
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustainable upgrading of glycerol into glycidol and its derivatives under continuous-flow conditions†
Alessandra
Sivo
 ,
Ilaria
Montanari
,
Mert Can
Ince
and
Gianvito
Vilé
*
,
Ilaria
Montanari
,
Mert Can
Ince
and
Gianvito
Vilé
*
Department of Chemistry, Materials, and Chemical Engineering “Giulio Natta”, Politecnico di Milano, Piazza Leonardo da Vinci 32, IT-20133 Milano, Italy. E-mail: gianvito.vile@polimi.it
First published on 23rd May 2024
Abstract
This study presents a continuous-flow process for the valorization of glycerol, a byproduct of the biofuel industry, into glycidol and its derivatives. The method ensures safety and allows for easy production of glycidol on a gram scale, even in the presence of hazardous substances such as hydrogen chloride and acetic acid. Moreover, this continuous-flow method can be easily integrated with downstream synthetic steps to produce value-added derivatives, which have potential applications in the fields of medicinal and polymer chemistry. The comprehensive evaluation of sustainability metrics, encompassing green indicators, a techno-economic analysis, and a life cycle assessment, substantiates the environmental benefits of the technology, and showed that the method is environmentally friendly and has the potential to enhance efficiency, safety, and sustainability in industrial processes.
Introduction
The development of industrial processes that make use of waste materials is a significant stride towards circularity and sustainability.1–4 For instance, glycerol (GLY, 1) is a byproduct generated in significant quantities during the transesterification of biodiesel,5,6 with limited value and market demand.7,8 Among the various applications of glycerol (Fig. 1a), one avenue is its potential use as a starting material for the preparation of pharmaceutically relevant epoxides, such as glycidol (GLD, 3). Glycidol has ubiquitous presence in the food, fine chemicals, and pharmaceuticals sectors, due to its double reactivity towards both nucleophilic and electrophilic species (Fig. 1b).9–17 The conventional methods for glycidol production rely on the glycerol carbonate route (Fig. 1c). Here, glycerol carbonate is first synthesized from glycerol through transesterification with dimethyl carbonate, in the presence of alkaline, acidic, or enzymatic catalysts. The subsequent conversion of glycerol carbonate to glycidol typically entails a ring-opening reaction conducted under mild conditions; during this step, CO2 is generated, contributing to environmental concerns regarding greenhouse gas emissions. Thus, addressing the CO2 generation issue remains a significant challenge for ensuring the sustainability of glycidol production.18–21 An alternative and less investigated approach involves the introduction of a chlorine group to activate glycerol for the intramolecular cyclization steps (Fig. 1c). The advantage of this approach lies in its ability to minimize CO2 generation, despite employing chlorine, which is used and released during the reaction, and that can potentially be recovered and reutilized in a more circular manner. Inherent challenges that have hampered the applicability of this method include extended reaction times, harsh operating conditions (such as high temperature and pressure, reaching up to 350 °C), and the formation of unstable intermediates (e.g., 2-chloropropanediol). Continuous-flow chemistry represents a breakthrough for the development of optimized processes, offering better control of reaction parameters,22–24 improved heat and mass transfer, along with the possibility to handle highly reactive intermediates in situ. Moreover, the reduction of reaction volumes generates lower waste production, with an overall reduction of the process's footprint. | ||
| Fig. 1 Proportional breakdown of glycerol applications in the main fields of interest based on the available literature (a).7 Examples of commercial active pharmaceutical ingredients derived from glycidol (b) and synthetic methods for the preparation of glycidol 3 (c) from glycerol. | ||
In this work, an optimized continuous-flow process for glycidol production has been studied, using commonly employed reagents, such as hydrogen chloride (HCl), acetic acid (AcOH), and glycerol. The gram-scale preparation of glycerol is then exploited to develop telescoped protocols for the preparation of value-added derivatives under integrated, multistep, continuous-flow conditions. These optimized processes were finally assessed in terms of green metrics, techno-economic, and life cycle evaluation.
