
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSetting benchmarks for ethylene and propylene oxidation via electrochemical routes: a process design and technoeconomic analysis approach†
Adam P.
Sibal
 a,
Richa
Ghosh
b,
David W.
Flaherty
*b and
Ashlynn S.
Stillwell
*a
a,
Richa
Ghosh
b,
David W.
Flaherty
*b and
Ashlynn S.
Stillwell
*a
aCivil and Environmental Engineering, University of Illinois Urbana-Champaign, 205 N Mathews Ave, MC-250, Urbana, IL 61801, USA. E-mail: ashlynn@illinois.edu
bSchool of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA. E-mail: dflaherty3@gatech.edu
First published on 6th August 2024
Abstract
The chemical industry must reduce greenhouse gas emissions to align with the Paris Agreement. In this study, we assess the economic feasibility of electrochemical oxidations (i.e., using water and electricity to create reactive oxygen species) as an alternative to current energy- and emissions-intensive industrial methods for producing ethylene oxide (EO) and glycol (EG) and propylene oxide (PO) and glycol (PG). Technoeconomic analyses reveal ethylene electrooxidations in a gas diffusion electrode assembly reach economic viability at single-pass conversions above 70% for EO and 40% for EG with overall Faradaic efficiencies (FE) above 20% and high carbon selectivities toward the desired products. Propylene electrooxidation achieves economic feasibility at a minimum single-pass conversion of 10% and overall FE greater than 10% for both PO and PG, while allowing variance in carbon selectivity. These electrochemical methods require current densities of at least 0.1 A cm−2 and lifetimes greater than 2 years for industrial scalability. We demonstrate that electrochemical oxidations can reduce emissions and operational hazards in the production of key chemical intermediates through benchmarking targets for the experimental advancement of alkene electrooxidations.
1. Introduction
According to the International Energy Agency (IEA), the chemical industry is not on track to meet the Paris Agreement target of preventing global warming above 1.5 °C.1 In tandem, data from the U.S. Environmental Protection Agency (EPA) show a continued increase in greenhouse gas (GHG) emissions from the U.S. petrochemical sector since 2011, accounting for 63 million metric tons (MT) of CO2e in 2021.2 These GHG emissions come from direct process emissions and the burning of fossil fuels needed to elevate the temperature and pressure of traditional thermochemical reactions used in industrial chemical processes, further contributing to global temperature rise.3 Replacing these processes with electrochemical reactions offers an attractive and more sustainable alternative, but current utilization and technology deployment remain low.4 Using an electrical driving force, many of these processes can operate with low-carbon electricity sources such as wind, solar, and hydroelectric power at modest pressures and near ambient temperature.5–8 The milder operating conditions of electrochemical processes reduce process costs and operational safety hazards compared to thermochemical processes. Processes that can benefit from electrification include the industrial production of epoxides and glycols from alkenes. Epoxides such as ethylene oxide (EO, 30 MMT globally in 2021) and propylene oxide (PO, 11.2 MMT globally in 2020) are key intermediates in the production of many useful, everyday goods, including pharmaceuticals, textiles, epoxy-resins, lubricants, antifreeze, and polyurethane foam.9 Required oxidants and elevated temperatures and pressures in current industrial production methods of epoxides and glycols lead to significant costs and emissions.10,11 Electrochemically oxidizing ethylene and propylene can decrease these emissions and decarbonize the chemical industry; however, academic literature on such reactions is limited, and the economics of scaling these reactions and their key experimental benchmarks remain understudied. Our work advances the field through process design and technoeconomic analysis of ethylene and propylene electrochemical oxidations.2. Background
2.1 Current industrial approaches for ethylene and propylene oxidation
Industrial epoxide production occurs through partial oxidation of alkenes. Direct and selective epoxidation with molecular oxygen occurs feasibly only for ethylene to EO.12 Undesired competing reactions include the formation of acetaldehyde by primary pathways, which subsequently combusts to form water (H2O) and carbon dioxide (CO2), and the combustion of EO to form H2O and CO2 by secondary pathways.13 Silver (Ag) remains the most effective ethylene epoxidation catalyst; however, unpromoted Ag catalysts only achieve an EO selectivity of ∼50%.14 Supported Ag/Al2O3 catalysts used in industry include promoters (chlorine, rhenium, and alkali metals) to increase EO selectivity to ∼90%.15–18 Additionally, the thermocatalytic nature of ethylene epoxidation to EO requires significant energy input to achieve temperatures and pressures elevated to 200–300 °C and 10–30 bar.12,13 Operating at low single pass conversions (<10%) minimizes undesired secondary pathways, but this approach necessitates large recycle streams. We estimate the U.S. EO production capacity was approximately 4.8 MMT per year in 2022.Epoxidation of propylene and other larger alkenes utilize halogenated or peroxide-containing oxidants.19 Industrial production of PO from propylene with varied byproducts occurs through one of five methods that include oxidation by ethylbenzene or tert-butyl hydroperoxide (global market shares of 27% and 15%, respectively, in 2015), the chlorohydrin route (41%), and oxygen atom transfer from either hydrogen peroxide or cumyl hydroperoxide (15% and 2%).19–24 We estimate that U.S. PO production capacity was approximately 2.4 MMT per year in 2022, with 42% of production from the tert-butyl hydroperoxide method, 19% from the ethylbenzene method, and 39% coming from the chlorohydrin method that coproduces 42 tonnes of alkaline hydrocarbon-laden brine per tonne of PO.25
Portions of EO and PO in the United States undergo energy-intensive thermal hydrolysis at 200 °C and pressures of 14–22 bar to produce ethylene glycol (EG) and propylene glycol (PG).26–29 Domestic production of EG and PG occurs at co-located facilities, as shown in the electronic supplementary information (ESI) (Tables S2 and S3†), to take advantage of the nearby available EO and PO feedstocks and minimize transport costs and risks. Annual U.S. production capacities of EG and PG are 7.6 MMT and 0.4 MMT, respectively.
Overall, costly oxidants, direct process emissions, and separation of reagents and undesired co-products lead to expensive and emissions- and energy-intensive industrial alkene oxidations.13,20 For example, the industrial production of EO yields the fifth-largest CO2 emissions of any petrochemical produced due to the co-production of CO2.30,31 We estimate that industrial ethylene epoxidation produces approximately 1.8 MMT of CO2 as direct process emissions in the United States alone. Drawbacks of industrial propylene epoxidation technologies include the use of environmentally harmful and costly reagents, low atom economy, and toxic stoichiometric co-products (dioxanes, hypochlorite).32,33 Industrial hydrolysis of epoxides to form glycols carry large carbon footprints due to the energy required to heat and pressurize reactors. Thus, the chemical industry must develop more efficient, sustainable, and economical alternatives to produce epoxides and glycols. Oxidizing alkenes with reactive oxygen species at electrode surfaces formed by electrocatalytic water (H2O) oxidation offers an attractive solution.
2.2 Electrochemical approach for ethylene and propylene oxidation
Alkenes do not react with H2O spontaneously; however, alkenes can directly undergo epoxidation with H2O as the sole oxygen source when surface reactions on transition metal or transition metal oxides at anodic potential (>0.8 volts versus the standard hydrogen electrode (VSHE)) facilitate the formation of pools of reactive monatomic and diatomic oxygen surface intermediates (e.g., O*, O2*, OOH*).34 H2O is a sustainable, abundant, and safe oxygen atom source for alkene epoxidations. This process can be driven with sustainably generated electricity to reduce carbon emissions, produces no hazardous side products, and instead can co-produce gaseous hydrogen (H2) by the hydrogen evolution reaction or co-catalyze the CO2 reduction reaction to form organic products. Thermodynamic calculations demonstrate that alkene electrooxidations with H2O can be driven using electrical power at ambient conditions.35 Oxidizing alkenes electrochemically offers an additional benefit, particularly for synthesizing the corresponding glycol (i.e., EG, PG), by removing the energy-intensive step required to create and isolate the epoxide and instead directly producing the glycol.Investigations of both indirect and direct electrochemical oxidations of alkenes appear in literature since the 1950s. Indirect oxidation routes involve the electrochemical generation of chemical oxidants in situ and the subsequent reaction of these species with alkenes in the bulk liquid-phase (i.e., off electrode). These oxidants include hydrogen peroxide,36–38 coordination complexes,39,40 or active halogens41–49 that exist as redox species in the electrolyte or form through oxidation or reduction of salts. Indirect electrooxidation routes achieve industrially relevant current densities and Faradaic efficiency (FE) levels; however, these methods require separation and neutralization of by-products derived from the oxidants. Furthermore, indirect epoxidation with active halogen requires stoichiometric or excess amounts of halides that corrode equipment and pose risk to environmental health. Direct electrooxidation routes, in which the alkene reacts with H2O- or O2-derived intermediates upon the electrode, allows for epoxide and glycol production without corrosive oxidants or spent oxidant by-products.
