
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBeyond 2,5-furandicarboxylic acid: status quo, environmental assessment, and blind spots of furanic monomers for bio-based polymers†
Mattia
Annatelli‡
a,
Julián E.
Sánchez-Velandia‡
b,
Giovanna
Mazzi‡
a,
Simão V.
Pandeirada
c,
Dimitrios
Giannakoudakis
 d,
Sari
Rautiainen
e,
Antonella
Esposito
f,
Shanmugam
Thiyagarajan
g,
Aurore
Richel
h,
Konstantinos S.
Triantafyllidis
d,
Tobias
Robert
i,
Nathanael
Guigo
j,
Andreia F.
Sousa
ck,
Eduardo
García-Verdugo
*b and
Fabio
Aricò
*a
d,
Sari
Rautiainen
e,
Antonella
Esposito
f,
Shanmugam
Thiyagarajan
g,
Aurore
Richel
h,
Konstantinos S.
Triantafyllidis
d,
Tobias
Robert
i,
Nathanael
Guigo
j,
Andreia F.
Sousa
ck,
Eduardo
García-Verdugo
*b and
Fabio
Aricò
*a
aDepartment of Environmental Sciences, Informatics and Statistics, Ca’ Foscari University of Venice, Scientific Campus Via Torino 155, 30170 Venezia Mestre, Italy. E-mail: fabio.arico@unive.it
bDepartment of Inorganic and Organic Chemistry, University Jaume I, Avda Sos Baynat s/n, E-12071-Castellon, Spain. E-mail: cepeda@uji.es
cCICECO – Aveiro Institute of Materials, Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal
dDepartment of Chemistry, Aristotle University of Thessaloniki, University Campus, P. O. Box 116, GR-54124 Thessaloniki, Greece
eVTT Technical Research Centre of Finland Ltd, P. O. Box 1000, FI-02044 VTT Espoo, Finland
fUniv. Rouen Normandie, INSA Rouen Normandie, CNRS, Groupe de Physique des Matériaux UMR 6634, F-76000, Rouen, France
gWageningen Food & Biobased Research, Wageningen University and Research, P. O. Box 17, Wageningen, 6700 AA, The Netherlands
hLaboratory of Biomass and Green Technologies, University of Liege – Gembloux Argo-Bio Tech, Passage des Desportés, 2, B-5030, Gembloux, Belgium
iFraunhofer Institute for Wood Research, Wilhelm-Klauditz Institute WKI, Bienroder Weg 54E, Braunschweig, Germany
jUniversité Côte d'Azur, CNRS, Institut de Chimie de Nice (ICN), UMR 7272, 06108 Nice Cedex 02, France
kCentre for Mechanical Engineering, Materials and Processes, Department of Chemical Engineering, University of Coimbra Rua Sílvio Lima – Polo II, 3030-790 Coimbra, Portugal
First published on 3rd May 2024
Abstract
Since 5-(hydroxymethyl)furfural (HMF) has been labelled as the “sleeping giant” of the bio-based platform-chemical realm, numerous investigations have been devoted to the exploitation of this versatile molecule and its endless chemical transformations into novel monomers for producing bio-based polymers. However, beyond 2,5-furandicarboxylic acid (2,5-FDCA), little attention has been devoted to key aspects that deserve being addressed before bringing forward other HMF-derivatives into the bio-based plastic market, i.e., procedures, scaling-up of the syntheses, products’ purification, physical–thermal properties, and above all green metrics (sustainability/greenness of procedures). This critical review focuses on the most investigated derivatives of HMF beyond 2,5-FDCA, assessing their exploitation as monomers for bio-based polymers. HMF-derived compounds have been classified according to their functionalities, i.e., aldehyde-, diol-, polyol-, amine-, acid-, ester-, carbonate-, acrylate-, and epoxy-based monomers. The related synthetic approaches are discussed, evaluating the sustainability of the procedures reported so far, based on green metrics such as the environmental factor (E-factor) and the process mass intensity (PMI). For each family of HMF derivatives, their use as monomers for the synthesis of bio-based polymers has been addressed, taking into consideration the efficiency of the polymerisation reactions, the physical–chemical and thermal properties of the resulting bio-based polymers, as well as their biodegradability if applicable. The overall picture that emerges is that much has been achieved for the synthesis of furan monomers; however, many obstacles still need to be overcome prior to massively introducing these compounds into the bio-based plastic market. Hopefully, the data reported in this review will shed light on the goals achieved so far, and on some critical issues that must still be tackled in the short- or medium-term for a more sustainable and however efficient industrial process.
 Mattia Annatelli | Mattia Annatelli obtained his Master's degree in Chemistry and Sustainable Technologies from Ca’ Foscari University of Venice (2019). Subsequently, he pursued and obtained a Ph.D. in Environmental Sciences (2024), under the supervision of Prof. Fabio Aricò focussing on new synthetic procedures for the synthesis of bio-based platform chemicals. He also spent a mobility period at Jaume I University (Spain). During this experience, he learned the basics of continuous flow reactions, further enriching his scientific background. Currently, he holds the position of post-doc researcher at Ca’ Foscari University, focusing on the valorization of crotonic acid. |
 Julián E. Sánchez-Velandia | Julián E. Sánchez-Velandia received his degree in Chemistry from Universidad Industrial de Santander (Colombia) in 2015 and completed his PhD thesis in Chemistry focusing on biomass valorization using heterogeneous catalysis in 2020 under supervision of Prof. Aída L. Villa-Holguín (Universidad de Antioquia). After two postdoctoral internships about one-pot transformations of biomass; computational chemistry; and condensation/oxidation strategies of terpenes using heterogeneous materials, he moved to Universidad Jaume I (Spain) where he is researcher in the field of biomass transformation, cascade reactions and development of new materials for valorization of CO2-derived compounds. |
 Giovanna Mazzi | Giovanna Mazzi has achieved her Master's degree in Chemistry at Ca’ Foscari University of Venice with a thesis focused on the large-scale synthesis of 5-hydroxymethylfurfural (HMF), its microwave-assisted functionalization and on the green evaluation of its synthesis using the green metrics. After one year as research fellow in analytical chemistry in the field of aerosol pollution, she won a PhD scholarship within the same topic and is currently working on Organic Contaminants of Emerging Concern in urban aerosol. |
 Simão V. Pandeirada | Simão Vidinha Pandeirada was born in Portugal, 1999. He received his bachelor's and master's degrees in Biochemistry in University of Aveiro, Portugal. He then joined the group of Prof. Andreia F. Sousa at the same University to start his PhD in 2023. He is currently working on polymers recycling trough greener processes using eutectic solvents and unveiling the reaction mechanisms with computational chemistry. |
 Dimitrios Giannakoudakis | Dr Dimitrios A. Giannakoudakis graduated as chemist from Aristotle University of Thessaloniki (AUTh). He received his Ph.D. at the City University of New York (full scholarship), 2017. He continued as postdoc and tutor at the City College of New York and Institute of Physical Chemistry in Warsaw. Currently, he is research associate at AUTh. His research focuses on R&D of nano-engineered materials for photo/thermo/sono-catalytic biomass valorization, environmental, and energy applications. He co-authored above 120 articles (>4500 citations), 1 monograph, 6 edited-books, 17 chapters, and 4 patents. DAG is included in the 2% top-world most influential scientists for 2021/2022, Stanford list. |
 Sari Rautiainen | Dr Sari Rautiainen is a senior scientist at VTT Technical Research Centre of Finland focusing on catalyst and process development for valorisation of biomass into sustainable chemicals and materials. She obtained her PhD from University of Helsinki in 2016 with focus on heterogeneous oxidation catalysis. Before joining VTT, she developed catalysts for lignin depolymerisation at Stockholm University during her postdoctoral studies. Her recent work includes the upcycling of agricultural pectin-containing waste streams into monomers for polyesters and polyamides. |
 Antonella Esposito | Antonella Esposito was born in 1979 (Terni, Italy). She got passionate about classical studies (ancient Greek, Latin, philosophy…) until 1998, then moved to modern science. She became a Materials Engineer (University of Perugia, 2005), then moved to France for her PhD (INSA Lyon, 2008). After two-year teaching in Oyonnax, she became Maître de Conférences in Université de Rouen Normandie (2010), Groupe de Physique des Matériaux (GPM). She investigates the relations between chemistry, microstructure and properties of (bio)polymers, and their evolution with time, accounting for molecular relaxations within complex two- and three-phase microstructures with different coupling between ordered and disordered phases. |
 Shanmugam Thiyagarajan | Shanmugam Thiyagarajan received his bachelor's and master's degrees in chemistry in India. He then joined the group of Prof. A. S. Nasar at the University of Madras in 2002, where he worked on synthesizing novel monomers suitable for preparing hyperbranched polymers, earning his Ph.D. in Organic-Polymer Chemistry. Later, he moved to the Netherlands to pursue his research career in the field of biomass research. Since 2009, he has been a senior scientist at Wageningen University and Research, where he is actively involved in synthesizing bio-based chemicals and materials for a wide range of applications. |
 Aurore Richel | Aurore Richel is a Full Professor at the University of Liège (Belgium), where she focuses on research and education in the chemistry of renewable resources and associated technologies. Dr Richel is actively engaged in over 15 national and international projects centered on bioenergy and bioproducts production. Her work encompasses innovative pretreatment protocols for lignocellulosic materials, extraction of platform chemicals for the polymer sector, formulation of novel biomass-energy solutions, and the enhancement of lignin for high-value applications. |
 Konstantinos S. Triantafyllidis | Konstantinos Triantafyllidis is Full Professor at the Department of Chemistry, Aristotle University of Thessaloniki, Greece, with undergraduate and PhD studies at the Department of Chemistry, University of Ioannina, Greece, postgraduate training at SHELL, Amsterdam, and post-doctoral work at Michigan State University, USA. His research interests focus on heterogeneous catalysis, porous and metal/oxides nanomaterials, polymer nanocomposites, conventional and bio/waste-refining to fuels and chemicals, and green chemistry and technology. He serves as delegate of Association of Greek Chemists to “EuChemS/Division of Green and Sustainable Chemistry” and is appointed as National Representative to “Interdivisional Committee on Green Chemistry for Sustainable Development” of IUPAC. |
 Tobias Robert | Tobias Robert is working as scientist at the Fraunhofer WKI in Braunschweig, Germany. His research interests involve the synthesis of bio-based polymers and polymer resins and the use of UV-curing polymers for inks and 3D-printing. In addition, he is interested in the material use of bio-based side-products. |
 Nathanael Guigo | Dr Nathanael Guigo is associate professor within the Institut de Chimie de Nice at Université Côte d'Azur (FR). He is specialized in physical chemistry of polymers with a peculiar interest on sustainable polymeric materials (e.g. preparation and characterization of biobased polymers) within a circular approach. N. Guigo has focused his research in the synthesis and characterization of furanic thermosets including resins and composites from polyfurfuryl alcohol and humins from biorefineries. He has also worked on understanding the behavior of furanoate polyesters. N. Guigo is the Vice-Chair of the COST Action FUR4Sustain. |
 Andreia F. Sousa | Andreia F. Sousa is an Assistant Researcher at CICECO, the University of Aveiro Institute of Materials, Portugal, since 2020, where she leads a multidisciplinary team on the design of sustainable polymers (renewable, biodegradable, recyclable & recycling), within a circular economy mind-set. Sousa is a distinguished scientist with >60 papers – 5 highly cited – accumulating over 3200+ citations. She is Associate Editor for Reactive and Functional Polymers (Elsevier) and very recently she was distinguished as an Early Career Board member of the ACS Sustainable Chemistry & Engineering journal. She coordinates/participates in several EU projects including FUR4Sustain COST Action (CA18220). |
 Eduardo García-Verdugo | Prof. Eduardo García-Verdugo received his degree in 1995 at the University of Valencia, and furthered his education with a PhD in Chemistry from the University Jaume I (Spain). He is a Full Professor in Organic Chemistry and leads the Supramolecular and Sustainable Research Group at the same university. His research focuses on the integration of catalysis, polymeric materials, continuous flow processes, microreactors, bio-catalysis, and neoteric solvents to design sustainable chemical processes under the umbrella of Green Chemistry. He co-founded Proyecto Kryptonite SL and Molecular Sustainable Solutions SL, two ventures that reflect his dedication to both innovation and environmental responsibility. |
 Fabio Aricò | Fabio Aricò is an associate professor of Organic Chemistry at the Department of Environmental Sciences and Informatics at Ca’ Foscari University of Venice (Italy) where he is leading a group working on Green Chemistry. His main research interests are green chemistry biorefinery (C5 and C6 bio-based platform chemicals) and chlorine-free reactions. He has published more than 80 scientific peer-reviewed papers and holds 3 granted patents. Fabio Aricò has also been recently awarded Experienced Chemist award by IUPAC-NHU International award for Advancement in Green Chemistry in recognition of his work on chlorine-free approaches for biorefinery development. |
1. Introduction
Synthetic polymers have greatly contributed to the welfare of mankind. However, the massive increase in plastic production over the last decades has led to serious environmental and health concerns, as plastic waste is being released into the environment in amounts larger than ever. As a result, microplastics can be found all over the world, even in the remotest areas, such as Antarctica. The recent finding that microplastics from our living environment eventually ended up in the human bloodstream is even more disturbing.1Several efforts have been advocated to reduce the waste generated from plastics, such as collecting and recycling. However, not all countries implemented efficient collecting systems, and the overall recycling rates are very low.2 In addition, the depletion of mineral oil reserves, the increasing demand for plastics, and the rising costs of petroleum-based raw materials associated with the climate and energy crises, have led the plastics industry to seek alternative feedstocks from renewable sources, with the additional aim to address circularity by re-designing the chemicals and the derived materials. Consequently, and unsurprisingly, the demand for polymers from renewable resources is significantly growing; this includes both (modified) biopolymers, such as starch, cellulose and lignin, as well as bio-based polymers synthesized from bio-based monomers.
Indeed, biorefineries can process different biomass feedstocks into a variety of suitable renewable monomers, commonly referred to as bio-based platform chemicals, such as polyols, lignin-derived monomers, fatty acids, aliphatic alkanes/alkenes or aromatics/phenolics, vegetable oils, terpenes, C5–C6 sugars and their furan-based derivatives.3 Over the last 20 years, these molecules have been exploited for potential applications in the polymer field, as plastics, composites, thermosets, coatings, etc. As an example, hemicellulose residues from crops (e.g., oat hulls, straws, bagasse, corncobs, husks, etc.) or forestry exploitation (e.g., birch, beech, eucalyptus, and balsa wood) contain large quantities of C5-sugar such as xylose,4 that upon dehydration, is converted into furfural.
The initial batch process of furfural production (i.e., cooking the pentosan-rich wastes with mineral acids such as sulfuric acid) was implemented by the Quaker Oats company in 1922 and still remains one the most employed industrial process, even though efforts for more sustainable and efficient processes are being made. The global annual production of furfural is more than 300 kilotons per annum and about 70% of this production is located in China. The worldwide production of furfural is massively driven by its derivatization in furfuryl alcohol (more than 2/3 of the entire furfural production), and the main use of furfuryl alcohol is for producing thermoset resins to prepare high-quality foundry moulds in the metal casting industry.
Both furfural and furfuryl alcohol can polymerize into highly cross-linked network and carbon-rich polymeric structures. Via gas-phase oxidation, furfural can also be derivatized into C4 building blocks for polymer applications such as maleic anhydride. Producing maleic anhydride from the furfural value chain could also pave the way to a bio-based route for key monomers such as succinic acid.5
Derivatizing furfural into monomeric compounds suitable for polymer synthesis implies an additional step leading to, at least, two functional groups necessary for step-growth polymerization.
Biomass-derived C6 carbohydrates offer the possibility to directly reach difunctional furanic building blocks. Among them, 5-(hydroxymethyl)furfural (HMF), firstly reported in 1895, has quickly become one of the bio-based platform chemicals archetype that has been extensively investigated due to its numerous potential applications as synthon of biofuels, chemical intermediates, and in particular monomers for bio-based polymers (Fig. 1).6
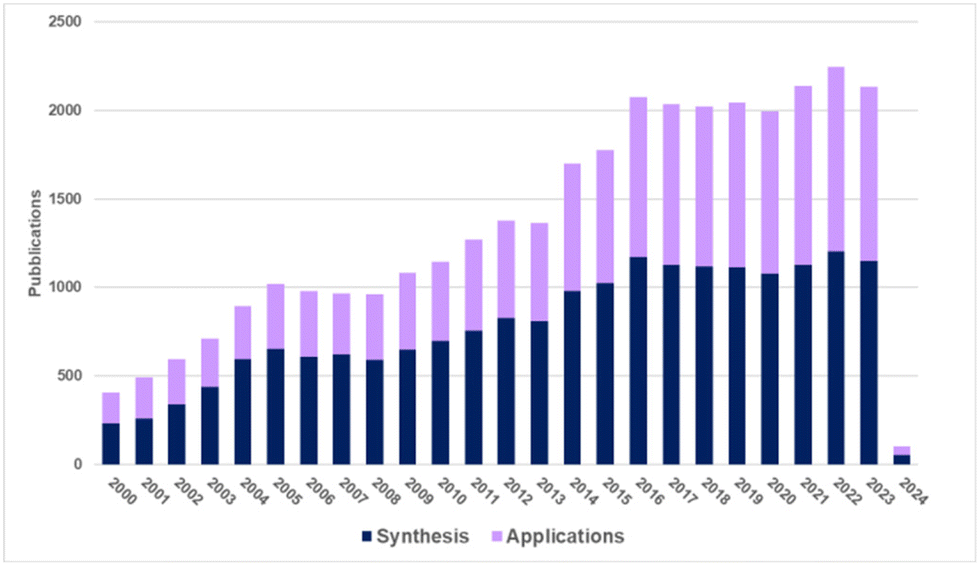 | ||
| Fig. 1 Publications dealing with the synthesis and applications of furanics (according to Reaxys data collected in January 2024). | ||
HMF can be synthesized in high yield via dehydration of carbohydrates, acid-catalysed hydrolysis/dehydration of cellulose, and also from food waste.7 This molecule incorporates two functional units (on the C2 and C5 furan ring position) and an aromatic structure that renders it a versatile substrate for numerous chemical transformations. Thus, HMF and its derivatives are particularly attractive for designing polymer architectures, as these bi-functional building blocks can lead to diols, diacids, diamines, etc. (Fig. 2). However, HMF also encompasses some issues, such as its hydrophilic and polar behaviour that prevents its recovery from aqueous media, and a fast degradation due to the formation of dimers, oligomers and humins.8 As a result, the efficient production of HMF is a hot topic, and continuous work has been devoted to the development of a cost-effective synthesis, as its use as a renewable commodity requires an estimated scale-up production of 100–1000 kilo metric ton per annum.
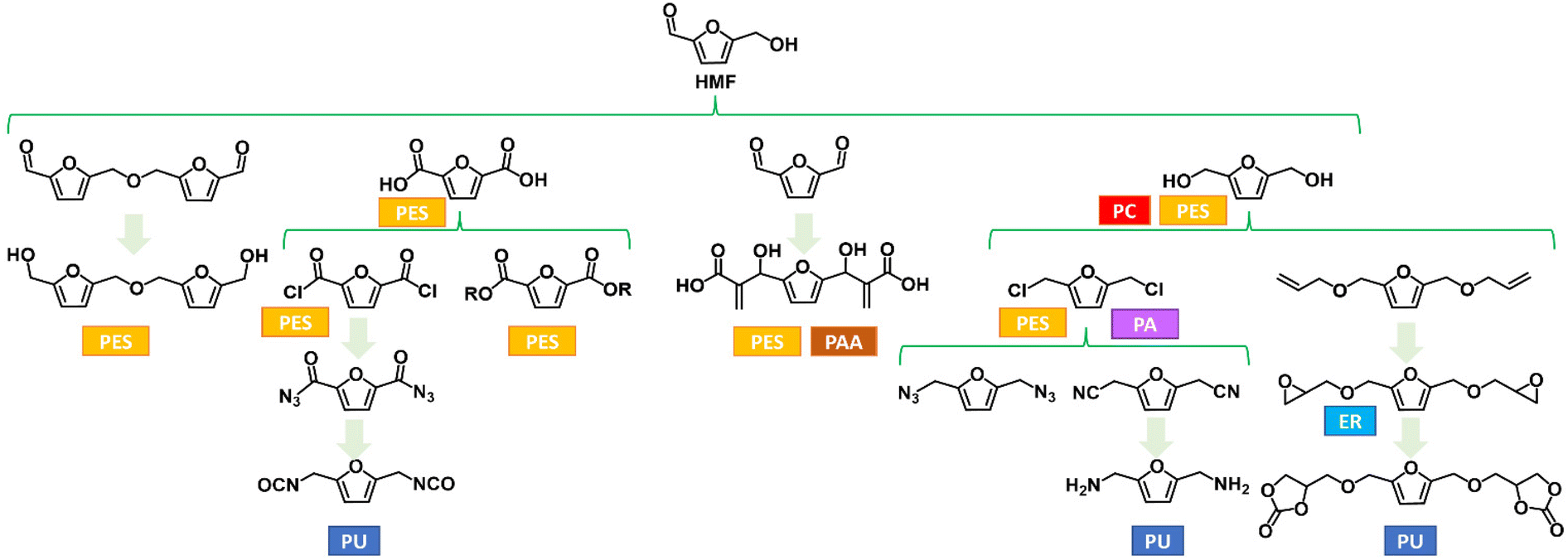 | ||
| Fig. 2 Selected furanic monomers and related bio-based polymers; PU = polyurethane; PES = polyester; PA = polyamides; PAA = polyacrylate; PC = polycarbonate; ER = epoxy resin. | ||
The great interest in the exploitation of HMF is justified by the fact that it can be easily converted by oxidation into 2,5-furandicarboxylic acid (2,5-FDCA), the key monomer for the synthesis of poly(ethylene 2,5-furandicarboxylate) (2,5-PEF), advocated as one of the most credible renewable alternatives to the widely used poly(ethylene terephthalate) (PET) for many applications including bottles, packaging and textiles.9
Although the main industrial interest in the exploitation of HMF is currently the production of 2,5-FDCA, numerous investigations have been conducted on other HMF derivatives with promising application as monomers for bio-based materials. Some examples so far reported in the literature are depicted in Fig. 2. The research effort in this field is of paramount importance, as these monomers could play a crucial role in the transition to a more sustainable bioeconomy by enlarging the scope of bio-based building blocks together with or even beyond 2,5-PEF. Even though thermoplastic polyesters have a huge range of potential applications, furan-based monomers are also valuable starting materials for other families of thermoplastic polymers (e.g., polyamides) and, even among polyesters, the furan-based building-block chemicals toolbox beyond FDCA can engineer more and innovative materials. Furthermore, their use for thermosets and resins has been generally overlooked.
For example, epoxidized mono- and bis-furans are promising substitutes for bisphenol-A-diglycidylether (DGEBA), which is used on large scale to produce two-components epoxy resins (see Section 5).
To the best of our knowledge, in the field of thermosetting polyurethanes, as well as polyurethane dispersions for coatings, there are no viable bio-based alternatives to cyclic diisocyanates, such as isophorone diisocyanate (IPDI), methylendi(phenyl isocyanate) (MDI) or toluene-2,4-diisocyanate (TDI). But cyclic building blocks could be interesting alternatives to phthalic and isophthalic acid also for other types of coatings, such as polyesters and alkyl resins, as cyclic structures significantly improve the properties of the coatings.
Despite the great effort of the scientific community in developing novel renewable monomers beyond 2,5-FDCA targeting their use for polymer synthesis or even as additives, most of them still have important issues to be addressed to reach their full potential for practical applications under the boundaries of green and sustainable chemistry.
From these premises, this critical review illuminates the flourishing field of bio-based polymers by focusing on the versatile molecule of HMF, often heralded as the “sleeping giant” within the bio-based platform-chemical domain. While considerable research has explored the transformation of HMF into novel monomers for bio-based polymers, particularly spotlighting 2,5-FDCA, this review shifts focus to other HMF-derived monomers that have not received as much attention. These derivatives are pivotal for advancing the bio-based plastic market but require thorough investigation beyond their synthesis such as scalability, purification, physical–thermal properties, and importantly, green metrics evaluation.
This review aims to methodically assess the most studied HMF derivatives beyond 2,5-FDCA, examining their potential as monomers for bio-based polymers. It categorizes HMF-derived compounds based on their functional groups—ranging from aldehydes, diols, and polyols to amines, acids, esters, carbonates, acrylates, and epoxies. The synthesis of these monomers is discussed, with a keen evaluation of the sustainability of the reported procedures using green metrics.
For specific bio-based monomers, i.e., HMF, DFF, BHMF, the large number of procedures reported in the literature obliged us to make a selection. Therefore, only the syntheses performed at least in gram scale (in some cases, also 0.5 gram scale) were reported. The other most representative synthetic approaches have been included in the ESI.†
It is noteworthy that most of the research focusing on the preparation of HMF and its chemical transformations, as it happens for many other bio-based platform chemicals, was mostly conducted on a small scale and/or in diluted experimental conditions. Developing multi-grams procedures to bio-based chemicals is mandatory to render these biorefinery approaches market-competitive.
Besides, the scientific community must be committed to pursue new synthetic approaches to these bio-based chemicals under the umbrella of Green and Sustainable Chemistry (GSC), for instance aiming at low E-factors10 and Process Mass Intensities (PMI),11 reducing the amount of (green) solvents and avoiding toxic reagents.
Briefly, the E-factor (or E-total) calculates the kilograms of waste produced per kilograms of product. Industrial (or bench-scale) procedures can be classified into 4 classes based on the related E-factor value: oil refining (0–0.1 kg kg−1), bulk chemicals (1–5 kg kg−1), fine chemical (5–50 kg kg−1) and pharmaceuticals (25–100 kg kg−1).
PMI represents the mass of materials (reagents, catalysts and solvents) employed to produce a certain amount of product(s), taking into account also their recover and reuse.
In this view, the synthetic approaches to the most investigated bio-based furanics have been evaluated also considering these green metrics in order to assess their greenness and indicate the road to eventual improvement.
Furthermore, for each class of HMF derivative, the review also addresses their utilization as monomers in bio-based polymer synthesis, considering the efficiency of polymerization reactions, the physical–chemical and thermal properties of the resulting bio-based polymers, and their biodegradability where relevant (ESI†). Although significant advances have been made in synthesizing furan monomers, the data collected underscore that numerous challenges remain to be surmounted before these innovative materials can be widely adopted in the bio-based plastic market.
Thus, this review aims to provide a comprehensive overview of the on the status quo of the furan-based monomers and polymers and highlights the critical challenges that need addressing in the short- to medium-term to go from the laboratory scale to the (pre)industrial scale. This approach seeks to pave the way for a more sustainable and efficient process, pushing the boundaries of current bio-based polymer production toward a greener future.
2. Aldehyde-based furanics
2.1. Syntheses of 5-hydroxymethylfurfural
As highlighted in Table 1 (see also ESI; Table S.2† for synthesis that have E-factor/PMI > 100), HMF is probably the most extensively studied bio-based platform chemical in the last 20 years, due to its wide range of applications.6 Over the years, this molecule has been exploited as versatile substrate to produce a large portfolio of furanic compounds that can be used as monomers for bio-based polymers.12| # | Substrate (g mmol−1) | Conc. (M) | Catalyst (mol% or wt%) | Reaction conditionsb | Cat. reuse | Yield (%) | E-f | PMI | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a The metrics do not consider preparation of the catalyst. All yields are isolated, excepted where otherwise specified. b A = autoclave, B = batch, C = continuous flow. c DMSO is partially recovered. d Excluding purification. e Non-isolated HMF. f Column chromatography is excluded from calculations. g Recovery of catalyst not included. h Amounts of work-up and/or purification materials are not reported. i A 0.56 M aqueous solution of fructose was pumped at a rate of 0.33 mL min−1 and mixed with MIBK, pumped at a rate of 1.00 mL min−1, in a second flow channel. j CeP3 = [(Ce(PO4)1.5(H2O)(H3O)0.5(H2O)0.5)]. | |||||||||
| 1c | D-Fructose (5/27.8) | 0.35 | [PPFPy][HSO4] (7.5 mol%) | B; DMSO; 100 °C, 0.5 h | 10 | 83 | 39.7 | 4.2 | 13 |
| 2d | D-Fructose (10/55.6) | 1.39 | Purolite CT275DR (5 wt%) | A; DMC/TEAB, 110 °C, 2 h | — | 72e | 27.5 | 5.1 | 14 |
| 3f | D-Fructose (5/27.8) | 1.54 | CO2 (7 MPa) | A; H2O; 90 °C, 168 h | — | 92 | 6.1 | 7.1 | 15 |
| 4g,h | D-Fructose (2.1/11.7) | 13.0 | Ti/Si500 (10 wt%) | B; H2O/TEAC; 80–100 °C, 0.75 h | — | 93 | 13.4 | 14.4 | 16 |
| 5 | D-Fructose (0.18/1) | 1.00 | SBA-15-SO3H (15 wt%) | B; DMSO; 120 °C; 1 h | — | 78 | 17.5 | 18.5 | 17 |
| 6 | D-Fructose (20/111.1) | 0.69 | Amberlyst-15 (10 wt%) | B; DMC/TEAB; 90 °C, 16 h | — | 70 | 24.6 | 25.6 | 18 |
| 7 | D-Fructose (10/55.6) | 0.37 | Amberlyst-15 (100 wt%) | B; ACN/TEAC; 100 °C, 2.5 h | — | 78 | 44.6 | 45.6 | 19 |
| 8h,i | D-Fructose (10/55.6) | 0.56 | HCl (0.25 M) | C; H2O/MIBK; 140 °C, 0.25 h | — | 74 | 45.5 | 46.5 | 20 |
| 9h,f | D-Fructose (10/55.6) | 0.56 | H2SO4 6.0 mol% (LiBr 0.2 mol%) | B; DMAc; 100 °C, 6 h | — | 45 | 56.3 | 57.3 | 21 |
| 10 | D-Fructose (0.4/2) | 0.13 | [Teim-PS] [AlCl4]4 (15 wt%) | A; H2O/1-octanol; 130 °C; 3 h | — | 84 | 63.7 | 64.7 | 22 |
| 11 | D-Glucose (0.4/2) | 0.13 | [Teim-PS] [AlCl4]4 (15 wt%) | A; H2O/1-octanol; 130 °C; 3 h | — | 71 | 75.2 | 76.2 | 22 |
| 12 | Sucrose (0.4/2) | 0.13 | [Teim-PS] [AlCl4]4 (15 wt%) | A; H2O/1-octanol; 130 °C; 3 h | — | 64 | 83.7 | 84.7 | 22 |
| 13j | D-Fructose (0.6/3.6) | 0.28 | CeP3 (15 wt%) | B; DMC/H2O; 150 °C, 3 h | 5 | 67 | 86.5 | 86.1 | 23 |
| 14g | D-Fructose (1/5.6) | 0.56 | FeCl3 (10 mol%) | B; NMP/TEAB; 90 °C, 2 h | — | 78 | 87.1 | 88.1 | 24 |
| 15f | D-Fructose (1.8/10) | 0.25 | HBr/silica (100 mol%) | B; THF; 30 °C, 24 h | — | 95 | 94.1 | 95.1 | 25 |
| 16 | D-Fructose (20/111.1) | 1.11 | Amberlyst-15 (10 wt%) | B; H2O/TEAB 100 °C, 0.25 h | — | 91 | 95.4 | 96.4 | 26 |
Although HMF is not directly used as monomer for bio-based polymers, it is employed as starting compounds for most of the herein discussed furanics, thus the greenness of HMF synthetic approaches is relevant for its further chemical transformations.
HMF is commonly produced through acid-catalysed triple dehydration of hexoses, such as D-glucose and D-fructose (Scheme 1), however it has also been synthesized from oligosaccharides such as, starch, cellulose, or inulin, or through the one-pot conversion of complex lignocellulosic, amylaceous, or free sugar rich raw materials.27,28
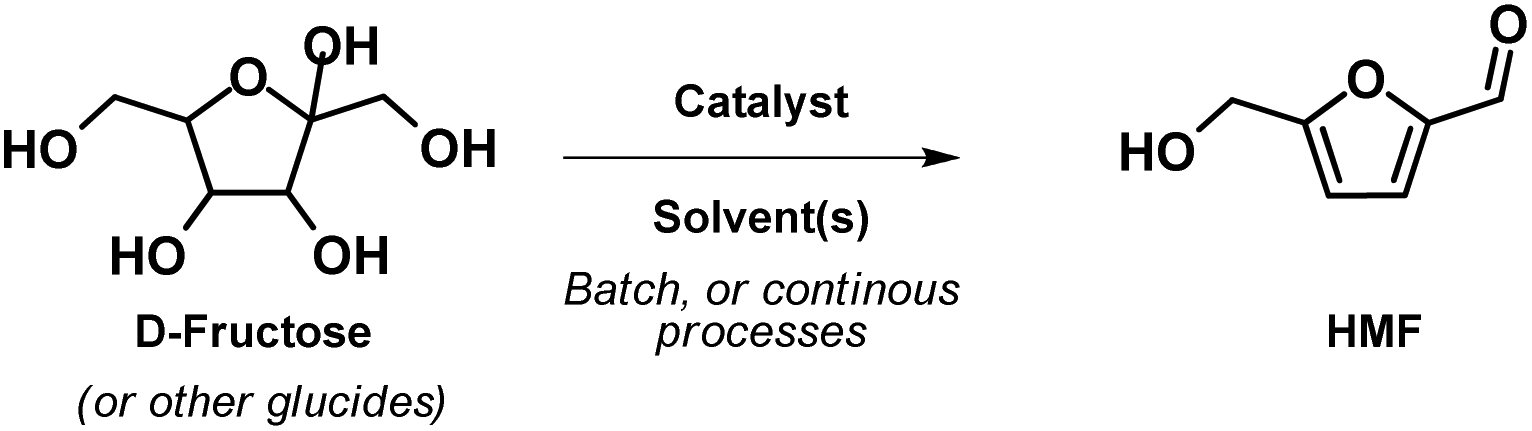 | ||
| Scheme 1 D-Fructose triple dehydration to form HMF. | ||
Numerous homogeneous or heterogeneous catalytic systems, combined with specific choices of solvents (single phase or biphasic media), have been systematically explored to increase HMF yield as the selectivity of the reaction is often low due to the concomitant formation of levulinic acid, formic acid and humins.29–31
Table 1 depicts some illustrative – although not exhaustive – approaches reported for the synthesis of HMF from different carbohydrates (D-fructose, D-glucose and sucrose), either in batch or under continuous flow conditions including lab-scale procedure and commercial/prototype-scale.32,33
High HMF yields were achieved starting from D-fructose when selecting an appropriate “catalyst–solvent” pair. The green efficiency of the synthesis (E-factor and PMI) is significantly variable, ranging from 6 to more than 600 when ionic liquids are used as solvents.
Generally speaking, this chemical transformation requires mild conditions and thus apparently the synthesis is quite sustainable, however in most of the reported trials the catalyst and solvent used were not recycled. Only in two examples, i.e., where ionic liquid [PPFPy][H2SO4] (#1, Table 1) and [BMIM][Cl]/sulfuric acid (#11 and #12, Table S2†) were used, it was investigated the stability and robustness (>10 reuses) of the catalytic system for the synthesis of HMF from D-fructose. As a result, the use of [PPFPy][H2SO4] led to the best PMI (4.2) among the reported procedure. Similar PMI value was achieved using commercially available Purolite as catalyst and a dimethyl carbonate (DMC) tetraethylammonium bromide (TEAB) solvent system (#2, Table 1).
Numerous procedures employed biphasic systems, since for laboratory scale reactions, represent the most convenient way to recover HMF from the reaction mixture (#4, #6–8, #10–13, #14 and #16, Table 1).
Among these procedures, the more efficient in term of E-factor (#3, Table 1) was achieved by Motokucho and co-workers when 7 MPa of CO2 was used. It is well-known that CO2 together with water at higher pressure and temperature can be converted into carbonic acidic capable to perform chemical transformations. The use of CO2 is very interesting since this molecule is the apparent catalyst of the reaction and its use represent a rational application of greenhouse gas for valorisation of biomass decreasing the apparent emissions. However, the greenness of the process may be compromised by the necessity of long reaction times and the energy-intensive and costly nature of high-pressure conditions.
The use of Ti/Si500 (#4; Table 1) and mesoporous silica (#5; Table 1) also resulted in good values of E-factor and PMI for the D-fructose conversion into HMF. In both cases, high yields of the desired product (>70%) were achieved in short reaction time (1 h). Although it should be mentioned that the use of DMSO as the solvent in the procedure of Gu and co-workers would affect the eventual recovery of the synthetized HMF.17
Considering the bench scale reaction E-factors is still in the range of fine chemical, i.e., 5–50 kg waste per kg of product. However, the values of PMI are significant lower indicating an increasing attention in recycling of the reagents and or solvents.
When considering the industrial production of HMF, it should be mentioned that multiple patents have been filed. Indeed, the industrial production of this molecule can be carried out either in batch or continuous mode, using reactors with specific configurations (tank, pipe, fluidized-bed reactors).
D-Fructose remains the most employed starting substrate for commercial-scale production, owing to its high yields of HMF. The choice of operating conditions, as well as the nature of catalyst and solvent, varies from one industry to another.32 Since 2014, AVA Biochem produces HMF at a commercial scale ca. 300 tons per year. However, due to the absence of commercial large-scale plants, the price of HMF remains quite high (Merck price being 5980 € per kg).
