
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCross-acyloin condensation of aldehydes catalysed by transketolase variants for the synthesis of aliphatic α-hydroxyketones†
Giuseppe
Arbia
,
Camille
Gadona
,
Hubert
Casajus
,
Lionel
Nauton
,
Franck
Charmantray
* and
Laurence
Hecquet
*
Université Clermont Auvergne, CNRS, Clermont INP, Institut de Chimie de Clermont-Ferrand (ICCF), F-63000 Clermont-Ferrand, France. E-mail: franck.charmantray@uca.fr; laurence.hecquet@uca.fr
First published on 30th May 2024
Abstract
We demonstrate that transketolase variants from Geobacillus stearothermophilus catalyse an acyloin condensation reaction involving two hydroxylated or not aliphatic aldehydes. This promiscuous TK-catalysed reaction offers an attractive alternative to the ketol transfer from α-ketoacids used as donor substrates in the usual TK mechanism, adding atom economy by avoiding carbon dioxide release. Transketolase variants H102L/L118I/H474G(S) showed a de novo activity towards the self-condensation of propanal, and to a lesser extent of ethanal, and a remarkable ability to control the selectivity of the more challenging cross-acyloin condensation reaction with propanal or iso-butanal used as nucleophiles, and different hydroxylated aldehydes (C2–C4) as electrophiles. The synthesis of seven aliphatic symmetrical and unsymmetrical α-hydroxyketones was performed from stoichiometric amounts of aldehydes, giving yields similar to those obtained with the common TK reaction based on α-ketoacid decarboxylation. This novel enzymatic cross-acyloin condensation reaction extends the toolbox for the synthesis of unsymmetrical aliphatic α-hydroxyketones while improving mass metrics of previous enzymatic and chemical strategies.
Introduction
α-Hydroxyketones (acyloins) are highly valuable compounds for numerous applications in fine chemicals,1 especially in pharmaceuticals,2 where they are used as building blocks for the synthesis of drugs and pro-drugs,3 such as olmesartan medoxomil,4 used to treat high blood pressure. Although symmetrical (aromatic or aliphatic) α-hydroxyketones can be prepared by chemo-benzoin-type condensation reactions catalysed by N-heterocyclic carbenes (NHCs) coupled with a base,5 this strategy is much less convenient for access to a broad range of unsymmetrical, aliphatic, and hydroxy-functionalised α-hydroxyketones,6 owing to the difficulty controlling both chemo- and regio-selectivity. Some organocatalytic methods are reported but they all use “chemical tricks” such as a large excess (typically 10-fold) of one of the reagents,7 or sterically-hindered substrates (e.g. ortho-substituted aromatics),8 an approach that inherently narrows the scope of applications and lowers overall efficiency. Biocatalysis is an interesting alternative for aliphatic α-hydroxyketone synthesis. Indeed, NHCs are chemical mimics of thiamine diphosphate (ThDP), a natural derivative of vitamin B1 which is a cofactor present in a variety of enzymes used for the synthesis of α-hydroxyketones. The biocatalytic strategies offer not only high chemo–regio- and stereoselectivities but also mild reaction conditions (neutral pH, room temperature, atmospheric pressure and water as solvent). The enzymatic mechanisms of ThDP-dependent enzymes can be a transfer of a ketol or acyl unit from an α-ketoacid to an aldehyde (pyruvate decarboxylase,9 pyruvate dehydrogenase,10 transketolase11) or an acyloin-type condensation reaction of two different aldehydes (benzaldehyde lyase,12 acetoin dehydrogenase,13 formolase14). This last strategy has the great advantage of adding to previous green criteria, atom economy by avoiding the release of carbon dioxide generated by the decarboxylation of α-ketoacids required by transferases but the best regio- and stereo-selectivities were obtained by cross-acyloin condensation of an aliphatic aldehyde with an aromatic or branched aldehyde.Here we found that acyloin-type condensation reaction can be catalysed by the ThDP-dependent enzyme transketolase (TK) which controls both the regio- and the stereoselectivity with two different aliphatic aldehydes (hydroxylated or not) leading to unsymmetrical, aliphatic, and hydroxy-functionalised α-hydroxyketones in one step. This unreported promiscuous cross-acyloin-type condensation reaction avoids carbon dioxide generation from α-ketoacids required by the common TK-catalyzed reaction and offers green advances compared to the chemical strategies catalysed by NHCs.
For biocatalytic applications, the common TK donor substrate is the α-ketoacid, hydroxypyruvate (HPA). The reaction being under kinetic control, the equilibrium is displaced toward the release of the α-hydroxyketone and carbon dioxide (Scheme 1). The TK-catalyzed reaction is stereospecific, and the newly-formed asymmetric centre of the product displays S absolute configuration. While HPA was exclusively used for biocatalytic applications, TKs offer more flexibility towards acceptor substrates than most other ThDP-dependent enzymes. Although TKs showed a preference for (2R)-hydroxylated aldehydes, differently substituted, non-phosphorylated alkyl, allyl, aromatic, or heterocyclic moieties have been demonstrated to be substrates, and many corresponding products have been isolated on a preparative scale.15–17 The mesophilic TKs from Saccharomyces cerevisiae18 (TKsce) and from Escherichia coli19 (TKeco) have been largely used for biocatalytic applications and altered by mutagenesis.15,16 More recently, we discovered the first thermostable TK from the thermophilic organism Geobacillus stearothermophilus (TKgst), offering significant tolerance to elevated temperatures and robustness towards non-conventional reaction conditions, whether with free or immobilised enzyme.17,20 Using HPA as donor substrate, we showed that selected TKgst variants were able to accommodate new acceptor substrates17a,c such as (2S)-hydroxyaldehydes of ranging carbon chain length (C4–C8), or aliphatic or arylated aldehydes.
 | ||
| Scheme 1 Transketolase-catalysed transfer of a ketol (or an acyl group) from an α-ketoacid to an aldehyde. | ||
We also reported that selected TKgst variants accepted pyruvate and some analogues as novel hydrophobic donors in place of HPA, offering interesting prospects.17e,f,h In the well-known TK mechanism, the activated ThDP (ylide) carries out a nucleophilic attack on the carbonyl of the donor substrate (α-ketoacid), followed by decarboxylation of the intermediate, yielding the α,β-dihydroxyethylthiamine carbanion (DHEThDP) as a ketol activated donor ready to condense with an aldehyde as an acceptor substrate. For example, this reaction leads to L-erythrulose with HPA as donor and glycolaldehyde as acceptor (Scheme 2, pathway A).
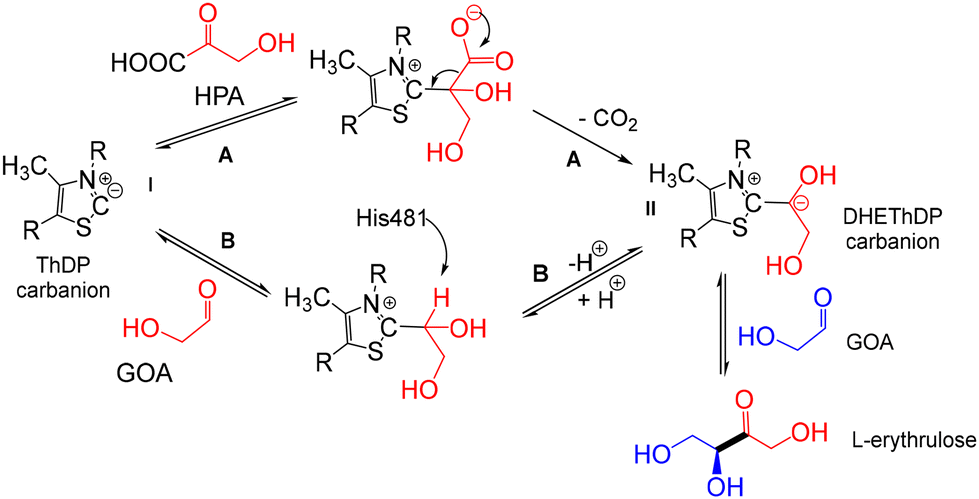 | ||
| Scheme 2 Proposed mechanisms catalysed by yeast TK for the formation of L-erythrulose from α,β-dihydroxyethylthiamine diphosphate carbanion (DHEThDP) generated with two different nucleophiles, hydroxypyruvate (HPA) by decarboxylation (pathway A) or from glycolaldehyde (GoA) by deprotonation (pathway B).21 | ||
Besides this commonly accepted ping-pong TK mechanism, a promiscuous acyloin-condensation reaction (Scheme 2, pathway B) has been reported with glycolaldehyde (GoA) only, this latter playing the role of both nucleophile and electrophile.21,22 In 2017, Hanefeld et al.21 suggested that the formation of the DHEThDP II intermediary occurred in two stages (Scheme 2). First, the activated ThDP I (ylide) carries out a nucleophilic attack on the carbonyl of a first GoA molecule (nucleophile) followed by a deprotonation reaction catalysed by His 481 in the active site of TK from Saccharomyces cerevisiae to generate the DHEThDP ThDP II common to both pathways A and B. In a second step, the carbonyl group of a second GoA molecule undergoes a stereospecific condensation with II to give L-erythrulose. Hanefeld et al.21 suggested that DHEThDP from pathway B should be catalysed more slowly than its generation by decarboxylation (Scheme 2, pathway A).
