
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic graphitization of pyrolysis oil for anode application in lithium-ion batteries†
Shaikat Chandra
Dey
 a,
Lillian
Lower
b,
Trevor
Vook
b,
Md. Nazrul
Islam
a,
William Joe
Sagues
b,
Sang-Don
Han
cd,
Mark R.
Nimlos
e,
Stephen S.
Kelley
a and
Sunkyu
Park
*a
a,
Lillian
Lower
b,
Trevor
Vook
b,
Md. Nazrul
Islam
a,
William Joe
Sagues
b,
Sang-Don
Han
cd,
Mark R.
Nimlos
e,
Stephen S.
Kelley
a and
Sunkyu
Park
*a
aDepartment of Forest Biomaterials, North Carolina State University, Campus Box 8005, Raleigh, NC 27695, USA. E-mail: sunkyu_park@ncsu.edu
bDepartment of Biological and Agricultural Engineering, North Carolina State University, 3110 Faucette Dr., Raleigh, NC 27695, USA
cDepartment of Chemistry, Sejong University, 209 Neungdong-ro, Seoul, 05006, Republic of Korea
dMaterials, Chemical, and Computational Science Directorate, National Renewable Energy Laboratory, 15013 Denver West Parkway, Golden, CO 80401, USA
eBioenergy Science and Technology Directorate, National Renewable Energy Laboratory, 15013 Denver West Parkway, Golden, CO 80401, USA
First published on 2nd July 2024
Abstract
Graphite demand is increasing rapidly due to the popularity of electric vehicles (EVs) and mobile devices. Lithium-ion batteries (LIBs) are the power source of EVs and mobile devices and the anodes of LIBs are mostly made of graphite, which is currently produced in an unsustainable and costly manner. For the first time, battery-grade biographite is produced from a renewable and sustainable precursor, biomass-derived pyrolysis oil, using iron as the graphitizing catalyst. Considering battery-grade graphite production at scale, pyrolysis oil offers several advantages as feedstock over biomass in terms of higher carbon density, low inorganics, easy handling, and low transportation cost. Catalytic graphitization of pyrolysis oil can be induced at low temperature (∼1100 °C). Herein, catalytic graphitization using iron powder was carried out under relatively moderate conditions (1500 °C) to reduce costs and environmental impact as compared to incumbent technologies. After graphitization, the residual catalyst was removed by refluxing the biographite + iron solid mixture with hydrochloric acid. The efficiency of iron removal was studied using the X-ray fluorescence (XRF) technique. The structural and morphological properties of biographites were assessed via X-ray diffraction (XRD), Raman spectroscopy, and high-resolution transmission electron microscopy (HR-TEM). The in situ XRD experiments provided a mechanistic understanding of the iron-catalyzed graphitization process. In addition, the effect of pyrolysis oil aging on the biographite crystalline structure was elucidated. The optimal biographite sample demonstrated impressive electrochemical performance with a reversible capacity of 350 mA h g−1, an initial coulombic efficiency of 90.2%, and minimal capacity loss over 100 cycles when applied as an LIB anode, thereby making pyrolysis oil-derived biographite potentially competitive with commercially available battery-grade graphite.
Introduction
Although lithium-ion batteries (LIBs) were initially developed for commercial use in portable electronic devices, their applications have since expanded to include a wide range of other products and technologies, including electric vehicles (EVs) and energy storage systems (ESSs). LIBs have generated significant interest as a very promising energy storage technology owing to their high energy density, high coulombic efficiency, minimal self-discharge, negligible memory effect, high open circuit voltage, and extended operational lifespan.1 The EV revolution has begun, which could potentially mitigate climate change by reducing greenhouse gas (GHG) emissions and LIBs are considered to be the major power source for EVs.2 Therefore, it is necessary to ensure a sustainable supply of LIB materials. The commercially used anode material for LIBs is graphite as it offers several advantages such as good gravimetric capacity (theoretical maximum is 372 mA h g−1), a low negative redox potential around 0.1 V (vs. Li/Li+, hereafter), long cycle life, and low-voltage hysteresis, resulting in high voltage and energy efficiency.3,4 Most of the demand for anode grade graphite is likely to come from the battery industry. For instance, Tesla is considered to be a prominent producer of LIBs within the United States (U.S.). In 2018, the Gigafactory located in Nevada successfully manufactured approximately 20 GWh of batteries, requiring an estimated 10 kilotons of graphite. The mass in question represents 50% of the whole graphite use in battery production inside the U.S. over the year 2018.5,6 Based on a projection by the World Bank, the anticipated demand for graphite in 2050 from energy storage batteries (particularly LIBs) is expected to be five-fold greater than the cumulative production of natural graphite in 2018. This forecast is based on a scenario that imposes restrictions on climate change, limiting the temperature to a maximum of two degrees. Due to the rapid growth of LIBs, natural and synthetic graphite production is predicted to double in 2028 globally.5 In the U.S., LIBs commonly consist of a combination of synthetic and natural graphite. Synthetic graphite exhibits dominance because of its superior uniformity, quality, and inherent properties.7 The utilization of synthetic graphite in the U.S. now stands at 83%, with no existing domestic mining operations for natural graphite.5 Moreover, graphite mining from geographically constrained natural deposits and its subsequent purification by toxic hydrofluoric acid (HF) make natural graphite less attractive for battery applications. The overall mining process for natural graphite is energy-intensive and time-consuming.8 Additionally, the recycling of LIB components in the U.S. is cathode-focused and it is assumed that there will be no graphite recycling in the near future at scale.9,10 As a result, synthetic graphite is receiving increased attention to meet demand. However, the production of synthetic graphite is also not a straightforward process. Synthetic graphite is produced commercially from low-sulfur petroleum distillation residues (petroleum coke and coal tar pitch) through several high temperature steps including delayed coking (∼500 °C), calcination (∼1200 °C), and graphitization (∼3000 °C).11 The slow production rate with the high cost and use of sulfur-containing petroleum residues as feedstock stimulates the search for alternative pathways and feedstocks for producing synthetic graphite.Graphitization is a process in which disordered carbon materials are converted into three-dimensional (3D) graphite through atomic displacement due to the application of heat. Heat treatment is the most important parameter for macromolecular rearrangement into a graphitic structure. The degree of crystallinity, i.e., the formation of an ordered carbon structure, increases with the increase in the heat treatment temperature. The graphitization reaction is carried out under the flow of an inert gas (mostly nitrogen). This process usually requires very high temperatures (∼3000 °C) and longer time when no catalyst is used. Therefore, catalytic graphitization has been developed to lower the graphitization temperature and shorten the production time without compromising graphite quality.12 An attractive feature of the catalytic process is that it can convert both the graphitizing and non-graphitizing carbons into graphite. Biomass materials are inherently non-graphitizing, and thus catalysts are typically needed to convert biomass into highly crystalline graphite.7 Numerous transition metals such as iron, cobalt, nickel, etc. can utilize their d-orbital electron vacancies to form metastable carbides that can promote the catalytic graphitization process. To adequately explain catalytic graphitization, two mechanisms have been proposed: (i) dissolution–precipitation mechanism and (ii) formation–decomposition mechanism. According to the first mechanism, the amorphous carbon (formed during the pyrolysis of biomass) first dissolves into molten metal or metal carbide and later precipitates as graphite. According to the second method, carbon forms carbide intermediates with metals, which decompose at certain temperatures to leave graphite. The quality of the product is strongly influenced by the type and amount of catalyst.13,14 To date, iron has been most commonly used for the catalytic graphitization of carbonaceous feedstocks as it offers several advantages. Iron is a prevalent element in the Earth's crust, constituting around 5% of its composition. This metal, together with its oxides and other salts, is widely accessible and economically affordable. Various naturally occurring oxides or ores have the potential to function as catalysts without necessitating alterations. Iron is currently regarded by regulatory authorities as a metal with minimal safety concerns due to its relatively low toxicity.15 Out of many possible forms of iron such as pure metal, salts, oxides, and organometallics, the highest degree of graphitization of biomass is achieved with a metallic iron powder catalyst. Frankenstein et al. graphitized coffee powder using four different catalysts (Fe, FeCl3, Fe(NO3)3, and Fe2O3), and tested the electrochemical performance of the corresponding biographites as anode active materials in LIB half-cells. Among the four catalysts used, Fe-catalyzed biographite demonstrated the highest reversible capacity, coulombic efficiency, and rate performance than the biographites derived with the other three catalysts.16 Irrespective of the iron precursor types, the catalytic graphitization is controlled by the dissolution–precipitation mechanism or formation–decomposition mechanism involving iron (Fe) and/or iron carbide (Fe3C). Iron salts are decomposed gradually into iron oxide, iron carbide, and metallic iron due to heat treatment. As a result, it is beneficial to introduce the iron catalyst in metallic powder form to accelerate the graphitization process. Moreover, the volatility of iron salts e.g., iron chloride (FeCl3) leads to a lower degree of biomass graphitization. Another notable advantage of using iron powder is that it can be dry-mixed physically and quickly with the biomass feedstock. On the other hand, iron salts are mixed via the wet-impregnation method using solvents, which requires a considerable period of time, such as hours or even days.
A wide variety of carbonaceous feedstocks have been catalytically converted into graphite at moderate temperatures. These feedstocks include petroleum coke,17,18 anthracite,19 hardwoods,20 softwoods,20 lignin,21 cellulose,22,23 bacterial cellulose,24 glucose,20 chitin,25 chitosan,26 algae,27 fiberboard,28 waste paper towel,29 oil palm frond,30 plant leaves,31etc. For a better understanding of the effects of feedstocks on the graphite properties including electrochemical performance as a LIB anode, the readers are referred to the recent review article published by Lower and Dey et al.7 To the best of our knowledge, there is no published study on the catalytic conversion of pyrolysis oil into LIB-grade graphite. The fast pyrolysis (FP) of biomass generates three main products: bio-char (solid), pyrolysis oil (liquid), and light gas. The fast quenching of the vapors produced during fast pyrolysis results in the formation of pyrolysis oil. Depending on the reaction conditions, the yield of pyrolysis oil varies from 50 to 70 wt% on a dry biomass basis. Higher lignin content in biomass results in a lower yield of pyrolysis oil, while higher cellulose content results in a higher yield. Pyrolysis oil is also known as bio-oil, bio-crude, wood oil, wood distillate, wood liquid, pyrolytic tar, pyroligneous acid, etc. Pyrolysis oil is a dark-brown liquid containing a wide variety of oxygenated compounds (alcohols, acids, aldehydes, ketones, sugars, esters, ethers, phenols, furans, etc.), which make it thermally unstable and less attractive as engine fuel due to its low heating value. The water content of pyrolysis oil is also very high, and it exists as a microemulsion. The characteristics of pyrolysis oil are determined by a wide variety of parameters, including the rate at which the reactor is heated, the amount of time that biomass spends inside the reactor, the temperature, the size of the biomass particles, and the type of biomass that is utilized. The high carbon content motivates research studies to valorize pyrolysis oil into various types of carbon materials. Considering battery-grade graphite production at scale, pyrolysis oil offers several advantages as feedstock over biomass such as high carbon density, low inorganics, easy handling, and low transportation cost.32–38
For the first time, pyrolysis oil was converted into biographite using reduced iron powder as the catalyst at a moderate temperature (1500 °C). The process was optimized and the biographite products were characterized for graphite crystallinity and electrochemical energy storage performance. In addition, in situ XRD provided insights into the mechanism of iron-catalyzed graphitization, and the effect of pyrolysis-oil aging was explored.
