
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCholine and lactic acid covalently incorporate into the lignin structure during deep eutectic solvent pulping†
Gijs
van Erven
 *ab,
Vincent J. P.
Boerkamp
b,
Johan W.
van Groenestijn
a and
Richard J. A.
Gosselink
a
*ab,
Vincent J. P.
Boerkamp
b,
Johan W.
van Groenestijn
a and
Richard J. A.
Gosselink
a
aWageningen Food and Biobased Research, Bornse Weilanden 9, 6708 WG, Wageningen, The Netherlands. E-mail: gijs.vanerven@wur.nl; Tel: +31 317 487010
bWageningen University & Research, Laboratory of Food Chemistry, Bornse Weilanden 9, 6708 WG, Wageningen, The Netherlands
First published on 15th May 2024
Abstract
Deep eutectic solvent (DES) pulping is a promising alternative to conventional pulping techniques, mainly owing to the favourable solvent properties and reduced environmental impact. DES lignin, however, still awaits complete structural characterisation, especially in terms of the potential incorporation of DES constituents. Here, we describe the structural modification of lignin during lactic acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :choline chloride DES pulping of Miscanthus biomass in unprecedented detail. We show that DES pulping induces β-O-4 aryl ether cleavage and extensive substitution by DES incorporation. The covalent incorporation of both lactic acid and choline was confirmed through saponification of the precipitated lignin and lignin model compound studies. Detailed multidimensional NMR analysis allowed us to validate aliphatic and benzylic lactic acid esterification as well as benzylic choline and lactic acid etherification of β-O-4 aryl ethers. We demonstrate that these reactions occur in all process phases by comprehensive analysis of the precipitated, residual and solubilised lignin fractions, together comprising 93% w/w of the initial lignin in Miscanthus. Covalent lactic acid and choline incorporation occurs independently of biomass type (grass, hardwood and softwood) and can be modulated by water content, reaction duration and temperature. Going forward, these new insights will offer ample opportunities for producing lignins with unique structural features and ultimately will open up new avenues for specific functional ingredient applications. Our work thus clearly advances the DES biorefinery concept and contributes to the valorisation of lignin in general.
:choline chloride DES pulping of Miscanthus biomass in unprecedented detail. We show that DES pulping induces β-O-4 aryl ether cleavage and extensive substitution by DES incorporation. The covalent incorporation of both lactic acid and choline was confirmed through saponification of the precipitated lignin and lignin model compound studies. Detailed multidimensional NMR analysis allowed us to validate aliphatic and benzylic lactic acid esterification as well as benzylic choline and lactic acid etherification of β-O-4 aryl ethers. We demonstrate that these reactions occur in all process phases by comprehensive analysis of the precipitated, residual and solubilised lignin fractions, together comprising 93% w/w of the initial lignin in Miscanthus. Covalent lactic acid and choline incorporation occurs independently of biomass type (grass, hardwood and softwood) and can be modulated by water content, reaction duration and temperature. Going forward, these new insights will offer ample opportunities for producing lignins with unique structural features and ultimately will open up new avenues for specific functional ingredient applications. Our work thus clearly advances the DES biorefinery concept and contributes to the valorisation of lignin in general.
Introduction
Deep eutectic solvents (DESs) are liquid mixtures of hydrogen-bond acceptors (HBAs) and hydrogen-bond donors (HBDs) with lower melting points than the individual components.1 DESs have been used in many fields due to their favourable features, including negligible vapour pressure, non-toxicity and biodegradability, easy preparation, and tuneable physicochemical properties.2,3 Because of these features, DESs are attracting increasing attention in the biorefinery context. As such, DES pulping is emerging as a more sustainable alternative to traditional pulping methods like Kraft and soda pulping, since it requires substantially less chemical and energy inputs and, in theory, is entirely biobased and circular.4–6A multitude of DES systems has thus far been evaluated in the literature, consistently showing that acidic DES are most promising and lactic acid:choline chloride (LA:ChCl) in particular.6–8 Many studies have been devoted to LA:ChCl biomass pretreatment in terms of HBD–HBA molar ratio, temperature, reaction duration and feedstock type. These studies typically point to a molar ratio of 10:1 LA:ChCl, 120 °C and 6 h as ‘optimum’ conditions, and these are therefore accordingly often considered the benchmark conditions.6–11 It should be noted, though, that this process optimization mainly focused on the carbohydrate valorisation perspective. Overall delignification extent and selectivity and ultimate enzymatic conversion of the residual carbohydrates have therefore been primary targets.
Optimal DES conditions from the lignin perspective, however, largely depend on the desired application outlet and thus require a compromise between yield, purity and structural integrity. Given the many ongoing research incentives focussing on DES pulping and status as emerging biorefinery concept, we argue that full structural characterization of the lignin fractionated through this process is imperative. Structural features are the main driver for functionality and concomitantly for application. Since total-use lignocellulose cascading through a DES biorefinery is pursued, lignin should obviously be utilized and conceivably to the highest value possible.
Despite advances in the structural characterisation of DES lignins in general, many structural features nonetheless still remain to be unravelled at the molecular level, next to how these structures relate to the conditions used.11–15 It has been well-established that DES pulping results in a reduced molecular weight due to acid-catalysed (partial) depolymerization of β-O-4 aryl ethers and a concomitant increased phenolic hydroxyl content.11–15 However, to date it remains largely elusive how DES extraction and conversion depends on specific substructures, and, more importantly, whether DES components are covalently incorporated into the lignin macromolecule during pulping. Covalent DES incorporation would have severe implications on the properties of lignin, and thus on its ultimate application. Previously, lactic acid and some unassigned signals attributed to DES constituents have been suggested in Heteronuclear Single Quantum Coherence Nuclear Magnetic Resonance (HSQC NMR) spectra of DES lignin,8,11,13 but the exact origin of these signals remained unidentified. It has not been determined whether these signals were the mere result of unbound, remaining DES impurities, or were in fact caused by chemically bound constituents. The esterification of cellulose and choline chloride by lactic acid during DES pulping, coined here DESterification, have previously been demonstrated.16,17
Next to the above, most studies dedicated to DES lignin characterisation have only focused on the precipitated lignin, i.e. the lignin obtained after anti-solvent addition to the black liquor. The water-soluble lignin fraction that does not precipitate, as well as the residual lignin fraction in the cellulosic pulp have largely been ignored. These fractions account for a substantial part of the lignin mass balance and are important for better understanding lignin modification during DES pulping. For example, these fractions could provide insight into the preferential extraction and conversion of structural moieties.