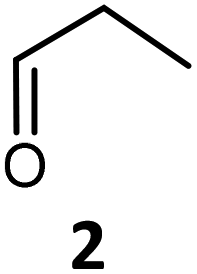
Here, our aim was first to exemplify the promiscuous self-acyloin-condensation reaction described with GoA, using three aliphatic aldehydes (ethanal 1, propanal 2, iso-butanal 3) in the presence of TKgst variants and to compare the results with the common TKgst reaction involving the corresponding α-ketoacids (pyruvate 1′, 2-oxobutyrate 2′ and 3-methyl-2-oxobutyrate 3′) that transfer the same acyl group after decarboxylation in the presence of ethanal 1, propanal 2, and iso-butanal 3 respectively as electrophiles.
Then, TKgst variants were selected to specifically control the cross-acyloin condensation to form mainly one α-hydroxyketone from two different aldehydes (R1–CHO acting as nucleophilic acyl donor and R2–CHO as an electrophilic acceptor) while the uncontrolled cross-acyloin condensation leads to four potential products (Scheme 3).
 | ||
| Scheme 3 Possible α-hydroxyketone product distribution from two different aldehydes by TKgst variant-catalysed cross-acyloin condensation or uncontrolled acyloin condensation. | ||
Finally, the cross-acyloin condensation was performed using two different aldehydes, propanal 2 and iso-butanal 3 as nucleophiles and aldehydes 4–6 (C2–C4) as electrophiles, giving unsymmetrical aliphatic α-hydroxyketones 9–13 (Scheme 4).
 | ||
| Scheme 4 Synthesis of α-hydroxyketones 7–13 catalysed by TKgst variants from α-hydroxy-β-(polyhydroxy)alkylthiamine diphosphate (acyl ThDP) carbanion II generated with two possible nucleophiles, aliphatic α-ketoacids 1′, 2′, 3′ (pathway A) or aliphatic aldehydes 1, 2, 3 (pathway B) in the presence of aldehydes 1–6 as electrophiles. | ||
Results and discussion
Analytical study of TKgst variants for self-acyloin condensation
In previous studies, we reported the improvement of TKgst activity towards aliphatic acyloins.17g We used a semi-rational engineering approach that focused on residues H102, L118, H474 as key positions. We screened five libraries, H102L/H474X/D470X (or L382X, or F435X or L191X or L118X), with aliphatic α-hydroxy ketone propioin 8, the product of the self-acyloin condensation reaction of propanal 2. During this work, when we performed the controls with propanal 2 only (R2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2–CH3), we observed that four variants H102L/L118I, H102L/H474S, H102L/L118I/H474S and H102L/L118I/H474G (selected here) were able to catalyse the self-acyloin condensation of this aldehyde to give propioin 8. This hitherto unreported finding with this type of aldehyde prompted us to study this phenomenon further. Indeed, this promiscuous self-acyloin-condensation reaction has been described with GOA only and wild-type TK by different groups (Scheme 2, pathway B) but the product L-erythrulose was obtained in quite low amounts.21,22
CH2–CH3), we observed that four variants H102L/L118I, H102L/H474S, H102L/L118I/H474S and H102L/L118I/H474G (selected here) were able to catalyse the self-acyloin condensation of this aldehyde to give propioin 8. This hitherto unreported finding with this type of aldehyde prompted us to study this phenomenon further. Indeed, this promiscuous self-acyloin-condensation reaction has been described with GOA only and wild-type TK by different groups (Scheme 2, pathway B) but the product L-erythrulose was obtained in quite low amounts.21,22
Following our previous results with propanal 2 and the best four variants H102L/L118I, H102L/H474S, H102L/L118I/H474S and H102L/L118I/H474, we set out to study the self-acyloin condensation of two other aldehydes, ethanal 1 and iso-butanal 3 (Scheme 3, pathway B). These variants were used earlier with the corresponding α-ketoacids (pyruvate 1′, 2-oxobutyrate 2′, 3-methyl-2-oxobutyrate 3′) as nucleophiles in the presence of the aldehydes 1, 2, and 3 as electrophiles to obtain the corresponding α-hydroxyketones.17d,f,h
For this study, TKgst variants were expressed in E. coli BL21(DE3)pLysS strain and purified by Ni2+ chelating affinity column chromatography. To quantify the formation over time of α-hydroxyketones (Fig. 1) produced by the self-condensation reaction of aldehydes 1, 2, 3 (Scheme 3, pathway B) and compare the results with the corresponding α-ketoacids bearing the same acyl group (pyruvate 1′, 2-oxobutyrate 2′, 3-methyl-2-oxobutyrate 3′) in the presence of aldehydes 1, 2, 3 (Scheme 3, pathway A), we used a spectropho tometric discontinuous assay based on the selective detection of α-hydroxyketone by reaction with 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-tetrazolium (WST-1) as a water-soluble tetrazolium salt. WST-1 was also used to determine the activity of other ThDP enzymes including pyruvic acid decarboxylase,23 TK,24 and transaminases.25 The principle of this assay consists in regularly drawing aliquots of TK-catalysed reactions assayed separately with WST-1 under basic conditions at 60 °C. The reaction generates a blue formazan colour, followed by measuring absorbance at 600 nm.26 We also determined the standard curves for acetoin 7 and propioin 8 to calculate the molar extinction coefficient ε of these two α-hydroxyketones 7 and 8 (ESI†).
 | ||
Fig. 1 Determination of α-hydroxyketone concentration using 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-tetrazolium (WST-1) assay following pathway A with pyruvate 1′ and ethanal 1 (a) 2-oxobutyrate 2′ and propanal 2 (b) or pathway B with ethanal 1 only (c) propanal 2 only (d). Control without TKgst wild-type TKgst wild-type TKgst H102L/L118I/H474S H102L/L118I/H474S  H102L/L118I/H474G H102L/L118I/H474G  . The reactions were performed at 37 °C in 200 μL phosphate buffer (50 mM, pH 7.0) containing ThDP (0.1 mM), MgCl2 (1 mM), aldehyde alone 1 or 2 (100 mM) or a mixture of aldehyde 1 or 2 (50 mM) with α-ketoacid 1′ or 2′ (50 mM) in the presence of wt-TKgst or TKgst variants (2.5 mg mL−1). . The reactions were performed at 37 °C in 200 μL phosphate buffer (50 mM, pH 7.0) containing ThDP (0.1 mM), MgCl2 (1 mM), aldehyde alone 1 or 2 (100 mM) or a mixture of aldehyde 1 or 2 (50 mM) with α-ketoacid 1′ or 2′ (50 mM) in the presence of wt-TKgst or TKgst variants (2.5 mg mL−1). | ||
After a first screening of the three aldehydes 1, 2 and 3 in the presence of the four variants and wild-type TKgst as control, H102L/L118I and H102LH474S were discarded because conversion was low whichever aldehyde was used, showing that the cooperative effect of the three mutations (H102L/L118I/H474S(G) was required to 1′, 2-oxobutyrate 2′, 3-methyl-2-oxobutyrate 3′) used in pathway A, an exchange of the two histidine H102 and H474 by smaller and less polar amino acids was required to make space for larger hydrophobic donor substrates compared with HPA (commonly used as donor), while keeping the catalytic mechanism intact. We expect similar results for the self-condensation of corresponding aldehydes 1, 2 and 3 in pathway B. In addition, as the Leu118 residue is involved in the stabilisation of the thiazolium cycle of ThDP, the mutation of Leu118 into an isoleucine (L118I) may also increase the stabilisation of these aliphatic substrates.