Experimental
Materials
Hardwood-derived pyrolysis oil, feedstock for biographite, was supplied by Ensyn (Ontario, Canada). Iron powder catalyst was purchased from Sigma-Aldrich (CAS: 7439-89-6, particle size <10 μm). Concentrated hydrochloric (HCl) acid (37 wt%) was obtained from Fisher Scientific. To compare the quality of biographite, several commercial graphite samples were purchased (Asbury, Sigma-Aldrich, Anovion, Thermo Scientific, Fasco Epoxies, Godora, Greatcolor, and raw graphite). For electrode preparation, conductive carbon black (C45, Timcal), polyvinylidine difluoride binder (Sigma-Aldrich), and N-mehyl-2-pyrrolidone solvent (Sigma-Aldrich) were used. All the chemicals were used without further purification.Determination of fixed carbon (FC) content of pyrolysis oil
The basis for the catalyst loading was the FC content of the pyrolysis oil as determined by heating the pyrolysis oil sample (∼3.5 g) to 1200 °C in a tubular furnace (GSL-1500X, MTI Corporation, Richmond, California, U.S.) under an inert nitrogen atmosphere (flow rate: 100 mL min−1). During a high temperature graphitization process, the pyrolysis oil gradually loses most of its fraction as volatiles and the remaining mass i.e., FC is available to interact with the catalyst to be converted into biographite. The sample holder used in the experiments was made of alumina (dimension: 100 × 20 × 20 mm).Catalytic graphitization of pyrolysis oil
Before catalytic graphitization, pyrolysis oil (∼7 g) was mixed uniformly with different proportions of iron powder to FC ratios at room temperature. To demonstrate the catalytic effect of iron, a control experiment was also conducted under the same pyrolysis conditions without any iron. A total of 12 catalytic experiments were performed with iron loading of 0.25–3.00 times the FC of pyrolysis oil. Catalytic graphitization was carried out in the same tubular furnace under inert nitrogen flow at 1500 °C using the following ramp rates: 25–600 °C (4 °C min−1); 600–1500 °C (2 °C min−1); 1500–600 °C (2 °C min−1); 600–25 °C (4 °C min−1). The biographite preparation method from pyrolysis oil using iron catalyst is schematically shown in Fig. 1. | ||
| Fig. 1 Steps in the preparation of biographite from pyrolysis oil using reduced iron powder catalyst. (a) Fresh pyrolysis oil, (b) mixing iron powder with pyrolysis oil, (c) uniformly mixed pyrolysis oil and iron powder, (d) pyrolysis oil + iron mixture before graphitization, (e) biographite + iron, (f) powdered sample (biographite + iron) before acid washing, (g) dissolution of iron into HCl as iron chloride, (h) separation of graphite from the liquid phase (rich in iron chloride) via filtration, (i) final biographite after drying. | ||
Biographite purification via acid washing
After graphitization, the residual catalyst was separated by washing the biographites + iron mixture with concentrated HCl (37 wt%) under reflux for 3 h. Before acid washing, the biographites + iron mixture was ground in a mortar to make fine powder for facilitating acid leaching. Acid washing converts the metallic iron into iron chloride with the co-production of hydrogen gas (H2). The solid biographite product was separated from the liquid phase (iron chloride + HCl) via vacuum filtration. The purified biographite was dried in an oven at 105 °C for 4 h before further analysis.X-ray diffraction (XRD)
XRD was used as the preliminary screening tool to analyze the the crystalline nature of biographites. In particular, the characteristic reflection at 2θ = ∼26.5° corresponding to the (002) crystallographic plane was analyzed using the diffraction patterns. Room temperature XRD patterns of the purified biographites (after acid washing) were recorded on a PANalytical Empyrean X-ray diffractometer fitted with a copper (anode) X-ray generator (λ = 1.5406 Å) in the continuous scanning mode (generator voltage: 45 kV, tube current: 40 mA, scan range: 20–90°, step size: 0.026°). High temperature (1200 °C) in situ XRD patterns were also recorded using a HTK1200N heating stage under a non-ambient environment (nitrogen atmosphere). A heating and cooling rate of 4 °C min−1 was maintained during in situ high temperature XRD experiments. To potentially prevent excessive mass loss during in situ XRD experiment, the pyrolysis oil + iron mixture was preheated to 500 °C for 1 h under nitrogen flow. The purpose of the preheating was to remove most of the volatiles as the extensive mass loss reduces the intensity of the XRD peaks.39 To measure the crystallite sizes (Lc and La) of biographites, the Debye–Scherrer formula (eqn (1)) was used and Line-Profile Analysis Software (LIPRAS) was used to calculate the full width at half maximum (FWHM) of the characteristic diffraction peaks of biographite i.e., β.29,40 To pinpoint the Lc and La values, instrumental broadening was subtracted from the desired signal before calculation. Another essential parameter i.e., the inter-planar distance (d002) was calculated using Bragg's law (eqn (2)).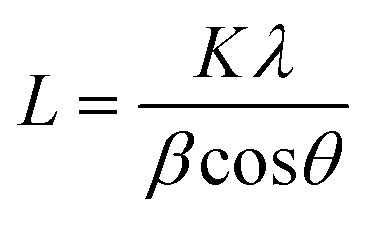 | (1) |
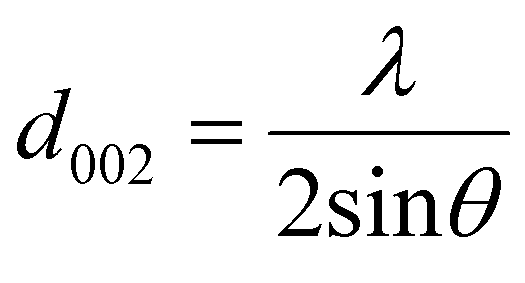 | (2) |
where θ indicates the Bragg angle in degrees, λ is the X-ray wavelength (1.5406 Å), and K is a constant called shape factor. For Lc, the value of K was considered to be 0.9 while for La the K value was 1.84. The characteristic (002) and (100) peaks at the respective 2θ positions of ∼26.5 and 42.3° were used to calculate Lc and La values, respectively. To quantify phase composition, Rietveld refinement analysis was applied to selected in situ XRD patterns using the GSAS software program.
Raman spectroscopy
Raman spectra of the biographite samples were acquired on a Horiba XploRA PLUS Confocal Raman Microscope using green laser radiation (532 nm, depolarized beam) at room temperature. Spectra were collected at different locations of the biographites and the average intensity ratio of D to G band (ID/IG) was calculated. Ten accumulations were acquired at each point and the exposure time was 30 s. The hole and slit sizes were 500 and 200 μm, respectively.Scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS)
The morphology of the biographites was analyzed by recording microphotographs on a FEI Quanta 650 SEM in the backscattered electron imaging mode. The biographite powder samples were sprinkled across ∼25 mm2 segments of double-sided carbon tape, which were fixed to a single Al sample plate. Coverage was >95% by eye. Sample coating to reduce charging was not required. The EDS data were acquired with an Oxford Instruments Silicon Drift Detector with 150 mm window. Images and spectra were acquired with 15 kV electrons.High-resolution transmission electron microscopy (HR-TEM)
HR-TEM images of the biographites were captured on a Titan G2 80–300 kV TEM operated at 200 kV. Simultaneous annular dark field (ADF) and bright field (BF) images were acquired in STEM mode with a convergence angle of around 19 mrad, and a beam current of ∼90 pA. The EDS maps were acquired under similar conditions but using beam currents of ∼400 pA. The TEM images were captured using a Gatan OneView camera in 4K resolution. Prior to recording the images, the samples were deposited onto a thin carbon TEM support grid.X-ray fluorescence (XRF)
Elemental concentrations were determined by loading the biographite material in a 28 mm sample cup with a thin (2.5 μm) Mylar foil. Analysis was done on a Malvern-Panalytical Epsilon 3× ED-XRF instrument using the OMNIAN (internal standards) methodology. Finite thickness effects and effects due to fluorescence volume geometry were considered in the analysis. The data were normalized to 100% using appropriate balance composition. Compton intensity was used to validate the analysis. It is worth noting that the Mylar foil has typical impurities at the ppm-level of Ca, P, Sb, Fe, and Zn.X-ray photoelectron spectroscopy (XPS)
The surface composition of oxygen (O) and carbon (C) was characterized using X-ray photoelectron spectroscopy (XPS) with an XPS/UVS-SPECS system featuring a PHOIBOS 150 analyzer under a pressure of approximately 3 × 10−10 mbar. The instrument utilized Mg Kα X-ray (hν = 1253.6 eV) and Al Kα X-ray (hν = 1486.7 eV) sources. The data were acquired using the Mg Kα X-ray source, which operated at 10 kV and 30 mA (300 W) and analyzed with the CasaXPS software. XPS spectra of the survey scan were recorded with a pass energy of 24 eV in a 0.5 eV step. The C 1s peak was used as an internal reference with a binding energy of 285 eV.Electrochemical performance testing
The electrochemical behavior and performance of the optimal biographite sample (2.50 × FC iron loading) were analyzed and evaluated as anode active material for LIBs using CR2032 coin-type half-cells. The electrode was prepared using 85 wt% biographite (active material), 5 wt% conductive carbon black, and 10 wt% polyvinylidine difluoride (PVDF) binder. For slurry preparation, N-mehyl-2-pyrrolidone (NMP) was used as a solvent. The well-mixed slurry was coated onto a current collector (copper foil), and then dried under vacuum for 12 h at 120 °C. The diameter of the punched electrodes was 14 mm having ∼3.3 mg cm−2 loading density of active material. The half-cells were assembled in an argon-filled glove box. Lithium metal (Li) was employed as the counter/reference electrode and 1.2 M lithiumhexafluorophosphate (LiPF6) in ethylene carbonate and ethyl methyl carbonate (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 wt% ratio) was used as the electrolyte. The electrodes were separated by a 19 mm diameter circular punch of Celgard 2325. Galvanostatic cycling tests were performed in the potential range of 0.005–1.5 V at different C-rates (1C corresponds to 350 mA g−1). Before cycling, the cells were allowed to rest at an open circuit voltage (OCV) for 12 h.
