
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic synthesis of carboxylic acids from oxygenated substrates using CO2 and H2 as C1 building blocks†
Matilde V.
Solmi
a,
Jeroen T.
Vossen
ab,
Marc
Schmitz
a,
Andreas J.
Vorholt
 b and
Walter
Leitner
*ab
b and
Walter
Leitner
*ab
aInstitute for Technical and Macromolecular Chemistry, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany
bMax Planck Institute for Chemical Energy Conversion, Stiftstraße 34–36, 45470 Mülheim an der Ruhr, Germany. E-mail: walter.leitner@cec.mpg.de
First published on 17th May 2024
Abstract
This work focuses on the development and investigation of a Rh/TPP catalytic system for the synthesis of saturated aliphatic carboxylic acids from readily available oxygenated substrates, H2 and non-toxic, renewable CO2. Optimization of the reaction conditions and reagents was carried out using 2-butanol and 1-butanol as typical secondary and primary alcohols. Afterwards, the reaction system was investigated in-depth with a separate investigation of the reverse water–gas-shift-reaction and the carbonylation reaction as key steps of the overall tandem transformation. Based on these results, a reaction network was established with two distinct main pathways. While the secondary alcohols are converted preferentially via acid catalysed dehydration to the corresponding olefins followed by hydroxycarbonylation, the primary alcohols react primarily via nucleophilic substitution to the iodide compound followed by a Monsanto-type carbonylation. Based on these results, a broad range of alcohols including bio-based substrates was converted to the corresponding C1 elongated carboxylic acids. Additionally, other oxygenated substrates such as aldehydes, ketones and industrially relevant substrate mixtures were applied successfully.
1. Introduction
Carboxylic acids and their derivatives are highly important for their synthetic utilization in the production of polymers, pharmaceuticals, solvents and food additives.1–5 In particular, aliphatic mono-carboxylic acids are employed in the polymer industry,6,7 in agrochemical applications, in the food and feedstock industry,1,8 as solvents1 and as biofuels.9 Industrially, aliphatic carboxylic acids (C4–C13) are mostly produced via an oxidation of the corresponding aldehydes, obtained from hydroformylation processes.1,10 Other types of carboxylic acids are produced by a carbonylation of alcohols or alkenes (acetic acid, pivalic acid and propionic acid).1,11–14 Further processes to obtain higher carboxylic acids from alcohols and carbon monoxide (CO) have been reported over the years by different research groups (from academia and industry) using both homogeneous and heterogeneous catalysts.11,15–34 Overall, the current production of saturated aliphatic carboxylic acids is limited to processes based mainly on toxic and mostly non-renewable CO.CO2 is a highly attractive renewable and non-toxic alternative to CO as a C1 building block. It can be used directly for carboxylations or in combination with “green” hydrogen as a reductant to provide in situ CO for carbonylations.35 Several substrate classes have been converted to carboxylic acids using CO2 as a C1 building block, such as boronic esters,36,37 organohalides,38–41 tosylates/mesylates,38,41 alkenes42,43 and dienes.44
Previously, our group reported a rhodium-based system, which produces carboxylic acids from simple alkenes with yields up to 91% using CO2 and H2 in a formal hydroxycarbonylation (Scheme 1). Mechanistic and labelling studies suggest the in situ formation of CO and H2O by a reverse Water–Gas Shift Reaction (rWGSR), which occurs in hydroxycarbonylation. Similar to the catalytic systems used in the Monsanto process for the carbonylation of methanol to acetic acid, the reaction requires acidic conditions and an iodide promoter and PPh3 as a ligand.45 The conversion of alcohols to carboxylic acids under these reaction conditions was also mentioned briefly in this preliminary study. Since then, Han et al. investigated the conversion of methanol to acetic acid with CO2/H2 using Rh/NHC catalytic systems with LiI as a promotor46,47 and Zhang et al. investigated the reaction with LiI as a promotor and iridium(III) acetate as a catalyst.48
 | ||
| Scheme 1 Formal hydroxycarbonylation of oxygenated substrates (primary and secondary alcohols, ketones, aldehydes) with CO2, H2 and Rh catalyst.45 | ||
Herein, we report the use of a Rh-based homogeneous catalytic system for the synthesis of carboxylic acids from various oxygenated substrates such as alcohols, ketones, aldehydes and mixtures of substrates with CO2 and H2. The two reactions involved, rWGSR and hydroxycarbonylation, will be studied in detail to investigate the reaction pathway and potential intermediates in the process. Based on the findings for the different substrate classes, a reaction network will be established, and the broad applicability will be demonstrated.
2. Results and discussion
Initial reactions were carried out with 2-butanol and 1-butanol as typical secondary and primary alcohols under a gas mixture of CO2 and H2 as carboxylating agents. The components of the catalytic system such as the Rh precursors, PPh3 as a ligand, and an acid (p-TsOH·H2O) from a previous study45 and CHI3 as an iodide compound were used as the starting point.2.1 Optimization of the reaction parameters
2-Butanol was used as a secondary alcohol for optimizing the reaction conditions (Table 1) with the initial reaction conditions of 140 °C in 1 mL acetic acid, 2.4 mol% [RhCl(CO)2]2 with TPP as a ligand (Rh![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P = 1:5), the Brønsted acid p-TsOH (Rh:p-TsOH = 1:3.5), an iodide source iodoform (Rh:CHI3 = 1:2.5), a hydrogen pressure of 10 bar and a CO2 pressure of 20 bar. The yield of target molecules, valeric acid (VA) and 2-methylbutanoic acid (2-MBA), was 45% with a VA-to-MBA ratio of 2:1 in the first experiment with over 99% conversion of the substrate alcohol (Table 1, entry 1). The obtained side products were the acetate, iodide, olefin and alkane.
:P = 1:5), the Brønsted acid p-TsOH (Rh:p-TsOH = 1:3.5), an iodide source iodoform (Rh:CHI3 = 1:2.5), a hydrogen pressure of 10 bar and a CO2 pressure of 20 bar. The yield of target molecules, valeric acid (VA) and 2-methylbutanoic acid (2-MBA), was 45% with a VA-to-MBA ratio of 2:1 in the first experiment with over 99% conversion of the substrate alcohol (Table 1, entry 1). The obtained side products were the acetate, iodide, olefin and alkane.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Volume of acetic acid (ml) | p-TsOH·H2O (mol molRh−1) | Temperature (°C) | H2-pressure (bar) | Carboxylic acid yield (%) |
| If not specified, conversion is over 99%, the VA/2-MBA ratio is about 2/1 and the mass balance is around or above 80%. Reaction conditions: 1.88 mmol 2-BuOH, 46 μmol [RhCl(CO)2]2, acetic acid as solvent, 2.5 mol molRh−1 CHI3, 5 mol molRh−1 PPh3, and 20 bar CO2. | |||||
| 1 | 1 | 3.5 | 140 | 10 | 45 |
| 2 | 2 | 3.5 | 140 | 10 | 67 |
| 3 | 3 | 3.5 | 140 | 10 | 8 |
| 4 | 2 | — | 140 | 10 | 2 |
| 5 | 2 | 3.5 | 160 | 10 | 77 |
| 6 | 2 | 3.5 | 180 | 10 | 66 |
| 7 | 2 | 3.5 | 160 | 5 | 26 |
| 8 | 2 | 3.5 | 160 | 20 | 26 |

