
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiocatalytic approaches for a more sustainable synthesis of sandalwood fragrances†
Maria C.
Cancellieri
 ,
Davide
Maggioni
,
Lorenzo
Di Maio
,
Daniele
Fiorito
,
Elisabetta
Brenna
,
Fabio
Parmeggiani
and
Francesco G.
Gatti
*
,
Davide
Maggioni
,
Lorenzo
Di Maio
,
Daniele
Fiorito
,
Elisabetta
Brenna
,
Fabio
Parmeggiani
and
Francesco G.
Gatti
*
Department of Chemistry, Materials and Chemical Engineering “Giulio Natta”, Politecnico di Milano, p.zza L. da Vinci 32, Milano, 20133, Italy. E-mail: francesco.gatti@polimi.it
First published on 16th April 2024
Abstract
The synthesis of campholenic-based fragrances requires the preservation of specific structural elements to capture the desired sandalwood scent. The most critical step of their preparation is the reduction of α,β-unsaturated carbonyl precursors while preserving the campholenic unsaturation. Classical reductions, especially hydrogenations, often lack complete chemoselectivity, leading to the formation of over-reduced byproducts. In addition, the stereochemistry plays a key role in the olfactory perception of these chiral fragrances. However, none of the current industrial syntheses are stereoselective, resulting in wasteful production of non-contributory isomers. Herein, we explore the untapped potential of biocatalytic reductions using ene-reductases (ERs) and alcohol dehydrogenases (ADHs) to enhance the sustainability of four commercial sandalwood fragrances (Brahmanol®, Firsantol®, Sandalore®, and Ebanol®), focusing on the stereoselective synthesis of their most odorant isomers. A comparison of green metrics, including E-factors and EcoScale, between bio- and chemo-based reductions is presented.
Introduction
The hydro-distillation of Santalum album wood yields one of the most renowned essential oils in fine perfumery. This essence, composed of a complex mixture of natural products, is highly appreciated by consumers for its unique sweet, balsamic, and exquisite woody scent notes.1 However, over the last century, the sandalwood oil demand increased to the point where its availability became increasingly difficult, reaching a critical level by the late 70s. Such high demand led to unsustainable cultivation practices and even to illegal harvesting in nature reserves. As a result of its intensive exploitation, in 1998 Santalum album was added to the red list (IUCN) of endangered species;2 this is a compelling example of how the label “natural” does not necessarily mean sustainable!Prompted by the rapid depletion of this raw material, many organic chemists were engaged in the challenging synthesis of its main components, i.e. the α- and the β-santalol sesquiterpenes (Scheme 1A), however, so far, none of these laboratory-scale syntheses appears to be exploitable on an industrial level.3 In this regard, very recently, Ventos has commercialized Isobionics® Santalol oil, which, in terms of composition and odorous properties, is very close to the natural one, but it is produced on a biotechnological basis from renewable raw materials (cornstarch).4 Alternatively, a new category of synthetic sandalwood-scented fragrances (1) like Brahmanol®, Firsantol®, Sandalore®, Ebanol®, and many others was developed.5 These compounds are efficiently synthesized starting from α-pinene, which is an abundant and renewable natural source (Scheme 1B). As a result, their production can be considered more environmentally sustainable than the sandalwood oil extraction from endangered Santalum trees.
 | ||
| Scheme 1 (A) α- and β-santalol are the major components of Indian sandalwood oil (around 65–75% w/w) and are responsible for of its characteristic scent. (B) Partial retrosynthesis of selected commercially available campholenic sandalwood fragrances. | ||
The carbon skeleton of alcohols 1 (C13–C14) is constructed from campholenic aldehyde 2 (obtained from α-pinene oxide) through well-established C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond forming reactions (crossed aldol condensation, Claisen rearrangement and Mannich methylene homologation, see ESI†). However, since the campholenic ring is a key structural element needed for the perception of sandalwood odour, the reduction of the carbonyl precursors, preserving the ring unsaturation, is undoubtedly the most challenging and delicate step of the overall synthesis. Currently, these reductive steps are accomplished either by hydrosilylation, hydrogen transfer or hydrogenation reactions, mostly catalysed by precious transition metals.5,6 Although these methodologies were preferred to the metal hydride-based procedures, mainly for practicality reasons, they are not yet fully chemoselective, leading to possible over-reduced products and to CC double bond positional isomerization.
C double bond forming reactions (crossed aldol condensation, Claisen rearrangement and Mannich methylene homologation, see ESI†). However, since the campholenic ring is a key structural element needed for the perception of sandalwood odour, the reduction of the carbonyl precursors, preserving the ring unsaturation, is undoubtedly the most challenging and delicate step of the overall synthesis. Currently, these reductive steps are accomplished either by hydrosilylation, hydrogen transfer or hydrogenation reactions, mostly catalysed by precious transition metals.5,6 Although these methodologies were preferred to the metal hydride-based procedures, mainly for practicality reasons, they are not yet fully chemoselective, leading to possible over-reduced products and to CC double bond positional isomerization.
In addition, preliminary olfactive evaluations on selected sandalwood fragrances have revealed significant differences in scent and odour thresholds among the stereoisomers.7 The correlation between stereochemistry and odour perception is not unusual among chiral odorant molecules,8 and, in some cases, even odourless enantiomers have been identified.9 Nevertheless, most commercial fragrances are still manufactured relying on non-stereoselective methodologies, resulting in a substantial waste of reagents, solvents and energy employed for the synthesis of all isomers that do not contribute to the scent of the fragrance.
To the best of our knowledge, biocatalytic reductions based on ene-reductases (ERs) and alcohol dehydrogenases (ADHs) enzymatic activities have never been applied to sandalwood fragrances synthesis.10 Indeed, based on safety, environmental but also on stereo- and chemoselectivity issues, such catalytic approach might significantly improve the sustainability of the productive process.
In more detail, the ERs catalyse the stereospecific reduction of CC double bonds conjugated with electron-withdrawing groups (typical EWGs are –COH, –COR, –CN, –NO2 or –CO2R), usually high stereoselectivities and conversions are achieved.11 Especially, many of the ERs belonging to the old yellow enzymes family (OYEs) are known to catalyse the reduction of prochiral (E)-α-methyl enones or enals with (S) stereoselectivity.12,13 Conversely, the reduction of their α-methylene regioisomers affords the (R) enantiomers (Scheme 2A).13 Thus, the stereochemical course of the reduction proceeds for these enzymes with unlike stereotopicity (proton addition from Re enantioface to give (S) products for α,β-unsaturated substrates and from Si face to give (R) products for α-methylene substrates, Scheme 2A).14 Only very recently, deazaflavin (F420) dependent ene-reductases (FDRs) have been found to be stereodivergent (like stereotopicity) to most common flavin mononucleotide (FMN) dependent ERs, including the OYEs.15
 | ||
| Scheme 2 (A) Stereoselectivity of flavin mononucleotide (FMN) cofactor dependent ERs like the old yellow enzymes (OYEs): (S) enantioselectivity is observed with α-methyl enals or enones, while for α-methylene isomers is (R). (B) The selection of an ADH with pro-R or pro-S stereoselectivity enables the access to either the erythro or to the threo alcohols. (C) ADH and ER enzymes can be combined in a one-pot two-step cascade process. | ||
ADHs, on the other hand, catalyse the reduction of carbonyl groups of ketones or aldehydes. These redox enzymes typically give excellent results in terms of conversion and stereoselectivity, and, as opposed to the ER enzymes, it is not rare to find ADHs with opposite stereoselectivity (Scheme 2B).16
Lastly, the combination of ER and ADH activities in a cascade process has proven to be highly effective for the reduction of several prochiral enones and enals.17 Chiral alcohols with up to two stereogenic centres are usually obtained in good yields and with a high optical purity (Scheme 2C).
