
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAgile synthesis and automated, high-throughput evaluation of diglycolamides for liquid–liquid extraction of rare-earth elements†
Lun
An‡
 a,
Yue
Yao‡
b,
Tyler B.
Hall
a,
Fu
Zhao
*bc and
Long
Qi
*a
a,
Yue
Yao‡
b,
Tyler B.
Hall
a,
Fu
Zhao
*bc and
Long
Qi
*a
aU.S. DOE Ames National Laboratory, Iowa State University, Ames, Iowa 50011, USA. E-mail: lqi@iastate.edu
bSchool of Environmental and Ecological Engineering, Purdue University, West Lafayette, IN 47907, USA. E-mail: fzhao@purdue.edu
cSchool of Mechanical Engineering, Purdue University, West Lafayette, IN 47907, USA
First published on 22nd May 2024
Abstract
Liquid–liquid extraction is one of the most scalable processes to produce rare-earth elements (REEs) from natural and recycled resources. Accelerating the research, development, and deployment (RD&D) of sustainable processes to manufacture REEs requires both facile synthesis of extractive ligands at scale and fast evaluation of process conditions. Here, we establish an integrated RD&D methodology comprised of agile ligand synthesis and automated high-throughput extraction studies. Using diglycolamides (DGAs) as an example, we first developed a method for DGA synthesis (scalable to 200 g) by directly coupling diglycolic acid and secondary amines via the solvent-free melt-amidation reaction. A substrate scope of the melt-amidation synthesis was demonstrated for 9 different DGAs with good yields (85–96%) and purities (88–96%) without any post-reaction workup or purification process. Life cycle assessment shows that our synthesis method outperforms the prior-art pathway in each environmental impact category, especially showing a 67% reduction in global warming potential. Furthermore, we investigate the structure–activity relationship of various alkyl-substituted DGAs using an automated, high-throughput workflow for liquid–liquid extraction, achieving over 180 runs in 48 hours. The acquired data enables the development of a promising flowsheet for separating light and heavy REEs. The integrated RD&D method of agile synthesis and automated, high-throughput extraction studies paves the way for future iterative development of sustainable production of REEs and other critical materials to meet the needs for clean energy transformation.
1. Introduction
Rare-Earth Elements (REEs) constitute a group of 17 metals that manifest unique physical and chemical properties,1 leading to their unsubstituted applications in modern society with increasing demands.2 Contrary to their name, REEs aren't rare when considering their abundance in the Earth's crust.3 For instance, cerium ranks as the 25th most abundant element, surpassing copper in prevalence. However, unlike the main-group and transition metals, REEs are dispersed and mixed in Earth's crust rather than concentrated in ore deposits.4 Furthermore, the similar physical properties of these REEs present additional challenges to separating them from one another, particularly among adjacent lanthanides (Ln) in the periodic table.5 Over the past decades, several techniques have been developed to achieve efficient REE separation,6 of which hydrometallurgical methods via liquid–liquid (L–L) extraction are extensively studied processes with large-scale industrial applications.7 In this process, REE minerals are first leached in inorganic acids and concentrated. The resulting aqueous metal solutions are mixed with an immiscible organic phase containing lipophilic extractants (ligands) and phase modifiers, which selectively bind and transfer REEs into the organic phase to realize separation. Among the diverse lipophilic ligands utilized in L–L extraction process,8–14 diglycolamides (DGAs) have recently received broad attention due to their chemical stability, high affinity to trivalent metal ions, neutral structure, and facile structural modifications.15,16In facing diverse natural and recycled feedstocks of varied REE compositions, fast development of new separation methods requires rapid evaluation of process parameters.17 These process parameters include but are not limited to types of ligands, concentrations of organic phase modifiers, types of inorganic acids, and pH of the aqueous phase. In a recent example, Schelter and co-workers demonstrated the high throughput optimization of acid concentrations for ligand-assisted precipitation of REEs.18 With the help of automated instrumentation, the screening workflow (dissolution, dilution, mixing, and sample taking) can be envisioned. However, the consumption of ligands is drastically increased and normally beyond gram scale. Therefore, it is critical to synthesize a series of separation agents at the scale of 10 g and above. In this context, developing an environment-friendly general method to synthesize a series of ligand derivatives with identical binding sites is highly desirable to minimize efforts for further scaling up and accelerate process development.
DGAs exemplify a promising candidate in further exploration and development for REE separation.19 However, their widespread application remains constrained because of limited commercial availability and high cost. For example, the well-established Schotten–Baumann approach requires pre-activating diglycolic acid to diglycolic chloride to promote the amide bond formation with alkyl amines (Scheme 1a).20 This multi-step method suffers from the use of moisture-sensitive reactants, including thionyl chloride (SOCl2), oxalyl chloride (COCl)2 and intermediate (diglycolic chloride), and anhydrous solvents with the emission of toxic side-products (SO2, CO, and HCl). To overcome the drawbacks of the Schotten–Baumann approach, Verboom and co-workers reported direct amidation of the diglycolic esters mediated by aluminum chloride (AlCl3) (Scheme 1b),21 allowing the one-step transformation of stable diesters into the desired DGAs and eliminating the preparation of the diglycolic chlorides. However, this method still requires an excessive amount of hygroscopic AlCl3. The direct amidation of carboxylic acids with amine, culminating in the exclusive generation of water as a byproduct, represents the most straightforward approach for amide synthesis owing to its simplicity and superior atom economy.22–24 In the seminal report, Filachione et al. presented the synthesis of lactamides by dehydration of lactic acid and amines.25 In the following work, Jansone-Popova and co-workers reported a one-step DGA synthesis by coupling diglycolic acid and secondary dialkyl amines through nucleophilic substitution (Scheme 1c).26 The use of xylenes as the reaction media, a high-boiling and toxic organic solvent,27 in conjunction with the Dean–Stark setup apparatus, may potentially impede its scalability. Moreover, direct amidation of carboxylic acids under solvent-free conditions has been reported under microwave irradiation28 and radiofrequency heating conditions.29 The reported direct amidation primarily focused on monocarboxylic acids with primary or secondary cyclic amines instead of dicarboxylic acids and secondary acyclic amines.30,31
 | ||
| Scheme 1 Comparison of different methods for DGA synthesis via (a) the Schotten–Baumann approach, (b) amidation of diglycolyl esters, (c) thermal amidation using Dean–Stark apparatus, and (4) the new solvent-free melt amidation method. | ||
Here, we report a new method for DGA ligand synthesis by directly coupling two readily available precursors, diglycolic acid and dialkyl amines, under melting conditions for one-step amidation (Scheme 1d). The two precursors are mixed to yield the ammonium carboxylate salt before being heated to melt and converted into DGAs under flowing nitrogen gas (N2). The current process is simple and readily scalable without any pre-activation step or post-reaction purification. The low-cost synthesis involving no solvent or catalyst is green. Life cycle analysis (LCA) was carried out to quantify the environmental benefit of this newly proposed synthetic method. Furthermore, we also established an automatic high-throughput L–L extraction workflow to accelerate the evaluation of the structure–activity relationship and the development of the REE separation process with the as-synthesized DGAs.