:7 wt% ratio) was used as the electrolyte. The electrodes were separated by a 19 mm diameter circular punch of Celgard 2325. Galvanostatic cycling tests were performed in the potential range of 0.005–1.5 V at different C-rates (1C corresponds to 350 mA g−1). Before cycling, the cells were allowed to rest at an open circuit voltage (OCV) for 12 h.
Results and discussion
Effect of temperature on biographite formation
The pyrolysis oil used in this study had a dark-brown appearance and was viscous in nature. Fig. 1 depicts various sequential steps in the formation of biographite starting from the raw feedstock (pyrolysis oil). The steps followed in the synthesis process were relatively straightforward, thereby demonstrating the simplicity and scalability of the process. The most important experimental variable that controls the graphite formation process is the temperature, as determined in our prior work.29 To understand the effect of temperature, preliminary catalytic graphitization experiments were carried out with 1.50 × FC iron loading at 1200, 1300, 1400, and 1500 °C. It was found that the crystallite size increased gradually with temperature (Fig. S1a†). To further investigate the effect of temperature, electrochemical performance of the biographites (1300, 1400, and 1500 °C) was tested as LIB anode in half-cells. The biographite prepared at 1200 °C was not tested for electrochemical performance due to low crystallite size. The reversible capacity of the three other biographite samples were found to be 285 (1300 °C), 325 (1400 °C), and 345 mA h g−1 (1500 °C) respectively (Fig. S1b†). The initial coulombic efficiency (ICE) values were calculated and found to be 60.6, 77.7, and 88.7% respectively (Fig. S1c†). Within the scope of these experiments, it can therefore be stated that 1500 °C was the optimal temperature for biographite synthesis. The tube furnace used in this study had a temperature limit of 1500 °C, and therefore, further experiments were only conducted at this temperature. To further improve the crystalline quality of biographite for battery applications, future studies should also consider running catalytic graphitization experiments at higher temperatures (>1500 °C).Optimization of iron loading and biographite characterization
For catalytic graphitization, it is important to study how the catalyst loading affects the graphite properties. As understood from Fig. 2, the control sample (no catalyst) demonstrated a broad diffraction pattern indicating disordered carbon formation. This control experiment supports the idea that the pyrolysis oil feedstock is non-graphitizable at 1500 °C without the help of a catalyst. Interestingly, the addition of a small proportion of iron helped sharpen the (002) peak and lower the degree of disorder. With further addition of iron, the overall diffraction patterns gradually became sharper. The intensity of the (002) peak enhanced significantly and progressively with the increase in catalyst loading. A closer inspection of the diffractogram at around 2.50 × FC iron loading indicates the formation of an optimal biographite similar to commercial graphite. Further addition of iron reduced the crystalline quality of biographites, especially at 3.00 × FC iron loading. Excessive iron loading negatively affected the graphitization process, as understood from the relatively broader diffraction patterns.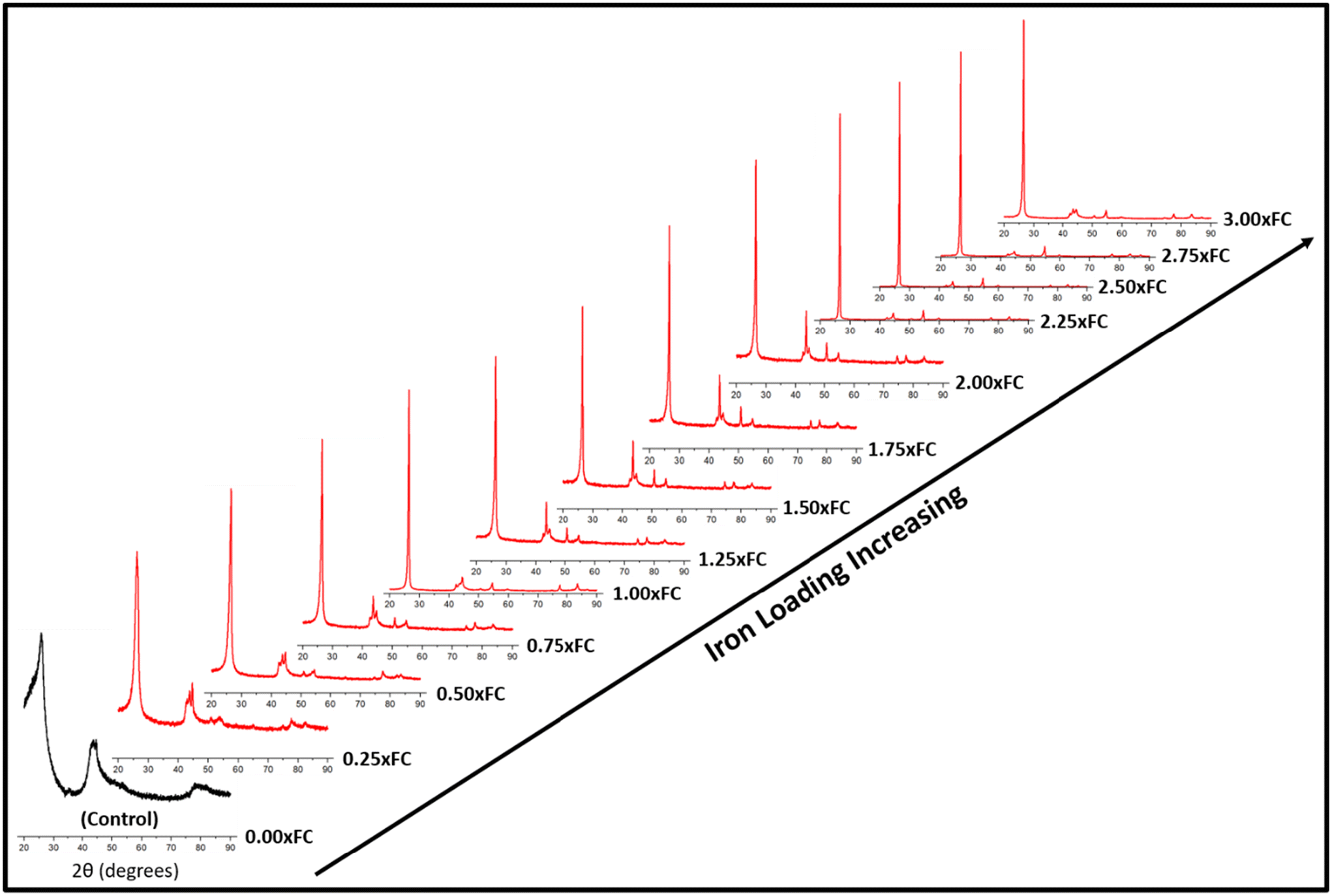 | ||
| Fig. 2 XRD patterns of biographites prepared at 1500 °C with different mass loading of iron (0–3 times the FC content of pyrolysis oil). | ||
To further explore the diffraction patterns, various important parameters were calculated, such as crystallite size along the c-direction (Lc), a-direction (La), and interlayer spacing (d002), which are listed in Table S1.† As understood from Table S1,† both Lc and La increased with increasing iron loading while d002 value decreased, which are the desired trends towards higher crystallinity. It is also worth noting that the intensity (I) of the (002) peak was the highest for the optimal biographite. To compare the crystalline structure of this biographite, XRD patterns of some commercial graphite samples were recorded, and the relevant parameters (I, Lc, La, and d002) were also calculated (Fig. S2†). The comparison demonstrates that the crystalline structure of the optimal biographite was similar to that of commercial graphite used for LIB anode applications (Sigma-Aldrich, Anovion, Asbury).
To support the XRD results, Raman spectra of the biographites were recorded, and the results are presented in Table S1† and Fig. 3. Graphitic carbon materials demonstrate two characteristic Raman peaks at ∼1350 cm−1 and ∼1581 cm−1. The peak at ∼1350 cm−1 (D-band) is assigned to the defects in the biographite (A1g symmetry) while the peak at ∼1581 cm−1 (G-band) corresponds to the ordered region in biographite. The G band is observed in all graphitic materials, serving as an indicator of the presence of sp2 hybridized carbon within the analyzed sample (E2g symmetry). The intensity ratio of these two bands (ID/IG) provided a good understanding of the degree of graphitization. For an ideal graphite, the D-band should be as minimum as possible. As understood from Table S1,† the ID/IG ratio declined with an increase in iron loading, and at 2.50 × FC iron loading the ratio showed a minimum, which supported the XRD observation.