In the following we show how the application of biocatalytic methods might significantly impact on the sustainability of sandalwood fragrances 1a–d.
Results and discussion
Epoxidation of α-pinene and synthesis of campholenic aldehyde
To begin, two literature procedures for the conversion of (−)-α-pinene ([α]D = −37.1, c = 1, CHCl3, ee = 82% by chiral GC, see ESI†) into pinene oxide 3 were evaluated, conversion and selectivity data are reported in Table 1. However, epoxide 3, due to its high ring-strain energy, underwent rearrangement already during the reaction, regardless of the method applied, affording a mixture of constitutional isomers: mainly campholenic aldehyde 2 and other terpenes18 (pinocamphone and trans-pinocarveol, detected by GC-MS). Only the non-campholenic terpenes must be considered as side-products of the reaction, since 2 is the product of the next step (Meinwald rearrangement). However, the product distribution was significantly correlated to the reaction temperature. Indeed, although the epoxidation with O2/acetaldehyde in the presence of a catalytic amount of N-hydroxyphtalimide (NHPI) at 60 °C is very appealing for its high atom economy,19 it yielded quantities of other terpenes considerably higher (28% vs. 10% by GC-MS) compared to the epoxidation with m-chloroperbenzoic acid (MCPBA),20 which instead was carried out at a lower temperature (5–10 °C). Nonetheless, the use of peracids has two main drawbacks: (a) safety concerns and (b) non-negligible production of carboxylic acid by-products (in this case m-chlorobenzoic acid).|
|
||||
|---|---|---|---|---|
| Reaction conditions | Conv.a (%) | Prod. distrib.a (%) | ||
| 3 | 2 | Other | ||
| a Not isolated yield, by GC-MS. b Bubbling. c 30% w/w. | ||||
| O2,b 3 eq. CH3CHO, 0.1 eq. NHPI, MeCN, 60 °C (ref. 19) | 62 | 55 | 17 | 28 |
| 1.3 eq. MCPBA, 1.0 eq. NaHCO3, CH2Cl2, 5–10 °C (ref. 20) | 91 | 82 | 8 | 10 |
| 1.1 eq. H2O2,c 1.8 eq. MeCN, MeOH, pH = 9 KHCO3 buffer, rt (ref. 23) | — | — | — | — |
| 1.1 eq. H2O2,c 13.3 MeCN, pH = 7 KPi, 60 °C (ref. 22) | 18 | 64 | 7 | 29 |
| 1.5 eq. H2O2,c 6.6 eq. PhCN, pH = 7 KPi, 60 °C (ref. 21) | — | — | — | — |
| 1.2 eq. H2O2,c 1.2 eq. CCl3CN, pH = 7 KPi, CH2Cl2, rt (ref. 21) | 100 | 86 | 5 | 9 |
| (i) O2,b cat. GOX, 3.0 eq. glucose, pH = 7 KPi, 30 °C; (ref. 27) (ii) 1.5 eq. CCl3CN, CH2Cl2, rt (ref. 21) | 65 | 68 | 17 | 15 |
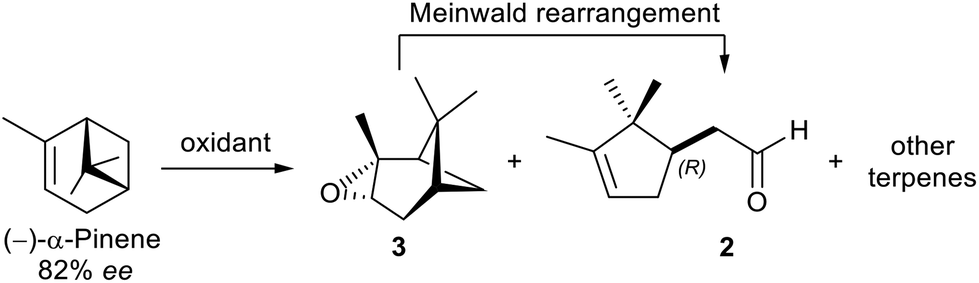
Surprisingly, the Payne epoxidation21 has never been applied to α-pinene. This procedure involves the addition of hydrogen peroxide (H2O2) to a nitrile (RCN) to produce a peroxycarboximic acid (RC(OOH)NH). Then, the acid oxidizes the olefin, resulting in the formation of epoxide and amide by-product (RCONH2).
Three different nitriles were tested (MeCN, PhCN and CCl3CN), conversions and chemoselectivity are reported in Table 1. Both MeCN and PhCN gave unsatisfactory conversions and required high reaction temperatures, that, as seen before, favour the rearrangement of pinene oxide into pinocamphone or carveol.22,23 In contrast, the higher reactivity of CCl3CN allowed to form 3 at room temperature with great benefit to the selectivity (86% by GC-MS). Even in this reaction, as seen for the MCPBA oxidation, we produced a stoichiometric amount of by-product (trichloroacetamide), quantitatively isolated by simple precipitation in hexane during the reaction work-up. But in this case, by heating the amide in presence of dehydrating phosphoric anhydride P4O10 in refluxing xylene,24 we could recover over 65% of the initial nitrile by distillation (Scheme 3, for more details see ESI†). The recovered nitrile was of sufficient purity (96% by quantitative 13C-NMR in presence of Cr(acac)3, see ESI†) to be reused for further epoxidations (Scheme 3).