In addition, much of the detailed understanding of lignin chemistry during DES pulping has been obtained by employing pre-isolated lignin preparations.11,13 Despite their usefulness, especially from the analytical point of view, these preparations do not capture the full complexity of the lignocellulose matrix in terms of mass transfer, extraction and covalent and physical entrapment; in fact the rationale of using pre-isolated lignins to begin with. Hence, interpretation of the yield and purity of the precipitated lignin and amount and structure of the residual lignin structure is not possible.
Here, we present the first comprehensive characterisation of all lignin-containing fractions following DES pulping of lignocellulosic biomass. We used Miscanthus as model lignocellulosic feedstock to show that β-O-4 aryl ether cleavage and extensive substitution by DES incorporation occurs in precipitated, residual and water-soluble lignin fractions. Through saponification of the precipitated lignin, lignin model compounds studies and multidimensional NMR analysis we firmly established that both choline and lactic acid covalently incorporate into the lignin macromolecule, and even more so through which exact structural motifs. In addition, we demonstrate that the structure of the resulting lignins, including the extent of DES incorporation, can be modulated by DES water content and reaction time and temperature. Our work thus highlights the truly unique character of DES lignin and strongly contributes to expediting targeted lignin valorisation.
Experimental section
Materials
Miscanthus × giganteus (Miscanthus, Cradle Crops, Westdorpe, The Netherlands), Pinus sylvestris (pine, Staatsbosbeheer, Amersfoort, The Netherlands) and Eucalyptus grandis (Eucalyptus, Klabin, Sao Paulo, Brazil) feedstocks were processed as described in the ESI.† Lactic acid (LA) (80%) and choline chloride (ChCl) (99%) were obtained from Sigma-Aldrich (St Louis, MO, USA). Guaiacylglycerol-β-guaiacyl ether (GBG), syringylglycerol-β-guaiacyl ether (SBG) and veratrylglycerol-β-guaiacyl ether (VBG) were respectively purchased from TCI Chemicals (Zwijndrecht, Belgium), BLDpharm (Reinbek, Germany) and abcr (Karlsruhe, Germany). All other chemicals were obtained from commercial suppliers and used without further purification. Water used in all experiments was purified via a Milli-Q water system (Millipore, Billerica, MA, U.S.A.).Deep eutectic solvent pulping
Lactic acid:choline chloride (10:1 molar ratio) was used as deep eutectic solvent, hereafter referred to as DES. DES pulping of extractive-free Miscanthus was performed in a Parr reactor at 120 °C for 6 h. After separation of the residue and dark liquor, the lignin was precipitated from the latter by antisolvent addition. Detailed procedures are reported in the ESI.†
Solid-phase extraction lignin precipitate filtrate
Solid-phase extraction (SPE) was used to isolate the solubilised lignin-derived fraction from the precipitation filtrate (S8, Fig. S1†). Experimental details are reported in the ESI.†Enzyme lignin isolation from Miscanthus and DES cellulose residue
Extractive-free Miscanthus (R2) and DES cellulose residue (R6) were planetary ball-milled and treated with (hemi)cellulases based on previously reported procedures to respectively yield ‘native lignin’ and residual lignin R11 (Fig. S1†).18,19 Experimental details are described in the ESI.†Saponification Miscanthus, residue, lignin precipitate and water-soluble lignin
Extractive-free Miscanthus (R2), DES cellulose residue (R6), DES lignin (R9) and DES solubilised lignin (S8) were saponified for organic acid quantification and structural characterisation of the alkali-resistant lignin fraction based on procedures described in the ESI.†DES lignin sequential solvent fractionation
Precipitated DES lignin (R9) was sequentially solvent extracted by ethyl acetate (EtOAc), ethanol (EtOH) and methanol (MeOH) as described in detail in the ESI.†Small scale DES pulping
To determine the effect of biomass type, small scale DES pretreatments were performed on Miscanthus, pine, and Eucalyptus biomass. Using the same small scale setup, Miscanthus biomass was treated to determine the effect of reaction duration and DES water content on overall outcome in terms of yield and lignin structure (Table S1†). Experimental details are described in the ESI.†DES treatment lignin model compounds
GBG, SBG and VBG were used as lignin model compounds for respectively understanding the conversion of phenolic guaiacyl- and syringyl and ‘internal’ nonphenolic substructures during DES pulping. Experimental details are reported in the ESI.†Analytical methods
Quantitative pyrolysis-gas chromatography mass spectrometry employing 13C lignin as internal standard (13C-IS pyrolysis-GC-MS), multidimensional NMR spectroscopy, 31P NMR spectroscopy, alkaline size-exclusion chromatography (SEC) and ultra-high performance liquid chromatography photodiode array high-resolution mass spectrometry (UHPLC-PDA-HR-MS) were all based on previously reported procedures as described in detail in the ESI.†20–25
Results & discussion
Given our commitment to contribute to the structural characterization of DES lignin to promote its ultimate application, we first established that the benchmark conditions reported in the literature could be replicated in terms of lignin yield and structure. This way representative lignin samples for detailed structural analysis were obtained.Lignin mass balance
Benchmark DES pulping conditions (120 °C, 6 h) resulted in 71.2% w/w delignification based on lignin contents of the residue and initial Miscanthus feedstock, and 81.2% w/w absolute delignification considering total dry matter recovered (Tables S2 and S3†), well in line with values previously reported.6 The residue (R6) contained 5.6% w/w lignin, corresponding to 19% w/w of the initial lignin. Of the total removed lignin, 62.5% w/w was recovered in the lignin precipitate (R9), which corresponds to 50.8% w/w of the initial lignin. This implies that more than 1/3 of the lignin removed remained in solution after precipitation. Indeed, solid phase extraction of this solution yielded another 23.6% w/w of the total lignin. Therefore, the lignin mass balance was close to quantitively accounted for by the residual, precipitated and water-soluble lignin fractions, with an excellent total recovery of 93% w/w. Structural characterisation of the lignin contained in these fractions was performed to map DES lignin in unprecedented detail. Additional discussion on the DES residue (R6) is described in the ESI and Fig. S2.†Structural characterisation DES lignin fractions