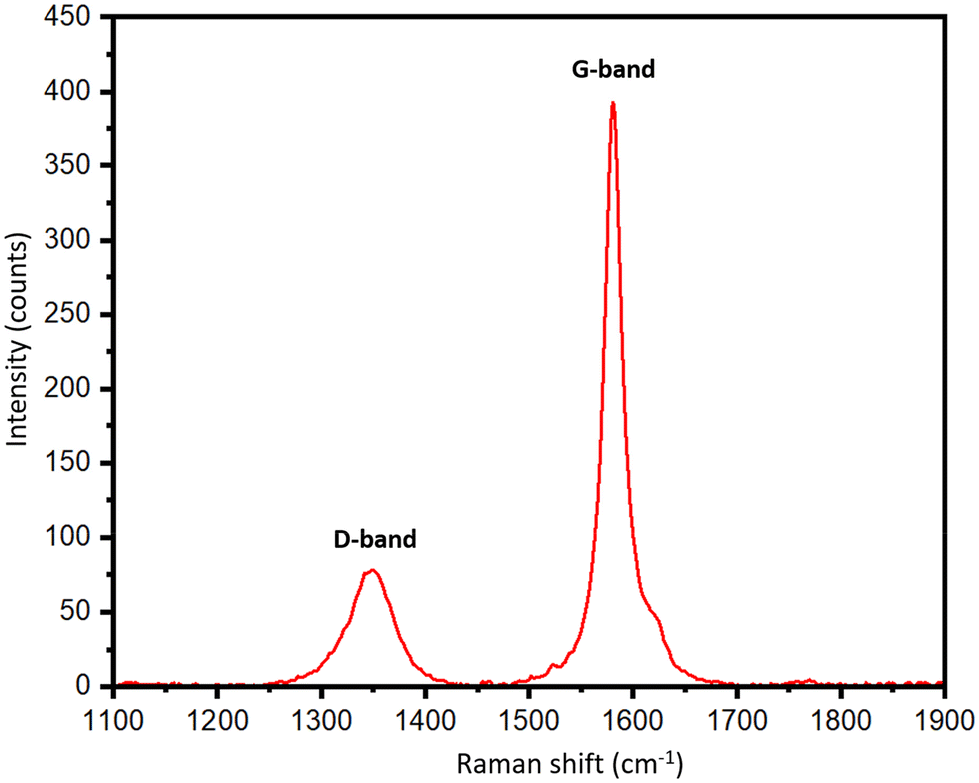 | ||
| Fig. 3 Raman spectrum of the optimal biographite made with 2.50 × FC iron loading. The result shown in the figure is an average of 10 different scans recorded at different positions of the biographite to get a representative idea of the defects and degree of graphitization. | ||
The surface morphology of the biographite was analyzed via recording the SEM images and the results are shown in Fig. 4a–c. As understood from Fig. 4a, the biographite showed platelet-like morphology and the size and shape distribution were not homogeneous. The surface structure was rugged and the particles were loosely packed. The sharp edge of the biographite particles was also seen (Fig. 4b). Fig. 4c reveals the presence of iron particles on the biographite surface. Iron showed bright spots on the SEM images as it has higher atomic mass than the lighter carbon (biographite) matrix. However, a closer inspection of the biographite surface revealed that the distribution of iron was not homogeneous. Iron did not seem bonded or aggregated in a particular manner to the biographite, but rather sprinkled in a very heterogeneous fashion. It should be emphasized that the size of the iron was found to be <200 nm while the reduced iron powder that was mixed with the feedstock (pyrolysis oil) was <10 μm. This significant reduction in particle size implied that iron powder underwent extensive phase change through melting and other possible interactions with carbon during the high temperature catalytic graphitization process. To further visualize the distribution of elements on biographite, EDS elemental mapping experiments were also performed (Fig. S3†). SEM imaging provided a good understanding of the external morphology of biographite, while it was required to record HR-TEM images to understand the internal structure. The layered parallel graphene sheet structure (onion-like) was clearly visible on the HRTEM images at high magnifications (Fig. 4d and e). The biographite sample was observed to consist of multilayer graphene. The orderly stacked graphene layers were clearly visible in the images and there was no strict core–shell type of structure. No defects were induced by iron during the catalytic graphitization process.41 The high vertical stacking indicated the formation of good quality crystalline biographite with layered structures suitable for better ion storage behaviors (e.g., ion intercalation/deintercalation) resulting in high reversible capacity. The TEM elemental mapping (Fig. 4f) showed the presence of trace amount of iron trapped inside the graphitic structure. Graphitic carbon matrix is shown in bright-green color while iron is shown as light-red color in the elemental mapping image. However, neither SEM nor TEM technique helped reveal the quantitative estimation of inorganics (including iron) in the biographite. Therefore, XRF analysis was conducted to have a clearer picture of the bulk elemental distribution in the structure of biographite (2.50 × FC iron loading) after acid washing. For a fair comparison, XRF analysis was also performed on the pyrolysis oil. The results of the XRF analyses are presented in Table S2.†
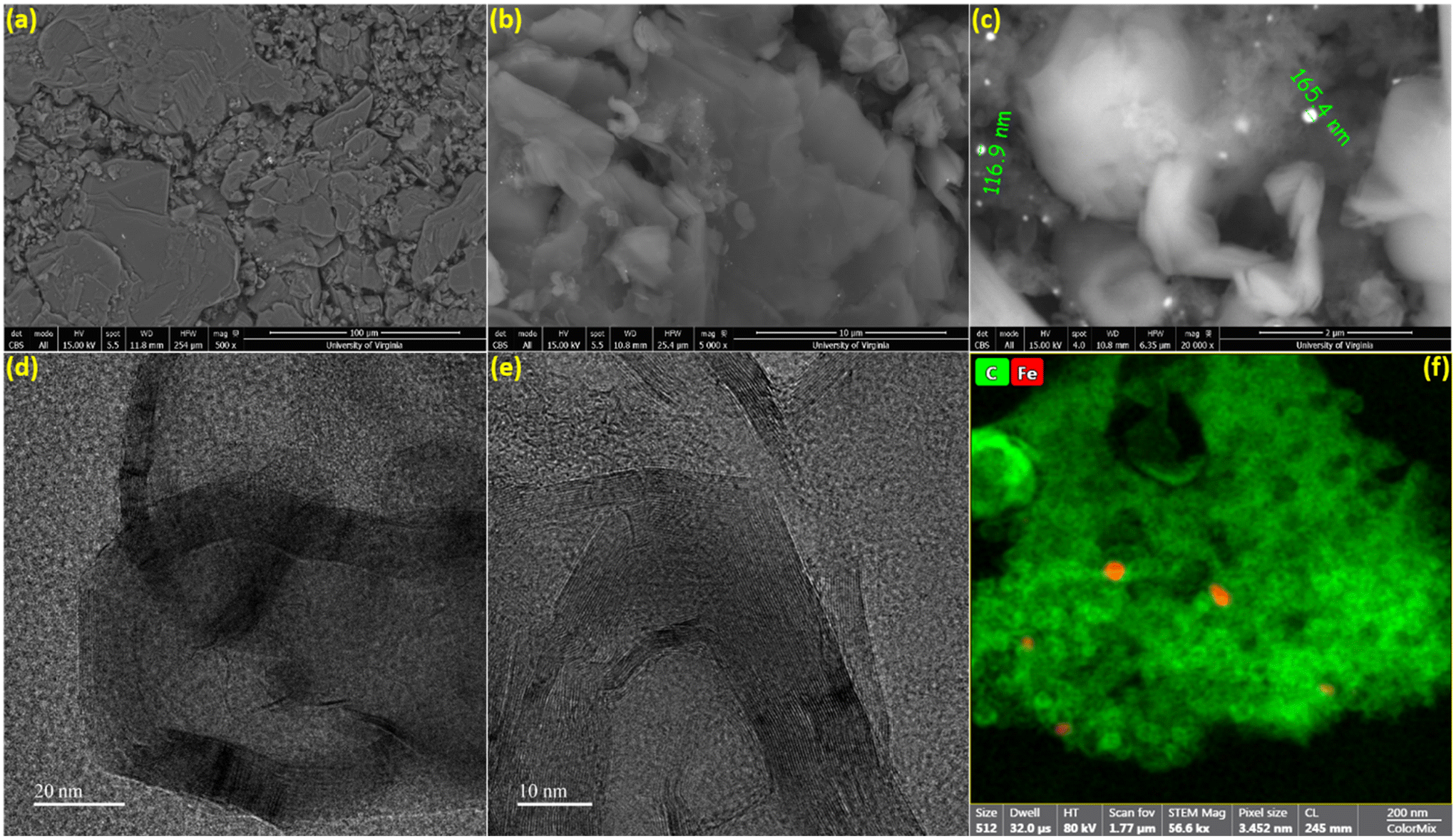 | ||
| Fig. 4 (a–c) SEM microphotographs and (d and e) HR-TEM images of biographite (2.50 × FC iron loading) at different magnifications, (f) HR-TEM elemental mapping of biographite (green: carbon; red: iron). | ||
Table S2† shows the presence of various inorganics in both pyrolysis oil and biographite. The XRF technique determined the composition of metals with confidence, while carbon was calculated as a balance component. Hydrogen and oxygen are also not detected by XRF. Therefore, the carbon fraction in the raw pyrolysis oil could be a combination of carbon, hydrogen, oxygen, nitrogen etc., i.e. organics. The pyrolysis oil contained trace amounts of heavier metallic elements, which are expected to have originated from the soil during the growth of plants, although the concentrations of metals in the pyrolysis oil are significantly lower than that in the raw biomass or pyrolysis-derived biochar. The low metals (ash) content of the pyrolysis oil makes it a particularly intriguing feedstock for battery-grade biographite production. During high temperature treatment, the volatiles are removed and the remaining carbon becomes biographite due to the action of heat and catalyst. For battery application, the carbon purity should be very high and the inorganics should be minimum. The final biographite had <0.15% inorganics, which originated from the pyrolysis oil as well as from the catalyst. Approximately 0.05% residual iron was found in the biographite after acid washing. For an improved electrode performance, future studies should attempt on ensuring a nearly complete removal of inorganics from biographite. This study performed only a single step acid refluxing. It has been shown in the literature that multiple step acid refluxing is helpful in efficient isolation of iron from the carbon matrix.42 To further explore the elemental distribution on the surface of the biographite, XPS analysis was performed and the results are shown in Fig. 5. The analysis depth of XPS is low typically <10 nm. The low-resolution survey spectrum (Fig. 5a) shows the presence of carbon (97.6%) and oxygen (2.4%) on the surface. The oxygen could potentially originate from the pyrolysis oil precursor or during the acid washing step. It has been shown that foreign elements influence the surface chemistry of carbon materials (e.g., graphite) and these foreign elements originate from the precursor or are introduced during the synthesis and post-synthesis steps. Hydrogen and oxygen are the most common foreign elements that are thought to predominately exist at the edges of graphene layers or at the dangling bonds of defects.43 The exact chemical nature of the carbon was investigated by fitting the high-resolution C 1s peak (Fig. 5b). The characteristic peaks were obtained at 284.44, 286.64, 287.94, and 290.85 eV respectively. The most prominent peak was obtained at 284.44 eV (95.3% peak area) corresponding to the sp2 hybridized graphitic carbon (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C). The other peaks such as 286.64 eV (C–O), 287.94 eV (CO), 290.85 eV (π–π* satellite bonds) are frequently present in the XPS spectrum of graphite. However, no peaks for C–H (285.0–285.2 eV) were recorded, which indicated that the biographite surface was free from C–H bonds.44 Additionally, no Fe 2p peak (>700 eV) was recorded in the spectrum, which implied that iron present on the surface was below the detection limit of the XPS analysis.45