2. Results and discussion
2.1. Melt-amidation synthesis of DGAs
We first established the viability of the melt amidation method utilizing diglycolic acid dihexylammonium salt (3a) as a representative model substrate, as delineated in Table 1. The ammonium salt was synthesized in quantitative yield by mixing diglycolic acid (1) and dihexylamine (2a) at room temperature using an environmentally friendly solvent such as water, ethanol, or 2-propanol. The solvent-free salt (3a) was transferred to a round bottom flask fitted with an air condenser (Fig. S1†). Industrial-grade N2 (99.9 vol%) was introduced into the system at a flow rate of 100 cubic centimetres per minute (ccm), ensuring a consistent N2 atmosphere. The flowing N2 prevents the oxidation of nitrogen-containing species at higher temperatures and continuously removes the by-product water to drive the reaction equilibrium toward the amide product.|
|
|||||
|---|---|---|---|---|---|
| Entry | Flowing Gas | Temperature | Time | Conversion, % | Purity, % |
| a Reaction conditions: 3a (505 mg, 1 mmol), N2 flow rate 50 ccm, all temperatures are the bath oil temperature; conversion and purity were determined by 1H NMR. b In a 25 mL sealed tube under N2 atmosphere, no gas flow. c Open to air, no gas flowing. d 50 mmol scale. | |||||
| 1 | N2 | 150 °C | 12 h | <2 | — |
| 2 | N2 | 180 °C | 12 h | 75 | — |
| 3 | N2 | 200 °C | 12 h | 100 | 91 |
| 4b | N2 | 200 °C | 12 h | 100 | 75 |
| 5c | Air | 200 °C | 12 h | 100 | 42 |
| 6d | N2 | 200 °C | 24 h | 100 | 92 |
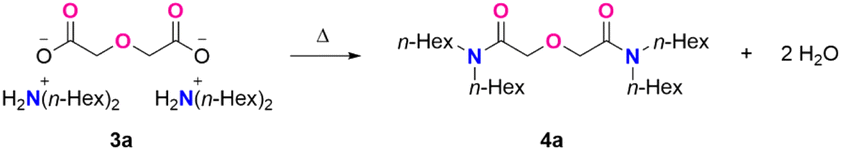
Initial screening efforts showed that the melt amidation reaction is sensitive to reaction temperatures. Less than 2% conversion of the starting material 3a was observed with no formation of product N,N,N′,N′-tetra(n-hexyl)diglycolamide (THDGA) 4a at 150 °C in 12 h (entry 1, Table 1). Increasing the reaction temperature to 180 °C resulted in a 75% conversion of 3a (entry 2). Full conversion was achieved at 200 °C, with the production of 4a in 91% purity (entry 3).
N2 flow was also essential to the reaction efficiency: inferior purity (75%) was observed under static instead of flowing N2 (entry 4). The purity of the product decreased further to 42% when the reaction was exposed to air (entry 5), resulting in a mixture of black color, suggesting a partial oxidation of the nitrogen-containing groups. Further efforts attempted to scale up the synthesis to 50 mmol (25.3 g) show no observable difference in the yield and purity of compound 4a (entry 6), thus demonstrating the scalability of the proposed method.
