
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemically driven green synthesis to unlock sustainable routes to β-keto spirolactones†
Ian
MacLean‡
 a,
Montaña J.
García‡
a,
Silvia
Cabrera
*bc,
Leyre
Marzo
*ac and
José
Alemán
*ac
a,
Montaña J.
García‡
a,
Silvia
Cabrera
*bc,
Leyre
Marzo
*ac and
José
Alemán
*ac
aOrganic Chemistry Department (Módulo 1) Universidad Autónoma de Madrid, 28049 Madrid, Spain. E-mail: jose.aleman@uam.es; leyre.marzo@uam.es
bInorganic Chemistry Department (Módulo 7) Universidad Autónoma de Madrid, 28049 Madrid, Spain. E-mail: silvia.cabrera@uam.es
cInstitute for Advanced Research in Chemical Sciences (IAdChem) Universidad Autónoma de Madrid, 28049 Madrid, Spain
First published on 18th April 2024
Abstract
In this work, we present a sustainable and environmentally benign electrochemical method for the synthesis of β-keto spirolactones. The reaction is carried out using green solvents such as acetone and water, and simple electrons as oxidants, instead of the stoichiometric oxidants used in classical approaches. The robustness of the method allows the functionalization of cyclic β-keto esters and a β-keto amide, the latter affording α-spiroiminolactone. The method also gives good results with double bonds bearing substituents of different electronic natures. Furthermore, this methodology can be easily scalable through a continuous flow electrochemical approach that improves the productivity of the reaction. Mechanistic investigations support the radicalic nature of the transformation, and the generation of a carbocation intermediate that is further trapped with the water employed as co-solvent in the reaction.
Introduction
The spirolactone moiety is present in a large number of pharmaceuticals and biologically active compounds and its unique structural and chemical features have recently aroused enormous interest from the scientific community.1–4 However, its preparation presents considerable challenges, such as the need for highly functionalized starting materials,5 the use of toxic reagents,6 or the necessity for transition metals as catalysts of the reaction.7–11 To date, there are only two approaches starting from simple β-keto esters and simple styrene derivatives to obtain α-keto spirolactones under oxidative conditions. Both methods require the use of a transition metal complex as a Lewis acid to promote the enolization of the β-keto ester upon coordination, prior to oxidation with one equivalent of a metallic oxidant (Scheme 1A).12,13 The resulting radical intermediate is stabilized by coordination to the Lewis acid and undergoes radical addition to the styrene, forming a stabilized benzylic radical, which is further oxidized by a second equivalent of a metallic oxidant. The final carbocation undergoes an