The selection of feedstock for the synthesis of HMF also holds considerable importance, particularly for potential industrial applications where the cost of the initial materials greatly influences the economic viability of the process. It is noteworthy that a significant portion of HMF synthesis research begins with D-fructose due to its more straightforward conversion pathway. However, the efficiency and the pathways of these reactions can vary substantially, presenting a range of challenges.
The process of converting glucose to HMF is notably more complex compared to that of D-fructose. It requires an initial step of isomerizing, followed by the dehydration of fructose to form HMF. This two-step method is generally less efficient due to the additional steps involved and the side reactions that can occur. Moreover, the isomerization of glucose to fructose is optimally performed with a basic catalyst, whereas the dehydration of fructose to HMF requires an acidic catalyst.
As a result, yields of HMF obtained from D-glucose, inulin, cellulose, or lignocellulosic feedstocks (such as sugarcane bagasse, wheat straws, and corn stover) are typically lower than those from fructose under similar conditions (#11 and #12; Table 1).
The moderate yields reported in these cases stem from the multi-step transformation that involves the isomerization of glucose into fructose, which is sometimes preceded by a preliminary hydrolysis step if the starting matrix is a polysaccharide, such as cellulose or inulin. Consequently, the E-factors for these processes are often higher than those calculated for the direct and one-step production of HMF from D-fructose under comparable conditions.
To address these challenges, it is necessary to develop more efficient systems capable of processing glucose at higher concentrations. This could involve several strategies, such as the innovation of catalyst development to discover more effective bifunctional catalysts capable of performing both the isomerization of glucose to fructose and the subsequent dehydration of fructose to HMF. Moreover, optimizing the reaction conditions and selecting appropriate solvent systems are critical for improving the overall efficiency and yield of HMF from glucose. By concentrating efforts in these areas, significant progress can be made towards enhancing the synthesis of HMF and related bio-based monomers, thereby increasing their economic viability for industrial applications.
2.2. Syntheses of 5,5′-[oxybis(methylene)]bis-2-furfural (OBMF)
5,5′-[Oxybis(methylene)]bis-2-furfural (OBMF) is an attractive molecule (see Table S.1† for thermal properties) for polymers, known for its ability to act as a precursor for some imine-based polymers exhibiting relatively high thermal properties and good electrical conductivity indicating a more stable compounds compared to HMF. However, up to date, its synthesis – mainly carried out from HMF in the presence of an acidic promoter – has been only scarcely reported40 and mostly conducted on milligram up to gram scale.Although some authors reported the formation of OBMF through Williamson reaction by combination of HMF and its chlorinated counterpart, the HMF self-etherification remains the most straightforward approach studied for OBMF preparation (Scheme 2).34Table 2 shows some of the most recent results obtained to produce OBMF through its self-etherification using an acid catalyst.
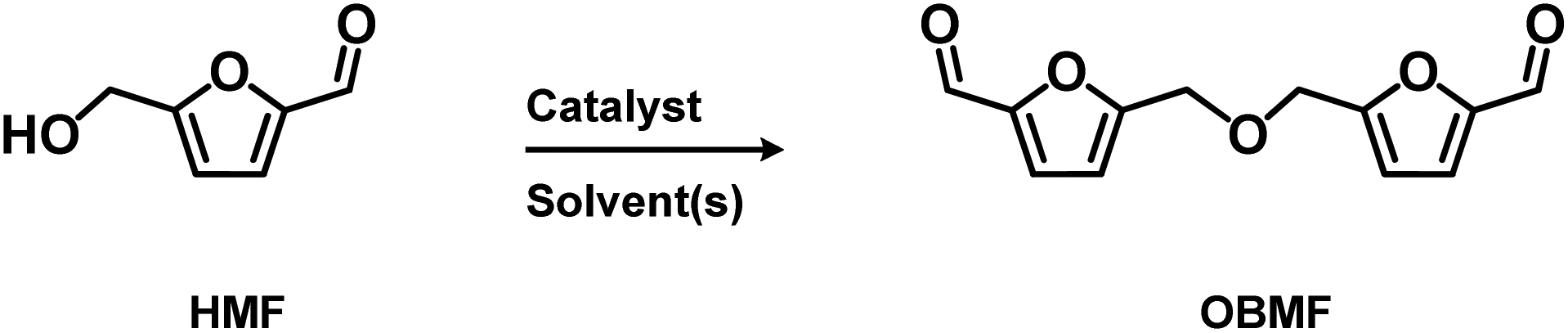 | ||
| Scheme 2 HMF self-etherification. | ||
| # | HMF (g mmol−1) | Conc. (M) | Catalyst (mol% or wt%) | Reaction conditions | Cat. reuse | Yield (%) | E-f | PMI | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Work-up materials not included. b Purification materials not included. c Reaction time not given. d Determined by analytical method. | |||||||||
| 1a,b | 0.2/1.6 | — | Amberlyst-15 (25 wt%) | Neat; 100 °C; 0.3 h | — | 51 | 1.6 | 2.6 | 34 |
| 2a,b | 0.06/0.5 | 1.00 | Graphene oxide (32 wt%) | CH2Cl2; 100 °C; 8 h | — | 88d | 13.5 | 14.5 | 35 |
| 3c | 1/7.94 | 0.66 | PTSA (6 mol%) | Toluene; 110 °C | 75 | 15.1 | 16.1 | 36 | |
| 4a,b | 1/7.94 | 0.44 | HMZ (50 wt%) | 4-Chlorotoluene; 100 °C; 2 h | — | 81 | 26.5 | 27.5 | 37 |
| 5a,b | 0.03/0.3 | 0.52 | Preyssler (HPAs) (60 wt%) | CH2Cl2; 40 °C; 2 h | — | 52d | 35.6 | 36.6 | 38 |
| 6a | 0.25/2. | 0.40 | Sn-Mont (40 wt%) | 1,2-Dichloroethane; 100 °C; 2 h | 5 | 89 | 37.1 | 37.6 | 39 |
| 7a,b | 5/39.7 | 0.24 | Fe2(SO4)3 (25 mol%) | DMC; 95 °C; 24 h | 3 | 81 | 48.3 | 48.5 | 40 |
| 8b | 15/120 | 0.67 | Amberlyst-15 (25 wt%) | Benzene; 80 °C; 7 h | — | 61 | 71.5 | 72.5 | 41 |
| 9a,b | 0.3/2.5 | 0.18 | Al-MCM-41 (10 wt%) | TFT; 100 °C; 10 h | — | 97d | 73.0 | 74.0 | 42 |
| 10a,b | 5/39.7 | 0.20 | PTSA (1 mol%) | Toluene; 110 °C; 12 h | — | 76 | 92.2 | 93.2 | 43 |
The most common catalyst used remains p-toluene sulfonic acid (PTSA), even if some acid resins, or mesoporous zeolites, are nowadays gaining interest (#3 and #10; Table 2).
The etherification is usually performed in anhydrous media or using an azeotrope to remove the water formed during the reaction. As a result, solvents employed for OBMF synthesis encompass benzene, toluene, chlorinated or trifluorinated media such as chlorotoluene, trifluorotoluene, or dichloroethane (#2–6; #8–10, Table 2). In one of the most recent procedures, dimethyl carbonate was employed as green media in combination with a Lewis acid catalyst resulting in moderate green metrics values (#7; Table 2).
Analysing the data reported in Table 2, it is evident the dominant use of common non green media as well as the moderate PMI (and E-factors) values that range from 14 to 93. The best value of PMI was achieved when the reaction was conducted in neat, although this procedure was carried out in very small scale.
The equivalent values of E-factors (13 to 92) highlight a general lack of investigations on solvent or catalyst recycling also pointing out that the status on the OBMF synthesis is still quite preliminary. In conclusion, despite the raising interest in OBMF further investigations in its synthesis should be focussed on developing greener procedures and also investigating multi-gram scale reaction. Furthermore, direct conversion of D-fructose to OBMF would be a very appealing synthetic approach to be investigated.
2.3. Synthetic approaches to 2,5-diformylfuran (DFF)
Besides the complete oxidation of HMF to FDCA, HMF partial oxidation of the hydroxyl-methyl group also leads to a value-added platform chemical i.e., 2,5-diformylfuran (DFF) or furan-2,5-dicarbaldehyde according to the IUPAC nomenclature.44 Over the last 10 years, DFF has been at the center of intensive research in consideration of its potential use for the synthesis of a wide-range of chemicals and materials such as antifungal agents, pesticides, pharmaceuticals/drugs, organic contactors and furan-containing polymers.45An important physicochemical difference between HMF and DFF is that the latter is hydrophobic due to its low-polarity, and hence insoluble in water but soluble in various organic solvents. This property renders DFF isolation/purification easier compared to HMF. In addition, DFF is stable upon long exposure to air or/and humidity without being oxidized or di-/polymerised. This is an important aspect for storage and transportation of the biomass derived furanics, especially considering HMF instability (ESI, Table S.1†). Hence, the direct production of DFF from biomass by one-pot approaches is among the most active research within furanics.
The formation of DFF starting from cellulose – or other carbohydrates/polysaccharides, like sucrose, D-fructose, D-glucose – by one-pot two-steps approaches using metal-based catalysts and acidic reagents have also been extensively studied.45 Most of the research has been focused on the hydrothermally catalytic conversion of D-fructose to DFF, where in the first step the acidic dehydration of fructose to HMF takes place (in inert atmosphere), followed by the selective oxidation to DFF under oxygen atmosphere.
As depicted in Scheme 3, D-fructose can be obtained by isomerization of glucose that is in turn produced from cellulose. Alternative carbohydrates/polysaccharides were also studied as feedstock for the one-pot conversion to DFF, such as sucrose, although to a limited extend.
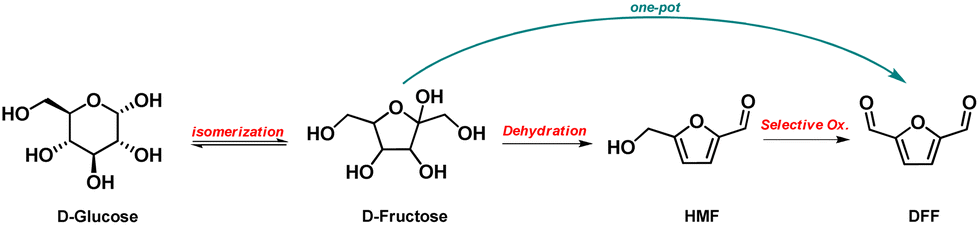 | ||
| Scheme 3 Synthesis of DFF. | ||
In general, DMSO is the predominately employed solvent for the one-pot conversion of D-fructose to DFF, since it can promote the formation of HMF and, in addition, it can also stabilize HMF. This was explained by Caratzoulas and co-workers based on the increase of HMF LUMO energy upon solvation with DMSO, and hence being less vulnerable to nucleophilic attack responsible for hydration and formation of humin.46 In addition, DMSO can play a key catalytic role as elucidated by Laugel et al., while using other solvents, like p-tolyl sulfoxide, DMF, di-n-butyl sulfoxide, or methyl phenyl sulfoxide, the HMF conversion was negligible or even zero under the same conditions.47
The synthesis of DFF starting from D-fructose was triggered in 2003, when Grushin et al. reported the first one-pot, two-steps approach for this chemical transformation (#5; Table 3).48 In this work, DFF was achieved in 41% yield starting from D-fructose (1.0 g) using a Dowex-type ion-exchange resin and DMSO as solvent in the presence of either a Brønsted acid catalysts (H3PO4 or H2SO4) or lanthanide salts for the HMF formation, while its subsequent oxidation to DFF was catalysed by vanadium-based catalysts at 150 °C. DFF was isolated by vacuum sublimation, followed with Soxhlet extraction using cyclohexane, reaching purity >99%. A crucial drawback was the formation of Me2S and MeSO2 that have an unpleasant smell. These by-products were neutralized/oxidized by passing the outgases through bleach (NaOCl). The E-factor of this process is very high (146.1) because of the purification procedures employed. Nevertheless, other parameters like the used amount of water and silica were not factored in since no details on the amounts were reported.
| # | D-Fructose (g mmol−1) | Conc. (M) | Catalyst (mol% or wt%) | Reaction conditions | Cat. reuse (%) | Yield (%) | E-f | PMI | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Isolated yield. b Analytical yield. c Amount of some work-up/purification materials not included due to missing amount in the article. d In parenthesis are reported the values without taking into consideration EtOAc and water for extraction/purification. | |||||||||
| One pot, hydrothermal | |||||||||
| 1 | 20/111.1 | 0.69 | Amberlite IR 120 H 25 wt% | KBr, DMSO; 120 °C, 12 h | 7 | 73a | 21.2 | 21.7 | 49 |
| 2 | 0.2/1.11 | 0.56 | V–g-C3N4 (H+) 50 wt% | (i) N2, DMSO; 120 °C, 2 h; (ii) O2 1 bar, 130 °C, 6 h | 6 | 45b | 39.3 | 38.7 | 50 |
| 3 | 0.05/0.3 | 0.28 | TFP-DABA 10 mol% | KBr, DMSO; 100 °C, 12 h | — | 65b | 55.1 | 56.1 | 51 |
| 4 | 0.4/2.22 | 1.31 | HPMoV 7 wt% | ChCl, DMSO (65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35); 120 °C, 14 h :35); 120 °C, 14 h |
— | 77b | 62.3 | 63.3 | 52 |
| 5 | 11.3/62.4 | 1.24 | (i) BioRad AG 50W-X8 44 wt%; (ii) VOHPO4, 0.5H2O 5 mol% | (i) DMSO, 110 °C, 5 h; (ii) air 1 bar, 150 °C, 13 h | — | 41a,c | 147.5 | 148.5 | 48 |
| 6 | 0.18/1.0 | 0.5 | NaBr 18 wt% | DMSO; 150 °C, 23 h | — | 67b,d, 50a | 1510.7 (28.3), 2024.7 | 1511.7 (29.3), 2025.7 | 47 |
| Two steps, hydrothermal followed by photocatalysis | |||||||||
| 7 | 0.3/1.7 | 0.3 | HBr 40 wt% | (i) DMSO, 120 °C, 2 h; (ii) O2 1 atm, 80 °C, 8 h, under visible light exposure | — | 80b | 120.2 | 121.2 | 53 |
More recently, Chauhan and co-workers reported a similar approach using up to 20.0 g of D-fructose in the presence of a commercial sulfonated polystyrene based Amberlite (IR120 H) alongside KBr and DMSO as media (#1; Table 3).49 DFF yield was 73% after extraction with ethyl acetate and diethyl ether solution. It should be mentioned that Amberlite was recovered and regenerated. Evaluation of the green metrics E-f and PMI indicated that this procedure is the most efficient among the one reported although it was not possible to evaluate the required volume of extraction medium since this information was not mentioned in the publication.
In other reported procedures custom-made catalysts were mostly used, i.e., vanadium-doped g-C3N4 (#2; Table 3),50 sulfonated 2-dimensional covalent organic framework (TFP-DAB) (#3; Table 3)51 and Mo–V containing Keggin HPA (#4; Table 3).52 Related PMI values were always quite high also considering that in some cases DFF was not isolated.
An interesting procedure employ a metal-free catalytic system (#6; Table 3)47 where DMSO/halide (as salts or acids) system can promote the formation of DFF; best results were achieved using NaBr.
The obtained DFF yield was of 67% (50% isolated DFF yield). Considering the DFF extraction/purification, the PMI and E-f value are ultimately very high. In this case optimization of the purification procedure might largely reduce these values, in fact when the amount of EtOAc and water used for DFF isolation was not considered in the calculation the resulting E-f value was 28.3.
The thermo-catalytic one-pot approaches discussed are intriguing, however they typically necessitate high temperatures, prolonged operational durations, and, in certain instances, elevated pressures and gas inputs. Given these considerations, future research on this synthetic methodology should aim to mitigate these challenges.
An alternative approach to increase the sustainability and economic feasibility of this chemical transformation could be employing photochemistry.
In fact, Hu et al. demonstrated that by using HBr in DMSO in a two-step process 80% DFF yield was achieved.53 Bromide and the photogenerated O2˙− anion radicals were responsible for the photocatalytic partial oxidation of HMF (#7; Table 3). The calculated E-f value is 120.2, but without evaluating separation/purification procedures. To the best of our knowledge, no further works for the one-pot photocatalytic conversion of D-fructose or other carbohydrates to DFF exist, however further investigations and optimization on this synthetic approach would be pivotal in pushing forward the greenness of this bio-based monomer.
On the other hand, the number of articles regarding the photocatalytic or photo-assisted conversion of HMF to DFF show an incremental trend in the last two decades. Some examples for best-performing procedures using water or different organics as solvents are collected in Table 4.
| # | HMF (g mmol−1) | Conc. (M) | Catalyst (wt%) | Light source | Reaction conditions | Cat. reuse | Yielda (%) | E-fb | PMIb | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a All yields are determined via analytical methods. b Values without considering possible isolation/purification of DFF, separation of the solid catalyst and separation/purification of solvent(s). c Unknown amount of O2 employed. | ||||||||||
| 1 | 0.25/2.0 | 0.1 | CdS QDs/Ch 20 | Light source 100 mW cm−2 | O2 10 mL min−1, ACN; 20 °C, 10 h | 5 | 79 | 83.7 | 84.4 | 54 |
| 2 | 0.06/0.5 | 0.05 | Au–Ru/rGO 63 | 300 W Xe lamp 0.5 W cm−2 | O2 5 bar, toluene; 80 °C, 8 h | 5 | 91 | 150.4 | 150.5 | 55 |
| 3 | 0.02/0.1 | 0.03 | HBr 40 | Tungsten-bromine lamp 35 W | O2 1 bar, DMSO; 80 °C, 8 h | — | 89 | 378.7 | 379.7 | 53 |
| 4c | 0.01/0.1 | 0.01 | CTF-Th@SBA-15 8 | Blue LED lamp 460 nm, 65 mW cm−2 | O2, H2O; r.t., 30 h | 4 | 56 | 1432.1 | 1431.7 | 56 |
| 5 | 0.006/0.05 | 0.005 | MXene/g-C3N4 1587 | Visible light | Air, TFT; r.t., 10 h | 4 | 90 | 2128.1 | 2111.1 | 57 |
| 6 | 0.001/0.01 | 0.0005 | MnO2 nanorods 1587 | InGaN based single chip LEDs 375 nm | CAN, r.t., 6 h | — | 98 | 12903.3 |
12904.3 |
58 |
| 7c | 0.009/0.08 | 0.0005 | g-C3N4 5291 | Solar light | O2, H2O; 39 °C, 4 h | 4 | 49 | 33614.5 |
33604.3 |
59 |
| 8 | 0.0001/0.0005 | 0.0001 | Bi2WO6/mpg-C3N4 79365 |
Xenon lamp 300 W, λ > 400 nm | O2 10 mL min−1 H2O; r.t., 8 h | 5 | 50 | 330672.3 |
329060.4 |
60 |
It can be observed that the initial HMF solution concentrations (CHMF,in) and volumes are in the range from 0.1 to 80 mM and from 5 to 150 mL, respectively (#1–8; Table 4). Hence, the used HMF initial mass is far away from the “gram” scale. It should be considered that in these selective photo-oxidation processes, no additional chemicals are required and in most cases no heating was applied. Hence, it is very easy either to repeat the experiments many times or to upscale them to increase the final production of DFF. Even more interesting is that these materials can be used for batch application but also for continuous flow setups.
Regarding the solvent selection, the use of acetonitrile (#1; Table 4), toluene (#2; Table 4), DMSO (#3; Table 4) and trifluorotoluene (TFT) (#5; Table 4) led to higher yield of DFF compared to the trials conducted in water (#4, #7 and #8; Table 4).
Regarding PMI green metric for the photo-oxidation of HMF to DFF, the best value was 84.4 while the highest was close ca. 330000 (#1 and #8; Table 4, respectively). This deviation depends predominately on the CHMF,in used in the above mentioned experiments. Considering that the solid catalyst can be separated by simple filtration while the main volume of the solvent can be also recovered by distillation, the very high values of the E-f can be potentially reduced.
In the overall evaluation of the process, it should be mentioned the very low energy consumption of the photocatalytic process compared to the thermocatalytic ones. Besides, the photocatalytic methods, various thermo-catalytic ones were also reported, while few works regarding electro-, sono-, and bio-catalytic as well as microwave-based approaches61 are also available in the literature although in the scale of milligram synthesis.62–67 Especially in the case of electrocatalytic HMF oxidation, the goal in all cases is to achieve full oxidation to FDCA and simultaneous H2 production by water splitting in the other electrode.62
In conclusion, the main drawback in most of presented research works related to DFF synthesis is that isolation/purification of the product as well as purification/regeneration of the used solvents and/or catalysts were rarely addressed. Furthermore, in some cases the use of additional reagents led to unclear side-effects. For instance, the use of strong acids like HCl or H2SO4 for the homogeneous conversion of D-fructose induce difficulties for purification of the products, solvents, and recycling/poisoning the catalyst. Analogues problems arisen using Amberlyst-15, that requires separation prior the oxidation step, while DMSO-derived compounds affect the process towards DMSO recycling and DFF purification.47
In general, further oxidation of DFF can also take place, leading to the formation of products mixture that complicate the DFF separation/purification process. The formation of DFF oxidized products is generally promoted at higher temperature and by tuning the solvent, hence these parameters among other should be always properly optimized.68 It should be mentioned that due to D-fructose dehydration to HMF, the water formed can lead to unselective reactions. In many cases it was also noted the gradual reduction of catalyst performance because of formation of side-products such as humins. The regeneration of catalysts requires high amount of energy, as mentioned by Yang et al. in the case of Ce–Mo mixed oxides catalyst, were regeneration required calcination at 600 °C.69
2.4. Bio-based polymers from aldehyde furanic monomers
Due to their high reactivity, aldehyde functions are very attractive for designing polymeric structures. As an example, under reductive coupling, DFF can lead to the formation of polypinacols.70 However, the stiffness of the furanic units does not allow reaching high molecular weight as it is also observed for the reductive coupling of terephthaldehyde.Bio-based furan dialdehydes have been exploited for preparing dynamic covalent polymeric networks based on imine bonds (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) obtained by condensing the corresponding aldehyde groups with different polyamine end-groups (Fig. 3).71 The synthesis of condensation polymers between DFF and different amines was first reported in the nineties.72,73 However, furan-based dialdehydes high cost and availability have hindered their development. The plethora of new synthetic methods (see Section 2.3) for the preparation of furan-based aldehydes may pave the way for developing further their use for the synthesis of novel polymers. In fact, materials based on furanic dialdehydes present a unique set of properties in comparison with traditional furanic polymers; the dynamic nature of imine functional groups allows them to be rearranged under external stimuli, such as temperature, pH or light irradiation.74 This dynamic nature has a deep implication not only in the intrinsic properties of the material but also in their reusability and recyclability enabling, under certain conditions, thermoset-like architectures to be reprocessed or recycled potentially increasing their sustainability and reducing end-of-life plastic waste. Thus, they can be classified as materials to bridge the gap between thermosets and thermoplastics by combining the advantages of both worlds.
N–) obtained by condensing the corresponding aldehyde groups with different polyamine end-groups (Fig. 3).71 The synthesis of condensation polymers between DFF and different amines was first reported in the nineties.72,73 However, furan-based dialdehydes high cost and availability have hindered their development. The plethora of new synthetic methods (see Section 2.3) for the preparation of furan-based aldehydes may pave the way for developing further their use for the synthesis of novel polymers. In fact, materials based on furanic dialdehydes present a unique set of properties in comparison with traditional furanic polymers; the dynamic nature of imine functional groups allows them to be rearranged under external stimuli, such as temperature, pH or light irradiation.74 This dynamic nature has a deep implication not only in the intrinsic properties of the material but also in their reusability and recyclability enabling, under certain conditions, thermoset-like architectures to be reprocessed or recycled potentially increasing their sustainability and reducing end-of-life plastic waste. Thus, they can be classified as materials to bridge the gap between thermosets and thermoplastics by combining the advantages of both worlds.
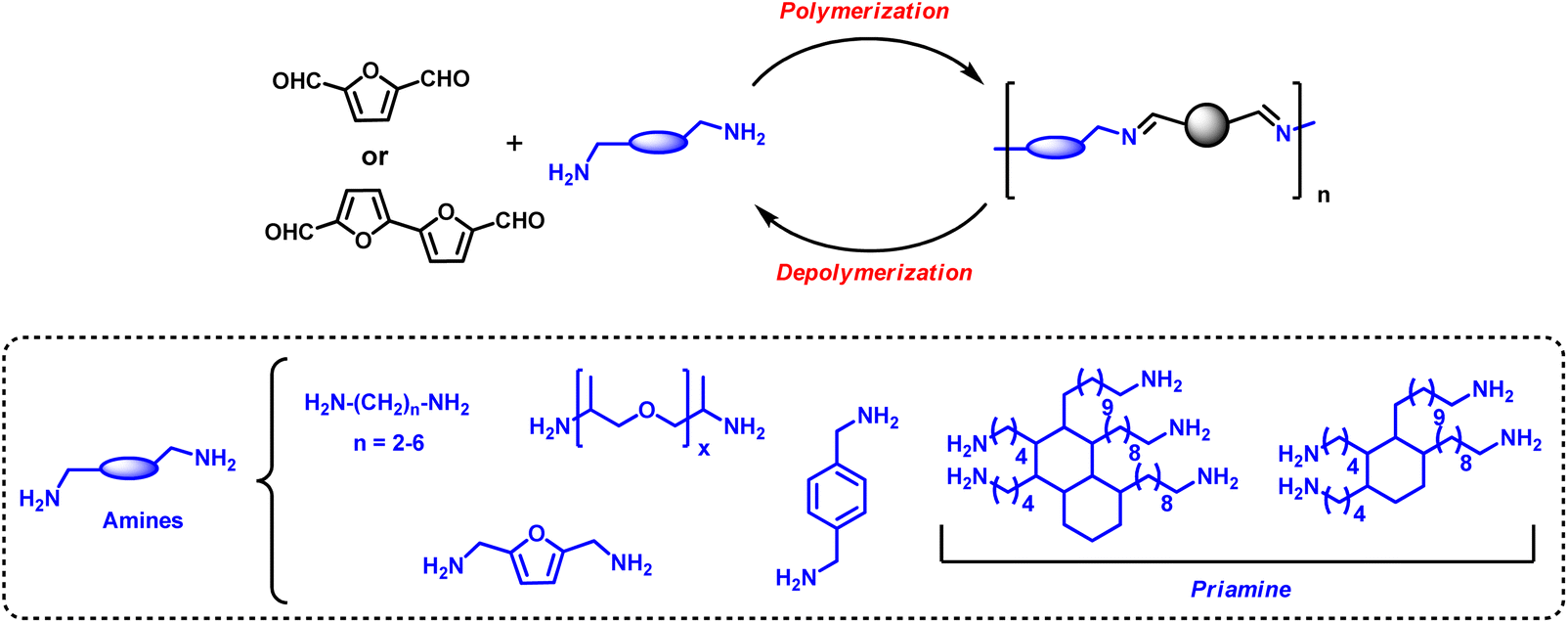 | ||
| Fig. 3 Bio-based poly-furanic imines using furan based dialdehydes. | ||
The condensation of the aldehydes and amines can be performed in solution generally yielding to the precipitation of the polymer or alternatively under solvent-free conditions (ESI; Table S.12†).
The nature of the polymers highly depends on the type of amine used, besides, by controlling the ratio of dialdehyde diamine, these Schiff base macromolecules can yield telechelic polymers with either –CHO or –NH2 functional groups. These reactive end-groups can also be exploited for the synthesis of more complex polymeric networks by post-functionalization or cross-linking.
It should be mentioned that the dynamic nature of the imine form results in the linear chains of different lengths as the thermally favoured products, but the condensation can also lead to a small fraction of cyclic oligomers.75
Kenichi Kasuya et al. has synthesised furan-based poly(Schiff base) by polycondensation of dimeric 2,2′-bifuran (BiFur) with different diamines (1,2-ethylenediamine, 1,3-propanediamine, 1,4-butanediamine, 1,5-pentanediamine, 1,6-hexanediamine and p-phenylenediamine) in absence of the solvent or using m-cresol as solvent at 140 °C for 6 h.76
Bifurfural – namely 2,2′-bifuran – was prepared from furfural in 60% yield by oxidative homocoupling catalysed by palladium acetate. The DSC traces reveal that the Tg of poly(Schiff base)s depended on the length of the alkyl of the aliphatic diamine decreasing with the increase of the alkyl length, whereas the material derived from aromatic amine did not exhibit any Tg. The 1H NMR, IR, and MALDI-TOF-MS spectra of the poly(Schiff base)s revealed the presence of thermally stable cyclic structures as a result of the dynamic nature of the imine group. These cyclic structures act as plasticizers that affect the mechanical properties differently. The polymers were moldable at 120 °C, and the molded films presented good mechanical properties being tough to bend and strong with Young's moduli around 1 GPa.
Avérous et al. have developed 100% renewable vitrimer elastomeric film materials obtained by casting and curing at 120 °C different mixtures of 2,5-furandicarboxaldehyde and different bio-based diamine and triamine prepared from fatty acids (commercialised as Priamine 1071 by Croda) in THF in the absence of a catalyst.77 An excess of amines with respect to the DFF 1:1.2 molar ratio was used to get faster stress relaxation times. The swelling of the films was tested by immersing them for two days at 25 °C in different solvents (THF, EtOH and DMF). The polyimine, although it swelled in the organic solvents, remained insoluble after a prolonged time of up to several months. The reversible nature of the polymeric network was probed by exposing the material to an excess of butylamine in THF at room temperature resulting in its complete dissolution and total depolymerisation of the network. This opens the possibility of recycling the polymer under controlled conditions.
Indeed, the properties of the polymer reprocess throughout the successive recycling steps by dissolution and repolymerisation remained almost unchanged with an elongation at break ranging from 24 to 21%, a Young's modulus from 4.4 to 5.1 MPa, and a stress at break ranging from 0.69 to 0.73 MPa.
Different vitrimers can be prepared by changing the 2,5-furandicarboxyaldehyde/amine ratio together with the type of diamine (Jeffamine D-230, D-400 or D-2000) or triamine (Jeffamine T-403) employed. By adjusting the composition, a set of polymeric networks having similar chemical structures, but varying crosslinking densities were obtained.78 The crosslinking density was found to have a drastic influence on the relaxation time, which can decrease by a 20-fold factor when it is reduced. This point is highly relevant because the control of the relaxation time of vitrimers is key to facilitating their recycling or processing by common industrial techniques such as extrusion. For the more crosslinking systems an increase in mechanical properties was also observed. This phenomenon could be attributed to an increase in the vitrimer crosslinking density, caused by the multiple processing cycles, which raises the stiffness after each healing step.
The furanic rings of the polymer-imine vitrimers obtained by condensation 2,5-furandicarboxaldehyde and oligo-ether-amine (Jeffamine D400, Mn ≈ 430 g mol−1) can also be exploited to design crosslinked covalent adaptable networks by Diels–Alder reactions.79 The two electron-donating imine groups provide the furan ring with suitable reactivity leading to extremely fast crosslinking with maleimide groups of 4,4′-methylenebis(N-phenylmaleimide) used as crosslinked agents. These materials exhibit tuneable physical properties depending on the amount of bismaleimide used for crosslinking. Increasing the crosslink density leads to an increase in the Tα, tensile strength, and Young's modulus. Besides, the combination of different types of dynamic linkages as well as their potential synergistic effect opens the possibility of a fast reprocessability of the polymeric network at low temperatures and a preservation of their mechanical properties.
Conjugated Organic Polymers (COPs) were also prepared by coupling 2,5-furandicarboxaldehyde with pyrrole. The resulting porphyrinic structure can particularly be efficient for removing CO2 or radioactive iodine (with a capture capacity adobe 650 mg g−1).80
3. Diol- and polyol-based monomers
This section focuses on the synthesis of diols and polyols primarily derived from HMF. These furanic-based alcohols can be utilized in the creation of innovative polymers, offering an alternative to traditional alcohols such as 1,4-butanediol or 1,6-hexanediol. The incorporation of the furanic ring may provide the resulting polymers with distinct characteristics in terms of enhanced structural properties.3.1. Synthetic approaches to 2,5-bis(hydroxymethyl)furan
2,5-Bis(hydroxymethyl)furan (BHMF) is one of the most investigated furanic monomer since it can be easily prepared via HMF hydrogenation. A number of synthetic strategies have been developed in order to selectively promote hydrogenation avoiding eventual hydrogenolysis, i.e., elimination of the oxygen-containing moiety, of this molecule.81,82Table 5 (see also ESI; Tables S.3 and S.4†) report the main procedures to BHMF published in scientific journals and patents categorized according to the synthetic approach used and ranked according to the best PMI values.
| # | Substrate (g mmol−1) | Conc. (M) | Catalyst (mol% or wt%) | Reaction conditions | Cat. reuse | Yield (%) | E-f | PMI | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Isolated product. b Amounts related to the purification of BHMF are not reported. c Determined product via analytical method. d Catalyst amount not included. e Time was not reported in the original procedure. | |||||||||
| Cannizzaro reaction | |||||||||
| 1 | HMF (0.1/0.79) | 0.2 | — | H2O; NaOH; Na2S2O4 0 °C, 1 h | — | 47a | 18.2 | 19.2 | 84 |
| 2 | HMF (12/95.16) | 0.48 | — | H2O; NaOH; r.t., 18 h, | — | 85a | 19.8b | 20.8b | 83 |
| Metal catalysed hydrogen-transfer | |||||||||
| 3 | HMF (5/39.65) | 0.42 | MgO (10 wt%) | i-PrOH; 170 °C, 5 h | — | 85c | 17.6 | 18.5 | 85 |
| 4 | HMF (0.50/3.97) | 0.40 | NC-MgAlO (20 wt%) | Cyclohexanol; 220 °C, 0.5 h | 5 | 100c | 19.1 | 19.9 | 86 |
| 5 | HMF (0.32/2.50) | 0.25 | ZrPN (30 wt%) | i-PrOH; 140 °C, 2 h | — | 98c | 25.5 | 26.2 | 87 |
| 6 | HMF (0.13/0.10) | 0.25 | Hf-MOF-808 (10 mol%) | i-PrOH; 100 °C, 1.5 h | — | 95c | 26.2d | 27.2d | 88 |
| 7 | HMF (0.13/1) | 0.20 | FDCA-Hf (16 mol%) | i-PrOH; 100 °C, 5 h | — | 95c | 33.0 | 33.4 | 89 |
| 8 | HMF (0.25/2) | 0.20 | m-PhPZr (56 wt%) | i-PrOH; 120 °C, 2 h | — | 93c | 33.8 | 34.8 | 90 |
| Hydrogenation reaction | |||||||||
| 9 | HMF (0.5/3.97) | 1.99 | Pt/MCM-41 (20 wt%) | H2, 8 bar; H2O; 35 °C, 2 h | 6 | 98c | 4.2 | 5.0 | 91 |
| 10 | HMF (10/79.37) | 1.59 | Cu/SiO2 (5 wt%) | H2, 25 bar; MeOH; 100 °C, 8 h | — | 97c | 4.1 | 5.1 | 92 |
| 11 | HMF (0.38/3) | 1.00 | Ir–ReOx/SiO2 (13 wt%) | H2, 8 bar; H2O; 30 °C, 6 h | — | 99c | 8.3 | 9.2 | 93 |
| 12 | HMF (0.13/1) | 0.52 | Au/SiO2 (1 mol%) | H2, 6 bar; i-PrOH; 2,4,6-collidine; 100 °C, 24 h | 5 | 99c | 12.8 | 13.8 | 94 |
| 13 | HMF (0.2/1.59) | 0.79 | NiRe2 (5 wt%) | H2, 50 bar; H2O; BuOH; 40 °C, 4 h | — | 72c | 15.0 | 16.0 | 95 |
| 14e | HMF (0.25/1.98) | 0.99 | RuCl2(NNN)(PPh3) (0.05 mol%) | H2, 10 bar; t-BuOK; THF; 80 °C | — | 52a | 18.7 | 19.7 | 96 |
| 15 | HMF (1.50/11.9) | 0.34 | Cu–ZnO (33 wt%) | H2, 15 bar; dioxane; 100 °C, 2 h | — | 99c | 24.5 | 25.5 | 97 |
| Integrated synthetic procedures | |||||||||
| 16 | D-Fructose (10/55.6) | (i) 0.70; (ii) 0.43 | Amberlyst-15 (10 wt%) | (i) TEAB, DMC; 90 °C, 16 h; (ii) NaBH4, THF; r.t., 16 h | — | 72 | 66.9 | 67.9 | 18 |
| 17 | D-Fructose (3.6/20) | 1.82 | Iridium catalyst (0.5 mol%) | (i) DMSO, THF, HCOOH; 150 °C, 5 h; (ii) THF; HCOOH, Et3N; 40 °C, flow = 2.5 mL h−1 | — | 71 | 121.9 | 122.9 | 98 |
BHMF main synthetic approaches include (i) Cannizzaro reaction (#1 and #2; Table 5); (ii) hydrogen transfer reaction (#3–8; Table 5) and (iii) metal catalysed selective hydrogenation (#9–15; Table 5). Other syntheses include integrated approaches starting from carbohydrates (mainly D-fructose, #16 and #17; Table 5) and some miscellaneous procedures such as DFF reduction (Scheme 4).