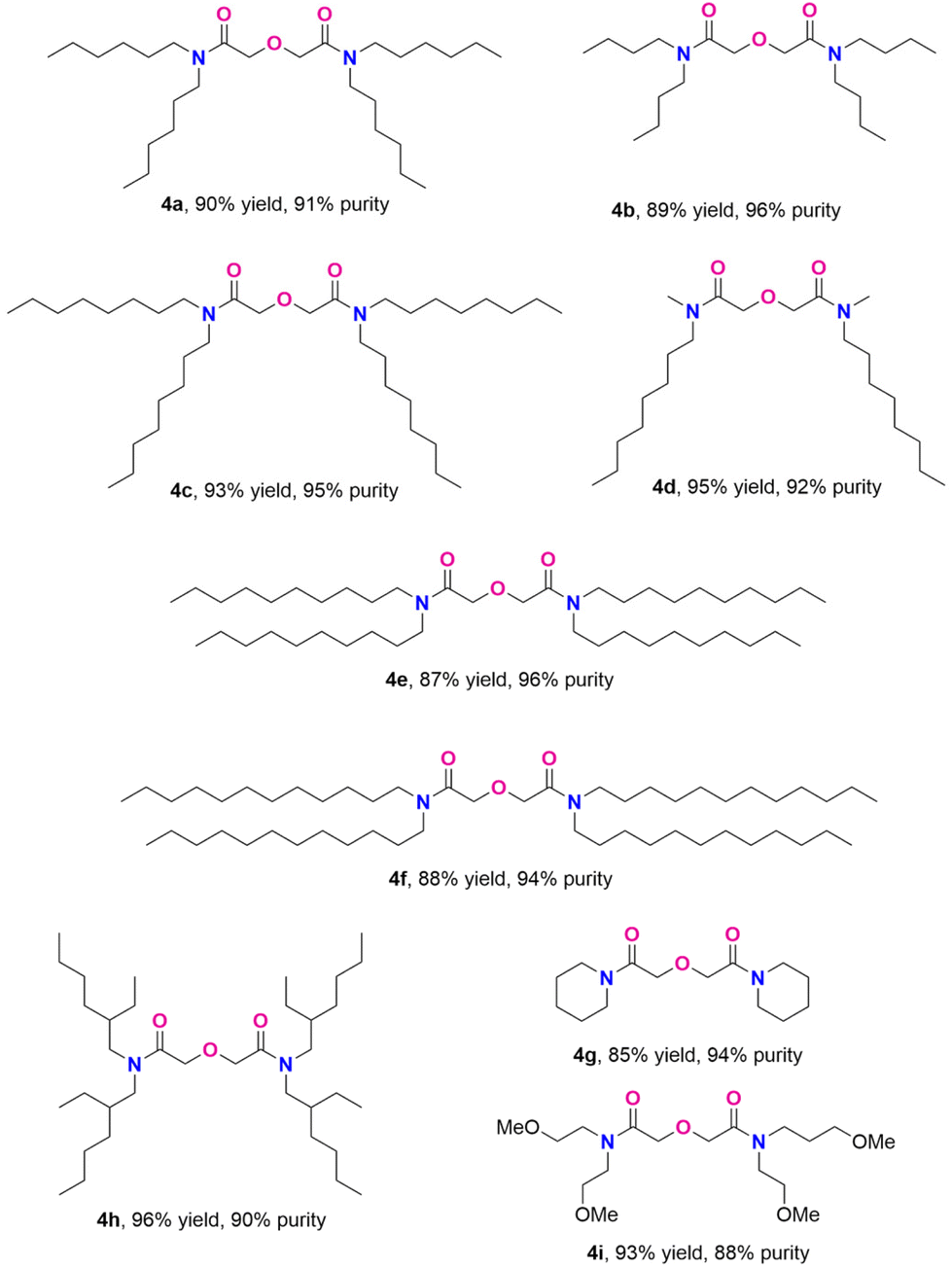
With the optimized conditions, we successfully explored the substrate scope of the melt-amidation reaction as shown in Scheme 2. Compared to 3a, the ammonium salt 3b, derived from the less sterically hindered dibutyl alkyl groups, yielded only 50% of DGA 4b with a purity of 70%, even though the starting material underwent quantitative conversion. As this amidation reaction involves equilibriums among free carboxylic acid, amine, and ammonium salt (Scheme S1†), we hypothesize that the diminished yield is attributed to the low boiling point of dibutyl amine (∼159 °C), causing its loss upon evaporation under the N2 flow at 200 °C. Indeed, the presence of dibutylamine in the N2 flow was verified through solution 1H NMR analysis of the collected condensed vapor. After condition optimization, we found that adding one more equivalent of dibutyl amine after 24 hours gave 4b in 89% yield with 96% purity.
 | ||
| Scheme 2 The substrate scope of the current melt-amidation method. Refer to part 2.4 of ESI† for reaction condition details. | ||
N,N,N′,N′-tetraoctyl diglycolamide (TODGA, 4c),16 one of the most studied DGA ligands, was obtained in 93% yield and 95% purity at a 300 mmol scale. Ammonium salts with longer carbon chains, derived from didecylamine and didodecylamine (3e and 3f, respectively), can be converted to the corresponding TDecDGA (4e) and TDodDGA (4f) in comparable yields with high purity. Secondary amines with two different alkyl groups also gave good yields and purities (4d). Besides linear secondary ammonium salts, a branched alkyl analogue also underwent the reaction smoothly, producing 4h in 96% yield with 90% purity. In addition to linear and branched chain dialkyl amines, cyclic alkyl amines were effectively converted to the desired product with high purity (4g). The current melt-amidation condition can be applied to synthesizing DGA 4i (93% yield and 88% purity), which has a secondary coordination sphere. Overall, the general synthetic method works well for both linear and branched secondary amines. Observed purities of 88–96% fall in the range for diglycolamides industrially produced at tonne scale.
To further highlight the versatility and scalability of the current melt-amidation approach, we integrate the two-step synthesis of DGA via diglycolate dialkylammonium salt into a one-pot process using THDGA as an example (Scheme S2†). This method further reduces the cost and process time for DGA synthesis with simple reaction set-up, no product purification, and feasible scale-up, indicating significant potential for industrial-scale DGA production. In comparison, prior art DGA synthesis is challenged by (1) pre-activation of diglycolic acid, (2) use of toxic organic solvents, (3) production of toxic side-products as waste, and (4) tedious purification steps.