Firstly, the influence of solvents and their volume on the reaction was studied. Neat conditions mainly lead to the formation of ethers in the presence of p-TsOH.49 After a solvent screening (ESI, Table S1†), acetic acid turned out to be the best of the tested solvents with a total yield of 67%. The amount of solvent also influenced the reaction with 2 ml of acetic acid showing better results than 1 or 3 mL (Table 1, entries 1–3). The influence of p-TsOH·H2O on the yield of carboxylic acids is related to the amount of solvent. In 1 ml of acetic acid, the presence of p-TsOH·H2O does not have any influence on the carboxylic acid yield (ESI, Table S1†). The best result was obtained using 2 mL of acetic acid in the presence of p-TsOH·H2O (Table 1, entry 2), while the absence of the additive only led to traces of the desired product (Table 1, entry 4). This demonstrates that sufficient Brønsted acidity of the reaction medium is vital for the conversion. The influence of temperature in the range between 140 °C and 180 °C was tested afterwards, with the best result obtained at a temperature of 160 °C (Table 1, entries 2, 5 and 6). The yield of VA and 2-MBA rises to 77%, due to a reduced production of side products. A further increase to 180 °C results in a drop in carboxylic acid yield, probably due to a deactivation of the catalyst via ligand dissociation from the metal centre.
Reducing the H2-pressure to 5 bar (Table 1, entry 7) or increasing it to 20 bar (Table 1, entry 8) caused a drastic reduction of the carboxylic acid yield. With the H2 pressure set at 10 bar, a screening of the CO2 pressure was carried out (10–30 bar, ESI, Table S1†). 20 bar of CO2 was chosen for further studies, because no significant improvement could be reported for any other pressure. With these reaction parameters further experiments on the CHI3 and PPh3 amounts and Rh precursor were performed which can be found in the ESI, Table S2.†50 Under the optimized reaction conditions, a yield of 77% of carboxylic acids with a 2:1 ratio of VA:2-MBA was obtained.
The corresponding primary alcohol 1-BuOH was also investigated to determine the influence of the electronic structure of the alcohol group (Table 2). Tables with solvent, precursor and parameter screening can be found in the ESI (Tables S3 and S4†). The solvent screening shows similar trends to that for 2-BuOH with the best yield obtained in acetic acid. In contrast to the secondary alcohol, however, higher carboxylic acid yields were obtained for the primary alcohols without p-TsOH·H2O (Table 2, entry 4). This became even more apparent when increasing the temperature from 140 °C to 160 °C: the yield of carboxylic acids decreased from 35% to 26% if p-TsOH·H2O was present, while it increased to 59% if p-TsOH·H2O was omitted (Table 2, entry 6–8). This may be due to an increased formation of side products, in particular n-butyl ester, at a higher temperature in the presence of p-TsOH·H2O.49 The pressure also showed a significant influence as a hydrogen pressure of 20 bar increased the yield, whereas it reduced the yield for 2-BuOH (Table 2, entry 9–11). The highest yield of 64% (VA:2-MBA = 2:1) was obtained with 1 mL acetic acid, 20 bar H2, without p-TsOH·H2O at 160 °C (Table 2, entry 10).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Volume of acetic acid (ml) | p-TsOH·H2O (mol molRh−1) | Temperature (°C) | H2-pressure (bar) | Carboxylic acid yield (%) |
| If not specified, conversion is over 99%, the VA:2-MBA ratio is about 2:1 and the MB is around or above 80%. Standard reaction conditions: 1.88 mmol 1-BuOH, 46 μmol [RhCl(CO)2]2, acetic acid as solvent, 2.5 mol molRh−1 CHI3, 5 mol molRh−1 PPh3, and 20 bar CO2. |
|||||
| 1 | 1 | 3.5 | 140 | 10 | 30 |
| 2 | 2 | 3.5 | 140 | 10 | 35 |
| 3 | 3 | 3.5 | 140 | 10 | 3 |
| 4 | 1 | — | 140 | 10 | 44 |
| 5 | 2 | — | 140 | 10 | 2 |
| 6 | 2 | 3.5 | 160 | 10 | 26 |
| 7 | 1 | — | 160 | 10 | 59 |
| 8 | 1 | — | 180 | 10 | 47 |
| 9 | 1 | — | 160 | 5 | 44 |
| 10 | 1 | — | 160 | 20 | 64 |
| 11 | 1 | — | 160 | 30 | 56 |