According to our experimental results at analytical scale (Fig. 1) in the presence of substrates and cofactor or in the presence only of wild-type TKgst, the self-acyloin condensation of aldehydes 1, 2, and 3 or ketol transfer from α-ketoacid 1′, 2′, 3′ did not take place, as confirmed by in situ NMR analysis of the reaction mixtures (ESI†). After 24 h, the largest amounts of α-hydroxyketone 8 (41–43 mM) were obtained in the presence of the TKgst variant H102L/L118I/H474G with 2-oxobutyrate 2′ and propanal 2 (pathway A) or with propanal 2 only (pathway B) (Fig. 1b–d), but this concentration level was achieved more slowly from pathway B. Pathway A (with 2′ and 2) gave the α-hydroxyketone 8 (43 mM) after 4 h (Fig. 1b), while pathway B catalysed by the same variant yielded a similar concentration (41 mM) after 24 h (Fig. 1d). This result confirms the hypothesis of Hanefeld21 suggesting a slower DHEThDP generation from pathway B (in the presence of GoA only) compared to its generation by decarboxylation of α-ketoacid from pathway A in the presence of HPA and GoA (Scheme 2).
We did not observe self-condensation of iso-butanal 3 or ketol transfer from 3-methyl-2-oxobutyrate 3′ to iso-butanal 3 (ESI†). These results show that TKgst variants H102L/L118I/H474S(G) greatly improved the formation of α-hydroxyketones particularly with propanal 2 compared to wild type TKgst. The concentrations of products 7 or 8 were similar whatever the pathway A (from decarboxylation of α-ketoacid 1′ or 2′) or pathway B (from self-condensation of aldehyde 1 or 2). These analytical results extend the self-acyloin condensation described only with GoA and wild type TK21,22 to non-hydroxylated aldehydes 1 and 2.
In silico study of acylThDP carbanion formation from pathway B
To propose a possible mechanism for acylThDP carbanion formation from aldehydes 1 and 2 (Scheme 3, pathway B), we conducted in silico studies using H102L/L118I/H474G(S) TKgst variant active site models (ESI†). The mechanism reported by Hanefeld for yeast TK21 cannot be applied in our case. His 481 in yeast TK identified as a possible residue for the deprotonation of the ThDP intermediate leading to DHEThDP corresponds to His 474 in TKgst replaced in the selected variants by a serine or glycine. This residue H474 in wild-type TKgst is responsible for ThDP carbanion formation, but in TKgst variants used in this work, His 474 was replaced by a glycine or serine, which are unable to capture the proton of the intermediate I-B. We explain the acylThDP carbanion formation in TKgst variant H102L/L118I/H474G with propanal 2 as nucleophile by an attack on the carbonyl of a first molecule of 2 followed by a proton transfer of I-Bvia a water molecule to His 263 (Fig. 2). This mechanism was confirmed by molecular dynamics studies (ESI†). | ||
| Fig. 2 Model of H102L/L118I/H474G TKgst active site with propanal 2 (cyan) as nucleophile and ThDP (light purple). Active site construction was based on 3M49 pdb structure with Chimera.27 The propanal 2 position was determined from a molecular dynamics study carried out by NAMD.28 | ||
In addition, the presence of leucine in place of histidine in the 102 position (H102L) near the methyl group of propanal 2 stabilised the molecule and favoured this mechanism. In a second step, the nucleophile and GoA 4 as electrophile. Both compounds compete for the same active site, but the well-known TK reaction proceeds according a ping-pong mechanism.29 Hence, with H102L/L118I/H474G TKgst, variant as aliphatic substrate, propanal 2 reacts firstly as nucleophile to give the DHEThDP carbanion, which carbonyl group of a second propanal molecule undergoes a condensation with acylThDP carbanion to give the acyloin product 8. The lower reactivity of 1 compared with 2 observed earlier (Fig. 1) could be explained by the smaller size of 1, leading to a lower stabilisation by the active site residues. The molecular dynamics studies also showed one main position of propanal 2, whereas iso-butanal 3 gave three possible positions, explaining the lower stereoselectivity in the presence of GoA 4 as acceptor substrate (ESI†). The absence of a self-condensation product in the presence of iso-butanal 3 may be due to steric hindrance preventing the condensation of the acylThDP carbanion on another iso-butanal molecule 3.
Preparative-scale synthesis and characterisation of α-hydroxyketones obtained by acyloin condensation
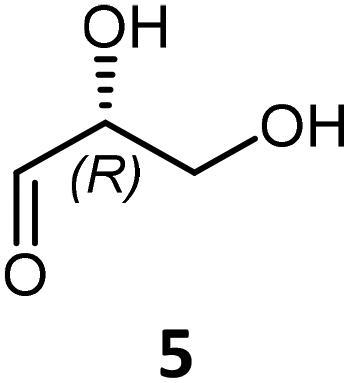
To study and identify the cross-acyloin condensation products (Scheme 3, pathway B), the three aldehydes (ethanal 1, propanal 2, iso-butanal 3) were chosen as nucleophiles in the presence of hydroxylated aldehydes (C2–C4) 4, 5 and 6 already reported as substrates of TKgst variants H102L/L118I/H474S and H102L/L118I/H474G according pathway A17h (Table 1).| Nucleophile | Electrophile | Product | Time (h) | TKgst variant | TKgst (mg mL−1) | In situ yieldb (%) | Isolated yield (%) | eec or deb (%) |
|---|---|---|---|---|---|---|---|---|
| a Addition of the total amount of 3 at initial time, sequential addition of stoichiometric amount of 4 or 5 for 20 h (2.5 mM h−1). b Determined by in situ1H NMR using TSP-d4 as an internal standard and calculated based on in situ product formation. c Determined by chiral GC-MS analysis after derivatization.17d | ||||||||
|

|
|
72 | H102L/L118I/H474G | 2.5 | 80 | 50 | 69 |

|
|
72 | H102L/L118I/H474S | 3 | 48 | 31 | >95 | |

|

|
48 | H102L/L118I/H474S | 2.5 | 75 | 50 | >95 | |

|

|

|
72 | H102L/L118I/H474G | 2 | 82 | 60 | 34 |
|

|
72 | H102L/L118I/H474S | 3 | 39 | 31 | >95 | |
In the common TKgst reaction based on decarboxylation of α-ketoacids as nucleophile (pathway A), we showed that H102L/L118I/H474G was selected in the presence of an aldehyde acceptor with short carbon chain (C2–C3) and H102L/L118I/H474S with hydroxylated and longer carbon chain aldehyde C3–C4.17g
The reactions were performed at 37 °C at 1 mmol scale, in the presence of 1.5 to 3 mg of purified TKgst variants in 20 mL phosphate buffer (50 mM) adjusted to pH 7.0. The substrates and products were quantified by in situ1H NMR analysis using 3-(trimethylsilyl) propionic-2,2,3,3-d4 acid (TSP-d4) as an internal standard from aliquots taken from the reaction mixtures over time, allowing th measurement of the final conversion levels (ESI†). The products were purified by silica gel chromatography and characterised by NMR.