3. Life cycle assessment studies
3.1 Goal and scope definition
To understand the potential advantages and/or disadvantages of the innovative system, an LCA study was carried out to identify the environmental hotspots in the new DGA synthesis method (Scheme 1d) compared to the prior art (Scheme 1a). The functional unit of both DGA synthesis systems is defined as 1 kg TODGA (95% purity) production using di-octylamine based on the data collected in the United States of America. The scope of this LCA study is defined as “cradle-to-gate”.3.2 Inventory analysis
Within the boundary of the novel TODGA synthesis system (Scheme 1d), we consider diglycolic acid production, di-octylamine production, and TODGA production. For the prior art TODGA synthesis system (Scheme 1a), we include the production of diglycolic chloride, di-octylamine, and TODGA in the assessment. All emissions related to the material production and energy consumption in the reaction are considered in the study. We assume the transportation of materials is the same for both synthetic pathways; thus, relevant emissions are ignored in this study. The production of the experiment apparatus (reaction vessel, magnetic stir bar, etc.) is cut off in the system boundary due to the small amount of use. The background data of unit processes are extracted from Ecoinvent v3.8 and US-EI 2.2, while the foreground data of the prior art and novel methods are collected from relevant literature and experimental studies, respectively. Each unit process of the product system is modeled by the Simapro software. Detailed data related to life cycle inventory (LCI) tables (Tables S1–S7†) and assumptions (Tables S8 and S9†) for 1 kg TODGA production (95% purity) using prior-art and novel methods are included in ESI.†3.3 Impact assessment
Ten impact categories from the TRACI 2.1 v1.07 (US 2008) method are analyzed, including ozone depletion, global warming, smog, acidification, eutrophication, carcinogenic, non-carcinogenic, respiratory effects, ecotoxicity, and fossil fuel depletion.32 The LCI results are automatically classified into these impact categories using Simapro software. The characterization factors from TRACI are applied to convert LCI results to impact scores. The contribution analysis is performed based on the impact scores after characterization for all the impact categories considered in the assessment. | ||
| Fig. 1 Comparative characterization results for two DGA synthesis methods: the new metal-amidation method and the traditional Schotten–Baumann approach. | ||
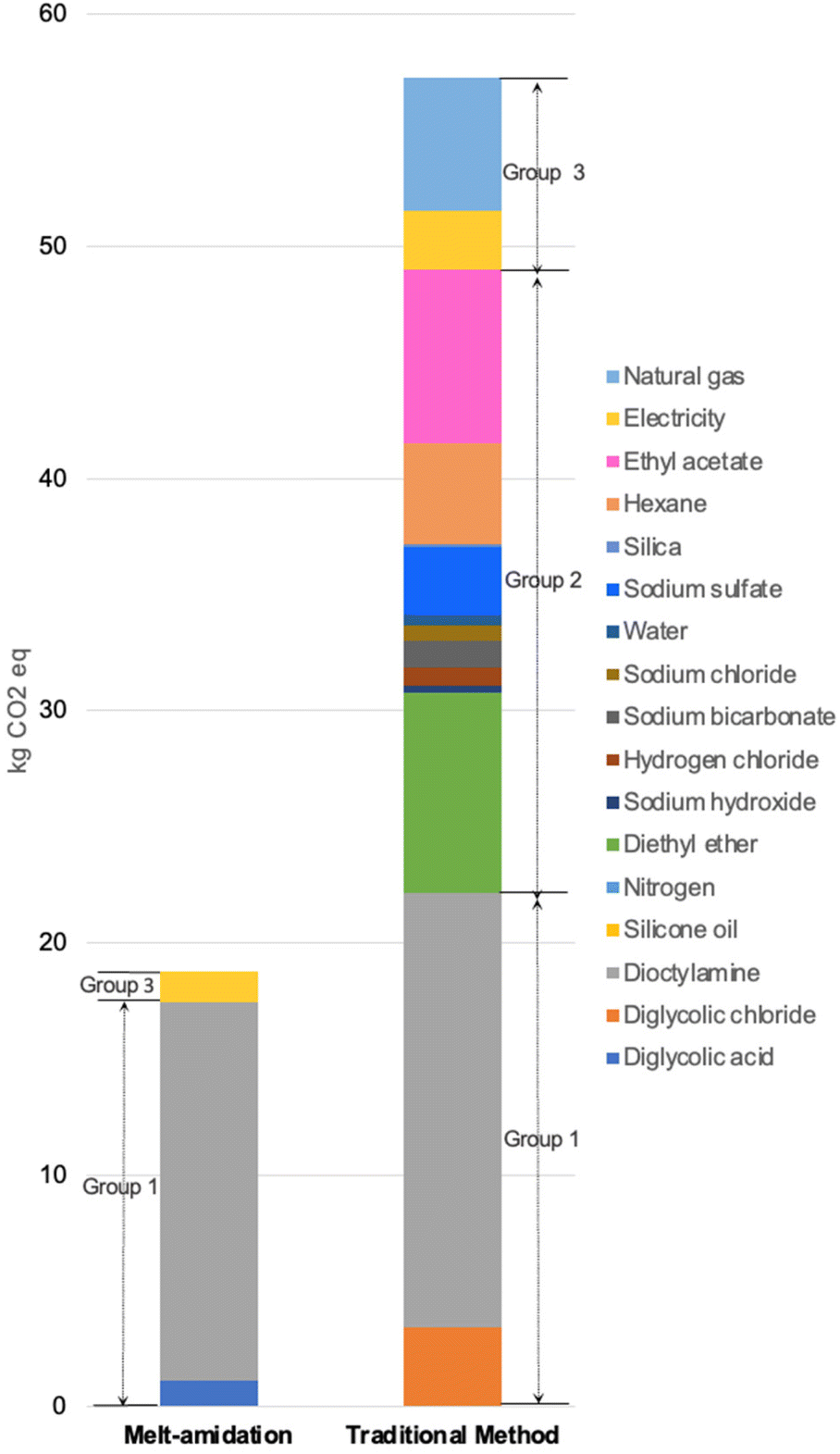 | ||
| Fig. 2 Comparative LCA contribution analysis between two DGA synthesis methods on global warming. | ||
3.4 Interpretation
As can be seen from the global warming hotspot analysis, the new TODGA synthesis pathway has a significant advantage over the prior art one regarding the production of reactants and other chemicals, and energy consumption. For the prior art, the use of toxic reagents (e.g., thionyl chloride) in diglycolic chloride manufacturing greatly increases the impacts of reactants production. At the same time, the use of organic solvents and inorganic additives/desiccants as supporting materials is responsible for a large proportion of the overall impacts. Furthermore, the energy consumption of complex purification and organic solvent regeneration steps occupies a noticeable part of the total results. Therefore, it is apparent that the novel pathway has preferable environmental performances as a result of the simple reaction set-up, solvent-free reaction condition, and easy product separation. In addition, it is worth noting the novel method still has the potential to further improve the environmental profiles for industrial-scale production since the higher efficiency of energy consumption will further reduce the environmental impacts.4. Rare-earth elements extraction studies
4.1. Automated high-throughput platform for REEs extraction
Our facile, scalable synthesis of DGAs provides abundant ligands in variety to accelerate the L–L extraction studies by systematic evaluation of process parameters using an automated high-throughput workflow. Using a Chemspeed Swing platform, we successfully implemented an automated workflow for solid dosing, liquid transfer, and on-demand sampling/dilution, constituting the three main experiment steps for REE separation studies (Fig. S4†). The extraction experiments were modified based on the literature method.35 In a typical experiment, isoparaffinic C11–C13 hydrocarbon (Isopar L) mixed with 30% (v/v) isotridecanol (Exxal 13) was used as the organic phase, and 3.0 M hydrochloric acid (HCl) or nitric acid (HNO3) containing the REE chlorides or nitrates salts as the aqueous phase. After mixing, a quantitative portion of the aqueous phase is sampled and diluted before concentration measurement with inductively coupled plasma optical emission spectroscopy (ICP-OES). The concentrations of REEs in the organic phase were further calculated by subtracting the amount of REEs remaining in the aqueous phase from the initial amount of REEs. Extraction efficiencies (E) are employed to evaluate the extractive capacity of DGAs, quantifying the ratio of REE concentrations extracted into the organic phase relative to the initial REE concentration in the aqueous phase. Distribution ratios (D) are utilized to describe the distribution of metal ions in the organic phase relative to their concentrations in the corresponding aqueous phase, which are plotted in their logarithmic form (Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D).