Results and discussion
Our investigation into the synthesis of glycidol 3 commenced with a preliminary study in a commercial batch reactor, namely the Carousel 12 Plus Reaction Station™, wherein multiple reactions could be conducted in parallel, in the presence of hydrochloric acid as chlorine source, and an organic acid catalyst (Fig. 2). The motivation to explore the performance of multiple organic acid catalysts and select the most appropriate one was based on the reaction mechanism, which involves a two-step process, in which glycerol 1 first undergo chlorination in presence of an organic acid as catalyst, producing a mixture of α-monochlorohydrin 2 (α-MCH) and α,γ-dichlorohydrin 2b (α,γ-DCH). Then, the reaction proceeds via Williamson intramolecular cyclization, in the presence of an inorganic base, forming the corresponding oxiranes 3 and 3b (see the details in the ESI†). According to the literature, the transient esterification of 1 over the organic acid catalyst is the rate limiting step of the overall process.25 A screening of different carboxylic acids was thus performed to study the formation of 3 (Fig. 2a). Due to the strong acidity of the reaction solution containing HCl, the acid strength of the individual organic acids was not a relevant parameter to be monitored. Compared to AcOH, 3 was obtained in slightly lower yield when adipic acid was used, due to the presence of the hindered dicarboxylic functionality on the aliphatic chain. This effect was confirmed by using the even bulkier citric acid, which provided 3 in only 10%. Moreover, uncomplete solubility and significant amount of precipitation of solids were observed in the presence of adipic and citric acids, limiting their applicability in flow conditions. Based on these results, and on the fact that a low glycidol yield was achieved in the absence of acid catalysts, we selected acetic acid for the follow up experiments. | ||
| Fig. 2 Influence of different organic acid catalysts (a) and reaction time (b, c) on the synthesis glycidol. Reaction conditions: 1 (12 mmol), HCl 13 M (48 mmol), organic acid catalyst (4 mmol), temperature = 120 °C. Differential scanning calorimetry of glycerol (d) and acetic acid (e). | ||
A kinetic evaluation in presence of different amounts of AcOH was performed (Fig. 2b). When the acid equivalents were set to 0.2 (corresponding to 2.5 mmol of AcOH), the product was formed in drastically low yield (25% after 24 h). Interestingly, increasing the equivalents of the acid to 0.7 (corresponding to 8 mmol of AcOH) led to a glycidol yield of approximately 50% after 24 h, with a prevalent formation of epichlorohydrin as main product, suggesting a shift of the reaction selectivity towards 2b and 3b. The best conditions were found at 0.3 equiv. of AcOH (corresponding to 4 mmol of AcOH). In terms of reaction time (Fig. 2c), the initial competitive formation of DCH 2b became less pronounced after 4 hours, thereby enabling the formation of glycidol.
It must be mentioned that numerous critical issues, related to the integrated management of the reaction, were observed during these tests. Among these, the safe handling of large amounts of hydrochloric acid, worsened by the high temperatures required for the chlorination step, and the exothermicity of the neutralization step were of particular relevance. To assess the thermal safety of both configurations, a differential scanning calorimetry analysis of the two main reaction components (glycerol 1 and acetic acid) was performed (Fig. 2d and e). The analysis revealed two distinct low temperature exotherms occurring at approximately 50 °C and 166 °C, corresponding to acetic acid and glycerol, respectively. Consequently, it is evident that the reaction can be safely executed in batch only at temperatures at least 100 °C below the first onset detected by DSC.26 In fact, it is worth mentioning that based on DSC data, many companies indicates a practice of 100 °C buffer between the maximum temperature of safe operation and the decomposition onset indicated by DSC.26 This approach, while conservative, indicated that when batch reactors are employed, the maximum temperature of safe operation is typically determined by reaction calorimetry.26 This widely-accepted ‘rule of thumb’ is valid for batch processes, but not for flow methods, as they does not pose the same level of danger due to superior heat management capabilities.26
Therefore, to overcome these drawbacks, we moved to the development of a flow protocol, in which the small volume and the high surface-to-volume ratio of the microreactor enabled a safer management of the highly reactive reagents and intermediates.26 Our optimization under continuous-flow conditions started with the evaluation of the reaction at different temperatures and residence times. Fixing the temperature at 120 °C, Fig. 3 shows the increment in the yield of glycidol when the residence time is increased from 0.5 to 1 h, followed by a plateau with longer residence times. This trend does not change at 80 and 100 °C. At this stage, fixing the temperature at 100 °C and the residence time to 2 h, we explored the possibility to decrease the concentration of the hydrochloric acid solution from 13 M to 6.5 M. The results revealed a drastic decrease in the yield of glycidol, from 83% to 2%, when the acid concentration was decreased from 13 M to 6.5 M, respectively. Further to the predictable dilution effect, hampering the mass transfer, the presence of water in the reactor shifted the thermodynamic equilibrium of the reaction favouring the hydrolysis of MCH, and limiting the glycerol conversion, in line with the literature.27,28