intramolecular lactonization reaction to afford the desired spirolactones. The first methodology employs In(OTf)3 as a Lewis acid, and 2 equivalents of silver oxide as the metallic oxidant.12 In addition, the reaction is performed in dichloroethane and requires high temperatures (120 °C) to take place. Later on, an approach was described using a nickel catalyst able to coordinate the β-keto ester.13 Nevertheless, superstoichiometric amounts of silver oxide were again required and dichloromethane was employed as the solvent. Other methods have also been used, such as the utilization of light-mediated reactions and the use of stoichiometric amount of iodine.14–18 Aiming to develop a greener synthetic approach for the preparation of β-keto spirolactones, we envisioned the use of electrochemistry as an excellent alternative, since two consecutive oxidation processes should take place to get the final product. | ||
| Scheme 1 Previous works (A) and our synthetic proposal (B). | ||
Electrochemistry has recently been reborn as an ecological and greener method for organic synthesis.19–26 Despite the enormous synthetic possibilities that it offers, the requirement for complex setups has limited its use in organic synthesis in previous decades. The emergence of simpler and more economical setups has led to its renaissance as one of the most appealing research lines in organic synthesis nowadays. Electrochemistry allows the generation of open shell radical intermediates under very mild reaction conditions using electrons as reagents. In addition, in comparison with photocatalysis,27–29 which also allows the generation of the open shell intermediates but with the limitation of the redox properties of the photocatalyst, electrochemistry gives access to transformations using reagents with higher redox potentials and to double oxidation or reduction steps in one reaction. In addition, electrochemical methods usually employ green solvents such as water or acetone. Therefore, we envisioned that electrochemistry would be an excellent synthetic alternative for the preparation of β-keto spirolactones under very mild conditions. Our proposal involves the electrochemical oxidation of the deprotonated β-keto esters in the presence of a base, to form the methylenic radical intermediate (Scheme 1B). This radical can subsequently engage with the double bond, producing the benzylic radical, which will then proceed through a second oxidation process, resulting in the formation of a carbocation, ultimately leading to the lactonization reaction. This electrochemical approach represents an environmentally friendly, safe and inexpensive method since (i) simple electrons applied through the electrical current are employed as oxidants, instead of the use of metallic oxidants; (ii) transition