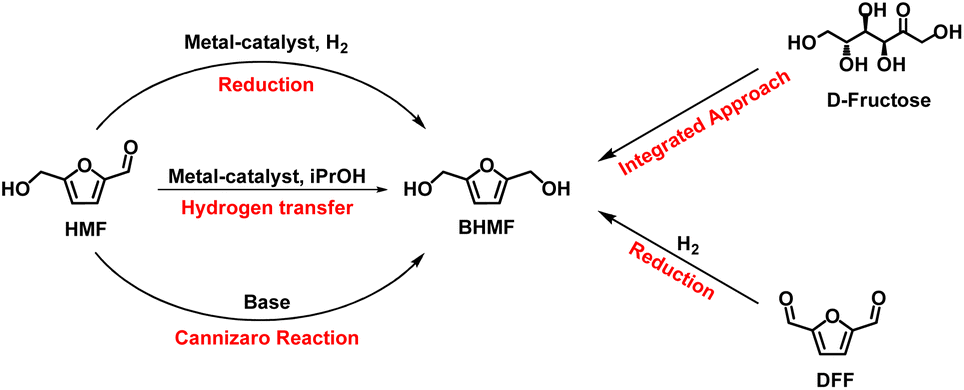 | ||
| Scheme 4 Synthetic pathways to BHMF. | ||
Cannizzaro reaction (Cann.) is a base-induced aldehyde disproportionation where an aldehyde acts as a hydride donor towards a second aldehyde molecule (acceptor). When HMF is used as substrate, BHMF and 5-hydroxymethylfuranoic acid (HMFA) (Scheme 4) form as products; the latter being also an interesting monomer for bio-based polymers.
In the procedure reported by Afonso and co-workers the reaction starting from 12.0 grams of HMF, BHMF was isolated via organic solvent/water extraction. PMI and E-factor were moderate (#2; Table 5) also considering that catalyst recycle was not viable.83
Metal catalysed hydrogen-transfer (CHT) reaction employs alcohols – generally isopropanol – as hydrogen donors. According to the catalyst used and to the reaction conditions, the reaction can occur via direct hydrogenation transfer or metal hydride route. In the first pathway, hydrogen transfer occurs directly from alcohols to carbonyl groups via a six-membered cyclic transition state (Meerwein–Ponndorf–Verley mechanism). Meanwhile in the metal hydride route, the dehydrogenation of alcohol occurs on metallic sites to form H atoms that are then added on the carbonyl moiety.81
CHT approach has been extensively investigated for the conversion of HMF into BHMF (#3–8; Table 5; ESI, Table S.3†). Most of the procedures are based on custom-made catalyst, required high temperatures (100–170 °C), relative short reaction time (<5 hours) and they all led to BHMF in very good yield.
Unfortunately, all these procedures assessed BHMF yield by analytical methods without isolating the product. Considering the related E-factor and PMI values, the best synthesis so far reported is the one proposed by Rode and co-workers based on a magnesium oxide (MgO) catalyst prepared by precipitation from magnesium nitrate and potassium carbonate (#3; Table 5).85 Even though in this work catalyst recycling was only tested on furfural reduction, E-factor and PMI resulted lower than the procedure developed by Li and coworkers where the nitrogen-doped carbon (NC)-decorated copper catalyst was recycled up to 5 times.86 It is also noteworthy that the synthesis based on MgO catalyst is the only one – among the procedure employing the CHT approach – conducted on 5.0 grams scale of HMF. As above stated BHMF was not isolated as pure product from the reaction mixture.
In conclusion, E-factor values are generally quite high and in any case within the ones related to fine chemical productions (5–50 kg kg−1).
Numerous metal-based catalytic systems have been investigated for the synthesis of BHMF via selective hydrogenation reaction (H); in most of the cases studied the reaction conditions are quite mild requiring a reaction temperature below 100 °C HMF (#9–15; Table 5).
Precious metal catalytic systems showed high efficiency in BHMF preparation. When Pt/MCM-41 was used as catalyst (#9; Table 5), the reaction was conducted in aqueous media in mild conditions (35 °C for 2 h). Although no information was reported on the isolation of BHMF as pure product, the catalyst was recycled 6 times. As a result, the related PMI and E-factor values were the best among the procedures so far reported (PMI = 5.0). Similar results in terms of green metrics were achieved by Mu and co-workers that employed a less expensive Cu/SiO2 catalytic system, obtaining BHMF in almost quantitative yields (#10; Table 5).
Few integrated synthetic procedures have been also reported (#16 and #17; Table 5) where starting from a simple carbohydrate is possible to achieve BHMF by a sequence of dehydration and reduction reaction. In these cases, as expected, the green metrics are quite high due to the more complex synthetic procedure.
In conclusion, without considering the lack of investigations on the isolation and or purification of BHMF, hydrogenation reaction seems to be the most convenient synthetic approach to this bio-based platform chemicals for their simplicity, mild reaction conditions, high yields and lower mass consumption of solvent and catalyst. Further investigations on integrated one-pot direct conversion of D-fructose into BHMF are highly desirable as they avoid all the stability issue related to HMF synthesis and/or purification.
3.2. Synthetic approaches to BHMTHF
Table 6 summarizes the synthetic procedure for the preparation of 2,5-bis(hydroxymethyl)tetrahydrofuran (BHMTHF) via total hydrogenation of HMF or BHMF (Scheme 5). This reaction generally requires harsher reaction conditions compared to the simple reduction of the HMF aldehyde moiety. As a result, to the best of our knowledge only one example of bio-based polymer was reported starting from this monomer (Section 3.5.3) although numerous investigations have been reported for the synthesis of this monomer.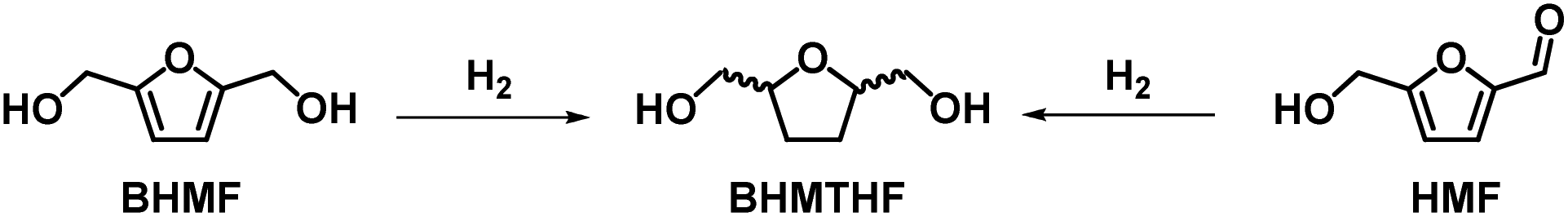 | ||
| Scheme 5 Synthetic pathways to BHMTHF. | ||
| # | Substrate (g mmol−1) | Conc. (M) | Catalyst (mol% or wt%) | Reaction conditions | Yield (%) | Cat. reuse | E-f | PMI | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Isolated product. b Determined via analytical method. | |||||||||
| 1 | HMF (1.8/14.28) | 1.43 | RANEY® Ni (6 wt%) | H2, 50 bar; EtOH; 120 °C, 3 h | 80a | — | 5.6 | 6.6 | 99 |
| 2 | HMF (0.63/5.0) | 0.56 | Pd–Ir/SiO2 (24 wt%) | H2, 80 bar; H2O; 2 °C, 4 h | 95b | 4 | 16.6 | 17.3 | 101 |
| 3 | HMF (0.63/5) | 0.56 | Ni–Pd/SiO2 (16 wt%) | H2, 80 bar; H2O, AcOH; 40 °C, 2 h | 96b | — | 17.9 | 18.9 | 102 |
| 4 | HMF (1.20/9.5) | 0.34 | Ru/CeOx (17 wt%) | H2, 28 bar; H2O, BuOH; 130 °C, 12 h | 91b | — | 18.9 | 19.9 | 103 |
| 5 | HMF (1/7.93) | 0.26 | Ru/MnCo2O4 (2 wt%) | H2, 82 bar; MeOH; 100 °C, 16 h | 97b | 4 | 24.5 | 25.1 | 104 |
| 6 | HMF (1.5/11.9) | 0.34 | RANEY® Ni (33 wt%) | H2, 15 bar; 1,4-dioxane; 100 °C, 30 h | 96b | — | 24.4 | 25.4 | 105 |
| 7 | HMF (1.5/11.9) | 0.34 | NiAl-450 (7 wt%) | H2, 60 bar; 1,4-dioxane; 60 °C, 6 h | 96b | — | 24.4 | 25.4 | 106 |
| 8 | HMF (0.25/2.0) | 0.40 | Ni/C (20 wt%) | H2, 20 bar; H2O; 140 °C, 5 h | 79b | — | 24.8 | 25.8 | 107 |
| 9 | HMF (0.3/2.38) | 0.25 | 1Ru1Pd/RGO (20 wt%) | H2, 10 bar; H2O; 20 °C, 8 h | 93b | 6 | 33.5 | 34.3 | 108 |
| 10 | HMF (0.12/1.0) | 0.5 | Ru/C (16 wt%) | H2, 40 bar; H2O; 140 °C, 2 h | 48b | 4 | 33.9 | 34.6 | 109 |
| 11 | HMF (1.0/7.93) | 0.20 | 10Ni15Ce/γ-Al2O3 (10 wt%) | H2, 50 bar; BuOH; 190 °C, 3 h | 88b | — | 35.8 | 36.8 | 110 |
| 12 | HMF (0.12/1) | 0.50 | Pd/C (0.2 mol%) | H2, 40 bar; H2O, AcOH; 120 °C, 1 h | 42b | — | 39.7 | 40.7 | 111 |
| 13 | HMF (0.5/3.97) | 0.13 | RANEY® Ni (10 wt%) | H2, 90 bar; EtOH; 100 °C, 14 h | 99b | — | 46.9 | 47.9 | 100 |
| 14 | HMF (0.06/0.5) | 0.17 | Pd@mpg-C3N4 (48 wt%) | H2, 10 bar; H2O; 60 °C, 4 h | 96b | 5 | 48.5 | 49.1 | 112 |
| 15 | HMF (1.0/7.94) | 0.16 | Ru/C (5 wt%) | H2, 70 bar; H2O; 100 °C, 4 h | 95b | 6 | 52.3 | 53.2 | 113 |
| 16 | D-Fructose (0.18/1) | 0.1 | Ru/SiO2-TM (17 wt%), Amberlyst-15 (22 wt%) | H2, 40 bar; cyclohexane, H2O; 130 °C, 4 h | 46 | 5 | 136.9 | 136.8 | 114 |
| 17 | D-Fructose (0.18/1) | 0.1 | Nb2O5-FP (22 wt%), Ru/SiO2-TM (17 wt%) | H2, 40 bar; cyclohexane, H2O; 160 °C, 4 h | 40 | — | 157.7 | 158.7 | 109 |
It should be mentioned that BHMTHF comprises three possible products due to the formation of cis/trans isomers. In some procedures the cis/trans ratio is indicated, meanwhile in other cases it is disregarded especially if BHMTHF was then directly used for other chemical transformations such as for the preparation of γ-butyrolactone.99
Table 6 report only the synthetic procedures that led to E-factor and PMI with values <55 (when HMF is the starting substrate); other relevant syntheses are reported in ESI (Table S.5†).
Precious metals, used as single metal catalyst (Ni, Ru, Pd) or in combination with other metals were investigated as promoters of the reaction resulting in very good yields. All the reactions were performed in a polar solvent at relatively low concentration.
In terms of green metrics, the procedure reported by Zhu and co-workers resulted in a PMI of 6.6 kg and thus a E-factor of 5.6 kg of waste for kg of product (#1; Table 6). Recycling of the catalyst or solvent was not investigated.
This procedure employs HMF as starting compounds and takes inspiration from a similar one previously published by Heeres and co-workers (#13; Table 6) indicating that the BHMTHF cis/trans ratio was of 98/2.100
In some cases, simple carbohydrates – mainly D-fructose – were employed as starting materials to achieve one-pot dehydration reaction followed by complete reduction to BHMTHF. As expected in these cases the related green metrics had quite high values (#16 and #17; Table 6).
It should be also mentioned that some interesting studies have been conducted on the conversion of BHMFTHF into 1,6-hexanediol a high value chemical extensively used as a monomer for polyesters and polyurethanes.120
Despite the extensive investigations conducted for this reduction and the high yields of the desired product(s) the main issue of these procedures is the small scale of the reactions. Furthermore, excluding one examples BHMTHF yields were always evaluated by analytical methods; isolation was only rarely addressed. In order to exploit this compound as monomer for bio-based polymers both purification procedure and large scale reaction should be properly addressed.
3.3. Acetals based diol- and polyol-furanic monomers
Furanic diols can be also easily achieved by formation of acetal between a polyol (glycerol, pentaerythritol, etc.) and a renewable aldehyde (i.e., vanillin or HMF) or ketone (i.e., levulinic acid).121aFive-membered acetals are usually formed as the kinetically preferred products while six-membered ring acetals are strongly affected by thermodynamic control.121b
Scheme 6 depicts the structure of different monomers obtained as acetals derivatives of HMF. In this regard, Table 7 shows some of the most relevant procedures and catalysts reported for the synthesis of acetals derivatives.
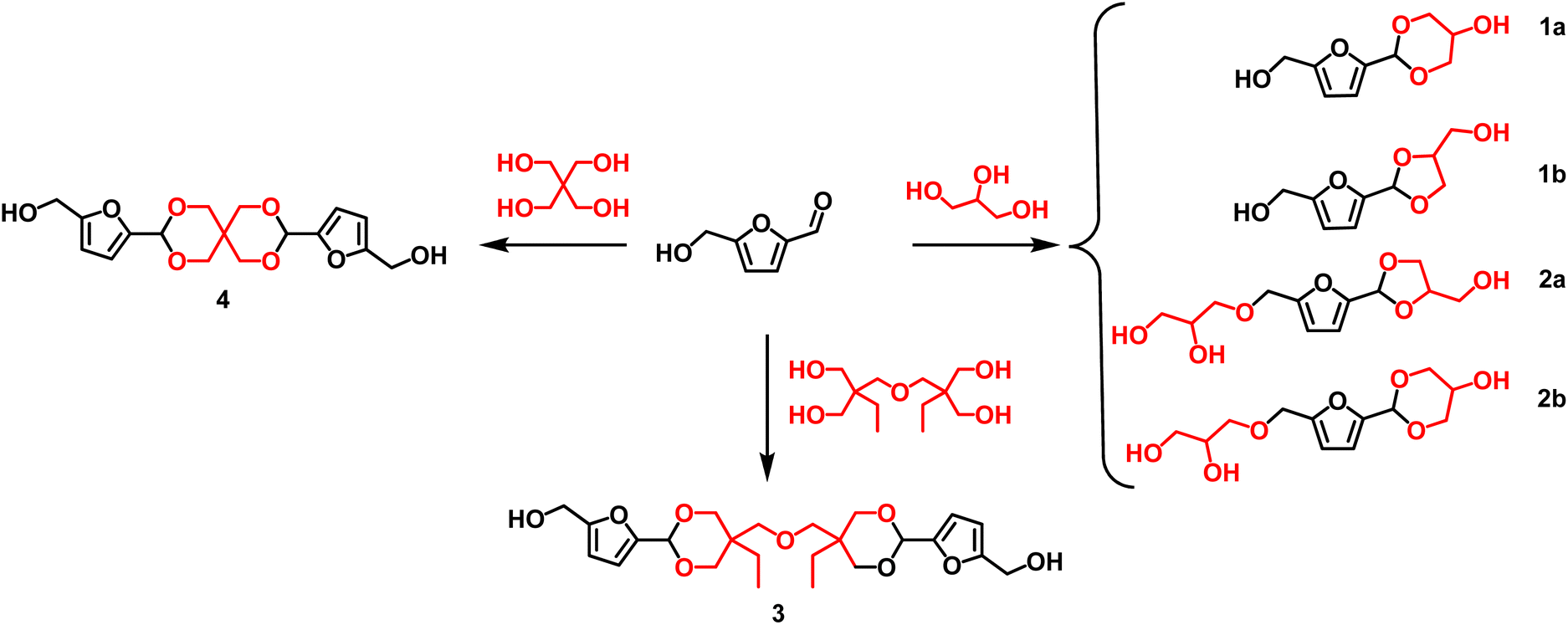 | ||
| Scheme 6 Cyclic acetals obtained from condensation of HMF and polyalcohols. | ||
| # | HMF (g mmol−1) | Polyalcohol (g mmol−1) | Conc. (M) | Catalyst (wt%) | Reaction conditions | Yield (%) | E-f | PMI | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Yield refers to the mixture of acetals 1a and 1b. b Yield and metrics refer to the amount of product obtained by the same reaction repeated twice. c Isolated yield. d Catalyst was recycled 4 times. e Yield refers to the mixture of acetals 2a and 2b. f Volume of solvents not mentioned in the original paper. | |||||||||
| 1 | 19.2/200.0 | Glycerol (18.4/200) | HMF (2.5), glycerol (2.5) | p-TsOH (0.03 wt%) | Benzene; reflux, 3 h | 1a + 1b, 75a | 3.2 | 4.2 | 115 |
| 2b | 10.1/80.2 | Pentaerythritol (5.1/37.2) | HMF (2.6), pentaerythritol (1.3) | p-TsOH (10 wt%) | i-PrOH; r.t., overnight | 4, 70c | 21.3 | 22.3 | 116 |
| 3d | 0.13/1.0 | Glycerol (0.18/2) | HMF (0.2), glycerol (0.4) | ITQ-2 (20 wt%) | TFT:CAN; 83 °C, 3 h | 2a + 2b, 99e | 25.6 | 26.5 | 117 |
| 4 | 11.5/91.2 | Di-TMP (11.4/45.6) | HMF (2.28) di-TMP (1.14) | p-TsOH (1.8 wt%) | i-PrOH; r.t., overnight | 3, 74c | 128.6 | 129.6 | 118 |
| 5f | 15.8/125.4 | Glycerol (23.2/250) | — | MCM-41 (20 wt%) | TFT:CAN; 95 °C, 24 h | 2, 65 | — | — | 119 |
As an example, the reaction of HMF and glycerol in 1:1 mol ratio in the presence of p-TsOH as a homogeneous catalyst led to acetals 1a and 1b (#1, Table 7; Scheme 6). The overall yield was 75% with relatively low values for both E-f and PMI. However, the use of benzene as media also impacts on the greenness of this procedure.
A similar approach was used (#2; Table 7) for the preparation of the spiro acetal diol 4 starting from HMF and pentaerythritol was obtained (Scheme 6) in a good yield (70%).116
The high values of E-f and PMI calculated can be ascribed to the poor recyclability of the catalyst, and the relatively low concentration of the reagents used. It should be however mentioned that the solvent of this reaction was selected among different media classified as green or non-green solvents according to CHEM21 solvent recommendation by testing the ability of HMF at different temperatures to identify the best reaction conditions for the synthesis of the targeted acetal.
Life cycle analysis of this acetal diol indicated that its production may generated significantly lower greenhouse gas emissions, compared with fossil-based and even bio-based 1,3-propanediol, which is a well-recognized green diol for polymer production. But the main disadvantage is related to the use of high catalyst loading, high dilution and solvent selection employed for the preparation of these acetals which are non-practical for large-scale synthesis.
Arias and co-workers reported the use of the acidic tridirectional large pore zeolite ITQ-2 and the mesoporous aluminosilicate MCM-41 as catalyst for the mutual valorisation of HMF and glycerol to produce diols monomers.117 Excellent yields towards the formation of acetals 2a and 2b (Scheme 6) were obtained at relative mild conditions (#3; Table 7). This methodology provides high yield (98%) and selectivity (100%) to the target compound due to the presence of mild Brønsted acid sites, large pores allowing an efficient diffusion of reactants and products. Green metrics values are still quite high, even though the catalyst was reused several times. The main issue is probably related to the extraction of glycerol required to isolate the desired acetal.
The synthesis of a tailor-designed spirocyclic acetal monomer 3 was also reported by reaction of HMF and a bio-based di-trimethylolpropane (di-TMP) using mild acid-catalysed p-TsOH in 2-propanol as acceptable green solvent with good yield (74%) after a straightforward purification by precipitation in water (#4; Table 7).116 The values of E-factor were much higher than the other strategies reported previously; the use of homogeneous catalyst and the numerous extractive procedures needed to remove both the starting material and the products surely affect the greenness of this procedure.
Finally, the commercial MCM-41 selectively leads at 95 °C to corresponding acetal 2 with a yield of 65% (#5; Table 7).119 The most important requirement of the catalyst for this reaction is to adjust the acidity to avoid the polymerization of HMF while still being able to catalyse the acetalization reaction. Missing information about extraction, purification did not allow us to calculate both the E-f and PMI being unable to compare with the previous synthetic strategies.
In consideration of the easy synthetic approach to bio-derived acetal diols 1–4 these compounds were recently reported as building blocks for epoxy resins, polyurethanes and polyesters. However, the synthetic approach to these monomers has to be considered preliminary. The use of homogenous acid (p-TsOH) and poor purification procedures affect the values of PMI and E-factor. However, it should be pointed out that most reactions were conducted on multi-gram scale and this is surely an advantage for their further exploitation as bio-based monomers for polymers.
3.4. Other furanic diols
Numerous other furanic diols with potential applications as monomers for bio-based polymers were reported in the literature. As an example 5,5′-oxybis(methylene)bis(furan-5,2-diyl)dimethanol (OBMF-H) was easily synthetized by reduction of OBMF with sodium borohydride at room temperature (Scheme 7 and Table 8). | ||
| Scheme 7 Synthesis OBMF-H. | ||
The syntheses reported for OBMF-H are very similar and the product is isolated in good yield by evaporating the reaction solvent or by water/organic solvent extraction of the reaction crude. Green metrics are still high (PMI > 25), although this could be ascribed to the scarce number of investigations so far reported for this potentially very interesting bio-based monomer.
Fig. 4 depicts a library of HMF derived monomers achieved via different type of condensation reaction. Among them furoins are interesting trifunctional building blocks for bio-based polymers and biofuels. These molecules were synthetized via cross-coupling of HMF with furfural using different types of organic N-heterocyclic carbene (NHC) catalysts.123
 | ||
| Fig. 4 Other furan-diols synthetized via several type of condensation reaction. | ||
In one of the procedures reported by Chen and co-workers the C-11 furoins were isolated in ca. 20% yield (#1; Table 9). According to the reaction conditions used it was possible to synthesised selectively C-11 Furoins 1–2. The homocoupled C12 furoin (Furoin-3) is always formed in smaller amount than cross-coupled Furoin-1 and Furoin-2. As a result of the poor yields the related green metrics have very high values.
| # | HMF (g mmol−1) | Conc. (M) | Catalyst/base (mol% or wt%) | Reaction conditions | Cat. reuse | Yield (%) | E-f | PMI | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Precatalyst B = 3-(2,6-diisopropylphenly)-4,5-dimethylthiazol-3-ium perchlorate. b Isolated product. c Work-up and/or purification materials not included. d Metrics referred to the global procedure, in brackets are reported the metrics for the single reactions. e Metrics could not be calculated due to missing data in the original procedure. f Determined product via analytical method; na = not applicable. | |||||||||
| 1 | 3.4/27 | 0.22 | Precatalyst Ba (15 wt%) | FF, Et3N, EtOH; 80 °C, 24 h | — | Furoin-1 22b,c, Furoin-2 20b,c | 79.5d (151.8, 169.3) | 80.5d (152.8, 170.3) | 123 |
| 2 | 3/24 | Neat | TPT-NHC (10 mol%), KOtBu (15 mol%) | 60 °C, 1 h | — | Furoin-3, 93b,c | 0.24 | 1.24 | 124 |
| 3 | 2.5/20 | Neat | TPT-NHC (1 mol%) | 60 °C, 1 h | — | Furoin-3, 95b | 1.9 | 2.9 | 125 |
| 4 | 0.12/1 | 18 | Et3N; MMT–[BHTM] (25 mol%) | 120 °C, 3 h | — | Furoin-3, 97b,c | 2.4 | 3.4 | 126 |
| 5 | 0.25/2 | 57 | DBU (0.01 mol%) | [BMIM]Cl; 80 °C, 12 h | — | Furoin-3, 19b,c | 5.6 | 6.6 | 127 |
| 6 | 1/8 | 0.66 | Silica-g-[BI] (10 mol%), DBU (20 mol%) | HCl, THF; r.t., 24 h | 10 | Furoin-3, 83b,c | 15.7 | 14.4 | 128 |
| 7 | 0.12/1 | 0.20 | TPT-NHC (5 mol%) | THF; 60 °C, 1 h | — | Furoin-3, 87b | 49.7 | 50.7 | 129 |
| 8e | 31.2/250 | — | BAL, ThDP | K3PO4, DMSO; r.t., 18 h | — | Furoin-3, 40b | na | na | 130 |
| 9 | 0.27/2.17 | 1.00 | NaOH(aq.) (25 mol%) | H2O, acetone; 35 °C, 1.25 h | — | DHMFA, 99f | 3.1 | 4.1 | 131a |
| 10 | 0.25/2 | 2.00 | NaOH(aq.) (25 mol%) | MeOH, acetone; 24 °C, 12 h | — | DHMFA, 42b | 17.4 | 18.4 | 127 |
| 11 | 0.25/2 | 1.00 | NaOH(aq.) (25 mol%) | MeOH, butanone; 24 °C for 12 h | — | DHMFA-2, 83b,c | 7.0 | 8.0 | 127 |
| 12 | 0.25/2 | 2.00 | NaOH(aq.) (25 mol%) | MeOH, CPO; 24 °C, 12 h | — | DHMFA-3, 91b,c | 6.9 | 7.9 | 127 |
| 13 | 0.25/2 | 2.00 | NaOH(aq.) (25 mol%) | MeOH, cyclohexanone; 24 °C, 12 h | — | DHMFA-4, 89b,c | 7.0 | 8.0 | 127 |
More extensive investigations were conducted on the preparation of the HMF homocoupled C12 Furoin-3 (#2–8; Table 9) namely the 5,5-dihydroxymethyl furoin (also named DHMF, Fig. 4). This compound was achieved from HMF via several (also supported) N-heterocycle carbene (NHC) catalysts124,125,128,129 (#2, #3 and #7; Table 9), a supported ionic liquid126 (#4; Table 9), via benzoin-type condensation promoted by an organocatalyst127 (#5; Table 9), and a thiamine diphosphate dependent-enzyme BAL130 (#8; Table 9).
The product was recovered in mediocre (#8; Table 9) to excellent yield (up to 97%), and NCH catalysts were in general the most efficient promoters of the benzoin condensation.
Green metrics were in some cases very impressive with values as low as 1.24 for PMI (0.24 E-factor) for Furoin-3 when the reaction was performed in neat in the presence of TPT-NHC as catalyst. Despite the small scale of the procedures, this is – so far – the procedure with the nearest E-factor values to oil refining process (0–0.1 kg kg−1) among the herein reported bio-based monomers. In this view future investigations should be focussing on the scalability of the procedure.
Finally, it should be mentioned that Furoin-4 can be synthesised by oxidation of Furoin-3 in THF in the presence of MnO2 in almost quantitative yield.132 Similarly Furoin-5 can be achieved by reduction of Furoin-3 in the presence of NaBH4. Both these monomers have been exploited as monomer for bio-based furanic polymers (Section 3.5.3).
Di-5-hydroxymethylfurfural acetone (DHMFA), another furan diol of interest was achieved in high yield by Dumesic and co-workers by a simple aldol condensation of HMF and acetone (Fig. 6) in the presence of sodium hydroxide as strong base (#9; Table 9).131 The product can be recovered by simple filtration in water, and this explains the relatively low E-factor value.
A similar procedure was reported by Ananikov and co-workers, although the product was recovered in lower yield (#10; Table 9).127 Furthermore, these authors employed the same procedure for the synthesis of similar bifuranic structures herein indicated for simplicity as DHMFA 2–4.127
These products were obtained by di-aldol condensation of HMF with butanone, cyclopentanone and cyclohexanone respectively. In all experiments the products were isolated with high yield (83–91%) via column chromatography (#11–13; Table 9). In all cases the relatively high values of the green metrics are an indication of the only preliminary research conducted on these furanic monomers; future studies should be focussed on the optimization of the reaction conditions and of the purification procedures.
It should be finally mentioned that DHMFA can be partially (double bonds in α to the ketone moiety) or completely (reduction of the furan to THF) hydrogenated to produce other aromatic (DHMFA-H) or aliphatic furanic diols that can be used as polymer precursors.131 Other synthetic approaches to DHMFA either miss data to allow metrics evaluation or have E-factor/PMI > 50 are reported in ESI (Table S.6†).
3.5. Bio-based polymers from furanic diols
Melting point, boiling point and eventual degradation temperature are key parameters to evaluate the use of the monomers in different type of polymerisation. In this view we have collected the physical chemical data of the furan diols herein discussed (ESI; Table S.1†). In some cases, data were not available in the literature which is an issue for their further exploitation as potential monomers to bio-based polymers.Numerous investigations have been reported on FDCA-based polyesters (PES) including most recent work on recyclability.9 On the other hand research on polyester from BHMF is still rather limited (Fig. 5). The main reason of the scarce number of reports can be ascribed to the low thermal stability of this monomer that start degrading at ca. 160 °C.
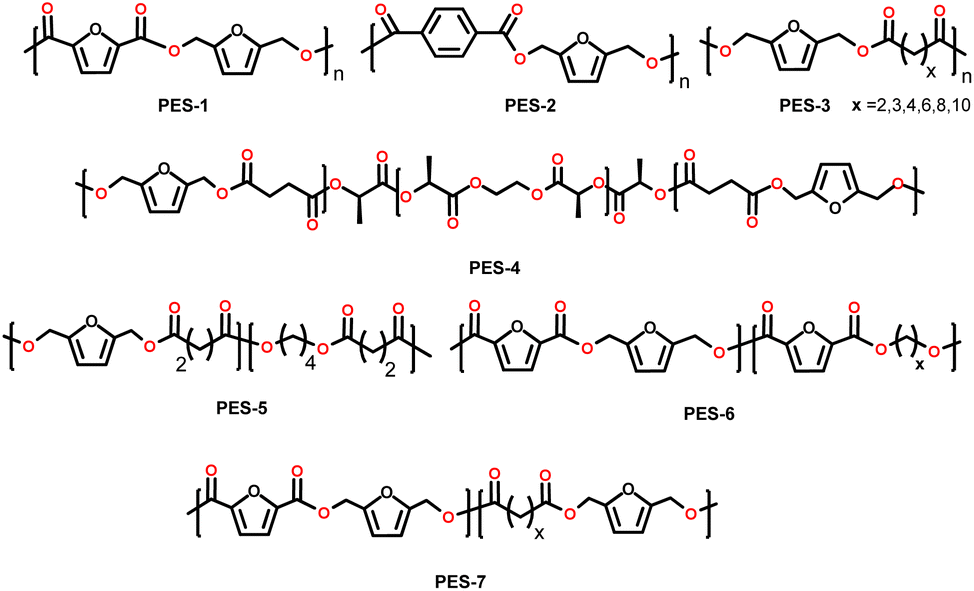 | ||
| Fig. 5 Bio-based co-polyesters achieved using BHMF as monomer. | ||
Most of the BHMF-derived PES are achieved by reaction with an aliphatic acid at low temperature via organo-catalysed solution polymerization or by enzymatic polymerization.133
Some examples of aromatic PES were also investigated as the first BHMF-derived copolyester that was reported in 1978 by Moore and co-workers via polymerization with 2,5-furandicarbonyl chloride in chloroform using triethylamine as a base.134 The resulting material PES-1 (Fig. 5) – namely poly(2,5-furandimethylene 2,5-furandicarboxylate) – had low intrinsic viscosity so it was not possible to prepare cohesive films. Furthermore, the polymer – as most of the materials reported in this work – when subjected to differential scanning calorimetry (DSC) analysis did not show a well-defined melting transition and decomposed to a black insoluble material at 200 °C.
In a different approach PES-1 was prepared via interfacial polycondensation at room temperature using a biphasic system constituted by (i) NaOH(aq.) containing BHMF and tetrabutylammonium bromide (TBAB) as the phase-transfer agent and (ii) dichloromethane containing the 2,5-furandicarbonyl chloride.135 The polyester precipitated from the solution and was then recovered by filtration. However, also in this case, the polymer had a low molecular weight and moderate thermal stability.
Ragno and co-workers prepared PES-1 type polymer employing N-heterocyclic carbene (NHC) catalysis for the polycondensation of diols and dialdehydes.136 BHMF and DFF were used as monomers to achieve a PES-1 structure although various fossil-based and bio-based monomers were also investigated by the authors. The polycondensation reaction was performed at room temperature in anhydrous THF with a catalytic dimethyl triazolium/1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and stoichiometric quinone oxidant.
Although the PES-1 prepared displayed the highest molecular weight among the polymers reported in this study, it also showed a rather poor thermal stability (<200 °C).
Similarly polymerization of BHMF with terephthalaldehyde led to a poly(2,5-furandimethylene terephthalate) (PES-2) with a Tg of 60 °C, low molecular weight and thermal degradation of ca. 247 °C.
Several investigations were reported on the preparation polyester via polycondensation by reaction of BHMF with an aliphatic diacid or the corresponding dialkyl ester. As an example, a bio-based self-healing polymers was prepared by reaction with succinic acid (SA) in the presence of N,N-dimethyl-4-aminopyridine (DMAP) and N,N′-diisopropylcarbodiimide (DIC), the latter used as condensation agent.137 Two polyesters (PES-3; x = 2) were achieved with a similar degree of polymerisation, i.e., n = 24 (Mn = 5100 g mol−1) and n = 27 (Mn = 5700 g mol−1). A subsequent Diels–Alder of the two polyesters with maleimide groups led to the formation of network polymers that showed a good self-healing ability without external stimulus.
A different approach to the poly(2,5-furandimethylene succinate) involved an integrated method where D-fructose was converted to HMF and thus reduced to BHMF using a CuSiO2 catalyst.138 Both reactions were performed in butanol from which BHMF could not be successfully isolated. As a result, it was decided to proceed into a polymerization reaction with succinic acid using once again DIC as condensation agent. The resulting polyester displayed a low molecular weight.
Similar aliphatic PESs were prepared by polymerization of BHMF with adipic acid (PE-3 x = 4) or pimelic acid (PE-3; x = 5) under mild conditions using first MgCl2 and then a Mg-based catalyst namely (TMP)MgCl·LiCl (TMP = 2,2,6,6-tetramethylpiperidyl), in the presence of di-tert-butyl dicarbonate (Boc2O) as a coupling agent.139 Using the latter sterically hindered catalyst, it was possible to achieve a PES-3 with a Mw value up to 16000 g mol−1 The polyester had Tg of −9 °C; this low value was ascribed to the more flexible nature of the pimelate backbone.
A BHMF-based block copolymers (PES-4) was also reported.140 This material namely poly(lactic acid)-block-poly(2,5-furandimethylene succinate) (PLA-b-PFS) was prepared in two-steps by (i) reacting L-lactide with ethylene glycol at 120 °C for 24 h; thus (ii) the recovered polymer was mixed with BHF and succinic acid in the presence of DMAP and DIC in dehydrated dichloromethane at room temperature for 24 hours. The so-formed polymer PES-4 was then purified by precipitation with excess methanol. The furan groups from PFS block were then crosslinked with bis(maleimido)triethylene glycol via a Diels–Alder reaction leading to a self-healing material.
A poly(2,5-furandimethylenesuccinate-co-butylene succinate) [P(FS-co-BS)] copolymers PES-5 was prepared by Yoshie and co-workers.141 PES-5 was achieved by polycondensation of BHMF, 1,4-butane-diol, and succinic acid once again in the presence of DMAP and DIC. These copolymers – that had a relatively low Mn (4300–7200 g mol−1) – were crosslinked via reversible Diels–Alder reaction with bis-maleimide to form network polymers.
BHMF-derived polyesters of general structure PES-3 were also prepared via enzymatic polymerization. Loos and co-workers firstly reported a polycondensation reaction of BHMF with several diethyl esters – i.e., diethyl succinate, diethyl glutarate, diethyl adipate, diethyl suberate, diethyl sebacate and diethyl dodecanedioate – promoted by Candida antarctica lipase B (CALB).142 In the optimized conditions the polycondensations were carried out at 80 °C in diphenyl ether and by employing a three stages procedure where the reaction mixture was firstly kept (1) for 2 h under atmospheric pressure, (2) then for 4 h at 350 mmHg and finally (3) the pressure was reduced to 2 mmHg while maintaining the same temperature for 66 h. All 2,5-bis(hydroxymethyl)furan-based polyesters were semicrystalline and thermally stable up to 250 °C, showing two decomposition steps with the major degradation process taking place at 276–332 °C. The Tg of the polyesters was in the range of −38 to 4 °C. The number-average molecular weights (Mn) were always around 2000 g mol−1 despite the different chain length of the aliphatic diester used. This result was ascribed to the formation of ether bonds via dehydration of the BHMF hydroxyl groups that hindered the polycondensation reactions.