 | ||
| Scheme 3 Reaction conditions: for steps (i) and (ii) see Table 1; (iii) 0.8 eq. P4O10, refluxing xylene. | ||
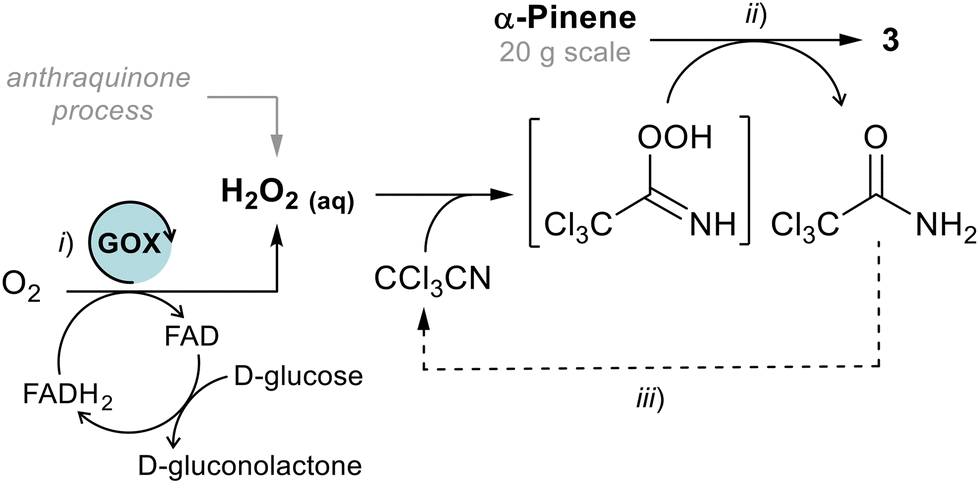
Although H2O2 is considered a “green” oxidant, we explored the possibility of generating and using it in situ in the Payne oxidation through a telescopic approach. Currently, H2O2 is produced by the anthraquinone process, which requires a Pd catalysed hydrogenation to regenerate the starting 2-alkyl anthrahydroquinone.25 Alternatively, H2O2 could be generated by a biocatalytic approach, avoiding the use of H2 and precious transition metals. For instance, glucose oxidase (GOX) catalyses the reduction of molecular oxygen to H2O2,26 using glucose as sacrificial substrate (Scheme 3). More recently, H2O2 was also bio-produced by oxidation of choline to trimethylglycine using choline oxidase enzymes and used in situ for limonene oxidation.27
In our case we succeeded to generate a dilute solution of H2O2 (≈1.2% w/w) by oxidation of glucose to gluconolactone (around 70% by 13C-NMR, see ESI†) in a phosphate buffer by bubbling O2 in presence of GOX from Aspergillus niger. Then, the solution was submitted to Payne oxidation of pinene, however both conversion and selectivity were not as good as those achieved using directly commercial H2O2 (65% vs. 100%, Table 1), in which case the oxidation was easily scaled-up on a multi-g scale, and on (+)-α-pinene ([α]D = +40.8, c = 1, CHCl3, 92% ee by chiral GC, ESI†) as well. Finally, the oxidation reaction mixture was submitted to the Meinwald rearrangement catalysed by ZnBr2 (7% w/w) in toluene to give the campholenic aldehyde 2 in a quantitative yield.18,28
Stereoselective one-pot two-step bioreduction
With the aldehyde 2 in our hands (purity 91% by GC-MS) we prepared the substrates for the bioreductions: the α,β-unsaturated carbonyl compounds 4a–c and 6 (Fig. 1A), and the α-methylene aldehydes 5a–b and ketones 5c–d (Fig. 1B). However, concerning their syntheses, we avoided as much as possible column chromatography purifications, more details on the synthetic routes are available in the ESI.† | ||
| Fig. 1 Substrates submitted to bioreduction: (A) α,β-unsaturated carbonyl compounds; (B) α-methylene isomers. | ||
The reduction of the conjugated CC double bond of substrates was catalysed by recombinant OYE2 from S. cerevisiae. This enzyme was selected from a panel of ERs from different sources for its optimal balance between selectivity and activity (for the screening see ESI†). For the regeneration of the reduced nicotinamide adenine dinucleotide phosphate (NADPH) cofactor, glucose dehydrogenase enzyme (GDH) from B. megaterium together with an excess of glucose, as sacrificial co-substrate,29 was used (Scheme 4). In most cases, the conversions were nearly quantitative (by GC-MS), usually within 10–12 hours, and occasionally, supplemental quantities of enzyme and cofactor was necessary to ensure the complete consumption of all starting material. Then, the commercially available Evo440 ADH (from evoxx technologies GmbH), known for its high activity on a broad range of substrates,17f–i was added to the reaction mixture containing the saturated aldehydes (I, R = H), yielding the primary alcohols 1a–b. After product removal from the reaction mixture by absorption on hydrophobic polyaromatic resins,17b,29,30 followed by column chromatography and/or bulb-to-bulb distillation purification procedures, each diastereoisomer of Brahmanol® and Firsantol® fragrances was isolated with a very high purity (>98% by GC-MS). This purification process ensured the elimination of all off-odours and contaminants typically arise during the biotransformation, rendering the fragrances suitable for the olfactory evaluation. For the reduction of ketone intermediates (I, R = Me), either a pro-R or a pro-S ADH, respectively Evo270 or Evo440, was employed, following the same work-up described above, affording secondary alcohols 1c–d.
 | ||
Scheme 4 Biocatalytic reduction of substrates 4a–c and 5a–d. Reaction conditions: substrate (3 mmol), pH = 7 KPi buffer (15 mL), i-PrOH cosolvent (1% v/v), 30 °C, 150 rpm; enzymes: for primary alcohols and secondary (2S) alcohols: (i) OYE2 (6 mL, ≈3 mg mL−1), GDH (≈200 U), NADP+ (20 mg), glucose (4.2 g); (ii) after 10–12 h Evo440 (30–40 mg) and NADP+ (10 mg) were added. For (2R) alcohols: same reaction conditions but in step (ii) Evo270 (30–40 mg) was used instead of Evo440. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Isolated yield after column chromatography followed by bulb-to-bulb distillation. bBy ratio of 13C-NMR signal integrations: C(1) for 1a–b, C(3) for 1c; C(2) for 1d. cIn CHCl3, c ≈ 1. dEstimated by mass balance taking in account the initial ee of pinene, see ESI.† Isolated yield after column chromatography followed by bulb-to-bulb distillation. bBy ratio of 13C-NMR signal integrations: C(1) for 1a–b, C(3) for 1c; C(2) for 1d. cIn CHCl3, c ≈ 1. dEstimated by mass balance taking in account the initial ee of pinene, see ESI.† | ||
In summary, the reaction conditions together with yields, diastereomeric ratios drs (measured by 13C-NMR) and optical rotatory values of alcohols 1a–d are reported in Scheme 4. In addition, since our attempts to determine the enantiomeric excess of either the carbonyl intermediates I or of the alcohols 1 by chiral GC or HPLC failed, we opted to quantify indirectly the stereoselectivity of OYE2 (Scheme 4) by a mass balance approach taking into account the initial ee of starting pinene, which was reasonably retained throughout the synthesis (see ESI†) and the dr of alcohols 1a–b (by 13C-NMR) and of ketone intermediates I (by GC-MS or by 13C-NMR).
Interestingly, even though Brahmanol® and Firsantol® differ only by a methylene group at C(4) position, the stereoselectivity of OYE2 in reducing the Firsantol® precursors 4b–5b was significantly higher than that of Brahmanol® homologues 4a–5a (Scheme 4), regardless of the type of substrate (methylene or α,β-unsaturated). A similar enhancement of selectivity was observed for the reduction of 5d (Ebanol® precursor) compared to the reduction of substrates 4c–5c (Sandalore® precursors), which differ from 5d solely by the absence of the CC unsaturation. Clearly, increasing the steric hindrance of substrates by adding a methylene group or by stiffening their structure proved beneficial to the stereoselectivity.
Lastly, in Fig. 2 we show, as representative example, the stacked 13C-NMR expansion spectra relative to the C(3) signal of each diastereoisomer of Sandalore® (1c), including the commercial mixture; integration of this signal allowed the determination of dr.
 | ||
| Fig. 2 Stacked 13C-NMR spectra relative to the C(3) signal of each diastereoisomer of 1c (CDCl3, 100 MHz). | ||
Determination of the absolute stereochemical configuration
The configurations initially assigned on the basis of the typical stereochemical courses commonly observed in the reductions catalysed by OYE2 (unlike stereotopicity),12,17 Evo270 (pro-S), and Evo440 (pro-R) enzymes,17h,i were confirmed by a combination of experiments on Sandalore® stereoisomers. Firstly, the absolute configuration at C(2) stereogenic centre was determined by analysis of 1H and 13C-NMR spectra of esters obtained by condensation of α-methoxy-α-trifluoromethylphenylacetic acid (MTPA) with 1c alcohols.31 In Fig. 3A is shown the expanded region of 13C-NMR spectra relative to C(1) signal of both (R)-MTPA and (S)-MTPA esters of the couple of alcohols 1c obtained from the bioreduction of 4c with OYE2 and Evo440, and with OYE2 and Evo270, respectively.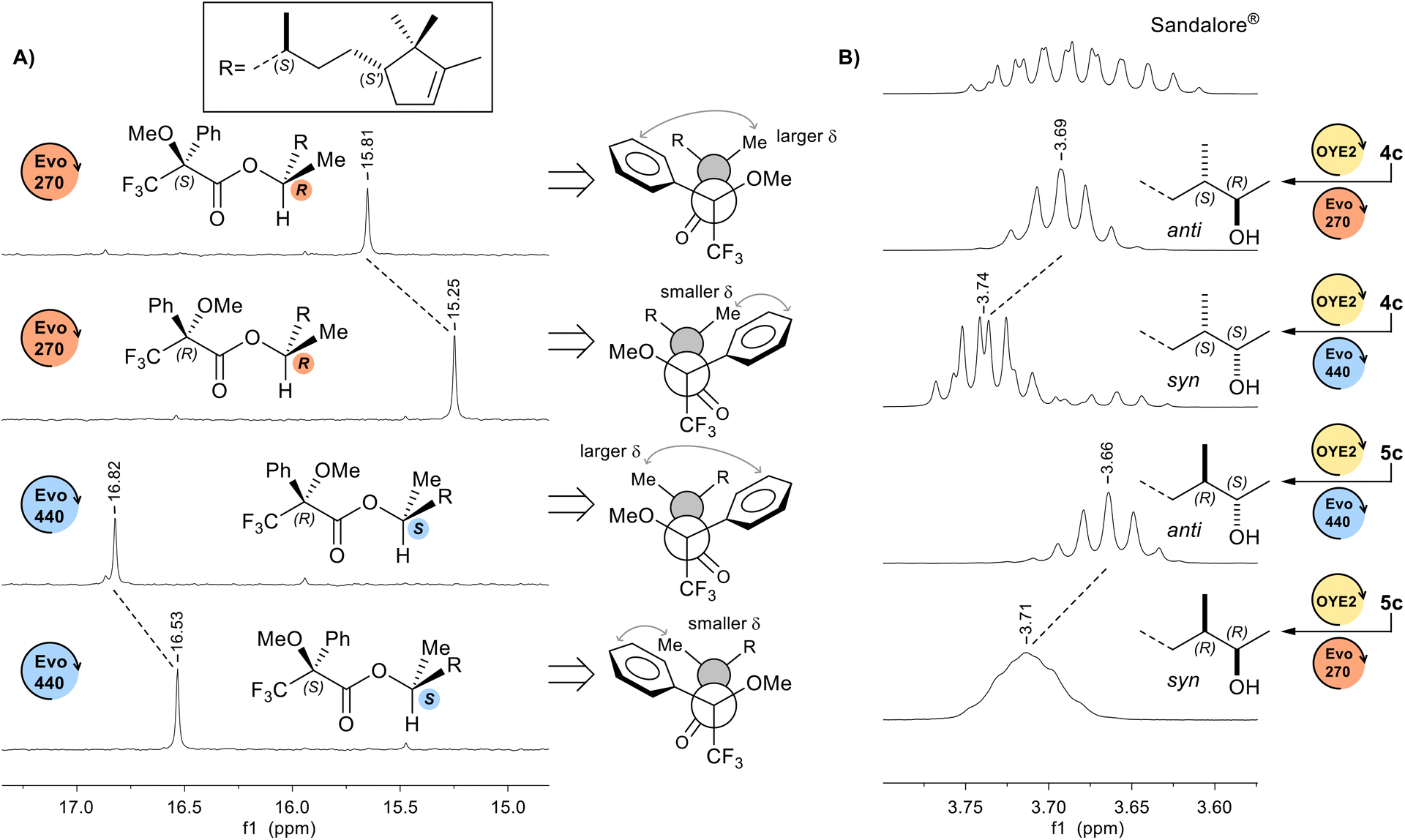 | ||
| Fig. 3 (A) 13C-NMR expansion spectra relative to C(1) signal of (R)-MTPA and (S)-MTPA esters of alcohols 1c obtained from the bioreduction of 4c (CDCl3, 100 MHz). (B) 1H-NMR expansion spectra relative to the CHOH signal of Sandalore® and its diastereoisomers (CDCl3, 400 MHz). | ||
According to Mosher's model, when the C(1) methyl group lies closer to the shielding region of phenyl ring, its 1H and 13C chemical shifts are up-field, conversely if it is distanced from the aromatic ring its protons are less deshielded, resulting in a downfield chemical shift. The differential chemical shift ΔδS,R (ΔδS,R = δMTPA(S) − δMTPA(R)) resulted +0.56 ppm for Evo270 and −0.29 ppm for Evo440, confirming the stereoselectivities of both ADHs. Then, we assigned the absolute configuration at C(3) centre knowing the C(2)–C(3) relative stereochemistry (syn or anti), which was attributed by 1H-NMR analysis of the chemical shift and multiplicity of CHOH signal. Indeed, it is known that for syn diastereoisomers this signal is a doublet of quartet (J = 6.3 and 4.4 Hz) downfield compared to that of its syn isomers (δ = 3.74 and 3.71 ppm for syn alcohols vs. 3.69 and 3.66 ppm for the couple of anti diastereoisomers, in CDCl3), for which instead it appears as a quintet (Fig. 3B).32
Notably, only by isolating ketone (Z)-7d, obtained by repeating the reduction without ADH, it was possible to confirm the (R) stereoselectivity of OYE2 in reducing α-methylene-β,γ-unsaturated substrates like dienone 5d (Fig. 4).
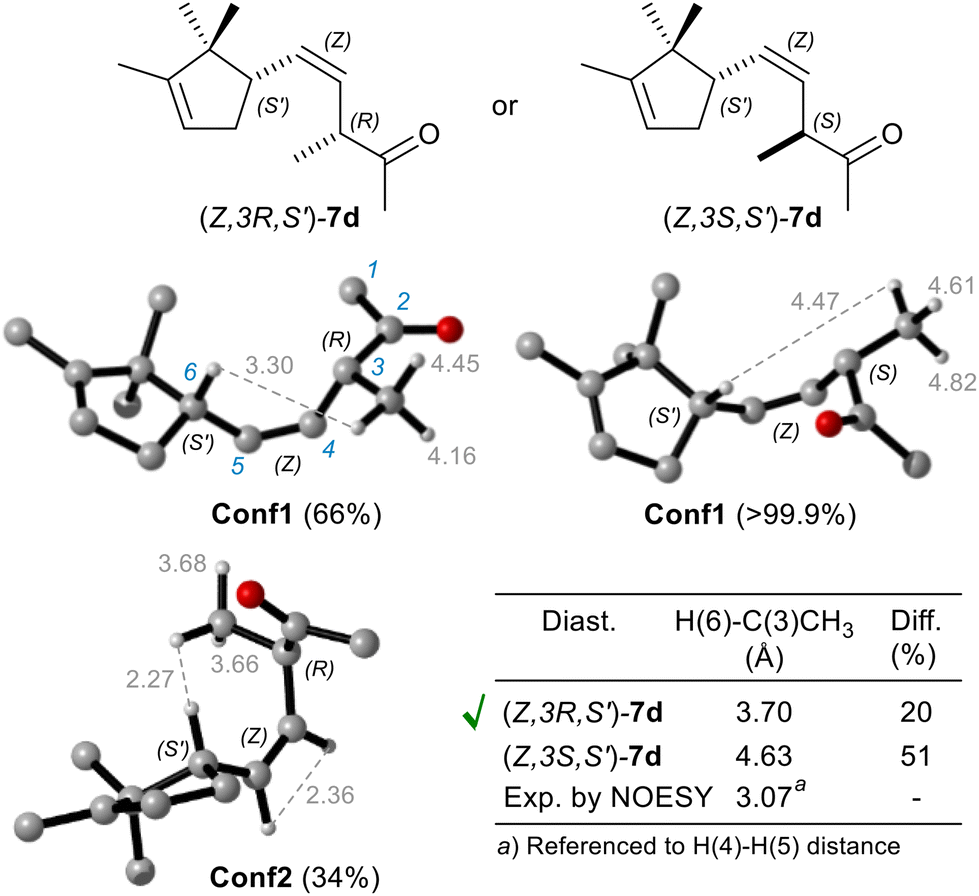 | ||
| Fig. 4 DFT computed H(6)–C(3)CH3 interproton distances (Å) of most stable conformers of C(3) epimers, compared to the experimental distance determined by quantitative NOE, for clarity some hydrogen atoms are omitted. In round brackets is reported the Boltzman conformer population. | ||
The measurement of Nuclear Overhauser Effect (NOE) through 2D NOESY experiment,33 between H(6) and C(3)CH3 protons allowed to determinate their average distance, which turned out to be crucial for the assignment of the relative configuration, and therefore of the absolute configuration as well. Indeed, a conformational study, conducted by density functional theory (DFT) computational chemistry (model chemistry: B3LYP/6-31+g(d,p), and SCRF = SMD, solvent = CHCl3, for more details see ESI†),34 revealed that the diastereoisomer with (Z,3R,S′) configuration predominantly exists as an equilibrium mixture of Conf1 (66%) and Conf2 (34%) conformers (Fig. 4), resulting in a weighted interproton distance of 3.7 Å, not so much different from that experimentally determined (3.1 Å). Conversely, the C(3) epimer is mainly populated by only one conformer Conf1 (99.9%), in which the H(6)–C(3)CH3 distance is much longer (4.6 Å) than the experimental one, approaching the typical detection limit (5 Å) of NOESY experiments.
Comparison of OYE2 chemoselectivity with other methodologies
Besides the stereoselective reduction of prochiral enones or enals (4–5),35 OYE2 has demonstrated its superiority in terms of chemoselectivity compared to other catalytic procedures.5b Indeed, in the synthesis of substrate 5c (Sandalore® precursor) we faced the problem of hydrogenating the α,β-unsaturated ketone 6 to ketone 8 preserving the campholenic double bond. Various reductive methodologies, known for their high chemoselectivity toward the reduction of conjugated CC double bonds were tested,36 the results of this screening are shown in Table 2. However, unlike in the case of OYE2 catalysed reduction, together with 8 a non-negligible quantity of over-reduced product (9) and other products was consistently present, regardless of the method applied. In addition, the GC-MS analysis of a commercially available Sandalore® sample revealed the presence of small amounts of completely saturated alcohols (around 3% by GC-MS, ESI†). This result highlights that even in the current industrial process, achieving complete chemoselectivity remains an unresolved issue.5
|
|
||||
|---|---|---|---|---|
| Reaction conditionsa | Conv.b (%) | Prod. distrib.b (%) | ||
| 8 | 9 | Otherc | ||
| a Catalyst/substrate ratio is in w/w. b Not isolated yield, by GC-MS. c Allylic alcohol, saturated alcohol, positional isomers. d Isolated yield without column chromatography. e 5% w/w. f Decomposition. g Pressure 1 atm. | ||||
| OYE2, (cond. of Scheme 4, without ADH) | 96 | >99(87)d | — | — |
| 0.15 eq. Pd/C,e NH4HCO2, MeOH, 30 °C (ref. 36a) | >99 | 95 | 4 | 1 |
| 0.2 eq. RANEY®-Ni, pyridine, EtOH, rt | 90 | 28 | 72 | — |
| 5 eq. Mg, MeOH, rt (ref. 36b) | — f | — | — | — |
| 0.25 eq. Lindlar cat., H2,g MeOH, 50 °C | 15 | 84 | — | 16 |
| 0.1 eq. Wilkinson cat., H2,g CH2Cl2, 36 °C (ref. 36c) | — | — | — | — |
| 3.0 eq. NaBH4, 5.0 eq. NiCl2·6H2O, MeOH/H2O, rt (ref. 36d) | 93 | 97 | 2 | 1 |

Olfactory evaluation of Sandalore® stereoisomers
The olfactory evaluation of Sandalore® revealed that the isomer with (S) configuration at C(2) stereogenic centre and (R) configuration at C(3) centre fits more effectively into the human olfactory receptors compared to the other isomers, since a stronger and more pleasant sandalwood odour was perceived (Table 3). These results agree with earlier evaluations carried out (detailed olfactory assessments are reported into ESI,† in Scheme 4 the most appealing stereoisomers are highlighted in grey) and confirm the postulated olfactophore model for the sandalwood fragrances. The latter consists of a bulky lipophilic moiety (such as the campholenic ring or the norbornane of santalol) separated by a spacer from the hydroxyl polar group.37 According to this model, the correct orientation of the osmophoric hydroxy group and the methyl substituent within the odour receptor is crucial for the perception of sandalwood scent, in contrast to the stereochemical configuration of campholenic ring that seems not to have influence.| Isomer | Odour description |
|---|---|
| (2R,3S,S′)-1c | Prominent amber-woody essence with a slightly creamy and tobacco notes. After few minutes the impact seems to be stronger (8/10) than the commercial Sandalore®.38 Very tenacious, it holds the scent consistently over time (7/10). |
| (2S,3S,S′)-1c | Unpleasant head impact (6/10). After a few minutes, although remaining unpleasant, the intensity of the note drops significantly compared to 1c-(2R,3S,S′), (3/10). |
| (2S,3R,S′)-1c | Impact 8/10. Creamy appearance reminiscent of sandalwood. Impressively tenacious even over time 9/10 (the most appealing of all diastereoisomers). |
| (2R,3R,S′)-1c | Delivering a notable impact at 7/10, it shares an olfactory resemblance to 1c-(2R,3S,S′), albeit slightly creamier and less dry. However, its tenacity falls short in comparison, scoring approximately 3/10 tenacity. |
Bio- vs. chemo-reduction process
Finally, we conducted a comparison between the Takasago reductive process for the stereoselective synthesis of the (S,S)-enantiomer of Brahmanol® and our bioreductive process, since both synthetic routes started from the common intermediate 4a.7b In Table 4 are reported the key technological and environmental factors and metrics for those two processes, including the simplified environmental factor (sEF),41 the complete environmental factor (cEF),42 and the EcoScale semi-quantitative analysis score.43|
|
|||
|---|---|---|---|
| Factors | Chemical processa | Bioreductionb | |
| LiAlH439 |
H27b |
OYE2 + Evo440 | |
| a Starting from pinene with 96% ee. b Starting from pinene with 82% ee. c Ice quenching, acidification, extraction, drying, concentration under vacuum. d Concentration under vacuum. e Extraction, drying, concentration under vacuum. f Not reported. | |||
| T (°C) | 0 to rt | 80 | 30 |
| P (atm) | 1 | 48 | 1 |
| Substrate conc. [M] | 0.4 | 1.3 | 0.2 |
| Time (h) | 3 | 24 | 24 |
| Catalytic | No | Yes | Yes |
| Solvent40 | Et2O hazardous | MeOH problematic | Water recommended |
| Technical set-up | Standard | High pressure | Standard |
| Work-up | Complexc | Very simpled | Simplee |
| Energy usage | Medium | Very high | Low |
| Resource depletion | — | Ru | — |
| Conversion (%) | —f | >99 | >99 |
| Yield (%) | 75 (84 and 85) | 84 | |
| Chemoselec. (%) | —f | >99 | |
| Stereoselectivity | 88(S):12(R) |
99(S):1(R) |
|
| Purification | Distillation ×2 | Distillation | |
| Waste | Non-biodegradable | Biodegradable | |
| Safety |

|

|
|
| sEF | 0.3 | 0.4 | 9.0 |
| cEF | 86 | 4 | 181 |
| EcoScale | 69/100 | 46/100 | 82/100 |
| Average EcoScale |

|

|
|
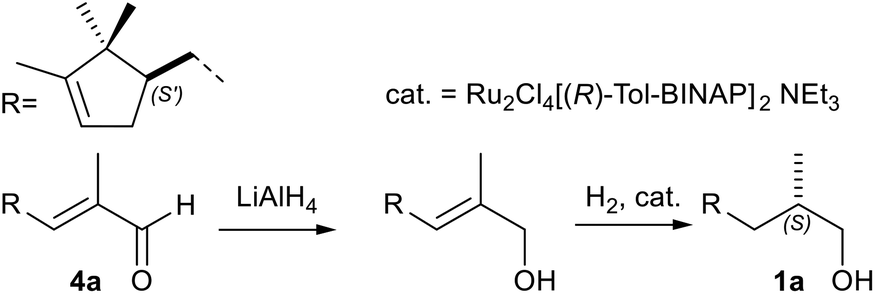
Regarding the chemical reduction, in the first step the α,β-unsaturated aldehyde 4a was transformed into the allylic alcohol intermediate by treatment with the very harsh reducing agent LiAlH4 (0 °C in Et2O).39,44 According to the patent, the allylic alcohol was then hydrogenated in presence of a Ru-based Noyori catalyst (48 atm, 80 °C, in MeOH).7b Although the overall yield and the stereoselectivity were acceptable, they were significantly lower than those achieved in the one-pot bioreductive process (98% ee vs. 76% ee).
Undeniably, both the sEF and cEF metrics reveal that the bioreduction generates a significantly higher amount of waste per kg of product compared to the chemical reductions, with a difference of one order of magnitude, irrespective of the environmental factor considered (Table 4). This result is not surprising, considering that the transition metal catalysed hydrogenations, among all catalytic transformations, are those operating at highest substrate concentrations with perfect atom-economy.
However, these metrics are strictly related to the yield and to the reagents concentration, but do not take in account other important parameters such as: (i) selectivity (chemo- and/or stereoselectivity), (ii) ease of workup/purification, (iii) safety, (iv) energy consumption, (v) technical set-up (pot economy and/or requirement for high pressure equipment), (vi) nature of waste (biodegradable vs. non-biodegradable, presence of heavy metals), (vii) resource depletion; all of which have an impact on the environmental sustainability of the process.
In this context, a semi-quantitative analysis based on the EcoScale approach could offer valuable insights for a more comprehensive and fair comparison of the two routes beyond what the cEF and the sEF had provided. Using this approach, a maximum of points is arbitrarily assigned to the ideal process (100/100), where all technological and environmental factors are optimized. For each parameter that deviates from the ideal process, penalty points are assigned accordingly. The higher the score, the more sustainable is the methodology applied.
Remarkably, the biocatalytic process scored very well, 82/100 on EcoScale, while both chemo-reductions had a significantly higher number of point penalties, 69/100 and 46/100 (detailed calculations are reported in the ESI†). The main penalties were ascribable to a lower stereoselectivity, to the use of hazardous and highly flammable reagents (LiAlH4 and H2) and solvents (Et2O and MeOH), to the need of a highly energy-consuming reaction set-up (cooling or heating, high-pressure), and to the production of non-biodegradable wastes.
In addition, our multi-enzymatic process is carried out in a single vessel, in accord with the principle of pot economy.45 Such set-up simplifies reaction work-up and product isolation, both in terms of energy consumption and quantity of solvents used. On the contrary, in the Takasago process the incompatibility between the two reductive steps prevents their combination in a more appealing and sustainable one-pot synthesis, and even the telescoped transformation results impracticable. To this regard biocatalysis maintains its primacy over other methodologies, including also the more recent and efficient homogenous metal-based stereoselective catalysts. Indeed, to our knowledge, these catalysts have never been combined in a one-pot two hydrogenations-step capable of stereoselectively reducing enals or enones into the corresponding alcohols.35
Lastly, the bioreduction is undoubtedly a safer process, as clearly indicated by the higher number of hazards and risks typically associated with the hydrogenations and the hydride-based reductions.
Experimental
A solution of substrate (3.0 mmol) in i-PrOH (0.8 mL) was added to a KPi buffer solution (pH 7.0, 50 mM, 15 mL) containing OYE2 (≈3 mg mL−1, 5 mL), GDH (200 U), glucose (4.2 g), NADP+ (20 mg). The mixture was incubated for 10–12 hours in an orbital shaker (150 rpm, 30 °C). The reaction was monitored by TLC until complete conversion. Eventually, more enzymes were added to increase the conversion. After 10–12 hours ADH (Evo270 or Evo440, 30–40 mg) and NADP+ (10 mg) were added to the reaction mixture. After 10–12 hours XAD-1180 resins (5.0 g) were added to the reaction mixture and left to shaker for 30 minutes. The mixture was filtered into a porous filter (porosity 0) and the resins were washed several times with EtOAc (15 mL × 4). The combined organic phase was washed with water (10 mL), dried over Na2SO4, and the solvent was removed under reduced pressure. The crude material was submitted to silica gel column chromatography purification and then distilled by bulb-to-bulb apparatus affording alcohol 1.Conclusions
Some of the most important commercial sandalwood fragrances were prepared without employing transition metal catalysed hydrogenations, and even the use of metal hydride based carbonyl reductions was very limited (preparation of 4b and 5b substrates). Such reductive methodologies were proficiently replaced by bioreductions, combing ER and ADH enzymatic activities in a one-pot two-step process. The yield and especially the chemo- and the stereoselectivity achieved through biocatalysis compare favourably with the actual industrial procedures.5a In addition, the most powerful and appealing stereoisomers were synthesized, with good diastereomeric ratios, by stereoselective reduction of methylene precursors 5.A fair comparison of the technical, safety and environmental factors, including the EcoScale semi-quantitative analysis, applied to the stereoselective synthesis of Brahamanol® clearly showed the superior eco-friendliness of the bioreductive approach with respect to the traditional chemo reductions. Lastly, such development should open an appealing perspective in the field of fragrance chemistry.
Author contributions
F. G. G. conceived and designed the experiments. M. C. C., D. M., L. D. M., F. P., F. G. G. and D. F. performed experiments and analysed data. F. G. G. prepared the manuscript, all authors equally contributed to the revision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Our sincere thanks go to Dr U. Grigoli (Novachem Aromatici S.r.l., Italy) for providing samples of commercial sandalwood fragrances, Dr P. Muñoz (Destilerías Muñoz Gálvez, S.A, Spain) for supplying both enantiomers of α-pinene and Dr P. Cerotti (CreaSens S.r.l., Italy) for the professional olfactory evaluations. MCC acknowledges MIUR for PhD grant (XXXVII Research Doctorate Cycle). DF and FP acknowledge the Agritech National Research Center and the European Union Next-GenerationEU (Piano Nazionale di Ripresa e Resilienza (PNRR) – Missione 4 Componente 2, Investimento 1.4 – D.D. 1032 17/06/2022, CN00000022).References
- (a) C. S. Sell, A Fragrant Introduction to Terpenoid Chemistry, The Royal Society of Chemistry, 2003 RSC; (b) K. Bauer, D. Garbe and H. Surburg, Common Fragrance and Flavour Materials, Wiley-VCH, 3rd edn, 1997 CrossRef.
- Asian Regional Workshop (Conservation & Sustainable Management of Trees, Vietnam August 1996), ‘Santalum album’, The IUCN Red List of Threatened Species, e. T31852A9665066, 1998.
- A. A. Birkbeck, Chimia, 2017, 71, 823–835 CrossRef CAS PubMed.
- M. J. Beekwilder, A. M. M. L. Houwelingen, H. J. Bosch, G. F. Lentzen, E. Melillo and H. W. Wisselink, US11390863 B2, 2022 (to Isobionics B.V.).
- (a) P. Dupau, Helv. Chim. Acta, 2018, 101, e18001 CrossRef; (b) E. Vrbkova, D. Šimackova, E. Vyskocilova and L. Cerventy, Perfum. & Flavor., 2021, 46, 39–47.
- L. A. Saudan, Acc. Chem. Res., 2007, 40, 1309–1310 CrossRef CAS PubMed.
- (a) Sandacore®: G. Buchbauer, P. Lebada, L. Wiesinger, P. Weiss-Greiler and P. Wolschann, Chirality, 1997, 9, 380–385 CrossRef CAS; (b) Brahmanol®: A. Takaschi, M. Hiroyuki and Y. Takeshi, JP-8268940, 1996 (to Takasago Perfumery Co LTD); (c) Firsantol®: C. Chapuis and P. A. Blanc, US-5696075, 1997 (to Firmenich SA); (d) Ebanol®: J. A. Bajgrowicz and G. Frater, EP-0841318A2, 1998 (to Givaudan-Roure SA).
- E. Brenna, C. Fuganti, F. G. Gatti and S. Serra, Chem. Rev., 2011, 111, 4036–4072 CrossRef CAS PubMed.
- E. Brenna, C. Fuganti, F. G. Gatti, L. Malpezzi and S. Serra, Tetrahedron: Asymmetry, 2008, 19, 800–807 CrossRef CAS.
- F. G. Gatti, F. Parmeggiani and A. Sacchetti, in Synthetic Methods for Biological Active Molecules: Exploring the Potential of Bioreductions, ed. E. Brenna, Wiley-VCH, 2013 Search PubMed.
- (a) H. S. Toogood and N. S. Scrutton, ACS Catal., 2018, 8, 3532–3549 CrossRef CAS PubMed; (b) C. K. Winkler, K. Faber and M. Hall, Curr. Opin. Chem. Biol., 2018, 43, 97–105 CrossRef CAS PubMed; (c) F. Parmeggiani, E. Brenna, D. Colombo, F. G. Gatti, F. Tentori and D. Tessaro, ChemBioChem, 2022, 23, e2021004 CrossRef PubMed.
- E. Brenna, S. Cosi, E. E. Ferrandi, F. G. Gatti, D. Monti, F. Parmeggiani and A. Sacchetti, Org. Biomol. Chem., 2013, 11, 2988–2996 RSC.
- E. Brenna, F. G. Gatti, D. Monti, F. Parmeggiani, A. Sacchetti and J. Valoti, J. Mol. Catal. B: Enzym., 2015, 114, 77–85 CrossRef CAS.
- D. Seebach and V. Prelog, Angew. Chem., Int. Ed. Engl., 1982, 21, 654–660 CrossRef.
- (a) S. Mathew, M. Trajkovic, H. Kumar, Q.-T. Nguyen and M. W. Fraaije, Chem. Commun., 2018, 54, 11208–11211 RSC; (b) S. W. Kang, J. Antoney, R. L. Frkic, D. W. Lupton, R. Speight, C. Scott and C. J. Jackson, Biochemistry, 2023, 62, 873–891 CrossRef CAS PubMed.
- (a) K. Nakamura, R. Yamanaka, T. Matsuda and T. Harada, Tetrahedron: Asymmetry, 2003, 14, 2659–2681 CrossRef CAS; (b) A. S. de Miranda, C. D. F. Milagre and F. Hollmann, Front. Catal., 2022, 2, 900554 CrossRef.
- (a) M. Korpak and J. Pietruszka, Adv. Synth. Catal., 2011, 353, 1420–1424 CrossRef CAS; (b) E. Brenna, D. Monti, F. G. Gatti, F. Parmeggiani and A. Sacchetti, Chem. Commun., 2012, 48, 79–81 RSC; (c) E. Brenna, D. Monti, F. G. Gatti, F. Parmeggiani and A. Sacchetti, ChemCatChem, 2012, 4, 653–659 CrossRef CAS; (d) E. Brenna, L. Malpezzi, D. Monti, F. G. Gatti, F. Parmeggiani and A. Sacchetti, J. Org. Chem., 2013, 78, 4811–4822 CrossRef CAS; (e) T. Classen, M. Korpak, M. Schölzel and J. Pietruszka, ACS Catal., 2014, 4, 1321–1331 CrossRef CAS; (f) E. Brenna, M. Crotti, F. G. Gatti, L. Marinoni, D. Monti and S. Quaiato, J. Org. Chem., 2017, 82, 2114–2122 CrossRef CAS PubMed; (g) E. Brenna, M. Crotti, M. De Peri, G. Manenti and D. Monti, Adv. Synth. Catal., 2018, 360, 3677–3686 CrossRef CAS; (h) S. Venturi, M. Trajkovic, D. Colombo, E. Brenna, M. W. Fraaije, F. G. Gatti, P. Macchi and E. Zamboni, ACS Catal., 2020, 10(21), 13050–13057 CrossRef CAS; (i) S. Venturi, M. Trajkovic, D. Colombo, E. Brenna, M. W. Fraaije, F. G. Gatti, P. Macchi and E. Zamboni, J. Org. Chem., 2022, 87, 6499–6503 CrossRef CAS.
- J. Kaminska, M. A. Schwegler, A. J. Hoefnagel and H. van Bekkum, Recl. Trav. Chim. Pays-Bas, 1992, 111, 432–437 CrossRef CAS.
- (a) F. Minisci, C. Gambarotti, M. Pierini, O. Porta, C. Punta, F. Recupero, M. Lucarini and V. Mugnaini, Tetrahedron Lett., 2006, 47, 1421–1424 CrossRef CAS; (b) R. Spadaccini, L. Liguori, C. Punta and H. R. Bjørsvik, ChemSusChem, 2012, 2, 261–265 CrossRef PubMed.
- J. K. Crandall and L. C. Crawley, Org. Synth., 1973, 53, 17 CrossRef CAS.
- G. B. Payne, P. H. Deming and P. H. Williams, J. Org. Chem., 1961, 26, 659–663 CrossRef CAS.
- (a) L. A. Arias, S. Adkins, C. J. Nagel and R. D. Bach, J. Org. Chem., 1983, 48, 888–890 CrossRef CAS; (b) R. D. Bach and J. W. Knight, Org. Synth., 1981, 60, 63 CrossRef CAS.
- W. C. Frank, Tetrahedron: Asymmetry, 1998, 9, 3745–3749 CrossRef CAS.
- D. B. Reisner and E. C. Horning, Org. Synth., 1950, 30, 22 CrossRef CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- B. O. Burek, S. Bormann, F. Hollmann, J. Z. Bloh and D. Holtmann, Green Chem., 2019, 21, 3232–3249 RSC.
- Y. Ma, P. Li, Y. Li, S. J.-P. Willot, W. Zhang, D. Ribitsch, Y. H. Choi, R. Verpoorte, T. Zhang, F. Hollmann and Y. Wang, ChemSusChem, 2019, 12, 1310–1315 CrossRef CAS PubMed.
- L. Lopez, G. Mele, V. Fiandanese, C. Cardellicchio and A. Nacci, Tetrahedron, 1994, 50, 9097–9106 CrossRef CAS.
- M. Bechtold, E. Brenna, C. Femmer, F. G. Gatti, S. Panke, F. Parmeggiani and A. Sacchetti, Org. Process Res. Dev., 2012, 16, 269–276 CrossRef CAS.
- J. T. Vicenzi, M. J. Zmijewski, M. R. Reinhard, B. E. Landen, W. L. Muth and P. G. Marler, Enzyme Microb. Technol., 1997, 20, 494–499 CrossRef CAS.
- T. R. Hoye, C. S. Jeffrey and F. Shao, Nat. Protoc., 2007, 2, 2451–2458 CrossRef CAS PubMed.
- (a) J. Zhao and K. Burgess, Org. Lett., 2009, 11, 2053–2056 CrossRef CAS PubMed; (b) E. Brenna, F. Dalla Santa, F. G. Gatti and D. Tessaro, Org. Biomol. Chem., 2019, 17, 813–821 RSC.
- M. J. Thrippleton and J. Keeler, Angew. Chem., Int. Ed., 2003, 42, 3938–3941 CrossRef CAS PubMed.
- For more details, see references into ESI.†.
- G. Lonardi, R. Parolin, G. Licini and M. Orlandi, Angew. Chem., Int. Ed., 2023, 62, e202216649 CrossRef CAS PubMed.
- (a) B. C. Ranu and A. Sarkar, Tetrahedron Lett., 1994, 35, 8649–8650 CrossRef CAS; (b) I. K. Youn, G. H. Yon and C. S. Pak, Tetrahedron Lett., 1986, 27, 2409–2410 CrossRef CAS; (c) A. Y. Hong, N. B. Bennett, M. R. Krout, T. Jensen, A. M. Harned and B. M. Stoltz, Tetrahedron, 2011, 67, 10234–10248 CrossRef CAS PubMed; (d) J. M. Khurana and P. Sharma, Bull. Chem. Soc. Jpn., 2004, 77, 549–552 CrossRef CAS.
- G. Fráter, J. A. Bajgrowicz and P. Kraft, Tetrahedron, 1998, 54, 7633–7703 CrossRef.
- GC-MS analysis of a commercially available sample of Sandalore® (Givaudan): 84% of 1c, 13.4% of 6-(2,2,3-trimethylcyclopent-3-en-1-yl)hexane-3-ol (regioisomer from crossed aldol condensation), 2.6% dihydro-Sandalore® (over-reduced product).
- I. Kosaku, T. Shigeyoshi and A. Takahiro, WO2007063703A1, 2007 (to Kao Corporation).
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehadad and P. J. Dunne, Green Chem., 2016, 18, 288–296 RSC.
- F. Roschangar, R. A. Sheldon and C. H. Senayanake, Green Chem., 2015, 17, 752–768 RSC.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- K. Van Aken, L. Strekowski and L. Patiny, Beilstein J. Org. Chem., 2006, 2 DOI:10.1186/1860-5397-2-3.
- Due to the lack of experimental details on both work-up and chemoselectivity, the reduction with LiAlH4 was replicated in our laboratory, see ESI.†.
- Y. Hayashi, Chem. Sci., 2016, 7, 866–880 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation and characterization of products, Mosher's ester analysis, optimization of multienzymatic protocols, olfactory evaluation; copies of 1H and 13C NMR spectra. See DOI: https://doi.org/10.1039/d4gc00746h |
| This journal is © The Royal Society of Chemistry 2024 |