metal Lewis acids are avoided; (iii) an environmentally benign solvent is used; and (iv) the reaction is performed at room temperature. In addition, this methodology circumvents the necessity of using an inert atmosphere, dry solvents, high temperatures or long reaction times, as traditionally required for the synthesis of spirolactones. Moreover, the productivity of the reaction can be easily enhanced under electrochemical flow conditions, enabling shorter reaction times and lower energy consumption.
Results and discussion
Optimization of the reaction conditions
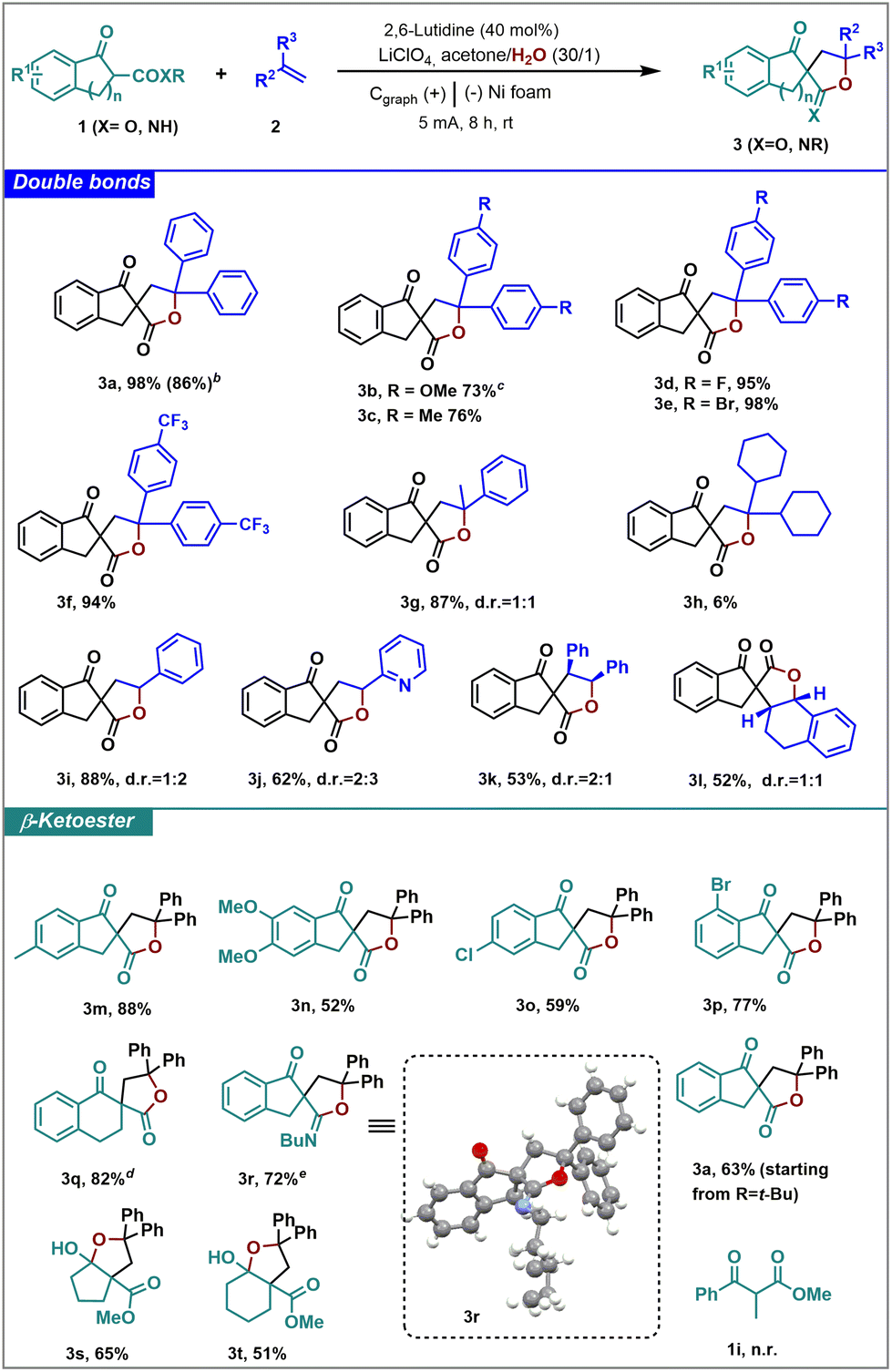
With this proposal in mind, we started studying the reaction between the cyclic β-keto ester 1a and diphenyl ethylene (2a). Starting from those reagents, the spirolactone 3a was isolated in 98% yield using 40 mol% of 2,6-lutidine, lithium perchlorate (0.1 M) as an electrolyte, a 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of acetone/water as the solvent, graphite electrode in the cathode and Ni electrode in the anode, under a 5 mA constant current for 8 hours in an undivided cell (Table 1, entry 1). Variations in the anode electrode to Zn or graphite electrodes afforded no conversion. Analogue results were achieved with the use of MeOH or CH2Cl2 as the solvent (Table 1, entries 3 and 4), whereas the use of THF afforded product 3a in 88% yield (Table 1, entry 5). The absence of base or water or shorter reaction times produced a decrease in the yield (Table 1, entries 6–8). Increasing or decreasing the intensity of the current gave lower conversion as well as the use of larger amounts of alkene or an increase of the reaction concentration (Table 1, entries 9–13). With these optimized conditions, a variety of β-keto esters and double bonds were studied under the electrochemical conditions in an undivided cell (Table 2).
:1 mixture of acetone/water as the solvent, graphite electrode in the cathode and Ni electrode in the anode, under a 5 mA constant current for 8 hours in an undivided cell (Table 1, entry 1). Variations in the anode electrode to Zn or graphite electrodes afforded no conversion. Analogue results were achieved with the use of MeOH or CH2Cl2 as the solvent (Table 1, entries 3 and 4), whereas the use of THF afforded product 3a in 88% yield (Table 1, entry 5). The absence of base or water or shorter reaction times produced a decrease in the yield (Table 1, entries 6–8). Increasing or decreasing the intensity of the current gave lower conversion as well as the use of larger amounts of alkene or an increase of the reaction concentration (Table 1, entries 9–13). With these optimized conditions, a variety of β-keto esters and double bonds were studied under the electrochemical conditions in an undivided cell (Table 2).