C). The other peaks such as 286.64 eV (C–O), 287.94 eV (CO), 290.85 eV (π–π* satellite bonds) are frequently present in the XPS spectrum of graphite. However, no peaks for C–H (285.0–285.2 eV) were recorded, which indicated that the biographite surface was free from C–H bonds.44 Additionally, no Fe 2p peak (>700 eV) was recorded in the spectrum, which implied that iron present on the surface was below the detection limit of the XPS analysis.45
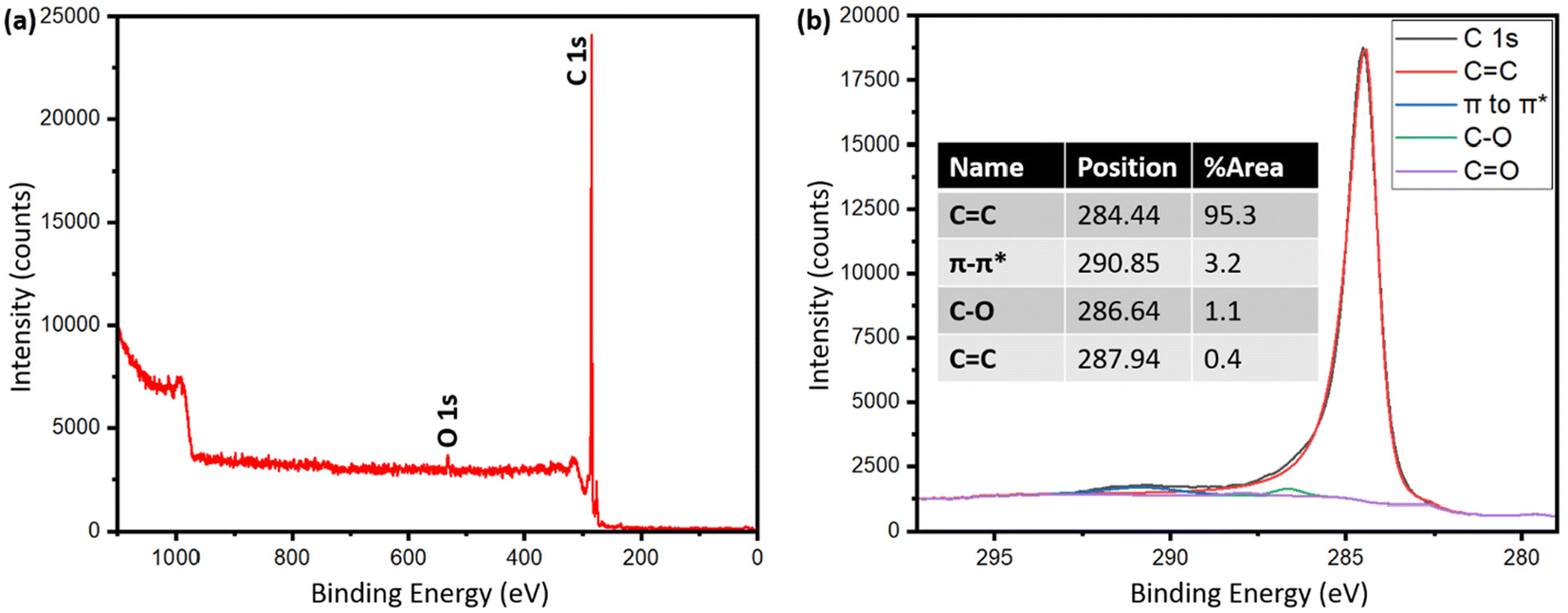 | ||
| Fig. 5 (a) XPS survey spectrum of biographite (2.50 × FC iron loading), and (b) high resolution C 1s peak after deconvolution. | ||
Mechanism of catalytic graphitization
The process of graphitization primarily involves the rearrangement of carbon atoms in the original substance, leading to the formation of a highly organized hexagonal lattice characteristic of graphite. The application of heat treatment at temperatures ∼3000 °C can induce these alterations by supplying sufficient energy to facilitate atomic rearrangement. However, addition of a catalyst can help form similar graphites at comparatively lower temperatures.46 To obtain an insight into the mechanism of catalytic graphitization, in situ XRD patterns were recorded during both the heating and cooling steps. As understood from Fig. 6a, the α-iron (bcc, ferrite) peak at ∼44.5° remained unaffected up to 700 °C during heating. At 800 °C, two new peaks at ∼43, and 50° appeared indicating the transformation of α-iron into γ-iron (fcc, austenite).47 For pure iron, this transformation occurs at 912 °C as understood from the iron-carbon phase diagram. The dissolution of carbon in iron as an interstitial solid solution helped the transition of iron from α to γ form at lower temperature. The γ-iron was stable until the heating temperature reached 1200 °C. At 1200 °C, γ-iron started melting partially, which was significantly lower than the melting point of pure iron (i.e., 1538 °C). This significant reduction in melting temperature strongly supported the presence of carbon in γ-iron in the form of solid interstitial solution.47 Interestingly, the biographite peak also emerged noticeably at 1200 °C, indicating that the biographite formation was predominantly controlled by the melting process.48 The molten iron dissolves the amorphous carbon from the pyrolyzed feedstock and later becomes supersaturated with carbon. As a result, carbon precipitates out of the molten iron as crystalline (ordered) graphite driven by the free energy difference (driving force for the process) between the disordered and ordered carbons. It is thermodynamically possible that both the carbon dissolution and precipitation from the molten iron as graphite occurs at the same temperature.48 The molten state of iron is an advantage to accelerate this process as iron melt can tunnel through the amorphous carbon matrix enhancing carbon dissolution. This mechanism is similar to the “kish” formation in steel making industry.46 The precipitation results in the encapsulation or coating of iron by graphitic carbon in the form of core–shell microstructure.41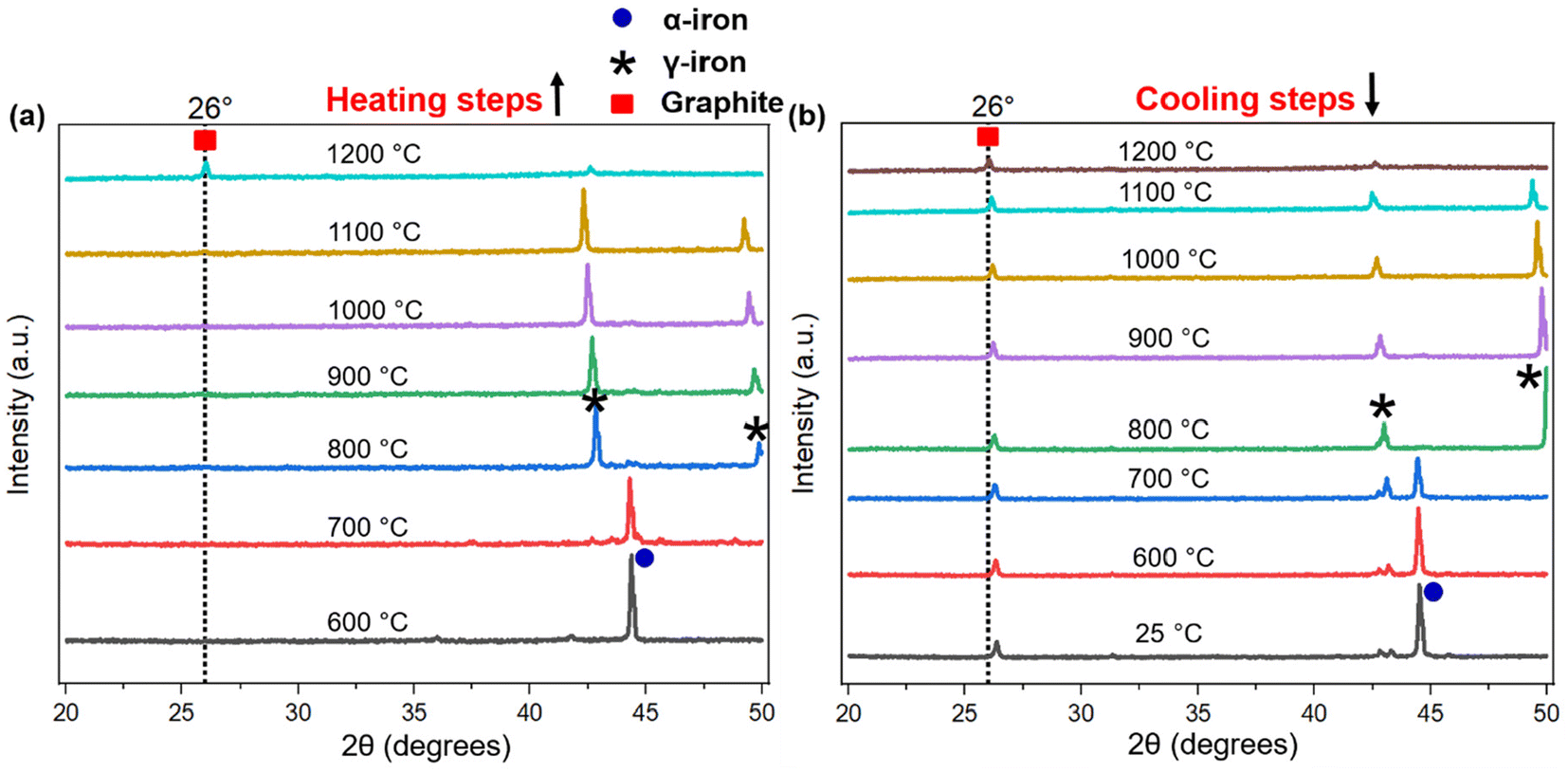 | ||
| Fig. 6 The in situ XRD patterns of biographite during (a) heating and (b) cooling steps. The in situ XRD experiments were conducted with the optimal iron loading (2.50 × FC), equivalent to ∼1:0.67 ratio of pyrolysis oil to iron. The associated symbols indicate the peak positions for biographite, α-iron, and γ-iron. | ||
During cooling (Fig. 6b), the γ-iron peak reappeared strongly indicating the crystallization of iron. The γ-iron peak position also remained unaffected during cooling until 800 °C. At temperatures below 800 °C, the γ-iron transformed into α-iron, a process just opposite to what was observed during the heating stage i.e., α → γ transition. The α-iron was very stable at room temperature (25 °C) as understood from Fig. 6b. On the other hand, the biographite peak (002) developed progressively during cooling and gradually shifted to higher 2θ positions indicating the formation of more ordered and crystalline graphitic structure (Table S3†). A closer inspection of the diffractogram at 1100 °C during heating (Fig. 6a), indicated the presence of a shoulder at ∼26°, which was more visible in Fig. S4d.† The appearance of this peak confirmed that the graphitization was initiated in the system at ∼1100 °C, which was before the melting of γ-iron at ∼1200 °C. The XRD peak of γ-iron was very strong at 1100 °C indicating its presence as a solid crystalline form. Therefore, the onset of graphitization at ∼1100 °C could not be explained with the help of γ-iron melting. The combined in situ XRD patterns collected during heating and cooling are shown in Fig. S5.† The stronger peaks corresponded to α-iron, γ-iron, and biographite. However, a group of low-intensity peaks was also visible which confirmed the presence of iron carbide (Fe3C, cementite).49 During heating, iron carbide formed (eqn (3)) due to the chemical interaction of carbon, from pyrolyzed feedstock, with iron. The formed iron carbide was noticeable up to 1000 °C as understood from Fig. S4a–c.† When the heating temperature reached 1100 °C, iron carbide melted completely and biographite formation initiated following the similar dissolution–precipitation mechanism discussed above for γ-iron. During cooling, the iron carbide peaks reappeared at ∼700 °C (Fig. 6b) confirming its crystallization from the melting phase. The overlap of the relatively weaker iron carbide peak with the intense γ-iron peak supported the possibility of iron carbide crystallization earlier than 700 °C during cooling. As the system was cooled further <700 °C, iron carbide decomposed forming carbon and iron (eqn (4)).41