In a similar study a series of aromatic–aliphatic polyesters based on the aliphatic diesters – i.e., succinic, adipic and sebacic ester derivatives – and aromatic diols including BHMF were prepared using Candida antarctica lipase B (CALB).143 The resulting polyesters with general structure PES-3 had Mw between up to 3000 g mol−1 with a Tg values ranging from −22 up to 10 °C. The lower Tg values were obtained when longer chain aliphatic diesters were used as monomers due to the greater flexibility of the resulting polyesters backbone. Thermal degradation of the co-polyesters was in line with the data previously reported for similar materials.
A series of furan-based co-polyesters with Mw up to 35000 g mol−1 was prepared still using CALB as biocatalyst.144 Furan-based co-polyesters PES-6 and PES-7 were prepared in the same three-steps procedure previously reported employing as different type of monomers. In a first investigation PESs were prepared starting from BHMF, FDME and aliphatic linear diol to achieve PES-4 type of co-polymers. In another approach, linear diacid ethyl esters were used instead of the diols and the resulting PEs had a PES-5 backbone.
The co-polyesters prepared from diols resulted to have the highest degree of polymerization and a higher thermal stability than diacid-ethyl-based polyesters, most probably as a result of higher molecular weight.
Examples of other polymers achieved from BHMF include polyurethanes, poly(silylether), polyethers and an example of humin-like material (Fig. 6; ESI Tables S.7 and S.8†).
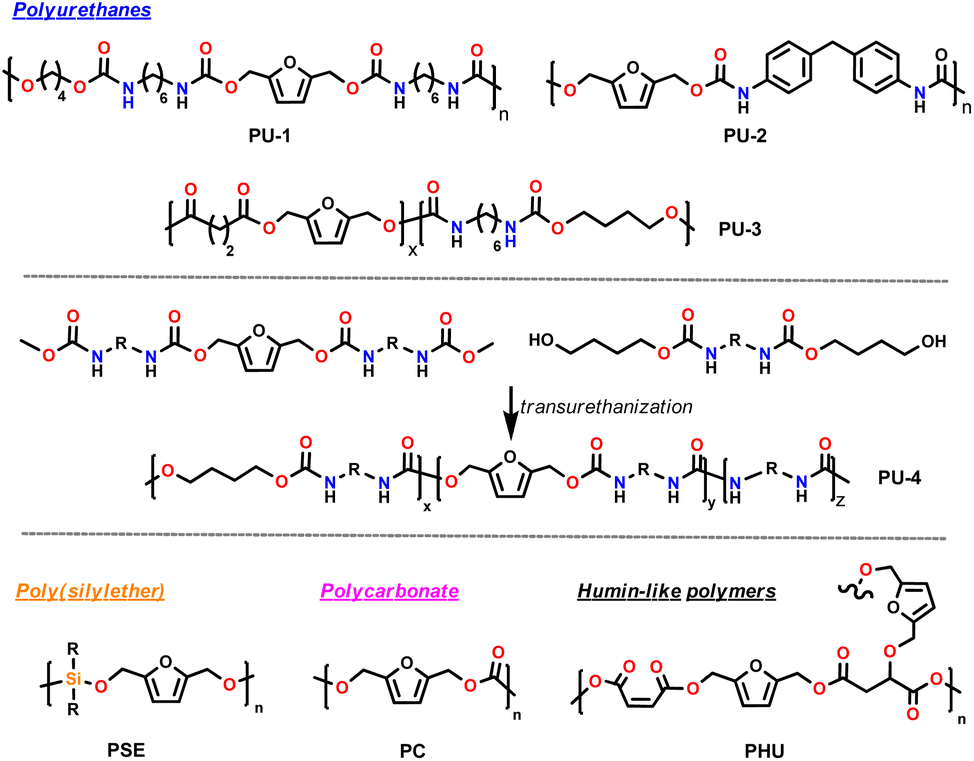 | ||
| Fig. 6 Other bio-based polymers achieved using BHMF as monomer. | ||
As an example, PU-1 was prepared by polycondensation of BHMF, 1,4-butandiol with hexamethylene diisocyanate in the presence of zirconium acetylacetonate at 70 °C.145 According to GPC analysis the co-polyurethane had Mn of 13000 g mol−1, however Tg and thermal degradation studies were not reported. PU-1 was then crosslinked with a bismaleimide via Diels–Alder reaction. The cured PU showed to possess thermo-reversibility and recyclability as proved by FTIR and DSC analysis.
In a different procedure, PU-2 was prepared employing solid-state mechanochemical approach via ball milling at room temperature by reaction of BHMF with methylene diphenyl diisocyanate in the presence of dibutyltin dilaurate (3 mol%).146 The PU so achieved had a high Mw up to 163300 g mol−1.
Ball milling enabled the synthesis of PU copolymers were several diols and/or diamines were added as co-monomers to change the polymer properties. The resulting PUs were flexible (Tg = 77–109 °C) and relatively thermally stable (Td = 163–220 °C).
Guo and co-workers reported the synthesis of bio-based multiblock polymers by reaction of two prepolymers diols with 1,6-hexamethylene diisocyanate.147 BHMF-based prepolymer was prepared by reaction of BHMF with succinic acid in dehydrated dichloroethane in the presence of DMAP under N2 atmosphere and DIC. The second prepolymer was achieved by reaction of succinic acid and 1,4-butanediol in the presence of tetra-n-butyl-titanate. Finally, the multiblock copolymer was prepared through chain extension reaction between these two prepolymer in the presence of 1,6-hexamethylene diisocyanate (HDI) at 130 °C. The so-formed materials had a negative Tg and resulted thermally stable above 250 °C.
In the latter procedures isocyanates have been used as monomers for the preparation of BHMF-based PUs. However, recently Co and co-workers reported an isocyanate-free route where a series of co-PUs (PU-3) were prepared via transurethanization between BHMF, 1,4-butandiol and a dicarbamate prepared by reaction of a diamines with dimethyl carbonate (DMC).148
In this synthetic approach, after preparing the desired carbamates, the polymer (PU-4) was prepared in three steps:
(i) BHMF was reacted with a dicarbamate (at different molar ratios) in the presence of K2CO3 so to achieve prepare a methoxycarbonyl-terminated prepolymer;
(ii) 1,4-Butandiol was reacted with a dicarbamate in the presence of K2CO3 in order to form a hydroxyl-terminated dicarbamates;
(iii) BHMF-based NIPUs were synthesized via transurethanization between the methoxycarbonyl-terminated prepolymers and hydroxyl-terminated dicarbamates. During the polycondensation a flow of nitrogen was continuously purged into the reactor to ensure methanol removal.
For all the PUs prepared the Tg was not visible in DSC analysis, while sharp melting peaks were observed (148–157 °C) due to their semicrystalline structures. The low value of Mn reported for these PUs (200–3000 g mol−1) was obtained via NMR spectroscopy, and it was ascribed to the low solubility of the polymers that did not allowed analysis via GPC of the samples. The NIPUs thermal stability was evaluated by TGA showing that these materials were thermally stable above 200 °C, with decomposition temperatures 214–224 °C. Also in this study the resulting PUs were crosslinked by addition of 1,10-(methylenedi-4,1-phenylene)bismaleimide.
A series of poly(silylether)s (PSE) have also been prepared by reaction BHMF (and other furan monomers) with diphenylsilane (Ph2SiH) catalysed by a Salen–Mn(V) complex.149 The PSE formed via dehydrogenative cross coupling had a Mn of 11000 g mol−1 and a Tg = 15.6 °C. The temperatures for 50% gravimetric loss (T−50%) used for assessment of thermal stability, was for this polymer ca. 453 °C.
A similar polymer was also achieved using by iron-catalysed polymerizations of BHMF and other 13 diols and phenylmethyl silane monomers.150 The resulting PSE had a Mn = 1800 g mol−1 and a Mw = 2900 g mol−1 meanwhile the value of Tg was not reported.
An example of polycarbonate (PC) BHMF-based polymer was successfully prepared by Song and co-workers.151 In a first attempt BHMF was reacted with green solvent and reagents dimethyl carbonate (DMC)152,153 or with diphenyl carbonate (DPC) via melt transesterification employing high vacuum (up to 0.3 torr) and a high temperature (200 °C). However, due to the high temperature BHMF decomposed forming humins. PC was instead achieved successfully by a two-step procedure where first BHMF is reacted with CDI and then the resulting furan bis(imidazolecarboxylate) was then coupled with BHMF at 40 °C in the presence of CsF. In these conditions it was possible to achieve a furan PC with a Mw = 5900 g mol−1. The polymer was thermally stable up to 150 °C. Obviously the main drawback of this procedure is the use of phosgene derivative CDI.
Finally, to increase the Tg, the linear furanic PC was reacted with N-substituted maleimide as reported in other studies so to obtain a crosslinked polymeric material.
An interesting humin-like resin (PHU, Fig. 5) was prepared by the polycondensation of BHMF with maleic anhydride in 2 min at 110 °C. Although no details were provided by the authors on the resin, this material was successfully used as a solid support for palladium immobilization.
Acetal monomers have been reported for the synthesis of novel materials bearing reversible acetal bonds on the main polymeric backbone allowing a controlled cleaved of the polymeric structure by the application of an acid (Fig. 7).156–160
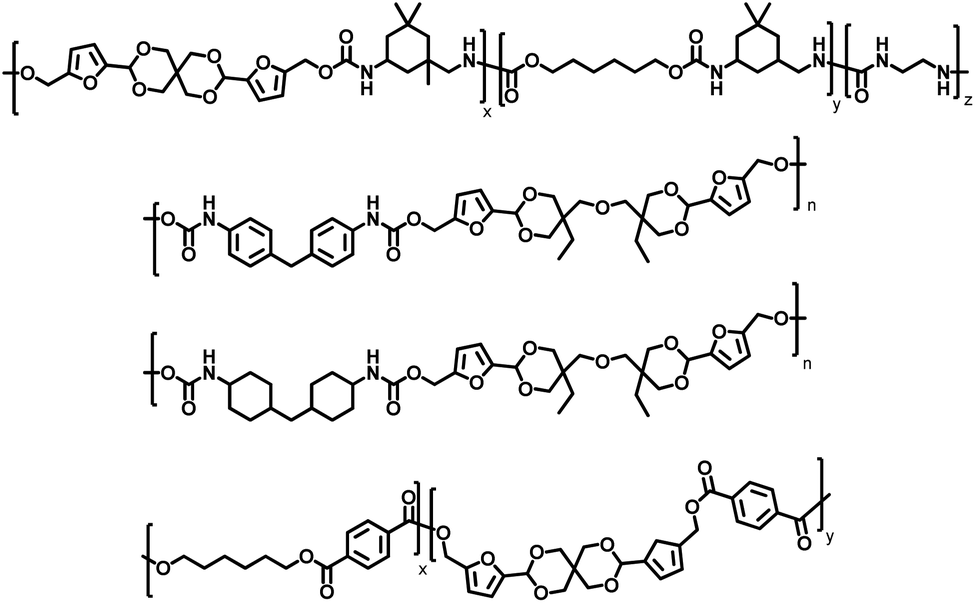 | ||
| Fig. 7 Polymers derived from acetal furanic based monomers. | ||
In this context, acetals monomer derived from furoic acid paved the way to novel acid-labile reversible functional bio-based polymers.
A small amount of acetal diol 1–2 (Section 3.3) can be used as debonding trigger units when are mixed into existing monomeric blends so that the properties of the resulting acid sensitive material do not change too much. Thus, different polyurethane films were synthesized containing none, 5 and 10% of acetal diol. These films presented similar properties to the acetal-free PUs by DSC and TGA. However, acidic treatment of the PU films led to very fast hydrolysis and dissolution of all urea cross-linked films.42
The properties of the obtained materials can be fine-tuned according to the structure of the acetal. A rigid diol III can copolymerize with potentially bio-based diisocyanates to prepare thermoplastic poly(cycloacetal–urethane) with relatively high molecular weights (Mn = 41500–98900 g mol−1). The acetal monomer 3 showed relatively high thermal stability among other hydroxyfuran-based monomers, which facilitated their polyurethane polymerization at elevated temperatures. Polymerizations of this rigid acetal monomer with isocyanate (MDI) and flexible diol polyTHF led to amorphous poly(cycloacetal–urethane)s with tuneable glass transition temperatures up to 104 °C and thermally stable up to 253 °C. Furthermore, these materials presented a relatively high pyrolysis char residue suggesting potential inherent flame resistance. The polymer can melt-spun into ∼150 meters of homogeneous fibres at 185 °C. The presence of the acetal units allows the hydrolysis of these fibres under acidic conditions, resulting in partial recovery of the original chemical building blocks.118
The incorporation of the HMF-based spirocyclic units in the monomer IV effectively increased the Tg of the copolyesters. On the other hand, a series of high molecular weights poly(urethane–urea) series were obtained containing up to 62 mol% of this monomer showing that the Tg value effectively increased from 79 to 131 °C upon the incorporation of the spirocyclic structures in the backbones.
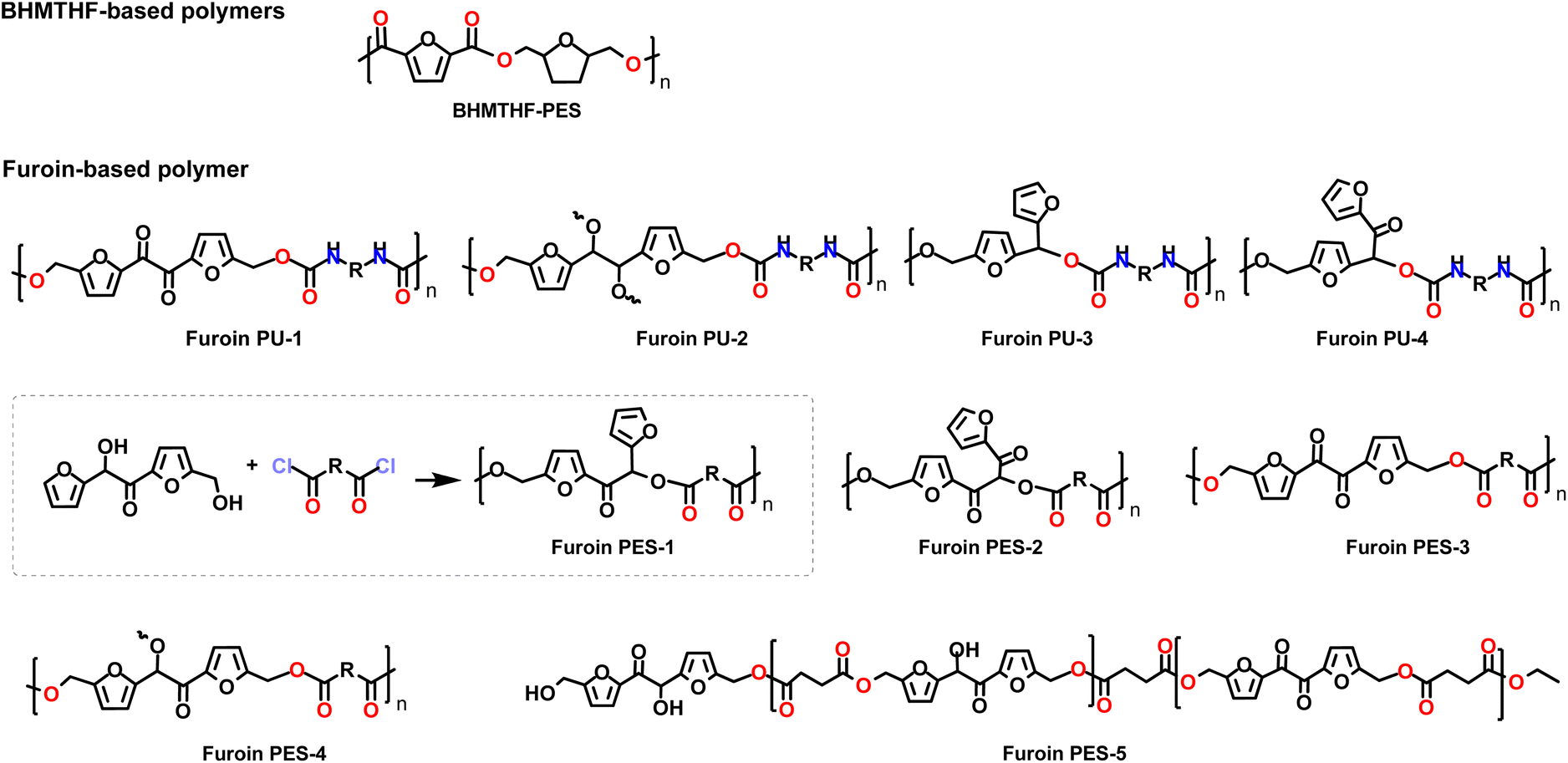 | ||
| Fig. 8 Bio-based polymers from furoin-based monomer. | ||
To the best of our knowledge BHMTHF exploitation in bio-based polymer is limited to one example reported by Moore and co-workers in 1978.134 This polymer was prepared employing two different procedures. In the first case a melt transesterification was carried out between cis-BHMFTHF was reacted with FDME at 230 °C in the presence of PbO. The mixture started to darken above 160 °C and the resulting polymer was described as a semisolid material with very low intrinsic viscosity. In a second attempt 2,5-furandicarbonyl chloride was reacted with cis-BHMTHF in the presence of triethylamine and 2-week polymerization time.
Although this procedure was somewhat more efficient the resulting polyester had once again a quite low inherent viscosity value. No other information on the characterization of this bio-based polymer were reported.
In 2016 Chen and co-workers reported the use of furoin monomers to prepare polyurethanes.132 5,5′-Bihydroxymethyl furil (Furoin-4), and tetraol, 5,5′-bihydroxymethyl hydrofuroin (Furoin-5) were reacted with various diisocyanates in the presence of dibutyltin dilaurate (DBTDL) to afford linear polyurethane Furoin-PU1 and cross-linked Furoin-PU2 respectively. The highest value of Mn of 39.8 kg mol−1 was achieved when Furoin-4 was reacted with diphenylmethane diisocyanate; the resulting polymer had an onset decomposition temperature of 234 °C, and a Tg of 140 °C. Linear polyurethanes based on rigid diisocyanates displayed higher Tg (>130 °C) than the ones derived from aliphatic ones (ca. 60 °C). The different PU thin films prepared starting from Furoin-4via solvent casting led to materials ranging from being brittle to flexible. Cross-linked PUs from tetraol Furoin-5 afforded very brittle materials that were not fully characterized. However, when studied via DSC their Tg's resulted to be a broad signal at ca. 80–100 °C.
Polycondensation of difuranic diols Furoin-1 and Furoin-2 with diacyl chlorides or diisocyanates led to linear polyesters and polyurethanes, respectively.161 These materials incorporated furan rings both in the polymer backbone and in pendent positions see Furoin PES-1, Furoin PES-2 and Furoin PU-3 as examples (Fig. 8).
Furoin-1 that encompasses a more electron-rich and flexible pendent furan, resulted more reactive in polycondensation reactions and led to polyesters with much higher molecular weight (Mn up to 20800 g mol−1; Tg up to 65 °C) compared to Furoin-2 that has with a more electron-deficient but rigid pendent furan ring. However, their thermal and mechanical properties were similar, being mainly influenced by the length of the alkyl bridge.
Same trend was observed for the Furoin-PUs. Interestingly the authors observed an inverse relationship between the Mn and the Tg of PUs, i.e., at higher values of Mn corresponded lower Tg's within the same diisocyanate series. This result was ascribed to hydrogen bonding which has a more pronounced effect on lower molecular weight polymers thus leading to higher Tg's.
The same research group investigated the use of bio-based difuranic polyol Furoin-3 and Furoin-4 for the preparation of a series of linear and cross-linked PES namely Furoin PES-3 and FuroinPES-4 as well as amorphous and semicrystalline poly(ester–urethane)s (PEUs).162
The polycondensation among Furoin-4 and various diacyl chlorides in THF led to linear Furoin PES-2 with Mn up to 5.5 kg mol−1, Tg = 103 °C and Td(5%) = 284 °C. On the other hands, when the bio-based triol Furoin-3 triol (DHMF) was reacted with diacyl chlorides, it resulted in the formation of cross-linked PESs Furoin PES-3 which resulted insoluble (Tg up to 89.1 °C and Td(5%) = 188 °C).
Furthermore, another diol monomer was also synthetized; in fact, reaction of adipoyl chloride with HMF in the presence of pyridine led to the corresponding difuranic dialdehyde with ester group linkage that was then reduced to form a new furan-based diol. The latter was used as monomer for both linear PES with Mn up to 8600 g mol−1 achieved by reaction with diacyl chlorides and linear PEUs by reaction with several commercially available diisocyanates. Among the PEUs, the one achieved by reaction with 2,4-toluenediisocyanate (TDI) showed Mn = 31400 g mol−1, Tg = 63.4 °C and Td(5%) = 231 °C.
An interesting approach for the polymerisation of furoin polyol scaffold was reported by Giovannini and co-workers that investigated the use of Candida antarctica lipase B (CALB) as biocatalyst.163 In this approach Furoin-3 was reacted with diethyl succinate in diphenyl ether at 50 °C employing the same three-stage procedure previously reported by Loos and co-workers.142 The recovered solid material analysed by NMR showed as expected the signals related to the Furoin-3 monomer esterified with the diethyl succinate, but also the presence of signals ascribed to the diketone monomer Furoin-4. From the spectrum the authors deduced that ca. 30% of the DHMF units incorporated in the oligomers were oxidized to the corresponding diketone form. Thus, the overall PES structure is reported in Fig. 6 (Furoin PES-5), this bio-based polyester has a Mn = 1150 g mol−1 evaluated via proton NMR. The same polycondensation was also carried out in the presence of diethyl esters of sebacic acid leading to the related linear PES with a Mn = 1000 g mol−1.
4. Amine-based monomers
4.1. Synthetic approaches to furfuryl amine
Furfurylamine (Fa) is a commercially available, monofunctional molecule employed for many purposes, i.e., corrosion inhibitor and monomer, but mainly as reaction intermediate as reported by the European Chemical Agency (ECHA) website.164Table S.10 in ESI† lists a series of chemical–physical properties of this monomer.
Scheme 8 indicates the principal class of reactions that lead to furfurylamine including (i) reductive amination of furfural, (ii) reduction of 2-furonitrile, (iii) alcoholysis of N-(furan-2-ylmethyl)formamide, (iv) amination of furfuryl alcohol, (v) hydrogenation of furfuryl oxime and (vi) hydroboration of furfurylamide (Table 10; ESI Table S.11†).
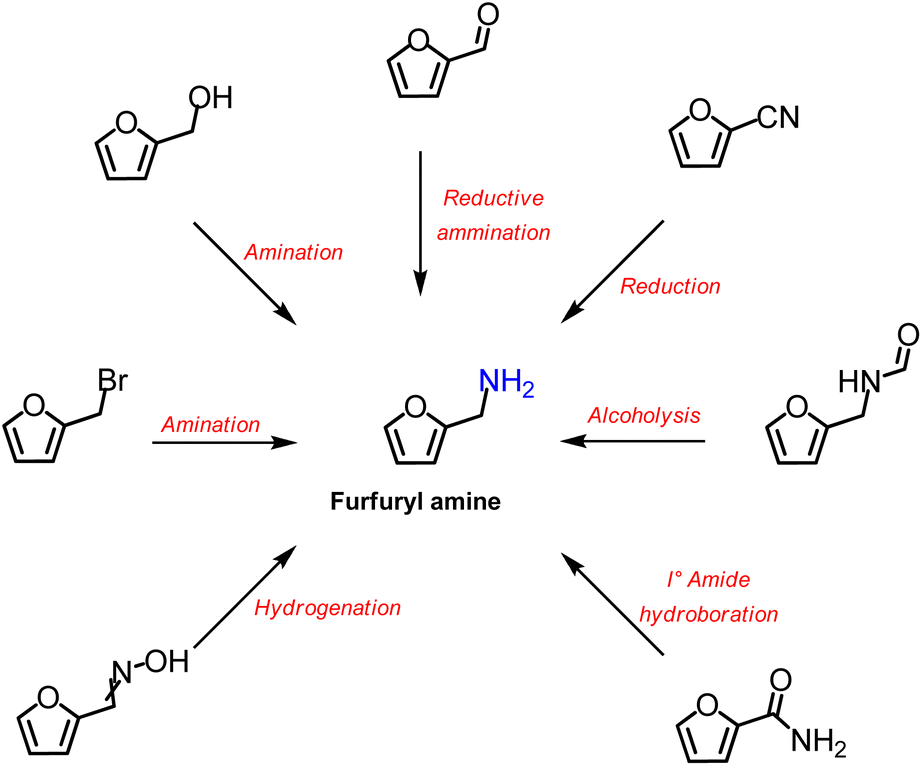 | ||
| Scheme 8 Principal reaction pathways to achieve furfurylamine from different starting substrates. | ||
| # | Furfural (g mmol−1) | Conc. (M) | Catalyst (wt%) | Reaction conditions | Cat. reuse | Yield (%) | E-f | PMI | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Determined product via analytical methods (1H-NMR, GC-MS, HPLC-MS). b Isolated product. c Amount of purification materials not reported. | |||||||||
| 1 | 3/31.2 | 1.50 | Ni–Al2O3 (3.3) | H2, 40 bar; NH3 25 wt% aq.; 60 °C, 1 h | — | 89a | 10.7 | 11.7 | 165 |
| 2 | 4.8/49 | 1.63 | Zn dust (166.7) | (i) NH3OHCl, EtOH, r.t., 35 min; (ii) HCl 12 M; r.t., 15 min; (iii) NH3, 30%, NaOH 6 M aq., r.t., 15 min | — | 99b | 19.7 | 20.7 | 166 |
| 3 | 0.2/2.1 | 0.68 | Rh/Al2O3 (1.0) | H2, 20 bar; NH3 28% aq.; 80 °C, 2 h | 5 | 92a | 23.5 | 24.5 | 167 |
| 4 | 0.5/5 | 0.35 | RANEY® Ni 50 | H2, 5 bar; NH3, 3.5 bar; THF; 80 °C, 2 h | — | 100a | 27.0 | 28.0 | 168 |
| 5 | 0.2/2 | 0.33 | 10Ni/Al2O3 (50) | H2, 20 bar; NH3 7 M in MeOH; 100 °C, 30 min | 5 | 99a | 29.3 | 29.8 | 169 |
| 6c | 0.2/2 | 0.29 | Ru/BN-e (15) | H2, 15 bar; NH3 25% aq.; MeOH; 90 °C, 2 h | — | 95b | 31.7c | 32.7c | 170 |
| 7 | 0.2/2 | 0.40 | Ru/HZSM-5 (75) | H2, 30 bar; NH3 7 M in MeOH; 100 °C, 15 min | 5 | 76a | 33.2 | 33.5 | 171 |
| 8 | 0.63/6.6 | 0.33 | Ru/γ-Al2O3 (8.0) | H2, 30 bar; NH3, 4 bar; MeOH; 80 °C, 2 h | — | 75a | 34.5 | 35.5 | 172 |
| 9 | 1.44/15 | 0.19 | Ru/Nb2O5·nH2O-300 (35) | H2, 30 bar; NH3, 10 bar; MeOH; 70 °C, 6 h | 3 | 87a | 50.7 | 51.3 | 173 |
| 10 | 0.05/0.5 | 0.25 | Co2P NRs (8.0) | H2, 5 bar; NH3 25% aq.; 100 °C, 10 h | — | 90a | 62.0 | 63.0 | 174 |
Other synthetic pathways starting from more complex substrates and/or using multistep reactions, were also reported.175–178 Since furfuryl amine is a commercially available product herein it is reported only a brief overview of the most promising new synthetic approaches of this bio-based molecule (yield > 70%).
All the catalyst employed in these synthetic procedures are noble or non-noble metal based, i.e., Ru, Rh, Pd, Pt, Co, Ni, Cu and Zn. Some of them are commercially available, such as RANEY® Ni, RANEY® Co, Rh/Al2O3 and Zn dust, however the majority are custom-made and tailored for this specific reaction. Although the latter category showed to be very efficient for this chemical transformation, they are also generally more time- and reagent-consuming.
For instance, for the synthesis of Ru/BN-e catalyst (#6; Table 10) it was required 24 h for the reaction, 12 h to dry and 2 h for the calcination.
Overall, almost all the procedures listed in Table 10 showed E-factors within the fine chemical sector range (#1–8; Table 10) (E-factor = 5–50 kg kg−1); only two procedures (#9 and #10; Table 10) are slightly over this range.
Zhang et al. procedure (#1; Table 10) has the lowest PMI, reaching 11.7.165 This is due to the high concentration of the starting furfural solution (1.5 M) and to the high yield of the desired product (89%). Nevertheless, the latter value was calculated by analytical method and therefore it is less indicative.
On the contrary, even if the second procedure listed (#2; Table 10) has almost a double PMI value (20.7), the yield is isolated (99%) thus representing a more solid data. Moreover, zinc dust was used as catalyst is very inexpensive (ca. 50 € per kg).194 The downside is that it is a multistep procedure, which requires many solvents and reagents.
The procedure with highest values of E-factor and PMI (#10; Table 10) used a custom-made catalytic system and most importantly the reaction is conducted at quite low furan concentration (0.25 M).
It should be finally mentioned that new synthetic approaches to furfuryl amine will have to compete with the actual market price of this compound (100 g, 35.9 € in the Merck catalogue) thus optimization of the reaction conditions, recycling of the catalyst and easy purification approach must be carefully addressed.
4.2. Synthetic approaches to 2,5-bis(aminomethyl)furan and 5-(aminomethyl)-2-furanmethanol
2,5-Bis(aminomethyl)furan (BAMF) and 5-(aminomethyl)-2-furanmethanol (AMF) are two examples of interesting monomers for bio-based polymers such as polyurethanes, polyamine, and polyurea. However, to date, studies on their synthesis are still at its early stage especially when compared to other furanic monomers, i.e., furan diols.Synthetic approaches to BAMF are summarized in Scheme 9 and Table 11; for simplicity reagents and materials employed for the preparation/purification of catalysts have not been accounted in the calculation of E-factor and PMI values.
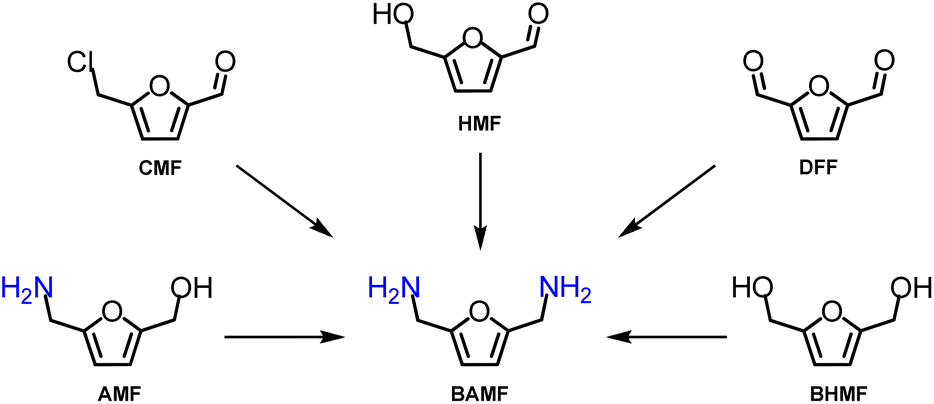 | ||
| Scheme 9 Synthetic pathways to AMF and BAMF. | ||
| # | Substrate (g mmol−1) | Conc. (M) | Catalyst (mol% or wt%) | Reaction conditions | Cat. reuse | Yield (%) | E-f | PMI | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a All reactions were conducted in an autoclave except entry 9. b Isolated yield. c Determined by analytical method. d AT = acid-treated. | |||||||||
| 1 | HMF (10/79.4) | 1.5 | Cu4Ni1Al4Ox (20 wt%) | NH3, Na2CO3, H2, 1,4-dioxane; 90–210 °C, 24 h | 3 | 86c | 9.1 | 9.9 | 179 |
| 2 | HMF (0.5/3.9) | 0.26 | 10NiMn (4:1)/γ-Al2O3 (80 wt%) |
NH3, H2, THF; 160 °C, 24 h | 4 | 82c | 33.7 | 33.7 | 180 |
| 3 | HMF (0.5/3.9) | 0.26 | RANEY® Ni (50 wt%) | NH3, H2, THF; 160 °C, 12 h | 4 | 82c | 33.3 | 33.7 | 181 |
| 4 | HMF (1.3/10) | 0.4 | RuHCl(acridine-iPr-PNP)(CO) (0.5 mol%) | NH3, H2, tamylalcohol; 140 °C, 11 h | — | 58b | 38.3 | 39.3 | 182 |
| 5 | HMF (0.5/3.9) | 0.26 | RANEY® Ni (50 wt%) | NH3, H2, THF; 160 °C, 12 h | 4 | 61c | 46.0 | 46.2 | 183 |
| 6 | HMF (0.06/0.5) | 0.1 | (i) Ru/Nb2O5 (33 wt%); (ii) Ru(CO)ClH(PPh3)3 10 mol, Xantphos (10 mol%) | (i) NH3, H2 MeOH, 90 °C, 4 h; (ii) tamylalcohol, NH3, 140 °C, 14 h | — | (i) 96b; (ii) 93c | 153.4 | 154.0 | 184 |
| 7 | HMF (1.0/7.8) | 0.52 | (i) CHF3O3S:P2O5 (3.0:3.0 mmol); (ii) RANEY® Ni (8 wt%) |
(i) CH3CN, 100 °C, 3 h; (ii) NH3, H2, MeOH, 120 °C, 3 h | — | (i) 67c; (ii) 60c | 305.8 | 306.8 | 185 |
| 8 | BHMF (3.1/25) | 0.5 | Ru(CO)ClH(C13H7N (CH2P–(Cy)2)2) (0.1 mol%) | THF, NH3; 140 °C, 21 h | 96c | 17.7 | 18.7 | 186 | |
| 9 | BHMF (0.06/0.5) | 0.1 | Ru–20MgO/TiO2 (>300 wt%) | NH3, PhMe; 110 °C, 20 h | — | 86c | 84.6 | 85.6 | 187 |
| 10 | BHMF (0.25/2) | 0.3 | (i) HCl | (i) CH2Cl2; (ii) (a) NaN3, DMF 65 °C, 16 h; (b) MeOH, PPh3, Boc2O, 16 h; (c) HCl, 1 h | — | 14b | 5189.6 | 5190.6 | 188 |
| 11 | DFF (0.12/1) | 0.14 | (ii) Rh/HZSM-5 (25 wt%) | (i) NH2OH, H2O; 110 °C, 2 h; (ii) NH3, H2, EtOH; 130 °C, 2 h | 4 | (i) 94c; (ii) 80b,c | 128.6 | 129.3 | 189 |
| 12 | DFF (0.3/2.5) | 0.08 | AT-Ni-RANEY®d (10 wt%) | THF:H2O, NH3, H2; 120 °C, 6 h |
— | 43c | 220.6 | 221.4 | 190 |
| 13 | DFF (0.03/0.25) | 0.08 | Co/ZrO2 (100 wt%) | ButNH2, NH3, MeOH; 100 °C, 10 h | 4 | 95b,c | 501.6 | 502.6 | 191 |
| 14 | AMF (0.5/3.9) | 0.2 | Ni2Al-600 (50 wt%) | THF, NH3, 160 °C, 18 h | — | 70c | 53.1 | 54.1 | 192 |
| 15 | CMF (0.5/3.5) | 1.1 | RANEY® Ni | THF, NH3, H2, 90 °C, 1 h | — | 62c | 33.8 | 34.8 | 193 |
Direct conversion of HMF into BAMF via combined alcohol oxidation followed by reductive amination was the most studied synthetic approach (#1–7; Table 11). This chemical transformation comprises of three steps: (i) oxidation of the alcoholic moiety to form DFF; (ii) imine formation condensation by reaction with NH3 and (iii) reduction of the imine to achieve BAMF. The formation of DFF via HMF dehydrogenation was indicated as the rate-controlling and most challenging step.181
BAMF preparations were mostly performed in an autoclave under ammonia and hydrogen pressure at temperatures above 140 °C in the presence of a variety of custom-made mono or bimetallic catalyst (#1–7; Table 11).
Considering the values of E-factors and PMI, the direct amination of HMF into BAMF developed by Yuan and co-workers that uses a bifunctional Cu4Ni1Al4Ox catalyst is the most convenient (#1; Table 11). The procedure was described as a two-stage temperature-controlled reaction process. At 90 °C the reductive amination of HMF aldehyde group takes place followed by hydrogen-borrowing reaction of hydroxyl of HMF at 210 °C. The reaction was performed in 10 grams scale and at high concentration (1.5 M); as a result, calculated E-factor and PMI were of ca. 9. Furthermore, the catalyst could be reused up to three times. Unfortunately, the authors did not report any details concerning the purification of the final compound that could be accounted in the metrics evaluation.
Two quite similar syntheses exploited the use of a commercially available catalyst, i.e., RANEY®-Ni (#3 and #5, Table 11). In the best conditions the value of E-factor was calculated to be 33 (#3, Table 11). Further optimization of this procedure might represent an interesting preparation of BAMF.
On the other hand, approaches employing two catalytic systems such as the ones reported by Li or by Lin (Ritter reaction followed by the reductive amination) – although quite efficient – required a larger amount of reagents/solvents resulting in higher valued of E-factor (#6 and #7; Table 11).
BHMF was also employed as starting material to BAMF (#8–10: Table 11). Among the reported procedures, the one proposed by BASF employing a custom-made ruthenium catalyst, resulted the most promising one with a E-factor of 17.7. However, it should be mentioned that BHMF is generally obtained via HMF reduction thus the use of additional reagents/solvents should be considered.