|
|
||
|---|---|---|
| Entry | Deviation from optimized conditions | Yieldb (%) |
| a General conditions: 1a (0.5 mmol), 2a (1.0 mmol), 2,6-lutidine (40 mol%) in a 0.1 M LiClO4 solvent mixture (3.0 mL) at room temperature. b Determined by 1H-NMR with an internal standard. c Isolated yield after flash chromatography purification. | ||
| 1 | None | 98c |
| 2 | C graphite (+) Zn (−) | 0 |
| 2 | C graphite (+) Cgraphite (−) | 0 |
| 3 | MeOH | 0 |
| 4 | CH2Cl2 | 0 |
| 5 | THF | 88c |
| 6 | Without H2O | 44 |
| 7 | Without base | 74 |
| 8 | 6 h | 88 |
| 9 | 10 mA | 65 |
| 10 | 3.5 mA | 84c |
| 11 | 3 equiv. 2a | 92 |
| 12 | 1.5 equiv. 2a | 0 |
| 13 | 0.2 M | 69 |

| a General conditions: 1a (0.5 mmol), 2a (1.0 mmol), 2,6-lutidine (40 mol%) in a 0.1 M LiClO4 solvent mixture (3.0 mL) at room temperature. b Reaction carried out at gram scale (6.0 mmol, 1.1 g of 1a. See the ESI†). c This reaction was carried out in 0.1 M concentration of 1a. d Cs2CO3 is used as the base instead of 2,6-lutidine. e 3r is obtained from the corresponding β-keto amide. |
|---|
|
Study of the scope of the reaction
Reactions performed with diphenyl ethylene with electron donating groups such as a methyl or methoxy group, and soft electron-deficient substituents such as halogens and the trifluoromethyl group afforded the desired spirolactones in very good yields (3a–f). Furthermore, the reaction can be carried out at gram scale (6 mmol), obtaining 3a with similar results (see the results in brackets). One alkyl substituent in the double bond is well tolerated and, thus, α-methylstyrene afforded 3g as a 1:1 mixture of diastereoisomers in 87% yield. However, the presence of at least one aromatic ring was necessary for the reaction to take place in good yield, since the reaction of 1a with dicyclohexyl ethylene afforded the desired product (3h) in only 6% yield. Monosubstituted double bonds such as styrene or the 2-vinylpyridine were well tolerated in the reaction, affording exclusively the corresponding spirolactone (3i, 3j) as a mixture of diastereoisomers in good to very good yields. Moreover, higher steric hindrance in the β position of the styrene did not affect the reactivity. Thus, the corresponding spirolactones 3k and 3l were obtained from stilbene and dihydronaphthalene as a 2:1 and 1:1 mixture of diastereoisomers, respectively, in good yields.
Then, we proceeded to study the scope of β-keto esters in the reaction. First, we studied the effect of different substituents in the aromatic ring. The presence of electron donating substituents (methyl or methoxy) in the aromatic ring of the indanone derivative did not affect the reactivity, obtaining the corresponding spirolactones in good to very good yields (3m and 3n). The reaction also tolerated the presence of halogen substituents, thus 3o and 3p were obtained with very good results. Remarkably, the method allowed the presence of aryl bromides, opening the door to further functionalizations of the molecule. The tetrahydronaphthalenone derivative 1q also underwent this reactivity, affording the spirolactone 3q in a very good yield using Cs2CO3 as the base instead of 2,6-lutidine. The presence of higher steric hindrance in the ester did not affect the reactivity, as with the tert-butyl derivative the spirolactone 3a was obtained in 63% yield. The reaction also proceeded starting from a β-keto amide, but instead of the expected lactone, spiroiminiolactone 3r was isolated in 72% yield, as corroborated by X-ray crystal analysis.30 This spiroiminolactone was formed upon nucleophilic addition of the oxygen of the amide group to the carbocationic intermediate. On the other hand, with five- and six-membered ring methyl oxocyclocarboxylates, the reaction also took place efficiently. However, for these substrates, the formation of the hemiketal was more favored than the lactonization reaction, giving access to tetrahydrofurane derivatives 3s and 3t in good yields. The main limitation of the method is the functionalization of non-cyclic β-keto esters (1i) that did not undergo this reactivity, even in the presence of different bases, recovering the starting materials unaltered. The non-cyclic nature of 1i compared to the other substrates and its lower acidity could be the reason for the null reactivity of this substrate. Cyclic diketones or alkyl-alkenes did not afford the final product.