The lignin in the various fractions was characterised and compared to native lignin enzymatically isolated from the same initial Miscanthus feedstock. Quantitative 13C-IS pyrolysis-GC-MS analysis substantiated that the isolated native lignin was representative of the lignin contained in the whole biomass, showing an identical subunit composition and comparable structural fingerprint (Table S4†), as expected.19 By comparing the HSQC NMR spectra of the DES lignin fractions to that of the native lignin, several new signals became apparent (Fig. 1). Besides (tentative) correlation peaks for substituted β-O-4 aryl ether substructures, lactic acid and choline signals were abundantly present in all spectra. This suggested that these DES components were responsible for the substitution observed. Semi-quantitative analysis showed substantial differences in subunit composition, an overall depletion in interunit linkages, increase in Cα-oxidized moieties, and preservation of p-coumaric acid moieties as compared to the Miscanthus native lignin (Table 1). In line with the cleavage of interunit linkages, Hibbert ketone substructures accumulated (Table 1) and the precipitated DES lignin showed a reduced molecular weight (Fig. S3†) and increase in phenolic OH content (Table S4†), all aligned with expectations from the literature.11,13 Hence, we concluded that a representative DES lignin sample was obtained for further detailed structural characterisation. | ||
| Fig. 1 Aliphatic (A) and aromatic (B) regions of HSQC NMR spectra (400 MHz, DMSO-d6) of native Miscanthus lignin, DES lignin R9 and saponified DES lignin R9. A: β-O-4; A′: β-O-4 γ-acyl; AR: β-O-4 α-ether; B: β-5; C: β–β; D′: β–β tetrahydrofuran (THF) γ-acyl; D: β–β THF; Ch: choline; LA: lactic acid; H: p-hydroxyphenyl; G: guaiacyl; Gox; guaiacyl Cα-ox; S: syringyl; Sox: syringyl Cα-ox; pCA: p-coumaric acid; FA; ferulic acid; HK; Hibbert ketone; I: cinnamyl alcohol; I′: cinnamyl alcohol γ-acyl. | ||
| Native | DES lignin R9 | DES lignin R9 saponified | DES lignin S10 | DES lignin S10 saponified | DES residue R11 | |
|---|---|---|---|---|---|---|
| a Relative distribution of lignin subunits (H + G + Gox + S + Sox = 100). b Relative volume integral of substructure versus volume integral of total lignin subunits. c Given the potential overlap of Cα–Hα and Cβ–Hβ correlations semi-quantified values might be overestimated. d Likely overlapped by carbohydrate-derived peak. e Only based on δC/δH 72.7/4.0 ppm correlation peak due to signal overlap. | ||||||
| Lignin subunitsa (%) | ||||||
| H | 2.0 | 1.4 | 1.5 | 1.7 | 2.2 | 1.5 |
| G | 60.9 | 55.2 | 53.4 | 55.1 | 56.0 | 60.4 |
| Gox | 0.0 | 0.7 | 2.3 | 6.1 | 3.4 | 0.8 |
| S | 37.1 | 41.9 | 41.3 | 35.1 | 34.3 | 36.4 |
| Sox | 0.0 | 0.7 | 1.6 | 2.0 | 4.0 | 0.9 |
| S/G | 0.61 | 0.76 | 0.77 | 0.61 | 0.65 | 0.61 |
| Hydroxycinnamatesb (per 100 ar) | ||||||
| p-Coumarate | 31.8 | 26.4 | 8.3 | 9.4 | 5.8 | 30.8 |
| Ferulate | 7.7 | 4.8 | 5.4 | 15.1 | 9.7 | 4.5 |
| Interunit linkagesb (per 100 ar) | ||||||
| β-O-4 aryl ether G + H | 20.2 | 10.3 | 12.0 | 5.8 | 13.9 | 13.1 |
| β-O-4 aryl ether S | 19.5 | 0.5 | 10.7 | 0 | 7.6 | 0 |
| β-O-4 aryl ether substitutedc | 0 | 25.4 | 12.9 | 26.2 | 8.7 | 21.1 |
| Total β-O-4 aryl ethers | 39.7 | 36.2 | 35.6 | 32.0 | 30.2 | 34.2 |
| β-5 phenylcoumaran | 5.8 | 4.4 | 4.9 | 5.6 | 6.2 | 3.9 |
| β–β resinol | 1.8 | 1.0 | 1.1 | 1.1 | 1.5 | 1.2 |
| β–β tetrahydrofuran acylated | 5.7d | 1.0 | 0 | 0.7 | 0 | 0 |
| β–β tetrahydrofuran nonacylated | 0 | 0 | 1.1 | 0 | 0 | 0 |
| Total | 52.9 | 42.7 | 42.7 | 39.3 | 37.9 | 39.3 |
| End-unitsb (per 100 ar) | ||||||
| Cinnamyl alcohol | 3.0 | 0 | 0 | 0 | 0 | 0 |
| Cinnamaldehyde | 1.5 | 0.8 | 0 | 0.4 | 1.4 | 0.9 |
| Benzaldehyde | 0 | 0 | 0.2 | 0.7 | 3.0 | 0.7 |
| Hibbert ketone | 0 | 2.5 | 0.4 | 1.6 | 0 | 3.2 |
| Diketone | 0 | 0.2 | 0.0 | 0.6 | 0.5 | 0.0 |
| DES incorporationb (per 100 ar) | ||||||
| Lactic acid ester | 0 | 19.6 | 0 | 186.8 | 0 | 11.0 |
| Lactic acid ether | 0 | 8.7 | 8.2 | 4.9e | 1.5e | 8.0 |
| Choline | 0 | 3.4 | 1.9 | 5.2 | 3.1 | 2.9 |
These observations were fully in line with complementary quantitative pyrolysis-GC-MS analysis (Table S5 and additional discussion†). Pyrolysis-GC-MS analysis further demonstrated a clear increase of diketones in the water-soluble lignin fraction (S10), products that have been shown to form from dihydroxypropiovanillone and syringone (DHPV/DHPS) substructures upon pyrolysis.26 Here, DHPV/DHPS substructures were absent, and diketone pyrolysis products originated instead from diketone moieties present in the lignin structure itself, based on their diagnostic correlation at (δC/δH 26.4/2.4 ppm) in the HSQC spectra (Fig. S4† and Table 1).26 Hong et al. have previously discussed monomeric diketone formation as a result of DES-induced lignin depolymerization.13 We now present the first evidence of the occurrence of lignin-bound diketone structures in actual DES lignin samples, albeit at relatively low levels.
Sharing very similar overall structural features (Table 1), the difference in extractability and solubility of the various DES fractions (precipitated, water-soluble, residual) likely was caused by differences in overall molecular weight (Fig. S3†) and/or the presence of lignin-carbohydrate complexes.
Despite the overall reduction in intact interunit linkages discussed above, DES pulping preserved most β-O-4 aryl ethers, presumably through substitution. Indeed, signals for substituted β-O-4 aryl ether substructures were abundantly present in the 1H–13C HSQC NMR spectra (δC/δH 81.7–78.0/4.9–4.4 ppm), amounting to 25 per 100 ar in the precipitated lignin (R9) (Table 1). The principle of stabilizing the benzylic position to preserve β-O-4 linkages has previously been demonstrated for mild alcoholic organosolv extractions27 and ternary DES systems containing ethylene glycol,28 and now can also be extended to benchmark lactic acid–choline chloride DES fractionation.