 | ||
| Fig. 3 General scheme for the preparation of glycidol under continuous-flow conditions. Influence of the glycidol yield as a function of the residence time and temperature (a), AcOH concentration (b), type of base (c), and concentration of base (d) used for the quenching step. Comparison of the batch and flow process in terms of green metrics (e). Environmental impact of glycidol production via batch and flow process (f). When not indicated otherwise, the continuous-flow experiments were conducted at the following conditions: 1 (12 mmol), HCl 13 M (48 mmol), AcOH (0.3 equiv., corresponding to 4 mmol), pressure = 2 bar. The analytical data was determined by GC-MS. | ||
The investigation proceeded with the optimization of the catalyst amount under flow conditions (Fig. 3b), at fixed reaction conditions (100 °C, 2 h). Decreasing the catalyst content to 0.2 equivalents resulted in a negative impact on the formation of 3, due to the insufficient presence of carboxylic functionalities relative to the equivalent amount of acetic acid. On the other hand, the equivalents of AcOH above a certain threshold (0.3 equiv.) did not significantly affect the yield. Finally, the quenching step after reaction was optimized by screening different inorganic bases (Fig. 3c). NaOH and KOH, with their similar pKa, showed a comparable ability towards the MCH deprotonation, crucial to initiate the ring closing reaction. Weaker bases, like CaCO3, required a much larger number of equivalents to achieve complete neutralization, with an overall reduction of the yield of 3. Thus, NaOH was selected as the base of choice, and a study on its concentration was conducted (Fig. 3d).
A comparative analysis between the optimized batch and flow processes was conducted by means of common green metrics (see also the details in the ESI†). Based on the results in Fig. 3e, the optimized flow process outlined in this study exhibited higher yield and reaction mass intensity which indicates a “greener” process due to the improvement of the reaction efficiency and resource utilization.29 The stoichiometric factor, atom economy, and mass recovery parameters show similar results. Additional advantages, that are not easily captured in the green metrics, are the use of glycerol as a bio-based starting material, along with the utilization of water as a solvent and the production of harmless sodium chloride as the only byproduct.
In order to investigate the economic and environmental effects comprehensively, techno-economic (TEA) and life cycle (LCA) analyses were also performed, comparing vis-à-vis both batch and flow conditions (Table S1† and Fig. 3f). In the context of the TEA (Table S1†), the flow process showed superior performance compared to the batch process in terms of total raw material cost, duration to reach batch capacity, and electricity usage amount per hour and grams of product. It was reported that the total raw material costs decreased more than one-third from 1.9 to 1.1 $ per h per gproductvia the implementation of the flow configuration. In addition to this, due to the significant minimization in process duration to reach the same capacity, from 24 h (batch) to 8.4 min (flow), the electricity expenses of the processes showed a 92.9% reduction from 22.4 to 1.6 W h−1 gproduct−1, respectively. To construct a fair comparison with respect to the literature, our results were compared with similar TEA studies investigating the performance improvement in API production, where typically a reduction in energy expenses of 25.6% have been observed through the implementation of flow conditions.30 In our case, with the observed 92.9% reduction in energy expenses, the study succeeded to achieve a performance beyond the literature. Overall, the TEA confirmed that the flow process demonstrated to be economically more feasible with respect to the conventional batch configuration.
In terms of LCA, the flow process led to a lower CO2 generation and radiative effects (with less negative effects on climate change). CO2 generation and radiative effects decreased by 65.0% and 59.4%, respectively, between batch and flow configurations. Also, the effects on global phosphorus (P) and nitrogen (N) cycles were halved through the flow process, denoting fewer negative effects on biogeochemical flows. In addition, via flow conditions, the impacts on freshwater and land system changes decreased of 55.1% and 53.8%, respectively, and ocean acidification dropped of 58.8%. In terms of air quality and ability to pursue cloud formation cycles efficiently, flow process showed an excellent performance due to a 60.7% reduction of fine particulate matter formations with respect to batch conditions.
Overall, the TEA and LCA investigations consistently showed that the differences between the configurations were majorly originated from the higher amounts of materials and electricity usage in batch conditions. Therefore, with respect to the Planetary Boundaries, flow conditions exhibited greener, safer, and more sustainable performance. Additional studies have been conducted to compare the utilized chlorination route with respect to the state-of-the-art glycidol synthesis on the 11 different environmental impact categories and to assess our process in terms of the 12 Principles of Green Chemistry (Fig. S1 and Table S6, ESI†).
