
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sustainable aviation fuel from prehydrolysis liquors†
Daria
Lebedeva
a,
Lars William
Schick
a,
Daniel
Cracco
a,
Withsakorn
Sangsuwan
a,
Gonzalo
Castiella-Ona
a,
Dagoberto O.
Silva
c,
Alessandro
Marson
d,
Erik
Svensson Grape
b,
A. Ken
Inge
 *b,
Liane M.
Rossi
*c,
Elena
Subbotina
*a,
Alessandro
Manzardo
*d and
Joseph S. M.
Samec
*a
*b,
Liane M.
Rossi
*c,
Elena
Subbotina
*a,
Alessandro
Manzardo
*d and
Joseph S. M.
Samec
*a
aStockholm University, Department of Organic Chemistry, Svante Arrhenius väg 16C, SE 106 91 Stockholm, Sweden. E-mail: joseph.samec@su.se
bDepartment of Materials and Environmental Chemistry, Stockholm University, Stockholm SE 10691, Sweden
cDepartamento de Química Fundamental, Instituto de Química, Universidade de São Paulo, Av. Prof. Lineu Prestes, 748, 05508-000, São Paulo, SP, Brazil
dCESQA (Quality and Environmental Research Center), University of Padova, Department of Civil, Environmental and Architectural Engineering, Via Marzolo 9, 35131 Padova, Italy
First published on 22nd May 2024
Abstract
Maximizing products of high value and minimizing incineration of side-streams is key to realize future biorefineries. In current textile production from forestry, hemicellulose is removed by prehydrolysis before delignification. The resulting prehydrolysis liquor is incinerated in the recovery boiler at low efficiency. This additional burden on the limiting recovery boiler reduces the pulp production. In this study, we demonstrate that prehydrolysis liquor can be upgraded, in 5 steps, to yield aviation fuels. Prehydrolysis liquors were dehydrated to furfural by zeolite catalysis. Furfural was selectively reduced to furfuryl alcohol by Au@NC. Rhenium-catalysed Achmatowicz rearrangement gave a C5 intermediate susceptible to self [2 + 2] cycloaddition to give the C10 oxygenated precursor. By using a combination of Ru/C and zeolites, full hydrodeoxygenation was achieved. The overall transformation from furfural to hydrocarbons resulted in a 48% carbon yield. The resulting hydrocarbons, containing an anticipated strained four-membered ring, are preferred aviation fuel components. This is an important step to show that aviation fuels can be produced sustainably from existing industrial side-streams. A comparative life cycle assessment was applied to evaluate the environmental impact of the proposed valorization approach, demonstrating benefits in the climate change impact category when implementing this technology in a pulp mill compared to the incineration of pre-hydrolysis liquor scenario.
Introduction
The increasing demand for sustainable aviation fuel (SAF) is driven by the depletion of fossil resources, awareness of carbon dioxide emissions, and political instability. Due to the extreme demands for propulsion in aviation, shifting towards electric engines is more challenging than for road transportation.1 For this reason, aviation will require liquid hydrocarbon fuels even in the future. SAF, by definition, requires the utilization of non-fossil-based feedstock; biogenic carbon sources (biomass) or CO2 are preferred.2 Currently, SAF:s are made from crops, which is not considered sustainable since this feedstock adversely affects land use and water scarcity. As follows, raw materials sourced from low value streams or wastes are preferred to avoid burdens on climate change and land use.3The generation of prehydrolysis liquor, from hemicelluloses, is a crucial wood pre-treatment step in the dissolving grade pulp production of the kraft process. Dissolving grade pulp is used for generation of textile fibers to substitute cotton production, which has lately been questioned for environmental concerns. The prehydrolysis liquor produced in this process, together with lignin, is currently incinerated to generate heat and power, however at low efficiencies. This brings an additional burden on the rate limiting recovery boiler, reducing the overall production capacity of pulp.4 Thus, there is a potential to simultaneously increase the value of prehydrolysis liquor beyond its heating value, and debottleneck pulp production (Fig. 1).
 | ||
| Fig. 1 Increasing the pulp production by emptying the recovery boiler. | ||
The state-of-the-art synthetic pathways for SAF:s includes pioneering research by Dumesic and co-workers, demonstrating the approach for utilizing sugars, such as glucose, to synthesise linear hydrocarbons within aviation boiling point range.5 Another approach was described by Corma and co-workers, who converted sugar-derived furanics into branched alkanes (Scheme 1, entries 1 and 2).6 Recent advancements have underscored the advantages of cyclic hydrocarbons in fuel applications, owing to their increased energy density and higher heat of combustion. The work by Li's research group demonstrated the synthesis of multicyclic hydrocarbons containing 5-membered rings from cellulose (Scheme 1, entry 3).7 Nevertheless, it's worth mentioning that cyclobutane's relative strain energy of 26.5 kcal mol−1 is notable when compared to cyclopentane (6.2 kcal mol−1) and cyclohexane (0 kcal mol−1).8 Thus, cyclobutane derivatives could be desired candidates for aviation fuel, as they afford a higher heat of combustion. Several research groups proposed synthetic pathways for preparing cyclobutane-based hydrocarbons from sources that are not as available as the sugars, vide supra. These approaches include a [2 + 2] cycloaddition to yield cyclobutane-derivatives from acetone derived isopherone followed by hydrodeoxygenation (HDO), when applicable (Scheme 1, entry 4).9–12 However, no reports disclose high energy 4-membered SAF:s produced from available side-streams of low value.
 | ||
| Scheme 1 The state-of-the-art synthesis of liquid hydrocarbons in aviation boiling point range. | ||
In this work, we report a novel value chain from industrially relevant prehydrolysis liquor to produce cyclobutane-containing hydrocarbons suitable for aviation fuel via furfural (Scheme 1, entry 5). Our approach involves the multi-step transformation of furfural into C5 precursors 3 and 4, susceptible to [2 + 2] cycloaddition, resulting in the formation of cyclobutane 5, that was successfully hydrotreated to produce high energy C10 SAF:s. To ascertain the sustainability of the novel route, a comparative LCA has been performed showing significant reduction in climate change, compared to conventional aviation fuels.