The design of efficient and sustainable methods for large scale production is one of the main challenges for the industry nowadays. Flow chemistry is gaining interest among scientists because it allows the development of simple, safe, highly efficient, and low waste generation methodologies.31,32 Given the relevance of the structures obtained above, we decided to develop this electrochemical methodology under flow conditions (Table 3). The flow system consists of a Vapourtec Ion electrochemical undivided-cell reactor with a fix reactor volume (see optimization of the reaction conditions in Table S1†). To our delight, under the optimized flow conditions, the electrochemical reaction presented a remarkable improvement of the efficiency, being possible to isolate 3a in 69% yield in only 30 minutes of residence reactor time. It is important to highlight the higher production of the flow process that allowed the preparation of 0.2768 mmol of 3a per hour compared to the 0.0613 mmol per hour obtained under batch conditions. To further demonstrate the applicability of the flow conditions, different β-keto esters (1) and alkene derivatives (2) were tested. The presence of a methyl or a CF3 group at the aryl alkene afforded the corresponding spirolactones (3c, 3f) in moderate conversions. In contrast, p-bromine and p-fluorine aryl substituents gave rise to 3d and 3e, respectively, in good yields. The modification of the β-keto ester core was also evaluated. Thus, product 3n was isolated in good yield starting from β-keto ester 1c. In general, the different spirolactone outcomes observed under flow electrochemical conditions are in consonance with the reactivity observed under batch conditions. In addition, we performed the reaction at a larger scale, isolating 3a with a slightly lower yield (see the results in brackets).
| a Reaction conditions: 1 (0.2 mmol), 2 (0.4 mmol), 2,6-lutidine (40 mol%), LiClO4 (0.1 M) at constant current (5.0 mA) in 3.6 mL of acetone/water mixture (90/1) at room temperature for 30 min of residence time, using Cgraph as an anode and Ni as a cathode in a recirculating system. b Reaction was carried out at 2.63 mmol scale. |
|---|

|
To prove the utility of the final products, derivatizations of products 3a and 3s were performed (Scheme 2). Starting from the spirolactone 3a, tetrahydro-2H-spirofuranoindene 4 was obtained in 58% overall yield, following a two reduction step pathway using DIBAL-H and Et3SiH to reduce the carbonyls and the resulting alcohols, respectively. Furthermore, the reduction of hemiketal 3s under triethylsilane conditions gave hexahydro cyclopentafuran carboxylate 5 in 52% yield.
 | ||
| Scheme 2 Derivatization of compounds 3a and 3s. | ||
Next, we compared qualitative and quantitative green metrics of our method to prepare β-keto spirolactones 3 from β-keto ester 1 and alkene 2 with the reported methods, that is, method a: iodine-mediated photochemical reaction,18 and method b: cooperative In(III)/Ag(I) cyclization12 (Table 4). While we can perform the reaction under batch or flow conditions, the previous methods were only performed under batch conditions. In addition, the previous methods require the use of stoichiometric additives such as iodine or Ag2O, or toxic halogenated solvents that our electrochemical method does not require. Moreover, previous reported methods present a higher E factor and a lower RME than the present synthesis making our synthetic approach a more attractive procedure for industrial applications.