2.2 Investigating the reaction network
After optimising the reaction conditions, a more detailed investigation of the reaction, its products and side products as well as the individual reaction steps combined in the reaction was performed. The catalytic system composed of a rhodium complex, PPh3 and CHI3 in acetic acid is known to catalyse the synthesis of carboxylic acids from olefins with CO2 and H2via a reverse water–gas-shift-reaction (rWGSR) step, followed by a hydroxycarbonylation of the olefin with CO and H2O produced in situ.45 Starting from alcohols as substrates, the carbonylation can be envisaged to occur alternatively at an alkyl iodide intermediate formed from the alcohol and the iodide source akin to the Monsanto process for methanol. Consequently, the following section is divided into three parts: (i) the rWGSR step, (ii) the subsequent carbonylation step and (iii) the overall reaction network.The rWGSR was tested by using the optimized conditions for either primary or secondary alcohols. The resulting gas phase was collected in a gas bag and analysed by GC. A CO yield of 2.6% was obtained for secondary alcohols and 3.1% for primary alcohols. The relatively high yield given the comparatively low reaction temperature for the rWGSR (Fig. 1) is largely due to the operation in the condensed phase. The same experiments were carried out in the absence of the precursor, demonstrating the catalytic role of the rhodium metal component (ESI, Table S9†).
 | ||
| Fig. 1 Influence of different amounts of CHI3 (left) and PPh3 (right) on the CO yield (rWGSR) using the optimized conditions for primary alcohols and secondary alcohols. Reaction conditions: 1.88 mmol butanol, 92 μmol Rh, 1 ml of acetic acid for primary alcohols, 2 mL acetic acid for secondary alcohols, 3.5 mol molRh−1p-TsOH·H2O for 2-butanol, CHI3, PPh3, 20 bar CO2, 20 bar H2, 160 °C, and 16 h. Yields were calculated on the total amount of CO2 pressurized in the reactor (22.7 mmol). | ||
The effect of the amount of CHI3 and PPh3 on the rWGSR was investigated (Fig. 1). CHI3 is required for the CO2 conversion to occur, as also reflected in the overall reaction system which barely shows any conversion without CHI3 (ESI, Fig. S4–S7†). However, large amounts reduce the CO yield, which may be due to the formation of inactive catalyst species at high I− concentrations. The phosphine ligand PPh3 has little influence on the rWGSR and presumably acts mainly as a stabilizing ligand for Rh species at low CO pressures.
:1 amount with respect to the 13CO used, representing the stoichiometry of the rWGSR. GCMS measurement showed that both 13C and 12C were incorporated into the acid product after 2 h of reaction. The 13C NMR spectra obtained starting from 1-butanol and the ratio between the areas is reported in the ESI (Fig. S8 and Table S8,† approximately COOH:CH = 25:1). The ratio between the integrals did not change from 2 h to 16 h of reaction time which implies that the added 13CO is incorporated immediately during the first 2 h of the reaction. This finding indicates clearly the integration of both catalytic cycles – rWGSR of CO2 into CO and carbonylation with the formed CO – in the overall formation of the carboxylic acids.
Varying the amount of iodide additive in the carbonylation step shows the same results already observed for the rWGSR with the best yield at small amounts (Fig. 1 and ESI, Fig. S2†). Similar experiments were performed by varying the amount of PPh3 (Fig. 1 and ESI, Fig. S3†). In a reaction using CO and H2O directly, the stabilizing ligand is not required to obtain the desired product as the CO stabilizes the complex, being readily available from the beginning of the reaction, unlike with CO2 and H2 where it has the stabilizing effect for the complex at low CO pressures. The maximum activity observed is obtained with 10 eq. of PPh3. Increasing the amount of phosphine to 20 eq. results in a decrease of activity.
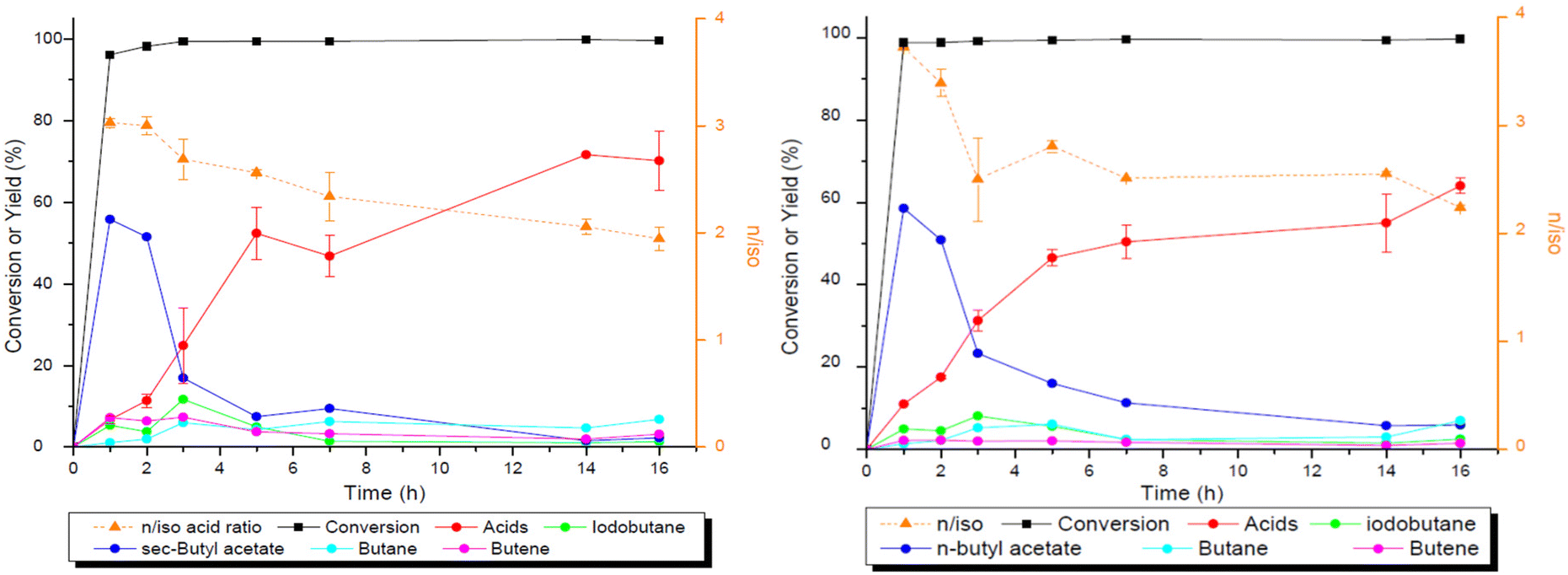 | ||
| Fig. 2 Time profile of the reaction of 2-BuOH (left) and 1-BuOH (right) to form VA and 2-MBA. Reaction conditions: 2-BuOH: 1.88 mmol of 2-BuOH, 92 μmol Rh, 2 ml of acetic acid, 2.5 mol molRh−1 of CHI3, 5 mol molRh−1 of PPh3, 3.5 mol molRh−1p-TsOH·H2O, 20 bar CO2, 10 bar H2, and 160 °C. Reaction conditions: 1-BuOH: 1.88 mmol of 1-BuOH, 92 μmol Rh, 1 ml of acetic acid, 2.5 mol molRh−1 of CHI3, 5 mol molRh−1 of PPh3, 20 bar CO2, 20 bar H2, and 160 °C. The mass balance for these reactions contains a gap due to the formation of highly volatile compounds such as butane and butene which are partially lost during the release of reaction pressure. | ||