Dahms and Bockris first demonstrated direct electrooxidation of alkenes with H2O with the oxidation of ethylene to CO2, acetone, and aldehydes on Au, Ir, Pd, Pt, and Rh in 1964.50 In 1975, Holbrook and Wise found ethylene formed ethylene glycol, and propylene formed propylene oxide under anodic oxidation conditions on an Ag electrode in an alkaline electrolyte.51 Studies on the electrooxidation of ethylene and propylene from the 1970s to the 2020s focused on identifying active catalysts for the chemistry and developing an understanding of the reaction mechanism. Multiple late transition metals including Pd,52–54 Au,55–57 Ag,51 Pt,58,59 and Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 60,61 oxidize ethylene to ethylene oxide and ethylene glycol electrochemically. The electrooxidation of propylene to propylene oxide and propylene glycol proceeds on a similar group of catalysts, such as Ag,51,62,63 Pd,64–70 Pt,69,71,72 Au,73 Rh70 and stainless steel.74 The majority of studies utilize batch or semi-batch three-electrode systems that differ from the flow systems necessary for industrial scale electrolysis; however, several published studies performed direct alkene electrooxidations in more industrially relevant electrolyzer systems. The Otsuka and Yamanaka groups investigated the electrooxidation of ethylene and propylene in flow fuel cells and solid-polymer-electrolyte electrolysis cells and achieved a PO production rate of 37 μmol h−1 cm−2 with an epoxidation FE of 7.4%.72,75,76 Recently, propylene epoxidation with propylene oxide formation exceeding 64 μmol h−1 with an epoxidation FE of 28.8% for over 72 hours has been demonstrated in a membrane electrode reactor with V-doped AgO catalysts.63 These electrolyzer systems that operate under continuous flow and utilize polymer membranes for ion transport between electrodes improve epoxidation yield due to increased control of reactant residence time, conductivity, and catalyst durability.72,75,76
60,61 oxidize ethylene to ethylene oxide and ethylene glycol electrochemically. The electrooxidation of propylene to propylene oxide and propylene glycol proceeds on a similar group of catalysts, such as Ag,51,62,63 Pd,64–70 Pt,69,71,72 Au,73 Rh70 and stainless steel.74 The majority of studies utilize batch or semi-batch three-electrode systems that differ from the flow systems necessary for industrial scale electrolysis; however, several published studies performed direct alkene electrooxidations in more industrially relevant electrolyzer systems. The Otsuka and Yamanaka groups investigated the electrooxidation of ethylene and propylene in flow fuel cells and solid-polymer-electrolyte electrolysis cells and achieved a PO production rate of 37 μmol h−1 cm−2 with an epoxidation FE of 7.4%.72,75,76 Recently, propylene epoxidation with propylene oxide formation exceeding 64 μmol h−1 with an epoxidation FE of 28.8% for over 72 hours has been demonstrated in a membrane electrode reactor with V-doped AgO catalysts.63 These electrolyzer systems that operate under continuous flow and utilize polymer membranes for ion transport between electrodes improve epoxidation yield due to increased control of reactant residence time, conductivity, and catalyst durability.72,75,76
Recent efforts in direct alkene electrooxidations mainly focus on improving epoxide and glycol selectivity and formation rates through catalyst development, specifically with promoter addition, crystal facet engineering, and metal alloying. Jirkovský et al. and Hong et al. combined computation and experiments to show that under ethylene electrooxidation conditions in acidic electrolytes the presence of Cl− promoters on RuO2 surfaces creates intermediates (*OCClO*) that poison the surface and isolates O* species, which leads to the prevention of overoxidation to CO2 and selective EO formation.77,78 Crystal facet engineering offers a route to exploit atomic coordination and electronic structure of materials to improve catalytic performance. Epoxidation of cyclohexene to cyclohexene oxide over α-MnO2 in aqueous-acetonitrile electrolytes exhibits facet selectivity as the (310), (110), and (100) facets lead to epoxide yields of 17.5%, 8.2%, and 6.1%, respectively. The (310) facet of α-MnO2 forms more oxygen vacancies, which weakens the Mn–O interaction and facilitates O atom transfer to cyclohexene.79 Propylene electrooxidation in neutral pH electrolytes on Ag3PO4 cubes with (100) facets demonstrates an epoxidation FE of ∼15%, in comparison ∼10% and 5% for the (111) and (100) facets. In addition, the Ag3PO4 cubes with (100) facets give a PO formation rate of 5.3 gPO m−2 h−1, a value 1.6 and 2.5 times that for the (111) and (100) facets. The promotion of epoxidation on the (100) facet results from required lower energies for propylene and OH adsorption and more conducive breaking of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond and formation of the C–O bond in comparison to the (111) and (100) facets, as suggested by density functional theory (DFT) calculations.62 Alloying of metals can enhance selectivity and activity through the tuning of electronic and geometric properties. Chung et al. increased the FE towards cyclooctene epoxidation on MnOx electrocatalysts in aqueous acetonitrile electrolytes from 32% to 46% by doping MnOx with Ir single atoms. The introduction of Ir to the MnOx with galvanic replacement leads to lattice vacancies, which increases highly electrophilic surface O atoms that participate in the epoxidation mechanism.80 Dendritic Pd doped with Au (3.2 at%) in neutral pH electrolyte catalyzes ethylene electrooxidation to EG with a glycol FE of 70% in comparison to 60% for pure dendritic Pd. DFT calculations reveal that the presence of Au reduces the energy of the limiting step (OH* addition of C2H4OH*) due to Au having a weaker OH binding energy in comparison to Pd. Furthermore, EG desorption becomes more facile on the Au-doped catalysts.52 Doping of dendritic Pd with Rh (5 at%) leads to similar promotions of propylene electrooxidation to PG in acidic electrolytes as the introduction of Rh increases PG FE from 45% to 75%. Rh lowers the energy to produce the final intermediate and makes the product desorption step spontaneous.70 Propylene epoxidation on Pd1Pt1Ox/C alloys in aqueous-acetonitrile electrolytes achieve PO FE of 66% with epoxidation partial current densities of 50 mA cm−2 over a period of 3 hours. The improved PO FE of Pd1Pt1Ox catalyst compared to PtOx (5%) or PdOx (18%) arises from the stabilization of Pt oxide species, which demonstrate high activity towards epoxidation, in Pd oxide.69 Recent work shows that doping Ag–O with V promotes propylene epoxidation in a membrane electrode assembly because V facilitates propylene adsorption onto the active center and accelerates Ag–O formation by decreasing the energy barriers for O* formation. The V–Ag–O electrocatalyst achieves a PO FE of 28.8% and a yield of 64.5 μmol h−1, whereas the Ag–O electrocatalyst achieves a PO FE of 21% and a yield of 50 μmol h−1.63 While recent efforts in catalyst development have led to promising FEs (>60%), direct comparisons between studies remain difficult because groups rarely use the same combinations of potential, electrolyte composition, and cell geometry and infrequently report the stability of observed rates and FE with time. In addition, the reported current densities (<100 mA cm−2) remain far below industrial benchmarks for the hydrogen evolution and CO2 reduction reactions (500–1000 mA cm−2).81 The current densities needed for viable electrocatalytic ethylene and propylene oxidation reactions, however, remain unknown.
C bond and formation of the C–O bond in comparison to the (111) and (100) facets, as suggested by density functional theory (DFT) calculations.62 Alloying of metals can enhance selectivity and activity through the tuning of electronic and geometric properties. Chung et al. increased the FE towards cyclooctene epoxidation on MnOx electrocatalysts in aqueous acetonitrile electrolytes from 32% to 46% by doping MnOx with Ir single atoms. The introduction of Ir to the MnOx with galvanic replacement leads to lattice vacancies, which increases highly electrophilic surface O atoms that participate in the epoxidation mechanism.80 Dendritic Pd doped with Au (3.2 at%) in neutral pH electrolyte catalyzes ethylene electrooxidation to EG with a glycol FE of 70% in comparison to 60% for pure dendritic Pd. DFT calculations reveal that the presence of Au reduces the energy of the limiting step (OH* addition of C2H4OH*) due to Au having a weaker OH binding energy in comparison to Pd. Furthermore, EG desorption becomes more facile on the Au-doped catalysts.52 Doping of dendritic Pd with Rh (5 at%) leads to similar promotions of propylene electrooxidation to PG in acidic electrolytes as the introduction of Rh increases PG FE from 45% to 75%. Rh lowers the energy to produce the final intermediate and makes the product desorption step spontaneous.70 Propylene epoxidation on Pd1Pt1Ox/C alloys in aqueous-acetonitrile electrolytes achieve PO FE of 66% with epoxidation partial current densities of 50 mA cm−2 over a period of 3 hours. The improved PO FE of Pd1Pt1Ox catalyst compared to PtOx (5%) or PdOx (18%) arises from the stabilization of Pt oxide species, which demonstrate high activity towards epoxidation, in Pd oxide.69 Recent work shows that doping Ag–O with V promotes propylene epoxidation in a membrane electrode assembly because V facilitates propylene adsorption onto the active center and accelerates Ag–O formation by decreasing the energy barriers for O* formation. The V–Ag–O electrocatalyst achieves a PO FE of 28.8% and a yield of 64.5 μmol h−1, whereas the Ag–O electrocatalyst achieves a PO FE of 21% and a yield of 50 μmol h−1.63 While recent efforts in catalyst development have led to promising FEs (>60%), direct comparisons between studies remain difficult because groups rarely use the same combinations of potential, electrolyte composition, and cell geometry and infrequently report the stability of observed rates and FE with time. In addition, the reported current densities (<100 mA cm−2) remain far below industrial benchmarks for the hydrogen evolution and CO2 reduction reactions (500–1000 mA cm−2).81 The current densities needed for viable electrocatalytic ethylene and propylene oxidation reactions, however, remain unknown.
While there has been promising work in alkene electrooxidation literature, such as high carbon selectivity for EG in ethylene oxidations (66–100%) and high carbon selectivity (73–80%) for PO in propylene oxidations,62 several key issues must be addressed before implementing these electrocatalytic reactions on an industrial scale. Most previous studies used batch and semi-batch systems rather than the flow systems required for continuous chemical manufacturing. Few studies investigated the effects of electrolyte composition, supporting ion identity, and pH, which all influence rates and selectivities of other electrochemical reactions. Reported current densities, oxidation FE toward the epoxide or glycol, and carbon selectivities remain low: all represent areas for improvement prior to scale-up. While several published studies provide guidelines for synthesizing active and selective catalysts,69,80,82 only a few of these studies report metrics to address catalyst durability and cycling tests.52,70 Furthermore, these studies do not include several key metrics necessary to improve rigor and reproducibility, such as normalized current densities with electrochemically active surface area (A cm−2), complete carbon balances that include reactant consumption and product formation, complete electron balances through quantification of products from all electrochemical reactions, and product analysis quantifying all organic products formed.
Limited work has investigated the economic feasibility of implementing alkene electrooxidations on an industrial scale or the experimental benchmarks needed to deploy this technology at scale.52,70 Here, we investigate the economic viability of direct alkene electrooxidation processes using process modeling and technoeconomic analyses, which have been shown to be effective analysis mechanisms for evaluating other electrochemical processes.83–85 Ethylene and propylene were selected as substrates of interest as their epoxidation products (EO and PO) possess the largest global production volume of epoxides. We utilize direct electrochemical alkene ethylene and propylene oxidation literature data (including catalyst utilized, potential applied, bulk electrolysis current density, and electron and carbon selectivity; see Fig. 1 and 2, and Table S1†) and scale these reactions to industrially relevant volumes to simulate electrochemical oxidation and assess the technoeconomic performance possibilities for U.S. EO, EG, PO, and PG production facilities under uncertainty. We conduct analysis using data only from reports that observed epoxide or glycol as a direct alkene oxidation product and that utilized product analysis methods (e.g., gas chromatography) to detect and quantify the amounts of other organic products formed. In particular, we modeled processes informed by parameters from three prior studies that met these criteria from approximately 45 investigations of direct ethylene or propylene electrooxidation. Finally, we use sensitivity analyses to provide benchmark values for key catalyst and electrolyzer attributes (current density, overall FE, electrolyzer durability, and electrolyzer cost) to guide experimental research on this topic.
 | ||
| Fig. 1 Reported reactant single pass conversion percentages and carbon selectivities for studied ethylene electrooxidation literature systems.52 | ||
3. Methodology
The three publications we investigated in detail (Lum et al.,52 Ke et al.,62 and Winiwarter et al.64) studied the oxidation of ethylene to EO and EG and propylene to PO and PG. Lum et al. investigated various Pd-based catalysts for ethylene oxidation primarily to EG and varied other products.52 Ke et al. investigated three facets of Ag3PO4 for electrochemical propylene oxidation to PO.62 Winiwarter et al. investigated the pathways of electrochemical propylene oxidation to PO with additional formation of allyl alcohol, acrolein, and acrylic acid.64 Detailed information summarized from each of these sources is in ESI Section S1.1.† From previously reported FE data and reaction condition information (reactant concentrations, electrolyte conditions, flow rates, etc.), when reported, we determined reactant single pass conversion and product carbon selectivities for four ethylene52 and four propylene electrooxidation62,64 reactions in Fig. 1 and 2, and are detailed in ESI Section S1.6.† | ||
| Fig. 2 Reported reactant single pass conversion percentages and carbon selectivities for studied propylene electrooxidation literature systems.62,64 | ||
Where total reported FEs were less than 100%, we assumed O2 production via the oxygen evolution reaction (OER) accounted for the difference. We also assumed only H2 was produced at the cathode. From this information, we calculated a stoichiometrically balanced full-cell reaction equation for the overall reaction per unit of reactant feed including hydrogen production at the cathode. Equations used to calculate the stoichiometrically balanced reaction are provided in the ESI Section S1.5.† Using SuperPro Designer process modeling software, we represented the stoichiometric reaction for each literature data set in a simulated electrolyzer reactor. SuperPro Designer process modeling software lacks electrolyzer units, so we developed an external mathematical model of this unit considering the stoichiometric reactions outlined in Fig. 1 and 2, respectively.84,86 The model assumes a steady-state reaction and leverages cost estimations from the DOE H2A model and updates key characteristics, such as the current and power requirements and sizing, to adjust cost estimates from the DOE H2A model in the external electrolyzer model.87
We modeled the carbon selectivities observed in literature where the feeds were saturated ethylene or propylene in an aqueous electrolyte (Section 4.1), along with modeled conditions with gaseous ethylene (Section 4.2) and propylene (Section 4.3) feeds using an electrolyzer system such as a gas diffusion electrode (GDE) assembly or membrane electrode assembly (MEA). Separation unit operations were designed around the reactor to recover all products and recycle unreacted ethylene or propylene to ensure at least 98% reactant stream purity. We created an additional set of process simulation models under varied theoretical scenarios with carbon selectivities for ethylene epoxidation to either EO or EG (Section 4.2) and propylene to either PO or PG (Section 4.3) without formation of side products. Detailed descriptions, process flow diagrams, and key input parameters for each model are provided in ESI (Sections S2–S4†).