Experimental
General considerations
All chemicals were purchased from Sigma Aldrich, if not stated otherwise. Zeolites were purchased from Zeolyst. Furfural was distilled prior to use. The light-driven reactions were performed using a Rayonet reactor equipped with eight high pressure mercury lamps (4 W each, maximum irradiation at 365 nm), stirring system, and air circulation. HDO reactions were performed in a stainless-steel reactor assembled from Swagelok units including a gas valve. NMR was performed on a Bruker Avance (400 MHz). Chemical shifts are expressed in parts per million (ppm, δ). GC-MS/FID was performed on a QP2020 system (SHIMADZU, Japan) equipped with two parallel HP-5MS columns (30 m × 0.25 mm × 0.25 μm).Prehydrolysis
Birch sawdust (0.5 g) and 5 mL of DI water were added to an autoclave and stirred at 200 °C. After 40 min, the reaction was stopped. The suspended residue was filtered off, washed with DI water, and dried at 60 °C overnight. The resulting residue was analysed by two-step hydrolysis to determine carbohydrate and lignin content (ESI†). Prehydrolysis liquor was analysed for monosaccharide content by HPLC (ESI†).Dehydration
D-(+)-Xylose (75 mg, 0.5 mmol) was dissolved in water (150 μL) in an autoclave. EtOAc (3 mL) was added, followed by H-beta zeolite (25 mg). The reaction was performed at 180 °C for 1 h. After completion, an internal standard (dodecane) was added. The reaction mixture was filtered and subjected to GC-MS/FID analysis. Yield was quantified by GC-FID (ESI†).Furfural reduction
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ligand = 1:2) were stirred in 20 mL of ethanol for approximately 5 min at 60 °C. The TiO2 support (1.0 g, titanium(IV) oxide, anatase) was then added, and the mixture was stirred for 30 min. After, the solvent was removed using a rotary evaporator. The obtained solid was ground to a fine powder and then pyrolyzed under N2 atmosphere in an oven (DEKEMA – Austromat 674i) at a heating rate of 20 °C per minute and held at 400 °C for 2 h. The percentage of gold in the fresh catalyst was determined by FAAS as 1.1 wt%.
:ligand = 1:2) were stirred in 20 mL of ethanol for approximately 5 min at 60 °C. The TiO2 support (1.0 g, titanium(IV) oxide, anatase) was then added, and the mixture was stirred for 30 min. After, the solvent was removed using a rotary evaporator. The obtained solid was ground to a fine powder and then pyrolyzed under N2 atmosphere in an oven (DEKEMA – Austromat 674i) at a heating rate of 20 °C per minute and held at 400 °C for 2 h. The percentage of gold in the fresh catalyst was determined by FAAS as 1.1 wt%.
Synthesis of 6-hydroxy-2H-pyran-3(6H)-one (Achmatowicz rearrangement)
Methyltrioxorhenium (12.5 mg, 50 μmol, 1 mol%) was combined with hydrogen peroxide (855.9 mL, 35%, 5 mmol) and cooled to 0 °C. The mixture was stirred for 15 min. A solution of furfuryl alcohol (490 mg, 5 mmol) in 2-propanol (2.6 mL) was added dropwise to the oxidation solution. The resulting mixture was stirred for 5 h, allowing the temperature to rise to room temperature. Molecular sieves (1.5 g) were then added, and the mixture was left to stir for an additional hour. The reaction mixture was then filtered through a Celite plug, condensed under reduced pressure, and left to crystallize in the freezer overnight. The following morning, the precipitate was filtered off and dried under high vacuum, yielding a colourless crystalline solid (559 mg, 98%).Synthesis of 6-methoxy-2H-pyran-3(6H)-one
6-Hydroxy-2H-pyran-3(6H)-one (790 mg, 6.92 mmol) was dissolved in dry DCM (10 mL). Trimethyl orthoformate (3.78 mL, 34.6 mmol) was added to the solution at 0 °C, followed by the addition of boron trifluoride (BF3·OEt2, 50 μL, 0.41 mmol 5.9 mol%). The reaction was stirred for 1 h at 0° C. Upon completion, additional DCM and saturated NaHCO3 solution were added, and the mixture was extracted three times with DCM. The solvent was removed under reduced pressure. The crude product was purified by flash column chromatography (20% EtOAc:pentane), yielding a colourless oil (608 mg, 69%). Methoxylation was performed to isolate the different stereoisomers of the product and this step was conducted for analysis purposes.
[2 + 2] cycloaddition
The light-driven [2 + 2] cycloaddition was performed in a Rayonet UV reactor. In a quartz tube, 6-methoxy-2H-pyran-3(6H)-one (154 mg, 1.2 mmol) was dissolved in MeCN (5 mL, 0.24 M). The reaction vessel was placed inside the Rayonet reactor, where the reaction was performed under UV irradiation (365 nm), continuous stirring, and cooling by air circulation. After 48 h, the reaction was stopped and solvent was removed under reduced pressure. The resulting residue was purified by column chromatography (40% EtOAc:pentane), yielding a white crystalline solid (88 mg, 57%). The isolated isomers were characterized by NMR (ESI†), recrystallized and analysed by single-crystal X-ray diffraction (ESI†).