Ethanal 1 gave only the self-condensation product 7 (ESI†) with good in situ (84%) and isolated yields (67%).
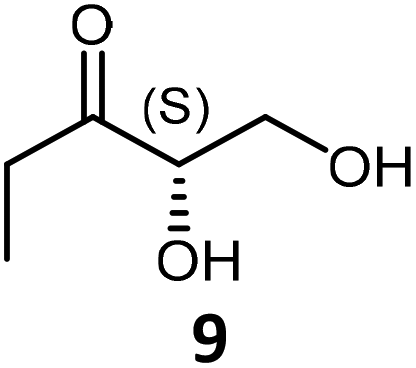
Propanal 2 alone or glycolaldehyde 4 alone led to the self-condensation products propioin 7 or L-erythrulose respectively (ESI†). When propanal 2 and glycolaldehyde 4 were together in the presence of TKgst variant, one main cross-acyloin condensation product 9 (80% in situ yield and 50% isolated yield) was obtained. Two minor products were observed: L-erythrulose from self-acyloin condensation of GoA 2 (8%) and propioin (8%) 8 from self-condensation of propanal 4. To explain the structure of the compound 9, propanal 2 should be considered as nucleophile and then attacks the carbonyl of glycolaldehyde 4 considered as electrophile (additional experiments in ESI† confirmed that glycolaldehyde 4 is a better acceptor substrate than propanal 2).
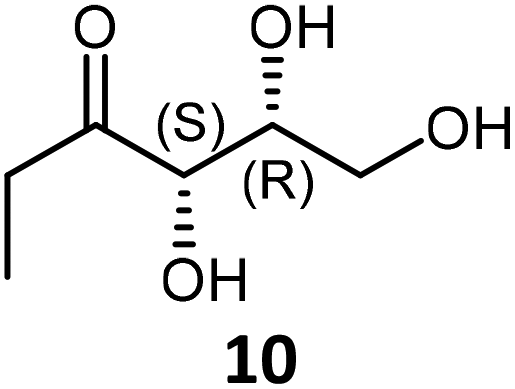
Propanal 2 with (2R)-hydroxylated aldehydes 5 and 6 bearing one and two carbon(s) more than GoA gave the corresponding α-hydroxyketones 10 and 11 respectively. 10 was obtained with lower in situ and isolated yields than 11 (48% and 31% after 72 h and 75% and 50% after 48 h respectively). In all cases, the analysis of the reaction mixtures by NMR showed a small amount of propioin 8 from the self-acyloin condensation of the nucleophile 2 (0–24%).
Although no self-acyloin condensation was observed with iso-butanal 3 only, probably owing to the steric hindrance of the isobutyl group, the cross-acyloin condensation with linear aldehydes 4 and 5 was efficient, the corresponding expected α-hydroxyketones 12 and 13 being isolated with excellent to fairly good in situ yields (82% for 12 and 39% for 13) after 72 h of reaction. To limit the formation of L-erythrulose resulting from the self-condensation of GoA 4 in the synthesis of α-hydroxyketones 12 and the chemical transformation of D-glyceraldehyde 5 into dihydroxyacetone in the synthesis of 13, aldehydes 4 and 5 were added to the reaction mixtures by syringe perfusion.
Overall, and following pathway B, the α-hydroxyketones 7–13 were obtained with yields and reaction times similar to those obtained following pathway A, except that the quantities of enzymes used were roughly 1.5 to 2 times greater.17h Overall, the results showed a greater ability of propanal 2 to catalyse self- and cross-condensation with a broader spectrum of electrophilic aldehydes. These results corroborate our previous findings showing that oxobutyrate 2′ was a better donor substrate than pyruvate 1′ or 3-methloxobutyrate 3′.17e,f,h
The selected TKgst variants were able to control the regioselectivity of the condensation, giving in all cases the targeted α-hydroxyketone. The stereoselectivity study conducted by chiral GC-MS chromatography after derivatization of products 9 and 12 (obtained with GoA 4 as eclectrophile and propanal 2 or iso-butanal 3 respectively as nucleophiles) gave the same enantiomeric excess (ee) values when products 9 and 12 were synthesised according pathway A (with α-ketocids 2′ and 3′) (ESI†). The higher ee for 9 (69%) than for 12 (34%) could be explained by the better stabilisation of the nucleophile propanal 2 in the active site compared to iso-butanal 3 as shown by molecular dynamics studies (ESI†). In the presence of chiral di- or tri-hydroxylated aldehyde 5 and 6 as electrophiles and propanal 2 or iso-butanal 3 as nucleophile, only one diastereoisomer of α-hydroxyketones 10, 11 and 13 was obtained as demonstrated by the in situ NMR analysis of the reaction mixtures. We considered that the new asymmetric carbon had mainly an S absolute configuration, given the NMR spectra identical to those obtained with the same compounds synthesised with α-ketoacids 2′ and 3′ and aldehydes 5 or 6 already reported in the literature.17d,h The high diastereoselectivities obtained with products 10, 11 and 13 (>95%) may be explained by a specific interaction of the C2 (R) hydroxyl group of the electrophiles 5 and 6 with Asp 470 as reported previously17 allowing a better stabilisation of these aldehydes compared to the mono-hydroxylated GoA 4, and favouring the formation of one diastereoisomer.
The characterisation of the cross-acyloin products 9–13 obtained with good yields confirms that pathway B can be an efficient and economical alternative to the decarboxylation of α-ketoacids required for the common pathway A. The determination of some mass metrics31 gave notably 100% of Atom Economy (AE) for pathway B against 60% for pathway A. This novel strategy can also be compared in terms of environmental impact to chemical approaches particularly those using NHCs. Triazolium or thiazolium Rovis salts were also reported to catalyse the cross-acyloin condensation with aliphatic aldehydes as nucleophiles such as ethanal 1, propanal 2 and iso-butanal 3 but only with aromatic aldehydes as electrophiles.32,33 The cross-products were obtained in good yields (55–80%) and ee (60–81%) but the reactions were performed in THF or m-xylene as solvents in the presence of a base (RbCO3 or CsCO3) and required often an excess of nucleophile (1.2 to 15 eq.) giving E factor values from 1.1 to 8.8 (ESI†). TKgst catalysed cross-acyloin reactions described in this paper with stoichiometric amounts of substrates gave lower E factor range of 0.6–1.9 (ESI†). In addition, E factor does not include the nature of catalysts important for our purpose since enzymes are biosourced while NHCs are synthetic and toxic.
Experimental
General
All chemicals were purchased from Sigma-Aldrich, Alfa-Aesar TCI chemicals and CarboSynth. Bradford reagent was from Bio-Rad. Ni-NTA resin was obtained from QIAGEN. Lyophilisation was carried out with Triad LABCONCO dryer. UV-visible absorbance was measured using a Spark control 10 microplate reader from TECAN and an Agilent Technologies. MARCHEREY-NAGEL GmbH & Co KG 60/40-63 mesh silica gel for Liquid Flash Chromatography and MARCHEREY-NAGEL GmbH & Co KG 60 F254 silica gel TLC plates with anisaldehyde stain for detection were used. Reaction pH for preparative synthesis was maintained using a TitroLine®7000 autotitrator. NMR spectra were recorded in D2O on 400 MHz Bruker Avance III HD and 500 MHz Avance III HD spectrometers. Chemical shifts are referenced to the residual solvent peak. The following multiplicity abbreviations are used: (s) singlet, (d) doublet, (dd) doublet of doublet, (dq) doublet of quartet, (ddq) doublet of doublet of quartet (dqd) doublet of quartet of doublet, (sep) septuplet, (t) triplet, (q) quadruplet, (m) multiplet.In silico studies of TKgst variants. Molecular modelling
The TKgst variants H102L/L118I/H474G(S) were modelled from the native TKgst structure deposited in the pdb (3M49). Each residue was mutated using the Chimera Rotamer module. The choice of the position of the side chains was made according to the steric bulk and the best possible choice in the Dunbrack positioning. Propanal was introduced into the active site and placed in such a way that the reactive carbons of ThDP and propanal were at a distance of 3.0 Å (determined in our previous studies),29 corresponding to an initial state. This construction was then solvated in a water box of 120 Å × 120 Å × 120 Å while retaining the crystallographic water molecules of 3M49 (38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 221 water molecules). The NaCl concentration was 0.15 M, i.e. 108 Cl− and 138 Na+ for charge equilibration of the system. A minimisation followed by a temperature rise to 300 K in NVT was carried out. Equilibration of the volume was done using a 4 ns dynamic simulation in NPT, and further 10 ns production dynamic simulation was run in NPT. One molecule of propanal was introduced in each site and the dynamics run under constraints for a distance of 3.2 Å between the reacting carbons to study the stabilisation of propanal.