Ethylene and propylene feed to the facility was modeled as approximately half of the median production volume for U.S. facilities (assuming ethylene and propylene producers would sell portions of these two products consistent with current markets) of 453 ktonnes per year and 181 ktonnes per year, respectively. Tables S2 and S3† provide a detailed breakdown of U.S. ethylene, propylene, and their oxide and glycol derivatives not previously reported in open literature.
We then performed an uncertainty analysis with 800000 trials to determine the potential net present value (NPV) ranges that could be obtained per kg of ethylene or propylene fed to an electrochemical process facility. The uncertainty analysis parameters are detailed in ESI (Section S.5†). The NPV considers all upfront plant costs (capital equipment, construction, installation, startup), operating costs (utilities, raw material, maintenance, labor, etc.), and revenues from product sales less taxes represented in 2023 U.S. dollars discounted at a 7% interest rate over a 15-year facility lifetime. The values obtained serve as a proxy for comparison to the traditional methods of producing EO, EG, PO, and PG. An ethylene or propylene producer could sell their product at market value. The electrochemical method will create a preferential economic return when compared to the traditional thermocatalytic production methods, however, if the NPV per kg of ethylene or propylene for a given process is greater than the reported market prices. Therefore, the results are presented in the NPV per kg of ethylene or propylene. We also performed a sensitivity analysis to determine feasible ranges of current density, overall FE, and other operational values that need to be achieved in electrooxidation research and development to compete with current industrial processes at scale.
4. Results and discussion
In this section, we first assess the economic feasibility of epoxidation reactions in a saturated liquid phase as has been done experimentally in literature. First, these analyses show that processes that saturate the liquid electrolyte with ethylene or propylene prior to entering the electrolyzer (i.e., gaseous reactants do not enter electrolyzer) can never be economically viable. Second, further analyses demonstrates that electrochemical epoxidations in gas diffusion electrode assemblies (or related membrane electrode assembly electrolyzers) can achieve economic viability and compete with incumbent technologies provided catalysis scientists and reactor engineers develop technologies that reach the minimum benchmarks for performance. Notably, current systems satisfy several of these benchmarks.4.1 Saturated aqueous alkene electrooxidations
This section examines the inherent limitations and scalability challenges of saturated aqueous electrochemical epoxidations of ethylene and propylene. Following protocols in electrooxidation literature, the saturated aqueous ethylene and propylene oxidation models assume all ethylene or propylene supplied to the facility is dissolved at standard temperature and pressure in the aqueous electrolyte mixture prior to introduction into the electrolyzer. Due to the relatively low solubility of these two gases at standard temperatures and pressures, 131 mgethylene L−188 and 200 mgpropylene L−1,89 respectively, large volumes of water and electrolyte are required to conduct the electrochemical epoxidations at scale, requiring billions of gallons of water per year. To screen the economic feasibility of saturated aqueous electrochemical epoxidations of ethylene and propylene at industrial scales, two processes were designed (ESI Section S.2†) that varied the single pass conversion of ethylene to ethylene oxide and propylene to propylene oxide at 100% overall FE, and 100% selectivity for the respective product as an ideal scenario. Post-reaction separation processes were designed to recover the products and recycle the unreacted reactants. Fig. 3 shows that saturated aqueous electrochemical epoxidations of both ethylene and propylene are limited at scale and economically infeasible, because the capital and operating costs of the extensive post-reaction separation process required to recover the extremely dilute (i.e., less than 0.001 mol%) unconsumed reactants and products are exorbitant.
 | ||
| Fig. 3 NPV of a facility utilizing saturated aqueous phase ethylene or propylene to form EO or PO, respectively, for varied single pass conversion rates. Circles represent the values observed in the model of each respective facility and the line represents the trend for single pass conversion percentages that were not modeled. | ||
4.2 Ethylene electrooxidations in a GDE reactor design
In this section, we present a process model design for each of the four electrochemical ethylene epoxidation reaction systems detailed in Fig. 1, assess their economic potentials at scale under observed and theoretical single pass conversion rates, and conduct uncertainty and sensitivity analysis to set benchmarks for overall FE, current density, and electrolyzer lifetime. This section ends with an economic comparison of theoretical scenarios in which only EO and/or EG are created without side product formation.Due to limitations of saturated aqueous phase electrooxidations of ethylene and propylene at industrial scales, we designed and modeled four facilities assuming a gas state ethylene stream is fed to an electrolyzer, such as a gas diffusion electrode assembly stack in a flow-by operation.90 A theoretical design of such a reactor is presented in Fig. 4.
 | ||
| Fig. 4 Theoretical gas diffusion electrode assembly in flow-by mode for ethylene electrooxidations. | ||
The facilities were modeled in SuperPro Designer with the same selectivities for ethylene electrooxidation observed in literature for each of the four reaction systems in Fig. 1. Fig. 5 shows a simplified schematic of the facility design for the ethylene electrooxidation System E1, which has the most extensive post-reaction separation processes due to the need to recover each of the five products separately. Systems E2–E4 require simpler separation processes as they produce only one to four products. The detailed SuperPro Designer models for each of the four systems can be found in Table S4† along with detailed explanations of the process design in ESI Section S.3.†
 | ||
| Fig. 5 General process flow diagram of the facility design for the electrooxidation of ethylene in a GDE electrolyzer with selectivities observed in literature System E1. Unless otherwise stated, a yellow lightning bolt symbolizes an electrified unit process. | ||
In this facility design and subsequent models, 453 ktonnes per year of ethylene is fed to the anode gas phase side of a GDE electrolyzer, where ethylene electrooxidation occurs, at standard temperatures and pressure to form the respective products at their respective carbon selectivities in either of the four systems observed in literature (Fig. 1). An aqueous electrolyte (0.1 M NaClO4) enters the cathode liquid side of the GDE electrolyzer and is recycled. In this reactor design, gases and liquids formed on the cathode side of the reactor are assumed to be separate from the anode and leave the electrolyzer in two separate streams. Only O2 gas that forms on the anode side is assumed to cross the membrane. The products from the cathode gaseous side of the reactor first enter an atmospheric flash vessel to separate the liquid and gas components. This process is limited by vapor liquid equilibrium (VLE) constraints. Liquids are further recovered from the gas stream via subsequent pressure increases and cooling. The remaining liquid stream components are passed through a series of distillation columns to recover glycolaldehyde, formic acid, and ethylene glycol formed during ethylene electrooxidation. The gaseous stream is compressed to 30 bar and passes through an absorption column fed with water at 25 °C, a current practice done in existing EO production facilities to take advantage of the high solubility difference of ethylene and produced EO and acetaldehyde.91 The liquid stream from the absorption column then passes through a series of distillation columns to remove the water and separate the EO and acetaldehyde streams. The gaseous stream from the absorber, containing unreacted ethylene, unabsorbed EO and acetaldehyde, H2, and O2, is heated to 200 °C via an electric heater. The heated stream then passes through a hollow fiber membrane system similar to those commercially available to recover 99% of the hydrogen.92 A purge stream is incorporated to ensure that the recycle stream back to the reactor maintains 98% purity of ethylene to not impede the electrochemical epoxidation reaction.
For all the ethylene electrooxidation reaction systems observed in literature, the single pass conversion of ethylene was estimated to be less than 0.01% for saturated aqueous systems. In SuperPro Designer, the reactor could not be modeled to demonstrate single pass conversions less than 0.01%, so 0.01% was used to model the literature single pass conversion. To understand the single pass conversion requirements that are required to compete with current industrial EO/EG production processes, we varied single pass conversions of ethylene from 0.01% to 90%. We did not model 100% single pass conversion rates as they are rarely seen industrially. Fig. 6 shows the ranges of possible NPVs per kg of ethylene for each of the four reaction systems under uncertainty ranges (ESI Section S.5.1†).
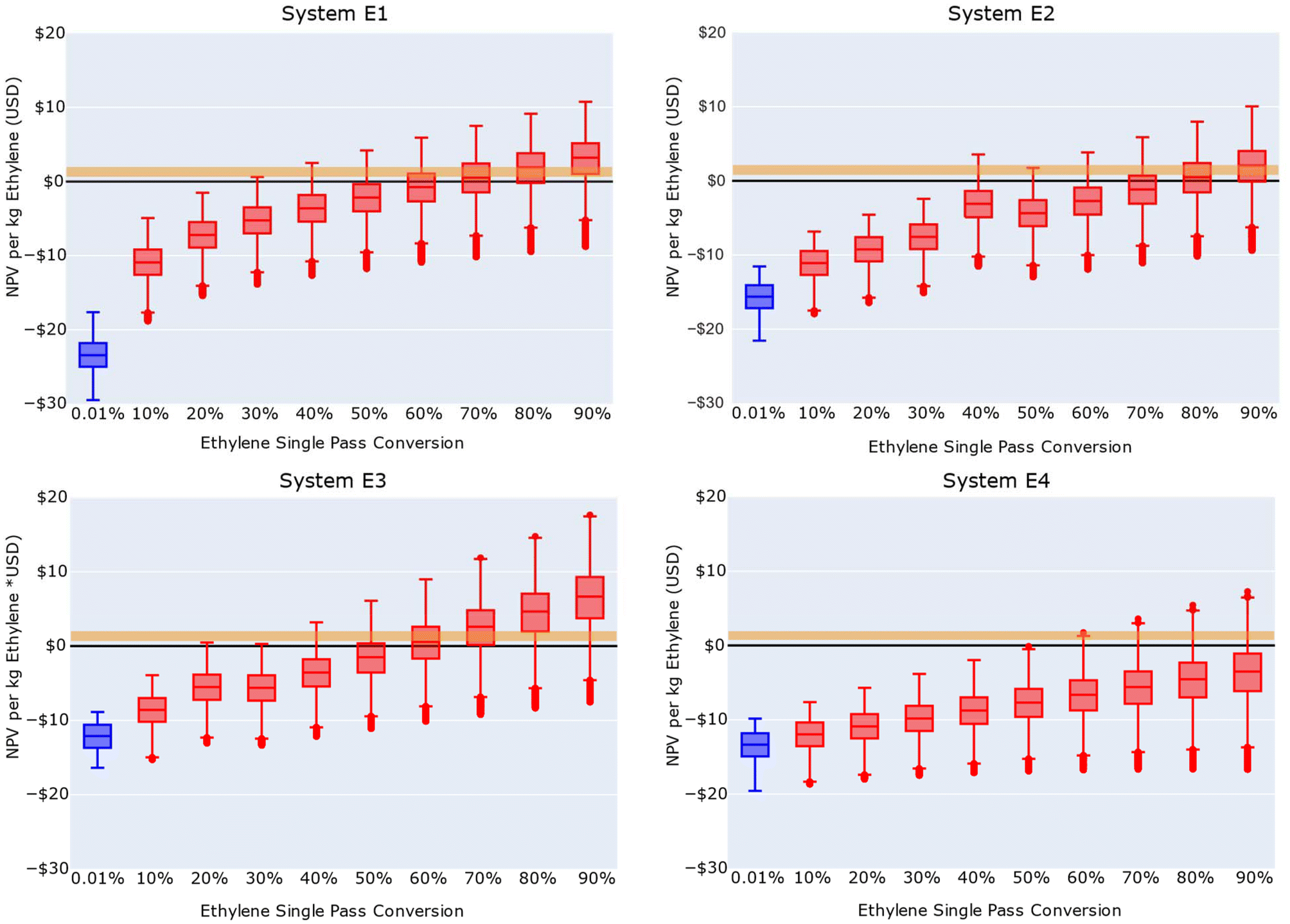 | ||
| Fig. 6 NPV per kilogram of ethylene for ethylene electrooxidation facility designs with observed literature reaction system carbon selectivities under varying single pass conversions. Literature single pass conversion is represented by a blue box plot, and theoretical single pass conversions are shown in red. The shaded box region of each box plot point contains the first, second, and third quartiles of the uncertainty analysis outcomes. The range of ethylene market values is shown in the orange-shaded region. | ||
Assuming a GDE assembly, electrochemical ethylene oxidation could be economically feasible when operated at single pass conversions of 70% or greater (Fig. 6). The conclusions from these analyses also depend on the market prices of reactants and products. Historic ethylene market prices range from $0.83–1.50 per kg,93 EO market prices range from $1.62 to $1.93 per kg,94 and EG market prices range from $0.83 to $1.00 per kg.95 Table S13† expands the findings from Fig. 6 and summarizes the highest economic outcomes for each of these four studied literature reaction systems.