| 3Fe + C = Fe3C (iron carbide formation) | (3) |
| Fe3C = 3Fe + C (iron carbide decomposition) | (4) |
Before this carbide decomposition, a vast majority of the biographites were formed following the dissolution–precipitation mechanism involving the reversible melting ↔ crystallization of Fe3C and γ-Fe. This was evident from the relatively intense (002) peak of biographite. It is worth noting that no extensive increases in (002) peak intensity of biographites were found at temperature <800 °C (Table S3†), which strongly supported that the carbide formation–decomposition mechanism was not applicable to the catalytic graphitization of pyrolysis oil by iron. It should also be noted that even at room temperature (25 °C), a tiny peak of iron carbide remained. It has been reported that the presence of residual iron carbide in the final biographite is dependent on the rate of cooling. A slow cooling rate accelerates the decomposition of iron carbide into iron and carbon, while a rapid cooling rate (quenching) interrupts the decomposition process resulting in a large quantity of residual iron carbide.41 The in situ XRD study was performed with a cooling rate of 4 °C min−1, which is why the quantity of iron carbide left was at a minimum as understood from the XRD patterns (Fig. 6b). To date, two types of mechanisms have been proposed to describe the catalytic graphitization process: (i) dissolution–precipitation mechanism, and (ii) formation–decomposition mechanism. From the in situ XRD experimental observation, it was evident that the dissolution–precipitation mechanism could adequately describe the catalytic graphitization of pyrolysis oil by iron. Both the γ-iron (Fe0) and iron carbide (Fe3C) contributed to the dissolution–precipitation mechanism. Graphitization was initiated by iron carbide melting and was later controlled by γ-iron melting. Overall, the γ-iron melting potentially controlled the catalytic graphitization process, while the contribution from iron carbide melting was relatively less.
For a quantitative understanding of the iron phase transitions, Rietveld refinement analysis was performed on the in situ XRD patterns recorded on heating at 700 °C (Fig. 7a) and 1000 °C (Fig. 7b). The phase proportions at 700 °C were calculated to be 74.4% α-Fe and 25.6% Fe3C, while at 1000 °C, the phase proportions were quantified to be 88.3% γ-Fe and 11.7% Fe3C. A comparatively higher percentage of Fe3C was present at 700 °C than 1000 °C. To get a further insight into the catalytic graphitization mechanism and the progressive improvement in graphitic structure during high temperature treatment of pyrolysis oil + iron mixture, in situ HR-TEM experiments could be very helpful. In addition to in situ HR-TEM imaging, molecular dynamics (MD) simulation would also be another theoretical approach to adequately study the catalytic graphitization process.
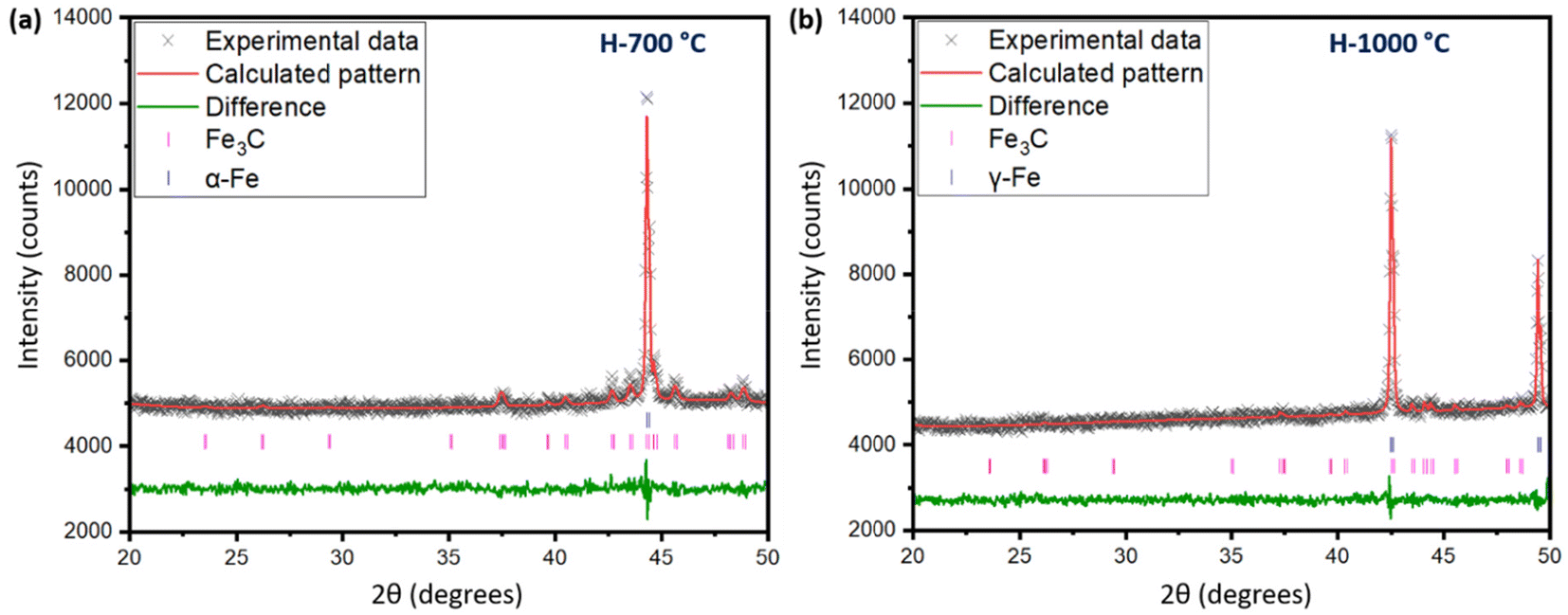 | ||
| Fig. 7 (a) Rietveld refinement of the in situ XRD pattern at 700 °C during heating. The criteria of fit parameters included Rp = 1.12%, wRp = 1.47%, and χ2 = 1.105, (b) Rietveld refinement of the in situ XRD pattern at 1000 °C during heating. The criteria of fit parameters included Rp = 1.10%, wRp = 1.39%, and χ2 = 0.92. | ||
Evaluation of electrochemical performance
The electrochemical behavior and performance of the pyrolysis oil-derived biographite is demonstrated in Fig. 8. The initial reversible capacity (RC) of the biographite was found to be ∼350 mA h g−1 as understood from two discharge–charge cycles (Fig. 8a). More importantly, the ICE was calculated to be 90.2%, which indicates that the pyrolysis oil derived biographite is promising for commercial production. However, the ICE value of the biographite was comparatively lower than commercial graphite (92–94%), which could happen due to the extent of surface reactivity of biographite for having different surface composition and area in comparison with the commercial graphite. Surface composition of the as synthesized biographite might have influenced the side reactions taking place between the graphite and the electrolyte leading to the formation of the solid electrolyte interface (SEI) that inherently influenced the ICE. Typically, low ICE results in poor cycling performance in a full cell that has finite lithium inventory from cathode and electrolyte, so it should be improved by employing strategies including surface modification and size and morphology control of particles. It is noteworthy that the CE values for the subsequent cycles quickly stabilized to ∼99%, indicating the formation of the stable SEI layer. The biographite demonstrated comparable cycling performance up to 90 cycles compared with a typical commercial graphite (around 350–360 mA h g−1) as shown in Fig. 8b. However, capacity fading started after 90 cycles which is worth exploring in future experiments. It can be assumed that the extent of electrolyte decomposition and undesirable SEI formation and evolution by the Li metal counter/reference electrode and/or Li dendritic growth could be possible factors.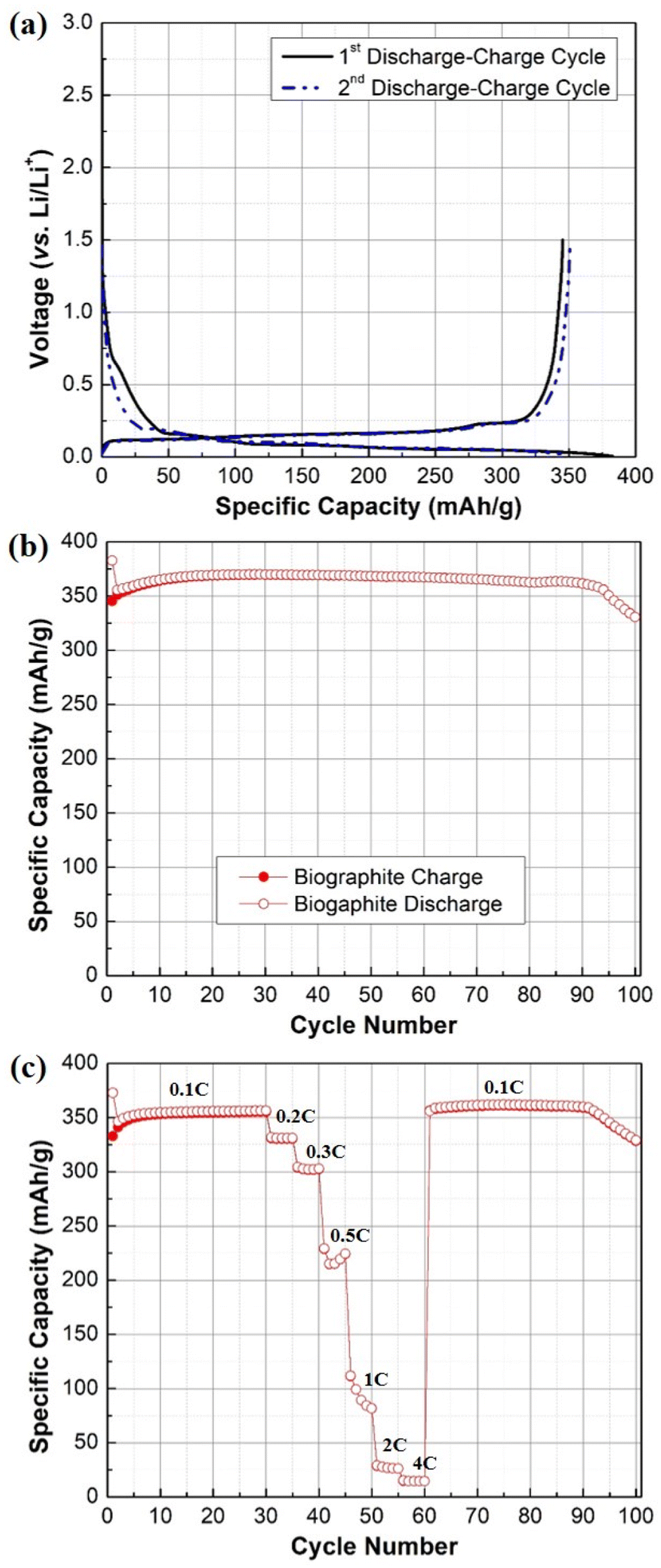 | ||
| Fig. 8 (a) Galvanostatic first and second discharge–charge profile, (b) cycling performance plot (0.1C rate), (c) rate capability plot of the biographite (2.50 × FC iron loading). | ||
The results of the rate capability experiments are shown in Fig. 8c. The rate capability of the biographite was compared at different current densities (e.g., 0.1C, 0.2C, 0.3C, 0.5C, 1C, 2C, and 4C). The capacity decreased with increasing C-rates, as expected from fast rate of cycling. However, the electrode could fully recover its initial capacity when the C-rate was returned to the initial value (i.e., 0.1C). Overall, the biographite demonstrated promising electrochemical performance as anode active material for LIBs. However, the performance could be improved further by tuning the surface morphology, size of the particles, and native surface species of the as synthesized biographite.