Blank tests without the catalyst were performed to see which reactions take place in the absence of the catalyst (Fig. 3). The corresponding iodide, acetate, alkane and olefin are formed in the absence of the transition metal catalyst. The olefin is formed preferentially over the iodide for 2-BuOH in comparison with that for 1-BuOH, reflecting that the dehydration of secondary alcohols is faster. 1-BuOH in turn forms more alkyl iodide, as the SN2 reaction is faster for primary alcohols in comparison with secondary alcohols.
 | ||
| Fig. 3 Comparison between the blank test and the results obtained with the pre-catalyst for the preliminary reaction conditions and the optimized conditions for 2-BuOH (left) and 1-BuOH (right). | ||
Based on these investigations and results from the previous sections, an overall reaction network is proposed in Scheme 2. The alcohol can react via a first path in an SN2 reaction to form the iodide compound with an iodide source such as CHI3 or HI. The iodide can then react similarly to the Monsanto process by carbonylation forming iodic acid initially which cannot be detected as it is hydrolysed to the acid in the presence of water right away forming the carboxylic acid and HI. This path is more likely for primary alcohols as the SN2 reaction is faster in comparison with that for secondary alcohols. The alternative path occurs via the olefin through an acid catalysed dehydration of the alcohol, with the acetic acid solvent, p-TsOH·H2O and/or HI generating the required Brønsted acid. This step is more likely for secondary alcohols as they are dehydrated more easily than primary alcohols. The olefin can react in the carbonylation to form each of the acid product isomers, and it can also isomerize in the presence of the rhodium catalyst. The internal olefin can only form iso-products. In the presence of hydrogen, the rhodium complex can also hydrogenate the olefin to the corresponding alkane.
 | ||
| Scheme 2 Reaction network of primary alcohols to carboxylic acid mainly via the iodides (blue top path) or the secondary alcohols mainly via the olefins (yellow bottom path) with CO from the rWGSR (green, centre). | ||
As observed in the screening experiments, without the iodide additive and without the acid additive p-TsOH·H2O or the acidic solvent acetic acid, the hydroxycarbonylation does not occur. This indicated that a direct reaction from the alcohols to the acids via a CO insertion is not possible and the reaction has to follow one of the two routes with iodoalkanes or olefins as intermediates. The acid-catalysed esterification to form acetates is considered as off-loop equilibrium as the direct conversion of acetates without hydrolysis to the alcohol has not been observed and, to the best of our knowledge, it is not known in the literature.
2.3 Alcohols as substrates
Based on the reaction network and the investigations so far, other substrate alcohols and their behaviour in the reaction were investigated. For some alkyl alcohols (C2 to C6), it was possible to reach yields from 45% to 80% (Table 3). Conversions were always complete with side products and intermediates being formed. The main side products in all cases are the corresponding alkane and n-acetate esters. Methanol resulted in the lowest yield of carboxylic acid among the tested alcohols (entry 1, Table 3). A higher amount of methane is formed as a side product compared to other substrates as it cannot form olefins and does not undergo SN reactions easily. For substrates which can form regio-isomers, the n-acid is consistently more abundant with a ratio of 2:1 compared to the iso-acid. In the case of tert-butanol, only one isomer is formed and no tertiary carboxylic acid is detected (Table 3, entry 10). The preferred regioselectivity for carbonylation at the less substituted carbon demonstrates that the C–C bond formation occurs in the coordination sphere of the rhodium catalyst (Keuleman's rule).51 In purely acid catalysed systems for the synthesis of carboxylic acids via CO and H2O, the more substituted carbon is preferentially carbonylated, due to a higher stability of the corresponding carbocation intermediate (Koch–Haaf-synthesis).52
| Entry | Substrate | Products | Yield (%) | Ratio 1:2:3 |
|---|---|---|---|---|
| Reaction conditions: 1.88 mmol substrate, 46 μmol [RhCl(CO)2]2, 1 ml acetic acid (primary alcohols) or 2 ml acetic acid (secondary alcohols), 3.5 mol molRh−1p-TsOH·H2O (secondary alcohols), 2.5 mol molRh−1 CHI3, 5 mol molRh−1 PPh3, 20 bar CO2, 20 bar H2 (primary alcohols) or 10 bar H2 (secondary alcohols), and 160 °C. For all substrates a conversion of 99% or higher was obtained.a 1 mL propionic acid.b Conditions for secondary alcohols applied. | ||||
| 1a | H3C-OH |
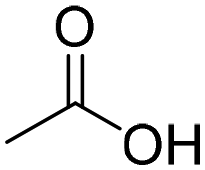
|
19 | 19 |
| 2 |
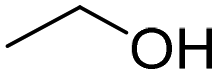
|
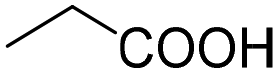
|
80 | 80 |
| 3 |