221 water molecules). The NaCl concentration was 0.15 M, i.e. 108 Cl− and 138 Na+ for charge equilibration of the system. A minimisation followed by a temperature rise to 300 K in NVT was carried out. Equilibration of the volume was done using a 4 ns dynamic simulation in NPT, and further 10 ns production dynamic simulation was run in NPT. One molecule of propanal was introduced in each site and the dynamics run under constraints for a distance of 3.2 Å between the reacting carbons to study the stabilisation of propanal.
Production of TKgst wild-type and variants
Expression was carried out in E. coli BL21(DE3) pLysS strain. This strain was transformed by heat shock with the following TKgst: artificial wild-type TKgst,17a H102L/L118I,17f H102LH474S,17c H102L/L118I/H474S17f and H102L/L118I/H474G.17f These strains were stored at −80 °C in glycerol 60% (10% final). A 100 μl culture aliquot of each of the clones was transferred to 50 ml liquid LB medium containing kanamycin (30 μg mL−1) and grown at 27 °C, 200 rpm, 12 h. The pre-culture (20 ml) was used to inoculate 1 L of culture medium containing kanamycin (30 μg mL−1) and grown at 37 °C, 200 rpm. Isopropyl β-D-1-thiogalactopyranoside (IPTG) at 0.5 mM was added when the OD600 nm range reached 0.6–0.8 A.U. Cells were then grown at 30 °C, 200 rpm, overnight. Cells were recovered by centrifugation (8000 rpm, 4 °C, 15 min), washed twice with phosphate buffer NaH2PO4·2H2O (50 mM), NaCl (300 mM) at pH 8.0, and finally harvested (4000 rpm, 4 °C, 15 min). The culture medium was removed and the bacterial pellets were stored at −25 °C (≈5 g of wet bacterial pellet per L).Purification of TKgst wild-type and variants
Harvested recombinant cells expressing TKgst wild-type and variants (H102L/L118I, H102LH474S, H102L/L118I/H474S and H102L/L118I/H474G) obtained from 1 L of culture were resuspended in 35 mL of phosphate buffer (50 mM) containing NaCl (300 mM) at pH 8. The cells were disrupted by sonication on ice for 30 min and the insoluble pellets were discarded after centrifugation at 8000 rpm for 15 min at 4 °C. Crude extracts were applied to a Ni-NTA column equilibrated with phosphate buffer for TKs and with phosphate buffer (50 mM) containing NaCl (1 M) and imidazole (20 mM). After washing each column with the same buffer, the His6-tagged TKs were finally eluted with phosphate buffer (50 mM) containing NaCl (300 mM) and imidazole (500 mM) at pH 8.0 for TKs. The fractions containing the eluted proteins were collected and dialysed against triethanolamine buffer (2 mM, pH 7.5) and then against water (pH 7.5) through dialysis tubing (cut-off 14 kDa) at 4 °C and twice against water (pH 7.5). These protein solutions were then lyophilised. Protein concentration was determined by the Bradford method.30 Bovine serum albumin (BSA) was used as the standard.Determination of TKgst activity
The pH-based colorimetric assay described earlier was performed in 96-well plates.13 One unit of TKgst activity was defined as the amount of enzyme that catalyses the formation of 1 μmol of product per minute at 37 °C in TEA buffer (2 mM, pH 6.8). TKgst enzymatic assay was performed in the presence in 96-well plates at 37 °C (200 μL per well). The assay mixture contained wild-type or variant TKgst (8 μg), acceptor (50 mM), ThDP (0.1 mM), MgCl2 (1 mM), TEA (2 mM, pH 7.0) and phenol red (0.084 mM). α-Ketoacid (50 mM) was added to start the reaction. Absorbance was measured at λ = 620 nm using a microplate reader. Absorbance data were converted into HCO3− concentrations according to the standard curve of the assay determined using a series of NaHCO3 concentrations (0–0.5 mM).α-Hydroxyketone quantification
The 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-tetrazolium (WST-1 assay)26 was performed in 96-well plates. Each well (200 μL) contained 90 μL of NaOH solution (0.22 M) 90 μL of water-soluble tetrazolium dye WST-1 solution (0.25 mM) and 20 μL of acyloin sample diluted in phosphate buffer (50 mM) at pH 7.0 containing ThDP (0.1 mM) and MgCl2 (1 mM). After incubation at 60 °C at 100 rpm and 1 h, the absorbances were recorded at 600 nm on a microplate reader.In situ 1H NMR measurements
Preparative-scale enzymatic reaction progress was monitored using quantitative in situ1H NMR relative to 3-(trimethylsilyl) propionic-2,2,3,3-d4 acid (TSP-d4) as internal standard. Aliquots of the reaction mixture were removed over time (450 μL) and mixed with 50 μL of TSP-d4 (50 mM in D2O) before 1H NMR analysis.General procedure of TKgst-catalysed reaction for α-hydroxyketone synthesis
In a 50 mL, round-bottom flask, ThDP (1 mg, 0.1 mM) and MgCl2·6H2O (4 mg, 1 mM) were dissolved in H2O (20 mL, total volume) in NaH2PO4 (50 mM) and the pH adjusted to 7.0. To this solution, the lyophilised TKgst variants H102L/H474S/L118I or H102L/H474G/L118I (0.5–3.5 mg mL−1) was added and the mixture stirred at 150 rpm at 37–50 °C. The reaction was initiated by aldehyde addition: propanal (58 mg, 50 mM, 1 eq.), ethanal (44 mg, 50 mM, 1 eq.) or iso-butanal (72 mg, 50 mM, 1 eq.). Reaction progress was followed by measuring aldehyde consumption by in situ1H NMR. After total substrate conversion (24–96 h), enzyme was discarded from the solution by precipitation with 15 ml of methanol followed by centrifugation at 4000 rpm for 10 min at 20 °C.Purification of α-hydroxyketone products
The pure products 9–13 were obtained after a two-step work-up consisting in the addition of 1 mL of silica gel in the reaction mixtures followed by evaporation to dryness under reduced pressure. The dry products adsorbed to silica were loaded onto a flash silica column pre-equilibrated with ethyl acetate/MeOH 95/5 for products 9, 10 and 11 and with ethyl acetate/MeOH/H2O 90/2.5/2.5 for products 12 and 13. The products 7 and 8 were isolated after liquid–liquid extraction. Product 7 was purified from the reaction mixture using salting out extraction, performed in 20 mL ethyl acetate and 20 ml reaction mixture containing K2HPO4 (3.2 M) for 4 h at 550 rpm. The process was repeated twice. After 4 h, the organic phase was dried over MgSO4 and the solvent evaporated under vacuum to give product 7. Product 8 was extracted with MTBE (3 × 150 mL). The organic phase was dried over MgSO4 and the solvent was removed in vacuo to afford product 8, isolated as a colourless oil.
3-Hydroxybutan-2-one
7 was isolated as a white powder (59 mg, 67% yield with TKgst variant H102L/L/118I/H474G) following the general procedure of the TKgst reaction. TLC: Rf 0.31 (cyclohexane/ethyl acetate, 8.5/1.5 v:v). The NMR data obtained for 7 matched precisely the commercial product reference. 1H NMR (400 MHz, D2O): δ = 1.38 (d, 3H, J = 7.2 Hz, H-4), 2.22 (s, 3H, H-1), 4.42 (q, 2H, J = 7.2, H-3); 13C NMR (101 MHz, D2O): δ = 18.2 (C-4), 24.9 (C-1), 73.0 (C-3), 215.4 (C-2), m/z HRMS ESI-MS calculated for C4H9O2: 89.0597; found [M + H]+ C4H9O2: 89.0603.