DES lignin saponification informs on substituent nature
Prompted by the substitution levels and intensities of lactic acid and choline signals observed, we saponified the precipitated lignin (R9) to better understand the nature of the substituents. The selectivity and effectiveness of the saponification procedure was confirmed by the identical subunit compositions and abundance of interunit linkages of the original and saponified lignins (Fig. 1 and Table 1). As expected, p-coumarate moieties were largely removed and acylated β–β tetrahydrofuran substructures were quantitatively converted to their nonacylated analogues (Table 1). Saponification released most lactic acid, indicating that these moieties were primarily incorporated through alkali-labile ester linkages. Some lactic-acid derived signals appeared resistant, as will be elaborated upon below. Excitingly, choline signals largely remained, strongly implying covalent incorporation through alkali-resistant linkages, presumably ethers.Overall, half of the substituted β-O-4 aryl ether substructures was found to be alkali labile, with numbers reducing from 25.4 to 12.9 per 100 ar (Table 1). Interestingly, β-O-4 aryl ethers linked to syringyl subunits (G-β-O-4-S/S-β-O-4-S) were found to be far more susceptible to lactic acid substitution than their guaiacyl linked counterparts, increasing from 0.5 to 10.7 per 100 ar and explaining more than 80% of the substituted β-O-4 aryl ethers converted upon saponification. Saponification of the water-soluble DES lignin (S10) provided the same insights (Table 1).
Lignin model compound studies confirm covalent DES incorporation
To further build on the DES incorporation observed by HSQC NMR, we performed lignin model compound studies in concert with detailed UHPLC-PDA-HR-MS and multidimensional NMR analysis. | ||
| Fig. 2 UHPLC-PDA280 nm chromatograms of GBG (A) and SBG (B) after DES pulping, annotated substructures based on HR-MS analysis (Table S6†) (C) and semi-quantification based on UV280 nm absorbance (D). Note for simplicity only one GBG/SBG-diLA structure is shown, while many are expected to be present. | ||
First of all, it is worth noting that the chromatographic gradient used allowed for the separation of two GBG/SBG diastereomers (erythro/threo) after DES conversion (Fig. 2) while the starting compounds were diastereomerically pure. This is in line with an acid-catalysed conversion mechanism and the formation of a prochiral benzylic carbocation intermediate, which upon water addition reforms the starting compound (Fig. 3A). Since water can be added at two faces two isomers are formed. Exact mass and MS2 fragmentation resolved the presence of various reaction products incorporating lactic acid and choline and even pinpointed reaction products containing combinations or multitudes thereof (Fig. 2, Fig. S5 and Table S6†). Choline-containing reaction products were readily identified by the presence of nitrogen in the calculated molecular formulas and the formation of cholinium ions (C5H14ON+, m/z 104.10731) upon fragmentation (Table S6†).
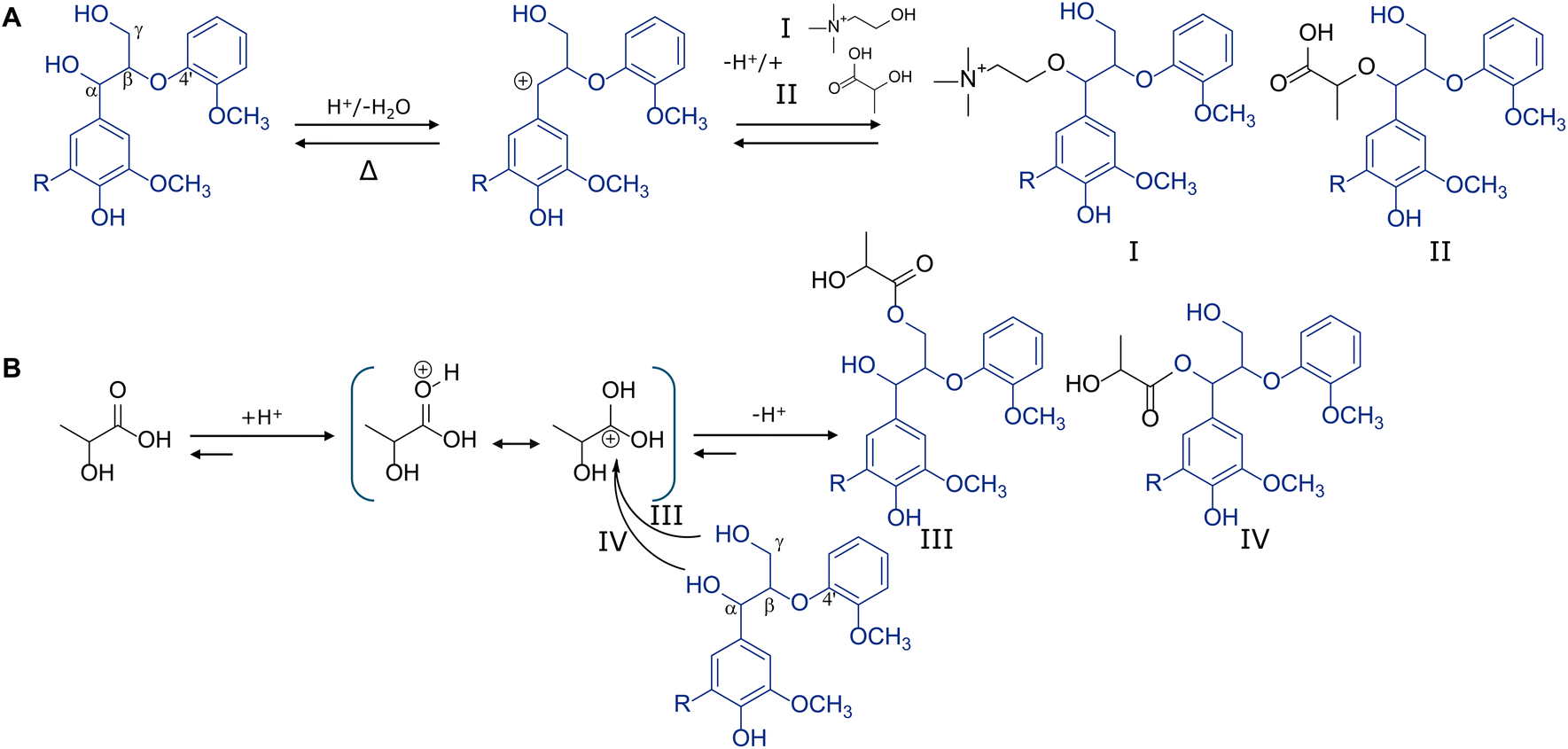 | ||
Fig. 3 Acid-catalysed benzylic etherification (A) of choline (I) and lactic acid (II) and lactic acid esterification (B) at the aliphatic (III) and benzylic (IV) positions of GBG (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H) and SBG (ROCH3). H) and SBG (ROCH3). | ||
The relatively wide retention time ranges of lactic acid containing reaction products suggested that many different isomers were formed. Indeed, many substructures incorporating LA can be envisaged, both on the Cα and Cγ hydroxyl groups. MS2 fragmentation, however, did not allow exact isomeric identification. The occurrence of GBG-triLA, SBG-triLA and VBG-triLa substructures furthermore implies that also LA-OH groups are susceptible towards (further) esterification, or that LA dimers and trimers can still esterify lignin hydroxyl groups. Condensation was only observed for the phenolic analogues GBG and SBG (Table S6†), suggesting that they are formed through nucleophilic addition of the 4-OH group to a benzylic carbocation intermediate. The low abundance of these dimerisation products indicates that such condensation is a minor reaction pathway only.