|
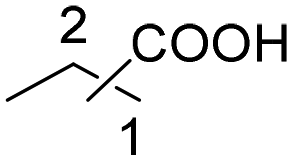
|
45 | 33:12 |
| 4 |

|
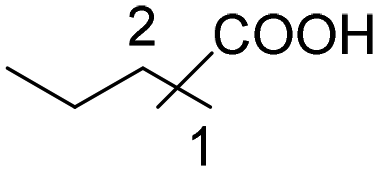
|
65 | 44:21 |
| 5 |
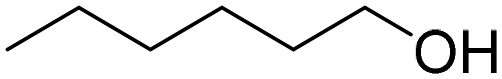
|

|
64 | 42:17:5 |
| 6 |
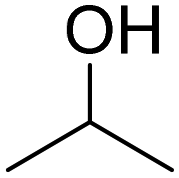
|
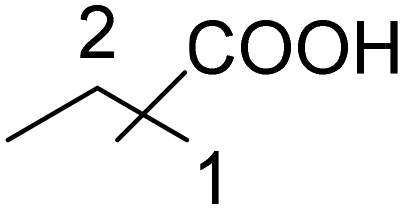
|
30 | 21:9 |
| 7 |
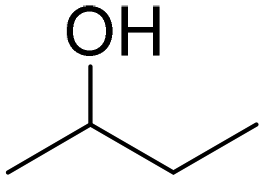
|

|
77 | 50:27 |
| 8 |

|

|
66 | 43:18:5 |
| 9 |

|

|
72 | 72 |
| 10b |

|

|
44 | 44 |
| 11 |

|

|
80 | 80 |
2.4 Ketones and aldehydes as substrates
Next, aldehydes and ketones were considered as possible substrates as they might be expected to be hydrogenated to the corresponding primary or secondary alcohols in the presence of H2, resulting in a fully integrated tandem hydrogenation/carbonylation network using CO2/H2 as the C1 building block (Scheme 3). | ||
| Scheme 3 Tandem reaction sequence of aldehydes and ketones to form carboxylic acids via a hydrogenation of the carbonyl group to the alcohols and subsequent carbonylation of the alcohol using CO2/H2 as the reducing atmosphere and the C1 building block. | ||
The reaction was investigated starting with cyclohexanone (CHN) due to its simplicity, since just one acid-product can be formed (cyclohexanecarboxylic acid, CA). The best conditions for 2-BuOH were used for the optimization of ketones because of the parallels between the two classes of substrates. Under these initial conditions, 68% of CA is formed at a conversion >99% (Table 4, entry 1). The most influential parameters (H2 pressure, volume of acetic acid and the reaction temperature) were studied as for the secondary alcohols to obtain high yields. An increase of H2 pressure from 10 to 20 bar increases the yield of carboxylic acid to 83% (Table 4, entries 1–3). A further increase up to 30 bar does not lead to any improvement, since the hydrogenolysis reaction towards cyclohexane becomes favoured. This finding agrees with the stoichiometry of the reaction as higher amounts of H2 are needed to convert the ketone into secondary alcohols before reacting to form carboxylic acids. In particular, it is reported that Rh/phosphine systems are active in the reduction of ketones to alcohols in the presence of H2O, which acts as a promoter.53–58
|
|
||||
|---|---|---|---|---|
| Entry | Volume of acetic acid (ml) | Temperature (°C) | H2-pressure (bar) | Yield (%) |
| If not specified, conversion is over 99% and the mass balance is around or above 80%. Reaction conditions: 1.88 mmol CHN, acetic acid as solvent, 46 μmol [RhCl(CO)2]2, 2.5 eq. CHI3, 5 eq. PPh3, and 20 bar CO2. | ||||
| 1 | 2 | 160 | 10 | 68 |
| 2 | 2 | 160 | 20 | 83 |
| 3 | 2 | 160 | 30 | 82 |
| 4 | 1 | 140 | 20 | 54 |
| 5 | 3 | 180 | 20 | 53 |
| 6 | 2 | 140 | 20 | 66 |
| 7 | 2 | 180 | 20 | 76 |

Afterwards, the effects of different amounts of solvents and the temperature were investigated. The best temperature and solvent for ketones were found to be the same as for the secondary alcohols with 160 °C and 2 mL of acetic acid (Table 4, entries 2, 4–7). Under optimised conditions, a yield of 83% of cyclohexane carboxylic acid was achieved starting from cyclohexanone (details in the ESI, Table S5†). To the best of our knowledge, this yield is the highest obtained from ketones with CO2 and H2.59
The sequential hydrogenation/carbonylation process is also reflected by the pressure drop as for example in the conversion of 2-butanone or 2-butanol to the C5 carboxylic acids (ESI, Fig. S10†). While the pressure-drop measured for 2-butanol is 7 bar, the pressure-drop for 2-butanone is 15 bar, in line with the theoretical pressure drop calculated for the ketone (details are reported in the ESI, Scheme S1†). Analysis of the reaction solution at different reaction times also clearly shows 2-butanol as an intermediate in the reaction sequence (ESI, Fig. S1†).
Various ketones could be converted to the C1 elongated carboxylic acids. Conversions of the ketones were always complete and the range of by-products was independent of the substrate used (corresponding acetate, iodide, olefin and alkane) with the alkane being the main side product in all reactions. On increasing the chain length from C3 to C6 ketones, the yield increases from 35% to 75%. Interestingly, the α,β-unsaturated ketone 2-cyclohexen-1-one led to the monocarboxylic acid also in high yield (Table 5, entry 5, 79%).
| Entry | Substrate | Products | Yield (%) | Ratio 1:2:3 |
|---|---|---|---|---|
| Reaction conditions: 1.88 mmol substrate, 46 μmol [RhCl(CO)2]2, 1 ml acetic acid (primary alcohols) or 2 ml of acetic acid (secondary alcohols), 3.5 mol molRh−1p-TsOH·H2O (secondary alcohols), 2.5 mol molRh−1 CHI3, 5 mol molRh−1 PPh3, 20 bar CO2, 20 bar H2 (primary alcohols) or 10 bar H2 (secondary alcohols), and 160 °C. For all substrates a conversion of 99% or higher was obtained.a Determined by NMR. | ||||
| 1 |

|

|
35 | 24:11 |
| 2 |

|

|
54 | 37:17 |
| 3 |

|
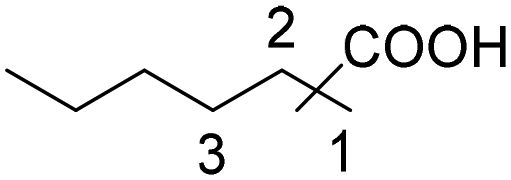
|
75 | 43:26:6 |
| 4 |

|

|
83 | 83 |
| 5 |

|
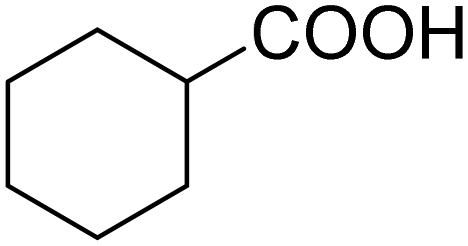
|
79 | 79 |
| 6 |
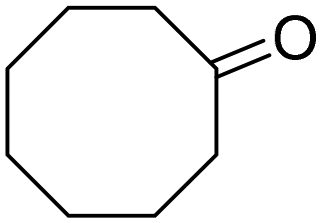
|