4-Hydroxyhexan-3-one
8 was isolated as a colourless oil (70 mg, 85% yield with TKgst variant H102L/L118I/H474G) following the general procedure of the TKgst reaction. TLC: Rf 0.46 (cyclohexane/ethyl acetate, 2/8 v:v). The NMR data obtained for 8 matched precisely the commercial product reference. 1H NMR (400 MHz, D2O): δ = 0.91 (t, 3H, J = 7.4 Hz, H-6), 1.03 (t, 3H, J = 7.4 Hz, H-1), 1.67 (m, 1H, H-5a), 1.86 (ddq, 1H, J1 = 4.3 Hz, J2 = 7.4 Hz, J3 = 14.5 Hz, H-5b) 2.61 (dqd, 2H, J1 = 3.6 Hz J2 = 7.4 Hz J3 = 14.5 Hz, H-2), 4.32 (dd, 1H, and J1 = 4.3 Hz and J2 = 7.4 Hz, H-4); 13C NMR (101 MHz, D2O): δ = 8.2 (C-1), 9.1 (C-6), 23.8 (C-5), 32.1 (C-2), 81.6 (C-4), 211.8 (C-3); m/z HRMS ESI-MS calculated for C6H13O2: 117.0910; found [M + H]+ C6H13O2: 117.0914.
1,2-Dihydroxypentan-3-one
9 was isolated as a white powder (72 mg, 60% yield with TKgst variant H102L/L118I/H474G) following the general procedure of the TKgst reaction. TLC: Rf 0.60 (methanol/ethyl acetate/H2O, 0.5/9/0.5 v:v). NMR data for 9 were identical to those previously described.17d1H NMR (400 MHz, D2O): δ = 0.95 (t, 3H J = 7.2 Hz, H-5), 2.56 (q, 2H, J = 7.2 Hz, H-4), 3.78 (dd, 1H, J1 = 3.8 Hz, J2 = 12.3 Hz, H-1a), 3.85 (dd, 1H, J1 = 3.8 Hz, J2 = 12.3 Hz, H-1b), 4.33 (t, 1H, J = 3.8 Hz, H-2); 13C NMR (125 MHz, D2O): δ = 6.6 (C-5), 32 (C-4), 62.8 (C-1), 77.4 (C-2), 215.4 (C-3); m/z HRMS ESI-MS calculated for C5H10O3Na: 141.05222; found [M + Na]+: 141.0524.
1,2-Dideoxy-D-threo-hex-3-ulose
10 was isolated as a pale yellow oil (50 mg, 34% yield with TKgst variant H102L/L118I/H474S) following the general procedure of the TKgst reaction. TLC: Rf 0.41 (methanol/ethyl acetate, 0.5/9.5 v:v). NMR data for 10 were identical to those previously described.17h Product ratio: α-furanose anomer (6%):β furanose anomer (14%):open chain (80%). 1H NMR (400 MHz, D2O): δ = 1.05 (t, 3H, J = 7.2 Hz, H-1), 2.67 (m, 2H, H-2), 3.64 (dd, 1H, J = 7.1 and 11.4 Hz, H-6a), 3.72 (dd, 1H, J = 5.8 and 11.4 Hz, H6b), 4.16 (m, 1H, H-5), 4.41 (d, J = 2.1 Hz, H-4); 13C NMR (101 MHz, D2O): δ = 6.7 (C-1), 31.9 (C-2), 62.2 (C-6), 71.7 (C5), 76.7 (C-4), 215.7 (C-3); β furanose anomer: 1 H NMR (400 MHz, D2O): δ = 0.96 (t, 3H, J = 7.5 Hz, H-1), 1.76 (m, 2H, H-2), 3.60 (dd, 1H, J = 4.8 and 9.7 Hz, H-6a), 3.90 (d, 1H, J = 5.4 Hz, H-4), 4.16 (m, 1H, H-6b), 4.34 (ddd, 1H, J = 5.4 and 10.9 Hz, H-5); 13C NMR (101 MHz, D2O): δ = 6.9 (C1), 30.1 (C-2), 69.4 (C-6), 75.2 (C-5), 78.8 (C-4), 104.5 (C-3); α-furanose anomer: 1H NMR (400 MHz, D2O): δ = 0.99 (t, 3H, J = 7.6 Hz, H-1), 1.76 (m, 2H, H-2), 3.86 (dd, 1H, J = 6.3 and 12.9 Hz, H-6a), 3.99 (d, 1H, J = 1.0 Hz, H-4), 4.24 (m, 2H, H-5 and H-6b); 13C NMR (101 MHz, D2O): δ = 7.2 (C-1), 26.8 (C-2), 72.0 (C-6), 76.3 (C-5), 79.6 (C-4), 108.2 (C-3); m/z HRMS ESI-MS calcd for C6H13O4: 149.0808; found [M + H]+: 149.0807.
1,2-Dideoxy-D-arabino-hept-3-ulose
11 was isolated as a pale yellow oil (90 mg, 50% yield with TKgst variant H102L/L118I/H474S) following the general procedure of the TKgst reaction. TLC: Rf 0.44 (methanol/ethyl acetate, 1/9 v:v). NMR data for 11 were identical to those previously described.17h Product ratio: open chain (7%), α-pyranose anomer (21%):β-pyranose anomer (58%). β-Pyranose anomer: 1H NMR (400 MHz, D2O): δ = 0.84 (m, 3H, H-1), 1.78 (m, 2H, H-2), 3.56 (dd, 1H, J = 2 Hz and 12.2 Hz, H-7a), 3.60 (d, J = 10.0 Hz, H-4), 3.77 (dd, 1H, J = 3.6 Hz and 10.0 Hz, H-5), 3.87–3.93 (m, 2H, H-6 and H-7b); 13C NMR (101 MHz, D2O): δ = 4.6 (C-1), 28.4 (C2), 61.4 (C-7), 67.3 (C-6), 67.6 (C-4), 67.8 (C-5), 97.8 (C-3); α pyranose anomer: 1H NMR (400 MHz, D2O): δ = 0.86 (t, 3H, J = 7.2 Hz, H-1), 1.68 (m, 2H, H-2), 3.66 (m, 1H, H-7a), 3.73–3.82 (m, 2H, H-6 and H-7b), 3.85 (d, J = 8 Hz, 1H, H-4), 3.97 (m, 1H, H-5); 13C NMR (101 MHz, D2O): δ = 5.0 (C-1), 28.3 (C-2), 60.6 (C-7), 72.9 (C-5), 76.0 (C-4), 78.5 (C-6), 101.3 (C3); open form: 1H NMR (400 MHz, D2O): 0.96 (t, 3H, J = 7.2 Hz, H-1), 2.56 (m, 2H, H-2), 3.62–3.65 (m, 1H, H-7a), 3.7–3.8 (m, 1H, H-6), 3.81–3.85 (m, 1H, H-7b), 4.0–4.05 (m, 1H, H5), 4.5 (d, J = 1.3 Hz, H-4); 13C NMR (101 MHz, D2O): δ = 5.0 (C-1), 30.0 (C-2), 61.0 (C-7), 68.8 (C-6), 69.2 (C-5), 74.5 (C4), 214.4 (C-3); m/z HRMS ESI-MS calculated for C7H14O5Na 201.0733; found [M + HCOO]− C7H14O5Na: 201.0736.