Reaction performed using DFF as starting compound are either multistep (#11 and #12; Table 11) or gave only a moderate yield (#13; Table 11) of the desired product.
Amination of the hydroxyl moiety of AMF via “hydrogen borrowing” or “hydrogen auto-transfer” mechanism was also reported in good yield although E-factor and PMI values are quite high.
Finally a special mention should be given to the procedure developed by Mitsubishi Gas Chemical Co. that uses 5-(chloromethyl)furfural (CMF) as starting substrate (#15; Table 11). CMF can be directly synthesized from cellulose or saccharide rendering this procedure quite interesting and viable to further interesting optimization.
As evident from the data collected in Table 11, little or no attention was so far given to BAMF purification/isolation as only in few cases the product was isolated as a pure and it was described as a viscous brown oil. Thus, further investigations in this sense are necessary for the exploitation of these amine derived furanics.
4.3. Bio-based polymers
2,5-Bis(aminomethyl)furan (BAMF), 5-hydroxymethyl-2-furfurylamine (AMF) and furfurylamine (Fa) are excellent candidates for subsequent derivatization and polymerization reactions.195–197 In fact, some of these compounds were already considered as a new type of amine monomer for the production of polybenzoxazine (PBPB) resins and polyamides, despite the fact that the number of studies is still moderate when compared to FDCA-based polyamides, and particularly considering polyesters counterparts.182,198–202Furfuryl amine-based PBs, synthetized trough benzoxazine monomers ring opening polymerization (ROP), is an emerging topic based upon Mannich condensation203 approach used to synthesise benzoxazine monomers taking advantage from the versatility of the structural design capacity they offer.198,203,204
Furthermore, these phenolic resins can have excellent thermal198–202 and dielectric properties, low moisture absorption, low surface energy and flammability and corrosion resistance,198,201,202 which makes them suitable for space, defence, and communication applications.199,200,205Table 12 summarizes some of the thermal features of previously reported PBs. It is reasonable to generalize that this class of polymers have enhanced thermal stability with a decomposition temperature at 5% weight as high as 395 °C.198 These cured polymers are very stiff materials exhibiting high glass transition temperature (typically > 300 °C).
| PBs | Co-monomers |
T
d,5%a (°C) |
T
d,maxb (°C) |
T
gc (°C) |
CY800d (%) | Ref. |
|---|---|---|---|---|---|---|
| a Decomposition temperature at 5 weight loss%. b T d,max: temperature at which the rate of weight loss is maximum. c Glass transition temperature. d Char yield at 800 °C. | ||||||
| PB(Do–Fa) |
|
351 | 380 | — | 37 | 199 |
| PB(Res–Fa) |
|
346 | 403 | 312 | 64 | 201 |
| PB(Ty–Fa) |
|
342 | — | 322 | 51 | 206 |
| PB(Th–Fa) |
|
306 | 401 | — | 29 | 202 |
| PB(Ap–Fa–Bz) |
|
395 | — | 283 | — | 198 |
| PB(Pp–Fa) |
|
319 | 393 | 176 | 47.3 | 200 |
| PB(Vp–Fa) |
|
325 | 424 | 226 | 51.6 | 200 |
Recently, there has been an effort to develop PBs with a greener profile in terms of renewable content and their ability to replace hazard reagents, such as formaldehyde.198 In this view, Mydeen et al. compared a thymol–furfurylamine-derived polybenzoxazine (Fig. 9) incorporating different long-chain diamines (thymol/dodecylamine and thymol/octadecylamine).202
 | ||
| Fig. 9 Thymol–furfurylamine-derived polybenzoxazine (PB(Th–Fa)) synthesis. | ||
The ensuing PB(Th–Fa) was just prepared by heating the Th–Fa monomers without catalyst and they show a better anticorrosion performance under moisture conditions and can be used as an effective coating material for different industrial applications under humid environments.202
Also, Zhang et al. synthesized a bio-based trioxazine PB resin using furfurylamine which performed as a flammability retardant.201 That same purpose was largely addressed in different polybenzoxazines studies.199,201,203 Consequently, these new furfurylamine bio-based benzoxazines and their corresponding polybenzoxazine materials possess excellent processability and thermal properties (Table 12), suggesting a great potential towards high-performance and fire-resistant materials.
Polybenzoxazines have also been shown to be able to have antibacterial properties, as in the study of Mohamed et al.,198 in which they synthesized a fully bio-based multifunctional benzoxazine (Ap–Fa–Bz) in high yield and purity from apigenin (Ap), furfurylamine, and benzaldehyde (Bz) (Fig. 10).
 | ||
| Fig. 10 Synthesis of the antibacterial PB(Ap–Fa–Bz) from furfurylamine, apigenin and benzaldehyde. | ||
This study showed the possibility of preparing these polymers under solventless conditions and, most importantly, of replacing hazard formaldehyde. Moreover, it was reported that in addition to the high thermal stability (Td,10 = 395 °C), a high char yield (52 wt%), and a high glass transition temperature (Tg = 283 °C), the resin was even capable to perform as an antibacterial agent against both Staphylococcus aureus and Escherichia coli.198
The first study on 2,5-bis(aminomethyl)furan based polymers dates back to 1991, where Gandini et al.,207 reported the polycondensation of BAMF and 2,5-bis(carboxyl chloride)furan for the preparation of bio-based furan polyamides. However, the hazard dichloride intermediate was involved and the solution polycondensation of 2,5-bis(carboxyl chloride)furan with BAMF results in polymeric products with low molecular weight, yields (not exceeding 60%) and intrinsic viscosity values (not surpassing the 0.5 dL g−1).207–209 Over the past years, the interest on these bio-based polyamides has been forgotten. In fact, only a few articles and patents have been published using this type of amino-based monomers.
It should be mentioned that BAMF can also be used as a hardener for epoxy resins applied to water resistant floor coatings or in polyimines.208 In spite of the fact that BAMF is a very interesting monomer to explore in the future for polymers, their systematic characterization, both in terms of mechanical and thermal properties, to evaluate its real potential is still lacking.
5. Epoxides-based monomers
5.1. Synthesis of epoxide-base furanics
HMF and furfural are suitable molecular platforms to be converted into a diversity of furan-based epoxide monomers. Fig. 11 depicts the structures of some of these epoxides that have been subsequently exploited for the synthesis of epoxy resins (Section 5.2). | ||
| Fig. 11 Examples of epoxides derived from furans chemicals. | ||
Two main synthetic strategies have been explored for the preparation of these derivatives. The first strategy involves the use of furanic derivatives that are prone to react with allylic compounds, such as allyl bromide, resulting in the formation of corresponding allylic ethers or ester derivatives (Fig. 12; Table 13; ESI Table S.13†). Subsequently, the allylic-derived furanic compounds are transformed into the corresponding epoxides through oxidation, often using reagents like m-chloroperoxybenzoic acid (m-CPBA).
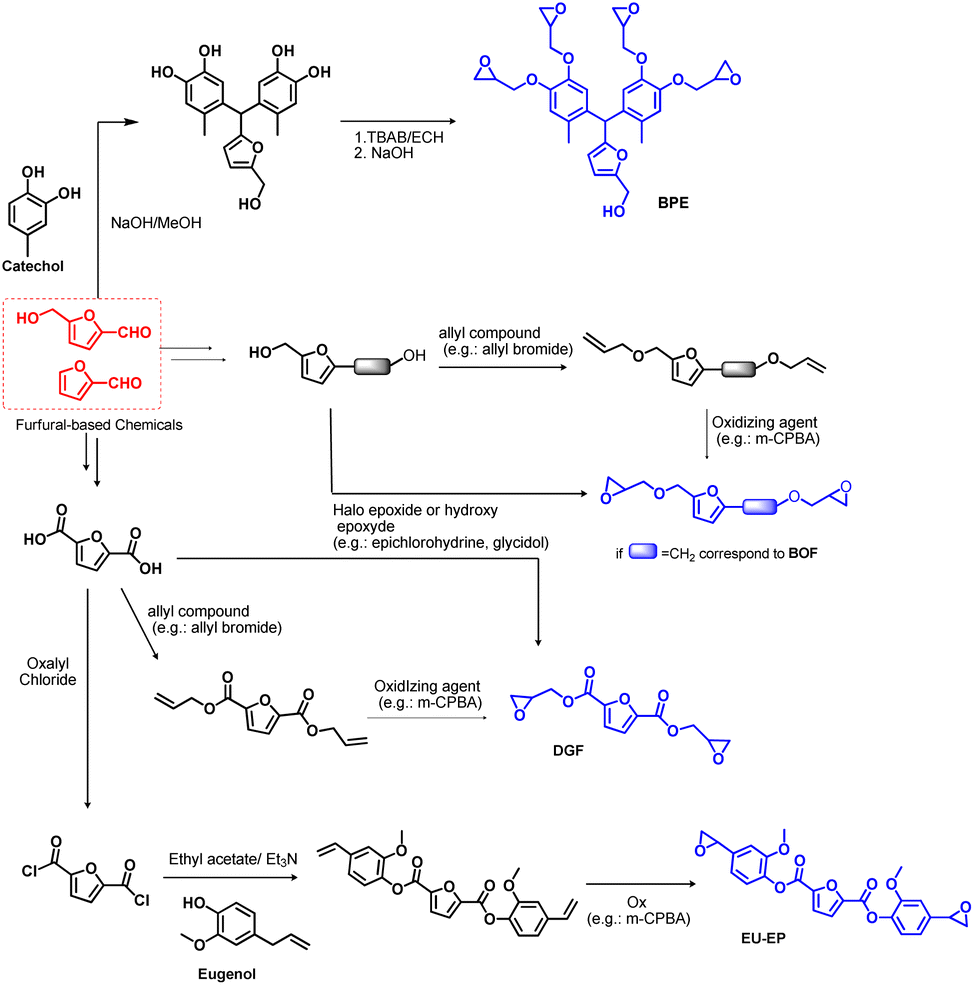 | ||
| Fig. 12 Synthetic pathways for the synthesis of epoxy derivatives from furan-based chemicals. | ||
| # | Substrate (g mmol−1) | Conc. (M) | Catalyst (mol% or wt%) | Reaction conditions | Yield (%) | E-f | PMI | Ref. |
|---|---|---|---|---|---|---|---|---|
| a For the second step. b Using HMF as the aldehyde and considering the tetraepoxide as the final product. Abbreviations can be found in the dedicated section. | ||||||||
| 1 | BHMF (128/1000) | 5.81 | TBHS (1.2 mol%) | ECH, NaOH; 65 °C, 4 h | BOF 60 | 3.1 | 4.1 | 210 |
| 2 | BHMF (51.3/400) | 1.28 | TBAB (2 mol%) | (i) ECH, 65 °C, 5 h; (ii) NaOH; 30 °C, 24 h | BOF 64 | 7.4 | 8.4 | 211 |
| 3 | BHMF (10.2/80) | 1.27 | TBAB (2 mol%) | ECH, NaOH; 65 °C, 10 h | BOF 62 | 8.4 | 9.4 | 212 |
| 4 | (i) HMF (50.4/400); (ii) BHMF (2.6/20) | 4 | TBAB (0.5 mol%) | (i) H2O, NaBH4; 0 °C, overnight; (ii) THF, ECH, NaOH, TBAB; 50 °C, 7 h | BOF 75 | 27.3 | 28.3 | 213 |
| 5 | BHMF (2.6/20) | 0.48 | TBAB (10 mol%) | THF, NaOH, ECH; 50 °C, 2 h | BOF 74 | 31.8 | 32.8 | 214 |
| 6 | (i) FDCA (50/320); (ii) FDCE (60/250) | FDCA 1.10; FDCE 0.56 | — | (i) Allyl bromide, DMF; reflux, 12 h; (ii) m-chloroperoxybenzoic, DCM; 40 °C, 3 days | DGF 57 | 9.1, 29.5a | 10.1, 30.5a | 215 |
| 7 | (i) FDCA (50/320); (ii) FDCE (60/250) | FDCA 2.28; FDCE 0.56 | — | (i) DMF, acetone, allyl bromide, Et3N; reflux, 24 h; (ii) m-chloroperoxybenzoic, DCM; 40 °C, 72 h | DGF 72 | 20.7 | 21.7 | 216 |
| 8 | (i) BHMF (15.6/100); (ii) FDCDCl (9.6/50); (iii) EUFU (8.4/20) | BHMF 0.88; FDCDCl 0.50; EUFU 0.20 | DMF (0.5 wt%) | (i) THF, oxalyl chloride; 25 °C, 3 h; (ii) eugenol, Et3N, AcOEt; 25 °C, 0.5 h; (iii) AcOEt, m-CPBA; 40 °C, 48 h | EU-EP 64 | 17.3 | 18.3 | 217 |
| 9 | (i) HMF (1.3/10); (ii) BPF (0.22/1) | HMF 5.00; BPF 0.13 | TBAB (40 mol%) | (i) MeOH, NaOH, catechol; 65 °C, 24 h; (ii) aq. NaOH, ECH; 60 °C, 3 h | BPE 22 | 46.0b | 47.0b | 218 |
Alternatively, the direct synthesis of furanic epoxide derivatives can be achieved by reacting furan-based intermediates with halo epoxides, such as epichlorohydrin (ECH), or hydroxy epoxides, like glycidol, to yield the desired ether or ester epoxide derivatives.
While these synthetic protocols are generally straightforward and scalable, they tend to generate a substantial amount of waste, primarily due to the frequent use of chlorine chemistry. Consequently, these methods are far from being environmentally efficient and optimized in terms of sustainability.
For example, the synthesis of 2,5-bis[(2-oxiranylmethoxy)-methyl]-furan (BOF; #1 and #2, Table 13) is typically carried out through the direct alkylation of BHMF. BOF is obtained by reacting BHMF with epichlorohydrin in the presence of NaOH, and utilizing tetra-n-butylammonium salts as a phase transfer catalyst. While this process achieves a 60% yield and demonstrates relatively good values in terms of green metrics (E-f and PMI), there is no reported reuse of the phase transfer catalyst. Importantly, these synthetic procedures, despite their scalability and reasonable yields, generate significant waste, with a production of 3 kg of waste per kg of epoxide due to the formation of inorganic salts during the etherification reaction. Efforts are needed to optimize these processes for greater environmental efficiency and sustainability.
In a similar fashion, BOF can be obtained using BHMF as the starting reactant with TBAB as the transfer catalyst (#2–5, Table 13). Also in these trials, transesterification of BHMF was performed with ECH to yield the desired BOF in moderate to good yields (62–75%). In all cases, the authors followed the same procedure (ECH/NaOH) with differences in the concentration of the initial reactant. In these procedures, the values of both E-f and PMI are higher because of the poor recyclability of the “apparent” catalyst and the larger amount of waste generated during the epoxidation.
The diglyceryl ester of 2,5-furandicarboxylic acid (DGF, #6, Table 13) was synthesized in multigram scale by esterification of FDCA with allyl bromide in mixture N,N-dimethylformamide/acetone and using triethylamine. The isolated ester was further oxidized to the corresponding epoxide with m-CPBA.
An alternative approach is based on the transesterification between DMF-FDCA and glycidol. Using this procedure, the values of E-f and PMI were 9.1 and 10.1, respectively for the first step while 29.5 and 30.5 for the second one.
Alternatively, the same protocol was applied to FDCA; in this case, overall yield of 72% was achieved however the green metrics show higher values (E-factor 20.7 and PMI = 21.7) in comparison to the previous synthetic protocol used for the preparation of the same product.
Transesterification does not require a large excess of reagents and is performed at lower temperature and for shorter time than the glycidylation.
The synthesis of aromatic glycidyl furan derivatives from FDCA and eugenol has also been reported (#8; Table 13).217 The authors highlight that the diepoxide monomer, bis(2-methoxy-4-(oxiran-2-ylmethyl)phenyl)furan-2,5-dicarboxylate (EU-EP), boasts a significant biomass content of up to 93%.
The synthetic route for EU-EP involves three key steps. In the first step, the acid chloride is obtained through the reaction of FDCA with oxalyl chloride. This intermediate is subsequently transformed into the corresponding ester through a reaction involving eugenol and triethylamine, achieving a high yield of 91%. Thus, the allyl group is oxidized to produce the corresponding epoxide using m-CPBA.
Interestingly, when compared to previous synthetic protocols for the preparation of epoxides derived from HMF, it appears that the values for both E-f and PMI are notably higher, reaching 17.3 and 18.3, respectively.
Similarly, lignin and carbohydrate-derived molecules, such as 4-methylcatechol, HMF, and furfural, can be employed for the synthesis of novel bisphenol-furan polyepoxides (#9, Table 13).218 Furfural and HMF were reacted with 4-methylcatechol in the presence of NaOH, yielding the corresponding bisphenol-furan polyalcohol under relatively mild conditions. This intermediate can then be further converted into the corresponding epoxides through glycidylation, using tetrabutylammonium bromide (TBAB) as a phase-transfer catalyst and epichlorohydrin as reagent.
The resulting renewable bio-based bisphenol-furan epoxy thermosets exhibit favourable thermal and mechanical performance. However, the environmental sustainability of this approach is compromised due to higher values of green metrics, with an E-factor of 46 and a PMI of 47. These high values can be ascribed to the limited reuse of the catalyst and the generation of a larger amount of waste during the procedure, rendering this alternative less environmentally sustainable compared to other methods.
In conclusion there are two main issues that should be addressed for epoxy-based furanics (i) avoid the use of chlorine reagents – despite these synthetic procedures results in good green metrics values; (ii) for transesterification-based procedures optimization of the reaction conditions is required in order to improve the actual PMI values.
5.2. Polymer based on epoxide furanic derivatives
Epoxide furanic derivatives have gained significant interest as a promising class of polymers due to their unique properties, which include Tg, rigidity, durability, and flame resistance. These compounds can be polymerized through an ionic mechanism, such as cationic polymerization, but this process typically exhibits a relatively slow reaction rate. Therefore, optimizing the curing time is essential to prevent damage to the final material resulting from prolonged exposure to UV light.214 Additionally, adjusting the initiator's percentage in the composition during curing can enhance the tensile-shear strength and improve the heat flow integrals, serving as indicators of the kinetic profile for cationic photo-curing of biomass-based furanic compounds.Indeed, an alternative approach to synthesize polymers based on epoxide furanic derivatives is through polyaddition reactions. Beginning with glycidyl furfuryl (DGF), cyclic carbonates can be synthesized and subsequently polymerized with diamines. As an example, bis(cyclic carbonate) derived from furandicarboxylic acid (FDCA) can react with various diamines, such as 1,6-hexanediamine, 1,8-diaminooctane, and isophorone diamine, under relatively mild conditions (in the range of 140–160 °C). This reaction leads to the production of non-isocyanate polyurethanes with characteristics comparable to traditional polyurethanes. This method offers a more environmentally friendly and sustainable route to produce polyurethane-like materials without the use of isocyanates, which are known for their potential health and environmental concerns.215
These novel furan-based epoxides open up new possibilities for the development of thermosetting resins with unique properties suitable for coatings, adhesives, composites, and electronic packaging materials.219 The inherent rigidity of these epoxide derivatives offers a promising path to creating bio-based renewable epoxy resins that outperform resins derived from long aliphatic chain epoxide monomers, particularly those obtained from plant oils as a renewable feedstock.
Bio-based polymers derived from the combination of epoxides and furans exhibit exceptional versatility, due to the reactivity of epoxides and the rigidity of furans within their structures. These materials hold significant potential for various applications, with a particular focus on advanced composites. When these biopolymers are appropriately cross-linked, they yield final materials with outstanding mechanical properties, characterized by high strength-to-weight ratios and resistance to fatigue. This makes them ideal for applications in high-performance environments, such as aerospace structures and the automotive industry. Besides, the incorporation of furan rings into the polymeric backbone enhances mechanical and thermal properties, such as higher glass transition temperatures, in comparison to petroleum-based analogues, like resins based on the diglycidyl ether of bisphenol A (DGEBA).214 These high values were attributed to the symmetry of the monomers and the presence of an oxygen atom in the furan ring, which enables the formation of inter-chain hydrogen bonding interactions. These interactions lead to distinct performance characteristics for furan-based epoxides when compared to their conventional counterparts, including relatively higher modulus and Tg.210,212,216
Both conventional diamines, such as 4,4′-methylenebiscyclohexanamine (PACM) or diethyl toluene diamine (EPI),220 and bio-based furanic diamines (Fig. 13) have been explored as curing agents for the synthesis of these innovative epoxy resins. This versatility allows for the tailoring of epoxy resin properties to meet specific application requirements and environmental considerations. When these biopolymers are appropriately cross-linked, they resulted in materials with outstanding mechanical properties, characterized by high strength-to-weight ratios and resistance to fatigue. This makes them ideal for applications in high-performance environments, such as aerospace structures and the automotive industry. Additionally, their excellent thermal stability renders them suitable for use in high-temperature settings. Furthermore, starting from different furan epoxides and employing various crosslinking agents and curing agents (as illustrated in Fig. 13), the properties of these materials can be tailored to achieve low dielectric constants and high electric insulation. This makes them invaluable in the production of printed circuit boards (PCBs) and the encapsulation of electronic components. Moreover, these polymers can be formulated to exhibit high adhesion properties, ensuring secure bonding in PCBs. This adhesion is crucial for maintaining signal integrity and preventing delamination, making them a preferred choice in the electronics industry for various applications.
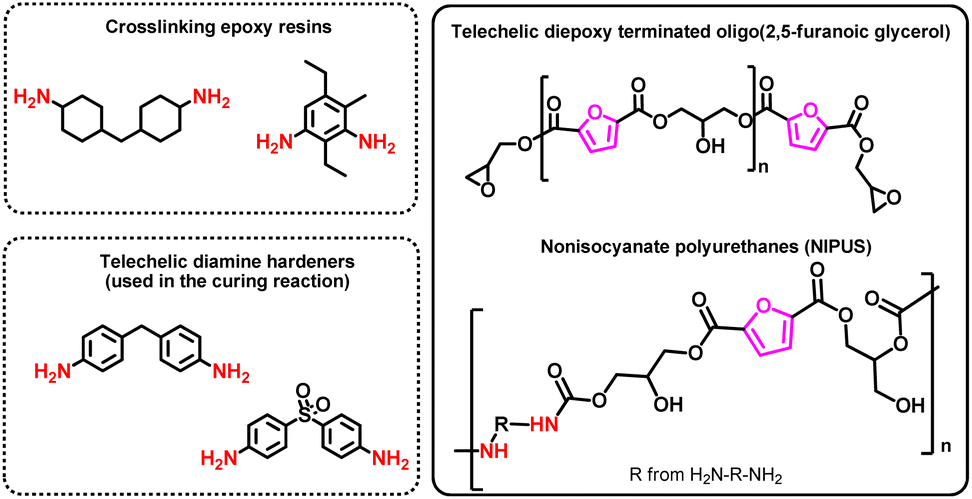 | ||
| Fig. 13 Some crosslinking epoxy resins, curing agents and oligomers/polymers from epoxide furoic derivatives. | ||
6. Alkyl carbonate-based furanics
6.1. Synthesis of alkyl carbonate furanic monomers
As above reported (Section 3), reduction of the aldehyde functionality of HMF and OBMF lead to diols the BHMF and OBMF-H2, respectively. These substrates can be easily converted into (bis)alkylcarbonate derivatives that represent another example of interesting monomers with potential applications for the preparation of bio-based polycarbonate and polyurethanes (Scheme 10).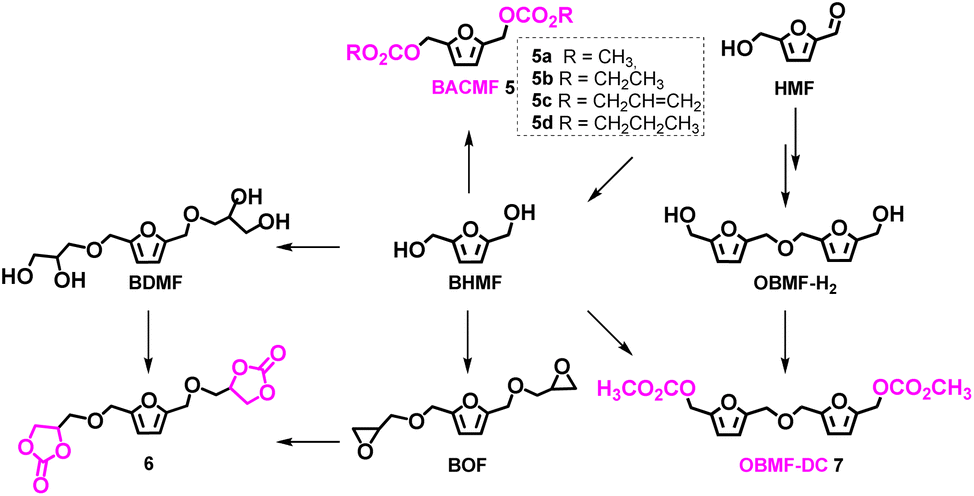 | ||
| Scheme 10 Synthetic pathways to alkylcarbonate-based monomers. | ||
Table 14 summarizes the synthetic procedures for the furan alkylcarbonates starting from BHMF and OBMF-H2.
| # | Substrate (g mmol−1) | Conc. (M) | Catalyst (mol%) | Reaction conditions | Yielda (%) | E-f | PMI | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Isolated yield. b Hünig's base. c Quantities not reported. d Experiment conducted in continuous-flow. | ||||||||
| 1 | BHMF (0.5/3.9) | 0.39 | K2CO3 (200) | DMC; 90 °C, 6 h | 5a, 87 | 13.6 | 14.6 | 221 |
| 2 | BHMF (0.5/3.9) | 0.28 | K2CO3 (200) | DEC; 140 °C, 6 h | 5b, 76 | 18 | 19 | 221 |
| 3 | BHMF (0.5/3.9) | 0.71 | K2CO3 (200) | All2C; 140 °C, 6 h | 5c, 70 | 7.8 | 8.8 | 221 |
| 4 | BHMF (0.1/0.78) | 0.64 | DIEAb (400) | DPC; 120 °C, 12 h | 5d, 10 | 75 | 76 | 222 |
| 5 | BOMF | Neat | La(III) scorpionate complexes (0.5) | TBAB; CO2, 70 °C, 4 h | 6, 85 | — | — | 223 |
| 6 | BOMF (1.2/5) | Neat | Al(III) scorpionate complexes (0.25) | CO2, TBAB; 80 °C, 3 h | 6, 98 | — | — | 224 |
| 7c,d | BDMF | 1.0 | TBAB (3.5) | DMSO, TBAB, DMC; 180 °C, residence time 9 min | 6, 69 | — | — | 225 |
| 8 | OBMF-H2 (1/4.2) | 0.08 | K2CO3 (220) | DMC; 90 °C, 6 h | 7, 97 | 37.7 | 38.7 | 40 |
BHMF was reacted with selected dialkylcarbonates (DACs) to achieve a library of bis-alkylcarbonate derivatives (BACMF), as shown in Scheme 10. As an example, the reaction of BHMF with dimethyl carbonate (DMC) gave the BACMF 5a namely 2,5-bis[(methoxycarbonyl)oxymethyl]furan.
Diethyl carbonate (DEC), diallyl carbonate (All2C) and dipropyl carbonate (DPC) were also used as both reagent and solvent to achieve the related BACMF 5b–5d in yield up to 87% (#2–4; Table 14) employing potassium carbonate in large excess as a base. It should be pointed out that for the calculations of the green metrics the total amount of DAC employed was split into two contributes. The first (as reagent) was calculated as the stoichiometric amount of DAC required to react with the reagent to form the final product. The second (as solvent) was calculated subtracting the moles of DAC “reagents” to the total.
Since K2CO3 was used in stoichiometric amounts, and because it was not recovered and reused, green metrics resulted quite high. PMI values ranged from 8.8 for BACMF 5c up to 76 for compound 5d.
The reaction of BHMF with DPC was conducted employing N,N-diisopropylethylamine (DIEA or Hünig's base) (#4; Table 14) The higher E-f and PMI values compared to the previously discussed procedures is ascribable to the large amount of base and the poor yield of the desired product 5d (10% yield).
Furanic cyclic carbonate 6 can be prepared by metal catalysed CO2 insertion into 2,5-bisepoxy furan BOF (Section 5) or by reaction of 3,3′-((furan-2,5-diylbis(methylene))bis(oxy))bis(propane-1,2-diol) (BDMF) with DMC (#5–7; Table 14). Unfortunately, E-f and PMI cannot be calculated for these procedures because technical specifications of the synthesis were not reported.
For what concerns OBMF-H2, the only trial so far reported with organic carbonates was performed with DMC; the related product OBMF-DC 7 was isolated in almost quantitative yield. The resulting high values of the green metrics have to be ascribed once again to the use of the excess of base (#8; Table 14).
Alkyl carbonates-based furanics are still only preliminary investigated, so future investigations are definitely desirable and should be focused on improving the reaction conditions (recovering and reuse of the reagents/catalysts) and purification methodologies.
6.2. Bio-based polymers from alkyl carbonate furanic monomers
Polyurethanes are typically synthesized using hazardous isocyanates, which can have serious health effects.226–228 An alternative approach would be the use as monomers bis-cyclic carbonates, obtained via CO2 insertion into bis-epoxides.These monomers can undergo step-growth polyaddition with diamines under mild reaction conditions yielding a wide range of non-isocyanate polyurethanes (NIPUs) that feature hydroxyl groups on the β-carbon of the urethane moieties.229–232
NIPUs can be easily produced using renewable resources as starting materials.233–235
In this view, Li and co-workers has reported the preparation of two furan-based NIPUs encompassing a different chain length in the diamine units (Scheme 11).236
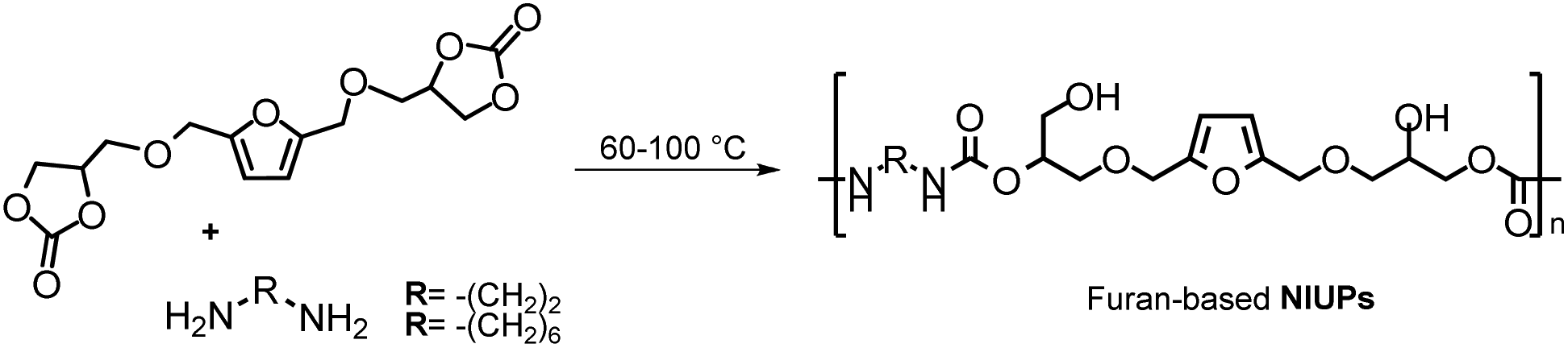 | ||
| Scheme 11 NIPUs from bis-cyclic carbonate furanic. | ||
Solvent-free and catalyst-free step-growth polymerizations of bis-cyclic carbonate furan were conducted in the presence of 1,2-ethanediamine (EDA) and 1,6-hexanediamine (HDA). Due to the volatility of EDA and HDA, a temperature program was chosen starting from 60 °C and gradually raise the temperature up to 100 °C. The so-formed NIPUs had a high molecular weight (Mn = 26700–34100 g mol−1). Furthermore since both the –OH and –NH units of the NIPUs are capable of forming hydrogen bonds with carbonate units, poly(propylene carbonate) (PPC) blends containing furan-based NIPUs as the second component were successfully prepared.
Glass transition temperatures, tensile strengths, and elongations at break have been significantly improved. In particular, with the addition of 5.0% by weight of furan-based NIPUs, the tensile strengths of the blends exceeded 30 MPa, approximately double that of neat PPC and equivalent to those of commercial polyethylene.
It is evident that NIPUs prepared from bis-cyclic furans have been so far only preliminary investigated.
Very recently a fully renewable polycarbonates (PCs) has been reported starting from cellulose-based platform molecules levoglucosenone (LGO) and BACMF 5a (Scheme 12). To promote the polymerization reaction, 2 mol% of Na2CO3 resulting in the bio-based PC with the highest Mn value (755000 g mol−1), as well as Td5% (197 °C).237
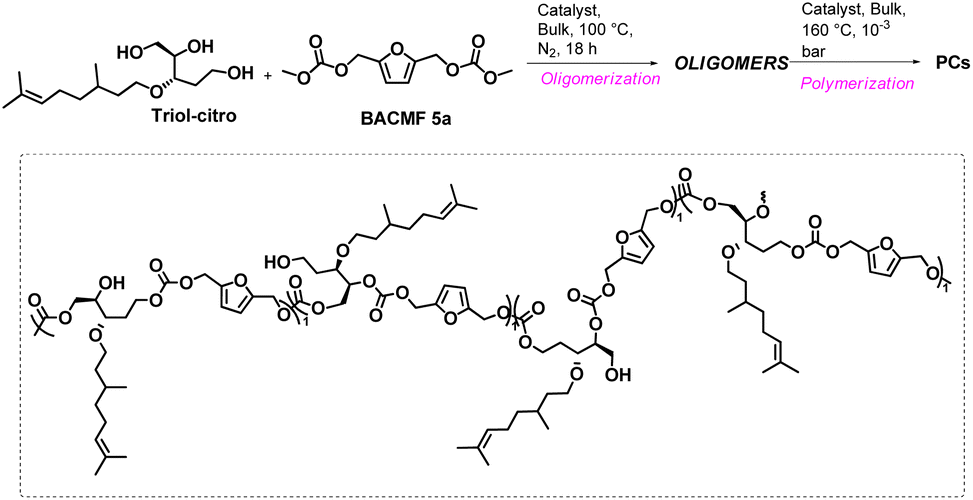 | ||
| Scheme 12 NIPUs from bis-cyclic carbonate furanic. | ||
7. Furan dicarboxylic acid monomers beyond 2,5-FDCA and polymers
7.1. Synthesis of 2,4-furan dicarboxylic acid
In 1901, Feist reported the first synthesis of 2,4-furandicarboxylic acid (2,4-FDCA).238 2,4-FDCA was synthetized in three steps: (i) bromination of methyl coumalate, followed by (ii) hydrolysis and (iii) cyclization with a strong acid (Scheme 13a). Numerous attempts to reproduce this method revealed that indeed 2,4-FDCA could only be recovered in poor yield.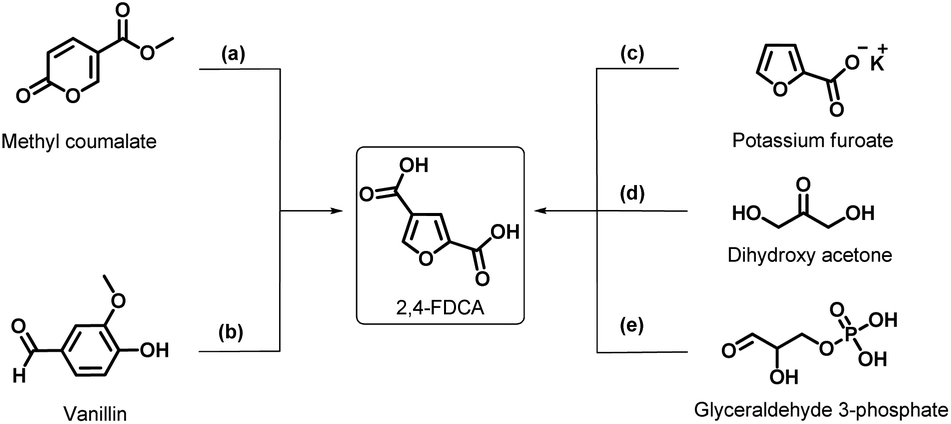 | ||
| Scheme 13 Synthesis of 2,4-FDCA from different substrates. | ||
Pearl and coworkers also described the use of vanillin (Scheme 13b) to synthesize 2,4-FDCA, but once again with very low isolated yield (3%).239 Later on, a few more strategies were also identified, requiring time-consuming or sequential steps; unfortunately, none of them succeeded in significantly increase the overall yield of 2,4-FDCA.240–242
In 1952, Raecke and co-workers reported a solid-state chemistry reaction, also known as disproportionation reaction, which involves the thermal rearrangement of monocarboxylate alkaline salt to synthesize symmetrical aromatic dicarboxylates in the presence of metallic salt (catalyst) and carbon dioxide.246
This process, known since then as the Henkel/Raecke process, was promptly used for the commercial production of terephthalic acid (TA) from potassium benzoate, but later on abandoned as the catalytic oxidation of p-xylene to produce TA was proven to be a more economically viable route.