The flow process in Fig. 3 was then scaled up, to obtain 1 g of glycidol within 1 h. Therefore, we explored the application of glycidol under continuous-flow mode to prepare, in a telescoped and integrated manner, a series of value-added derivatives, for application in the preparation of agrochemicals, pharmaceuticals, and food additives. A protocol for the aminolysis of glycidol was first developed, using ammonium hydroxide to obtain 3-aminopropane-1,2-diol (4a) as target product in gram quantities. After a preliminary experiment in batch (Table S3†), the optimization in a continuous-flow setup showed that elevated temperatures and reduced residence times accelerated the reaction speed and improved mixing efficiency, resulting in higher yields of the desired product (Fig. 4a and b). The versatility of the developed reaction protocol was evaluated also for the generation of secondary amines, under optimized conditions (125 °C, 2 h) yielding 4b in 44% yield. We also investigated the preparation of hyperbranched polyglycerol 5 from glycidol in the presence of trimethylol propane (TMP) as initiator.31 After a preliminary examination in batch conditions (Table S4†), the development of a continuous-flow polymerization protocol was achieved (Fig. 4c), evaluating polymerization degree and GLD conversion by 1H NMR analysis (ESI†). Finally, a tosylation reaction (Fig. 5) was investigated, producing valuable derivatives bearing an activated hydroxyl group.32p-Toluensulfonyl chloride (TsCl) and 3, in equimolar amounts, were combined with a mixture of H2O/CHCl3 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to address solubility differences (Table S5†). Continuous-flow conditions were employed to assess residence time, TsCl concentration, and temperature effects (Fig. 5a and b). Given the use of glycidol as a building block for a variety of compounds, a complete process that combines the production of glycerol 1 from the transesterification of sunflower oil (for further details, see the ESI†), and the functionalization of glycidol 3 to produce glycidyl tosylate 6 was evaluated in batch and flow configurations, performing TEA and LCA (Fig. 5c and Table S2†).
:1) to address solubility differences (Table S5†). Continuous-flow conditions were employed to assess residence time, TsCl concentration, and temperature effects (Fig. 5a and b). Given the use of glycidol as a building block for a variety of compounds, a complete process that combines the production of glycerol 1 from the transesterification of sunflower oil (for further details, see the ESI†), and the functionalization of glycidol 3 to produce glycidyl tosylate 6 was evaluated in batch and flow configurations, performing TEA and LCA (Fig. 5c and Table S2†).