1,2-Dihydroxy-4-methylpentan-3-one
12 was isolated as a pale yellow oil (79 mg, 60% yield with TKgst variant H102L/L118I/H474G) following the general procedure of the TKgst reaction. TLC: Rf 0.41 (methanol/ethyl acetate, 1/9 v:v). NMR data for 12 were identical to those previously described.17d β-Pyranose: 1H NMR (400 MHz, D2O): δ = 1.07 (d, 3H, J = 7 Hz, H-5a), 1.12 (d, 3H, J = 7 Hz, H-5b), 3.03 (sep, 1H, J = 7 Hz, H-4), 3.89 (dd, 1H, J = 4 and 12.3 Hz, H1-b), 3.94 (dd, 1H, J = 4 and 12.3 Hz, H-1a), 4.59 (t, 1H, J = 4 Hz, H-2); 13C NMR (101 MHz, D2O): δ = 16.6 (C-5a), 18.2 (C-5b), 36.5 (C-4), 62.6 (C-1), 76.3 (C-2), 218.6 (C-3), m/z HRMS ESI-MS calculated for C6H13O3: 133.0589; found [M + H]+: 133.0857.
4,5,6-Trihydroxy-2-methylhexan-3-one
13 was isolated as a white powder (51 mg, 31% yield with TKgst variant H102L/L118I/H474S). TLC: Rf 0.39 (methanol/ethyl acetate, 1/9 v:v). NMR data for 13 were identical to those previously described.17d Product ratio: open-chain form (80%), β-anomer (15%), α-anomer (5%). Open-chain form: 1H NMR (400 MHz, D2O): δ = 4.59 (d, 1H, J = 2 Hz, H-4), 4.19 (m, 1H, H-5), 3.73 (dd, 1H, J = 11.5 and 6 Hz, H-6a), 3.67 (dd, 1H, J = 11.5 and 6 Hz, H-6b), 3.08 (sep, 1H, J = 6.9 Hz H-2), 1.12 (d, 3H, J = 2 Hz, H-1a), 1.09 (d, J = 1.5 Hz, 3H, H-1b) 13C NMR (101 MHz, D2O): δ = 16.8 (C-1α), 18.4 (C-1b), 36.2 (C-2), 62.3 (C-6), 71.4 (C-5) 75.5 (C-4), 218.7 (C-3) β-anomer: 1H NMR (400 MHz, D2O): δ = 4.32 (m, 1H, H-5), 4.14 (dd, J = 6.3 and 9.7 Hz, H-6b), 4.01 (d, 1H, J = 5 Hz, H-4), 3.60 (dd, 1H, J = 4.1 and 9.9 Hz, H-6a), 1.96 (m, 1H, H-2), 0.99 (d, 3H, J = 1.8 Hz, H-1a), 0.97 (d, 3H, J = 1.8 Hz, H-1b); 13C NMR (101 MHz, D2O): δ = 15.8 (C-1a), 16.0 (C-1b), 34.9 (C-2), 69.3 (C-6), 75.8 (C-5), 77.8 (C-4), 106.0 (C-3), m/z HRMS ESI-MS calculated for C7H15O4: 163.0965; found [M + H]+: 163.0964.
Conclusions
Here we show that TKgst variants H102L/L118I/H474G(S) catalysed a de novo self-(R1CHO = R2CHO) or cross-(R1CHO ≠ R2CHO) acyloin condensation of aldehydes 1–3 as nucleophiles on identical or different aldehydes 4–6 as electrophiles for the synthesis of aliphatic, and hydroxy-functionalised α-hydroxyketones 7–13.The self-condensation of ethanal 1 and especially propanal 2 was proved with variant H102L/L118I/H474G leading to the expected α-hydroxyketones 7 and 8 respectively. According to the analysis of TKgst active site of the variant H102L/L118I/H474G, pathway B seems to be based on the same acylThDP carbanion as pathway A, which could be formed by the proton transfer via a water molecule to His 263.
We found that this promiscuous TKgst-catalysed reaction could be extended to the challenging cross-acyloin condensation with propanal 2 or iso-butanal 3 as nucleophiles and aldehydes 4, 5, 6 as electrophiles, leading to the α-hydroxyketones 9–13 with good to excellent yields comparable to those obtained with pathway A by increasing enzyme quantity to a factor of 1.5–2 only. In addition, the TKgst variants H102L/L118I/H474G(S) were able to control the regio and stereoselectivity, giving mainly one stereoisomer of the targeted α-hydroxyketones 9, 10, 11 and 13. This hitherto unreported application of TKgst acyloin-condensation reaction improves the green chemistry metrics of the common strategy avoiding the release of carbon dioxide from α-ketoacids, which could add instability and/or cost, and extends the toolbox of ThDP enzyme-catalysed acyloin condensation with non-aromatic aldehydes as nucleophiles. This novel strategy performed in mild conditions with natural biocatalysts and stoichiometric amounts of substrates reduces the environmental impact compared to the chemical ways catalysed by synthetic and toxic NHCs coupled with a base and performed in organic solvents with often an excess of nucleophile.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by MSCA-ITN-ETN-2020 CC-TOP-ID: 956931 and by ANR-22-CE07-0038-01 (grants to L. H.). We thank Martin Leremboure for the determination of enantiomeric excess by chiral GC analysis and Dr Muriel Joly for the expression of TKgst variants in E. coli BL21(DE3) pLysS strain.References
- F. Neuser, H. Zorn and R. G. Berger, J. Agric. Food Chem., 2000, 48, 6191–6195 CrossRef CAS PubMed.
- P. Hoyos, J.-V. Sinisterra, F. Molinari, A. R. Alcántara and P. Domínguez de María, Acc. Chem. Res., 2010, 43, 288–299 CrossRef CAS PubMed.
- R. P. Dash, T. Tichý, V. Veeravalli, J. Lam, J. Alt, Y. Wu, L. Tenora, P. Majer, B. S. Slusher and R. Rais, Mol. Pharm., 2019, 16, 4292–4301 CrossRef CAS PubMed.
- J. Lu, X. Li, S. Yuan, Y. Wang, H. Sun, W. Weng, Y. Shi, X. Wang, K. Huang, X. Sun and T. Wu, Org. Process Res. Dev., 2022, 26, 1247–1257 CrossRef CAS.
- R. S. Menon, A. T. Biju and V. Nair, Beilstein J. Org. Chem., 2016, 12, 444–461 CrossRef CAS PubMed.
- N. Gaggero and S. Pandini, Org. Biomol. Chem., 2017, 15, 6867–6887 RSC.
- M. Y. Jin, S. M. Kim, H. Han, D. H. Ryu and J. Woon Yang, Org. Lett., 2011, 13, 880–883 CrossRef CAS PubMed.
- I. Piel, M. D. Pawelczyk, K. Hirano, R. Fröhlich and F. Glorius, Eur. J. Org. Chem., 2011, 5475–5484 CrossRef CAS.
- (a) D. Meyer, L. Walter, G. Kolter, M. Pohl, M. Müller and K. Tittmann, J. Am. Chem. Soc., 2011, 133, 3609–3616 CrossRef CAS PubMed; (b) H. Li, N. Liu, X. Hui and W.-Y. Gao, RSC Adv., 2017, 7, 32664–32668 RSC.
- J. Yi, N. Nemeria, A. McNally, F. Jordan, R. S. Machado and J. R. Guest, J. Biol. Chem., 1996, 271, 33192–33200 CrossRef CAS PubMed.
- (a) Y. Kobori, D. C. Myles and G. M. Whitesides, J. Org. Chem., 1992, 57, 5899–5907 CrossRef CAS; (b) G. A. Sprenger, Arch. Microbiol., 1995, 164, 324–330 CrossRef CAS PubMed; (c) M. Sundström, Y. Lindqvist, G. Schneider, U. Hellman and H. Ronne, J. Biol. Chem., 1993, 268, 24346–24352 CrossRef; (d) N. J. Turner, Curr. Opin. Biotechnol., 2000, 11, 527–531 CrossRef CAS PubMed.