The carbon selectivity of System E3 exhibits the best economic performance potential of the four literature carbon selectivities studied, which can be attributed to its 100% carbon selectivity for EG, minimizing the required post-reaction separation process in both the gas and liquid product streams. While Systems E1 and E2 formed significantly more products, they exhibited less promising economic outcomes in part due to the capital and operation impacts of separating the high number of products. The selectivity of System E4 did not demonstrate any scenarios between the first and third quartiles that would be economically competitive with current industrial ethylene epoxidation processes.
We conducted a sensitivity analysis to understand which electrooxidation reaction system and reactor design parameters possess the greatest influence on the economic viability of electrooxidations of ethylene (with comparisons to current industrial ethylene epoxidation methods) and to provide benchmark targets for advancing research on these reactions. The key benchmarks analyzed were Faradaic efficiency (overall FE, %), current density (A cm−2), electrolyzer cost ($ per m2), and electrolyzer lifetime (years). We explored these parameters for the best performing literature reaction system, System E3, at varied single pass conversions with representations of current literature values (Fig. 7), and a benchmark of values seen in commercially advanced hydrogen-producing electrolysis in polymer electrolyte membrane (PEM) electrolyzer systems (Fig. 7 and 8). We also found that System E3 was not highly sensitive to the applied voltage or electricity price (ESI Section S3.7†).
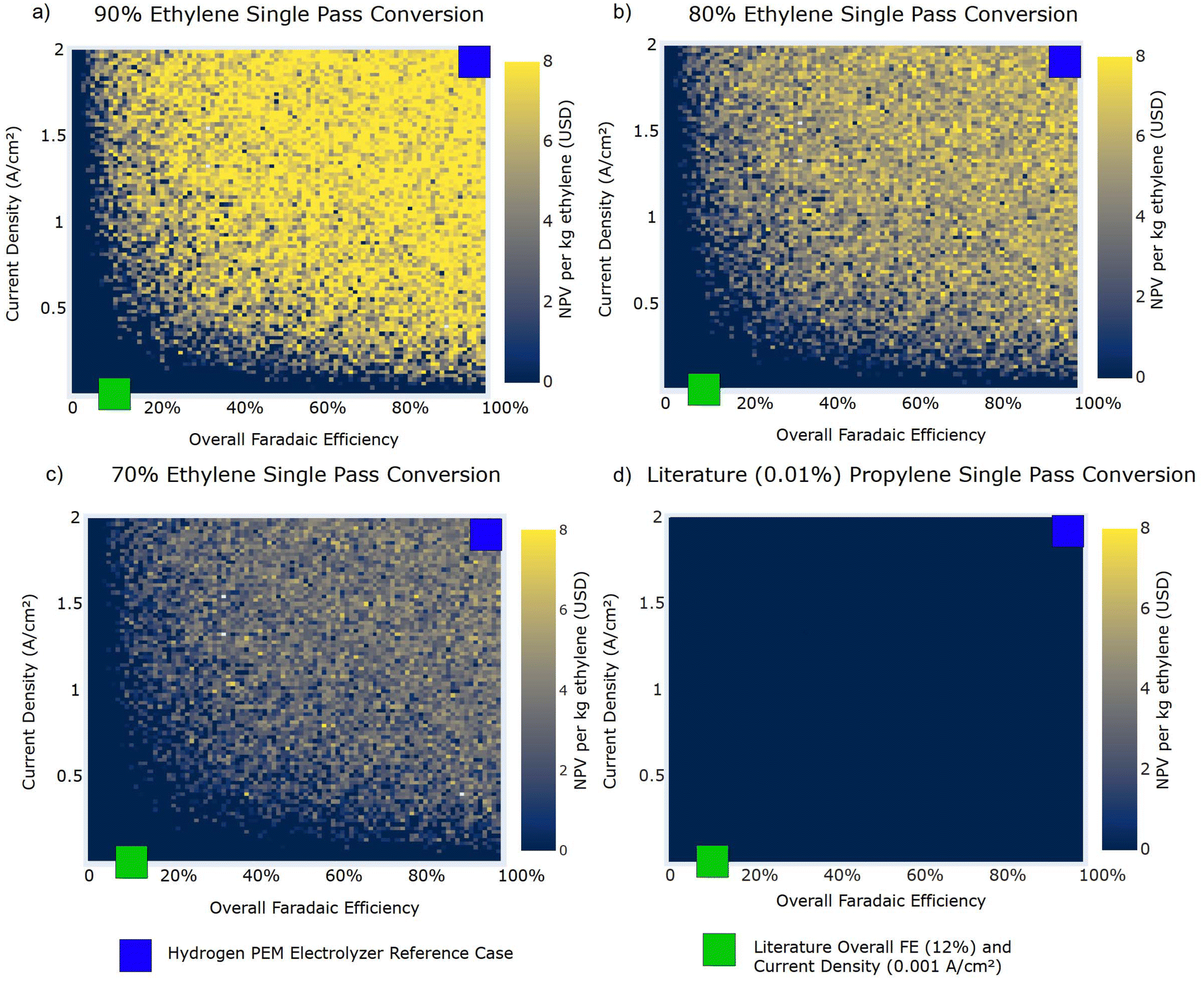 | ||
| Fig. 7 NPV per kg of ethylene for System E3 carbon selectivities against overall FE and current density at observed literature single pass conversion and modeled single pass conversions. Uncertainty is incorporated in the heat map by utilizing data from all 800000 simulations and taking the mean value in each pixel to signify the region in which, under uncertainty, the electrochemical process is economically feasible. Overall FE and current density values observed in literature for this reaction system are represented in each plot with a not-to-scale green square. Current overall FE (near 100%)96 and current densities (1–2 A cm−2)97 observed in commercial PEM hydrogen electrolyzers are demonstrated in not-to-scale blue squares. | ||
 | ||
| Fig. 8 NPV per kg of ethylene for System E3 carbon selectivities against the lifetime of the electrolyzer and the electrolyzer cost (USD per m2) at observed literature single pass conversion and modeled single pass conversions. Uncertainty is incorporated in the heat map by utilizing data from all 800000 simulations and taking the mean value in each pixel to signify the region in which, under uncertainty, the electrochemical process is economically feasible. Current electrolyzer lifetimes (8 years)98 and cost per m2 ($12000 per m2)87 observed in commercial PEM hydrogen electrolyzers are demonstrated in not-to-scale blue squares. | ||
Fig. 7 shows that ethylene electrooxidation reactions are not economically feasible at an industrial scale when the overall FE is less than 10%. Additionally, any current density less than approximately 0.1 A cm−2 is not economically feasible at industrial scales. In Fig. 7, our results show that current density and overall FE have a dynamic relationship in contributing to economic viability. As overall FE increases, the required current density to move into the economically feasible region decreases. For example, at a 90% single pass conversion of ethylene (Fig. 7a) and a 20% overall FE, the minimum required current density for economic feasibility is nearly 0.5 A cm−2, which is 5 times the baseline of 0.1 A cm−2. Electrode current densities must be increased significantly from values observed in literature for ethylene electrooxidations, where the highest current density observed was 0.007 A cm−2 in aqueous batch electrolyzer systems. Overall FEs have been observed in ethylene electrooxidation literature that exceed the minimum 10% threshold. Values of the FE for System E3 (12%) would require both a 90% single pass conversion and a current density of nearly 1 A cm−2.
Fig. 8 shows a less distinct trend comparing electrolyzer lifetime and cost per m2; however, electrolyzer lifetimes that exceed 5 years tend to correlate with economically feasible ethylene electrooxidations, even at higher electrolyzer costs. Where current hydrogen PEM electrolyzer systems are at approximately $12000 per m2,87 the electrolyzer lifetime must be at least 2.5 years to ensure economic feasibility. As current ethylene electrooxidations reported in literature have all been conducted at short times in batch systems with gas-saturated aqueous electrolytes, future studies must investigate operation of this reaction in continuous flow electrolyzers (e.g., GDE or MEA) to assess stability of the system over extended periods and to develop accelerated durability testing protocols as used for hydrogen electrolyzers.
Improvements to the reactivity of electrocatalysts and adaptation of continuous flow GDE-type electrolyzers will increase performance in multiple categories needed for economic feasibility (single pass conversion, electrode currents, FE) but will likely reveal carbon selectivities that differ from those reported in literature (e.g., concentrations of off product pathways may rise above detection limits). Current literature reports production of EG predominately. To provide further research direction on the targets for ethylene electrooxidations, we developed additional process models (ESI Section S3.5†) to assess hypothetical scenarios in which the carbon selectivity among EO and EG varies. For simplicity, we assumed 100% overall FE towards ethylene electrooxidation products and negligible formation of side products (e.g., 100% EO in comparison to 20% EO with 80% EG). Fig. 9 presents the results of these simulations.
 | ||
| Fig. 9 NPV per kg of ethylene at varied carbon selectivities for EO and EG at varied single pass conversions of ethylene in a GDE electrode. Uncertainty is incorporated in the heat map by utilizing data from all 800000 simulations and taking the mean value in each pixel to signify the region in which the electrochemical process is economically feasible. | ||
Fig. 9 shows that higher single pass ethylene conversions generally improve the process economics; however, ethylene electrooxidations with greater carbon selectivities for EG (and hence lower EO selectivities) reach economic feasibility at significantly lower single pass conversions (∼30%) than reactions that only form EO, which require single pass conversions of at least 70%. For a 100% EO carbon selectivity, the overall process ethylene conversion peaks at 90%, because the separation between gaseous EO and unreacted ethylene in the electrolyzer effluent poses significant challenges. For example, absorption of EO from the gas stream recovers about 53% of the EO per absorption column.99 As also observed in current thermocatalytic EO production processes as a result of the challenging EO and ethylene separations, a purge stream is required to operate at steady-state, which loses a fraction of EO and C2H4. In both the proposed electrochemical and current industrial thermocatalytic processes, there are economic tradeoffs associated with increasing the recovery of EO in the gas stream that must be balanced with diminishing CAPEX and OPEX associated with increasing that recovery. For the proposed electrochemical design, however, the overall conversion of ethylene in the system increases with increasing single pass conversion, thereby improving the economic potential to alleviate the need for more extensive and costly recycle loops.
The coproduction of EG with EO improves economics, however, for two reasons. First, the overall process ethylene conversion reaches up to 94% compared to a 90% maximum for a 100% EO carbon selectivity. As the separation of EO and ethylene is limiting to the economic potential and overall conversion of ethylene, when EG is created in tandem, less unreacted ethylene can potentially be lost through the EO recovery and required purge system as ethylene is converted to EG. Second, EO has a higher market value than EG leading to greater overall value delivered per kg of ethylene. Although our analysis compares tradeoffs introduced by variations in the selectivity for the conversion of ethylene to either EO or EG, more deeply oxidized products will likely emerge as an increasingly large fraction of the product distribution when investigators operate electrolyzers at increasing ethylene conversions. The plausible products (e.g., formaldehyde, formic acid, carbon monoxide, and carbon dioxide) will only reduce the NPV per kg of ethylene. Consequently, the results in Fig. 9 represent maxima values for each combination of conversion and EO or EG selectivity.