|
48 | 48 |
| 7 |
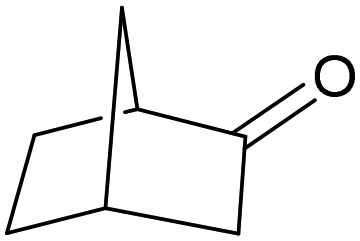
|

|
38 | endo: 1 |
| exo: 37a | ||||
For aldehydes the conditions optimized for primary alcohols were used as the starting point. A moderate yield of 37% of the C5 carboxylic acids was obtained starting from butanal (n:iso = 2.3). Increasing the H2 pressure to 30 bar increased the yield up to 45%, reflecting the additional need for hydrogen during the reduction. While full optimization was not carried out in this case, the system clearly shows potential for the direct conversion of aldehydes to C1 elongated carboxylic acids as well.
2.5 Industrially relevant mixtures as substrates
As the catalytic transformation reported here allows the conversion of ketones and alcohols in the same way, it was evaluated for the transformation of mixtures to their corresponding carboxylic acids. For example, the mixture of cyclohexanol and cyclohexanone (so-called KA-Oil) is industrially produced from the oxidation of cyclohexane. Using the conditions optimized for cyclohexanol alone, cyclohexylcarboxylic acid was obtained as a product with a yield of 79% (Table 6, entry 2).| Entry | Substrate | Products | Yield (%) | Ratio |
|---|---|---|---|---|
| Conversions: >99%.a Cyclohexanone:cyclohexanol = 1:1. Reaction conditions: 1.88 mmol substrate, 46 μmol [RhCl(CO)2]2, 2 mL acetic acid, 3.5 mol molRh−1p-TsOH·H2O, 2.5 mol molRh−1 CHI3, 5 mol molRh−1 PPh3, 20 bar CO2, 20 bar H2, and 160 °C.b Reaction conditions: 1.88 mmol substrate, 46 μmol [RhCl(CO)2]2, 2 mL acetic acid, 2.5 mol molRh−1 CHI3, 5 mol molRh−1 PPh3, 20 bar CO2, 20 bar H2, and 160 °C. |
||||
| 1a | KA oil (ketone alcohol oil) |