As already indicated by the HSQC NMR analyses discussed above, quantification based on UV280 nm absorbance demonstrated that lactic acid incorporation by far exceeded choline incorporation. Nonetheless, these results constitute the first evidence that choline is covalently incorporated into the lignin structure upon DES pulping. Having established that choline and lactic acid indeed incorporate into lignin model compounds during DES pulping, the reaction products were further characterised by detailed NMR analysis to elucidate the exact bonding motifs.
To definitively assign the appearing HSQC signals and unambiguously annotate the substructures present Heteronuclear Multiple Bond Correlation (HMBC) and HSQC-TOCSY (Total Correlation Spectroscopy) NMR spectra were recorded. These experiments respectively establish correlations between hydrogens and carbons two to three bonds apart and correlations between hydrogens in the same proton–proton coupling network.
HMBC NMR analysis confirmed the presence of α-esterified lactate substructures, with a diagnostic HSQC NMR correlation peak at δC/δH 74.4/5.90 ppm and consistent HMBC correlation peaks to β-O-4β, β-O-4γ, G2, G6, G1 and the LA1 carboxyl group (Fig. S8†). The more commonly reported esterification of γ-OH groups11 was also confirmed through connectivity analysis for the first time, with diagnostic HSQC NMR correlation peaks at δC/δH 62.9/4.27 and 63.2/3.84 ppm (Fig. S9†).
Besides α- and γ-esterified lactate substructures, HMBC NMR analysis convincingly demonstrated the presence of α-etherified lactate substructures. Spectra materialized the complete coupling network, with diagnostic and isolated HSQC NMR correlation peaks at δC/δH 73.1/3.97 and 72.2/3.93 ppm (Fig. S10†), peaks that also appeared in the DES lignin and indeed resisted saponification as to be expected from the ether motif (Fig. 1).
HMBC NMR analysis also surfaced obvious correlations between various choline-derived peaks (Fig. 4), i.e. between –N(CH3)3 and Ch2 and between Ch2 and Ch1, which allowed us to annotate the Ch1 correlation peaks at δC/δH 62.2/3.69 and 62.1/3.61 ppm in the HSQC spectra. These Ch1 signals further correlated with the substituted β-O-4 aryl ether region at δC/δH 79.1/4.62 ppm (β-O-4α) and importantly could be discerned from neighbouring β-O-4γ correlation signals.
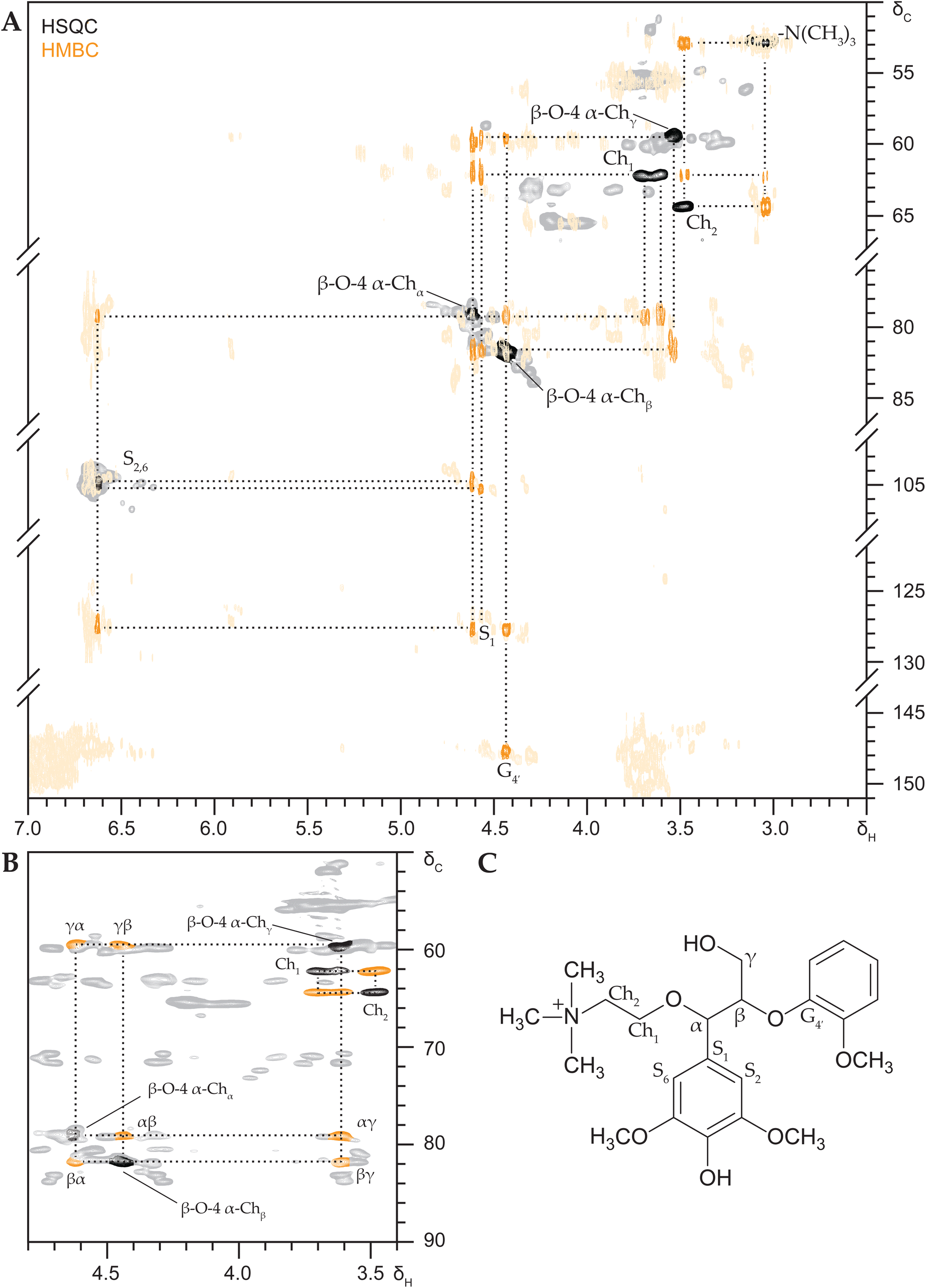 | ||
| Fig. 4 Two-dimensional NMR analysis (600 MHz, DMSO-d6) of SBG after DES reaction. Overlayed HSQC and HMBC NMR spectra (A) with annotated HSQC signals in black and unannotated in grey, annotated HMBC signals in orange and unannotated in light orange; HSQC-TOCSY NMR spectrum (B) with annotated HSQC signals in black, TOCSY correlation peaks in orange and unannotated in grey; structure of benzylic choline etherified SBG with annotations (C). | ||
The β-O-4α signal in turn also strongly correlated to the β-O-4β position at 81.9/4.44 ppm and additionally correlated to signals at δC/δH 59.5/3.61 ppm (β-O-4γ), δC/δH 105.3/6.95 ppm (S2,6) and δC 127.7 ppm (S1). The β-O-4β signal correlated to β-O-4γ, S1 and G4′ at δC 147.7 ppm, all consistent with a syringyl-β-O-4-guaiacyl substructure that is etherified by choline at the benzylic position.