These findings demonstrate the importance of developing ethylene electrooxidation reactions that have a high carbon selectivity for either EO or EG, with limited creation of low-value side products such as formic acid or CO2 that increase the cost and complexity of the downstream separation processes, thereby reducing the economic feasibility of electrooxidations. Formation of other side products will not diminish the economic potential due to the process gains in the epoxidation reaction and the relatively similar sale price of glycolaldehyde and acetaldehyde to EG as observed in Systems E1 and E2.
In summary, electrochemical processes for ethylene oxidation in gaseous ethylene reactor systems can be effectively scaled up if numerous experimental parameters can be achieved as outlined in Table 1.
| Ethylene oxidation reaction parameter | Experimental literature best result in aqueous batch electrolysis | Benchmark for economically feasible GDE-type reactor system |
|---|---|---|
| Single pass conversion | 0.0095% | 70% |
| Overall FE | 94% | 20% |
| Current density (A cm−2) | 0.007 | 0.1 |
| Electrolyzer lifetime (years) | Unknown | 2.5 |
The benchmark values of single pass conversion and current density necessary for ethylene electrooxidation economic feasibility greatly exceed values reported in contemporary research articles, which is due in part to an emphasis on reporting on materials that display high overall FE and carbon selectivities. Consequently, many groups utilize semi-batch reactors in which most gas bypasses the catalyst or that operate at minimal contact times such that alkene conversions rarely achieve values greater than 0.01%.
4.3 Propylene electrooxidations in a GDE reactor design
Here, we present a process model design for four electrochemical propylene epoxidation reaction systems (Fig. 2), assess their economic potentials at scale under observed and theoretical single pass conversion rates, and conduct uncertainty and sensitivity analysis to set benchmarks for overall FE, current density, and electrolyzer lifetime. This section ends with an economic comparison of theoretical scenarios in which PO and PG form without side products to determine the economic impact of product selectivity.Saturated aqueous propylene electrooxidations, like ethylene, were not economically feasible at industrial scales. We designed and modeled four GDE system facilities in SuperPro Designer with carbon selectivities for propylene electrooxidations observed in literature for four reaction systems (Fig. 2). The process design differs from the ethylene oxidation reaction systems in that PO is a liquid at standard temperature and pressure (STP) and separates more easily from the unreacted propylene than EO from ethylene. Fig. 10 presents a simplified schematic of the process and facility design for propylene electrooxidation Systems P1–P3, which created PO, acetone, and acetic acid at varied carbon selectivities (Fig. 2). SuperPro Designer model details for each of the four reaction systems can be found in ESI Section S4.†
 | ||
| Fig. 10 General schematic of the facility design for propylene electrooxidation in a GDE electrolyzer with carbon selectivities observed in literature Systems P1–P3. Unless otherwise stated, a yellow lightning bolt symbolizes an electrified unit process. | ||
In this facility design and subsequent model, 181 ktonnes per year of propylene contacts the anode side of a GDE electrolyzer at STP. Here the electrooxidation of propylene occurs to form the products at their respective carbon selectivities in the reaction systems observed in literature. Similar to the ethylene epoxidation reaction systems, we model a gas diffusion electrode assembly stack in flow-by operation.90 A theoretical design of such a reactor is presented in Fig. 11.
 | ||
| Fig. 11 Theoretical gas diffusion electrode assembly in flow-by mode for propylene electrooxidations. | ||
Water containing 0.1 M of a phosphate buffer, used in Systems P1–P3, is fed and recycled on the liquid phase cathode side of the GDE. In this reactor design, gases and liquids formed on the cathode side of the reactor remain separate from the anode and leave the electrolyzer in two separate streams. Only the gaseous O2 that forms on the cathode side is assumed to cross the membrane. The products from the gaseous side of the reactor first enter an atmospheric flash vessel to separate the liquid and gaseous components but are limited by the vapor liquid equilibrium (VLE) constraints. The gaseous stream containing primarily unreacted propylene, H2, O2, and vapor phase reaction productions is compressed and cooled via a condenser to recover unreacted propylene and condense the reaction products. The remaining liquid stream components are passed to a series of distillation columns to recover PO, acetone, and acetic acid.
From the condenser, the propylene stream is reheated in an electric heater and vaporized to recycle back to the electrolyzer. The remaining gas stream (H2, O2, and unrecovered propylene) from the condenser is used to provide interstage cooling to the compression system before being reheated to 200 °C and passes through a hollow fiber membrane system similar to those commercially available in industry to recover 99% of the hydrogen92 and O2 and propylene as the retentate. The remaining propylene and O2 stream are purged to prevent buildup of O2 at varied rates as detailed in ESI Section S.4† and recycled back to the electrolyzer.
Propylene electrooxidation System P4 differs from Systems P1–P3, because System P4 forms CO2 as a gaseous product along with the liquid products PG, allyl alcohol, acetone, and acrylic acid. Notably, this reaction proceeded at acidic conditions and utilized 0.1 M HClO4 in the electrolyte. A detailed SuperPro Designer process flow diagram for this system is provided in ESI Section S4.2.†
For the propylene electrooxidation reaction systems observed in literature, the single pass conversion of propylene in saturated aqueous systems far exceeds those of the ethylene oxidations for Systems P1–P3 at 11.4%, 14.1%, and 12.4%, respectively, while System P4 achieves less than 0.01%. As demonstrated in Fig. 12, the single pass conversion of propylene observed for System P4 in literature (0.01%) is not economically viable, and this system leads to negative NPV per kg of propylene. At the single pass conversions observed in literature for Systems P1–P3 in saturated aqueous propylene electrooxidation (11.4–14.1%), most cases exhibit economically viable oxidations. To understand the single pass conversion necessary to better compete with current industry PO and PG processes, we varied single pass conversions of propylene from 10% to 90%. For comparison, historic propylene market prices range from $0.52 to $1.64 per kg,100 PO market prices range from $2.60 to $3.72 per kg,101 and PG market prices range from $1.28 to $3.60 per kg,102 where a potential significant profit margin exists in production of these epoxides. Fig. 12 shows the ranges of possible NPVs per kg of propylene for each of the four reaction systems under the uncertainty ranges.
 | ||
| Fig. 12 NPV per kilogram of propylene for propylene electrooxidation facility designs with observed literature reaction system carbon selectivities under single pass conversions observed in literature and theoretical single pass conversions. Single pass conversion observed in literature is represented by a blue box plot, and theoretical single pass conversions are shown in red. The shaded box region of each box plot point contains the first, second, and third quartiles of the uncertainty analysis outcomes. The range of market values for propylene appears in the orange-shaded region. | ||
Propylene electrooxidations become increasingly economically feasible when single pass conversion increases in the electrolyzer system. Further assessing propylene electrooxidation literature Systems P1–P3, System P1 exhibits the highest carbon selectivity for PO (80.8%) and also performs the best in both carbon selectivity (79.5%) and economic performance, followed by System P2. System P3 shows the worst economic viability of the three scenarios and gives a carbon selectivity for PO (73.2%). This result implies that higher carbon selectivity for PO improves potential economic outcomes. Although System P1 exhibits the largest PO carbon selectivity of the three, System P2 shows the greatest single pass conversion in literature at 14.1% compared to 11.4% for System P1. System P2 shows the best economic potential at its demonstrated single pass conversion. Carbon selectivity in System P4 leads to the worst performance of the four reaction systems, partly attributed to the 16.5% carbon selectivity for CO2 and the extensive separations required to extract six different liquid products. This result highlights the important role that single pass conversion plays on an industrial scale, a metric that often is overlooked in electrooxidation literature.
We conducted a sensitivity analysis for propylene electrooxidations, similar to that of ethylene, where the overall FE (%), current density (A cm−2), electrolyzer cost ($ per m2), and electrolyzer lifetime were varied to identify key benchmarks for achieving economic feasibility. These parameters are explored for System P2 presented in Fig. 13 and 14. We also found that System P2 was not highly sensitive to the applied voltage or electricity price (ESI Section S4.4†).
 | ||
| Fig. 13 NPV per kg of propylene for System P2 carbon selectivities against overall FE and current density at observed literature single pass conversion and modeled single pass conversions. Uncertainty is incorporated in the heat map by utilizing data from all 800000 simulations and taking the mean value in each pixel to signify the region in which, under uncertainty, the electrochemical process is economically feasible. Overall FE and current density values observed in literature for this reaction system are represented in each plot with a not-to-scale green squares. Current overall FE (near 100%)96 and current densities (1–2 A cm−2)97 observed in commercial PEM hydrogen electrolyzers are demonstrated in not-to-scale blue squares. | ||
 | ||
| Fig. 14 NPV per kg of propylene for System P2 carbon selectivities against the lifetime of the electrolyzer and the electrolyzer cost per m2 at observed literature single pass conversion and modeled single pass conversions. Uncertainty is incorporated in the heat map by utilizing data from all 800000 simulations and taking the mean value in each pixel to signify the region in which, under uncertainty, the electrochemical process is economically feasible. Current electrolyzer lifetimes (8 years)98 and cost per m2 ($12000 per m2)87 observed in commercial PEM hydrogen electrolyzers are demonstrated in not-to-scale blue squares. | ||
Fig. 13 demonstrates that positive NPV values can be achieved down to a single pass conversion of 14.1% (observed in literature) for a broad range of overall FE and current densities, which falls below the minimum single pass conversion required for ethylene oxidation reactions (70%). If single pass conversions exceed the literature value at 14.1%, the overall FE must rise above 10% to maintain economic viability for the electrooxidation of propylene. However, as the conversion rate approaches 90% and current density rises, the necessary overall FE can decrease to 5%. Moreover, current densities must remain above roughly 0.1 A cm−2 to attain economic viability at large scales. Fig. 13 also suggests that the required current density to reach economic viability decreases as overall FE rises. For instance, with a 90% propylene conversion and a 26% overall FE, the lowest viable current density is about 0.25 A cm−2. A significant gap exists between the current densities reported in literature for these reactions and the minimum required for economic viability, given the highest current density in literature was 0.002 A cm−2 in saturated aqueous batch systems. For Systems P1–P3, overall FEs reached 26%, and up to 77% for System P4. If Systems P1–P3 can achieve higher overall FEs, increase conversion to at least 20%, and boost current densities to a minimum of 0.1 A cm−2, then the electrooxidation process might become economically viable at large scales.
Fig. 14 compares impacts of electrolyzer lifespan and cost per m2 and does not reveal clear patterns. Electrolyzers over 2 years often align with economically viable propylene electrooxidations. Since current studies use batch systems with gas-saturated aqueous electrolytes, research studies should move to studying these reactions in flow systems that do not require gas saturation in electrolyte, such as a GDE, MEA, or similar setups.
A similar analysis to ethylene electrooxidation was conducted in which we varied the carbon selectivity among PO and PG, assuming no other products are created. The results of these theoretical carbon selectivity simulations are presented in Fig. 15.
 | ||
| Fig. 15 NPV per kg of propylene at varied carbon selectivities for PO and PG at varied single pass conversion percentages of propylene in a GDE electrode assembly for propylene electrooxidation. Uncertainty is incorporated in the heat map by utilizing data from all 800000 simulations and taking the mean value in each pixel to signify the region in which the electrochemical process is economically feasible. | ||
Fig. 15 illustrates that propylene electrooxidations that favor PG production show a marginally higher NPV per kg of propylene than those that favor PO. These findings emphasize that if reaction carbon selectivities or subsequent separation methods can be optimized to limit side product formation to only PO or PG, then such electrooxidations might become economically viable at industrial levels with single pass conversions exceeding 10%. Current studies, such as Systems P1–P3, meet these conversion rates already. Unlike in an ethylene epoxidation system where co-production of the oxide and glycol are advantageous to minimize unreacted ethylene loss associated with challenges around gaseous ethylene and EO separations, in a propylene epoxidation system, high selectivity for one product over the other is preferred to minimize the extent of the liquid–liquid separations.