In recent years, Thiyagarajan et al. performed a solvent-free Henkel-type disproportionation of potassium furoate that starts with low food-value agro-residues to synthesize 2,5-FDCA (Scheme 13c).243 In addition to this symmetrical compound, two isomers (2,4-FDCA and 3,4-FDCA) were also obtained (Scheme 14).243
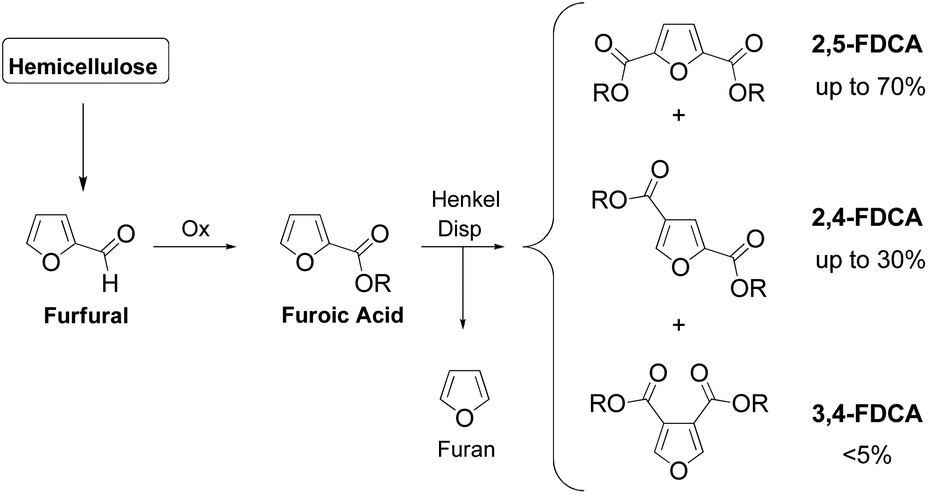 | ||
| Scheme 14 Production of three FDCA isomers through the Henkel disproportion. | ||
The authors reported that the selectivity towards 2,4-FDCA (up to 30%; 70% 2,5-FDCA) depended on the type of catalyst employed and on selected experimental condition (#1; Table 15). 3,4-FDCA was only obtained in <5% yield.
| # | Reaction type | Substrate (g mmol−1) | Conc. (M) | Catalyst (g) | Reaction conditions | Yieldb (%) | E-f | PMI | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Dis = disproportion, Mul = multistep. Work-up and purification material not included in the metrics calculations. b All yields are isolated. c Amounts of the first step normalized with respect to the second step. | |||||||||
| 1 | Dis | Potassium-2-furanoate (10/66.7) | Neat | CdI2 (53 wt%) | 260 °C, 5.5 h | 27 | 40.2 | 41.2 | 243 |
| 2 | Dis | Potassium-2-furanoate (0.3/1.7) | Neat | CuI (13 wt%) | CO2, 40 bar; 300 °C, 16 h | 7 | 54.3 | 55.3 | 244 |
| 3c | Mul | 1,2-DHA (0.2/2.7) | 0.67 | (i) Amberlite® IRA-900 (100 wt%); (ii) Amberlyst-15 (50 wt%) | (i) H2O; 0 °C, 24 h; (ii) NaOH, KMnO4, H2O, 0 °C, 15, pH 1 | 66 | 112.3 | 113.3 | 245 |
A recent patent application from Iowa State University describes the use of copper(I) iodide (CuI) as a catalyst in the disproportionation of heteroaromatic carboxylate (potassium furoate) and reports 7% selectivity to 2,4-FDCA compared to the main isomer 2,5-FDCA (93%) (#2; Table 15).244 Both these syntheses showed a quite high values of PMI and E-factor.
A similar procedure to the well-known method for synthesizing 2,5-FDCA through the catalytic oxidation of HMF was reported allowing the synthesis of 2,4-FDCA from the oxidation of 4-hydroxymethyl furfural (4-HMF).245
Under mild oxidation conditions, glycerol gives dihydroxy acetone with high selectivity, which then undergoes a self-condensation reaction resulting in branched chain hexoses. In the presence of an acid catalyst, the branched chain hexoses undergo a dehydration reaction that eliminates three molecules of water, producing the stable 4-HMF. It has been demonstrated that this produces the 2,4-FDCA with an overall isolated yield of 66% (#3; Table 15) when it is oxidized with potassium permanganate from dihydroxy acetone (Scheme 13d).245 In this case, as well, green metrics show very high values compared to several other furanic monomers.
Alexandrino and coworkers at Braskem, a Brazilian petrochemical company, recently developed the direct fermentation method to produce 2,4-FDCA in a recombinant microorganism from glyceraldehyde-3-phosphate (G3P).247,248 The latter is effectively produced from carbon feedstock, such as glucose, xylose, and glycerol, which is catalysed into [5-formylfuran-3-yl]methyl phosphate by methyl phosphate synthase. The phosphatase catalyses the conversion into 4-HMF, which is then oxidized to produce the final product, 2,4-FDCA, either directly or by producing a number of intermediates, such as furan-2,4-carbaldehyde (Scheme 13e). Unfortunately, in this case it was not possible to evaluate the green metrics.
It is evident that the methods developed so far for the synthesis of 2,4-FDCA are significantly meagre compared to its analogue 2,5-FDCA. This is viewed as a barrier to further exploration and realization of the potential of this unsymmetrical unique monomer in a variety of applications. Nevertheless, there are reports that describe the structural features of 2,4-FDCA and its impact on the polymer properties, compared to the well-researched 2,5-FDCA and its benzene analogue TA. It is noteworthy that the combination of this monomers with 2,5-FDCA appears full of promises even when used in small amounts. Future investigations should focus on the synthesis of 2,4-FDCA in large scale including optimization of the reaction conditions.
7.2. Bio-based polyesters from 2,4-FDCA
In terms of linearity, the structure of 2,4-FDCA is more similar to TA compared to 2,5-FDCA.243 The comparison is made based on (a) the distance between carboxylic acids, and (b) the angle between the carbon atom and the carboxylic functionality attached to it, either in the furan or in the benzene ring (Fig. 14). This insight encouraged to investigate the structure–property relationships of homo-polyesters derived from 2,4-FDCA to compare furan and benzene analogues.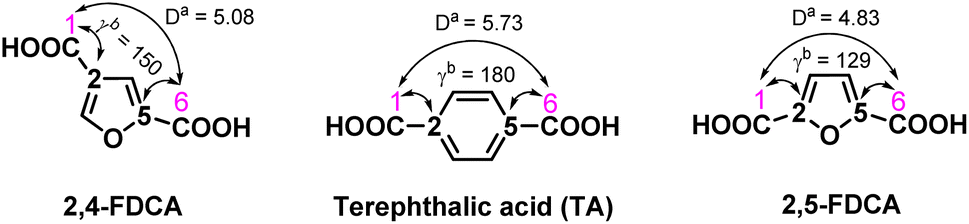 | ||
| Fig. 14 Atom labelling and structural parameters for 2,4-FDCA and 2,5-FDCA compared to TA. Da is the interatomic distance (Å) between C1–C6 (carboxylic acid groups) and γb is the projected angle (°) between the C1–C2 bond and C5–C6 bond. | ||
Similar to the classical procedure used to synthesize poly(ethylene terephthalate) (PET) and poly(ethylene furanoate) (2,5-PEF), the melt-polycondensation of 2,4-FDCA dimethyl ester in combination with ethylene glycol (EG) provided high molecular weight polyethylene furanoate (2,4-PEF) polyester, while in combination with other linear diols (1,3-propane diol and 1,4-butane diol) 2,4-PPF and 2,4-PBF are respectively obtained (Scheme 15).249 The high molecular weight obtained from the 2,4-FDCA monomer further confirmed that carboxylic esters have similar reactivity for the synthesis of both benzene and furan analogues. However, position isomerism and random distribution of polymer repeating unit resulted in a considerable effect on the aptitude to crystallize, because 2,5-PEF samples appeared opaque after cooling from the melt, whereas 2,4-PEF samples appeared translucent. Zaidi et al. studied the optical properties of 2,4-PEF highlighting the high transmittance in the visible region (400–700 nm) and thus its optically transparency.250 Thermogravimetric analysis (TGA) showed that thermal stability is not affected by FDCA position isomerism, and that 2,4-FDCA-based polyesters have an even better thermal stability compared to the 2,5-FDCA-based analogues.249
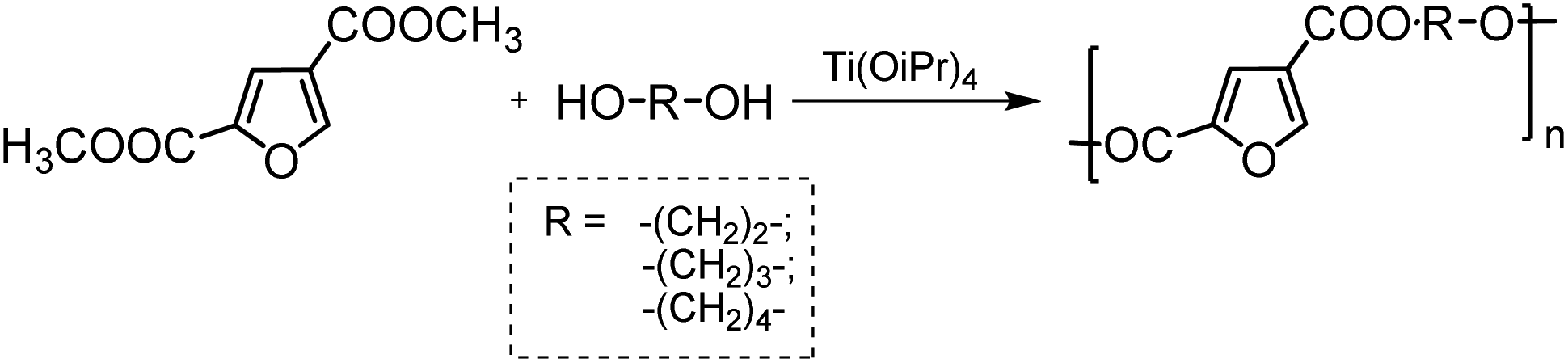 | ||
| Scheme 15 Synthesis of FDCA isomers (2,5- and 2,4-) based polyesters. | ||
The Tg of 2,4-FDCA-derived polyesters are slightly lower compared to 2,5-FDCA-derived analogues for all the three diols investigated. The difference in symmetry of the FDCA isomers also impacts the crystallization and melting behaviours; no melting point is detected for 2,4-PEF even after annealing at 90 °C for 5 h. Wide-angle X-ray diffraction (WAXD) analyses further showed that the homopolyesters synthesized from the 2,4-FDCA isomer are amorphous in nature, whereas the use of 2,5-FDCA produces semi-crystalline materials.249
A series of copolyesters using different mixtures of 2,5- and 2,4-FDCA (physical blends and the mixtures directly obtained from Henkel reaction) in combination with different diols were also reported.251 Detailed investigations disclose that, in most cases, the mixture of FDCA-isomers obtained during the Henkel reaction can be in principle directly used to synthesize copolyesters with significantly good properties, without any downstream processing to separate and purify the isomers. This conclusion is interesting since it may considerably reduce the time and costs for biomass valorization. In terms of properties, it was shown that the incorporation of the 2,4-FDCA isomer into 2,5-PEF has a significant effect on Tg, Tm and the overall crystallization process. In particular, an amount of 2,4-FDCA-based repeating units as little as 5–10 mol% is sufficient to disrupt PEF crystallization.
Bourdet et al. compared two copolymers obtained with ethylene glycol and different ratios of 2,5- and 2,4-FDCA to the homopolymers 2,5-PEF and 2,4-PEF.252 They observed that the incorporation of 2,4-FDCA-based units into a polymer backbone mainly constituted of 2,5-FDCA-based units is responsible for longer β relaxation times, but has no striking effects on the fragility index m, on cooperativity, and on the activation energy for the α relaxation process in the liquid state (T > Tg), however they observed a decrease in the activation energy in the glassy state (T < Tg), which suggests a possible effect on physical aging.
Zaidi et al. could develop transparent PET-co-2,4-PEF films by carefully controlling the amount of 2,4-FDCA introduced in the 2,5-FDCA-based copolyesters, taking advantage of the resistance of 2,4-FDCA-based repeating units to crystallize.250 This feature, that clearly opposes 2,4-FDCA and 2,5-FDCA, was later on explained by Nolasco et al. via a combination of vibrational spectroscopy techniques and ab initio calculations.253 In particular, the films developed by Zaidi et al. were found to be semi-crystalline for contents of 2,4-FDCA up to 15 mol%, fully amorphous and perfectly transparent for contents of 50 mol% and above.250 The use of 2,4-FDCA as a co-monomer to improve the transparency of polyester films may still need some improvements, but it is promising for applications such as transparent packaging, plastic windows, headlight lenses, contact lenses, etc. The properties of the 2,4-FDCA isomer have also been found to be more suitable than the properties of TA in the synthesis of nematic liquid crystal materials for LCD displays.245
Bianchi et al. used 2,4-FDCA to synthesize poly(butylene 2,4-furanoate) (2,4-PBF) in combination with 1,4-butanediol and compared it to its analogue 2,5-PBF.254 Both the homopolyesters were processed into films by compression molding. Thermal analysis showed a reduction of both Tg and Tm, and a decrease in the aptitude to crystallize. Interestingly, 2,4-PBF is thermally more stable than 2,5-PBF, is less hydrophilic (likely due to the lower value of the average dipolar moment),255 and therefore less prone to hydrolysis, is more flexible and tough, and has even better barrier properties compared to 2,5-PBF. Even more interestingly, the gas barrier properties were measured as outstanding both in dry and humid conditions, but only for amorphous films.
Bourdet et al. recently focused on the effects of isothermal crystallization in homo- and random co-polyesters containing different amounts of 2,4-FDCA-based units (0, 10 and 15 mol%, i.e. in the composition range where crystallization is still possible, even though disrupted).256 They concluded that the partial replacement of 2,5-FDCA by its position isomer could help controlling crystallinity analogously to what happens when ethylene glycol is partially replaced by cyclohexane dimethanol in glycol-modified polyethylene terephthalate (PETg).
Almost at the same time, Braskem filed a patent application proving that 2,4-FDCA can be used to synthesize copolyesters with increased crystallization control, in amounts as low as 0.1 to 10 mol% with respect to the total molar content of the dicarboxylic acid component.257 They reported a slower crystallization rate and higher gas barrier properties to CO2 and O2 due to the incorporation of 2,4-FDCA-based units in PET. The same company had previously filed another patent suggesting to use the 2,4-isomer of FDCA diesters for the production of plasticizers, aiming at better controlling physical properties such as flexibility, durability and processability.258 Braskem explored several routes to synthesize 2,4-FDCA and use it to produce molecules possibly interesting as plasticizers, in variable amounts and for a large panel of different polymers, with or without the addition of other components (e.g. stabilizers, fillers and reinforcements, expansion agents, lubricants, pigments, flame retardants, etc.).
8. Ester based furanics
The low solubility, high melting point and boiling point of FDCA hinder its use and purification.259 Therefore, ester derivatives of FDCA (FDME), such as furan-2,5-dimethylcarboxylate (FDMC), have gained an increasing attention as alternatives for FDCA. FDMC has lower boiling point compared to FDCA (ca. 270 °C vs. 420 °C, respectively), which enables its efficient purification by vacuum distillation.260 Furthermore, the ester derivatives are more soluble in common solvents, enabling more efficient synthesis and purification.The so far reported synthetic routes to FDCA esters from biomass can be divided into two pathways based on the intermediate molecule used (Scheme 16) (i) oxidative esterification and (ii) acid catalysed cyclisation starting from galactaric acid.
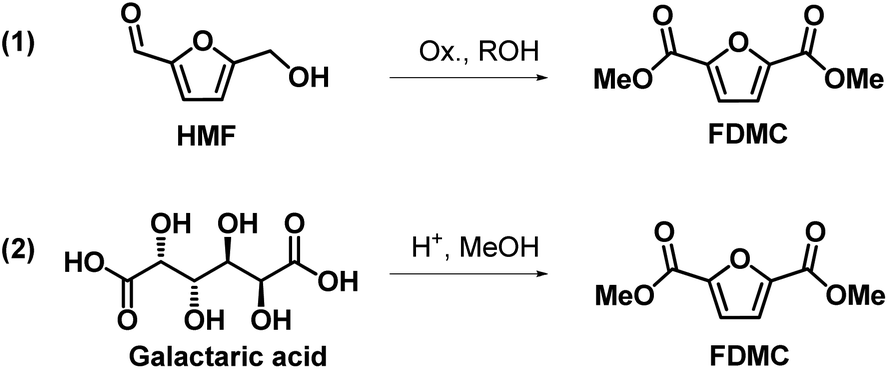 | ||
| Scheme 16 The two pathways to FDCA esters from carbohydrates (1) oxidative esterification; (2) acid catalysed cyclisation from galactaric acid. | ||
8.1 Ester based furanics by oxidative esterification
The advances in oxidative esterification of HMF into FDMC have been covered by several dedicated reviews.259–261Table 16 reports several FDCA esters syntheses employing this approach. Unfortunately, for most of the procedures present in literature (see also ESI; Table S.14†) it was not possible to calculate the green metrics due to missing information such as autoclave capacity, pressure of O2 employed, etc. Nevertheless, a brief explanation of the synthesis procedure is given in the main text.| # | Substrate (g mmol−1) | Conc. (M) | Catalyst (mol% or wt%) | Reaction conditions | Yield (%) | Cat. reuse | E-f | PMI | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Recovery of work-up solvent. b Isolated yield. c Catalyst was recycled. | |||||||||
| 1a | HMF (0.25/2) | 2.0 | Co@CN (3.8 mol%) | O2 1 bar, MeOH; 80 °C, 24 h | FDMC 84b | 5 | 17.8 | 2.9 | 262 |
| 2 | HMF (0.5/4) | 0.3 | Au/TiO2 (50 wt%); MeONa (7.5 mol%) | O2 4 bar, MeOH; 130 °C, 3 h | FDMC98 | — | 16.5 | 17.5 | 263 |
| 3 | HMF (0.25/2) | 0.1 | Au/HAP (1 mol%) | Air 24 bar, MeOH; 130 °C, 6 h | FDMC 89 | — | 55.5 | 56.2 | 264 |
| 4c | HMF (0.2/1.5) | 0.08 | Au/CeO2 (25 wt%) | N2 10 bar, O2 0.33 mL s−1 MeOH; 130 °C, 5 h | FDMC 99 | 5 | 55.7 | 56.5 | 265 |
| 5 | HMF (0.03/0.25) | 0.1 | CoxOy–N@C (39 wt%); K-OMS-2 (39 wt%); K2CO3 (23 wt%) | O2, 10 bar, MeOH; 100 °C, 6 h | FDME 96 | 37.6 | 38.3 | 266 | |
In a typical approach HMF oxidation was carried out employing nitrogen-doped catalysts obtained using metal–organic frameworks (MOF) as template.
Sun et al. used cobalt-containing ZIFs and a protective silica layer to produce a hollow yolk–shell Co@CN catalyst able to efficiently oxidised HMF into FDM at high 2 M concentration with 84% isolated yield using 1 bar O2 (#1; Table 16).262 After five reuses, the catalyst showed only minor loss of activity as a result the calculated PMI is very low.
In 2008, Christensen et al. reported the first example of oxidative esterification of HMF by oxidation via a gold catalyst (#2; Table 16).263 Addition of a catalytic amount (8 mol%) of sodium methoxide was needed to reach high conversion and yield (98% FDMC). Green metrics values are quite high most probably because of the very low concentration of substrate used.
The requirement of additional base is a major drawback in HMF oxidation, as it increases waste, complicates purification and is conflicting with the principles of green chemistry. The role of base has been attributed to promoting the reaction of aldehyde into hemiacetal which is then oxidized into the ester.260
Mishra et al. used gold supported on hydroxyapatite (HAP) for base-free oxidative esterification HMF; 89% FDMC was obtained using air as oxidant (#3; Table 16).264 The strong basic sites of the support could function as the base. Stability of the catalyst was shown in six consecutive runs with small loss of activity, which could be restored by calcination in air to remove organic contaminants.
Interestingly, gold supported on nanoparticulated ceria (Au/CeO2) oxidized HMF into FDMC with 99% yield even in the absence of base (#4; Table 16).265 According to this study, defects on the ceria support were able to activate oxygen and stabilize the gold particles. After five reuses, the catalyst gave over 90% FDMC yield even though the reaction time was increased. Unfortunately, both these synthetic approaches led to quite high PMI and E-f values.
Kim et al. showed an effective strategy to increase substrate concentration using HMF acetals instead of the less stable HMF.272
Oxidation of 1 wt% HMF in methanol with Au/CeO2 and Na2CO3 gave 91% yield of FDMC (#1; Table S.14†), whereas increasing the concentration to 10 wt% gave only 4% yield due to side reactions. However, protection of the HMF aldehyde group using diols, such as 1,3-propanediol (PD), enabled up to 20 wt% substrate concentrations; 92% FDMC was produced from 10 wt% solution of PD-HMF (#2; Table S.14†). Furthermore, changing the solvent to DMF and ethylene glycol gave 91% yield of bis(2-hydroxyethyl)furan-2,5-dicarboxylate (HEFDC) (#3; Table S.14†), which could directly be polymerized into PEF by self-condensation.
Gold catalysts have shown exceptional selectivity in oxidation reactions. Interestingly, commonly used noble metal catalysts like Pt, Ru, Pd showed little activity or selectivity towards oxidative esterification of HMF.273 The high price of noble metal catalysts has driven the development of catalysts based on cheap metals to increase the feasibility of the processes in industrial scale. Cobalt-based catalysts especially on nitrogen-doped carbon supports have proven efficient catalysts for various organic transformations, such as reductions and oxidations.274 As a result, several cobalt based catalysts were investigated resulting in good to excellent yield in the oxidation of HMF (#5, Table 16 and #4–11, Table S.14†). A bimetallic catalyst composed by cobalt and ruthenium deposited on carbon particles was developed so to work under flow conditions.275 FDMC was produced at 57% selectivity and 98% conversion with only 10 min residence time (#5; Table S.14†). Compared to the batch reaction, over 15-fold increase in production of FDMC was obtained (from 0.03 mmol h−1 to 0.47 mmol h−1 FDMC), showing promise in transferring the FDMC synthesis to industrially applicable flow conditions.
In a recent study, a combination of cobalt and iron on N-doped carbon showed high performance in HMF oxidation; 93% FDMC was obtained in a dilute HMF solution (#10; Table S.14†), and 92% FDMC in a 10 wt% solution at a prolonged reaction time.276 The Co7Fe3-NC catalyst was further applied to continuous flow conditions showing high 91% FDMC yield and good stability over the 80-hour experiment (#11; Table S.14†).
Pyrolysis of a cobalt complex bearing a nitrogen-containing ligand, e.g. 1,10-phenanthroline, onto carbon support generates cobalt nanoparticles stabilized by nitrogen in the graphitic carbon surface.266 Together with a porous manganese oxide and base as co-catalysts, CoxOy–N@C produced total of 96% FDCA mono- and dimethyl esters (FDME) in oxidative esterification of HMF (#5; Table 16). Once again, the green metrics resulted quite high.
A above reported several examples of bimetallic gold catalysts have been studied in oxidation reactions to enhance activity and stability of the catalysts.277 Du et al. showed that copper-doping improved the selectivity of alumina-supported gold catalysts; 48% and 98% FDMC were obtained with Au/Al2O3 and AuCu/Al2O3, respectively (#12; Table S.14†).278 The catalyst was used in five consecutive reactions with minimal loss of activity.
8.2. Synthesis of ester based furanics from sugar acid
Aldaric acids like galactaric acid (GalA) are attractive bio-based chemicals for food, pharmaceutical and chemicals industries.279 The most crucial benefit of aldaric acids is their high stability compared to HMF, which allows to circumvent the challenges with HMF synthesis, purification and storage.The first synthesis of FDCA employing GalA was reported in 1876 by using an excess HBr.280 Subsequently, a handful of reports have described FDCA synthesis in moderate yields using an excess of strong acids.281,282
Very recently van Strien et al. used a catalytic amount of solid acid catalyst to produce furan-2,5-dicarboxylate butyl esters (mono- and diester, FDBE) in up to 76% yields using dibutyl galactarate (GABE) as the substrate (#1; Table 17).267 Esterification prior to the dehydration increased the solubility of GalA (up to 20 wt% concentration was used) and enhanced the yields. In terms of green metrics this is so far the best performing reaction among the one starting from sugar acids.
| # | Substrate (g mmol−1) | Conc. (M) | Catalyst (mol% or wt%) | Reaction conditions | Yield (%) | E-f | PMI | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Metrics include purification. b Isolated yield. Abbreviations can be found in the dedicated section. | ||||||||
| 1 | GABE (2.0/6.2) | 0.62 | Si-tosic acid (5 wt%) | n-BuOH; 220 °C, 4 h | FDBE 76 | 7.6 | 8.6 | 267 |
| 2 | GalA (2.0/9.6) | 0.81 | H2SO4 (21 mol%); Si-tosic acid (5 wt%) | n-BuOH; (i) 120 °C, 24 h; (ii) 220 °C, 4 h | FDBE 62 | 10.4 | 11.4 | 267 |
| 3 | GalA (0.2/1.0) | — | MSA (200 mol%) | (i) Neat; 160 °C, 0.5 h; (ii) EtOH; 90 °C, 16 h | FDEC 30 | 67.3 | 68.3 | 268 |
| 4a | GalA (1.0/4.8) | 0.14 | Amberlyst-36 (50 wt%) | DMC; 200 °C, 2 h | FDMC 70b | 85.6 | 86.6 | 269a |
| 5a | GalA (5.0/23.8) | 0.5 | H2SO4 (200 wt%) | n-BuOH; 118 °C, 7 h | FDBC 64b | 185.4 | 186.4 | 270 |
| 6 | GAME (2.0/8.4) | 0.84 | Si-tosic acid (1 mol%) | MeOAc, 220 °C, 4 h | FDME 52 | 13.8 | 14.8 | 271 |
| 7 | GAME (700/2800) | 0.56 | Si-tosic acid (1 mol%) | MeOAC, 235 °C | FDME 49 | 20.7 | 21.7 | 271 |
A one-pot process including both the esterification and dehydration steps gave 62% yield of FDBE (#2; Table 17). To expand the substrate scope to glucose-based feedstocks, glucaric acid dibutyl ester (GlBE) was also shown to give similar yields of FDBE.
Zhao et al. performed a two-step synthesis of furan-2,5-diethylcarboxylate (FDEC); the dehydration of GalA into FDCA was carried out with 2 equivalents of methanesulfonic acid (MSA) without solvent. Addition of ethanol produced the ester FDEC in overall 30% yield (#3; Table 17) and quite high values of green metrics.268
Trapasso et al. employed dimethyl carbonate as a solvent and esterification agent in GalA dehydration with Amberlyst-36 catalyst.269a More recently also Fe2(SO4)3 have been also used as catalyst for the same chemical transformation.269b FDMC was isolated in high 70% yield, although the solvent decomposed during the reaction leading to increased pressure (#4; Table 17).
In 2008, Taguchi et al. reported the synthesis of furan-2,5-dibutylcarboxylate (FDBC) by refluxing GalA in n-butanol with excess H2SO4 (#5; Table 17).270 Sulfuric acid catalysed both the esterification of GalA as well as the dehydration reaction to form the furan ring, giving FDBC in 64% yield.
Although an interesting synthetic approach, the absence of recycling of catalyst and/or solvent led to high values in the metrics surely improving the reaction conditions would be beneficial for further exploitation in larger scale trails for instance employing continuous flow apparatus.
Very recently FDCA methyl esters (FDMC) were prepared using silica-supported tosic acid (#6 and #7; Table 17).271 The procedure led to FDMC in 52% yield starting from 2 grams, but interestingly it showed to the scalable up to 700 grams of GalA. Purification of the crude product by vacuum distillation and precipitation gave FDMC with 98%purity. One of the main advantages of these two procedures is that they are carried out at high concentrations, leading to very similar PMI values (14.8 and 21.7 respectively).
Alternative synthetic pathways to FDCA and its esters are of pivotal importance for the future development of this bio-based monomers. Some significant advances have already been made especially in the use of galactaric acid as starting compounds. In this view, future work should particularly focus on exploiting new and more efficient catalytic system so to perform this reaction in milder reaction conditions.
8.3. Bio-based polymers from furanic esters
FDCA esters have similar potential applications of FDCA thus research on new synthetic pathways for this bio-based monomers is increasing in the last years. Despite the interest in these molecules their exploitation for the preparation of novel bio-based monomer is still limited. To the best of our knowledge so far only an article has been reported focussing on a solvent-free preparation of glycerol-based polyesters.268In particular after the synthesis of FDCE from mucic acid different furan-based polyesters were prepared i.e., polyethylene-2,5-furan dicarboxylate (PEF) and polyhydropropyl-2,5-furan dicarboxylate (PHPF). In a typical reaction FDCE was reacted with a diglycerol or a diol furan-based prepolymer in the presence of Co(Ac)2·4H2O (Scheme 17). No details were reported on the polymer molecular weight.
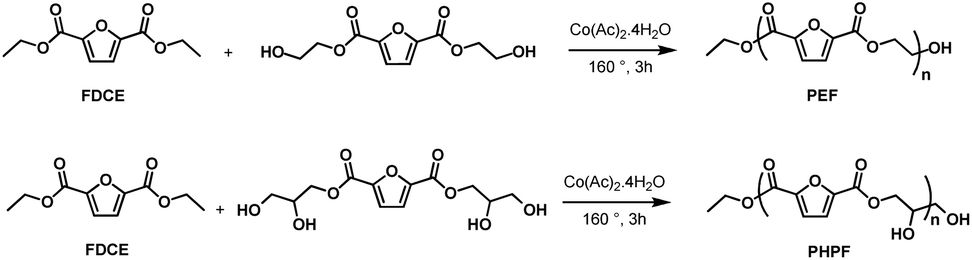 | ||
| Scheme 17 Examples of furan-based polyesters from FDCE. | ||
9. Bi-cyclic furanic monomers derived from 5-HMF and furfural
9.1. Bi-cyclic furanic monomers
The [4 + 2] cyclo-addition reaction between a conjugated diene and dienophile, also known as Diels–Alder reaction has been known since 1928.The reaction is very efficient yielding the product (DA adduct) in near quantitative yield while using an appropriate diene containing electron withdrawing group (EWG), for example, substituted furan, and a dienophile with electron donating group (EDG), for example, maleic anhydride.
An important feature of this reaction is, the fact that the DA adduct formed is thermally reversible, i.e., the equilibrium is tuneable and shifted toward the starting substrates by heating the DA adduct in the presence (or) absence of solvents and catalysts, this reversible reaction is called as retro Diels–Alder reaction (r-DA). This interesting characteristic prompted many researchers to construct thermally reversible cross-links for example, thermoset-like material, using oligomeric/polymeric dienes and dienophiles.283
Very recently, Thiyagarajan et al., demonstrated a slightly modified approach to synthesize functionalized aromatic compounds either in the presence of a solvent or under neat conditions (Table 18).284,285
| # | Substrate (g mmol−1) | Conc.b (M) | Cat. (wt%) | Reaction conditions | Yield (%) | E-f | PMI | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Starting amount of the first step were normalized with respect to the second; work-up and purification material not included. b Concentration of furan-based reagent with respect to the solvent of the second step. | ||||||||
| 1 | 2-Methylfuran (42.1/511.7) | 1.1 | Pd/C (2.0) | (i) Maleic anhydride; 15 °C, 3 h; (ii) H2, 80 bar; THF; 10–25 °C, 8 h | 12 96 | 5.7 | 6.7 | 286 |
| 2 | 2,5-Dimethylfuran (5/49.5) | 1.6 | Pd/C (2.0) | (i) Maleic anhydride; 15 °C, 3 h; (ii) H2, 80 bar; THF; 10–25 °C, 5 h | 13 94 | 5.7 | 6.7 | 286 |
| 3 | Furfural (4/58.2) | 1.2 | Pd/C (2.0) | (i) Maleic anhydride; 15 °C, 3 h; (ii) H2, 80 bar; THF; 10–25 °C, 8 h | 11 96 | 5.9 | 6.9 | 286 |
The oxabicyclic adduct (Scheme 18, compound 10), in principle, is subsequently dehydrated to the desired aromatic compound (Scheme 1, step 2). However, the acid-catalysed dehydration is often critical due to the thermally reversible nature of the intermediate adduct 10 that is prone to retro-Diels–Alder reaction giving 8 and 9 and very low yield to desired aromatic compound 12.
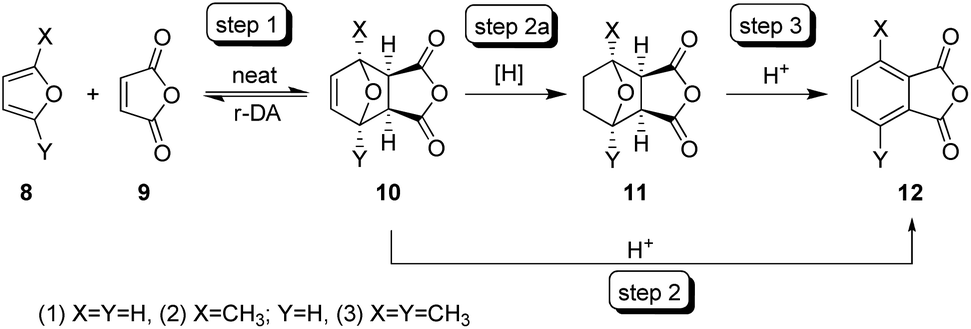 | ||
| Scheme 18 Synthesis of functionalized aromatic compounds using DA reaction. | ||
In the modified approach, the thermal stability of DA adduct 10 has been increased by hydrogenating the double bonds making the adduct a more stable intermediate (step 2a, compound 11) which efficiently undergoes dehydration reaction giving aromatic compounds in relatively high yields. The bicyclic structure of the hydrogenated DA adducts 11–13 (Fig. 15) have a rigid structure compared to its corresponding aromatic analogues, which is beneficial for improving the physical and thermal properties of the polymers thereof.
 | ||
| Fig. 15 Structure of rigid-bicyclic hydrogenated DA adducts. | ||
Given its ease in the synthesis of these hydrogenated DA adducts from lignocellulosic biomass and the relative low values of the related green metrics (PMI = 6.7–6.9), it is surprising that the significance of these building blocks is overlooked in the polymer synthesis given the very low number of publications recorded to date.
9.2. Bio-based polymers
Although the oxabicyclics (cycloaddition Diels–Alder reactions) have a great potential to be used as monomer in the polycondensation polymers, to the best of our knowledge, there are no publications reporting their use, except one but starting from the anhydride counterpart instead of the diester.287Tachibana et al. reported the preparation of a bio-based oxabicyclic dicarboxylic anhydride and its polymerization with several linear α,ω-alkanediols (1,2-ethanediol, 1,3-propanediol, 1,4-butanediol, 1,6-hexane-diol, 1,8-octanediol, 1,10-decanediol) to synthesize polyoxabicyclates (POBC) (Fig. 16).287
 | ||
| Fig. 16 Preparation of polyoxabicyclate. | ||
The ensuing polyesters had a very interesting range of mechanical properties. Indeed, whereas POBC-p, prepared from 1,3-propanediol, showed to be a soft, highly elastic material, POBC-b (derived from a longer alkyl-chain diol, 1,4-butanediol) was a tough, elastic material. Besides, POBC-b films display transparency in the visible region within 1100 nm to 350 nm.
These set of properties make polyoxabicyclates possible alternatives to replace some commercially available polymers. POBC-b has a tensile strength and strain at break very similar to linear low density polyethylene (LDPE) (10.2 ± 0.8 N mm−2 and 555 ± 41%, respectively).287 Some interest was also dedicated to the oxabicyclic related oxanorbornene monomers (the equivalent alkenes from oxabicyclic monomers). Studies have been conducted on their ROMP catalyzed with generation II Grubbs catalysts.288–290
N'Guyen et al. used that type of aromatic furan derived monomers (oxanorbornene monomers) as macromolecular building blocks in the co-monomerization with two poly(ε-caprolactone) (PCL) side chains.290 In this work, high grafting density poly(oxa)norbornene–PCLs were successfully obtained with a backbone length between 10 and 100 repeating units and grafts length between 10 and 48, while retaining a narrow distribution of molecular weights, using the ROMP as well catalysed with generation II Grubbs catalysts.288–290
Poly(oxa)norbornene-g-PCLs were obtained with different number average molecular weight caprolactone (CL) repeating units achieving acceptable weight-average molecular weight (35900–74900 g mol−1). Moreover, DSC thermograms showed a higher stability of the bottle-brush poly(oxa)norbornene-g-PCLs compared to PCL-based (oxa)norbornene macromonomers together, with a weaker crystallinity, resulting from steric hindrance.