 | ||
| Fig. 4 Application of glycidol to prepare derivatives 4a or 4b under continuous-flow conditions. Influence of reaction temperatures (a) and residence time (b) on the formation of 4a. Reaction conditions: 3 (10 mmol), amine, MeOH (100 mL), pressure = 10 bar. Application of glycidol to prepare the polymer 5 under continuous-flow conditions. Reaction conditions: 3 (150 mmol), TMP (13 mmol), DMF (10 mL), pressure = 2 bar. The analytical data was determined by 1H NMR. | ||
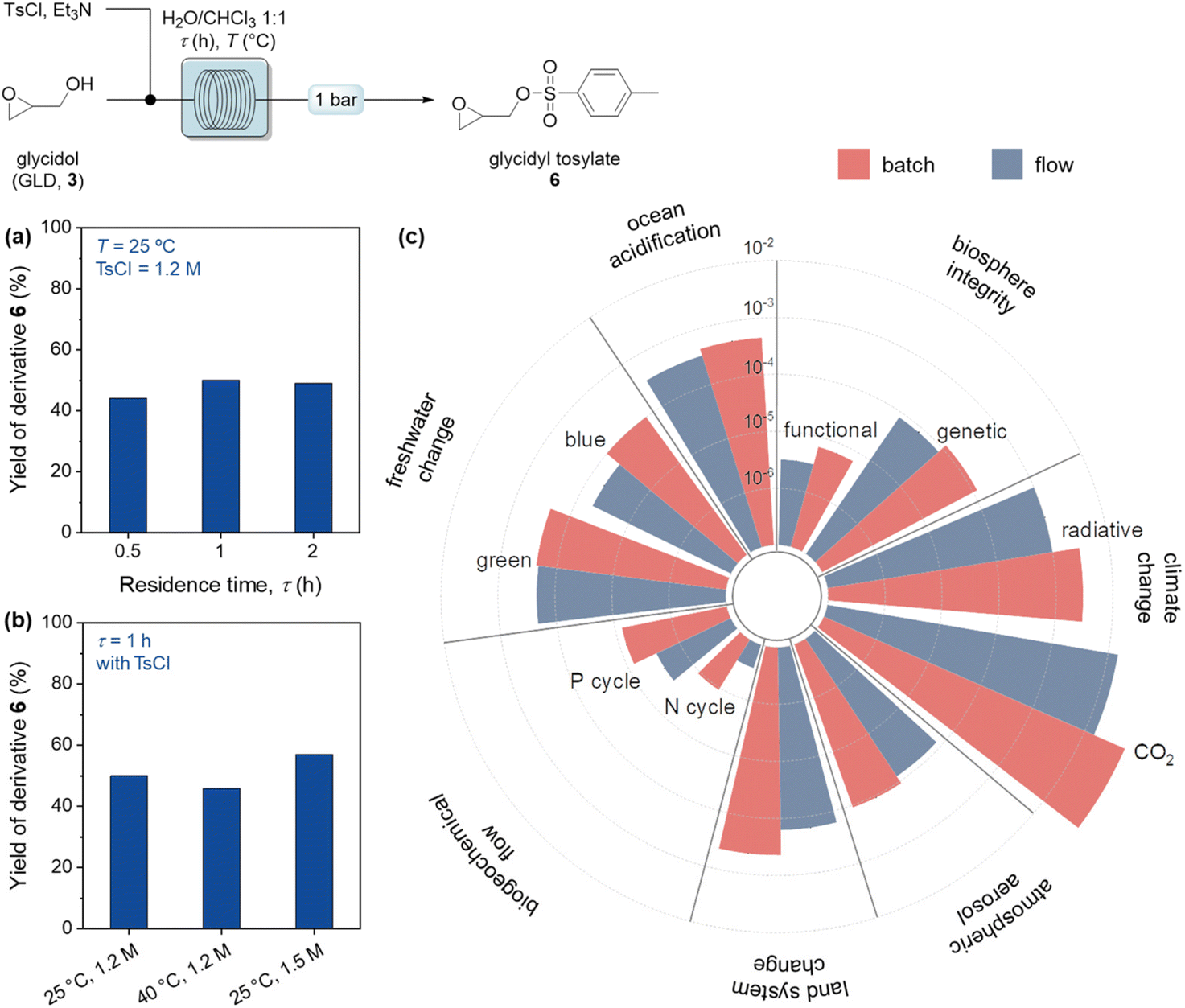 | ||
| Fig. 5 Application of glycidol to prepare derivative 6 under continuous-flow conditions. Influence of residence time (a) and temperature and TsCl concentration (b) on the formation of 6. Environmental impact of glycidyl tosylate 6 production via batch and flow process (c). Reaction conditions: 3 (18 mmol), Et3N (23 mmol), H2O (15 mL), CHCl3 (12 mL), pressure = 1 bar. The analytical data was determined by 1H NMR. | ||
Through the implementation of flow conditions, a 58.3% reduction of the total raw material cost of the process was achieved, from 17.3 (batch) to 7.2 $ per h per gproduct (flow). The duration to reach the batch capacity was also examined and it is noted that flow conditions reached the target capacity in 7.8 min, which is much less compared to the 1 h required for the batch reaction. This reduction led to a noteworthy decrease (94%) in electricity usage, from 56.1 to 3.4 W h−1 gproduct−1 between batch and flow, respectively. These values were again compared with the literature and the effect was outstanding compared to the typical annual operational reduction in expenses in the range of 9 to 40% for continuous-flow processes.33 In the context of environmental impacts (Fig. 5c), radiative forces and CO2 generation were reduced of 66.7% and 74.6% moving from batch to flow, respectively, leading to a minimized impact on climate change. It was also observed that the effects on land system change decreased by 63.6% when flow conditions were used instead of batch processes. In addition, atmospheric aerosol loads, freshwater change, and ocean acidification were moderately in favor of flow configuration, with 28.1%, 29.1% and 24.4% reductions. This positive trend was also perceived on both functional and genetic biosphere integrities. Furthermore, the impacts on biogeochemical flows such as those on the P and N cycles were lowered by 63.1% and 75.5% moving from batch to flow. These differences predominantly originate from the combination of high electricity usage and larger reagent consumption in the batch process. Therefore, considering the TEA and LCA results, continuous flow conditions were determined to be environmentally more sustainable compared to the batch conditions.