Since the choline-substituted β-O-4α and β-O-4β signals appeared in rather crowded areas of the spectra we recorded an HSQC-TOCSY NMR experiment to also provide proof from another coupling perspective. The resulting spectrum convincingly showed the full α,β,γ-proton coupling network (Fig. 4B), corroborating the HMBC-based annotations and thus unambiguously confirming the presence of benzylic choline etherified SBG.
HMBC and HSQC-TOCSY NMR spectra were also recorded for GBG after DES reaction, highlighting identical correlations, which affirmed our annotations and provided definitive proof of benzylic choline etherification of the guaiacyl analogue as well (Fig. S11†).
Proof of covalent DES incorporation into actual lignin
As already recognized in the HSQC NMR spectra of reacted lignin model compounds, various signals for substituted β-O-4 aryl ether substructures coincided in the δC/δH 81.7–78.0–4.9/4.4 ppm region and this overlap was obviously even worse for complex lignin samples (Fig. 1), hence complicating unambiguous annotation. To facilitate annotation and confirm covalent choline incorporation into actual lignin, the saponified DES lignin was selected given the fact the 2D spectra of this sample were undoubtedly substantially reduced in interfering peaks, while the signals corresponding to Ch2 and substituted β-O-4 aryl ethers remained (Fig. 1 and Fig. S12†).We opted for recording 13C band-selective HSQC and HMBC NMR spectra of the regions of interest to improve spectral resolution and sensitivity. The selective HMBC spectrum (Fig. 5) materialized clear correlations between the choline-related signals and excitingly also between the Ch1 signal and a signal at δC/δH 79.0/4.62 ppm, indeed identical to the signal we previously assigned to the Cα–Hα signal of α-Ch β-O-4 aryl ethers for DES reacted model compounds (Fig. 4 and Fig. S11†). An HMBC NMR correlation peak between the β-O-4α and β-O-4β (81.7/4.43 ppm) signals further substantiates the annotation. We can thus be positive, choline covalently incorporates into lignin during DES pulping via etherification of β-O-4 substructures.
 | ||
| Fig. 5 Selected regions of overlayed selective HSQC and HMBC NMR spectra (600 MHz, DMSO-d6) of saponified DES lignin R9, with annotated HSQC signals in black and unassigned in grey, and annotated HMBC signals in orange and unassigned in light orange. | ||
Following the annotation of α-lactate esterified β-O-4 aryl ethers in DES treated lignin model compounds (Fig. S8†), the well-isolated diagnostic peak (δC/δH 74.0/5.94) ppm could be readily observed in the HSQC spectrum of the DES lignin R9 as well (Fig. 1) and semi-quantified at 2.1 per 100 ar. Likewise, α-lactate etherified β-O-4 aryl ether substructures could be clearly identified in the HSQC spectra at δC/δH 73.0/3.98 and 72.2/3.98 ppm. Chemical shifts were indeed identical to those annotated for the lignin model compounds (Fig. S10†) and further confirmed by HMBC correlations in the band selective spectra.
Being the most abundant interunit linkage, we initially focused our characterisation efforts on the β-O-4 aryl ether substructures. Nonetheless, HSQC-TOCSY NMR spectra before and after saponification proved extremely insightful with regard to β-5 phenylcoumaran substructures, as others have also demonstrated.30 These spectra allowed definitive annotation and unambiguous assignment of γ-lactate esterified β-5 phenylcoumaran motifs in DES lignin (Fig. S13†).
Covalent DES incorporation depends on molecular weight population
Having unambiguously demonstrated that DES covalently incorporates into the lignin structure, we further looked into the distribution of the structural motifs of DES lignin across the molecular weight range. Thereto, we quantitatively fractionated the precipitated DES lignin (R9) by sequential solvent extraction (Table S7†). Together, the sequential EtOAc, EtOH, MeOH and residual fractions made up 99.2% of the input mass (Table S7†). The obtained residue was entirely soluble in 80% aqueous acetone, and hence this common final extraction step was omitted.As expected, the molecular weight and dispersity of the lignin populations increased as fractionation proceeded (Fig. S14†), ranging from an average molecular weight (Mw) of 1220 (EtOAc) up to 10330 (residue) g mol−1. Besides molecular weight, the fractions also diverged in subunit composition, interunit linkage content and composition, hydroxycinnamates and DES components incorporated (Table S7†). Sequential solvent fractionation of Kraft lignin has shown that the lower molecular weight fractions accumulate most pulping-induced substructures and concomitantly are most depleted in original, native structural features.31–33 DES lignin, however, did not show such an obvious pattern, with for example intact β-O-4 aryl ethers and Hibbert ketone substructures being fairly uniformly distributed across the molecular weight range (Table S7†). Interestingly though, the highest molecular weight fraction showed the highest abundance of both lactic acid and choline incorporated, and the incorporation of the latter showed a clear increasing trend with molecular weight. In fact, the incorporation of choline was found to increase with increasing polarity of the extraction solvent used. Obviously, upon choline incorporation the lignin structure becomes more polar, which therefore could have driven the fractionation rather than the molecular weight (alone). Following the same line of thought, being more hydrophilic, choline-incorporating structures, especially those of lower molecular weight, are expected to be less prone to precipitate upon anti-solvent addition. Indeed, the lignin fraction that remained soluble (S10) showed a higher abundance of choline (Table 1).