In summary, in GDE-type systems, propylene electrooxidation can be achieved at economically feasible industrial levels with notably lower single-pass conversion rates, overall FEs, and electrolyzer lifetimes compared to requirements for ethylene electrooxidation. This finding can be attributed to the ease of separations relative to ethylene oxidations as the products of propylene oxidations are generally liquids at STP and thereby also increase the overall process propylene conversion to well over 90% even at single pass conversion rates of 50%, compared to around 70% for ethylene oxidation at the same single pass conversion rate. Promisingly, several of the key benchmarks for propylene oxidation have already been achieved. Recent work has explored alloying palladium with other metals to improve propylene electrooxidation selectivity towards PO and PG, respectively. Using an oxidized Pd–Pt catalyst, Chung et al. demonstrated 70% overall FE towards PO at a current density of 0.05 A cm−2.69 Huang et al. alloyed palladium with various other metals and found that Rh-doped Pd leads to 75% overall FE towards PG at a current density of 0.005 A cm−2.70 While these studies improve propylene electrooxidation overall FE and current density, the single pass conversion remains far below 1%. Experimental improvements are required to achieve economic scale-up, as further summarized in Table 2.
| Propylene oxidation reaction parameter | Experimental literature best result in aqueous batch electrolysis | Economic feasibility benchmark for GDE-type reactor system |
|---|---|---|
| Single pass conversion | 14.1% | 10% |
| Overall FE | 77% | 10% |
| Current density (A cm−2) | 0.002 | 0.1 |
| Electrolyzer lifetime (years) | Unknown | 2 |
5. Conclusion
Our simulation and technoeconomic analysis results show that electrooxidations of ethylene and propylene have potential to compete with current EO, EG, PO, and PG production methods. However, operating these electrooxidations using batch reactors that require saturated aqueous electrolytes leads to these reactions not being economically feasible at industrial scales. Research focused on implementing such reactions in GDE-type systems that reduce post-reaction separation processes would improve economic feasibility.Economically viable ethylene electrooxidations occur in such industrial-scale reactor systems at relatively high single pass conversion percentages (above 70%) and high EO carbon selectivity (above 90%). High EG carbon selectivity (above 80%) reactions are potentially economically viable at single pass conversions above 40%. Utilizing GDE-type stack assemblies that can increase the residence time can significantly improve reported single pass conversion in literature, which currently stands at less than 0.01% in saturated aqueous batch electrolyzers. Current literature on ethylene electrooxidations has, in at least one case, reported a 94% overall FE, well above the 20% threshold observed in this study, but the current densities observed have been around 0.007 A cm−2 in saturated aqueous batch systems and need to achieve at least 0.1 A cm−2 at scale to compete with current ethylene epoxidation processes. Industrial scale electrolyzer systems will also need to achieve minimum durability of 2.5 years to support such systems.
Propylene electrooxidations appear to be more economically viable than those for ethylene given reported performance metrics and the larger difference between market prices of propylene and either PO or PG. Notably, electrooxidations of propylene are feasible in GDE-type systems at industrial scales at single pass conversions of 10% and greater. The potential NPV per kg of propylene also exhibits up to three times the value of ethylene electrooxidations, which can largely be attributed to the ease of separating liquid products, whereas ethylene epoxidations inherently face more challenging gaseous product separations. Propylene electrooxidation literature has achieved overall FEs greater than 20%, well above the demonstrated desired 10%. Significant improvement must be made to increase current densities from 0.002 A cm−2 in saturated aqueous batch systems to at least 0.1 A cm−2 in GDE-type electrolyzers. These electrolyzers must also exhibit a lifetime of at least 2 years.
In addition to these metrics, alkene electrooxidation literature should focus on providing transparent data on the single pass conversions achieved. Single pass conversion is under-reported in literature and could provide a crucial benchmark to achieving industrially relevant production. Literature often focuses exclusively on reporting overall FE, lacking valuable information on current density, single pass conversion, and carbon selectivity. Additionally, the literature analysis was potentially limited by the formation of unquantified products such as CO2, where only one of the reactions reported its formation. Correct and demonstrated carbon accounting and full quantification of products are crucial to setting a benchmark for the state-of-the-art electrochemical research and identifying areas of improvement.
We demonstrate that electrochemical ethylene and propylene oxidations are economically feasible under different conditions and process advancements. Reported literature on such reactions has already demonstrated promising progress in many areas, such as carbon selectivity and overall FE. Further research in catalysts, kinetics, and reactor design is paramount to bridging the gap and achieving industrially-competitive ethylene and propylene electrooxidations.
Author contributions
A. P. S. formulated the analysis, conducted the modeling, and analyzed the results; R. G. conducted the literature review, assisted in formulating the analysis, and assisted in analyzing the results; D. W. F. assisted with formulating the analysis and acquired a portion of funding; A. S. S. assisted with formulating the analysis and acquired a portion of funding; all authors contributed to writing.Data availability
All data supporting the findings of this study are available within the ESI.† Specifically, ESI 1† includes additional reaction mechanism summaries, reactions and equations, details of the SuperPro Designer models, additional discussion figures, U.S. ethylene and propylene and their oxide/glycol production capacity data, and uncertainty/sensitivity modeling inputs (PDF).Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Science Foundation, grant 2029326; the opinions, findings, and conclusions or recommendations are those of the authors and do not necessarily reflect the views of the National Science Foundation. We gratefully acknowledge the members of the University of Illinois Urbana-Champaign EFRI DCheM research team (Paul J. A. Kenis, Andrew G. Gewirth, and Joaquín Rodríguez-López) for their feedback through the development of this analysis. R. G. acknowledges support from the National Science Foundation Graduate Research Fellowship Program (DGE-2039655).Notes and references
- IEA, Tracking Clean Energy Progress 2023, Paris, 2023, https://www.iea.org/reports/tracking-clean-energy-progress-2023.
- U.S. EPA, Greenhouse Gas Reporting Program, U.S. EPA, 2022, https://www.epa.gov/ghgreporting/ghgrp-chemicals.
- Intergovernmental Panel on Climate Change, Climate Change 2014: Mitigation of Climate Change: Working Group III Contribution to the IPCC Fifth Assessment Report, Cambridge University Press, Cambridge, 2015. DOI:10.1017/CBO9781107415416.
- G. G. Botte, Electrochemical Manufacturing in the Chemical Industry, Electrochem. Soc. Interface, 2014, 23(3), 49, DOI:10.1149/2.F04143if.
- Z. J. Schiffer and K. Manthiram, Electrification and Decarbonization of the Chemical Industry, Joule, 2017, 1(1), 10–14, DOI:10.1016/j.joule.2017.07.008.
- E. J. Biddinger and M. A. Modestino, Electro-Organic Syntheses for Green Chemical Manufacturing, Electrochem. Soc. Interface, 2020, 29(3), 43, DOI:10.1149/2.F06203IF.
- E. J. Horn, B. R. Rosen and P. S. Baran, Synthetic Organic Electrochemistry: An Enabling and Innately Sustainable Method, ACS Cent. Sci., 2016, 2(5), 302–308, DOI:10.1021/acscentsci.6b00091.
- R. Xia, S. Overa and F. Jiao, Emerging Electrochemical Processes to Decarbonize the Chemical Industry, JACS Au, 2022, 2(5), 1054–1070, DOI:10.1021/jacsau.2c00138.
- W. Yan, G. Zhang, H. Yan, Y. Liu, X. Chen, X. Feng, X. Jin and C. Yang, Liquid-Phase Epoxidation of Light Olefins over W and Nb Nanocatalysts, ACS Sustainable Chem. Eng., 2018, 6(4), 4423–4452, DOI:10.1021/acssuschemeng.7b03101.
- M. Ghanta, T. Ruddy, D. Fahey, D. Busch and B. Subramaniam, Is the Liquid-Phase H2O2-Based Ethylene Oxide Process More Economical and Greener Than the Gas-Phase O2-Based Silver-Catalyzed Process?, Ind. Eng. Chem. Res., 2013, 52(1), 18–29, DOI:10.1021/ie301601y.
- T. A. Nijhuis, M. Makkee, J. A. Moulijn and B. M. Weckhuysen, The Production of Propene Oxide: Catalytic Processes and Recent Developments, Ind. Eng. Chem. Res., 2006, 45(10), 3447–3459, DOI:10.1021/ie0513090.
- J. P. Dever, M. A. Nunley, J. R. Rader and G. Proulx, Ethylene Oxide, Kirk-Othmer Encyclopedia of Chemical Technology, 2022, pp. 1–40. DOI:10.1002/0471238961.0520082504052205.a01.pub3.
- S. Rebsdat and D. Mayer, Ethylene Oxide, Ullmann's Encyclopedia of Industrial Chemistry, 2001. DOI:10.1002/14356007.a10_117.
- M. O. Ozbek, I. Onal and R. A. van Santen, Why Silver Is the Unique Catalyst for Ethylene Epoxidation, J. Catal., 2011, 284(2), 230–235, DOI:10.1016/j.jcat.2011.08.004.
- H. Van Milligen, B. VanderWilp and G. J. Wells, Enhancements in Ethylene Oxide/Ethylene Glycol Manufacturing Technology, Shell Global Solutions, 2016 Search PubMed.
- M. Matusz, Process for Preparing Ethylene Oxide Catalysts, US Pat., 5739075, 1998 Search PubMed.
- W. Diao, C. D. DiGiulio, M. T. Schaal, S. Ma and J. R. Monnier, An Investigation on the Role of Re as a Promoter in AgCsRe/α-Al2O3 High-Selectivity, Ethylene Epoxidation Catalysts, J. Catal., 2015, 322, 14–23, DOI:10.1016/j.jcat.2014.11.007.
- J. T. Jankowiak and M. A. Barteau, Ethylene Epoxidation over Silver and Copper–Silver Bimetallic Catalysts: II. Cs and Cl Promotion, J. Catal., 2005, 236(2), 379–386, DOI:10.1016/j.jcat.2005.10.017.
- H. Baer, M. Bergamo, A. Forlin, L. H. Pottenger and J. Lindner, Propylene Oxide, Ullmann's Encyclopedia of Industrial Chemistry, 2012. DOI:10.1002/14356007.a22_239.pub3.
- D. L. Trent, Propylene Oxide, Kirk-Othmer Encyclopedia of Chemical Technology, 2000. DOI:10.1002/0471238961.1618151620180514.a01.
- K. Weissermel and H. Arpe, Industrial Organic Chemistry, Wiley, 1st edn, 2003. DOI:10.1002/9783527619191.
- J. Teržan, M. Huš, B. Likozar and P. Djinović, Propylene Epoxidation Using Molecular Oxygen over Copper- and Silver-Based Catalysts: A Review, ACS Catal., 2020, 10(22), 13415–13436, DOI:10.1021/acscatal.0c03340.
- C. P. Gordon, H. Engler, A. S. Tragl, M. Plodinec, T. Lunkenbein, A. Berkessel, J. H. Teles, A.-N. Parvulescu and C. Copéret, Efficient Epoxidation over Dinuclear Sites in Titanium Silicalite-1, Nature, 2020, 586(7831), 708–713, DOI:10.1038/s41586-020-2826-3.
- V. Russo, R. Tesser, E. Santacesaria and M. Di Serio, Chemical and, Technical Aspects of Propene Oxide Production via Hydrogen Peroxide (HPPO Process), Ind. Eng. Chem. Res., 2013, 52(3), 1168–1178, DOI:10.1021/ie3023862.
- KNAK, po-world-n, https://www.knak.jp/munikai/japan/po-world-n.htm, accessed 2024-04-08.