Conclusions
In conclusion, this study has successfully demonstrated the development and optimization of a continuous-flow process to produce glycidol from glycerol. The transition from batch to flow conditions addressed challenges such as extended reaction times, harsh operating conditions, and the formation of unstable intermediates. The optimized flow process not only showed higher yields and improved reaction mass intensity but also offered advantages in terms of green metrics. The investigation further expanded to the telescoped and integrated preparation of value-added derivatives of glycidol under continuous-flow conditions. Protocols for aminolysis, polymerization, and tosylation reactions were successfully developed, demonstrating the versatility and scalability of the continuous-flow approach. The assessment of techno-economic and life cycle metrics confirmed the superior performance of the flow process over traditional batch methods, considering factors such as raw material cost, duration, electricity usage, and environmental impact. Overall, this study presents an efficient and sustainable route for glycerol valorization and highlights the broader potential of continuous-flow chemistry in the synthesis of valuable derivatives. The environmentally friendly nature of the continuous-flow process, along with its economic feasibility, positions it as a promising approach for the synthesis of various compounds with applications in agrochemicals, pharmaceuticals, and food additives.Experimental methods
Batch preparation of glycidol (GLD, 3)
The batch studies were conducted using an automated multi-station batch reactor system, namely the Carousel 12 Plus Reaction Station™. This advanced reactor features twelve reaction stations, each equipped with independent temperature and stirring control. Its compact design allows simultaneous operation of multiple reactions, offering precise control over reaction parameters, including temperature, pressure, and agitation speed, and ensuring reproducibility and accuracy during high-throughput experimentation. HCl 13 M (48 mmol, 4 equiv.) was added dropwise to a stirred solution of GLY 87 wt% (12 mmol, 1 equiv.) and AcOH (4 mmol, 0.3 equiv.) at 120 °C. After the indicated reaction time, the obtained crude mixture was quenched with an aqueous solution of NaOH 9.5 M. 20 μL of the resulting solution were diluted with 4 mL of EtOH and the reaction yield was evaluated by GC-MS and NMR. In particular, GC-MS data were collected on an Agilent 5977C GC/MSD™ equipped with a HP-5MS Ultra Inert fused silica GC Column coupled with a mass spectrometer. 1H NMR and 13C NMR spectra were recorded on a Bruker 400 MHz spectrometer (see the ESI† for more details). 1H NMR (400 MHz, D2O) δ 3.90 (dd, J = 12.8, 2.7 Hz, 1H), 3.49 (dd, J = 12.9, 5.8 Hz, 1H), 3.25 (ddt, J = 5.7, 4.3, 2.9 Hz, 1H), 2.91 (t, J = 4.4 Hz, 1H), 2.76 (dd, J = 4.3, 3.0 Hz, 1H); 13C NMR (101 MHz, D2O) δ 61.8, 53.1, 45.1.Flow preparation of glycidol (GLD, 3)
The continuous-flow reactions were conducted in a modular systhem consisting in a PFA tubing (1/16′′ o.d.) equipped with PEEK/ETFE connectors and ferrules. Liquid feeds were handled using the Harvard Apparatus PHD ULTRA™ syringe pumps, equipped with gastight Hamilton syringes or with HPLC pumps (UNIQSIS FlowLab unit™). Due to the potential hazardousness of the reaction, an accurate control of the temperature was essential to ensure safe operating conditions. Therefore, the reactor temperature was managed with a HotCoil heater reactor station (UNIQSIS FlowLab unit™). Furthermore, to operate a strict regulation of the downstream pressure, a back pressure valve regulator (1–10 bar) was used. HCl 13 M (48 mmol, 4 equiv.) was added dropwise to a stirred solution of GLY (12 mmol, 1 equiv.) and AcOH (4 mmol, 0.3 equiv.). The resulting solution was introduced in a 19 mL PFA coil reactor by a syringe pump at 2 bar, using the indicated residence times and temperatures. The collected solution was quenched with an aqueous solution of NaOH 9.5 M. 20 μL of the collected solution were diluted with 4 mL of EtOH, and the reaction yield was evaluated by GC-MS and/or NMR (ESI†).Techno-economic analysis
