrossRef CAS.
- C. D. Baertsch, K. T. Komala, Y.-H. Chua and E. Iglesia, J. Catal., 2002, 205, 44–57 CrossRef CAS.
- J. Macht, C. D. Baertsch, M. May-Lozano, S. L. Soled, Y. Wang and E. Iglesia, J. Catal., 2004, 227, 479–491 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc03390f |
| This journal is © The Royal Society of Chemistry 2024 |