HDO of cycloadducts
The mixture of cycloadduct isomers (100 mg, 0.39 mmol) was loaded into a stainless-steel reactor (25 mL), followed by the addition of solvent (3 mL, 0.13 M). Both catalysts Ru/C (25 mg, 5% Ru content) and HY zeolites (50 mg) were then added. The reactor was sealed and subjected to three cycles of nitrogen gas flushing through the valve. Following this, hydrogen gas was added to the system. The reaction was conducted in an oil bath maintained at 220 °C for 16 h with continuous stirring. Upon completion, the reactor was cooled down, and the pressure was slowly released. The resulting reaction mixture was filtered to separate solid catalysts, and an aliquot of the filtrate was analysed by GC-MS/FID with dodecane as a standard (ESI†). Due to the complexity of the product mixture, individual components were identified indirectly using GC-MS (ESI†).Life cycle assessment
To enhance the understanding of environmental impacts related to the extraction and conversion of prehydrolysis liquor into cyclobutane-based aviation fuels, a comparative Life Cycle Assessment (LCA) was conducted. The assessment included two scenarios: S1-Burn depicts the kraft pulping process with an initial prehydrolysis step for extracting hemicellulose, followed by combustion in a co-generation facility to produce heat and electricity; S2-AF considers the conversion of hemicellulose, obtained in the prehydrolysis phase, into cyclobutane-based aviation fuels while the residual hemicellulose is combusted in a co-generation plant to supply heat and electricity.A consequential LCA approach is adopted as it more effectively captures the environmental implications of decision-making in prehydrolysis liquor management.15
A functional unit of 1 kg of unbleached kraft pulp was chosen to focus on the determining product of the system,16,17 treating all the co-products by substitution. A cradle to gate approach was chosen, thereby excluding the bleaching process, usage phase, and end-of-life of dissolving grade pulp from our analysis. Since these life cycle stages are identical for both scenarios being compared, our chosen approach does not compromise the comparative validity. This assessment is contextualized within Northern Europe, with a subsequent extension to a broader European context examined during the sensitivity analysis. Detailed information on the choices made for scope definition and a comprehensive review of both primary and secondary data sources (Table S8) can be found in ESI.†
The impact assessment results are displayed using 16 midpoint impact categories from the Environmental Footprint (EF) method 3.0.18 In the main text, only five categories are presented and discussed: climate change (GWP-total), land use (SQP), water use (WDP), resource use of fossils (ADP-fossil), and mineral and metal resources (ADP-min&met). The complete set of indicators and results is available in ESI.†
Aspen Plus
As explained in the previous section, many solvents play a major role in the pathway to prepare cyclobutane-based aviation fuels. According to the principles of the green chemistry, recyclable environmentally benign solvents should be exploited to minimize the burden to eco-systems. In building the inventory phase for the LCA study, an assumption of 2% loss of solvent every 5 runs was assumed and tested through simple simulations with Aspen Plus. The separation of the EtOAc and water mixture from furfural after de-hydration was tested through distillation (Radfrac column) followed by a decanter to separate the EtOAc/water mixture, achieving a loss of EtOAc < 1%. In the same way, 2-propanol and acetonitrile separations from the products were tested through distillation, achieving almost complete separation. These preliminary results confirmed the assumption of small solvent loss and recyclability to minimize the associated environmental impacts.Results and discussion
Prehydrolysis of birch sawdust
Birch sawdust with hemicellulose (xylan) content of 25% (ESI, Table S1†) was pre-treated using previously reported autogenous hydrolysis conditions.19 Optimized prehydrolysis (40 min, 200 °C, autogenous pressure) gave 97% removal of hemicelluloses. Only negligible amount of cellulose (0.4 wt% based on the raw material) was solubilized under these conditions. The xylose content in prehydrolysis liquor was measured to be 32% of the initial hemicellulose content (ESI, Table S2†).Dehydration of xylose
Currently, furfural is industrially produced from C5 sugars by homogeneous catalysis with mineral acids. The use of mineral acids causes technical challenges such as equipment corrosion and environmental issues.20 Recent research demonstrated benefits of solid acid catalysis as more environmentally friendly furfural production.21 Among solid catalysts, inorganic oxides such as zeolites demonstrated their catalytic activity and high selectivity in xylose dehydration into furfural.22Previously we reported a successful transformation of prehydrolysis liquors to furfural using beta zeolites in a dioxane–water mixture as solvent.19,23 In this work, we explored EtOAc, a more sustainable solvent for the transformation of xylose into furfural.24
Dehydration to furfural 1 was performed through catalysis by beta zeolites at 180 °C in EtOAc giving 74% yield under optimized conditions (ESI, Table S3†), slightly lower than using dioxane (83%). It was found that addition of small amounts of water was important for obtaining high yields. Stopping the reaction after 1 h was also crucial to avoid humin formation.
Selective hydrogenation of furfural to furfuryl alcohol
The next step is reduction of furfural 1 to furfuryl alcohol 2. Selective reduction of the aldehyde moiety in furfural is not trivial due to the competing decarbonylation, hydrogenolysis, and hydrogenation of the furan ring (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds).25,26
C bonds).25,26
Hydrogenation catalysed by Ru/C and Pd/C were tested using 2-propanol as the hydrogen donor (Table 1, entries 1 and 2). Reactions catalysed by Ru/C showed higher conversion of furfural and higher selectivity towards furfuryl alcohol, however, the maximum yield observed was only 53% (Table 1, entry 1).27 Among the hydrogen donors tested (2-propanol, EtOH, MeOH, HCOOH), 2-propanol showed the best performance. When molecular hydrogen was used as a hydrogen source, full conversion was achieved with Ru/C, although selectivity dropped significantly (Table 1, entry 3). Thus, seemingly straight forward reduction proved challenging. Selectivity issues are known in most traditional heterogeneous hydrogenation catalysts, e.g., ruthenium, palladium, and nickel, and we decided to explore a less active metal, such as gold. Gold has been considered catalytically inactive towards H2, but it can develop into a very selective catalyst once H2 heterolytic activation occurs across a metal–ligand interface.28,29 Base ligands, such as ammonia and pyridine, were suggested to be used as additives to improve hydrogenation of aldehydes over gold-based catalysts.30 DFT calculations suggested that the base allows the chemical adsorption of H2 and its cleavage via an heterolytic dissociation mechanism, forming NH4+ and H− that will be adsorbed on the active metal site. Finally, H+ (of NH4+) and H− will be transferred to the aldehyde and produce the corresponding alcohol. Later, Rossi and co-workers developed a fully heterogenous gold embedded on N-doped carbon (Au@NC/TiO2) catalyst, which was active and selective for alkynes semihydrogenation.13 It was suggested that the heterolytic cleavage of molecular H2 occur at the basic nitrogen species on the N-doped carbon material in close vicinity of Au nanoparticles. Later, this catalyst was tested for the hydrogenation of various aldehydes to alcohols, showing high selectivity.14 A similar mechanism was also proposed by Nagpure et al.,31 for hydrogenation of cinnamaldehyde, based on the heterolytic dissociation of H2, followed by the transfer of nucleophilic H− ion to electrophilic carbonyl carbon of the aldehyde, whereas the electrophilic H+ ion transfers to nucleophilic carbonyl oxygen, leading to the desired alcohol.