- M. R. Nanda, Z. Yuan, W. Qin and C. Xu, Recent Advancements in Catalytic Conversion of Glycerol into Propylene Glycol: A Review, Catal. Rev., 2016, 58(3), 309–336, DOI:10.1080/01614940.2016.1166005.
- S. Rebsdat and D. Mayer, Ethylene Glycol, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2000. DOI:10.1002/14356007.a10_101.
- C. J. Sullivan, Propanediols, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2000, p. a22_163. DOI:10.1002/14356007.a22_163.
- M. W. Forkner, J. H. Robson, W. M. Snellings, A. E. Martin, F. H. Murphy and T. E. Parsons, Glycols, Kirk-Othmer Encyclopedia of Chemical Technology, Wiley, 2004. DOI:10.1002/0471238961.0520082506151811.a01.pub2.
- Anne Trafton, Shrinking the carbon footprint of a chemical in everyday objects, MIT News|Massachusetts Institute of Technology, https://news.mit.edu/2019/synthesizing-epoxides-0409, accessed 2024-05-13.
- EPA, Inventory of U.S. Greenhouse Gas Emissions and Sinks: 1990–2020, EPA 430-R-22-003, U.S. Environmental Protection Agency, 2022, https://www.epa.gov/ghgemissions/draft-inventory-us-greenhouse-gas-emissions-and-sinks-1990-2020.
- Nexant, Propylene Oxide Process Technologies, 2009.
- C. Kim, T. G. Traylor and C. L. Perrin, MCPBA Epoxidation of Alkenes: Reinvestigation of Correlation between Rate and Ionization Potential, J. Am. Chem. Soc., 1998, 120(37), 9513–9516, DOI:10.1021/ja981531e.
- K. Jin, J. H. Maalouf, N. Lazouski, N. Corbin, D. Yang and K. Manthiram, Epoxidation of Cyclooctene Using Water as the Oxygen Atom Source at Manganese Oxide Electrocatalysts, J. Am. Chem. Soc., 2019, 141(15), 6413–6418, DOI:10.1021/jacs.9b02345.
- Y. Chung, Developing Predictive Tools for Solvent Effects on Thermodynamics and Kinetics, PhD thesis, Massachusetts Institute of Technology, 2023 Search PubMed.
- L. Espinal, S. L. Suib and J. F. Rusling, Electrochemical Catalysis of Styrene Epoxidation with Films of MnO2 Nanoparticles and H2O2, J. Am. Chem. Soc., 2004, 126(24), 7676–7682, DOI:10.1021/ja048940x.
- M. Guan, L. Dong, T. Wu, W. Li, G. Hao and A. Lu, Boosting Selective Oxidation of Ethylene to Ethylene Glycol Assisted by In Situ Generated H2O2 from O2 Electroreduction, Angew. Chem., 2023, 135(19), e202302466, DOI:10.1002/ange.202302466.
- A. Zimmer, D. Mönter and W. Reschetilowski, Catalytic Epoxidation with Electrochemically in Situ Generated Hydrogen Peroxide, J. Appl. Electrochem., 2003, 33(10), 933–937, DOI:10.1023/A:1025881213041.
- H. Wu, Y. Xu, P. Guo, Y. Xu, Z. Huang and L. Zhang, Electrochemical Epoxidation of Alkene with High Faradaic Efficiencies Using Water as an Oxygen Source, Green Chem., 2024, 26(5), 2922–2927, 10.1039/D3GC05126A.
- P. K. Roy, K. Amanai, R. Shimizu, M. Kodera, T. Kurahashi, K. Kitayama and Y. Hitomi, Electrochemical Epoxidation Catalyzed by Manganese Salen Complex and Carbonate with Boron-Doped Diamond Electrode, Molecules, 2023, 28(4), 1797, DOI:10.3390/molecules28041797.
- A. Herman, J.-L. Mathias and R. Neumann, Electrochemical Formation and Activation of Hydrogen Peroxide from Water on Fluorinated Tin Oxide for Baeyer–Villiger Oxidation Reactions, ACS Catal., 2022, 12(7), 4149–4155, DOI:10.1021/acscatal.1c06013.
- C. Tian, X.-Y. Li, V. E. Nelson, P. Ou, D. Zhou, Y. Chen, J. Zhang, J. E. Huang, N. Wang, J. Yu, H. Liu, C. Liu, Y. Yang, T. Peng, Y. Zhao, B.-H. Lee, S. Wang, E. Shirzadi, Z. Chen, R. K. Miao, D. Sinton and E. H. Sargent, Paired Electrosynthesis of H2 and Acetic Acid at A/cm2 Current Densities, ACS Energy Lett., 2023, 4096–4103, DOI:10.1021/acsenergylett.3c01327.
- C. Lucky, T. Wang and M. Schreier, Electrochemical Ethylene Oxide Synthesis from Ethanol, ACS Energy Lett., 2022, 7(4), 1316–1321, DOI:10.1021/acsenergylett.2c00265.
- M. Chung, K. Jin, J. S. Zeng and K. Manthiram, Mechanism of Chlorine-Mediated Electrochemical Ethylene Oxidation in Saline Water, ACS Catal., 2020, 10(23), 14015–14023, DOI:10.1021/acscatal.0c02810.
- X. Liu, Z. Chen, S. Xu, G. Liu, Y. Zhu, X. Yu, L. Sun and F. Li, Bromide-Mediated Photoelectrochemical Epoxidation of Alkenes Using Water as an Oxygen Source with Conversion Efficiency and Selectivity up to 100%, J. Am. Chem. Soc., 2022, 144(43), 19770–19777, DOI:10.1021/jacs.2c06273.
- H. Tang, J. R. Vanhoof and D. De Vos, Olefin Epoxidation Using Electricity as Renewable Power in a Bromide-Mediated Electrochemical Process, Green Chem., 2022, 24(24), 9565–9569, 10.1039/D2GC02883B.
- W. R. Leow, Y. Lum, A. Ozden, Y. Wang, D.-H. Nam, B. Chen, J. Wicks, T.-T. Zhuang, F. Li, D. Sinton and E. H. Sargent, Chloride-Mediated Selective Electrosynthesis of Ethylene and Propylene Oxides at High Current Density, Science, 2020, 368(6496), 1228–1233, DOI:10.1126/science.aaz8459.
- W. Jud, C. O. Kappe and D. Cantillo, One–pot Multistep Electrochemical Strategy for the Modular Synthesis of Epoxides, Glycols, and Aldehydes from Alkenes, Electrochem. Sci. Adv., 2021, 1(3), e2100002, DOI:10.1002/elsa.202100002.
- Y. Zhao, M. Duan, C. Deng, J. Yang, S. Yang, Y. Zhang, H. Sheng, Y. Li, C. Chen and J. Zhao, Br−/BrO−-Mediated Highly Efficient Photoelectrochemical Epoxidation of Alkenes on α-Fe2O3, Nat. Commun., 2023, 14(1), 1943, DOI:10.1038/s41467-023-37620-8.
- H. Dahms and J. O. Bockris, The Relative Electrocatalytic Activity of Noble Metals in the Oxidation of Ethylene, J. Electrochem. Soc., 1964, 111(6), 728, DOI:10.1149/1.2426221.
- L. L. Holbrook and H. Wise, Electrooxidation of Olefins at a Silver Electrode, J. Catal., 1975, 38(1), 294–298, DOI:10.1016/0021-9517(75)90090-1.
- Y. Lum, J. E. Huang, Z. Wang, M. Luo, D.-H. Nam, W. R. Leow, B. Chen, J. Wicks, Y. C. Li, Y. Wang, C.-T. Dinh, J. Li, T.-T. Zhuang, F. Li, T.-K. Sham, D. Sinton and E. H. Sargent, Tuning OH Binding Energy Enables Selective Electrochemical Oxidation of Ethylene to Ethylene Glycol, Nat. Catal., 2020, 3(1), 14–22, DOI:10.1038/s41929-019-0386-4.
- K. Otsuka and I. Yamanaka, Electrochemical Cells as Reactors for Selective Oxygenation of Hydrocarbons at Low Temperature, Catal. Today, 1998, 41(4), 311–325, DOI:10.1016/S0920-5861(98)00022-4.
- F. Goodridge and C. J. H. King, Oxidation of Ethylene at a Palladium Electrode, Trans. Faraday Soc., 1970, 66, 2889, 10.1039/tf9706602889.
- J. W. Johnson, S. C. Lai and W. J. James, Anodic Oxidation of Ethylene on Gold Electrodes, Electrochim. Acta, 1970, 15(9), 1511–1518, DOI:10.1016/0013-4686(70)80071-8.
- E. Pastor and V. M. Schmidt, Electrochemical Reactions of Ethene on Polycrystalline Au Electrodes in Acid Solution Studied by Differential Electrochemical Mass Spectrometry and Isotope Labelling, J. Electroanal. Chem., 1995, 383(1), 175–180, DOI:10.1016/0022-0728(94)03736-M.
- J. Šebera, H. Hoffmannová, P. Krtil, Z. Samec and S. Záliš, Electrochemical and Density Functional Studies of the Catalytic Ethylene Oxidation on Nanostructured Au Electrodes, Catal. Today, 2010, 158(1–2), 29–34, DOI:10.1016/j.cattod.2010.05.025.
- U. Müller, U. Schmiemann, A. Dülberg and H. Baltruschat, Adsorption and Hydrogenation of Simple Alkenes at Pt-Group Metal Electrodes Studied by DEMS: Influence of the Crystal Orientation, Surf. Sci., 1995, 335, 333–342, DOI:10.1016/0039-6028(95)00458-0.
- T. Löffler and H. Baltruschat, Temperature Dependent Formation of Multiple Adsorption States from Ethene at Polycrystalline Pt and Pt(111) Electrodes Studied by Differential Electrochemical Mass Spectrometry, J. Electroanal. Chem., 2003, 554–555, 333–344, DOI:10.1016/S0022-0728(03)00259-6.
- A.-Z. Li, B.-J. Yuan, M. Xu, Y. Wang, C. Zhang, X. Wang, X. Wang, J. Li, L. Zheng, B.-J. Li and H. Duan, One-Step Electrochemical Ethylene-to-Ethylene Glycol Conversion over a Multitasking Molecular Catalyst, J. Am. Chem. Soc., 2024, 146(8), 5622–5633, DOI:10.1021/jacs.3c14381.
- Y.-J. Chu, C.-Y. Zhu, X.-Y. Zuo, C.-G. Liu, Y. Geng, Z.-M. Su and M. Zhang, Dispersed Cu (Ni, Co) in MN3 Moiety on Graphene as Active Site via Electrolytic Water towards Electro-Epoxidation of Ethylene, Appl. Surf. Sci., 2024, 652, 159362, DOI:10.1016/j.apsusc.2024.159362.
- J. Ke, J. Zhao, M. Chi, M. Wang, X. Kong, Q. Chang, W. Zhou, C. Long, J. Zeng and Z. Geng, Facet-Dependent Electrooxidation of Propylene into Propylene Oxide over Ag3PO4 Crystals, Nat. Commun., 2022, 13(1), 932, DOI:10.1038/s41467-022-28516-0.
- Z. Li, Y. Lin, X. Miao, Y. Sun, H. Li, H. Ren, W. Cui and M. Wu, V Activated Ag-O Catalytic Sites for Direct Electro-Epoxidation to Successive Producing Propylene Oxide, Research Square 2024, preprint, DOI:10.21203/rs.3.rs-4390560/v1.
- A. Winiwarter, L. Silvioli, S. B. Scott, K. Enemark-Rasmussen, M. Sariç, D. B. Trimarco, P. C. K. Vesborg, P. G. Moses, I. E. L. Stephens, B. Seger, J. Rossmeisl and I. Chorkendorff, Towards an Atomistic Understanding of Electrocatalytic Partial Hydrocarbon Oxidation: Propene on Palladium, Energy Environ. Sci., 2019, 12(3), 1055–1067, 10.1039/C8EE03426E.