|
79 | 79 |
| 2b | ABE mixture (acetone–butanol–ethanol) 3:6:1 |

|
C4: 28 | 20:8 |
| C5: 43 | 30:13 |
|||
| C3: 99 | 99 | |||
Bio-based feedstocks are also often complex mixtures, as for example acetone, butanol, and ethanol from the fermentation of carbohydrates (ABE process). Notably, the mixture obtained from ABE fermentation is transformed successfully to C3 (99%), C4 (28%), and C5 (43% yield) carboxylic acids using the optimized conditions for primary alcohols (Table 6, entry 1). This single step transformation from renewable alcohols is particularly interesting for the platform chemical valeric acid, which is currently produced in a two-step process via hydroformylation and oxidation from butene obtained from fossil resources (Scheme 4).1
 | ||
| Scheme 4 Production of valeric acid via the current industrial two-step approach based on fossil resources vs. the single step renewable approach presented in this work. | ||
3. Conclusion
The application of CO2 as a C1 building block is an important topic in green chemistry.60 Carboxylic acids have many important applications, making their synthesis from CO2 and intermediates of the current value chain or bio-based feedstocks such as alcohols and other oxygenated substrates highly desirable. In this work, a homogeneous catalytic system for the synthesis of carboxylic acids from alcohols, CO2 and H2 was developed. The carboxylic acid yield was optimized in parameter screening experiments with the highest yields obtained for secondary alcohols of 80% and for primary alcohols of 64%.After the optimization, the reaction network was investigated in depth. The role of the individual components of the catalytic system was elucidated in detail. It was shown that the overall transformation is a tandem process comprising the reverse water–gas-shift-reaction of CO2 and H2 to CO and H2O, integrated with a subsequent carbonylation step. Both steps are catalysed by organometallic rhodium complexes formed from a single precursor under the reaction conditions. The carbonylation of the alcohols occurs either via the olefin formed by acid-catalysed dehydration or via the iodoalkane formed by nucleophilic substitution. Primary alcohols follow preferentially the substitution pathway, while secondary alcohols react mainly through the olefin path.
The scope of substrates of this reaction was extended from butanols used for the mechanistic investigation to a range of primary and secondary alcohols. Furthermore, it was shown that ketones and aldehydes can be hydrogenated in situ to alcohols before they undergo the hydroxycarbonylation reaction. Mixtures of substrates as applied in industry were successfully used in the reaction. In particular, the ABE fermentation mixture of acetone, butanol and ethanol is very promising, as it allows the conversion of biomass-derived C2 to C4 oxygenates to value-added products in a single reaction step.
In summary, the current study substantiates the potential of CO2/H2 as C1 building blocks for the general synthesis of carboxylic acids starting from alcohols, ketones, and aldehydes, and even mixtures thereof. This opens novel synthetic pathways to carboxylic acids based on renewable feedstocks as “short cuts” compared to the conventional sequence of hydroformylation and oxidation starting from fossil-based olefins.
4. Experimental
4.1 General considerations
All reactions were performed and compounds were handled under an inert gas atmosphere (Argon 4.8 Messer, Germany) if not stated otherwise, using the Schlenk technique or were handled in a glovebox (MBraun LabMaster SP).4.2 Solvents and chemicals
Acetic acid and other solvents used were dried and stored over molecular sieves (4 Å) and degassed by bubbling argon through a frit for at least 1 h. All liquid substrates were degassed by at least three freeze–pump–thaw cycles and stored over molecular sieves (4 Å) under argon. Cyclohexane oxide was stored under argon and the molecular sieves used for drying were removed after 24 h. Deionized water was taken from a reverse-osmosis purification system (Werner EasyPure II) and degassed by bubbling argon with a frit for at least 1 h. The water contents of all organic solvents and substrates were measured by a Karl-Fischer titration (Metrohm 756 F Coulometer). All reagents were commercially supplied and used as received, unless stated otherwise.4.3 Mass spectrometry
Mass spectroscopy measurements were performed on a Varian 1200L Quadrupole Ms/ms using the ESI ionization method. The detected masses are given in m/z and correlated with calculated masses of the respective species.4.4 Gas chromatography
GC analyses of the liquid phases were performed on a Trace GC Ultra (Thermo Scientific) using a packed CP-WAX-52-CB column (length = 60 m, diameter = 0.25 mm) and a flame ionization detector (FID) or mass spectroscopy detector (MS). Analysis of butane and butene gases was performed on a Sichromat using a capillary PLOT Al2O3 column (length = 50 m) equipped with an FID. GC analysis of CO, CO2 and H2 gases was performed on an HP6890 using a capillary Chem Carbon ST column (length = 2 m) and a thermal conductivity detector (TCD). Examples of the different types of chromatograms are reported in the ESI (Fig. S11–S13†).Liquid substances were analysed using (±)-1-phenylethanol and/or n-dodecane as the standard. Acetone was used as a solvent for the work-up procedure (for cyclohexanol reactions acetone was substituted by dichloromethane). The correction factors were calculated preparing solutions with known amounts of substances and standard. The gaseous substances were quantified using ethane as a standard. As for the liquid samples, the correction values were obtained from self-made gas solutions with known amounts of gases.
4.5 NMR analysis
NMR measurements were conducted on a Bruker AVIII-300 spectrometer (300 MHz) at ambient temperature. The 2H-NMR measurements were conducted on a Bruker AVIII HD-600 spectrometer (600 MHz) at ambient temperature. For 1H, 2H and 13C chemical shifts are given in ppm relative to tetramethyl silane. For 31P{1H} NMR spectra, chemical shifts are given in ppm relative to H3PO4.4.6 Autoclave reactions
The catalytic runs were performed in 10 ml stainless steel autoclaves. The autoclaves were equipped with glass inlets in which the reactions took place. The autoclaves containing iodoform (CHI3), triphenylphosphine (PPh3) and para-toluensulfonic acid monohydrate (p-TsOH·H2O) were evacuated under high vacuum for at least one hour and then charged with an argon atmosphere. Stock solutions of the catalyst precursor were prepared and transferred to the autoclave, which was then pressurized with CO2 and H2 (or CO, 13CO and D2 in the labelling experiments). The mixture was heated and stirred for the desired reaction time after which the autoclave was cooled down and the pressure was relieved. The obtained reaction mixture was analysed via gas chromatography with a flame ionization detector (GC FID) and a mass spectroscopy detector (GC MS).Catalytic tests were repeated two or more times. The corresponding error bars are shown in the graphs or tables or indicated in the text. Errors for side products are not indicated for simplicity reasons, but they are usually around ±2%.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank RWTH Aachen for funding this research. We would like to thank the Verband der Chemischen Industrie e.V. for the stipend provided to Jeroen T. Vossen and for financing this research. Open Access funding was provided by the Max Planck Society.References