Gratifyingly, the Au@NC/TiO2 catalyst showed very high selectivity in the conversion of furfural to furfuryl alcohol, reaching full conversion and no other side-products (Table 1, entry 4). It is worth to mention that the heterogenous catalyst can be recycled and reused, as demonstrated in the hydrogenation of benzaldehyde.14
Achmatowicz rearrangement of furfuryl alcohol into compound 3
The classical Achmatowicz rearrangement requires the use of reagents such as m-chloroperoxybenzoic acid (m-CPBA)32 or brominating agents (bromine33 or N-bromosuccinmide (NBS)34). In these reactions, stoichiometric amounts of waste are generated (Scheme 2), which lowers the sustainability of the rearrangement. More benign approaches have been reported. Examples include, oxidative rearrangement using hydrogen peroxide, catalyzed by homogeneous transition metal complexes such as methyltrioxorhenium (MTO)35 and vanadyl acetylacetonate (VO(acac)2).36 Unfortunately, these novel more benign approaches have not been reported for unsubstituted furfuryl alcohol, that is a more challenging substrate.37,38 Substituted furfuryl alcohols were also needed for the photolytic rearrangement described by Gilmore and co-workers achieved yields up to 89%.39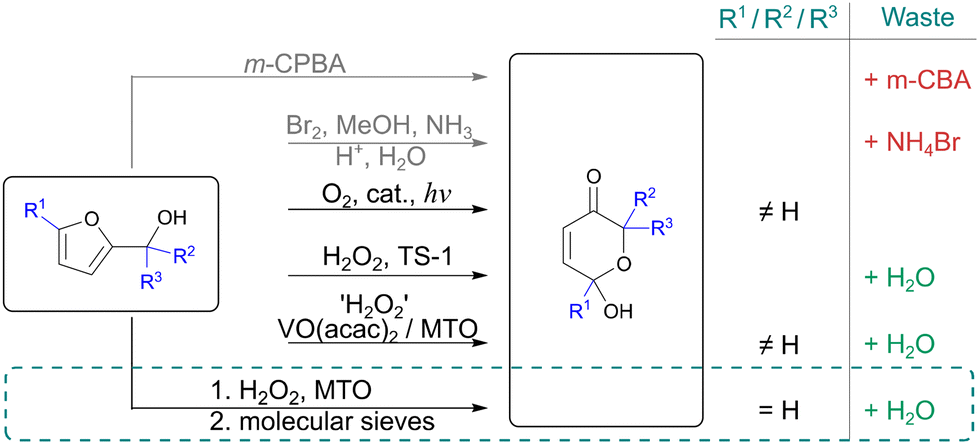 | ||
| Scheme 2 Achmatowicz rearrangement. | ||
Heterogeneous titanium silicalite (TS-1) has been reported in this transformation.40 Attempts to use TS-1 catalyst, as previously reported, were unsuccessful. Only 48% yield could be observed, compared to the reported 94%.40,41 A possible explanation could be the difference in the crystal structure of TS-1 that can significantly influence the activity of the catalyst.42,43 Even though several different procedures were used to prepare TS-1, higher yields were not achieved, showing challenges with this approach.
We were able to successfully transform unsubstituted furfuryl alcohol into compound 3via epoxidation by hydrogen peroxide using MTO catalyst. MTO together with hydrogen peroxide forms active peroxo-metal species able to catalyse double bond epoxidation.44 The reaction gave full conversion of the starting material within 5 h (ESI, Table S4†). After 6 h, selectivity towards the product 3 dropped. If the reaction was left for 2 days, the yield of product 3 decreased to 48% (ESI, Table S4†). This is not surprising as product 3 also contain a double bond that can undergo further reactions.
To confirm product degradation, a stability test was performed. After 3 h using standard reaction conditions, half of the product 3 was degraded, and after 12 h, only 28% of the product remained. Therefore, quenching the reaction after 5 h was essential to prevent side-reactions. Attempts to stop the reaction by simple work-up after 5 h were unsuccessful. Perhaps product degradation in the presence of oxidative agents occurred. We proposed to quench the reaction using molecular sieves once the reaction was completed. The reaction and quenching procedure were optimized, and it was shown that 1 h treatment with molecular sieves after 5 h reaction gave 98% isolated yield of compound 3.
To prevent waste generation at each stage of our transformations, we hypothesized the possibility of recycling the molecular sieves used. When the optimized reaction was completed and quenched, molecular sieves (4 Å) were recovered and dried at 70 °C under reduced pressure for 30 h to release solvent and moisture. Dried molecular sieves were re-used twice, without significant loss in the yield of product 3 (Fig. 2).
 | ||
| Fig. 2 Recycling of the molecular sieves. Reaction conditions: furfuryl alcohol (10 mmol), 2-propanol, addition at 0 °C, reaction at room temperature, H2O2 (1 equiv.), MTO (1 mol%), dried molecular sieves (4 Å) were added after 5 h, the reaction was continued for 1 h more. | ||
Light-driven [2 + 2] cycloaddition
The enone functionality of compound 3 makes it susceptible to light-driven intermolecular [2π + 2π] additions. The self-cycloaddition of enones can occur at longer wavelengths (>300 nm) eliminating the need for a photosensitizer.45In this work, we report, for the first time, the cycloaddition of enone-containing substrate 4 for creating 4-membered adducts (Scheme 2). The resulting adducts are desired precursors for C10 cyclobutane-containing aviation fuels.
The substrate 4 was selected for [2 + 2] cycloaddition due to its more convenient handling and the ease of isolating of the isomeric cycloadducts compared to substrate 3. Compound 4 was synthesised through the methylation of substrate 3 using the procedure described in the Experimental section.
The light absorption spectrum of compound 4 was analysed. In accordance with the anticipated behaviour of the conjugated π-system, the compound showed π–π* absorption band in the range of 300–400 nm, which allows dimerization to occur (ESI, Fig. S1†).
The substrate 4 was irradiated at 365 nm with eight high-pressure mercury lamps (4 W each), resulting in the formation of cycloadducts. The photoreactions were conducted for a total of 48 h, although shorter reaction times can be achieved by employing more powerful lamps. The photoreactions were performed in solution and as neat reactions. The neat reaction gave lower conversion of starting material 4 than the reaction performed in acetonitrile where 79% and 96% conversion, respectively, were observed.