- A. Winiwarter, M. J. Boyd, S. B. Scott, D. C. Higgins, B. Seger, I. Chorkendorff and T. F. Jaramillo, CO as a Probe Molecule to Study Surface Adsorbates during Electrochemical Oxidation of Propene, ChemElectroChem, 2021, 8(1), 250–256, DOI:10.1002/celc.202001162.
- X.-C. Liu, T. Wang, Z.-M. Zhang, C.-H. Yang, L.-Y. Li, S. Wu, S. Xie, G. Fu, Z.-Y. Zhou and S.-G. Sun, Reaction Mechanism and Selectivity Tuning of Propene Oxidation at the Electrochemical Interface, J. Am. Chem. Soc., 2022, 144(45), 20895–20902, DOI:10.1021/jacs.2c09105.
- R. P. H. Jong, E. Dubbelman and G. Mul, Electro-Oxidation of Propylene by Palladium Functionalized Titanium Hollow Fibre Electrodes, J. Catal., 2022, 416, 18–28, DOI:10.1016/j.jcat.2022.10.007.
- S. Koroidov, A. Winiwarter, O. Diaz-Morales, M. Görlin, J. Halldin Stenlid, H.-Y. Wang, M. Börner, C. M. Goodwin, M. Soldemo, L. G. M. Pettersson, J. Rossmeisl, T. Hansson, I. Chorkendorff and A. Nilsson, Chemisorbed Oxygen or Surface Oxides Steer the Selectivity in Pd Electrocatalytic Propene Oxidation Observed by Operando Pd L-Edge X-Ray Absorption Spectroscopy, Catal. Sci. Technol., 2021, 11(10), 3347–3352, 10.1039/D0CY02134B.
- M. Chung, J. H. Maalouf, J. S. Adams, C. Jiang, Y. Román-Leshkov and K. Manthiram, Direct Propylene Epoxidation via Water Activation over Pd-Pt Electrocatalysts, Science, 2024, 383(6678), 49–55, DOI:10.1126/science.adh4355.
- J. E. Huang, Y. Chen, P. Ou, X. Ding, Y. Yan, R. Dorakhan, Y. Lum, X.-Y. Li, Y. Bai, C. Wu, M. Fan, M. G. Lee, R. K. Miao, Y. Liu, C. O'Brien, J. Zhang, C. Tian, Y. Liang, Y. Xu, M. Luo, D. Sinton and E. H. Sargent, Selective Electrified Propylene-to-Propylene Glycol Oxidation on Activated Rh-Doped Pd, J. Am. Chem. Soc., 2024, 146(12), 8641–8649, DOI:10.1021/jacs.4c00312.
- J. O. Bockris, H. Wroblowa, E. Gileadi and B. J. Piersma, Anodic Oxidation of Unsaturated Hydrocarbons on Platinized Electrodes, Trans. Faraday Soc., 1965, 61, 2531, 10.1039/tf9656102531.
- S. Iguchi, M. Kataoka, R. Hoshino and I. Yamanaka, Direct Epoxidation of Propylene with Water at a PtOx Anode Using a Solid-Polymer-Electrolyte Electrolysis Cell, Catal. Sci. Technol., 2022, 12(2), 469–473, 10.1039/D1CY01888D.
- V. M. Schmidt and E. Pastor, Electro-Oxidation of Propene on Gold in Acid Solution Studied by DEMS and FTIRS, J. Electroanal. Chem., 1996, 401(1), 155–161, DOI:10.1016/0022-0728(95)04299-7.
- C. Tse-Chuan and C. Jang-Chyang, Anodic Oxidation of Propylene on a Screen Electrode, Chem. Eng. Sci., 1980, 35(7), 1581–1590, DOI:10.1016/0009-2509(80)80051-0.
- I. Yamanaka, Electrolytic Synthesis of Propene Oxide from Propene and Water in the Gas Phase, Electrochem. Solid-State Lett., 1999, 2(3), 131, DOI:10.1149/1.1390758.
- K. Otsuka, T. Ushiyama, I. Yamanaka and K. Ebitani, Electrocatalytic Synthesis of Propylene Oxide During Water Electrolysis, J. Catal., 1995, 157(2), 450–460, DOI:10.1006/jcat.1995.1310.
- J. S. Jirkovský, M. Busch, E. Ahlberg, I. Panas and P. Krtil, Switching on the Electrocatalytic Ethene Epoxidation on Nanocrystalline RuO2, J. Am. Chem. Soc., 2011, 133(15), 5882–5892, DOI:10.1021/ja109955w.
- J.-C. Hong, T.-C. Kuo, G.-L. Yang, C.-T. Hsieh, M.-H. Shen, T.-H. Chao, Q. Lu and M.-J. Cheng, Atomistic Insights into Cl−-Triggered Highly Selective Ethylene Electrochemical Oxidation to Epoxide on RuO2: Unexpected Role of the In Situ Generated Intermediate to Achieve Active Site Isolation, ACS Catal., 2021, 11(21), 13660–13669, DOI:10.1021/acscatal.1c03574.
- Y. Tian, B. Li, J. Wang, Y. Ge, W. Gao, L. Yu, L. Ma, Y. Li, L. Wang, Z. Liu and J. Chen, Facet Engineering of α-MnO2 for Directly Electrocatalytic Oxygen Atom Transfer from Water toward Epoxidation, Chem. Eng. J., 2024, 490, 151704, DOI:10.1016/j.cej.2024.151704.
- M. Chung, K. Jin, J. S. Zeng, T. N. Ton and K. Manthiram, Tuning Single-Atom Dopants on Manganese Oxide for Selective Electrocatalytic Cyclooctene Epoxidation, J. Am. Chem. Soc., 2022, 144(38), 17416–17422, DOI:10.1021/jacs.2c04711.
- Y. Luo, L. Tang, U. Khan, Q. Yu, H.-M. Cheng, X. Zou and B. Liu, Morphology and Surface Chemistry Engineering toward pH-Universal Catalysts for Hydrogen Evolution at High Current Density, Nat. Commun., 2019, 10(1), 269, DOI:10.1038/s41467-018-07792-9.
- H. Li, C. S. Abraham, M. Anand, A. Cao and J. K. Nørskov, Opportunities and Challenges in Electrolytic Propylene Epoxidation, J. Phys. Chem. Lett., 2022, 13(9), 2057–2063, DOI:10.1021/acs.jpclett.2c00257.
- A. Somoza-Tornos, O. J. Guerra, A. M. Crow, W. A. Smith and B.-M. Hodge, Process Modeling, Techno-Economic Assessment, and, Life Cycle Assessment of the Electrochemical Reduction of CO2: A Review, iScience, 2021, 24(7), 102813, DOI:10.1016/j.isci.2021.102813.
- J. Na, B. Seo, J. Kim, C. W. Lee, H. Lee, Y. J. Hwang, B. K. Min, D. K. Lee, H.-S. Oh and U. Lee, General Technoeconomic Analysis for Electrochemical Coproduction Coupling Carbon Dioxide Reduction with Organic Oxidation, Nat. Commun., 2019, 10(1), 5193, DOI:10.1038/s41467-019-12744-y.
- H. J. Kim, Y. Kim, D. Lee, J.-R. Kim, H.-J. Chae, S.-Y. Jeong, B.-S. Kim, J. Lee, G. W. Huber, J. Byun, S. Kim and J. Han, Coproducing Value-Added Chemicals and Hydrogen with Electrocatalytic Glycerol Oxidation Technology: Experimental and Techno-Economic Investigations, ACS Sustainable Chem. Eng., 2017, 5(8), 6626–6634, DOI:10.1021/acssuschemeng.7b00868.
- M. Jouny, W. Luc and F. Jiao, General Techno-Economic Analysis of CO2 Electrolysis Systems, Ind. Eng. Chem. Res., 2018, 57(6), 2165–2177, DOI:10.1021/acs.iecr.7b03514.
- D. Desantis, B. James and G. Saur, Future (2040) Hydrogen Production from Distributed Grid PEM Electrolysis, 2022, https://www.nrel.gov.
- International Labor Organization, ICSC 0475 – ETHYLENE, https://www.ilo.org/dyn/icsc/showcard.display?p_card_id=0475&p_version=2&p_lang=en, accessed 2024-04-07.
- PubChem, Propylene, https://pubchem.ncbi.nlm.nih.gov/compound/8252, accessed 2024-04-07.
- S. Hernandez-Aldave and E. Andreoli, Fundamentals of Gas Diffusion Electrodes and Electrolysers for Carbon Dioxide Utilisation: Challenges and Opportunities, Catalysts, 2020, 10(6), 713, DOI:10.3390/catal10060713.
- A. P. Sánchez, J. G. B. García and J. R. Montalván, Simulation of the Ethylene Oxide Production Process in ChemCAD® Simulator, Rev. Cienc. Tecnol., 2022, 37, 15–31 Search PubMed.
- Air Liquide, Hydrogen Membrane Overview: Advanced Membrane Technology for Hydrogen Purification and Recovery, 2022.
- K. Hays , US ethylene exports down as producers focus on restocking: Navigator CEO|S&P Global Commodity Insights, S&P Global Commodity Insights, https://www.spglobal.com/commodityinsights/en/market-insights/latest-news/petrochemicals/081721-us-ethylene-exports-down-as-producers-focus-on-restocking-navigator-ceo, accessed 2023-07-16.
- ChemAnalyst, Ethylene Oxide Price Trend and Forecast, ChemAnalyst, https://www.chemanalyst.com/Pricing-data/ethylene-oxide-1102#:~:text=FOB-US%20Ethylene%20Oxide%20prices,higher%20demand%<?pdb_no 20in?>20in<?pdb END?>%20the%20region, accessed 2023-07-16.
- ChemAnalyst, Mono Ethylene Glycol (MEG) Price Trend and Forecast, ChemAnalyst, https://www.chemanalyst.com/Pricing-data/mono-ethylene-glycol-4, accessed 2023-07-16.
- H. Zhou, F. Yu, Q. Zhu, J. Sun, F. Qin, L. Yu, J. Bao, Y. Yu, S. Chen and Z. Ren, Water Splitting by Electrolysis at High Current Densities under 1.6 Volts, Energy Environ. Sci., 2018, 11(10), 2858–2864 RSC.
- M. A. Khan, T. Al-Attas, S. Roy, M. M. Rahman, N. Ghaffour, V. Thangadurai, S. Larter, J. Hu, P. M. Ajayan and M. G. Kibria, Seawater Electrolysis for Hydrogen Production: A Solution Looking for a Problem?, Energy Environ. Sci., 2021, 14(9), 4831–4839, 10.1039/D1EE00870F.
- B. Yang, R. Zhang, Z. Shao and C. Zhang, The Economic Analysis for Hydrogen Production Cost towards Electrolyzer Technologies: Current and Future Competitiveness, Int. J. Hydrogen Energy, 2023, 48(37), 13767–13779, DOI:10.1016/j.ijhydene.2022.12.204.
- H. Perzon, A Simulation Model of a Reactor for Ethylene Oxide Production, Masters Thesis, Lund University, 2015 Search PubMed.
- ChemAnalyst, Propylene Price Trend and Forecast, ChemAnalyst, https://www.chemanalyst.com/Pricing-data/propylene-51, accessed 2023-07-16.
- ChemAnalyst, Propylene Oxide Price Trend and Forecast, ChemAnalyst, https://www.chemanalyst.com/Pricing-data/propylene-oxide-58, accessed 2023-07-16.
- ChemAnalyst, Propylene Glycol Price Trend and Forecast, ChemAnalyst, https://www.chemanalyst.com/Pricing-data/propylene-glycol-1095, accessed 2023-07-16.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc02672a |
| This journal is © The Royal Society of Chemistry 2024 |