D).
Prior to the high throughput method, it is critical to validate that the DGAs produced using the new melt-amidation method have similar REE extraction capabilities as DGAs from other sources; thus we first manually carried out side-by-side experiments comparing TODGAs from three sources: (1) synthesized through our melt-amidation method (TODGA-1), (2) synthesized using the Schotten–Baumann method (TODGA-2), and (3) acquired from a commercial vendor (TODGA-3). As illustrated in Fig. 3a, all three TODGAs gave similar extraction efficiencies for all lanthanides(III) under manual conditions, demonstrating the high quality of DGAs using our synthetic method.
 | ||
| Fig. 3 (a) Validation of the extraction efficiency of TODGAs from different sources, synthesized through the current melt-amidation method (TODGA-1), synthesized using the Schotten–Baumann method (TODGA-2), and commercially available TODGA-3 in 3.0 M HCl and Isopar L/Exxal 13(7/3). (b) Comparison of the LogD value of manual and robotic experiments using TODGA and DMDODGA in 3.0 M HCl and Isopar L/Exxal 13 (7/3). (c) Comparison of the LogD values of sequential and direct sampling methods. (d) The effects of Isopar L and Exxal 13 ratio on the extraction experiments of TODGA in 3.0 M HCl. (e) The concentration of HCl on the extraction efficiency of TODGA. (f) The effect of acid media and counter anion on the extraction experiments of TODGA. All experiments are triplicated by repeating the whole liquid–liquid extraction process, and uncertainty is, in general, too small to be shown (see ESI Tables S14–S19† for details). | ||
We then establish the automated high-throughput workflow for L–L extraction by automating the solid dosing and liquid transfer through the gravimetric dispensing unit and liquid dispensing tool. As shown in Fig. 3b, extraction data obtained through automated workflow using TODGA and DMDODGA as ligands are consistent with those by manual operations (see ESI† for details), which substantiates the reliability of our automated workflow.
For the ICP sample preparation after the extraction step, two distinct sampling approaches were tested: direct sampling and sequential sampling. The direct sampling involves the use of a sampling needle inserted by the robotic arm directly through the upper organic layer into the lower aqueous phase for sample collection. For the sequential sampling, the upper organic layer was removed before sampling the aqueous phase. Fig. 3c illustrates the results obtained from these experiments, wherein it can be observed that sequential sampling generally gave better and more consistent results than direct sampling. The plausible explanation for this observation is that the sample solution obtained via direct sampling may have been contaminated when the needle head passed through the upper organic layer, which typically contains a higher concentration of rare earth elements.
4.2. Optimization of extraction parameters
Introducing automated high-throughput platform for REE extraction has considerably accelerated the process evaluation, thus allowing us to optimize various extraction process parameters (such as solvent composition, counter anions, and acid type) within 24 h.Notably, the compositions of both organic and aqueous phases play a pivotal role in influencing extraction efficiencies and distribution factors.36 Aliphatic alcohols (such as Exxal 13) are often included in the organic phase as a phase modifier to preclude the formation of an emulsion phase.37 As the volumetric ratio of Isopar L to Exxal 13 was adjusted at 5:5, 7:3, and 9:1, the highest extraction efficiency was observed at the medium Exxal 13 concentration (Fig. 3d); thus the solvent with Isopar L to Exxal 13 of 7:3 volumetric ratio was chosen for future studies. Even at a 9:1 ratio, no phase disengagement or emulsion formation was observed. Consequently, the enhanced extraction observed with higher Exxal 13 ratios probably arises from other causes, such as solvent modification38,39 and kinetic enhancement.40,41
We further varied the concentrations and types of inorganic acids. Conventionally, for REE extraction experiments with DGAs, both HCl and HNO3 have been used as acidic media.42,43 First, hydrochloric acid was examined at concentrations of 0.1, 1.5, and 3.0 M. As illustrated in Fig. 3e, higher acid concentration in the aqueous phase facilitates the migration of metal ions from the aqueous to the organic phase and drastically increases the distribution ratios.44,45 For example, the D value for Dy is increased from 3.1 to 94.1 by doubling the HCl concentration from 1.5 M to 3.0 M.
Counter anions in the metal salt and acid media, as ligands, can influence the phase transfer of metal cations. We then compared HCl and HNO3 as acid media with the respective Ln chloride or nitrate salts, showing that HNO3 outperformed HCl in both distribution ratios and extraction efficiencies (Fig. 3f). For TODGA, the extraction efficiency of La increased from 22% to 94% when the counter anion in both metal salts and acid media was switched from chloride to nitrate, at the acid concentration of 3.0 M. Compared to REE chlorides, REE nitrates typically display higher ionization potential46,47 but weaker hydration energy,48 thereby improving the extraction efficiency of the nitrate complex into the organic phase.43 Interestingly, TEHDGA, found to be an ineffective ligand for all 14 Ln chloride salts in HCl, was significantly improved when switched to nitrate as the counter anion (Fig. 3f). For instance, the extraction efficiency of La increased from 5% to 52%, that of Nd rose from 9% to 74%, and the E for Dy boosted from 27 to 97%, when switching from Cl− to NO3− in both metal salts and acid media. This observation underscores the salient role of extraction media in the extraction process. Such key insights suggest that simple adjustments to extraction parameters, such as aqueous media and acid concentration, could yield extraction and separation improvements on par with or surpassing those achieved through structural modifications of DGAs, which often require extensive synthetic efforts. Moreover, the former approach may also lead to reduced process development costs. It is worth noting that, in the current study, the improvement in extraction efficiencies by varying the acid media is general to all REEs.