| Criterion | This electrochemical method | Method a: photochemical18 | Method b12 |
|---|---|---|---|
| a All data have been estimated excluding the work-up and purification steps, as they will be the same in the three methods. b E factor calculated using the equation E = mg of waste/mg of product. c The reaction mass efficiency (RME) was calculated using the equation RME = mg product × 100/total mg of reagents. DCE: 1,2-dichloroethane. | |||
| Qualitative metrics | |||
| Additional stoichiometric additives | H2O | I2, Na2CO3 | Ag2O |
| Type of reactor | Batch/flow | Batch | Batch |
| Temperature | rt | rt | 120 °C |
| Solvent | Acetone | tBuOH/H2O | DCE |
| Quantitative metrics | |||
| Yield (%) | 98 | 70 | 67 |
| E factorb | 3.28 | 9.35 | 11.14 |
| RMEc (%) | 36.6 | 21.1 | 18.1 |
Finally, the mechanism of the reaction was examined. Our mechanistic proposal (Scheme 3a) starts by the formation of the enolate upon deprotonation of the β-keto ester 1 by the base, that is further oxidized under oxidative electrochemical conditions to form the radical intermediate I. To obtain evidence on the formation of this intermediate, the reaction was performed in the presence of 1.0 equivalent of TEMPO (Scheme 3b). Under these conditions, we were able to isolate compound 6 in 89% yield, which evidences the formation of the α-keto radical intermediate I. After the formation of intermediate I, this reacts with alkene 2 to form the benzylic radical intermediate II. The formation of this latter intermediate is further evidenced by the detection by mass spectrometry of the TEMPO adduct 7 (Scheme 3b). Intermediate II is again oxidized to form the benzylic carbocation III, which is further trapped by water to generate the alcohol IV. Next, IV undergoes lactonization to afford the observed β-keto spirolactone 3. At this point, there are two mechanistic possibilities to explain the formation of the spirolactone. The first involves the intramolecular reaction of the ester of III to form IV′via carbocation, followed by hydrolysis to yield 3 (mechanism B, Scheme 3a). The second consists of the intermolecular attack of a water molecule to form intermediate alcohol IV, and finally lactonization to form 3 (mechanism A, Scheme 3a). To prove that in this transformation the carbocation is trapped by water and that the resulting OH (IV) is responsible for the lactonization process we performed