To perform the techno-economic analysis, Aspen Plus® V11 software was used. The simulations were run in a stoichiometric reactor model, which was based on fractional conversions in order to mimic the exact experimental performance of the processes at steady-state conditions. During the simulations the convergence tolerance was set to 0.0001. The physico-chemical properties of the various components were obtained from relevant databases (i.e., APV110, APESV110 and NISTV110) integrated with the Aspen Plus® V11 software. Temperature, pressure, and molar flow rates were directly taken from the experimental reaction conditions. Electricity was also used as a utility. Equipment cost was computed by considering the real reactor volumes in this study, using the Aspen Process Economic Analyzer (APEA) suite. To analyse the raw material cost, the price of the chemical components was acquired from Merck.Life cycle assessment
The life cycle assessment was conducted in the context of cut-off system model on mass allocation and cradle-to-gate.34 Productivity capacities were selected as basis of the configurations to compare the environmental impacts of the processes per grams of final product. ReCiPe 2016 Midpoint (H) method35 and Environmental Footprint 3.1 were selected to assess the processes based on planetary boundaries. In particular, the environmental impacts were computed as follows: climate change was linked with global warming and ionizing radiation categories (ReCiPe), atmospheric aerosol loading was linked with fine particulate matter category (ReCiPe), land system change was linked with land use category (ReCiPe), biogeochemical flows were linked with freshwater and marine eutrophication categories (ReCiPe), freshwater change were linked with freshwater and marine ecotoxicity (ReCiPe), functional and genetic biosphere integrities were linked with carcinogenic toxicity; mineral and fossil resource scarcities, terrestrial acidification, ozone formation respectively (ReCiPe and EF). Ocean acidification was linked with acidification category (EF). Ecoinvent-3 datasets used for mass and energy flows which were adopted from the life-cycle inventory database. The required energy for the processes were obtained via Aspen Plus® V11 simulations as mentioned previously on the techno-economic analysis section of this study.Author contributions
AS and GV designed the study. AS and IM collected catalytic data and performed data analysis. MCI performed the techno-economic assessment. AS and GV wrote the manuscript with contributions from all co-authors. All authors gave approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
AS thanks the European Commission for funding (grant agreement 101057430, SusPharma). MCI has received funding from the European Commission's Horizon Europe research and innovation program under the Marie Skłodowska-Curie doctoral network GreenDigiPharma (grant agreement 101073089). The authors would like to thank Politecnico di Milano for providing open access funding.References
- T. Keijer, V. Bakker and J. C. Slootweg, Nat. Chem., 2019, 11, 190–195 CrossRef CAS PubMed.
- T. L. Chen, H. Kim, S. Y. Pan, P. C. Tseng, Y. P. Lin and P. C. Chiang, Sci. Total Environ., 2020, 716, 136998 CrossRef CAS PubMed.
- R. A. Sheldon and M. Norton, Green Chem., 2020, 22, 6310–6322 RSC.
- A. Sivo, R. de S. Galaverna, G. R. Gomes, J. C. Pastre and G. Vilé, React. Chem. Eng., 2021, 6, 756–786 RSC.
- S. M. Gade, V. B. Saptal and B. M. Bhanage, Catal. Commun., 2022, 172, 106542 CrossRef CAS.
- Y. Zheng, X. Chen and Y. Shen, Chem. Rev., 2010, 108, 5253–5277 Search PubMed.
- G. Bagnato, A. Iulianelli, A. Sanna and A. Basile, Membranes, 2017, 7, 17–47 CrossRef PubMed.
- M. Checa, S. Nogales-Delgado, V. Montes and J. M. Encinar, Catalysts, 2020, 10, 1279–1319 CrossRef CAS.
- G. S. Singh, K. Mollet, M. D′hooghe and N. De Kimpe, Chem. Rev., 2013, 113, 1441–1498 CrossRef CAS PubMed.
- M. O. Sonnati, S. Amigoni, E. P. Taffin de Givenchy, T. Darmanin, O. Choulet and F. Guittard, Green Chem., 2013, 15, 283–306 RSC.
- M. Ricciardi, F. Passarini, I. Vassura, A. Proto, C. Capacchione, R. Cucciniello and D. Cespi, ChemSusChem, 2017, 10, 2291–2300 CrossRef CAS PubMed.
- J. Seiwert, D. Leibig, U. Kemmer-Jonas, M. Bauer, I. Perevyazko, J. Preis and H. Frey, Macromolecules, 2016, 49, 38–47 CrossRef CAS.
- F. Della Monica, A. Buonerba, A. Grassi, C. Capacchione and S. Milione, ChemSusChem, 2016, 9, 3457–3464 CrossRef CAS PubMed.