- J. Kubitschke, H. Lange and H. Strutz, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2014, pp. 1–18 Search PubMed.
- M. Hoelscher, C. Guertler, W. Keim, T. E. Mueller, M. Peters and W. Leitner, Z. fur Naturforsch. B, 2012, 67, 961–975 CrossRef CAS.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- E. A. Quadrelli, G. Centi, J. L. Duplan and S. Perathoner, ChemSusChem, 2011, 4, 1194–1215 CrossRef CAS PubMed.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711 RSC.
- K. J. Edgar, in Encyclopedia of Polymer Science and Technology, John Wiley & Sons, Inc., 2002, pp. 129–158 Search PubMed.
- Z. W. Wicks, in Encyclopedia of Polymer Science and Technology, John Wiley & Sons, Inc., 2007 Search PubMed.
- M. Szilagyi, in Patty's Toxicology, John Wiley & Sons, Inc., 2012, pp. 471–532 Search PubMed.
- J. P. Lange, R. Price, P. M. Ayoub, J. Louis, L. Petrus, L. Clarke and H. Gosselink, Angew. Chem., Int. Ed., 2010, 49, 4479–4483 CrossRef CAS PubMed.
- F. Röhrscheid, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2000 Search PubMed.
- H. Koch and W. Haaf, Angew. Chem., 1958, 70, 311–311 Search PubMed.
- W. Reppe and H. Kröper, Liebigs Ann., 1953, 582, 38–71 CrossRef CAS.
- U.-R. Samel, W. Kohler, A. O. Gamer, U. Keuser, S.-T. Yang, Y. Jin, M. Lin and Z. Wang, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2014, pp. 1–20 Search PubMed.
- A. Haynes, Adv. Catal., 2010, 53, 1–45 CAS.
- T. W. Dekleva and D. Forster, Mechanistic aspects of transition-metal-catalyzed alcohol carbonylations, Vol. 34, 1986 Search PubMed.
- J. Hjortkjaer and J. C. Aerbo Jørgensen, J. Mol. Catal., 1978, 4, 199–203 CrossRef CAS.
- J. Hjortkjaer and J. C. E. Jorgensen, J. Chem. Soc., Perkin Trans. 2, 1978, 763–766 RSC.
- S. B. Dake, D. S. Kolhe and R. V. Chaudhari, J. Mol. Catal., 1984, 24, 99–113 CrossRef CAS.
- B. R. Sarkar and R. V. Chaudhari, Catal. Surv. Asia, 2005, 9, 193–205 CrossRef CAS.
- R. S. Ubale, A. A. Kelkar and R. V. Chaudhari, J. Mol. Catal. A: Chem., 1997, 118, 9–19 CrossRef CAS.
- C. Carlini, M. Di Girolamo, M. Marchionna, A. M. R. Galletti and G. Sbrana, Stud. Surf. Sci. Catal., 1998, 119, 491–496 CrossRef CAS.
- B. J. Daniel, B. P. Gracey and J. G. Sunley, WO2009077726A1, GB, 2009.
- K. Matsushita, T. Komori, S. Oi and Y. Inoue, Tetrahedron Lett., 1994, 35, 5889–5890 CrossRef CAS.
- Q. Cao, N. L. Hughes and M. J. Muldoon, Chemistry, 2016, 22, 11982–11985 CrossRef CAS PubMed.
- D. C. Roe, R. E. Sheridan and E. E. Bunel, J. Am. Chem. Soc., 1994, 116, 1163–1164 CrossRef CAS.
- E. Amadio, Z. Freixa, P. W. N. M. van Leeuwen and L. Toniolo, Catal. Sci. Technol., 2015, 5, 2856–2864 RSC.
- L. Wu, X. Fang, Q. Liu, R. Jackstell, M. Beller and X.-F. Wu, ACS Catal., 2014, 4, 2977–2989 CrossRef CAS.
- M. Sakamoto, I. Shimizu and A. Yamamoto, Bull. Chem. Soc. Jpn., 1996, 69, 1065–1078 CrossRef CAS.
- K. Dong, R. Sang, J. Liu, R. Razzaq, R. Franke, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 6203–6207 CrossRef CAS PubMed.
- N. Tsumori, Q. Xu, Y. Souma and H. Mori, J. Mol. Catal. A: Chem., 2002, 179, 271–277 CrossRef CAS.
- B. R. Sarkar and R. V. Chaudhari, Catal. Today, 2012, 198, 154–173 CrossRef CAS.
- E. Iglesia, J. G. Sunley, D. J. Law and A. Bhan, US20080146833A1, The University of California, USA; BP Chemicals Ltd. USA, 2008, p. 5.
- Z. Zhang, J.-H. Ye, T. Ju, L.-L. Liao, H. Huang, Y.-Y. Gui, W.-J. Zhou and D.-G. Yu, ACS Catal., 2020, 10, 10871–10885 CrossRef CAS.
- Z. Fan, Y. Yi and C. Xi, Asian J. Org. Chem., 2022, 11, e202200207 CrossRef CAS.
- W. Leitner and J. Klankermayer, Science, 2015, 350, 629–630 CrossRef PubMed.
- X. Zhang, W.-Z. Zhang, L.-L. Shi, C.-X. Guo, L.-L. Zhang and X.-B. Lu, Chem. Commun., 2012, 48, 6292–6294 RSC.
- M. Juhl, S. L. R. Laursen, Y. Huang, D. U. Nielsen, K. Daasbjerg and T. Skrydstrup, ACS Catal., 2017, 7, 1392–1396 CrossRef CAS.
- Y. Liu, J. Cornella and R. Martin, J. Am. Chem. Soc., 2014, 136, 11212–11215 CrossRef CAS PubMed.
- F. Atsushi, G. Naotaka, K. Norikazu, H. Masafumi and K. Sanshiro, Chem. Lett., 1995, 24, 567–568 CrossRef.
- F. Juliá-Hernández, T. Moragas, J. Cornella and R. Martin, Nature, 2017, 545, 84–88 CrossRef PubMed.
- R. Martin Romo, F. Julia Hernandez and J. Cornella, Catalytic carboxylation of activated alkanes and/or olefins with carbon dioxide, WO2018010932A1, 2018 Search PubMed.
- A. L. Lapidus, S. D. Pirozhkov and A. A. Koryakin, Catalytic Synthesis of propionic acid by carboxylation of ethylene with carbon dioxide, Plenum Publishing Corporation, 1979 Search PubMed.
- H. Hoberg, Y. Peres, C. Krüger and Y.-H. Tsay, Angew. Chem., 1987, 99, 799–800 CrossRef CAS.
- A. Tortajada, R. Ninokata and R. Martin, J. Am. Chem. Soc., 2018, 140, 2050–2053 CrossRef CAS PubMed.
- T. G. Ostapowicz, M. Schmitz, M. Krystof, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2013, 52, 12119–12123 CrossRef CAS PubMed.
- M. Cui, Q. Qian, J. Zhang, C. Chen and B. Han, Green Chem., 2017, 19, 3558–3565 RSC.
- Q. Qian, J. Zhang, M. Cui and B. Han, Nat. Commun., 2016, 7, 11481 CrossRef PubMed.
- Y. Zhang, Y. Wang, Q. Qian, Y. Li, B. B. Asare Bediako, J. Zhang, J. Yang, Z. Li and B. Han, Green Chem., 2022, 24, 1973–1977 RSC.
- J. T. Vossen, A. J. Vorholt and W. Leitner, ACS Sustainable Chem. Eng., 2022, 10, 5922–5931 CrossRef CAS.
- B. G. Frederick, G. Apai and T. N. Rhodin, J. Am. Chem. Soc., 1987, 109, 4797–4803 CrossRef CAS.
- A. I. M. Keulemans, A. Kwantes and T. van Bavel, Recl. Trav. Chim. Pays-Bas Belg., 1948, 67, 298–308 CrossRef CAS.
- H. Koch and W. Haaf, Liebigs Ann., 1958, 618, 251–266 CrossRef CAS.
- R. Noyori and O. Takeshi, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS PubMed.
- Y. Chi, W. Tang and X. Zhang, in Modern Rhodium-Catalyzed Organic Reactions, WILEY-VCH Verlag GmbH & Co. KGaA, 2005, pp. 1–31 Search PubMed.
- M. J. Burk, T. G. P. Harper, J. R. Lee and C. Kalberg, Tetrahedron Lett., 1994, 35, 4963–4966 CrossRef CAS.
- R. R. Schrock and J. A. Osborn, J. Chem. Soc. D, 1970, 567–568 RSC.
- V. Polo, R. R. Schrock and L. A. Oro, Chem. Commun., 2016, 52, 13881–13884 RSC.
- P. Etayo and A. Vidal-Ferran, Chem. Soc. Rev., 2013, 42, 728–754 RSC.
- Y. Zhang, J. Zhang, Y. Wang, Y. Li, J. He, Y. Wang, L. Zhang, Z. Wang, Q. Qian and B. Han, Organometallics, 2023, 42, 2312–2318 CrossRef CAS.
- M. Poliakoff, W. Leitner and E. S. Streng, Faraday Discuss., 2015, 183, 9–17 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional graphics, detailed experimental procedures and analytical data. See DOI: https://doi.org/10.1039/d4gc01732c |
| This journal is © The Royal Society of Chemistry 2024 |