In the concerted light-driven [2 + 2] cycloaddition, the substrate 4 can form two regioisomers: head-to-head (HH) and head-to-tail (HT). Further complexity arises from the possibility of syn- or anti-addition, leading to syn-HH, anti-HH, syn-HT, and anti-HT isomers.46 In this study, we successfully separated and isolated two anti-HH diastereomers (Scheme 3, cycloadducts 1 and 2, both were racemic mixtures) and one anti-HT isomer (Scheme 3, cycloadduct 3). The structures as well as relative stereoisomers of isolated cycloadduct were confirmed by single-crystal X-ray analysis (ESI, Fig. S2–S4†).
 | ||
| Scheme 3 Isolated cycloadducts. The reaction was performed using the following conditions: In a quartz tube, 6-methoxy-2H-pyran-3(6H)-one (154 mg, 1.2 mmol) was dissolved in MeCN (5 mL, 0.24 M) and irradiated using UV lamps (365 nm) for 48 h. The cycloadducts were isolated by column chromatography (40% EtOAc:pentane), recrystallized and analysed by single-crystal X-ray diffraction (ESI†). | ||
Hydrodeoxygenation
As the cycloadducts are highly oxygenated, HDO is required to convert these intermediates to desired aviation fuels.Before optimizing the HDO conditions for cycloadduct transformation, catalyst screening was performed using substrate 3. In our initial experiments, we employed Pd/C while running the reaction at 220 °C at 20 bar of hydrogen pressure using cyclohexane as carrier liquid. After 16 h treatment, we observed full conversion of the starting material and the formation of hydroxy-substituted tetrahydropyran (THP-OH) as a major product (Fig. 3). This confirms that Pd/C facilitates hydrogenation, but not full deoxygenation.
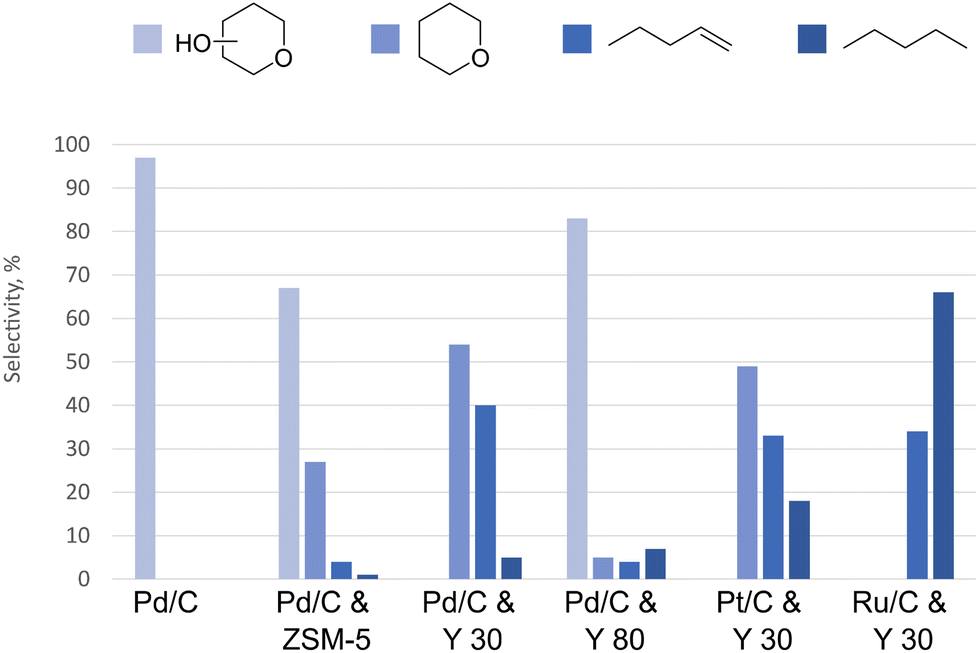 | ||
| Fig. 3 The distribution of products after hydrotreating the compound 3. Reaction conditions: compound 3 (0.88 mmol), cyclohexane (3 mL), 220 °C, 20 bar of hydrogen pressure, 16 h. | ||
When zeolites were introduced together with Pd/C, a shift in the product selectivity towards hydrocarbon formation was observed (Fig. 3). Zeolite screening revealed that the addition of HZSM-5 zeolites (Si/Al ratio of 23) still yielded THP-OH as a major product, while HY zeolites (Si/Al ratio of 30) shifted selectivity towards THP and C5 hydrocarbons. It is likely that larger channels in HY type of zeolites facilitate better diffusion of the substrates through zeolites, making active sites more accessible. Changing the Si/Al ratio of zeolites from 30 to 80 shifted the selectivity back to THP-OH. This shift is connected to the decreased Brønsted acidity in HY with a Si/Al ratio of 80, compared to a Si/Al of 30.
We then tested other noble metals in tandem with HY zeolites (Si/Al of 30). The performance of Pt/C was similar to Pd/C. The more oxophilic Ru/C demonstrated the capacity to hydrodeoxygenate the THP ring, resulting in selective transformation to hydrocarbons (Fig. 3).
With an optimized catalytic system in hand, we performed HDO on the dimeric cycloadducts. The reaction showed full conversion of the starting material and high selectivity towards fully deoxygenated molecules, resulting in a 51% yield of detected hydrocarbons, which included 48% of C7–C10 compounds. Notably, C9 hydrocarbons were the major product, followed by C8, suggesting that C–C bond cleavage took place in the form of decarbonylation of reaction intermediates (Table 2, entry 1).1 Further improvements were attempted by varying carrier liquid (Table 2, entry 2), pressure (Table 2, entries 3 and 4), and reaction temperature (Table 2, entries 5 and 6).
However, no improvement in yields of hydrocarbons in aviation fuel range (C8–C10) was observed. Increasing the pressure to 80 bar resulted in a shift in selectivity towards C8 and C7 products, possibly due to cracking reactions. A similar trend was observed when the temperature was increased to 245 °C. In reactions conducted at lower pressure (Table 2, entry 4) and temperature (Table 2, entry 6), complete deoxygenation was not achieved.
Mass balance
The carbon balance and mass balance were calculated for the proposed value chain. Total carbon yield for the transformation starting from furfural was 48% (Fig. S16, ESI†).The mass balance of the complete value chain was assessed for two distinct scenarios (Fig. 4). In one scenario (S1-Burn), prehydrolysis liquor, generated during a wood pretreatment step, is utilized as a source of energy through combustion in an external co-generation plant. In the second scenario (S2-AF), the prehydrolysis liquor is utilized to produce furfural with residual side-streams contributing to energy co-generation. Subsequently, furfural undergoes a multi-step transformation process to yield hydrocarbons suitable for aviation fuel applications (S2-AF). Quantitative values, expressed in kilograms, were calculated relative to the functional unit, defined as the kilogram of unbleached pulp.