4.3. Experimental sidechain effect on extraction studies
With the optimized process conditions, our research further delved into understanding structure–activity relationships on the extraction performance of DGAs. It has been hypothesized that the coordination affinity of DGAs is pivotal in modulating their REE extraction efficiency.35 This affinity can be adjusted by altering the chain length and branching of N-alkyl substituents in DGAs, thereby affecting the first coordination sphere. TODGA (4c) was selected as a representative to examine the impact of sidechain variations on REE extraction. When comparing 4c with its branched isomer TEHDGA (4h), a marked reduction in the D value for all 14 Ln was observed, Fig. 4. This decrease is attributed to the steric hindrance caused by ethyl groups at the β position, which undermines the interaction with the Ln ions. | ||
| Fig. 4 Comparison of the extraction efficiency of 4c, 4d, 4g, 4h, and 4i in 3.0 M HCl and Isopar L/Exxal (7/3). See Table S20† for details. | ||
Conversely, substituting one octyl group on each N atom in 4c with a methyl group (4d) significantly enhances D for all 14 lanthanides, which is attributable to the reduced steric impact. A deviation was noted with 4g, where shrinking the N substituents to a cyclic hexyl group surprisingly exhibited slightly better extraction capability than 4h. This lower extraction efficiency in 4g compared to 4c and 4d is hypothesized to stem from increased aqueous solubility due to shorter hydrophobic alkyl chains, which exert a higher impact on the extraction capability relative to the steric effect.49 The solubility factor also plays a crucial role in the performance of 4i. Originally designed to incorporate additional coordination sites in the secondary coordination sphere beyond the primary sphere, 4i was aimed to enhance interaction with Ln(III) and improve extraction efficiency. However, 4i demonstrated lower REE extraction efficiency among the substrate scope, most likely also caused by increased aqueous solubility because of its hydrophilic methyloxyl substituents.
Overall, the separation study of different N-substituted DGAs suggests that decreasing steric hindrance around the primary coordination sphere (4d < 4c < 4h) leads to a significant enhancement in extraction strength (4d > 4c > 4h), as illustrated in Fig. 4. Aqueous solubility also plays a substantial role in determining the REE extraction efficacy of DGAs.50
4.4. Stripping studies
The stripping and recovery of REEs from the organic phase is a crucial step to complete the separation process, as recovery of the separated REE metals allows repeated use of the valuable ligands.19,51–53 Using the automated workflow, we evaluated the stripping of the lanthanides in the organic phase with an aqueous solution of varied acid concentrations. The stripping efficiency (ES) is defined as the ratio of Ln(III) concentration in the aqueous stripping solution to that in the organic phase. When TODGA was used in the extraction study (Fig. 3f), moderate extraction efficiency was achieved for light lanthanides (La to Sm), with E values of over 90% for the medium and heavy ones. In contrast to the Ln(III) extraction, we found that optimal stripping occurred at low acid concentrations, as shown in Fig. 5. Specifically, at a 0.1 M HCl condition, we achieved over 80% ES for all 14 Ln(III) elements in one hour of mixing time. Under 1.5 M HCl conditions, we achieved over 70% ES for La–Sm, while Ho–Lu exhibited approximately 30% ES. The highest selectivity was attained at 3.0 M HCl, with over 95% ES for La and moderate ES (45–60%) for Ce, Pr, and Nd while stripping efficiencies were significantly less than 10% for the heavy Ln(III) elements. | ||
| Fig. 5 Stripping 14 lanthanides from the organic phase to the aqueous phase under different HCl concentration conditions. | ||
Integrating selective extraction and stripping using aqueous solutions of optimal acid concentrations enables a new flowsheet to separate light and heavy REEs (Scheme 3). The residual aqueous solution from the extraction process and the used stripping solution are rich in light REE and can be combined. The light REEs can be recovered by further coupling with selective precipitation.54 Similarly, the heavy REE in the organic phase can be stripped using aqueous solutions of low acid concentration and precipitated. Thus, aqueous solutions can be recycled without neutralization/reacidification, and organic ligands can be reused without purification. It becomes possible to achieve selective separation of heavy and light Ln(III) elements by fine-tuning the acid media while maintaining maximum extraction efficiency.
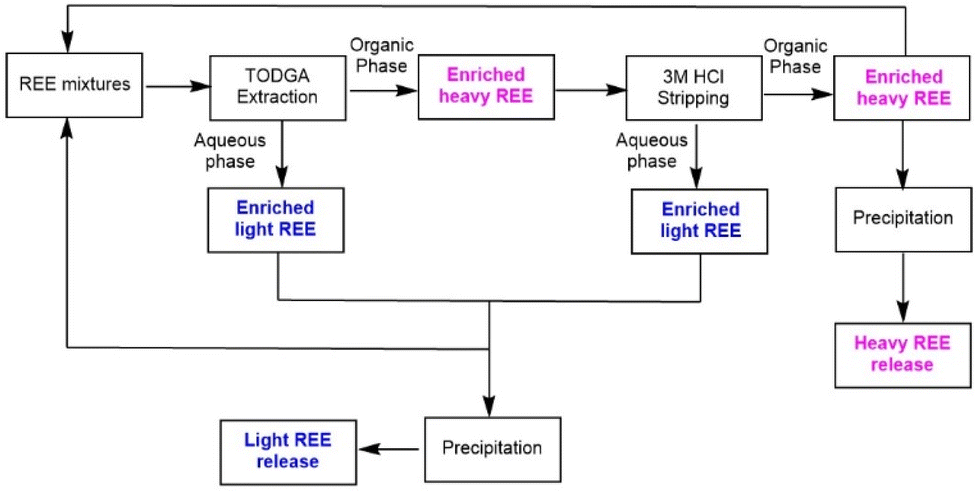 | ||
| Scheme 3 Selective separation of heavy and light Ln(III) elements via cascade extraction and stripping. The recycling of the organic and aqueous phases is not illustrated. | ||
5. Conclusions
We have demonstrated a scalable and sustainable approach to accelerate the liquid–liquid extraction of REEs through the integration of agile ligand synthesis and automated high-throughput extraction studies. The novel green synthesis of diglycolamides via solvent-free melt-amidation reaction is marked as a significant advancement with its simplicity, high yield, and purity. The ability to scale this synthesis up to 200 grams without compromising yield or purity highlights its industrial feasibility, offering a practical solution for large-scale REE extraction processes. This aspect is further complemented by the sustainability of the process, as evidenced by a significant 67% reduction in global warming potential based on a comprehensive life cycle assessment. Furthermore, a high-throughput workflow for liquid–liquid extraction is demonstrated to accelerate the understanding of the impact of process parameters and the structure–activity relationship of ligands on liquid–liquid separation. Consequently, this integrated approach enables the rapid determination of optimal conditions for REE separation and stripping studies, leading to a simple workflow for separating light and heavy REEs. Our approach can be readily applied to the research, development, and deployment of other critical materials, including Li, Co/Ni/Mn, and others associated with natural or reclaimed materials, to meet the growing needs of clean energy transformation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Critical Materials Innovation Hub funded by the U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy, Advanced Materials and Manufacturing Technologies Office (AMMTO). The synthesis and extraction studies were performed in Ames National Laboratory, operated for the U.S. Department of Energy by Iowa State University of Science and Technology under Contract No. DE-AC02-07CH11358. The life cycle assessment was performed at the University of Purdue under Subcontract SC-24-599 with the Ames National Laboratory. T. B. H. acknowledges the DOE Office of Science's Office of Workforce Development for Teachers and Scientists (WDTS) for the Science Undergraduate Laboratory Internships (SULI) program. The ICP-OES analysis was supported by the U.S. Department of Energy (DOE), Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences, Catalysis Science program. We thank Chemspeed Technologies AG for the kind support.References
- T. Cheisson and E. J. Schelter, Science, 2019, 363, 489–493 CrossRef CAS PubMed.
- V. Balaram, Geosci. Front., 2019, 10, 1285–1303 CrossRef CAS.
- A. Kumari, R. Panda, M. K. Jha, J. R. Kumar and J. Y. Lee, Miner. Eng., 2015, 79, 102–115 CrossRef CAS.
- N. Dushyantha, N. Batapola, I. M. S. K. Ilankoon, S. Rohitha, R. Premasiri, B. Abeysinghe, N. Ratnayake and K. Dissanayake, Ore Geol. Rev., 2020, 122, 103521 CrossRef.
- J. H. L. Voncken, in The Rare Earth Elements: An Introduction, ed. J. H. L. Voncken, Springer International Publishing, Cham, 2016, ch. Physical and Chemical Properties of the Rare Earths, pp. 53–72. DOI:10.1007/978-3-319-26809-5_3.
- Z. Y. Chen, Z. Li, J. Chen, P. Kallem, F. Banat and H. D. Qiu, J. Environ. Chem. Eng., 2022, 10, 107104 CrossRef CAS.
- A. M. Wilson, P. J. Bailey, P. A. Tasker, J. R. Turkington, R. A. Grant and J. B. Love, Chem. Soc. Rev., 2014, 43, 123–134 RSC.
- N. A. Thiele, D. J. Fiszbein, J. J. Woods and J. J. Wilson, Inorg. Chem., 2020, 59, 16522–16530 CrossRef CAS PubMed.
- A. H. Hu, M. E. Simms, V. Kertesz, J. J. Wilson and N. A. Thiele, Inorg. Chem., 2022, 61, 12847–12855 CrossRef CAS PubMed.
- A. Kovács, C. Apostolidis and O. Walter, Inorganics, 2019, 7, 26 CrossRef.
- S. Jansone-Popova, A. S. Ivanov, V. S. Bryantsev, F. V. Sloop, R. Custelcean, I. Popovs, M. M. Dekarske and B. A. Moyer, Inorg. Chem., 2017, 56, 5911–5917 CrossRef CAS PubMed.
- E. A. Mowafy and H. F. Aly, Solvent Extr. Ion Exch., 2006, 24, 677–692 CrossRef CAS.
- J. Sulakova, R. T. Paine, M. Chakravarty and K. L. Nash, Sep. Sci. Technol., 2012, 47, 2015–2023 CAS.
- M. Simonnet, T. Kobayashi, K. Shimojo, K. Yokoyama and T. Yaita, Inorg. Chem., 2021, 60, 13409–13418 CrossRef CAS PubMed.
- S. A. Ansari, P. Pathak, P. K. Mohapatra and V. K. Manchanda, Chem. Rev., 2012, 112, 1751–1772 CrossRef CAS PubMed.
- Y. Sasaki, Y. Sugo, S. Suzuki and S. Tachimori, Solvent Extr. Ion Exch., 2001, 19, 91–103 CrossRef CAS.
- D. S. Sholl and R. P. Lively, Nature, 2016, 533, 316–316 CrossRef PubMed.
- J. J. M. Nelson, T. Cheisson, H. J. Rugh, M. R. Gau, P. J. Carroll and E. J. Schelter, Commun. Chem., 2020, 3, 7 CrossRef CAS PubMed.
- D. Whittaker, A. Geist, G. Modolo, R. Taylor, M. Sarsfield and A. Wilden, Solvent Extr. Ion Exch., 2018, 36, 223–256 CrossRef CAS.
- L. Li, C. J. Xu, X. J. Peng, M. M. Zhang, S. Zeb, X. Jiang, Y. Liu, Y. Cui and G. X. Sun, Sep. Purif. Technol., 2021, 277, 119479 CrossRef CAS.