the electrochemical reaction under 18O labelled water (Scheme 3c). The final β-keto spirolactone containing 18O was further reduced to the tetrahydro spirofuranindene 184. The presence of 18O by mass spectrometry analysis of 4 evidences that the oxygen in the lactone comes from the water and not from the initial ester (mechanism A). This is also in accordance with the formation of hemiketals 3s and 3t, whose generation can take place when the carbocation is trapped by the water present in the reaction, followed by intramolecular ketal formation.
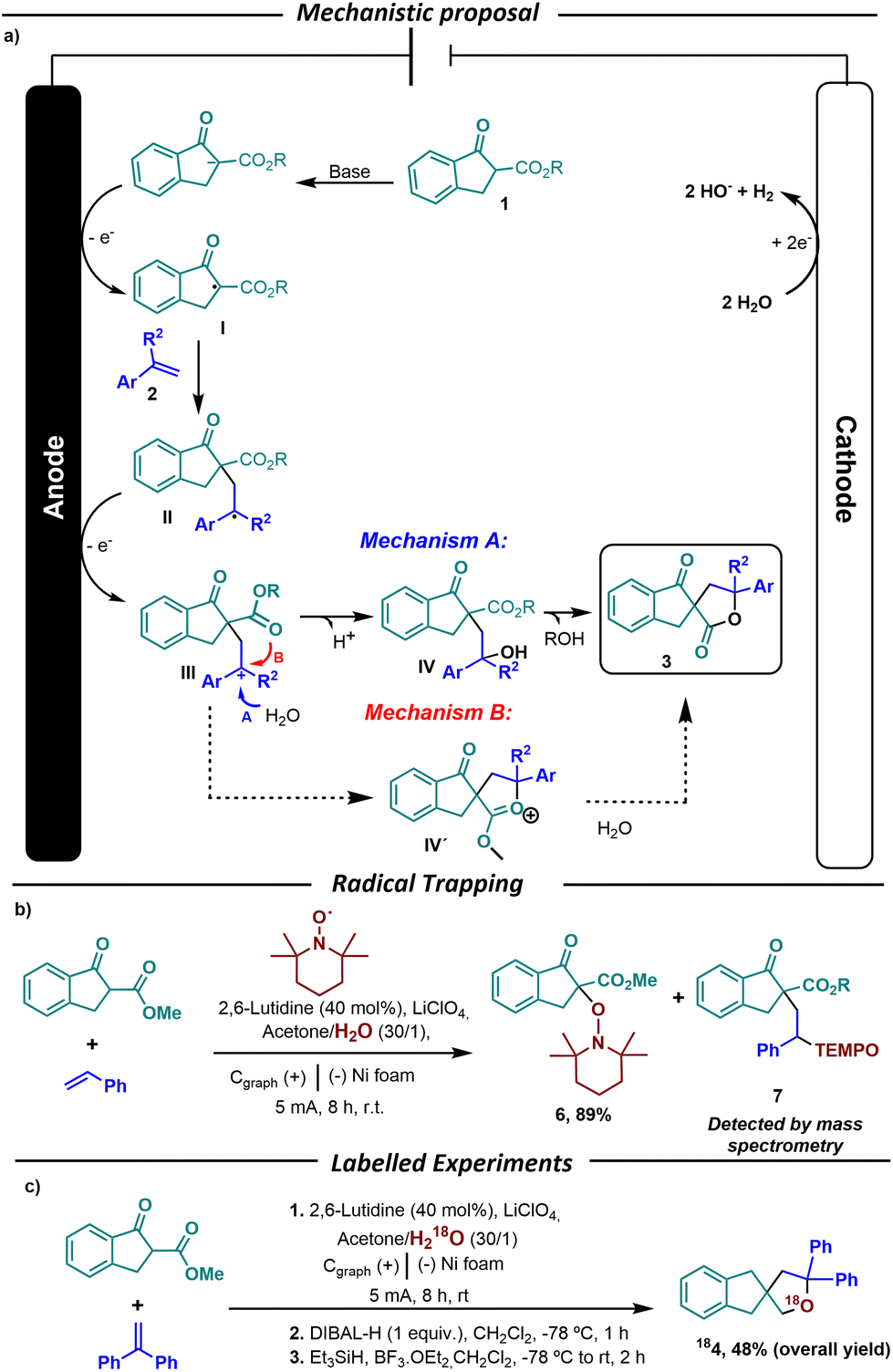 | ||
| Scheme 3 Reaction mechanism proposal and mechanistic proofs. | ||
Conclusions
To sum up, we have developed a sustainable synthesis of β-keto spirolactones under mild reaction conditions using simple electrons as oxidants, avoiding stoichiometric metallic oxidants. This electrochemical approach allows the use of green solvents and presents broad scope and functional group tolerance. In addition, further derivatizations of the final products allow the preparation of spirofurane derivatives that are difficult to prepare under other reaction conditions. Moreover, the implementation of the method under continuous flow conditions has allowed the productivity of the reaction to be increased in an efficient manner, being possible to prepare 0.2768 mmol of products per hour. Finally, studies corroborate the proposed mechanism that involves two consecutive oxidation steps to afford a carbocation, which is further trapped by the water employed as co-solvent. The resulting alcohol undergoes a lactonization reaction to afford the observed spirolactones.Author contributions
I. M., M. J. G., S. C., L. M. and J. A. designed the experiments and analyzed the data. I. M. and M. J. G. performed the experiments. I. M. and M. J. G. described the ESI.† S. C., L. M. and J. A. wrote the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the Spanish Government (PID2021-122299NB-I00, TED2021-130470B-I00, and TED2021-129999B-C32), European Structural Funds (S2018/NMT-4367), Proyectos Sinérgicos I + D (Y2020/NMT6469) and ‘Comunidad de Madrid’ (SI1/PJI/2019–00237). M. J. G. thanks the Spanish Government for a FPI contract (PRE2019-090203) and L. M. thanks the Spanish Government for a Ramón y Cajal contract (RYC2021-031590-I).References
- A. Quintavalla, Curr. Med. Chem., 2018, 25, 917 CrossRef CAS PubMed.