Effect of pyrolysis oil aging on biographite crystalline structure
To scale up the process, it is essential to understand how aging time affects the catalytic graphitization process because aging is very common as pyrolysis oil is chemically and thermally unstable.50–53 To study this effect, pyrolysis oil was aged in a refrigerator for months and catalytic graphitization was carried out at four different time periods (0, 6, 8, and 9 months). These repeated experiments were conducted following the optimal graphitization conditions i.e., iron loading of 2.50 × FC. To pinpoint the true effects of the aging time, XRD patterns of the biographites were recorded after aged pyrolysis oils (0, 6, 8, and 9 month/s) were graphitized. The results of the aging study are shown in Table S4.† As understood, the crystallite size along c-direction, Lc (also known as stacking height), decreased with aging time. Similarly, the interlayer spacing i.e., d002 value was also negatively affected. The characteristic (002) peak intensity at ∼26.5° decreased significantly with time. To add, the overall XRD patterns (Fig. S6†) seemed comparatively less sharper (less crystalline). Therefore, it can be stated that inferior quality biographite crystals were formed when aged pyrolysis oil was used as a feedstock in comparison with the fresh oil. It was understood that there was a strong correlation between the crystalline structures of biographites with the electrochemical performance (Fig. S1†). Therefore, it was expected that the inferior quality biographite crystals, formed from the aged pyrolysis oil, would show poor electrochemical performance as active material in LIB anodes. The fresh oil (month 0) had a low viscosity (thin) while the viscosity increased significantly with aging and it was very difficult to handle the thick viscous oil after aging. Due to low viscosity, it was much easier to mix the iron catalyst with the fresh oil. Prior to mixing iron catalyst, it was required to heat up the aged oil to reduce viscosity. Aged oil would not only make poor quality biographite but also increase the handling cost. The observation from this aging study implies that pyrolysis oil cannot be stored for a long time when it is considered as a feedstock for commercial production of biographite. The dark-brown viscous oil obtained from the condensation of biomass pyrolysis vapor exhibits promising economic prospects for conversion into LIB anodes. However, pyrolysis oil is unstable as it contains relatively higher proportion of oxygen, water, and numerous organic compounds such as alcohols, aldehydes, ketones, acids, etc. During aging, certain pyrolysis oil components undergo condensation reactions leading to the formation of high molecular weight polymers and therefore the oil becomes highly viscous and sticky.54 Meng et al. thoroughly explored pyrolysis oil aging mechanism using solvent-based oil fractionation approach.51 It was found that the low pH (higher acidity) of pyrolysis oil accelerated several polymerization reactions including phenol-formaldehyde resin formation. The pyrolysis oil used in this study had a low pH value (3.2) and it is believed that similar polymerization reactions occurred during the 9-month storage of the oil. These polymerization reactions, especially the formation of highly cross-linked phenol-formaldehyde resin due to pyrolytic lignin condensation, are believed to be a potential hurdle for converting the aged pyrolysis oil into battery-grade biographite. This is because more energy is required to break the highly cross-linked carbon–carbon bonds before the amorphous carbons can be reorganized into graphitic crystalline structures.55Conclusions
For the first time, the thermocatalytic conversion of pyrolysis oil into a battery-grade biographite anode was successfully demonstrated. Reduced iron powder exhibited excellent performance as a graphitization catalyst at moderate temperature (1500 °C). One of the key highlights of this study was the understanding of the mechanism of catalytic graphitization. The in situ XRD investigation found the dissolution–precipitation mechanism helpful in describing the conversion of amorphous carbon into ordered biographite in the presence of the iron catalyst. Under the experimental conditions, the optimal biographite was formed when the catalyst loading was 2.5 times the fixed carbon (FC) content of the pyrolysis oil, thereby highlighting the importance of iron loading. The optimal biographite sample demonstrated promising electrochemical performance in terms of coulombic efficiency, reversible capacity, cyclability, and rate capability. It was also found that pyrolysis oil aging negatively affected the formation of high quality biographite. Pyrolysis oil is a renewable biocarbon feedstock which could be a reliable and sustainable precursor for the mass-scale production of bio-based graphite. Once proven at scale, it will enhance the supply of high-quality graphite to meet the global demand for LIB anode materials in EVs. Based on the observation from this study several future research directions can be highlighted. (i) The process described in this study holds tremendous potential for commercialization; however, the use of metallic iron powder in the process seems to create some concerns, which should be adequately explored via conducting experiments at a relatively larger scale. To make a fair comparison of this technology with the current synthetic graphite production process, it is essential to explore the direct conversion of pyrolysis oil into biographite at high temperature (∼3000 °C) without using any catalyst. (ii) This study considered only a single particle size reduced iron powder (particle size <10 μm). Future studies should examine how the iron particle size affects graphitization. Additionally, oxidized iron such as iron oxide or iron salts should be tested as a graphitization catalyst for pyrolysis oil. As the catalytic graphitization was initiated by iron carbide (Fe3C) melting, future research should also explore the use of iron carbide as a catalyst. (iii) Refluxing a biographite + iron mixture with concentrated HCl (37 wt%) for 3 h was unable to completely remove residual iron from biographite. Iron is undesired in an electrode and therefore future studies should apply extended refluxing time and refluxing steps with fresh acid to increase the extent of iron removal from biographite. Continuous acid refluxing using a Soxhlet extractor should be explored in the future studies. The acid refluxing step could be coupled with other possible techniques such as magnetic separation. Acid refluxing converts iron into acid soluble iron chloride. Iron chloride and spent HCl should be separated and recycled back to the process. Additionally, during the acid reflux experiments, hydrogen gas (H2) is produced in pure form, which could be captured to improve the economic potential of the process. (iv) The in situ XRD experiments were very helpful to understand the catalytic graphitization mechanism. Future studies should also apply in situ HR-TEM imaging to monitor the progress of graphitization. (v) Considering the possible scale-up of the demonstrated process, it is essential to explore pathways to address the aging so that pyrolysis oil could be stored for an extended period of time avoiding undesired physicochemical changes. (vi) This study used hardwood-derived pyrolysis oil only as a biographite feedstock; however, it is worth exploring how the softwood-derived pyrolysis oil affects the catalytic graphitization process as the chemistry of the two oils is expected to be different. (vii) Pyrolysis oil modification could also be considered e.g., the oil can be doped with certain proportions of petroleum coke (current commercial feedstock for synthetic graphite) or other feedstocks. (viii) The pyrolysis vapors coming out during the process have the potential to be converted into sustainable aviation fuel (SAF) after appropriate treatment (e.g., hydrotreating) (ix). Understanding the chemical composition of pyrolysis oil is very challenging using the traditional chromatographic techniques due to the complex nature of pyrolysis oil. Advanced analytical techniques such as Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR MS) or molecular beam mass spectrometry (MBMS) could be very helpful in exploring the chemical composition. (x) This study demonstrates the electrochemical performance of biographite as an anode active material in a Li-ion half-cell. Evaluation of the performance of biographite electrodes in full cells using a real cathode should be explored in the future. (xi) Other future research needs are scaling up the process to measure mass balances and technoeconomic and life cycle analyses.Author contributions
Shaikat Chandra Dey: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft, and writing – review and editing. Lillian Lower: visualization, and writing – review and editing. Trevor Vook: investigation, and writing – review and editing. Md. Nazrul Islam: validation, and writing – review and editing. William Joe Sagues: funding acquisition, resources, supervision, and writing – review and editing. Sang-Don Han: investigation, visualization, and writing – review and editing. Mark R. Nimlos: funding acquisition and writing – review and editing. Stephen S. Kelley: funding acquisition and writing – review and editing. Sunkyu Park: conceptualization, funding acquisition, project administration, resources, supervision, and writing – review and editing.Data availability
All data generated or analyzed during this study are included in this published article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the BioEnergy Technologies Office (BETO), U.S. Department of Energy (DOE) (award number: DE-EE0009260). This work was also supported by the faculty research fund of Sejong University in 2023. The pyrolysis oils were cordially supplied by Ensyn, Ontario, Canada. Utilization of the XRF and SEM (with EDS) instruments within the University of Virginia's Nanoscale Materials Characterization Facility (NMCF) was fundamental to this work, and the authors acknowledge the assistance of Catherine Dukes for performing experiments and analysis of the data. The room temperature XRD, in situ XRD, Raman, XPS, and TEM analyses were carried out at the Analytical Instrumentation Facility (AIF) at the North Carolina State University. The authors gratefully thank Dr Jennifer S. Forrester at AIF for helping with the in situ XRD experiments and Rietveld refinement.References
- T. Kim, W. Song, D. Y. Son, L. K. Ono and Y. Qi, J. Mater. Chem. A, 2019, 7, 2942–2964 RSC.