The T5% (higher than 300 °C) and the Tmax (higher than 520 °C) temperatures of the poly(oxa)norbornene–PLCs were also higher than the corresponding temperatures for polynorbornene–PLCs, reflecting also their higher thermal stability.290
10. Acrylate furanic monomers
10.1. Synthesis of acrylate furanic monomers
Furan-derived acrylates and methacrylates represent a compelling avenue for the development of UV-curable monomers, offering a viable alternative to conventional thermal curing methods.299 These monomers can be effectively synthesized using alcohols derived from furfural and HMF. Typically, the preparation of acrylic monomers involves either an esterification reaction employing acid chlorides or anhydrides with the corresponding furanic alcohols or alternatively an etherification followed by the ester formation (Fig. 17a). An overview of the reactions is given in Table 19. | ||
| Fig. 17 General synthetic protocols for the functionalized furanic (meth)acrylate from furanic alcohols and diols. | ||
| # | Substrate (g mol−1) | Base (mol%) | Additive (mol%) | Reaction conditions | Yield (%) | E-f | PMI | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Work-up not included. b Ar atmosphere not included. c Purification not included. | ||||||||
| 1a | MMA (25/0.25) | KOH (30) | Hydroquinone 0.4 mol% | Cyclohexane; 86 °C, 7 h | 60 | 5.1 | 6.1 | 291 |
| 2b | MCCh (40/0.38) | Et3N (203) | — | Ar, DCM; 0–25 °C, overnight | 58 | 8.4 | 9.4 | 292 |
| 3a | MMA (113/1.13) | KOH (12) | — | Cyclohexane; 110 °C, 12 h | 60 | 10.2 | 11.2 | 293 |
| 4a | MAA (3.3/0.02) | Et3N (143) | DMAP 1 mol% | AcOEt; 25–55 °C, 24 h | 89 | 14.9 | 15.9 | 294 |
| 5c | MMA (61/0.61) | KOH (17) | — | Cyclohexane; 95 °C, 5 h | 96 | 17.3 | 18.3 | 295 |
| 6c | MCCh (15.6/0.15) | Et3N (100) | — | DCM; 0–25 °C, 12 h | 82 | 33.5 | 34.5 | 296 |
| 7 | MCCh (4.7/0.04) | Et3N (100) | — | DCM; 10 °C, 24 h | 72 | 34.5 | 35.5 | 297 |
| 8a | AC (10.1/0.11) | Et3N (113) | DCM; 0–25 °C, 2 h | 94 | 59.3 | 60.3 | 298 | |
The synthesis of acrylic esters from furfuryl alcohol with methyl methacrylate (MMA) derivatives can yield moderate to good yields, typically ranging from 60–90% (#1, #3, and #5; Table 19). These procedures result in moderate values of both E-f and PMI.
The alternative use of other active acid derivatives, such as (methy)acryloyl chloride (MMCCh) (#2, #6 and #7; Table 19) or methacrylic anhydride (MAA) with furfuryl alcohol (#4; Table 19), can lead to the production of desired acrylates although accompanied by the generation of significant amount of waste.
In these cases, higher values of both E-f and PMI can be ascribed to the formation of secondary products, as well as the required quantities of reactants and the purification/separation processes involved.
Acrylic derivatives can be also obtained using acryloyl chloride (AC) with good yield, although once again with high values E-f and PMI (#8; Table 19). Noteworthy, in addition to the waste generated during the synthesis of these acrylic monomers, it is clear that substituting chlorinated solvents (or other hazardous solvents) with eco-friendly alternatives has the potential to substantially enhance the sustainability of the process, aligning with the principles of green and sustainable chemistry.
In the search for greener synthetic methods for acrylic monomers, the use of epoxide derivatives from BHMF and furfuryl alcohol or 2-furan methanol (FM) has been explored as an alternative (Fig. 17b; #2 and #3; Table 20). The process for these monomers involves glycidylation with an excess of epichlorohydrin and 50% aqueous NaOH as a base to neutralize the generated HCl, with tetrabutylammonium bromide (TBAB) as a phase transfer catalyst (Section 5).301 The introduction of methacrylate groups is achieved by ring opening of the epoxides using excess equivalents of methacrylic acid and triethylamine as a base. Metrics reported in the Table 20 regard the overall process.
| # | Substrate (g mmol−1) | Conc. (M) | Catalyst (mol% or wt%) | Reaction conditions | Yield (%) | E-f | PMI | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Metrics refer to the global process. Quantities of the first and second step were normalized. b Some/all the work-up materials were not included in the metrics calculations. c The original procedure reports only reagent concentrations. Metrics were calculated on a generic 10 mL synthesis. d Metrics could not be calculated due to unknown solvent volume employed. e Metrics could not be calculated due to unknown yield in the original paper. | ||||||||
| 1a,b | FDCA (0.53/3.4) | 0.5 | — | (i) DMF, SOCl2, DCM; 10 h, reflux. (ii) TEA, glycidol, DCM; 0 °C to r.t., 1 h. (iii) MAA, TEA, DCM; 70 °C, 20 h | (i) 89; (ii) 86 | 42.5 | 43.5 | 300 |
| 2a,b | FA (0.74/7.5) | 2.4 | TBAB (2 mol%) | (i) NaOH, ECH, THF; r.t., 2 h. (ii) MAC, TEA, diphenyl picryhydrazyl; 100 °C, 2 h | (i) 86; (ii) 68 | 51.7 | 52.7 | 301 |
| 3a,b | BHMF (2.56/2.0) | 1.0 | TBAB (10 mol%) | (i) NaOH, ECH, THF; 50 °C, 2 h. (ii) MAA, TEA, diphenyl picryhydrazyl; 100 °C, 2 h | (i) 74; (ii) 86 | 89.5 | 90.5 | 301 |
| 4c | DFF (0.62/5.0) | 0.1 | — | Grignard reactant, trisodium citrate, THF; 0 °C, 2 h | 80 | 283.6 | 284.6 | 302 |
| 5c | HMF (0.63/5.0) | 0.1 | — | Grignard reactant, trisodium citrate, THF; 0 °C, 2 h | 88 | 316.3 | 317.3 | 302 |
| 6d | BHMF (0.13/1.00) | — | — | TEA, DCM, methacryloyl chloride; 0 °C, overnight | >99 | — | — | 303 |
| 7e | FCA (11.2/100) | Neat | — | Antioxidant A71010, GMA, p-hydroxyanisole, TPP; 95 °C, 3 h | na | — | — | 304 |
The methodology reported by Webster, Sibi et al. for the direct synthesis of acrylic monomers from asymmetric and symmetric diols, obtained using DFF, HMF and BHMF as feedstock (Fig. 17c; #4–6; Table 20) represents an alternative route.302,303
In the case of using BHMF as the starting reagent, the acrylate monomer is obtained with a quantitative yield (99%), and the process results in very low values for both E-f and PMI. However, it's worth noting that to convert the corresponding aldehydes (HMF or DFF) to substituted secondary alcohols, an excess of Grignard reagents is required.
An alternative synthetic protocol has been devised for the production of acrylic monomers from furan dicarboxylic acid derivatives, serving as substitutes for bisphenol A in the preparation of partially bio-based dimethacrylate resins (Fig. 18). The process involves the synthesis of furan-based dimethacrylates using their diglycidyl esters as intermediates, which are subsequently reacted with methacrylic acid. The overall yield for this method is approximately 77%. However, this process results in relatively high values of E-f and PMI (#1; Table 20).300
 | ||
| Fig. 18 Overview over the synthetic protocols for the furamic di(meth)acrylate from furanic di carboxylic acids. | ||
Liu and colleagues have presented a direct and environmentally friendly method for synthesizing a related acrylic derivative. This method involves the direct reaction of 2-furoic acid (FCA) with commercially available glycidyl methacrylate (GMA) to produce the desired monomer (#7; Table 20).304
This reaction is conducted without the use of solvents, and the target product is obtained with high purity, eliminating the need for further purification steps. As a result, these protocols meet the criteria for a straightforward and environmentally friendly synthesis that can be applied on a large scale. However, in this latter case the green metrics could not be evaluated due to some missing data.
From the data collected it is evident that future investigations on these monomers should be focus on optimizing the reaction conditions (such as avoiding the use of halogenated compounds). Some scale-up experiments have already been conducted although once again mostly employing chlorine-based chemistry.
10.2 Polymers derived from furanic acrylic derivatives
UV-light curing technologies based on biomonomers have been steadily gaining popularity in various large-scale industries, particularly in the realms of coatings, adhesives, printing inks, and medical materials.305 These curing methods offer the advantage of rapid curing processes, energy efficiency, and the elimination of solvents, opening up exciting possibilities across a wide range of applications, including emerging techniques like 3D printing, such as digital light processing (DLP) and stereolithography (SLA).306In this context, acrylic furanic derivative can provide UV-light curing formulations, presenting innovative opportunities. For example, biomonomers derived from furans, such as 2,5-dicarboxylic acid and 2,2′-bifuran-5,5′-dicarboxylic acid, have emerged as promising alternatives to traditional bisphenol A acrylic monomers. Bio-based dimethacrylate thermoset resins have been prepared through radical polymerization, employing combination of furan-based acrylates and additional bio-based monomers, such as methacrylated eugenol, in a 60:40 mass ratio.300 These resulting polymers exhibit properties comparable to resins based on bisphenol A, with high Tg in the range of 200 °C and complete thermal decomposition occurring at approximately 410–430 °C. This development represents a significant step toward sustainable and high-performance materials in UV-curing applications.
Similarly, a range of acrylate and methacrylate monomers has been developed based on both symmetric and asymmetric furan diols.302 These acrylic derivatives have been employed in the formulation of UV-cured coatings, in combination with a commonly used urethane acrylate resin.303 The resulting UV-cured coatings, incorporating furan-based diluents, exhibit outstanding solvent resistance, favourable hardness, and high modulus. This study underscores the potential of furan-based diacrylates and dimethacrylates as reactive diluents for producing high-quality UV-curable coatings. Notably, the coatings generated using these furan-based diluents exhibit superior properties when compared to materials prepared using commercially available 1,6-hexanediol diacrylate (HDDA) as a reactive diluent. This development holds promise for enhancing the performance and sustainability of UV-curable coatings in various applications.
Additionally, 2,5-furanfuran acrylate has been identified as an effective difunctional stiffener for creating innovative bio-based crosslinked polymer networks through UV photopolymerization.307 This process involves the use of other bio-based acrylate materials like acrylated epoxidized soybean oil (AESO), castor oil (ACO), and 7,10-dihydroxy-8(E)-octadecenoic acid (ADOD). The resulting films exhibit a significant enhancement in tensile strength, with improvements ranging from 1.4 to 4.2 times compared to films produced without the incorporation of bisacryl-furanic monomers. This development shows the potential for creating robust and sustainable bio-based polymer materials with superior mechanical properties.
Photo-polymerizable 2-hydroxypropyl methacrylate furan monomers have been utilized in the production of thermoset materials through UV light photocuring. The properties of these resulting materials, including their adhesive capability and surface hardness, were assessed and compared to polymers produced from bisphenol A-glycerolate dimethacrylate and triethylene glycol dimethacrylate. Notably, the materials exhibited similar values for tensile shear strength, ranging from 0.2 to 0.6 MPa, and pencil hardness ratings in the range of 2H to 3H. This suggests that the 2-hydroxypropyl methacrylate furan monomers can be used to create thermoset materials with comparable performance characteristics to those obtained using conventional dimethacrylate materials.301
Furfuryl methacrylate finds application as a component in UV-curable anticorrosive coatings, especially when reinforced with hexagonal boron nitride.295 Bio-based epoxidized soybean oil methacrylate (ESOM) coatings, containing 0.25–0.75 wt% of furanic monomer along with hexagonal boron nitride as reactive diluents, have demonstrated remarkable corrosion resistance properties for carbon steels. These coatings exhibited superior performance in resisting corrosion during 240-hour immersion test in a 3.5 wt% NaCl solution. This development suggests the potential for using furfuryl methacrylate and hexagonal boron nitride to enhance the protective properties of coatings in corrosive environments.
Self-healing materials can be achieved through the UV curing of two bio-based UV-curable systems, employing different combinations of acrylic comonomers derived from itaconic acid and 2-furoic acid (Fig. 19).
 | ||
| Fig. 19 Schematic representation of the synthesis of polymers coming from furans-derived acrylates. | ||
The resulting polymers exhibit comparable or even superior properties compared to petroleum-based UV-curing coatings. Depending on the composition, these materials can have Tg in the range of 66.8–148.3 °C, tensile strengths ranging from 32.1 to 46.4 MPa, pencil hardness values of 4–6H, and thermal decomposition temperatures reaching up to 328.4 °C. Moreover, these bio-based UV-curable coatings influence the dynamic transesterification within their cross-linked network, enabling them to demonstrate good healing, remoulding, and repairability under moderate thermal conditions. This capability is attributed to the presence of sufficient hydroxyl and ester groups in both monomers. As a result, the molecular structure of the bio-based monomer can be tailored and explored to design materials with exceptional repairability properties, offering promising applications in self-healing materials and sustainable coating technologies.
11. Conclusions and future perspectives
Over the last 15 years, investigations on the synthesis of furanics derived from renewable resources to be employed as monomers for bio-based polymers have been incrementally expanding as demonstrated by the number of publications within this field of biorefinery.According to data collected in this review we can draw some conclusion on the most investigated furanics herein categorized according to their main functional groups.
Aldehyde-based furanics are key monomers employed as starting materials of subsequent numerous chemical transformations reported in this review. Their syntheses have been extensively investigated resulting in good values of PMI and E-factor as these compounds can be easily achieved by controlled oxidation of HMF or by its self-etherification. The resulting bio-based materials are mostly centred on dynamic covalent imine bond showing properties between thermosets and thermoplastics. As a result of the intrinsic thermodynamic nature of this bond these polymers can be reprocessed or recycled reducing their end-of-life plastic waste. However, the scope of the possible bio-based polymers deriving from the aldehyde-based furanics are so far quite limited.
Diols and polyol monomers are evidently the most investigated monomers due to their relatively easy preparation. Green metrics evaluation showed in some cases – BHMF, BHMTHF, acetal furanics, Furoin-3 – quite promising synthetic approaches although in some cases purification and or isolation of the products is not fully addressed. Numerous types of bio-based polymers have been reported starting from these furanic monomers, the most common ones being polyesters, polyethers and polyurethanes. Some examples of polycarbonates, poly(silylether) and humin-like polymers have been also investigated. Unfortunately, data collected showed that the thermal properties of the starting furanic diols or polyols have not been reported or studied in detail despite these being a fundamental information for the synthesis of novel materials. As an example, it is now well known that BHMF, easily achieved via HMF reduction, showed very low thermal stability that render the possibility to achieve high molecular weight polymers quite difficult.
Among the discussed monomers a special mention should be made for acetal furanic based polyesters which can be easily achieved from the related bio-based monomers (PMI lowest value 4.2) and resulting in some fascinating acid-labile reversible functional bio-based polymers.
In any case the above discussed diols and polyol furanics are surely interesting building blocks for bio-based polymers also as potential hazard bisphenol-A substitutes.
Besides furfuryl amine – that is a commercially available compound – the synthetic procedures reported to amine-based furanic must be considered still quite preliminary as catalysts used are custom made and reactions are conducted on small scale. While they have the potential to be monomers for bio-based polymers such as polyurethanes, their practical usage has been surprisingly scarce.
Epoxides derived monomers has been also reported; their syntheses although efficient, still relies on the use of chlorine or halogenated chemistry. New greener synthetic approaches would definitely be beneficial, especially in consideration of their use for the preparation of thermosetting resins.
Acrylates derivatives of furanics are also partially achieved via chlorine chemistry although in general the procedures reported have a quite low E-factor and PMI. These monomers are highly interesting especially for the preparation of thermosets via UV-light curing technologies with potential applications in the field of coatings, adhesives, printing inks, and medical materials.
Alkyl carbonate – including cyclic carbonates – furanic monomers are so far only scarcely reported. Similar consideration can be drowned for carboxylic acid (different from FDCA) and Diels–Alder furanic derivatives. Evaluation on the applications of these monomers is still premature, further investigations must be conducted so to assess their potential applications.
Direct approaches to ester derivatives of FDCA, such as furan-2,5-dimethylcarboxylate (FDMC), have gained an increasing amount of attention as alternatives to the FDCA synthesis via oxidation of HMF. Important features of FDCA esters are also their enhanced solubility and lower boiling point compared to the 2,5-furandicarboxylic acid. These monomers would give access to the same bio-based polymers so far reported for FDCA most probably employing milder reaction conditions. The synthesis of FDCA esters still needs to be implemented, but it is one of the most interesting monomers reported in this review.
Overall, the green evaluation on the synthetic procedures of furanic monomers has evidently highlighted some downsides and blind spots, i.e. very few synthetic approaches focus on isolating the products, gram or multi gram synthesis are still scarcely investigated, studies on thermal properties should be implemented as routine reported analysis.
Acknowledging the significance of Process Mass Intensity (PMI) and E-factor values in evaluating the greenness of chemical reactions, it is crucial to regard these metrics as indicative, recognizing their limitations. Many reported synthetic methods do not comprehensively address the purification of products or the recycling of catalysts, suggesting that these values could potentially be improved through further research aimed at optimizing these aspects.
The choice of catalyst also warrants careful consideration. Catalysts of the same mass may differ significantly in both cost and environmental impact, highlighting the need for a thorough evaluation of the catalyst's nature in addition to its mass. Other important considerations include the energy required for heating, pressurization, or light irradiation during reactions, as these factors directly influence the sustainability of the process. Lower energy consumption is preferable for greener synthesis.
Moreover, the costs associated with the synthesis and purification of furanic monomers are crucial for assessing the economic viability of new synthetic approaches, which, in turn, influences their potential for industrial application. While the E-factor primarily accounts for the mass of reagents and solvents, it is important to also consider their toxicity and the feasibility of reusing or disposing of them, as these factors significantly affect the overall greenness of the process.
For a comprehensive assessment of a chemical transformation's sustainability, a life cycle analysis (LCA) is essential. An LCA provides a holistic view of the environmental impacts associated with all stages of a product's life cycle, from raw material extraction through to processing, use, and disposal. This approach offers valuable insights into the sustainability of chemical processes at an industrial scale.
By addressing these considerations, the evaluation and enhancement of the sustainability of synthetic approaches for furanic monomers can be more effectively achieved. This broader assessment of environmental and economic impacts encourages advancements toward more sustainable practices in chemical design and implementation of new bio-based monomers.
Indeed, the development of new monomers should go hand in hand with innovative strategies that ensure the resulting materials not only possess the necessary properties for their intended applications but are also designed with recyclability in mind. This approach underscores the importance of integrating both bio-based monomers and their related polymers into a circular economy framework.
In this context, designing for sustainability involves several key considerations:
(i) Monomers should be designed to facilitate the easy recycling of polymers without significant loss of quality. This could involve incorporating reversible chemical bonds or designing polymers that can be depolymerized under mild conditions;
(ii) The physical properties of materials, such as durability, should not compromise their ability to be recycled. Materials should be designed to maintain integrity over their use life but still allow for efficient recycling processes;
(iii) By prioritizing biodegradability and tackling related challenges, we have the opportunity to harness essential properties that significantly mitigate the environmental impacts of materials. This approach is particularly effective in reducing bioaccumulation and decrease the generation and persistence of microplastics. Through strategic emphasis on biodegradability, we can ensure that materials break down in an environmentally friendly manner, thus addressing critical issues associated with plastic pollution and its adverse effects on ecosystems. This focus not only contributes to a cleaner environment but also emphasizes the importance of sustainable material innovation in combating the pressing issue of microplastic pollution;
(iv) Early-stage LCA of new monomers and polymers can help identify potential environmental impacts throughout the material's life cycle, including end-of-life. This can inform design choices to minimize negative impacts and enhance recyclability. Incorporating circular economy principles into the design and development phase means considering the end-of-life scenario from the beginning.
We should also point out that the development of the new biobased monomers and polymers require a collaborative effort across the entire value chain, from chemists, material scientists and product designers to manufacturers, recyclers, and policymakers. This collaborative approach ensures that materials are designed not only for performance and sustainability but also for practical and efficient recycling.
The insights gathered from this review underscore the remarkable variety of bio-based polymeric materials, showcasing a breadth that extends well beyond the current scope of materials derived from petrochemical sources. The distinctive properties of polymers originating from furanic monomers stand as a testament to the potential within this domain, serving as a beacon for both industrial and academic researchers to devise efficient strategies for synthesizing these novel molecules. It's noteworthy that the focus of the scientific community has primarily been on FDCA-based polymers, which are typically synthesized through step-growth polymerization. Expanding the range of available monomers opens the door to the synthesis of chain-growth polymers, in addition to the conventional polyesters or polyamides, thereby broadening the spectrum of potential bio-based polymeric materials.
When embarking on the journey of compiling this review, the objective was to delineate a clear landscape of the most promising platform chemicals derived from renewable resources beyond FDCA for the creation of bio-based polymers. The pivotal question that emerges is: have we now acquired enough knowledge and technology to effectively replace petrochemical-derived monomers in the fabrication of materials that either match or surpass their properties? The quick answer is not yet!
Nonetheless, the information presented in this review should serve as a foundation, guiding further exploration and refinement of the identified challenges. It underscores the imperative to pursue and enhance green synthetic methodologies, marking a path towards a sustainable future in material science where bio-based alternatives play a pivotal role in replacing petrochemical-derived materials.
Data availability
Besides data reported in the ESI,† data for this paper, including green metrics calculations are available at Open Science Framework https://doi.org/10.17605/OSF.IO/G368Y.Author contributions
Conceptualization, supervision and writing – review & editing: E. García-Verdugo and F. Aricò; data curation and writing – original draft: M. Annatelli, J. E. Sánchez-Velandia and G. Mazzi; data curation and writing – original draft (on specific parts of the review): S. V. Pandeirada, D. Giannakoudakis, S. Rautiainen, A. Esposito, S. Thiyagarajan, A. Richel, K. S. Triantafyllidis, T. Robert, N. Guigo and A. F. Sousa.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors want to also acknowledge FUR4Sustain—European network of FURan based chemicals and materials FOR a Sustainable development, CA18220, supported by COST (European Cooperation in Science and Technology). The University of Aveiro wants to acknowledge the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020 (https://doi.org/10.54499/UIDB/50011/2020), UIDP/50011/2020 (https://doi.org/10.54499/UIDP/50011/2020) & LA/P/0006/2020 (https://doi.org/10.54499/LA/P/0006/2020), financed by national funds through the FCT/MCTES (PIDDAC). The FCT is acknowledged for the research contract under Scientific Employment Stimulus to AFS (CEECIND/02322/2020) and for a PhD grant to SVP (2020.04495.BD).Notes and references
- H. A. Leslie, M. J. M. van Velzen, S. H. Brandsma, A. D. Vethaak, J. J. Garcia-Vallejo and M. H. Lamoree, Environ. Int., 2022, 163, 107199 CrossRef CAS PubMed.
- Global Commitment 2022 Progress Report|Shared by New Plastics Economy, https://emf.thirdlight.com/link/f6oxost9xeso-nsjoqe/@/#id=2, accessed January 22, 2024.
- B. Annevelink, L. G. Chavez, R. van Ree and I. V. Gursel, Global biorefinery status report, 2022.
- K. J. Zeitsch, The Chemistry and Technology of Furfural and its Many By-Products, Elsevier, 2000 Search PubMed.
- C. Xu, E. Paone, D. Rodríguez-Padrón, R. Luque and F. Mauriello, Chem. Soc. Rev., 2020, 49, 4273–4306 RSC.
- Q. Hou, X. Qi, M. Zhen, H. Qian, Y. Nie, C. Bai, S. Zhang, X. Bai and M. Ju, Green Chem., 2021, 23, 119–231 RSC.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. L. Granados, Energy Environ. Sci., 2016, 9, 1144–1189 RSC.
- K. I. Galkin, E. A. Krivodaeva, L. V. Romashov, S. S. Zalesskiy, V. V. Kachala, J. V. Burykina and V. P. Ananikov, Angew. Chem., Int. Ed., 2016, 55, 8338–8342 CrossRef CAS PubMed.
- B. Agostinho, A. J. D. Silvestre and A. F. Sousa, Green Chem., 2022, 24, 3115–3119 RSC.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- D. J. C. Constable, A. D. Curzons and V. L. Cunningham, Green Chem., 2002, 4, 521–527 RSC.
- R. Bielski and G. Grynkiewicz, Green Chem., 2021, 23, 7458–7487 RSC.
- X.-L. Shi, M. Zhang, Y. Li and W. Zhang, Green Chem., 2013, 15, 3438–3445 RSC.
- G. Trapasso, G. Mazzi, B. Chícharo, M. Annatelli, D. Dalla Torre and F. Aricò, Org. Process Res. Dev., 2022, 26, 2830–2838 CrossRef CAS PubMed.
- S. Motokucho, H. Morikawa, H. Nakatani and B. A. J. Noordover, Tetrahedron Lett., 2016, 57, 4742–4745 CrossRef CAS.
- L. Capuzzi, F. Digioia and G. Carotenuto, US Pat., US9409877B2, 2016 Search PubMed.
- X. Dong, K. Sheng, Z. Chen, C. Guo, J. Huang and Y. Gu, ChemSusChem, 2023, 16, e202201755 CrossRef CAS PubMed.
- M. Musolino, J. Andraos and F. Aricò, ChemistrySelect, 2018, 3, 2359–2365 CrossRef CAS.
- D. W. Brown, A. J. Floyd, R. G. Kinsman and Y. Roshanhyphen Ali, J. Chem. Technol. Biotechnol., 2007, 32, 920–924 CrossRef.
- M. Brasholz, K. von Känel, C. H. Hornung, S. Saubern and J. Tsanaktsidis, Green Chem., 2011, 13, 1114–1117 RSC.
- C. S. Kovash, Jr., E. Pavlacky, S. Selvakumar, M. P. Sibi and D. C. Webster, ChemSusChem, 2014, 7, 2289–2294 CrossRef PubMed.
- J. Zhao, C. Li, X. Fan, H. Liu, Z. Liu, J. Zhang, Z. Sun and W. Chu, Appl. Catal., A, 2023, 649, 118981 CrossRef CAS.
- A. Dibenedetto, M. Aresta, L. di Bitonto and C. Pastore, ChemSusChem, 2016, 9, 118–125 CrossRef CAS PubMed.
- X. Tong, M. Li, N. Yan, Y. Ma, P. J. Dyson and Y. Li, Catal. Today, 2011, 175, 524–527 CrossRef CAS.
- R. Rajmohan, S. Gayathri and P. Vairaprakash, RSC Adv., 2015, 5, 100401–100407 RSC.
- S. P. Simeonov, J. A. S. Coelho and C. A. M. Afonso, ChemSusChem, 2012, 5, 1388–1391 CrossRef CAS PubMed.
- Y. Zhao, K. Lu, H. Xu, L. Zhu and S. Wang, Renewable Sustainable Energy Rev., 2021, 139, 110706 CrossRef CAS.
- S. Fulignati, D. Licursi, N. Di Fidio, C. Antonetti and A. M. Raspolli Galletti, Catalysts, 2022, 12, 1664 CrossRef CAS.
- T. Istasse and A. Richel, RSC Adv., 2020, 10, 23720–23742 RSC.
- M. A. A. B. A. Rani, N. A. Karim and S. K. Kamarudin, Int. J. Energy Res., 2022, 46, 577–633 CrossRef CAS.
- J. H. Teles, ChemSusChem, 2019, 12, 338–339 CrossRef CAS PubMed.
- C. Rosenfeld, J. Konnerth, W. Sailer-Kronlachner, P. Solt, T. Rosenau and H. W. G. van Herwijnen, ChemSusChem, 2020, 13, 3544–3564 CrossRef PubMed.
- C. Megías-Sayago, S. Navarro-Jaén, F. Drault and S. Ivanova, Catalysts, 2021, 11, 1395 CrossRef.
- N. Stanev, J. C. M. Bordado, C. A. M. Afonso and S. P. Simeonov, ChemCatChem, 2018, 10, 5406–5409 CrossRef CAS.
- H. Wang, Y. Wang, T. Deng, C. Chen, Y. Zhu and X. Hou, Catal. Commun., 2015, 59, 127–130 CrossRef CAS.
- M. P. Sibi, S. Sermadurai, N. Zimmermann, E. Serum, G. Ma, R. Moorthy and K. Kalliokoski, US Pat., US2017233325A1, 2017 Search PubMed.
- P. Zhang, J. Yang, H. Hu, D. Hu, J. Gan, Y. Zhang, C. Chen, X. Li, L. Wang and J. Zhang, Catal. Sci. Technol., 2020, 10, 4684–4692 RSC.
- A. Páez, H. A. Rojas, O. Portilla, G. Sathicq, C. A. M. Afonso, G. P. Romanelli and J. J. Martínez, ChemCatChem, 2017, 9, 3322–3329 CrossRef.
- S. Shinde and C. Rode, Catal. Commun., 2017, 88, 77–80 CrossRef CAS.
- M. Annatelli, G. Trapasso, D. D. Torre, L. Pietrobon, D. Redolfi-Bristol and F. Aricò, Adv. Sustainable Syst., 2022, 6, 2200297 CrossRef CAS.
- G. M. Averochkin, E. G. Gordeev, M. K. Skorobogatko, F. A. Kucherov and V. P. Ananikov, ChemSusChem, 2021, 14, 3110–3123 CrossRef CAS PubMed.
- O. Casanova, S. Iborra and A. Corma, J. Catal., 2010, 275, 236–242 CrossRef CAS.
- D. Chundury and H. H. Szmant, Ind. Eng. Chem. Prod. Res. Dev., 1981, 20, 158–163 CrossRef CAS.
- D. A. Giannakoudakis, F. F. Zormpa, A. G. Margellou, A. Qayyum, R. F. Colmenares-Quintero, C. Len, J. C. Colmenares and K. S. Triantafyllidis, Nanomaterials, 2022, 12, 1679 CrossRef CAS PubMed.
- J. Dai, Green Energy Environ., 2021, 6, 22–32 CrossRef CAS.
- G. Tsilomelekis, T. R. Josephson, V. Nikolakis and S. Caratzoulas, ChemSusChem, 2014, 7, 117–126 CrossRef CAS PubMed.
- C. Laugel, B. Estrine, J. Le Bras, N. Hoffmann, S. Marinkovic and J. Muzart, ChemCatChem, 2014, 6, 1195–1198 CrossRef CAS.
- G. A. Halliday, R. J. Young and V. V. Grushin, Org. Lett., 2003, 5, 2003–2005 CrossRef CAS PubMed.
- A. S. Chauhan, A. Kumar, R. Bains and P. Das, Green Chem., 2022, 24, 6125–6130 RSC.
- J. Chen, Y. Guo, J. Chen, L. Song and L. Chen, ChemCatChem, 2014, 6, 3174–3181 CrossRef CAS.
- Y. Peng, Z. Hu, Y. Gao, D. Yuan, Z. Kang, Y. Qian, N. Yan and D. Zhao, ChemSusChem, 2015, 8, 3208–3212 CrossRef CAS PubMed.
- W. Ghezali, K. De Oliveira Vigier, R. Kessas and F. Jérôme, Green Chem., 2015, 17, 4459–4464 RSC.
- W. Hu, J. She, Z. Fu, B. Yang, H. Zhang and D. Jiang, RSC Adv., 2021, 11, 23365–23373 RSC.
- C. Yuan, Q. Liu, M. Wei, S. Zhao, X. Yang, B. Cao, S. Wang, A. El-Fatah Abomohra, X. Liu and Y. Hu, Fuel, 2022, 320, 123994 CrossRef CAS.
- B. Ma, Y. Wang, X. Guo, X. Tong, C. Liu, Y. Wang and X. Guo, Appl. Catal., A, 2018, 552, 70–76 CrossRef CAS.
- C. Ayed, W. Huang, G. Kizilsavas, K. Landfester and K. A. I. Zhang, ChemPhotoChem, 2020, 4, 571–576 CrossRef CAS.
- X.-X. Wang, S. Meng, S. Zhang, X. Zheng and S. Chen, Catal. Commun., 2020, 147, 106152 CrossRef CAS.
- D. A. Giannakoudakis, V. Nair, A. Khan, E. A. Deliyanni, J. C. Colmenares and K. S. Triantafyllidis, Appl. Catal., B, 2019, 256, 117803 CrossRef CAS.
- I. Krivtsov, E. I. García-López, G. Marcì, L. Palmisano, Z. Amghouz, J. R. García, S. Ordóñez and E. Díaz, Appl. Catal., B, 2017, 204, 430–439 CrossRef CAS.
- L. Cheng, D. Huang, Y. Zhang and Y. Wu, Appl. Organomet. Chem., 2021, 35, e6404 CrossRef CAS.
- T. Su, D. Zhao, Y. Wang, H. Lü, R. S. Varma and C. Len, ChemSusChem, 2021, 14, 266–280 CrossRef CAS PubMed.
- D. A. Giannakoudakis, J. C. Colmenares, D. Tsiplakides and K. S. Triantafyllidis, ACS Sustainable Chem. Eng., 2021, 9, 1970–1993 CrossRef CAS.
- P. Hoang Tran, ChemSusChem, 2022, 15, e202200220 CrossRef CAS PubMed.
- R. Patel, P. Dhar, A. Babaei-Ghazvini, M. Nikkhah Dafchahi and B. Acharya, Bioresour. Technol. Rep., 2023, 22, 101463 CrossRef CAS.
- X. Tong, Y. Ma and Y. Li, Appl. Catal., A, 2010, 385, 1–13 CrossRef CAS.
- L. Yang, L. Shao, Z. Wu, P. Zhan and L. Zhang, Polymers, 2023, 15, 2630 CrossRef CAS PubMed.
- Y. Meng, S. Yang and H. Li, ChemSusChem, 2022, 15, e202102581 CrossRef CAS PubMed.
- Z. Yang, W. Qi, R. Su and Z. He, Energy Fuels, 2017, 31, 533–541 CrossRef CAS.
- Z. Yang, W. Qi, R. Su and Z. He, ACS Sustainable Chem. Eng., 2017, 5, 4179–4187 CrossRef CAS.
- A. W. Cooke and K. B. Wagener, Macromolecules, 1991, 24, 1404–1407 CrossRef CAS.
- A. Liguori and M. Hakkarainen, Macromol. Rapid Commun., 2022, 43, 2100816 CrossRef CAS PubMed.
- Z. Hui and A. Gandini, Eur. Polym. J., 1992, 28, 1461–1469 CrossRef CAS.
- C. Méalares and A. Gandini, Polym. Int., 1996, 40, 33–39 CrossRef.
- M. J. Webber and M. W. Tibbitt, Nat. Rev. Mater., 2022, 7, 541–556 CrossRef.
- T. Xiang, X. Liu, P. Yi, M. Guo, Y. Chen, C. Wesdemiotis, J. Xu and Y. Pang, Polym. Int., 2013, 62, 1517–1523 CrossRef CAS.
- Y. Tachibana, S. Hayashi and K. Kasuya, ACS Omega, 2018, 3, 5336–5345 CrossRef CAS PubMed.
- S. Dhers, G. Vantomme and L. Avérous, Green Chem., 2019, 21, 1596–1601 RSC.
- R. Hajj, A. Duval, S. Dhers and L. Avérous, Macromolecules, 2020, 53, 3796–3805 CrossRef CAS.
- M. A. Lucherelli, A. Duval and L. Averous, ACS Sustainable Chem. Eng., 2023, 11, 2334–2344 CrossRef CAS.
- L. Jiang, Z. Yin, J. Chu, C. Song and A. Kong, J. Solid State Chem., 2022, 306, 122771 CrossRef CAS.
- W. Fang and A. Riisager, Green Chem., 2021, 23, 670–688 RSC.
- F. Aricò, Pure Appl. Chem., 2021, 93, 551–560 CrossRef.
- S. Subbiah, S. P. Simeonov, J. M. S. S. Esperança, L. P. N. Rebelo and C. A. M. Afonso, Green Chem., 2013, 15, 2849–2853 RSC.
- R. F. A. Gomes, Y. N. Mitrev, S. P. Simeonov and C. A. M. Afonso, ChemSusChem, 2018, 11, 1612–1616 CrossRef CAS PubMed.
- N. S. Biradar, A. M. Hengne, S. S. Sakate, R. K. Swami and C. V. Rode, Catal. Lett., 2016, 146, 1611–1619 CrossRef CAS.
- Z. Gao, C. Li, G. Fan, L. Yang and F. Li, Appl. Catal., B, 2018, 226, 523–533 CrossRef CAS.
- H. Li, J. He, A. Riisager, S. Saravanamurugan, B. Song and S. Yang, ACS Catal., 2016, 6, 7722–7727 CrossRef CAS.
- S. Rojas-Buzo, P. García-García and A. Corma, ChemSusChem, 2018, 11, 432–438 CrossRef CAS PubMed.
- H. Li, T. Yang and Z. Fang, Appl. Catal., B, 2018, 227, 79–89 CrossRef CAS.
- H. Li, Z. Fang, J. He and S. Yang, ChemSusChem, 2017, 10, 681–686 CrossRef CAS PubMed.
- M. Chatterjee, T. Ishizaka and H. Kawanami, Green Chem., 2014, 16, 4734–4739 RSC.
- Q. Cao, W. Liang, J. Guan, L. Wang, Q. Qu, X. Zhang, X. Wang and X. Mu, Appl. Catal., A, 2014, 481, 49–53 CrossRef CAS.
- M. Tamura, K. Tokonami, Y. Nakagawa and K. Tomishige, Chem. Commun., 2013, 49, 7034–7036 RSC.
- R. J. M. Silva, J. L. Fiorio, P. Vidinha and L. M. Rossi, J. Braz. Chem. Soc., 2019, 30, 2162–2169 CAS.
- J. J. Wiesfeld, M. Kim, K. Nakajima and E. J. M. Hensen, Green Chem., 2020, 22, 1229–1238 RSC.
- R. Sole, M. Bortoluzzi, A. Spannenberg, S. Tin, V. Beghetto and J. G. de Vries, Dalton Trans., 2019, 48, 13580–13588 RSC.