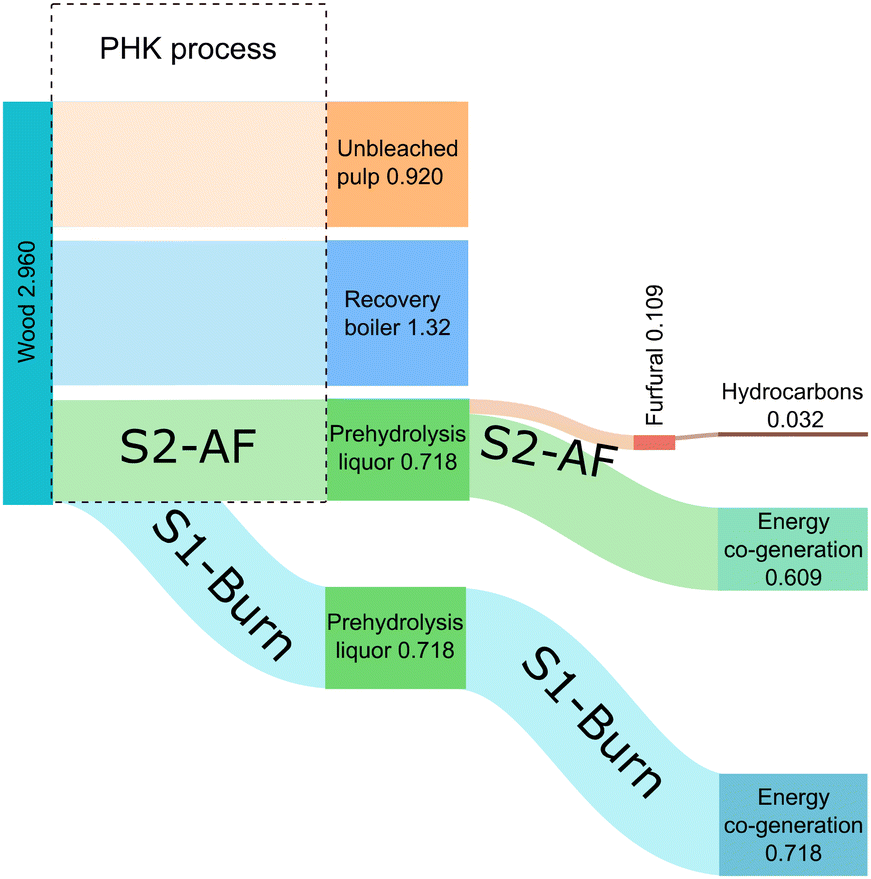 | ||
| Fig. 4 Mass balance. Values are expressed in kilograms. | ||
Life cycle assessment
This section presents the findings of the LCA study. A cradle to gate approach was chosen excluding from our scope the bleaching step, the use phase, and the end life of the dissolving grade pulp. These the life cycle phases are equal for both scenarios (vide supra) to be compared; thus, this approach won't affect the comparison according to ISO 14044. To model the consumption and production electricity, an average of the marginal electricity of Sweden and Finland was considered, then for heat the substitution of biomass and coal were adopted for Sweden and Finland according to the procedure described by Marson et al.Detailed representations of the process model with the system boundaries are available in ESI (Fig. S6–S9†).
To determine the carbon content [kgC kg−1] in cellulose, hemicellulose and lignin and their Low Heating Value [MJ kg−1] to be used in the model secondary data from Ecoinvent 3.9.1 consequential were exploited. From the values retrieved the theoretical energy that can be produced from incineration phases was calculated and the efficiency of the recovery boiler was calculated to be 13% and used in the modelling of the scenarios. From the same source, secondary data for the values of efficiencies to heat and electricity from a co-generation plant were retrieved and are 45% and 15% respectively.
Fig. 5 graphically illustrates the Life Cycle Impact Assessment outcomes for the primary five categories, with comprehensive data detailed in ESI (Tables S10–S13†). S2-AF emerged as the superior performer in the GWP-total (−5.63 × 10−2 kg CO2eq kgUSP−1) and ADP-min&met categories, while S1-Burn was the most performing in SQP, WDP and ADP-fossil categories.
 | ||
| Fig. 5 Contribution of different product system stages comparing S1-Burn and S2-AF. Raw materials: wood chips production. Pulping and incineration: pulping process (including chemicals involved) and all the emissions from the hemicellulose combustion. Chemicals and solvents: chemicals and solvents used in the aviation fuel production chain. Catalysts: catalysts used in the production of aviation fuels. Avoided products: avoided production of pentane and kerosene. Avoided CO2: avoidance of fossil CO2 from the combustion of fossil aviation fuels. Energy: Consumption and avoidance (by production from combusted biomass) of energy from the grid. | ||
In GWP-total for S2-AF, the principal factor contributing to reduced impacts was the avoidance of fossil CO2 emissions relative to conventional aviation fuel, decreasing emissions by 0.57 kg CO2eq kgUSP−1 (USP stands for unbleached sulfate pulp). The efficacy of S1-Burn in the GWP-total, which utilizes prehydrolysis liquor as an energy source in a cogeneration plant, is highly contingent on the replaced energy mix. It should be noted that prehydrolysis liquors are currently burnt in the recovery boiler. When this scenario was calculated, the environmental sustainability was worse in all impact categories; mainly, because the recovery boiler has a lower efficiency. Thus, to study the effect of valorization, incineration in an external co-generation plant was modelled.
Sensitivity analysis reveals that substituting this with a European energy mix enhances environmental benefits, outstripping S2-AF with a GWP-total of −0.634 kg CO2eq.
Conversely, in the SQP category, S1-Burn outperforms S2-AF, attributable to the increased combustion of hemicellulose for heat production, thereby supplanting biomass-derived heat prevalent in Northern Europe's energy mix. This substitution mitigates wood chip consumption and reduces land use impacts. S2-AF's intensified use of solvents and catalysts leads to greater WDP impacts than S1-Burn.
In terms of ADP-fossil, while biobased aviation fuel production substantially reduces impacts, it does not suffice for an overall benefit in this category due to solvent usage in the value chain upgrade. S1-Burn prevails due to enhanced energy yield from hemicellulose. For ADP-min&met, S2-AF outstrips S1-Burn since metal extraction for hydrogenation catalyst concurrently yield other metals concurrently yield other metals, diminishing the need for their primary production, as per the Ecoinvent marginal background data. A full discussion of the results in the other impact categories is available in ESI.†
Four sensitivity analyses assessed the effects of various modeling and operational parameters on the outcomes. The most significant involved an average European energy mix scenario for the integrated pulp mill, resulting in notable shifts for both S1-Burn and S2-AF, particularly in GWP-total, WDP, and ADP-fossil. The impact on GWP-total is attributed to the higher emission factor of the European marginal energy mix (0.218 kgCO2eq kW−1 h−1) compared to the Northern mix (0.07 kgCO2eq kW−1 h−1), suggesting a net reduction in greenhouse gas emissions when replacing an energy mix dominated by fossil fuels. A separate sensitivity analysis evaluated the impact of a 10% lower yield to aviation fuels in the hydrodeoxygenation (HDO) section on S2-AF. Although the GWP-total and ADP-fossil indicators deteriorated, as indicated in Table S15,† S2-AF still maintains a climate change advantage over S1-Burn, albeit with increased impacts on fossil resource use.
Another analysis for S2-AF assessed the effects of producing aviation fuels in an isolated facility, predicting increased GWP-total and ADP-fossil due to higher energy demands. However, the results showed a decrease, contrary to expectations, due to varying efficiencies between recovery boilers and co-generation plants in hemicellulose combustion.
Lastly, altering the assessment to the ReCiPe 2016 (midpoint), H method in the final sensitivity analysis did not yield significant differences in the relevant impact categories. ReCiPe 2016 (midpoint) is a Life-Cycle Impact Assessment method which name stands for the initials of the institutes that were the main contributors to the design and development of the method: RIVM and Radboud University, CML, and PRé Consultants. This method calculates 18 midpoint indicators which focus on single environmental issues and express their relative severity.
Conclusions
Prehydrolysis liquors from forestry textile production are generated in kraft mills. As environmental sustainability concerns of cotton production has increased, it has been projected that wood textile production will increase in the future.47,48 Thus, prehydrolysis liquors will become more available. Currently, these liquors are incinerated in the recovery boiler, which is a bottleneck in pulp mills. This additional load hampers pulp production. This work describes a way to valorize this side-stream beyond its heating value, and at the same time debottleneck pulp production.Herein, a full value chain from an industrial side-stream to high energy content aviation fuel is demonstrated. The prehydrolysis liquors were transformed to furfural using zeolites. Furfural was reduced using homogeneous catalysis, selectively yielding furfuryl alcohol by transfer hydrogenation. Furfuryl alcohol was then rearranged to the Achmatowicz product using MTO and hydrogen peroxide. The resulting adduct was dimerized using a photo [2 + 2] cycloaddition. HDO of the cycloadducts gave desired hydrocarbons in aviation fuel range. Catalyst screening revealed that Ru/C and zeolites were required to cleave all C–O bonds, including the challenging tetrahydropyran bonds. LCA showed benefits in climate change when implementing this technology in a pulp mill instead of incineration.
This study makes progress in mild and selective HDO of challenging C–O bonds. We hope that this inspires researchers to further development of value chains from available side streams.
Author contributions
Daria Lebedeva conducted investigations and wrote the original draft of the article. Lars William Schick conducted investigations and contributed to the writing process. Withsakorn Sangsuwan conducted investigations. Gonzalo Castiella-Ona participated in the investigation phase. Erik Svensson Grape and A. Ken Inge performed single crystal X-ray analysis to determine the absolute configuration of the [2 + 2] isomers. Dagoberto O. Silva, Liane M. Rossi conducted selective hydrogenation of furfural. Daniel Cracco, Alessandro Marson, and Alessandro Manzardo conducted LCA. Elena Subbotina contributed to the conceptualization of the study. Joseph S. M. Samec provided conceptualization, supervised the research, and contributed to the writing process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JSMS acknowledges Swedish Research Council VR (2020-04143) and Swedish Energy Agency (grant 45903-1). LMR acknowledges King Carl XVI Gustaf 50th Anniversary Fund for Science, Technology and Environment for the King Carl XVI Gustafs professorship in environmental science (Konung Carl XVI Gustafs professur i miljövetenskap).References
- J. A. Muldoon and B. G. Harvey, ChemSusChem, 2020, 13, 5777–5807 CrossRef CAS PubMed
.
- C. G. Okoye-Chine, K. Otun, N. Shiba, C. Rashama, S. N. Ugwu, H. Onyeaka and C. T. Okeke, J. CO2 Util., 2022, 62, 102099 CrossRef CAS
- A. Singh, S. I. Olsen and P. S. Nigam, J. Chem. Technol. Biotechnol., 2011, 86, 1349–1353 CrossRef CAS
- V. Lundberg, J. Bood, L. Nilsson, E. Axelsson, T. Berntsson and E. Svensson, Clean Technol. Environ. Policy, 2014, 16, 1411–1422 CrossRef CAS
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS PubMed
- A. Corma, O. de la Torre and M. Renz, Energy Environ. Sci., 2012, 5, 6328–6344 RSC
- Y. Liu, G. Li, Y. Hu, A. Wang, F. Lu, J.-J. Zou, Y. Cong, N. Li and T. Zhang, Joule, 2019, 3, 1028–1036 CrossRef CAS
- T. Dudev and C. Lim, J. Am. Chem. Soc., 1998, 120, 4450–4458 CrossRef CAS
- C. F. Ryan, C. M. Moore, J. H. Leal, T. A. Semelsberger, J. K. Banh, J. Zhu, C. S. McEnally, L. D. Pfefferle and A. D. Sutton, Sustainable Energy Fuels, 2020, 4, 1088–1092 RSC
- J. Xie, X. Zhang, C. Shi, L. Pan, F. Hou, G. Nie, J. Xie, Q. Liu and J.-J. Zou, Sustainable Energy Fuels, 2020, 4, 911–920 RSC
- L. Pan, G. Nie, J. Xie, Y. Liu, C. Ma, X. Zhang and J.-J. Zou, Green Chem., 2019, 21, 5886–5895 RSC
- D. M. Morris, R. L. Quintana and B. G. Harvey, ChemSusChem, 2019, 12, 1646–1652 CrossRef CAS PubMed
- J. L. Fiorio, R. V. Gonçalves, E. Teixeira-Neto, M. A. Ortuño, N. López and L. M. Rossi, ACS Catal., 2018, 8, 3516–3524 CrossRef CAS
- R. J. M. Silva, J. L. Fiorio, P. Vidinha and L. M. Rossi, J. Braz. Chem. Soc., 2019, 30, 2162–2169 CAS
- T. Schaubroeck, S. Schaubroeck, R. Heijungs, A. Zamagni, M. Brandão and E. Benetto, Sustainability, 2021, 13, 7386 CrossRef CAS
- C. Culbertson, T. Treasure, R. Venditti, H. Jameel and R. Gonzalez, Nord. Pulp Pap. Res. J., 2016, 31, 30–40 CrossRef CAS
- A. Marson, J. S. M. Samec and A. Manzardo, Sci. Total Environ., 2023, 882, 163660 CrossRef CAS PubMed
- L. Zampori and R. Pant, Suggestions for updating the Product Environmental Footprint (PEF) method, https://publications.jrc.ec.europa.eu/repository/handle/JRC115959, (accessed February 9, 2024).
- D. Lebedeva and J. S. M. Samec, Sustainable Energy Fuels, 2023, 7, 3637–3643 RSC
- A. S. Mamman, J. Lee, Y. Kim, I. T. Hwang, N. Park, Y. K. Hwang, J. Chang and J. Hwang, Biofuels, Bioprod. Biorefin., 2008, 2, 438–454 CrossRef CAS
- R. Karinen, K. Vilonen and M. Niemelä, ChemSusChem, 2011, 4, 1002–1016 CrossRef CAS PubMed
- S. B. Kim, S. J. You, Y. T. Kim, S. Lee, H. Lee, K. Park and E. D. Park, Korean J. Chem. Eng., 2011, 28, 710–716 CrossRef CAS
- E. Subbotina, A. Velty, J. S. M. Samec and A. Corma, ChemSusChem, 2020, 13, 4528–4536 CrossRef CAS PubMed
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC
- H.-Y. Zheng, Y.-L. Zhu, B.-T. Teng, Z.-Q. Bai, C.-H. Zhang, H.-W. Xiang and Y.-W. Li, J. Mol. Catal. A: Chem., 2006, 246, 18–23 CrossRef CAS
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. L. Granados, Energy Environ. Sci., 2016, 9, 1144–1189 RSC
- F. Li, W. Zhu, S. Jiang, Y. Wang, H. Song and C. Li, Int. J. Hydrogen Energy, 2020, 45, 1981–1990 CrossRef CAS
- J. L. Fiorio, N. López and L. M. Rossi, ACS Catal., 2017, 7, 2973–2980 CrossRef CAS
- D. Ren, L. He, L. Yu, R.-S. Ding, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, J. Am. Chem. Soc., 2012, 134, 17592–17598 CrossRef CAS PubMed
- I. Cano, A. M. Chapman, A. Urakawa and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2014, 136, 2520–2528 CrossRef CAS PubMed
- A. S. Nagpure, P. Gogoi and S. V. Chilukuri, Chem. – Asian J., 2021, 16, 2702–2722 CrossRef CAS PubMed
- L. Zhu, L. Song and R. Tong, Org. Lett., 2012, 14, 5892–5895 CrossRef CAS PubMed
- O. Achmatowicz, P. Bukowski, B. Szechner, Z. Zwierzchowska and A. Zamojski, Tetrahedron, 1971, 27, 1973–1996 CrossRef CAS
- E. A. Couladouros and M. P. Georgiadis, J. Org. Chem., 1986, 51, 2725–2727 CrossRef CAS
- J. Finlay, M. A. McKervey and H. Q. N. Gunaratne, Tetrahedron Lett., 1998, 39, 5651–5654 CrossRef CAS
- J. Robertson, C. North and J. E. R. Sadig, Tetrahedron, 2011, 67, 5011–5023 CrossRef CAS
- J. Deska, D. Thiel and E. Gianolio, Synthesis, 2015, 3435–3450 CrossRef CAS
- A. K. Ghosh and M. Brindisi, RSC Adv., 2016, 6, 111564–111598 RSC
- M. B. Plutschack, P. H. Seeberger and K. Gilmore, Org. Lett., 2017, 19, 30–33 CrossRef CAS PubMed
- S. P. Simeonov, M. A. Ravutsov and M. D. Mihovilovic, ChemSusChem, 2019, 12, 2748–2754 CrossRef CAS PubMed
- S. P. Simeonov, H. I. Lazarova, M. K. Marinova and M. D. Popova, Green Chem., 2019, 21, 5657–5664 RSC
- V. Smeets, E. M. Gaigneaux and D. P. Debecker, ChemCatChem, 2022, 14, e202101132 CrossRef CAS
- B. Wang, Y. Guo, J. Zhu, J. Ma and Q. Qin, Coord. Chem. Rev., 2023, 476, 214931 CrossRef CAS
- W. A. Herrmann, R. W. Fischer, W. Scherer and M. U. Rauch, Angew. Chem., Int. Ed. Engl., 1993, 32, 1157–1160 CrossRef
- Y.-J. Chen, H.-L. Wang, N. R. Villarante, G. J. Chuang and C.-C. Liao, Tetrahedron, 2013, 69, 9591–9599 CrossRef CAS
- M. Sakamoto, F. Yagishita, M. Kanehiro, Y. Kasashima, T. Mino and T. Fujita, Org. Lett., 2010, 12, 4435–4437 CrossRef CAS PubMed
- A. Adler, I. Kumaniaev, A. Karacic, K. R. Baddigam, R. J. Hanes, E. Subbotina, A. W. Bartling, A. J. Huertas-Alonso, A. Moreno, H. Håkansson, A. P. Mathew, G. T. Beckham and J. S. M. Samec, Joule, 2022, 6, 1845–1858 CrossRef CAS
- K. Witthayolankowit, A. Marson, K. R. Baddigam, D. Lebedeva, M. Shaikh, A. Kane, D. Gupta, M. I. Wide, A. P. Mathew, D. Kubička, A. Manzardo and J. S. M. Samec, Chem. Eng. J., 2023, 470, 144179 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2307840, 2307841 and 2308513. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4gc01257g |
| This journal is © The Royal Society of Chemistry 2024 |