- X. Xia, P. Li, Z. Xia, R. Wu and Y. Cheng, Sep. Purif. Technol., 2022, 301, 122063 CrossRef.
- J. Asenbauer, T. Eisenmann, M. Kuenzel, A. Kazzazi, Z. Chen and D. Bresser, Sustainable Energy Fuels, 2020, 4, 5387–5416 RSC.
- P. Meister, H. Jia, J. Li, R. Kloepsch, M. Winter and T. Placke, Chem. Mater., 2016, 28, 7203–7217 CrossRef.
- J. Zhang, C. Liang and J. B. Dunn, Environ. Sci. Technol., 2023, 57, 3402–3414 CrossRef.
- D. W. Olson, R. L. Virta, M. Mahdavi, E. S. Sangine and S. M. Fortier, Spec. Pap. – Geol. Soc. Am., 2016, 520, 67–77 Search PubMed.
- L. Lower, S. C. Dey, T. Vook, M. Nimlos, S. Park and W. J. Sagues, ChemSusChem, 2023, 16, e202300729 CrossRef.
- A. D. Jara, A. Betemariam, G. Woldetinsae and J. Y. Kim, Int. J. Min. Sci. Technol., 2019, 29, 671–689 CrossRef.
- L. Gaines, Sustainable Mater. Technol., 2018, 17, e00068 CrossRef.
- L. Gaines, Q. Dai, J. T. Vaughey and S. Gillard, Recycling, 2021, 6, 1–18 CrossRef.
- C. E. Burgess-Clifford, D. L. Narayanan, D. T. Van Essendelft, P. Jain, A. Sakti and A. D. Lueking, Fuel Process. Technol., 2009, 90, 1515–1523 CrossRef.
- A. S. Kamal, R. Othman and N. H. Jabarullah, Syst. Rev. Pharm., 2020, 11, 881–894 Search PubMed.
- F. J. Derbyshire, A. E. B. Presland and D. L. Trimm, Carbon, 1975, 13, 111–113 CrossRef.
- A. Ōya and H. Marsh, J. Mater. Sci., 1982, 17, 309–322 CrossRef.
- H. Du, F. Deng, R. R. Kommalapati and A. S. Amarasekara, Renew. Sustain. Energy Rev., 2020, 134, 110292 CrossRef.
- L. Frankenstein, P. Glomb, J. Ramirez-Rico, M. Winter, T. Placke and A. Gomez-Martin, ChemElectroChem, 2023, 10, e202201073 CrossRef.
- A. Nugroho, E. B. Nursanto, S. A. Pradanawati, H. S. Oktaviano, H. Nilasary and H. Nursukatmo, Mater. Lett., 2021, 303, 130557 CrossRef.
- L. Wang, C. Du, Z. Li, Y. Han, N. Feng and J. Yang, J. Alloys Compd., 2023, 960, 170949 CrossRef.
- L. Tang, Q. Mao, Z. You, Z. Yao, X. Zhu, Q. Zhong and J. Xiao, Carbon, 2022, 188, 336–348 CrossRef CAS.
- W. J. Sagues, J. Yang, N. Monroe, S. D. Han, T. Vinzant, M. Yung, H. Jameel, M. Nimlos and S. Park, Green Chem., 2020, 22, 7093–7108 RSC.
- M. A. A. Farid, A. L. T. Zheng, T. Tsubota and Y. Andou, J. Anal. Appl. Pyrolysis, 2023, 173, 2–4 CrossRef.
- S. Xia, N. Cai, W. Lu, H. Zhou, H. Xiao, X. Chen, Y. Chen, H. Yang, X. Wang, S. Wang and H. Chen, J. Clean. Prod., 2021, 329, 129735 CrossRef CAS.
- L. M. R. Millán, A. C. Ghogia, C. E. White and A. Nzihou, ACS Appl. Nano Mater., 2023, 6, 3549–3559 CrossRef.
- M. P. Illa, C. S. Sharma and M. Khandelwal, Mater. Today Chem., 2021, 20, 100439 CrossRef CAS.
- R. D. Hunter, E. C. Hayward, G. J. Smales, B. R. Pauw, A. Kulak, S. Guan and Z. Schnepp, Mater. Adv., 2023, 4, 2070–2077 RSC.
- B. Li, Y. Zhang, J. Xiong, Y. Gui, T. Huang, J. Peng, H. Liu, F. Yang and M. Li, J. Mater. Sci., 2022, 57, 9939–9954 CrossRef CAS.
- X. Wu, K. Chen, Z. Lin, Y. Zhang and H. Meng, Mater. Today Energy, 2019, 13, 100–108 CrossRef.
- A. Gomez-Martin, J. Martinez-Fernandez, M. Ruttert, M. Winter, T. Placke and J. Ramirez-Rico, ACS Omega, 2019, 4, 21446–21458 CrossRef CAS.
- T. Vook, S. C. Dey, J. Yang, M. Nimlos, S. Park, S. D. Han and W. J. Sagues, J. Energy Storage, 2023, 73, 109242 CrossRef.
- A. S. Kamal, N. H. Jabarullah and R. Othman, Mater. Today Proc., 2020, 31, 211–216 CrossRef.
- Y. Liu, Q. Liu, J. Gu, D. Kang, F. Zhou, W. Zhang, Y. Wu and D. Zhang, Carbon, 2013, 64, 132–140 CrossRef CAS.
- W. N. R. W. Isahak, M. W. M. Hisham, M. A. Yarmo and T. Y. Yun Hin, Renewable Sustainable Energy Rev., 2012, 16, 5910–5923 CrossRef CAS.
- A. V. Bridgwater, S. Czernik and J. Piskorz, Prog. Thermochem. Biomass Convers., 2001, 977–997 CAS.
- X. Hu and M. Gholizadeh, J. Energy Chem., 2019, 39, 109–143 CrossRef.
- D. Lachos-Perez, J. C. Martins-Vieira, J. Missau, K. Anshu, O. K. Siakpebru, S. K. Thengane, A. R. C. Morais, E. H. Tanabe and D. A. Bertuol, Analytica, 2023, 4, 182–205 CrossRef CAS.
- R. Muruganantham, T. H. Hsieh, C. H. Lin and W. R. Liu, Mater. Today Energy, 2019, 14, 100346 CrossRef.
- D. C. P. Vélez, W. L. E. Magalhães and G. Capobianco, Sci. Technol. Mater., 2018, 30, 16–22 Search PubMed.
- N. Jendoubi, F. Broust, J. M. Commandre, G. Mauviel, M. Sardin and J. Lédé, J. Anal. Appl. Pyrolysis, 2011, 92, 59–67 CrossRef CAS.
- S. Yoo, C. C. Chung, S. S. Kelley and S. Park, ACS Sustainable Chem. Eng., 2018, 6, 9113–9119 CrossRef CAS.
- G. Esteves, K. Ramos, C. M. Fancher and J. L. Jones, LIPRAS: Line Profile Analysis Software, 2017. DOI:10.13140/RG.2.2.29970.25282/3.
- A. Gutiérrez-Pardo, J. Ramírez-Rico, R. Cabezas-Rodríguez and J. Martínez-Fernández, J. Power Sources, 2015, 278, 18–26 CrossRef.
- N. A. Banek, D. T. Abele, K. R. McKenzie and M. J. Wagner, ACS Sustainable Chem. Eng., 2018, 6, 13199–13207 CrossRef CAS.
- A. Ashraf, S. A. Dastgheib, G. Mensing and M. A. Shannon, J. Supercrit. Fluids, 2013, 76, 32–40 CrossRef CAS.
- X. Chen, X. Wang and D. Fang, Fullerenes, Nanotubes Carbon Nanostruct., 2020, 28, 1048–1058 CrossRef CAS.
- L. Li, P. Ma, S. Hussain, L. Jia, D. Lin, X. Yin, Y. Lin, Z. Cheng and L. Wang, Sustainable Energy Fuels, 2019, 3, 1749–1756 RSC.
- H. Marsh and A. P. Warburton, J. Appl. Chem., 1970, 20, 133–142 CrossRef CAS.
- G. J. Long and H. P. Leighly, J. Chem. Educ., 1982, 59, 948–953 CrossRef CAS.
- H. Li, H. Zhang, K. Li, J. Zhang, M. Sun and B. Su, Fuel, 2020, 279, 118531 CrossRef CAS.
- J. Hoekstra, A. M. Beale, F. Soulimani, M. Versluijs-Helder, J. W. Geus and L. W. Jenneskens, J. Phys. Chem. C, 2015, 119, 10653–10661 CrossRef CAS.
- J. Meng, A. Moore, D. C. Tilotta, S. S. Kelley, S. Adhikari and S. Park, Energy Fuels, 2015, 29, 5117–5126 CrossRef CAS.
- J. Meng, A. Moore, D. Tilotta, S. Kelley and S. Park, ACS Sustainable Chem. Eng., 2014, 2, 2011–2018 CrossRef CAS.
- J. Meng, J. Park, D. Tilotta and S. Park, Bioresour. Technol., 2012, 111, 439–446 CrossRef CAS.
- J. Meng, T. I. Smirnova, X. Song, A. Moore, X. Ren, S. Kelley, S. Park and D. Tilotta, RSC Adv., 2014, 4, 29840–29846 RSC.
- J. Cai, M. M. Rahman, S. Zhang, M. Sarker, X. Zhang, Y. Zhang, X. Yu and E. H. Fini, Energy Fuels, 2021, 35, 11665–11692 CrossRef CAS.
- S. I. Talabi, A. P. da Luz, V. C. Pandolfelli, V. H. Lima, V. R. Botaro and A. de Almeida Lucas, Mater. Res., 2020, 23, e20190686 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc01647e |
| This journal is © The Royal Society of Chemistry 2024 |