- Y. Zhu, X. Kong, H. Zheng, G. Ding, Y. Zhu and Y.-W. Li, Catal. Sci. Technol., 2015, 5, 4208–4217 RSC.
- T. Thananatthanachon and T. B. Rauchfuss, ChemSusChem, 2010, 3, 1139–1141 CrossRef CAS PubMed.
- R. Zhu, G. Zhou, J. Teng, W. Liang, X. Li and Y. Fu, Green Chem., 2021, 23, 1758–1765 RSC.
- T. Buntara, S. Noel, P. H. Phua, I. Melián-Cabrera, J. G. de Vries and H. J. Heeres, Angew. Chem., Int. Ed., 2011, 50, 7083–7087 CrossRef CAS PubMed.
- Y. Nakagawa, K. Takada, M. Tamura and K. Tomishige, ACS Catal., 2014, 4, 2718–2726 CrossRef CAS.
- Y. Nakagawa and K. Tomishige, Catal. Commun., 2010, 12, 154–156 CrossRef CAS.
- R. Alamillo, M. Tucker, M. Chia, Y. Pagán-Torres and J. Dumesic, Green Chem., 2012, 14, 1413–1419 RSC.
- D. K. Mishra, H. J. Lee, C. C. Truong, J. Kim, Y.-W. Suh, J. Baek and Y. J. Kim, Mol. Catal., 2020, 484, 110722 CrossRef CAS.
- X. Kong, Y. Zhu, H. Zheng, F. Dong, Y. Zhu and Y.-W. Li, RSC Adv., 2014, 4, 60467–60472 RSC.
- X. Kong, R. Zheng, Y. Zhu, G. Ding, Y. Zhu and Y.-W. Li, Green Chem., 2015, 17, 2504–2514 RSC.
- S. Zhang, H. Ma, Y. Sun, Y. Luo, X. Liu, M. Zhang, J. Gao and J. Xu, Green Chem., 2019, 21, 1702–1709 RSC.
- J. Tan, J. Cui, Y. Zhu, X. Cui, Y. Shi, W. Yan and Y. Zhao, ACS Sustainable Chem. Eng., 2019, 7, 10670–10678 CrossRef CAS.
- Y. Duan, J. Zhang, D. Li, D. Deng, L.-F. Ma and Y. Yang, RSC Adv., 2017, 7, 26487–26493 RSC.
- B. Pomeroy, M. Grilc and B. Likozar, Green Chem., 2021, 23, 7996–8002 RSC.
- Y. Yang, D. Yang, C. Zhang, M. Zheng and Y. Duan, Molecules, 2020, 25, 2475 CrossRef CAS PubMed.
- J. Chen, Y. Ge, Y. Guo and J. Chen, J. Energy Chem., 2018, 27, 283–289 CrossRef.
- S. Fulignati, C. Antonetti, D. Licursi, M. Pieraccioni, E. Wilbers, H. J. Heeres and A. M. Raspolli Galletti, Appl. Catal., A, 2019, 578, 122–133 CrossRef CAS.
- Y. Yang, Z. Du, J. Ma, F. Lu, J. Zhang and J. Xu, ChemSusChem, 2014, 7, 1352–1356 CrossRef CAS PubMed.
- E. V. Gromachevskaya, F. V. Kvitkovsky, E. B. Usova and V. G. Kulnevich, Chem. Heterocycl. Compd., 2004, 40, 979–985 CrossRef CAS.
- N. Warlin, M. N. G. Gonzalez, S. Mankar, N. G. Valsange, M. Sayed, S.-H. Pyo, N. Rehnberg, S. Lundmark, R. Hatti-Kaul, P. Jannasch and B. Zhang, Green Chem., 2019, 21, 6667–6684 RSC.
- K. S. Arias, A. Garcia-Ortiz, M. J. Climent, A. Corma and S. Iborra, ACS Sustainable Chem. Eng., 2018, 6, 4239–4245 CrossRef CAS.
- N. Warlin, E. Nilsson, Z. Guo, S. V. Mankar, N. G. Valsange, N. Rehnberg, S. Lundmark, P. Jannasch and B. Zhang, Polym. Chem., 2021, 12, 4942–4953 RSC.
- S. Kirchhecker, A. Dell'Acqua, A. Angenvoort, A. Spannenberg, K. Ito, S. Tin, A. Taden and J. G. de Vries, Green Chem., 2021, 23, 957–965 RSC.
- J. He, S. P. Burt, M. Ball, D. Zhao, I. Hermans, J. A. Dumesic and G. W. Huber, ACS Catal., 2018, 8(2), 1427–1439 CrossRef CAS.
- (a) S. Ma and D. C. Webster, Prog. Polym. Sci., 2018, 76, 65–110 CrossRef CAS; (b) F. J. A. G. Coumans, Z. Overchenko, J. J. Wiesfeld, N. Kosinov, K. Nakajima and E. J. M. Hensen, ACS Sustainable Chem. Eng., 2022, 10, 3116–3130 CrossRef CAS.
- J. M. Timko, S. S. Moore, D. M. Walba, P. C. Hiberty and D. J. Cram, J. Am. Chem. Soc., 1977, 99, 4207–4219 CrossRef CAS.
- J. Wilson and E. Y.-X. Chen, ACS Sustainable Chem. Eng., 2016, 4, 4927–4936 CrossRef CAS.
- D. Liu and E. Y.-X. Chen, ACS Catal., 2014, 4, 1302–1310 CrossRef CAS.
- D. Liu and E. Y.-X. Chen, ChemSusChem, 2013, 6, 2236–2239 CrossRef CAS PubMed.
- B. Yan, H. Zang, Y. Jiang, S. Yu and E. Y.-X. Chen, RSC Adv., 2016, 6, 76707–76715 RSC.
- K. I. Galkin and V. P. Ananikov, ChemSusChem, 2019, 12, 185–189 CrossRef CAS PubMed.
- L. Wang and E. Y.-X. Chen, ACS Catal., 2015, 5, 6907–6917 CrossRef CAS.
- D. Liu, Y. Zhang and E. Y.-X. Chen, Green Chem., 2012, 14, 2738 RSC.
- J. Donnelly, C. R. Müller, L. Wiermans, C. J. Chuck and P. D. de María, Green Chem., 2015, 17, 2714–2718 RSC.
- (a) H. Chang, A. H. Motagamwala, G. W. Huber and J. A. Dumesic, Green Chem., 2019, 21, 5532–5540 RSC; (b) H. Chang, I. Bajaj, G. W. Huber, C. T. Maravelias and J. A. Dumesic, Green Chem., 2020, 22, 5285–5295 RSC; (c) H. Chang, E. B. Gilcher, G. W. Huber and J. A. Dumesic, Green Chem., 2021, 23, 4355–4364 RSC.
- Z. Mou, S. Feng and E. Y. X. Chen, Polym. Chem., 2016, 7, 1593–1602 RSC.
- C. Post, D. Maniar, V. S. D. Voet, R. Folkersma and K. Loos, ACS Omega, 2023, 8, 8991–9003 CrossRef CAS PubMed.
- J. A. Moore and J. E. Kelly, Macromolecules, 1978, 11, 568–573 CrossRef CAS.
- M. Gomes, A. Gandini, A. J. D. Silvestre and B. Reis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3759–3768 CrossRef CAS.
- D. Ragno, G. Di Carmine, A. Brandolese, O. Bortolini, P. P. Giovannini, G. Fantin, M. Bertoldo and A. Massi, Chem. – Eur. J., 2019, 25, 14701–14710 CrossRef CAS PubMed.
- C. Zeng, H. Seino, J. Ren, K. Hatanaka and N. Yoshie, Macromolecules, 2013, 46, 1794–1802 CrossRef CAS.
- P. P. Upare, Y. K. Hwang and D. W. Hwang, Green Chem., 2018, 20, 879–885 RSC.
- L. Guillaume, A. Marshall, N. Niessen, P. Ni, R. M. Gauvin and C. M. Thomas, Green Chem., 2021, 23, 6931–6935 RSC.
- S. Cai, Z. Qiang, C. Zeng and J. Ren, Mater. Res. Express, 2019, 6, 045701 CrossRef.
- T. Ikezaki, R. Matsuoka, K. Hatanaka and N. Yoshie, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 216–222 CrossRef CAS.
- Y. Jiang, A. J. J. Woortman, G. O. R. Alberda van Ekenstein, D. M. Petrović and K. Loos, Biomacromolecules, 2014, 15, 2482–2493 CrossRef CAS PubMed.
- A. Pellis, S. Weinberger, M. Gigli, G. M. Guebitz and T. J. Farmer, Eur. Polym. J., 2020, 130, 109680 CrossRef CAS.
- D. Maniar, Y. Jiang, A. J. J. Woortman, J. van Dijken and K. Loos, ChemSusChem, 2019, 12, 990–999 CrossRef CAS PubMed.
- T. T. Truong, H. T. Nguyen, M. N. Phan and L.-T. T. Nguyen, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1806–1814 CrossRef CAS.
- C. Oh, E. H. Choi, E. J. Choi, T. Premkumar and C. Song, ACS Sustainable Chem. Eng., 2020, 8, 4400–4406 CrossRef CAS.
- Y. Zhang, T. Li, Z. Xie, J. Han, J. Xu and B. Guo, Ind. Eng. Chem. Res., 2017, 56, 3937–3946 CrossRef CAS.
- L. Zhang, F. C. Michel and A. C. Co, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1495–1499 CrossRef CAS.
- S. Vijjamarri, S. Streed, E. M. Serum, M. P. Sibi and G. Du, ACS Sustainable Chem. Eng., 2018, 6, 2491–2497 CrossRef CAS.
- M. A. Farcaş-Johnson, S. H. Kyne and R. L. Webster, Chem. – Eur. J., 2022, 28, e202201642 CrossRef PubMed.
- E. H. Choi, J. Lee, S. U. Son and C. Song, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1796–1800 CrossRef CAS.
- (a) P. Tundo, M. Musolino and F. Aricò, Green Chem., 2018, 20, 28–85 RSC; (b) G. Sandra, F. Aricò and P. Tundo, Pure Appl. Chem., 2012, 84, 695–705 Search PubMed.
- (a) F. Aricò and P. Tundo, Russ. Chem. Rev., 2010, 79, 479 CrossRef; (b) F. Aricò and P. Tundo, ChemSusChem, 2023, 16, e202300748 CrossRef PubMed.
- A. J. R. Amaral and G. Pasparakis, Polym. Chem., 2017, 8, 6464–6484 RSC.
- Y. Liu, Z. Yu, B. Wang, P. Li, J. Zhu and S. Ma, Green Chem., 2022, 24, 5691–5708 RSC.
- B. Wang, S. Ma, Q. Li, H. Zhang, J. Liu, R. Wang, Z. Chen, X. Xu, S. Wang, N. Lu, Y. Liu, S. Yan and J. Zhu, Green Chem., 2020, 22, 1275–1290 RSC.
- S. Ma, J. Wei, Z. Jia, T. Yu, W. Yuan, Q. Li, S. Wang, S. You, R. Liu and J. Zhu, J. Mater. Chem., 2019, 7, 1233–1243 RSC.
- H. Matsukizono and T. Endo, J. Am. Chem. Soc., 2018, 140, 884–887 CrossRef CAS PubMed.
- B. Wang, S. Ma, X. Xu, Q. Li, T. Yu, S. Wang, S. Yan, Y. Liu and J. Zhu, ACS Sustainable Chem. Eng., 2020, 8, 11162–11170 CrossRef CAS.
- W. Yuan, S. Ma, S. Wang, Q. Li, B. Wang, X. Xu, K. Huang, J. Chen, S. You and J. Zhu, Eur. Polym. J., 2019, 117, 200–207 CrossRef CAS.
- J. F. Wilson and E. Y.-X. Chen, ACS Sustainable Chem. Eng., 2019, 7, 7035–7046 CrossRef CAS.
- Z. Mou and E. Y.-X. Chen, ACS Sustainable Chem. Eng., 2016, 4, 7118–7129 CrossRef CAS.
- S. Baraldi, G. Fantin, G. Di Carmine, D. Ragno, A. Brandolese, A. Massi, O. Bortolini, N. Marchetti and P. P. Giovannini, RSC Adv., 2019, 9, 29044–29050 RSC.
- Brief Profile – ECHA, https://echa.europa.eu/brief-profile/-/briefprofile/100.009.580, accessed March 23, 2023.
- J. Zhang, J. Yang, J. Tian, H. Liu, X. Li, W. Fang, X. Hu, C. Xia, J. Chen and Z. Huang, New J. Chem., 2021, 45, 4236–4245 RSC.
- M. A. Ayedi, Y. Le Bigot, H. Ammar, S. Abid, R. E. Gharbi and M. Delmas, Synth. Commun., 2013, 43, 2127–2133 CrossRef CAS.
- M. Chatterjee, T. Ishizaka and H. Kawanami, Green Chem., 2016, 18, 487–496 RSC.
- K. Zhou, H. Liu, H. Shu, S. Xiao, D. Guo, Y. Liu, Z. Wei and X. Li, ChemCatChem, 2019, 11, 2649–2656 CrossRef CAS.
- C. Dong, Y. Wu, H. Wang, J. Peng, Y. Li, C. Samart and M. Ding, ACS Sustainable Chem. Eng., 2021, 9, 7318–7327 CrossRef CAS.
- M. Gao, X. Jia, J. Ma, X. Fan, J. Gao and J. Xu, Green Chem., 2021, 23, 7115–7121 RSC.
- C. Dong, H. Wang, H. Du, J. Peng, Y. Cai, S. Guo, J. Zhang, C. Samart and M. Ding, Mol. Catal., 2020, 482, 110755 CrossRef CAS.
- B. Dong, X. Guo, B. Zhang, X. Chen, J. Guan, Y. Qi, S. Han and X. Mu, Catalysts, 2015, 5, 2258–2270 CrossRef CAS.
- D. Deng, Y. Kita, K. Kamata and M. Hara, ACS Sustainable Chem. Eng., 2019, 7, 4692–4698 CrossRef CAS.
- M. Sheng, S. Fujita, S. Yamaguchi, J. Yamasaki, K. Nakajima, S. Yamazoe, T. Mizugaki and T. Mitsudome, JACS Au, 2021, 1, 501–507 CrossRef CAS PubMed.
- Q.-W. Sun, J.-D. Xing, Y.-H. Qin, X.-W. Yin and Y. Zhou, J. Chem. Res., 2018, 42, 181–183 CrossRef CAS.
- E. M. Carter, F. Subrizi, J. M. Ward, T. D. Sheppard and H. C. Hailes, ChemCatChem, 2021, 13, 4520–4523 CrossRef CAS PubMed.
- Thieme E-Journals – Synthesis/Abstract, https://www.thieme-connect.com/products/ejournals/abstract/10.1055/s-2008-1067040, accessed January 25, 2024.
- L. Zhao, C. Hu, X. Cong, G. Deng, L. L. Liu, M. Luo and X. Zeng, J. Am. Chem. Soc., 2021, 143, 1618–1629 CrossRef CAS PubMed.
- Z. Yuan, B. Liu, P. Zhou, Z. Zhang and Q. Chi, J. Catal., 2019, 370, 347–356 CrossRef CAS.
- Z. Wei, Y. Cheng, H. Huang, Z. Ma, K. Zhou and Y. Liu, ChemSusChem, 2022, 15, e202200233 CrossRef CAS PubMed.
- Z. Wei, Y. Cheng, K. Zhou, Y. Zeng, E. Yao, Q. Li, Y. Liu and Y. Sun, ChemSusChem, 2021, 14, 2308–2312 CrossRef CAS PubMed.
- D. Pingen, J. B. Schwaderer, J. Walter, J. Wen, G. Murray, D. Vogt and S. Mecking, ChemCatChem, 2018, 10, 3027–3033 CrossRef CAS.
- K. Zhou, H. Liu, H. Shu, S. Xiao, D. Guo, Y. Liu, Z. Wei and X. Li, ChemCatChem, 2019, 11, 2649–2656 CrossRef CAS.
- T. Komanoya, T. Kinemura, Y. Kita, K. Kamata and M. Hara, J. Am. Chem. Soc., 2017, 139, 11493–11499 CrossRef CAS PubMed.
- X. Wang, W. Chen, Z. Li, X. Zeng, X. Tang, Y. Sun, T. Lei and L. Lin, J. Energy Chem., 2018, 27, 209–214 CrossRef.
- T. Schaub, B. Buschhaus, M. K. Brinks, M. Schelwies, R. Paciello, J. Melder and M. Merger, (BASF SE, Ludwigshafen), WO2012119929A1, 2012.
- Y. Kita, M. Kuwabara, S. Yamadera, K. Kamata and M. Hara, Chem. Sci., 2020, 11, 9884–9890 RSC.
- A. Dunbabin, F. Subrizi, J. M. Ward, T. D. Sheppard and H. C. Hailes, Green Chem., 2017, 19, 397–404 RSC.
- Y. Xu, X. Jia, J. Ma, J. Gao, F. Xia, X. Li and J. Xu, Green Chem., 2018, 20, 2697–2701 RSC.
- N.-T. Le, A. Byun, Y. Han, K.-I. Lee and H. Kim, Green Sustainable Chem., 2015, 05, 115–127 CrossRef CAS.
- H. Qi, F. Liu, L. Zhang, L. Li, Y. Su, J. Yang, R. Hao, A. Wang and T. Zhang, Green Chem., 2020, 22, 6897–6901 RSC.
- K. Zhou, R. Xie, M. Xiao, D. Guo, Z. Cai, S. Kang, Y. Xu and J. Wei, ChemCatChem, 2021, 13, 2074–2085 CrossRef CAS.
- A. Ryo and K. Tomoaki, (Mitsubishi Gas Chemical Co., Tokyo), US Pat., US2020181105A1, 2017 Search PubMed.
- Zinc dust, = 98 7440-66-6, https://www.sigmaaldrich.com/, accessed March 28, 2023.
- P. W. Roesky, Angew. Chem., Int. Ed., 2009, 48, 4892–4894 CrossRef CAS PubMed.
- K. Krüger, A. Tillack and M. Beller, ChemSusChem, 2009, 2, 715–717 CrossRef PubMed.
- O. Kreye, H. Mutlu and M. A. R. Meier, Green Chem., 2013, 15, 1431 RSC.
- M. G. Mohamed, C.-J. Li, M. A. R. Khan, C.-C. Liaw, K. Zhang and S.-W. Kuo, Macromolecules, 2022, 55, 3106–3115 CrossRef CAS.
- C. Zhao, Z. Sun, J. Wei, Y. Li, D. Xiang, Y. Wu and Y. Que, Polymers, 2022, 14, 1597 CrossRef CAS PubMed.
- Z. Feng, J. Mater. Sci., 2022, 57, 4895–4913 CrossRef CAS.
- K. Zhang, M. Han, Y. Liu and P. Froimowicz, ACS Sustainable Chem. Eng., 2019, 7, 9399–9407 CrossRef CAS.
- K. M. Mydeen, J. P. Kanth, A. Hariharan, K. Balaji, S. Rameshkumar, G. Rathika and M. Alagar, J. Polym. Environ., 2022, 30, 5301–5312 CrossRef CAS.
- Y. Lu, Y. Zhang and K. Zhang, Chem. Eng. J., 2022, 448, 137670 CrossRef CAS.
- J. Cao, H. Duan, J. Zou, J. Zhang, C. Wan, C. Zhang and H. Ma, Polym. Degrad. Stab., 2022, 198, 109878 CrossRef CAS.
- S. Ren, S. Zhang, W. Zhao and W. Wang, J. Mater. Sci., 2020, 55, 806–816 CrossRef CAS.
- S. Ren, F. Tian, S. Zhang, W. Zhou and Y. Du, J. Polym. Sci., 2022, 60, 1492–1500 CrossRef CAS.
- A. Mitiakoudis and A. Gandini, Macromolecules, 1991, 24, 830–835 CrossRef CAS.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed.
- V. M. Chernyshev, O. A. Kravchenko and V. P. Ananikov, Russ. Chem. Rev., 2017, 86, 357–387 CrossRef CAS.
- F. Hu, J. J. La Scala, J. M. Sadler and G. R. Palmese, Macromolecules, 2014, 47, 3332–3342 CrossRef CAS.
- X. Shen, X. Liu, J. Wang, J. Dai and J. Zhu, Ind. Eng. Chem. Res., 2017, 56, 8508–8516 CrossRef CAS.
- X. Shen, X. Liu, J. Dai, Y. Liu, Y. Zhang and J. Zhu, Ind. Eng. Chem. Res., 2017, 56, 10929–10938 CrossRef CAS.
- J. Meng, Y. Zeng, G. Zhu, J. Zhang, P. Chen, Y. Cheng, Z. Fang and K. Guo, Polym. Chem., 2019, 10, 2370–2375 RSC.
- J. K. Cho, J.-S. Lee, J. Jeong, B. Kim, B. Kim, S. Kim, S. Shin, H.-J. Kim and S.-H. Lee, J. Adhes. Sci. Technol., 2013, 27, 2127–2138 CrossRef CAS.
- L. Zhang, X. Luo, Y. Qin and Y. Li, RSC Adv., 2017, 7, 37–46 RSC.
- J. Deng, X. Liu, C. Li, Y. Jiang and J. Zhu, RSC Adv., 2015, 5, 15930–15939 RSC.
- J.-T. Miao, L. Yuan, Q. Guan, G. Liang and A. Gu, ACS Sustainable Chem. Eng., 2017, 5, 7003–7011 CrossRef CAS.
- Y. Jiang, D. Ding, S. Zhao, H. Zhu, H. I. Kenttämaa and M. M. Abu-Omar, Green Chem., 2018, 20, 1131–1138 RSC.
- R. Auvergne, S. Caillol, G. David, B. Boutevin and J.-P. Pascault, Chem. Rev., 2014, 114, 1082–1115 CrossRef CAS PubMed.
- N. Eid, B. Ameduri and B. Boutevin, ACS Sustainable Chem. Eng., 2021, 9, 8018–8031 CrossRef CAS.
- A. G. Sathicq, M. Annatelli, I. Abdullah, G. Romanelli and F. Aricò, Sustainable Chem. Pharm., 2021, 19, 100352 CrossRef CAS.
- K. Stensrud and P. Venkitasubramanian, US Pat., US9890131B2, 2018 Search PubMed.
- J. Martínez, J. Fernández-Baeza, L. F. Sánchez-Barba, J. A. Castro-Osma, A. Lara-Sánchez and A. Otero, ChemSusChem, 2017, 10, 2886–2890 CrossRef PubMed.
- F. de la Cruz-Martínez, M. Martínez de Sarasa Buchaca, J. Martínez, J. Fernández-Baeza, L. F. Sánchez-Barba, A. Rodríguez-Diéguez, J. A. Castro-Osma and A. Lara-Sánchez, ACS Sustainable Chem. Eng., 2019, 7, 20126–20138 CrossRef.
- R. Gérardy, J. Estager, P. Luis, D. P. Debecker and J.-C. M. Monbaliu, Catal. Sci. Technol., 2019, 9, 6841–6851 RSC.
- G. T. Howard, Int. Biodeterior. Biodegrad., 2002, 49, 245–252 CrossRef CAS.
- A. Das and P. Mahanwar, Adv. Ind. Eng. Polym. Res., 2020, 3, 93–101 Search PubMed.
- K. M. Zia, H. N. Bhatti and I. Ahmad Bhatti, React. Funct. Polym., 2007, 67, 675–692 CrossRef CAS.
- J. Guan, Y. Song, Y. Lin, X. Yin, M. Zuo, Y. Zhao, X. Tao and Q. Zheng, Ind. Eng. Chem. Res., 2011, 50, 6517–6527 CrossRef CAS.
- A. Gomez-Lopez, F. Elizalde, I. Calvo and H. Sardon, Chem. Commun., 2021, 57, 12254–12265 RSC.
- H. Khatoon, S. Iqbal, M. Irfan, A. Darda and N. K. Rawat, Prog. Org. Coat., 2021, 154, 106124 CrossRef CAS.
- N. L. Tai, M. Ghasemlou, R. Adhikari and B. Adhikari, Carbohydr. Polym., 2021, 265, 118029 CrossRef CAS PubMed.
- M. Bähr, A. Bitto and R. Mülhaupt, Green Chem., 2012, 14, 1447–1454 RSC.
- L. Zhang, X. Luo, Y. Qin and Y. Li, RSC Adv., 2017, 7, 37–46 RSC.
- X. Xi, A. Pizzi, C. Gerardin and G. Du, J. Renewable Mater., 2019, 7, 301–312 CAS.
- X.-J. Li, Y.-F. Wen, Y. Wang, X.-P. Zhou and X.-L. Xie, Chin. J. Polym. Sci., 2023, 41, 1069–1077 CrossRef CAS.
- A. Kayishaer, M. Annatelli, C. M. Hansom, L. M. M. Mouterde, A. A. M. Peru, F. Aricò, F. Allais and S. Fadlallah, Macromol. Rapid Commun., 2024, 45, 2300483 CrossRef CAS PubMed.
- F. Feist, Ber. Dtsch. Chem. Ges., 1901, 34, 1992–1996 CrossRef CAS.
- I. A. Pearl and J. S. Barton, J. Am. Chem. Soc., 1952, 74, 1357–1357 CrossRef CAS.
- H. Gilman, N. O. Calloway and E. W. Smith, J. Am. Chem. Soc., 1934, 56, 220–221 CrossRef CAS.
- C. H. Eugster and P. G. Waser, Helv. Chim. Acta, 1957, 40, 888–906 CrossRef CAS.
- C. A. Citron, P. Rabe and J. S. Dickschat, J. Nat. Prod., 2012, 75, 1765–1776 CrossRef CAS PubMed.
- S. Thiyagarajan, A. Pukin, J. van Haveren, M. Lutz and D. S. van Es, RSC Adv., 2013, 3, 15678–15686 RSC.
- A. D. Sadow, Z. B. Weinstein and G. A. Kraus, US Pat., US10844031B2, 2020 Search PubMed.
- M.-S. Cui, J. Deng, X.-L. Li and Y. Fu, ACS Sustainable Chem. Eng., 2016, 4, 1707–1714 CrossRef CAS.
- B. Raecke, Angew. Chem., 1958, 70, 1–5 CrossRef CAS.
- I. E. Gouvea, M. R. Simoes, M. T. Marchesin, A. S. Romao Dumaresq and A. Silva, US Pat., US2022064683A1, 2022 Search PubMed.
- P. M. R. Alexandrino, I.E. Gouvea and V. L. Queiroz, US Pat., US2020277639A1, 2020 Search PubMed.
- S. Thiyagarajan, W. Vogelzang, R. J. I. Knoop, A. E. Frissen, J. van Haveren and D. S. van Es, Green Chem., 2014, 16, 1957–1966 RSC.
- S. Zaidi, S. Thiyagarajan, A. Bougarech, F. Sebti, S. Abid, A. Majdi, A. J. D. Silvestre and A. F. Sousa, Polym. Chem., 2019, 10, 5324–5332 RSC.
- S. Thiyagarajan, M. A. Meijlink, A. Bourdet, W. Vogelzang, R. J. I. Knoop, A. Esposito, E. Dargent, D. S. van Es and J. van Haveren, ACS Sustainable Chem. Eng., 2019, 7, 18505–18516 CrossRef CAS.
- A. Bourdet, S. Araujo, S. Thiyagarajan, L. Delbreilh, A. Esposito and E. Dargent, Polymer, 2021, 213, 123225 CrossRef CAS.
- M. M. Nolasco, C. F. Araujo, S. Thiyagarajan, S. Rudić, P. D. Vaz, A. J. D. Silvestre, P. J. A. Ribeiro-Claro and A. F. Sousa, Macromolecules, 2020, 53, 1380–1387 CrossRef CAS.
- E. Bianchi, M. Soccio, V. Siracusa, M. Gazzano, S. Thiyagarajan and N. Lotti, ACS Sustainable Chem. Eng., 2021, 9, 11937–11949 CrossRef CAS PubMed.
- A. Bourdet, A. Esposito, S. Thiyagarajan, L. Delbreilh, F. Affouard, R. J. I. Knoop and E. Dargent, Macromolecules, 2018, 51, 1937–1945 CrossRef CAS.
- A. Bourdet, C. Fosse, M.-R. Garda, S. Thiyagarajan, L. Delbreilh, A. Esposito and E. Dargent, Polymer, 2023, 272, 125835 CrossRef CAS.
- F. C. Fernandes, I. E. Gouvea and B. Mano, US Pat., US2022243006A1, 2022 Search PubMed.
- F. C. Fernandes, I. E. Gouvea, A. Rodolfo and M. S. Garcez Lopes, US Pat., US20210371622A1, 2021 Search PubMed.
- D. Zhao, T. Su, C. Len, R. Luque and Z. Yang, Green Chem., 2022, 24, 6782–6789 RSC.
- H. Liu, X. Tang, X. Zeng, Y. Sun, X. Ke, T. Li, J. Zhang and L. Lin, Green Energy Environ., 2022, 7, 900–932 CrossRef CAS.
- Z.-M. Xu, J.-Y. Luo and Y.-B. Huang, Green Chem., 2022, 24, 3895–3921 RSC.
- K. Sun, S. Chen, Z. Li, G. Lu and C. Cai, Green Chem., 2019, 21, 1602–1608 RSC.
- E. Taarning, I. S. Nielsen, K. Egeblad, R. Madsen and C. H. Christensen, ChemSusChem, 2008, 1, 75–78 CrossRef CAS PubMed.
- D. K. Mishra, J. K. Cho, Y. Yi, H. J. Lee and Y. J. Kim, J. Ind. Eng. Chem., 2019, 70, 338–345 CrossRef CAS.
- O. Casanova, S. Iborra and A. Corma, J. Catal., 2009, 265, 109–116 CrossRef CAS.
- J. Deng, H.-J. Song, M.-S. Cui, Y.-P. Du and Y. Fu, ChemSusChem, 2014, 7, 3334–3340 CrossRef CAS PubMed.
- N. van Strien, S. Rautiainen, M. Asikainen, D. A. Thomas, J. Linnekoski, K. Niemelä and A. Harlin, Green Chem., 2020, 22, 8271–8277 RSC.
- D. Zhao, F. Delbecq and C. Len, Molecules, 2019, 24, 1030 CrossRef PubMed.
- (a) G. Trapasso, M. Annatelli, D. D. Torre and F. Aricò, Green Chem., 2022, 24, 2766–2771 RSC; (b) G. Trapasso, B. Chícharo, T. Gherardi, D. Redolfi-Bristol and F. Aricò, Catalysts, 2023, 13, 1114 CrossRef CAS.
- Y. Taguchi, A. Oishi and H. Iida, Chem. Lett., 2008, 37, 50–51 CrossRef CAS.
- N. van Strien, J. Niskanen, A. Berghuis, H. Pöhler and S. Rautiainen, ChemSusChem, 2024, 17, e202300732 CrossRef CAS PubMed.
- M. Kim, Y. Su, T. Aoshima, A. Fukuoka, E. J. M. Hensen and K. Nakajima, ACS Catal., 2019, 9, 4277–4285 CrossRef CAS.
- A. Salazar, P. Hünemörder, J. Rabeah, A. Quade, R. V. Jagadeesh and E. Mejia, ACS Sustainable Chem. Eng., 2019, 7, 12061–12068 CAS.
- R. V. Jagadeesh, H. Junge, M.-M. Pohl, J. Radnik, A. Brückner and M. Beller, J. Am. Chem. Soc., 2013, 135, 10776–10782 CrossRef CAS PubMed.
- A. Salazar, A. Linke, R. Eckelt, A. Quade, U. Kragl and E. Mejía, ChemCatChem, 2020, 12, 3504–3511 CrossRef CAS.
- W. Xie, B. Chen, W. Jia, H. Liu, Z. Li, S. Yang, X. Tang, X. Zeng, Y. Sun, X. Ke, T. Li, H. Fang and L. Lin, J. Energy Chem., 2022, 75, 95–108 CrossRef CAS.
- A. Cho, S. Byun, J. H. Cho and B. M. Kim, ChemSusChem, 2019, 12, 2310–2317 CrossRef CAS PubMed.
- J. Du, H. Fang, H. Qu, J. Zhang, X. Duan and Y. Yuan, Appl. Catal., A, 2018, 567, 80–89 CrossRef CAS.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass: Volume I – Results of Screening for Potential Candidates from Sugars and Synthesis Gas, National Renewable Energy Lab. (NREL), Golden, CO (United States), 2004.
- R. Fittig, Ber. Dtsch. Chem. Ges., 1876, 9, 1189–1199 CrossRef.
- J. Lewkowski, Pol. J. Chem., 2001, 1943–1946 CAS.
- W. N. Haworth, W. G. M. Jones and L. F. Wiggins, J. Chem. Soc., 1945, 1–4 RSC.
- B. Briou, B. Améduri and B. Boutevin, Chem. Soc. Rev., 2021, 50, 11055–11097 RSC.
- S. Thiyagarajan, H. C. Genuino, J. C. van der Waal, E. de Jong, B. M. Weckhuysen, J. van Haveren, P. C. A. Bruijnincx and D. S. van Es, Angew. Chem., 2016, 128, 1390–1393 CrossRef.
- H. C. Genuino, S. Thiyagarajan, J. C. van der Waal, E. de Jong, J. van Haveren, D. S. van Es, B. M. Weckhuysen and P. C. A. Bruijnincx, ChemSusChem, 2017, 10, 277–286 CrossRef CAS PubMed.
- K. I. Galkin and V. P. Ananikov, Int. J. Mol. Sci., 2021, 22, 11856 CrossRef CAS PubMed.
- Y. Tachibana, M. Yamahata and K. Kasuya, Green Chem., 2013, 15, 1318–1325 RSC.
- R. H. Grubbs, Tetrahedron, 2004, 60, 7117–7140 CrossRef CAS.
- J. B. Binder and R. T. Raines, Curr. Opin. Chem. Biol., 2008, 12, 767–773 CrossRef CAS PubMed.
- D. Anh N'Guyen, F. Leroux, V. Montembault, S. Pascual and L. Fontaine, Polym. Chem., 2016, 7, 1730–1738 RSC.
- C. Xiao, Y. Zhu, J. Chen and S. Zhang, Polymer, 2017, 110, 74–79 CrossRef CAS.
- T. Engel and G. Kickelbick, Polym. Int., 2014, 63, 915–923 CrossRef CAS.
- L. Ye, S.-F. Zhang, Y.-C. Lin, J.-K. Min, L. Ma and T. Tang, Chin. J. Polym. Sci., 2018, 36, 1011–1018 CrossRef CAS.
- R. Techie-Menson, C. K. Rono, A. Etale, G. Mehlana, J. Darkwa and B. C. E. Makhubela, Mater. Today Commun., 2021, 28, 102721 CrossRef CAS.
- H. Zhao, J. Ding and H. Yu, ChemistrySelect, 2018, 3, 11277–11283 CrossRef CAS.
- Z. Tang, X. Lyu, A. Xiao, Z. Shen and X. Fan, Chem. Mater., 2018, 30, 7752–7759 CrossRef CAS.
- A. K. Padhan and D. Mandal, Polym. Chem., 2018, 9, 3248–3261 RSC.
- C. S. Lancefield, B. Fölker, R. C. Cioc, K. Stanciakova, R. E. Bulo, M. Lutz, M. Crockatt and P. C. A. Bruijnincx, Angew. Chem., Int. Ed., 2020, 59, 23480–23484 CrossRef CAS PubMed.
- L. Pezzana, G. Melilli, N. Guigo, N. Sbirrazzuoli and M. Sangermano, React. Funct. Polym., 2023, 185, 105540 CrossRef CAS.
- T. P. Kainulainen, P. Erkkilä, T. I. Hukka, J. A. Sirviö and J. P. Heiskanen, ACS Appl. Polym. Mater., 2020, 2, 3215–3225 CrossRef CAS.
- J. Jeong, B. Kim, S. Shin, B. Kim, J.-S. Lee, S.-H. Lee and J. K. Cho, J. Appl. Polym. Sci., 2013, 127, 2483–2489 CrossRef CAS.
- C. A. Sutton, A. Polykarpov, K. Jan van den Berg, A. Yahkind, L. J. Lea, D. C. Webster and M. P. Sibi, ACS Sustainable Chem. Eng., 2020, 8, 18824–18829 CrossRef CAS.
- J. Wu, Y. Qian, C. A. Sutton, J. J. La Scala, D. C. Webster and M. P. Sibi, ACS Sustainable Chem. Eng., 2021, 9, 15537–15544 CrossRef CAS.
- S. Wang, Y. Wu, J. Dai, N. Teng, Y. Peng, L. Cao and X. Liu, Eur. Polym. J., 2020, 123, 109439 CrossRef CAS.
- L. Pezzana, E. Malmström, M. Johansson and M. Sangermano, Polymers, 2021, 13, 1530 CrossRef CAS PubMed.
- C. Mendes-Felipe, J. Oliveira, I. Etxebarria, J. L. Vilas-Vilela and S. Lanceros-Mendez, Adv. Mater. Technol., 2019, 4, 1800618 CrossRef.
- N. R. Jang, H.-R. Kim, C. T. Hou and B. S. Kim, Polym. Adv. Technol., 2013, 24, 814–818 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc00784k |
| ‡ These authors have equally contributed to the preparation of the manuscript and have to be considered as first authors. |
| This journal is © The Royal Society of Chemistry 2024 |