Covalent DES incorporation occurs independent of biomass botanical origin
To further highlight the relevance of our findings, small scale DES pulping was performed on Miscanthus, Eucalyptus and pine as respective models for grass, hardwood and softwood feedstocks, at reaction temperatures of 120, 140 and 160 °C. As expected, woody biomass proved more recalcitrant towards delignification, requiring higher pulping temperatures to obtain decent yields of precipitated lignin (Fig. S15†).34 As a consequence of the higher temperatures required, the content of intact interunit linkages severely dropped, approaching absolute depletion at 160 °C (Fig. S15 and Table S8†). Importantly, these experiments emphasized that the content of lactic acid and choline containing substructures remained stable even when native features started to decline as an effect of increased pulping temperatures (until 140 °C) (Table S8†). Our characterisation efforts highlighted that DES incorporation, both in terms of lactic acid and choline, is not limited by biomass type. Nonetheless, incorporation into Miscanthus lignin was double than both Eucalyptus's and pine's. Thus, if lignin with DES incorporation and related functionalities are strived for, grass biomass would remain the feedstock of choice.DES incorporation can be tuned by reaction conditions
Given the presumed acid-catalysed etherification and esterification mechanisms (Fig. 3), DES incorporation was expected to be influenced by water content. Indeed, an effect of DES water content has previously been suggested for the conversion of a lignin model compound.35 Hence, by relying on the same small scale experiments as used for the various biomass types above, the effect of DES water content, and reaction duration, on the resulting precipitated lignins was further scrutinized. Indeed, DES water content proved to be a major determinant for the structural features of the resulting lignins, in terms of subunit composition, β-O-4 aryl ether and p-coumarate preservation, β-O-4 aryl ether substitution, Hibbert ketone formation and the incorporation of lactic acid and choline (Fig. S16†). On the one hand, if native-like lignin is pursued, DES incorporation ‘side-reactions’ can be reduced by water addition, but this comes at a yield penalty. Conversely, ‘dry’ DES could be used to specifically produce modified lignins.Targeted DES lignin application and outlook
The new structural understanding is anticipated to lead to substantial environmental and green impact, at least on three axes, as elaborated below.1. The structural features established have profound implications for the application of DES lignin, directly increasing its valorisation options and thus contributing to getting closer to the pursued total-use lignocellulose cascading through the DES concept. Lactic acid incorporation is expected to improve compatibility in polylactic acid-lignin blends for producing biobased composites and coatings.36 We further reckon that specifically exploiting the quaternary nitrogen functionality incorporated through choline is exciting to pursue, for example for flocculant or surfactant applications. The flocculation and surfactant properties are worthwhile to explore over the pH range since the carboxylic acid of etherified lactate and quaternary nitrogen of choline govern the lignin zwitterionic behaviour.
2. Importantly, one-step lignin extraction and modification through DES pulping could constitute a clear advance compared to separate isolation and derivatization strategies for targeted lignin functionalization for the addressed applications. This combination could be more efficient in terms of processing, the number of unit operations and resources required.
3. Once optimized in terms of covalent DES constituent incorporation, amongst others by the handles provided above, the advertised functionalization through lactic acid and choline chloride could result in a substantially reduced environmental impact and reagent toxicity compared to traditional derivatization agents used for lignin esterification (anhydrides37) and cationisation (epoxides38).
Future work will explore said targeted functionalization and application strategies, accompanied by assessments of technoeconomic feasibility and energy, water and carbon footprints. Given the extensive incorporation of DES constituents into lignin and cellulose demonstrated here, solvent recovery and recyclability should be closely evaluated. Unproductive lactic acid oligomerization and coupling between lactic acid and choline chloride should furthermore be minimized to reduce solvent consumption. Logically, the gained value of the modified biopolymers produced needs to outweigh the solvent cost to make this concept technoeconomically viable.
Conclusions
By performing comprehensive structural characterisation in concert with model compound studies we have unambiguously demonstrated that lactic acid and choline covalently incorporate into the lignin structure during DES pulping and established the exact binding motifs. Moreover, we have identified that and how ultimate DES lignin structure depends on biomass type and pulping conditions in terms of water content, temperature and duration. By pinpointing which buttons to push, our results provide access to the unique features DES lignins have to offer for high-value, functional ingredient applications, thus expediting the realization of the overall DES biorefinery concept.Author contributions
GvE: conceptualization, data curation, formal analysis, investigation, methodology, visualisation, writing – original draft, writing – review & editing. VB: data curation, formal analysis, visualisation, writing – review & editing. JvG: funding acquisition, project administration, supervision, writing – review & editing. RG: funding acquisition, supervision, writing – review & editing. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing interest.Acknowledgements
Matthijs van Lint, Daan S. van Es, Jacinta van der Putten, Martijn van Walsem and Guus E. Frissen (Wageningen Food & Biobased Research) are acknowledged for valuable discussion and performing alkaline SEC, compositional and 31P NMR analyses. Pauline Damhof and Sarah van Dinteren (Laboratory of Food Chemistry, Wageningen University) are thanked for assisting UHPLC-PDA-HR-MS analysis. Yanzhang Luo (MAGNEFY) is acknowledged for technical assistance on the 600 MHz NMR analyses. The MAGNEFY facility at Wageningen University (part of the Dutch national uNMR-NL facility) is gratefully acknowledged for the access to the NMR instruments. This work was supported by Topconsortium voor Kennis en Innovatie (TKI, grant number LWV20.136) and Dutch Food Initiative (DFI, grant number DFI-AF-18002).References
- A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. K. Rasheed, J. Am. Chem. Soc., 2004, 126, 9142–9147 CrossRef CAS PubMed.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- M. Francisco, A. Van Den Bruinhorst and M. C. Kroon, Green Chem., 2012, 14, 2153–2157 RSC.
- X. Tang, M. Zuo, Z. Li, H. Liu, C. Xiong, X. Zeng, Y. Sun, L. Hu, S. Liu and T. Lei, ChemSusChem, 2017, 10, 2696–2706 CrossRef CAS PubMed.
- D. J. G. P. van Osch, L. J. B. M. Kollau, A. van den Bruinhorst, S. Asikainen, M. A. A. Rocha and M. C. Kroon, Phys. Chem. Chem. Phys., 2017, 19, 2636–2665 RSC.
- Z. Chen, A. Ragauskas and C. Wan, Ind. Crops Prod., 2020, 147, 112241 CrossRef CAS.