- R. Villa, R. Porcar, S. Nieto, A. Donaire, E. Garcia-Verdugo, S. V. Luis and P. Lozano, Green Chem., 2021, 23, 4191–4200 RSC.
- L. Chiummiento, M. Funicello, P. Lupattelli, F. Tramutola and P. Campaner, Tetrahedron, 2009, 65, 5984–5989 CrossRef CAS.
- K. Pieper, R. Bleith, C. Köhler, R. Mika and A. Gansäuer, Angew. Chem., Int. Ed., 2024, 63, e2023175 CrossRef PubMed.
- C. Muzyka, S. Renson, B. Grignard, C. Detrembleur and J.-C. M. Monbaliu, Angew. Chem., Int. Ed., 2024, 63, e2023190 CrossRef PubMed.
- S. Gade, M. Munshi, B. M. Chherawalla, V. H. Rane and A. A. Kelkar, Catal. Commun., 2012, 27, 184–188 CrossRef CAS.
- A. Dibenedetto, A. Angelini, M. Aresta, J. Ethiraj, C. Fragale and F. Nocito, Tetrahedron, 2011, 67, 1308–1313 CrossRef CAS.
- C. L. Bolívar-Diaz, V. Calvino-Casilda, F. Rubio-Marcos, J. F. Fernández and M. A. Bañares, Appl. Catal., B, 2013, 129, 575–579 CrossRef.
- Y. K. Endah, M. S. Kim, J. Choi, J. Jae, S. D. Lee and H. Lee, Catal. Today, 2017, 293–294, 136–141 CrossRef CAS.
- D. V. Silva-Brenes, S. K. Reyes-Vargas, J. Duconge, C. Vlaar, T. Stelzer and J.-C. M. Monbaliu, Org. Process Res. Dev., 2024, 28, 1704–1712 CrossRef CAS.
- A. Sivo, T. K. Kim, V. Ruta, R. Luisi, J. Osorio-Tejada, M. Escriba-Gelonch, V. Hessel, M. Sponchioni and G. Vilé, React. Chem. Eng., 2022, 7, 2650–2658 RSC.
- A. Kostyniuk, D. Bajec, P. Djinović and Blaž Likozar, Chem. Eng. J., 2020, 394, 124945–124958 CrossRef CAS.
- R. Tesser, E. Santacesaria, M. Di Serio, G. Di Nuzzi and V. Fiandra, Ind. Eng. Chem. Res., 2007, 46, 6456–6465 CrossRef CAS.
- A. D. Allian, N. P. Shah, A. C. Ferretti, D. B. Brown, S. P. Kolis and J. B. Sperry, Org. Process Res. Dev., 2020, 24, 2529–2548 CrossRef CAS.
- E. Santacesaria, R. Vitiello, R. Tesser, V. Russo, R. Turco and M. Di Serio, Ind. Eng. Chem. Res., 2014, 53, 8939–8962 CrossRef CAS.
- F. Tollini, A. Occhetta, F. Broglia, V. Calemma, S. Carminati, G. Storti, M. Sponchioni and D. Moscatelli, React. Chem. Eng., 2022, 7, 2211–2223 RSC.
- C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef CAS.
- C. Sampat, L. Kotamarthy, P. Bhalode, Y. Chen, A. Dan, S. Parvani, Z. Dholakia, R. Singh, B. J. Glasser, M. Ierapetritou and R. Ramachandra, J. Adv. Manuf. Process., 2022, 4, e10136 CrossRef CAS.
- R. K. Kainthan, E. B. Muliawan, S. G. Hatzikiriakos and D. E. Brooks, Macromolecules, 2006, 39, 7708–7717 CrossRef CAS.
- T. Devadoss, J. Mol. Struct., 2023, 1289, 135850 CrossRef CAS.
- S. D. Schaber, D. I. Gerogiorgis, R. Ramachandran, J. M. B. Evans, P. I. Barton and B. L. Trout, Ind. Eng. Chem. Res., 2011, 50, 10083–10092 CrossRef CAS.
- “ISO 14040 Environmental management, life cycle asssessment, principles and framework”, 2006; https://www.iso.org/standard/37456.html (accessed Jan 2024).
- M. A. J. Huijbregts, Z. J. N. Steinmann, P. M. F. Elshout, G. Stam, F. Verones, M. Vieira, M. Zijp, A. Hollander and R. van Zelm, Int. J. Life Cycle Assess., 2017, 22, 138–147 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc01565g |
| This journal is © The Royal Society of Chemistry 2024 |