- (a) Z. Ju, J. Xu, Z. Li, J. Fang, M. Li, D. C. Howell and F. Chen, Green Synth. Catal., 2022, 3, 317–326 CrossRef CAS; (b) A. S. Demir, Ö. Şeşenoglu, E. Eren, B. Hosrik, M. Pohl, E. Janzen, D. Kolter, R. Feldmann, P. Dünkelmann and M. Müller, Adv. Synth. Catal., 2002, 344, 96–103 CrossRef CAS; (c) T. Stillger, M. Pohl, C. Wandrey and A. Liese, Org. Process Res. Dev., 2006, 10, 1172–1177 CrossRef CAS; (d) M. Beigi, E. Gauchenova, L. Walter, S. Waltzer, F. Bonina, T. Stillger, D. Rother, M. Pohl and M. Müller, Chem. – Eur. J., 2016, 22, 13999–14005 CrossRef CAS PubMed.
- (a) G. Bernacchia, O. Bortolini, M. De Bastiani, L. A. Lerin, S. Loschonsky, A. Massi, M. Müller and P. P. Giovannini, Angew. Chem., 2015, 127, 7277–7281 CrossRef; (b) P. P. Giovannini, L. A. Lerin, M. Müller, G. Bernacchia, M. D. Bastiani, M. Catani, G. Di Carmine and A. Massi, Adv. Synth. Catal., 2016, 358, 2767–2776 CrossRef CAS; (c) G. Di Carmine, O. Bortolini, A. Massi, M. Müller, G. Bernacchia, G. Fantin, D. Ragno and P. P. Giovannini, Adv. Synth. Catal., 2018, 360, 4132–4141 CrossRef CAS.
- S. Güner, V. Wegat, A. Pick and V. Sieber, Green Chem., 2021, 23, 6583–6590 RSC.
- (a) J. L. Galman, D. Steadmann, S. Bacon, P. Morris, M. E. B. Smith, J. Ward, P. A. Dalby and H. C. Hailes, Chem. Commun., 2010, 46, 7608–7610 RSC; (b) F. Subrizi, M. Cárdenas-Fernández, G. J. Lye, J. M. Ward, P. A. Dalby, T. D. Sheppard and H. C. Hailes, Green Chem., 2016, 18, 3158–3165 RSC; (c) H. Yu, R. Icken Hernandez Lopez, D. Steadman, D. MendezSanchez, S. Higson, A. Cazares-Korner, T. D. Sheppard, J. M. Ward, H. C. Hailes and P. A. Dalby, FEBS J., 2020, 9, 1758–1776 CrossRef PubMed.
- A. Ranoux, S. K. Karmee, J. Jin, A. Bhaduri, A. Caiazzo, I. W. Arends and U. Hanefeld, ChemBioChem, 2012, 13, 1921–1931 CrossRef CAS PubMed.
- (a) J. Abdoul Zabar, I. Sorel, V. Hélaine, F. Charmantray, T. Devamani, D. Yi, V. de Berardinis, D. Louis, P. Marlière, W. D. Fessner and L. Hecquet, Adv. Synth. Catal., 2013, 355, 116–128 CrossRef CAS; (b) J. Abdoul Zabar, M. Lorillière, D. Yi, S. Thangavelu, T. Devamini, L. Nauton, F. Charmantray, V. Hélaine, W.-D. Fessner and L. Hecquet, Adv. Synth. Catal., 2015, 357, 1715–1720 CrossRef CAS; (c) D. Yi, T. Saravanan, T. Devamani, F. Charmantray, L. Hecquet and W.-D. Fessner, Chem. Commun., 2015, 51, 480–483 RSC; (d) T. Saravanan, S. Junker, M. Kickstein, J. Hegen, S. Hein, M. K. Link, S. Witt, M. Lorillière, L. Hecquet and W.-D. Fessner, Angew. Chem., Int. Ed., 2017, 56, 5358–5362 CrossRef CAS PubMed; (e) M. Lorillière, R. Dumoulin, M. L'Enfant, A. Rambourdin, V. Thery, L. Nauton, W.-D. Fessner, F. Charmantray and L. Hecquet, ACS Catal., 2019, 9, 4754–4763 CrossRef; (f) H. Casajus, A. Lagarde, M. Leremboure, T. De Dios Miguel, L. Nauton, V. Thery, W.-D. Fessner, N. Duguet, F. Charmantray and L. Hecquet, ChemCatChem, 2020, 12, 5772–5779 CrossRef CAS; (g) H. Casajus, A. Lagarde, L. Nauton, N. Ocal, M. Leremboure, W.-D. Fessner, N. Duguet, F. Charmantray and L. Hecquet, ACS Catal., 2022, 6, 3566–3576 CrossRef; (h) N. Ocal, G. Arbia, A. Lagarde, M. Joly, S. Gittings, K. M. Graham, F. Charmantray and L. Hecquet, Adv. Synth. Catal., 2023, 365, 78–87 CrossRef CAS.
- M. Sundström, Y. Lindqvist, G. Schneider, U. Hellman and H. Ronne, J. Biol. Chem., 1993, 268, 24346–24352 CrossRef.
- J. Littlechild, N. Turner, G. Hobbs, M. Lilly, A. Rawas and H. Watson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1995, 6, 1074–1076 CrossRef PubMed.
- G. Ali, T. Moreau, C. Forano, C. Mousty, V. Prevot, F. Charmantray and L. Hecquet, ChemCatChem, 2015, 7, 3163–3170 CrossRef CAS.
- S. R. Marsden, L. Gjonaj, S. J. Eustace and U. Hanefeld, ChemCatChem, 2017, 9, 1808–1814 CrossRef CAS PubMed.
- I. A. Sevostyanova, O. N. Solovjeva and G. A. Kochetov, Biochem. Biophys. Res. Commun., 2004, 313, 771–774 CrossRef CAS PubMed.
- M. Breuer, M. Pohl, B. Hauer and B. Lingen, Anal. Bioanal. Chem., 2002, 374, 1069–1073 CrossRef CAS PubMed.
- (a) M. E. B. Smith, U. Kaulmann, J. M. Ward and H. C. Hailes, Bioorg. Med. Chem., 2006, 14, 7062–7065 CrossRef CAS PubMed; (b) M. Lorillière, M. de Sousa, F. Bruna, E. Heuson, T. Gefflaut, V. de Beradinis, T. Saravanan, D. Yi, W.-D. Fessner, F. Charmantray and L. Hecquet, Green Chem., 2017, 19, 425–435 RSC.
- T. Sehl, R. C. Simon, H. C. Hailes, J. M. Ward, U. Schell and M. Pohl, J. Biotechnol., 2012, 159, 188–194 CrossRef CAS PubMed.
- M. Bommer M and J. M. Ward, Anal. Biochem., 2016, 15, 493–511 Search PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 2, 1605–1612 CrossRef PubMed.
- M. C. R. Melo, R. C. Bernardi, T. Rudack, M. Scheurer, C. Riplinger, J. C. Phillips, J. D. C. Maia, G. B. Rocha, J. V. Ribeiro, J. E. Stone, F. Neese, K. Schulten and Z. Luthey-Schulten, Nat. Methods, 2018, 15, 351–354 CrossRef CAS PubMed.
- (a) L. Nauton, V. Helaine, V. Théry and L. Hecquet, Biochem, 2016, 2144–2152 CrossRef CAS PubMed; (b) L. Nauton, L. Hecquet and V. Théry, J. Chem. Inf. Model., 2021, 61, 3502–3515 CrossRef CAS PubMed; (c) G. A. Kochetov and O. N. Solovjeva, Biochim. Biophys. Acta, 2014, 1844, 1608–1618 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- M. G. T. C. Ribeiroa and A. A. S. C. Machado, Green Chem. Lett. Rev., 2013, 6, 1–18 CrossRef.
- S. E. O'Toole, C. A. Rose, S. Gundala, K. Zeitler and S. J. Connon, J. Org. Chem., 2011, 76, 347–357 CrossRef PubMed.
- M. Y. Jin, S. M. Kim, H. Han, D. H. Ryu and J. W. Yang, Org. Lett., 2011, 13, 880–883 CrossRef CAS PubMed , 7–357.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc01373e |
| This journal is © The Royal Society of Chemistry 2024 |