- C. Alvarez-Vasco, R. Ma, M. Quintero, M. Guo, S. Geleynse, K. K. Ramasamy, M. Wolcott and X. Zhang, Green Chem., 2016, 18, 5133–5141 RSC.
- X.-J. Shen, J.-L. Wen, Q.-Q. Mei, X. Chen, D. Sun, T.-Q. Yuan and R.-C. Sun, Green Chem., 2019, 21, 275–283 RSC.
- M. Jablonský, A. Škulcová, L. Kamenská, M. Vrška and J. Šima, BioResources, 2015, 10, 8039–8047 Search PubMed.
- C.-W. Zhang, S.-Q. Xia and P.-S. Ma, Bioresour. Technol., 2016, 219, 1–5 CrossRef CAS PubMed.
- X.-J. Shen, T. Chen, H.-M. Wang, Q. Mei, F. Yue, S. Sun, J.-L. Wen, T.-Q. Yuan and R.-C. Sun, ACS Sustainable Chem. Eng., 2019, 8, 2130–2137 CrossRef.
- Z. Chen, X. Bai, H. Zhang and C. Wan, ACS Sustainable Chem. Eng., 2020, 8, 9783–9793 CrossRef CAS.
- S. Hong, X.-J. Shen, B. Pang, Z. Xue, X.-F. Cao, J.-L. Wen, Z.-H. Sun, S. S. Lam, T.-Q. Yuan and R.-C. Sun, Green Chem., 2020, 22, 1851–1858 RSC.
- L. Das, M. Li, J. Stevens, W. Li, Y. Pu, A. J. Ragauskas and J. Shi, ACS Sustainable Chem. Eng., 2018, 6, 10408–10420 CrossRef CAS.
- W.-X. Li, W.-Z. Xiao, Y.-Q. Yang, Q. Wang, X. Chen, L.-P. Xiao and R.-C. Sun, Ind. Crops Prod., 2021, 170, 113692 CrossRef CAS.
- E. S. Morais, A. M. Da Costa Lopes, M. G. Freire, C. S. Freire and A. J. Silvestre, ChemSusChem, 2021, 14, 686–698 CrossRef CAS PubMed.
- N. Rodriguez Rodriguez, A. van den Bruinhorst, L. J. Kollau, M. C. Kroon and K. Binnemans, ACS Sustainable Chem. Eng., 2019, 7, 11521–11528 CrossRef CAS.
- G. van Erven, A. F. Kleijn, A. Patyshakuliyeva, M. Di Falco, A. Tsang, R. P. de Vries, W. J. H. van Berkel and M. A. Kabel, Biotechnol. Biofuels, 2020, 13, 1–12 Search PubMed.
- G. van Erven, P. Hendrickx, M. Al Hassan, B. Beelen, R. op den Kamp, E. Keijsers, K. van der Cruijsen, L. M. Trindade, P. F. H. Harmsen and A. F. van Peer, ACS Sustainable Chem. Eng., 2023, 11, 6752–6764 CrossRef CAS.
- G. van Erven, R. de Visser, D. W. H. Merkx, W. Strolenberg, P. de Gijsel, H. Gruppen and M. A. Kabel, Anal. Chem., 2017, 89, 10907–10916 CrossRef CAS PubMed.
- G. van Erven, R. de Visser, P. de Waard, W. J. H. van Berkel and M. A. Kabel, ACS Sustainable Chem. Eng., 2019, 7, 20070–20076 CrossRef CAS.
- S. D. Mansfield, H. Kim, F. Lu and J. Ralph, Nat. Protoc., 2012, 7, 1579–1589 CrossRef CAS PubMed.
- H. Nishimura, A. Kamiya, T. Nagata, M. Katahira and T. Watanabe, Sci. Rep., 2018, 8, 1–11 CAS.
- R. J. A. Gosselink, J. E. G. van Dam, E. de Jong, E. L. Scott, J. P. M. Sanders, J. Li and G. Gellerstedt, Holzforschung, 2010, 64, 193–200 CAS.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. Van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen and R. J. A. Gosselink, Green Chem., 2016, 18, 2651–2665 RSC.
- G. van Erven, R. Hilgers, P. de Waard, E.-J. Gladbeek, W. J. H. van Berkel and M. A. Kabel, ACS Sustainable Chem. Eng., 2019, 7, 16757–16764 CrossRef CAS.
- D. S. Zijlstra, C. W. Lahive, C. A. Analbers, M. B. Figueirêdo, Z. Wang, C. S. Lancefield and P. J. Deuss, ACS Sustainable Chem. Eng., 2020, 8, 5119–5131 CrossRef CAS.
- Y. Liu, N. Deak, Z. Wang, H. Yu, L. Hameleers, E. Jurak, P. J. Deuss and K. Barta, Nat. Commun., 2021, 12, 5424 CrossRef CAS PubMed.
- N. Bryant, C. G. Yoo, Y. Pu and A. J. Ragauskas, ChemistrySelect, 2020, 5, 3359–3364 CrossRef CAS.
- J. Rencoret, G. Marques, O. Serrano, J. Kaal, A. T. Martínez, J. C. del Río and A. Gutiérrez, ACS Sustainable Chem. Eng., 2020, 8, 12521–12533 CrossRef CAS.
- C. S. Lancefield, H. L. J. Wienk, R. Boelens, B. M. Weckhuysen and P. C. A. Bruijnincx, Chem. Sci., 2018, 9, 6348–6360 RSC.
- N. Giummarella, P. A. Lindén, D. Areskogh and M. Lawoko, ACS Sustainable Chem. Eng., 2019, 8, 1112–1120 CrossRef.
- N. Giummarella, I. V. Pylypchuk, O. Sevastyanova and M. Lawoko, ACS Sustainable Chem. Eng., 2020, 8, 10983–10994 CAS.
- M. Zhou, O. A. Fakayode, A. E. A. Yagoub, Q. Ji and C. Zhou, Renewable Sustainable Energy Rev., 2022, 156, 111986 CrossRef CAS.
- A. M. da Costa Lopes, J. R. B. Gomes, J. A. P. Coutinho and A. J. Silvestre, Green Chem., 2020, 22, 2474–2487 RSC.
- K. Shi, G. Liu, H. Sun and Y. Weng, Polymers, 2023, 15, 2807 CrossRef CAS PubMed.
- X. Zhao, Y. Zhang, M. Yang, Z. Huang, H. Hu, A. Huang and Z. Feng, Polymers, 2018, 10, 907 CrossRef PubMed.
- R. Wahlström, A. Kalliola, J. Heikkinen, H. Kyllönen and T. Tamminen, Ind. Crops Prod., 2017, 104, 188–194 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc00909f |
| This journal is © The Royal Society of Chemistry 2024 |