- A. Leoncini, J. Huskens and W. Verboom, Synlett, 2016, 2463–2466 CAS.
- R. M. Lanigan and T. D. Sheppard, Eur. J. Org. Chem., 2013, 7453–7465 CrossRef CAS.
- N. Martín and F. G. Cirujano, Catal. Commun., 2022, 164, 106420 CrossRef.
- H. Charville, D. Jackson, G. Hodges and A. Whiting, Chem. Commun., 2010, 46, 1813–1823 RSC.
- M. L. Fein and E. M. Filachione, J. Am. Chem. Soc., 1953, 75, 2097–2099 CrossRef CAS.
- S. Jansone-Popova and I. Popovs, US Pat., US20230002311A1, 2023 Search PubMed.
- W. Y. Duan, F. P. Meng, F. F. Wang and Q. Q. Liu, Ecotoxicol. Environ. Saf., 2017, 145, 324–332 CrossRef CAS PubMed.
- A. Ojeda-Porras, A. Hernández-Santana and D. Gamba-Sánchez, Green Chem., 2015, 17, 3157–3163 RSC.
- T. K. Houlding, K. Tchabanenko, M. T. Rahman and E. V. Rebrov, Org. Biomol. Chem., 2013, 11, 4171–4177 RSC.
- B. S. Jursic and Z. Zdravkovski, Synth. Commun., 1993, 23, 2761–2770 CrossRef CAS.
- L. J. Goossen, D. M. Ohlmann and P. P. Lange, Synthesis, 2009, 160–164 CrossRef CAS.
- J. Bare, Clean Technol. Environ. Policy, 2011, 13, 687–696 CrossRef CAS.
- M. H. Cheng, J. J. K. Sekhon, K. A. Rosentrater, T. Wang, S. Jung and L. A. Johnson, Food Bioprod. Process., 2018, 108, 58–68 CrossRef CAS.
- M. L. Richardson and S. Gangolli, The Dictionary of Substances and Their Effects, Royal Society of Chemistry, 1992 Search PubMed.
- D. Stamberga, M. R. Healy, V. S. Bryantsev, C. Albisser, Y. Karslyan, B. Reinhart, A. Paulenova, M. Foster, I. Popovs, K. Lyon, B. A. Moyer and S. Jansone-Popova, Inorg. Chem., 2020, 59, 17620–17630 Search PubMed.
- F. Xie, T. A. Zhang, D. Dreisinger and F. Doyle, Miner. Eng., 2014, 56, 10–28 CrossRef CAS.
- D. M. Brigham, A. S. Ivanov, B. A. Moyer, L. H. Delmau, V. S. Bryantsev and R. J. Ellis, J. Am. Chem. Soc., 2017, 139, 17350–17358 CrossRef CAS PubMed.
- A. W. Knight, B. F. Qiao, R. Chiarizia, G. Ferru, T. Forbes, R. J. Ellis and L. Soderholm, Langmuir, 2017, 33, 3776–3786 CrossRef CAS PubMed.
- M. Spadina, K. Bohinc, T. Zemb and J. F. Dufrêche, ACS Nano, 2019, 13, 13745–13758 CrossRef CAS PubMed.
- P. Sun, K. Huang and H. Z. Liu, Langmuir, 2018, 34, 13155–13161 CrossRef CAS PubMed.
- M. Spadina, J. F. Dufrêche, S. Pellet-Rostaing, S. Marcelja and T. Zemb, Langmuir, 2021, 37, 10637–10656 CrossRef CAS PubMed.
- E. A. Mowafy and D. Mohamed, Sep. Sci. Technol., 2017, 52, 1006–1014 CrossRef CAS.
- M. R. Healy, A. S. Ivanov, Y. Karslyan, V. S. Bryantsev, B. A. Moyer and S. Jansone-Popova, Chem. – Eur. J., 2019, 25, 6326–6331 CrossRef CAS PubMed.
- K. Mokoena, L. S. Mokhahlane and S. Clarke, Int. J. Coal Geol., 2022, 259, 104037 CrossRef.
- Y. Zhou, J. X. Liu, G. J. Cheng, X. X. Xue and H. Yang, Hydrometallurgy, 2022, 208, 105782 CrossRef CAS.
- A. R. Finney, S. Lectez, C. L. Freeman, J. H. Harding and S. Stackhouse, Chem. – Eur. J., 2019, 25, 8725–8740 CrossRef CAS PubMed.
- C. Bonal, J. P. Morel and N. MorelDesrosiers, J. Chem. Soc., Faraday Trans., 1996, 92, 4957–4963 RSC.
- F. Aydin, M. R. Cerón, S. A. Hawks, D. I. Oyarzun, C. Zhan, T. A. Pham, M. Stadermann and P. G. Campbell, Nanoscale, 2020, 12, 20292–20299 RSC.
- Y. Sasaki, Y. Sugo, K. Morita and K. L. Nash, Solvent Extr. Ion Exch., 2015, 33, 625–641 CrossRef CAS.
- H. Du, X. Peng, Y. Cui and G. Sun, IOP Conf. Ser.: Earth Environ. Sci., 2019, 310, 042023 CrossRef.
- H. Narita and M. Tanaka, Solvent Extr. Res. Dev., Jpn., 2013, 20, 115–121 CrossRef CAS.
- Y. Sasaki, Y. Sugo, Y. Kitatsuji, A. Kirishima, T. Kimura and G. R. Choppin, Anal. Sci., 2007, 23, 727–731 CrossRef CAS PubMed.
- Y. Sasaki, Z. X. Zhu, Y. Sugo and T. Kimura, J. Nucl. Sci. Technol., 2007, 44, 405–409 CrossRef CAS.
- D. Prodius, M. Klocke, V. Smetana, T. Alammar, M. P. Garcia, T. L. Windus, I. C. Nlebedim and A. V. Mudring, Chem. Commun., 2020, 56, 11386–11389 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc01146e |
| ‡ These authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2024 |