- A. Bartoli, F. Rodier, L. Commeiras, J.-L. Parrain and G. Chouraqui, Nat. Prod. Rep., 2011, 28, 763 RSC.
- C. M. Marson, Chem. Soc. Rev., 2011, 40, 5514 RSC.
- J.-H. Xie and Q.-L. Zhou, Acc. Chem. Res., 2008, 41, 581 CrossRef CAS PubMed.
- A. Voituriez, A. Marinetti and M. Gicquel, Synlett, 2015, 26, 142 CrossRef CAS.
- S. Sternativo, A. Calandriello, F. Costantino, L. Testaferri, M. Tiecco and F. Marini, Angew. Chem., Int. Ed., 2011, 50, 9382 CrossRef CAS PubMed.
- N. R. Ball-Jones, J. J. Badillo and A. K. Franz, Org. Biomol. Chem., 2012, 10, 5165 RSC.
- R. Rios, Chem. Soc. Rev., 2012, 41, 1060 RSC.
- S. Kotha, A. C. Deb, K. Lahiri and E. Manivannan, Synthesis, 2009, 165 CrossRef CAS.
- M.-E. Sinibaldi and I. Canet, Eur. J. Org. Chem., 2008, 4391 CrossRef CAS.
- R. Pradhan, M. Patra, A. K. Behera, B. K. Mishra and R. K. Behera, Tetrahedron, 2006, 62, 779 CrossRef CAS.
- T. Y. Ko and S. W. Youn, Adv. Synth. Catal., 2016, 358, 1934–1941 CrossRef CAS.
- X. Zhang, W. Wu, W. Cao, H. Yu, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2020, 59, 4846 CrossRef CAS PubMed.
- S. Maejima, E. Yamaguchi and A. Itoh, J. Org. Chem., 2019, 84, 9519 CrossRef CAS PubMed.
- S. Maejima, E. Yamaguchi and A. Itoh, ACS Omega, 2019, 4, 4856 CrossRef CAS PubMed.
- S. Maejima, E. Yamaguchi and A. Itoh, Adv. Synth. Catal., 2017, 359, 3883 CrossRef CAS.
- K. Oe, M. Goto, S. Maejima, E. Yamaguchi and A. Itoh, Asian J. Org. Chem., 2020, 9, 571 CrossRef CAS.
- S. Maejima, E. Yamaguchi and A. Itoh, J. Org. Chem., 2020, 85, 10709 CrossRef CAS PubMed.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594 CrossRef CAS PubMed.
- C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Green Chem., 2020, 22, 3358 RSC.
- T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484 CAS.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS PubMed.
- E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302 CrossRef CAS PubMed.
- D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386 RSC.
- H. He, Y. Lv, J. Hu, Z.-W. Hou and L. Wang, Green Chem., 2024, 26, 2157–2161 RSC.
- Z.-W. Hou, H.-C. Xu and L. Wang, Curr. Opin. Electrochem., 2024, 44, 101447 CrossRef CAS.
- J. Liu, L. Lu, D. Wood and S. Lin, ACS Cent. Sci., 2020, 6, 1317 CrossRef CAS PubMed.
- R. H. Verschueren and W. M. De Borggraeve, Molecules, 2019, 24, 2122 CrossRef PubMed.
- H. Huang, K. A. Steniger and T. H. Lambert, J. Am. Chem. Soc., 2022, 144, 12567 CrossRef CAS PubMed.
- CCDC 2337523.†.
- C. A. Hone and O. Kappe, Chem.: Methods, 2021, 1, 454 CAS.
- Z. Dong, Z. Wen, F. Zhao, S. Kuhn and T. Noël, Chem. Eng. Sci.: X, 2021, 10, 100097 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2337523. For ESI see DOI: https://doi.org/10.1039/d4gc01127a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |