
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light-swing CO2 capture: photoirradiation-based chemical CO2 release based on photoisomerization of azobenzene-amine/guanidine derivatives†
Ryo
Murakami
 *,
Keitaro
Shiota‡
,
Ayaka
Uchida‡
and
Fuyuhiko
Inagaki
*
*,
Keitaro
Shiota‡
,
Ayaka
Uchida‡
and
Fuyuhiko
Inagaki
*
Faculty of Pharmaceutical Sciences, Kobe Gakuin University, 1-1-3 Minatojima, Chuo-ku, Kobe, Hyogo 650-8586, Japan. E-mail: finagaki@pharm.kobegakuin.ac.jp; rmurakami@pharm.kobegakuin.ac.jp
First published on 30th May 2024
Abstract
The world is committed to reducing CO2 emissions, and research on CO2 capture and effective utilization is being actively studied. Among the methods in development, direct air capture (DAC) is classified as a negative emission technology and has attracted significant study. The current problem with CO2 capture technologies for decarbonization is their cost due to the high separation energy required to release CO2. We have developed a new light-swing method that can potentially utilize a natural source of energy, i.e., sunlight, as an alternative to temperature- and pressure-swing methods. Herein, we report photoirradiation-based CO2 capture based on photoisomerization of azobenzene-amine and guanidine derivatives. The visible light-swing CO2 absorption and release system using azobenzene-guanidine has shown potential in DAC systems owing to its reusability. A plausible mechanism for CO2 release under light irradiation involves photoisomerization from trans- to cis-azobenzene in which steric repulsion with other molecules is the driving force, and CO2 is released due to the functional disruption of intermolecular interactions. This concept demonstrates the potential of using various photokinetic molecules as a driving force for light-swing CO2 capture.
1. Introduction
CO2 capture,1–6 a technology that captures CO2 directly from the atmosphere or from exhaust gases is one of the most important methods for decarbonization. In the well-studied chemical absorption method using amines, CO2 and an absorbent are combined through a chemical reaction. There are several types of CO2 capture methods, including carbon capture and storage (CCS),7,8 which captures CO2 from the exhaust gases from thermal power plants, direct air capture (DAC),9–22 which captures atmospheric CO2, and bioenergy with carbon capture and storage (BECCS),23,24 which captures CO2 produced in biomass power generation. In all of these methods, low-concentration CO2 [exhaust gas: ca. 1–15 vol%, air: ca. 400 ppm] is recovered with absorbent/adsorbent (amines, etc.), and then CO2 is released and concentrated by heating or decompression.In the chemical absorption method using amines,25 the reaction proceeds exothermically through a neutralization reaction between the base, amine, and the acid (CO2). On the other hand, when CO2 is released, the reaction becomes endothermic, requiring energy from the outside to release/concentrate CO2. This enormous separation energy is a major issue for general CO2 capture technologies. One of the reasons for this large separation energy is the water content. For example, we found that monoethanolamine (MEA),14 which is a benchmark for CO2 absorbents, absorbed CO2 and moisture in air to form 1(CO2)·3(MEA)·3(H2O). Theoretically, two molecules of amine should react with one molecule of CO2, providing 1(CO2)·2(amine) [e.g., 2 × R-NH2 + CO2 → R-NH-COO−·+H3N-R]. Thus, the CO2 absorption efficiency of the amine is reduced due to the water content, resulting in an increase in the separation energy. In addition, extra thermal energy for moisture content during CO2 release/concentration is required. It was thought that the presence of water was unavoidable because amines are hydrophilic groups and CO2 is soluble in water, as exemplified by carbonated water. However, we26–28 recently found that aralkyl amines with hydrophobic phenyl groups near the amine can selectively absorb CO2 in air without moisture. This technology eliminates the necessity for extra energy related to water content when releasing CO2.
The ultimate challenge in CO2 capture by chemical absorption is how to avoid wasting energy during the dissociation of amines and CO2. As far as we know, there are currently three methods of dissociating CO2: (a) a thermal-swing method by heating,29 (b) a pressure-swing method30,31 by depressurization and (c) an electro-swing method32–35 using an electrochemically activated redox compound (Scheme 1, top). In the thermal-swing method, the use of existing thermal energy sources such as industrial waste heat and geothermal heat has been actively studied. This is attractive in that it does not require generation of the energy necessary for CO2 dissociation. However, it is limited because CO2 must be recovered near the heat source. It also has the problem of sacrificing the efficiency of the absorption phase to improve the efficiency of CO2 emission. The pressure-swing method requires CO2 to be recovered under reduced pressure. However, in general, recovered CO2 should be under pressurized conditions since it is expected to be stored and utilized. Therefore, re-pressurizing CO2 after decompression recovery is necessary, resulting in energy consumption. The electro-swing method also requires that significant energy is supplied by renewable electricity. To satisfy these energy requirements, light is one of the most accessible energy sources in an energized environment, and sunlight is a natural source of energy. The Liu group recently reported a CO2 release method using a light-induced pH swing approach36 with spiropyran compounds. However, this method is limited to absorption of CO2 with a concentration of 100%. From the perspective of green processes, it is necessary to capture low-concentration CO2 such as that contained in air or industrial exhaust gas.
 | ||
| Scheme 1 Types of CO2 release in chemical absorption. | ||
Thus, we attempted to develop a new direct light-swing method that is applicable to low-concentration CO2 capture. Herein, we report a photoirradiation-based CO2 capture method involving photoisomerization of azobenzene-amine or guanidine derivatives (Scheme 1, bottom).
2. Results and discussion
2.1. Photoirradiation-based CO2 release of azobenzene-amine
It is well known that photoirradiation of azobenzene37–41 causes geometric isomerization. Several conventional azobenzene-introduced CO2 absorbers have been reported,42–44 but they are not adapted to visible light or CO2 enrichment. Thus, we considered that the trans form of azobenzene bearing an aminomethyl group should selectively absorb CO2, and the photoisomerized cis-form of aminomethyl-substituted azobenzene should release the absorbed CO2 upon the structural change. Based on this hypothesis, azobenzene derivatives with an aminomethyl group were synthesized (Fig. 1a). Protection of the benzyl amino group in 2-aminomethylaniline with Boc2O followed by dehydration condensation with nitrosoamine afforded N-protected 2-aminomethylazobenzene 2a in 60% overall yield. Deprotection of the Boc group provided the desired 2-aminomethylazobenzene o-3a in 97% yield. 3-Aminomethylazobenzene m-3b was generated from 3-aminomethylaniline in similar yield using the same procedure. | ||
| Fig. 1 Synthesis of and CO2 absorption by azobenzene-amines. (a) Syntheses of azobenzene-amine derivatives. (b) Increase in mass of azobenzene-amines (10 mmol, 2.11 g) in air at room temperature upon absorbing aerial CO2. (c) Composition ratio by elemental analysis of azobenzene-amines in air. (d) Composition ratio by elemental analysis of azobenzene-amines in 1% CO2. | ||
With the required aminomethylated azobenzene derivatives in hand, we next examined their ability to absorb CO2. For this experiment, 10 mmol of the aminomethylated azobenzene derivative (o-3a or m-3b) was placed on a scale exposed to air (ca. 400–600 ppm CO2) and weighed over time. The results are shown in Fig. 1b. In both cases, the mass gradually increased, reaching equilibrium after about 10 hours. To determine the composition of the mixture of substrate (o-3a and m-3b) and air, elemental analyses were conducted (Fig. 1c). Samples collected after 1 week revealed that the mixture derived from o-3a contained CO2 and o-3a at a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5. The composition ratio derived from m-3b was the same. Although general alkylamines absorbed a large amount of H2O from aerial moisture, these amines absorbed aerial CO2 only. Thus, the introduction of a hydrophobic azobenzene was effective for selective absorption of CO2. As described in the Introduction, the ratio of amine to CO2 should be 2:1. However, the relative amount of CO2 was lower when using o-3a and m-3b. Because the concentration of CO2 in the atmosphere is very low (ca. 0.03–0.06 vol%), the total amount of absorbed CO2 may not reach the theoretical value.
:5. The composition ratio derived from m-3b was the same. Although general alkylamines absorbed a large amount of H2O from aerial moisture, these amines absorbed aerial CO2 only. Thus, the introduction of a hydrophobic azobenzene was effective for selective absorption of CO2. As described in the Introduction, the ratio of amine to CO2 should be 2:1. However, the relative amount of CO2 was lower when using o-3a and m-3b. Because the concentration of CO2 in the atmosphere is very low (ca. 0.03–0.06 vol%), the total amount of absorbed CO2 may not reach the theoretical value.
Therefore, the same experiments were conducted in a 1 vol% CO2 atmosphere (99 vol% N2) with a higher CO2 concentration (Fig. 1d). Under these conditions, the ratio of amine to CO2 was 2:1, which matches the theoretical ratio.
We next focused on CO2 release upon photoirradiation. In order to select the appropriate wavelength for the irradiating light, UV-vis measurements of CO2-absorbed aminomethylazobenzene (amine:CO2 = 2:1) were performed (Fig. 2a). For both amines (o-3a and m-3b), the maximum absorption wavelength was around 300 nm (o-3a: λmax = 313 nm, m-3b: λmax = 315 nm).
 | ||
| Fig. 2 CO2 desorption from CO2-absorbed azobenzene-amines under photoirradiation. (a) UV-vis measurements of CO2-absorbed o-3a and m-3b (amine:CO2 = 5:2). (b) Device for CO2 release from CO2-absorbed amines under UV irradiation (λ = 302 nm). (c) CO2 concentration in outlet gas using device under UV irradiation. | ||
To confirm CO2 release under light irradiation, the device in Fig. 2b was designed and used. A test tube containing CO2-absorbed aminomethylazobenzene (o-3a and m-3b; amine:CO2 = 5:2; derived from 10 mmol of amine) was placed on a UV lamp (λ = 302 nm), and nitrogen gas was flowed into the UV-irradiated test tube at a constant flow rate (200 mL min−1) using a mass flow controller (MFC). The CO2 concentration in the outlet gas was measured over time using a CO2 densitometer (Fig. 2c). As expected, CO2 release was observed for both compounds, and the maximum release concentrations were around 70–80 ppm.
2.2. Photoirradiation-based CO2 release from azobenzene-guanidine
While aniline, which has an amine directly bonded to the benzene ring, has lower basicity than normal alkyl amines, it has problems with CO2 absorption. To avoid the introduction of an amine at the benzylic position, a new structure was designed with a more basic guanidino group21 on the phenyl group. To increase the basicity of the guanidino group, guanidine 7 was selected as a candidate compound, in which a dimethylamino group was introduced opposite the phenyl group of azobenzene. Synthesis of guanidine 7 is shown in Fig. 3a. After reduction of the nitro group to an amino group to give 6 in 78% yield, the reaction of 6 with cyanamide gave the desired azobenzene-guanidine derivative 7 in 53% yield. In the initial trial, guanidine 7 did not absorb CO2. Although the reason is unclear, solid state guanidine 7 might not be able to absorb CO2 due to the specific surface area. After several screenings, we found that guanidine 7 in MeOH absorbed 1% CO2. Thereafter, CO2 absorption was conducted in MeOH solution. A mixed gas containing 1% CO2 and 99% N2 was flowed into guanidine 7 (3.0 mmol) in MeOH (100 mL) at a rate of 20 mL min−1 using an MFC, and the resulting CO2 concentration on the outlet side was measured over time (Fig. 3b). The flow rate was reduced from 200 to 20 mL min−1 due to concerns about solvent volatilization. After 45 min, the CO2 concentration decreased to a minimum of 1756 ppm, and then increased slowly (Fig. 3c). The amount of absorbed CO2 was calculated to be about 2.9 mmol, and the 7:CO2 ratio was 1:1. After absorbing CO2, CO2-absorbed 7 was precipitated as a solid and could be filtered out from the mixture.
 | ||
| Fig. 3 Synthesis of and CO2 absorption by azobenzene-guanidine 7. (a) Synthesis of azobenzene-guanidine derivative 7. (b) Device for measuring CO2 absorption of 7 under 1% CO2 flow. (c) CO2 concentration in outlet gas. | ||
Next, UV measurements were performed in a EtOH solution using the filtered solid (Fig. 4a). The maximum absorption was found at a wavelength of 419 nm, which is in the visible region and is longer than that for azobenzene-amine 3. Based on this result, a 440 nm blue LED light source was selected to investigate CO2 emission. The device is shown in Fig. 4b. The desiccator containing CO2-absorbed guanidine 7 was irradiated with LED light under a N2 gas flow of 20 mL min−1 controlled by an MFC, and the CO2 concentration on the outlet side was measured over time. As shown in Fig. 4c, CO2 release occurred immediately after irradiation, and up to 3644 ppm of CO2 was released at 50 min. In this system, heat was generated by the light source, and the temperature in the desiccator was about 40 °C. To confirm that the release of CO2 was not simply due to the heat from the light source, CO2 release under heating conditions using the same solid was investigated. Using the same equipment as in Fig. 4b, the desiccator was gradually heated (10 °C per 30 min) with an oil bath instead of being irradiated by light, and the CO2 concentration in the outlet gas was measured (Fig. 4d). CO2 was gradually released at 70 °C, and high levels of CO2 emission were found at temperatures above 90 °C. However, no CO2 release was observed when heating at around 40 °C. Therefore, it is clear that CO2 release under light irradiation is caused by light energy rather than by thermal energy from the light source.
 | ||
| Fig. 4 CO2 desorption from CO2-absorbed azobenzene-guanidine 7 under photoirradiation. (a) UV-vis measurements of CO2-absorbed 7. (b) Device for CO2 release from CO2-absorbed 7 under photoirradiation (440 nm) (c) CO2 concentration in outlet gas using device under visible light irradiation. (d) CO2 desorption under thermal conditions without photoirradiation. | ||
As a final challenge for light-swing CO2 capture, DAC (lower CO2 concentration) and recyclability were examined (Scheme 2). For the first CO2 absorption, air was flowed (100 mL min−1) into guanidine 7 (10 mmol) in EtOH (50 mL) using an MFC, and the resulting CO2 concentration on the outlet side was measured over time. In this examination, EtOH instead of MeOH was chosen for practical recycling studies due to its lower volatility and toxicity. After 40 h, the air flow was stopped. For the first CO2 desorption cycle, CO2-absorbed guanidine 7 in EtOH was irradiated with LED light (440 nm) under flowing N2 gas (50 mL min−1) using an MFC, and the CO2 concentration on the outlet side was measured over time. Subsequently, CO2 absorption/release operations were performed in the same manner, and each CO2 concentration was measured three times in total. No significant change in atmospheric CO2 absorption performance was found for the recycled (2nd and 3rd cycle) guanidine 7. In addition, under irradiation by visible light, CO2 was released during all three cycles, with the highest release concentration recorded in the 3rd cycle. Although the reason is unclear, some EtOH volatilization may have increased the efficiency of CO2 release.
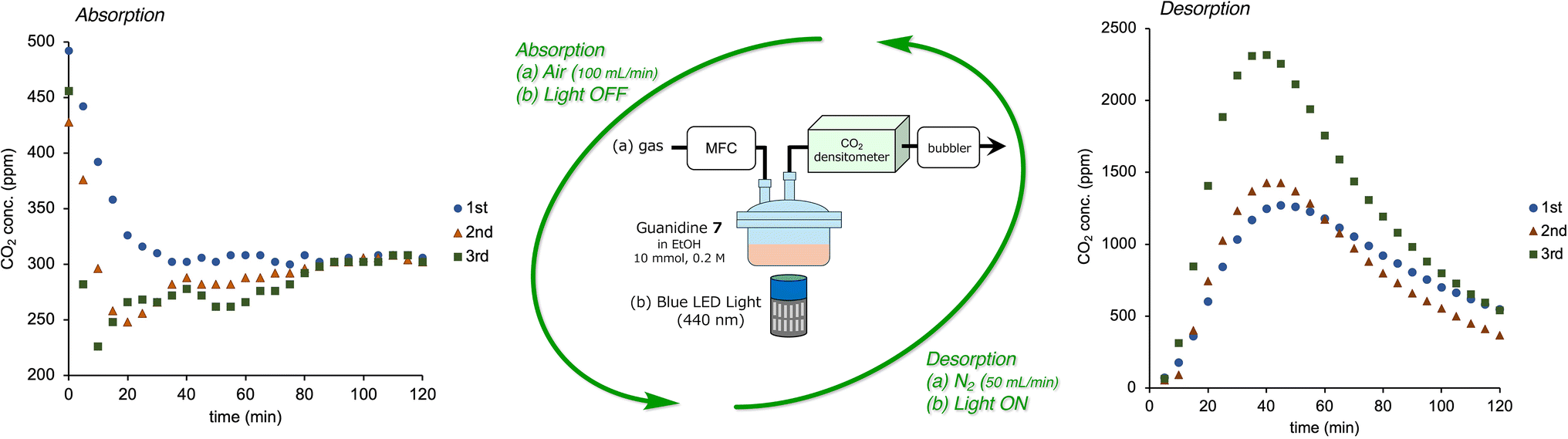 | ||
| Scheme 2 Recyclable visible light-swing CO2 capture using azobenzene-guanidine 7 in air. | ||
To elucidate the mechanism for CO2 release under photoirradiation, a structural analysis of CO2-absorbed azobenzene-guanidine 7 in EtOH was performed. A single crystal was obtained from an EtOH solution of 7 at room temperature in air. An X-ray crystal structure analysis of this crystal revealed that the product was 7·(CO2)·(EtOH) (Fig. 5a), which is consistent with the elemental analysis results for the solids observed in Fig. 3. Neutralization between guanidine 7 and ethyl hydrogen carbonate (EtOC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)OH) derived from CO2 and EtOH formed ionic guanidine 7·(CO2)·(EtOH) composed of ethyl carbonate (EtOC(O)O−) and guanidinium (R-NHC(NH2+)NH2). Next, we focused on intermolecular interactions. As shown in Fig. 5b, a hydrogen bonding network involving R-
O)OH) derived from CO2 and EtOH formed ionic guanidine 7·(CO2)·(EtOH) composed of ethyl carbonate (EtOC(O)O−) and guanidinium (R-NHC(NH2+)NH2). Next, we focused on intermolecular interactions. As shown in Fig. 5b, a hydrogen bonding network involving R-![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) C(NH2+)NH2 and EtOC(O)
C(NH2+)NH2 and EtOC(O)![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) − in adjacent molecules was identified. Also, p–p interactions between benzene moieties in the overlapped azobenzenes were observed (Fig. 5c). Based on these results, a plausible mechanism for CO2 release under photoirradiation is proposed as shown in Fig. 5d. Under photoirradiation, trans-azobenzene is converted to cis-azobenzene. The formation of cis-azobenzene causes steric repulsion with other molecules, disrupting the intermolecular interactions. As a result of the dissociation of cis-azobenzene from the aggregated molecules, CO2 is released along with the production of EtOH.
− in adjacent molecules was identified. Also, p–p interactions between benzene moieties in the overlapped azobenzenes were observed (Fig. 5c). Based on these results, a plausible mechanism for CO2 release under photoirradiation is proposed as shown in Fig. 5d. Under photoirradiation, trans-azobenzene is converted to cis-azobenzene. The formation of cis-azobenzene causes steric repulsion with other molecules, disrupting the intermolecular interactions. As a result of the dissociation of cis-azobenzene from the aggregated molecules, CO2 is released along with the production of EtOH.
 | ||
| Fig. 5 X-ray crystallographic analysis of 7·(CO2)·(EtOH). (a) ORTEP drawing. (b) Intermolecular hydrogen bonding network. (c) Intermolecular π–π stacking interaction. (d) Plausible mechanism for CO2 release under photoirradiation. | ||
3. Conclusion
In summary, a light-swing CO2 capture method based on photoisomerization reactions of azobenzene derivatives has been developed. In the case of azobenzene-amine 3, it was possible to absorb CO2 not only at 1% concentration but also at atmospheric concentration (about 400 ppm). In addition, CO2 in air was selectively captured without moisture contamination. CO2-absorbed 3 was found to release CO2 under UV irradiation. Although problems remain in terms of reusability, 3 showed that light energy has the potential to become a driving force for CO2 release. Based on these results, azobenzene-guanidine 7 was newly designed to overcome the issue of recyclability. In EtOH solution, 7 selectively absorbed CO2 in air to form 7·(CO2)·(EtOH) without moisture content. Desorption of CO2 from 7·(CO2)·(EtOH) occurred upon irradiation with visible light (440 nm). The results of CO2 release experiments under heating conditions indicated that light energy, and not thermal energy, caused the release of CO2. A plausible mechanism for CO2 release under photoirradiation was proposed. From the results of an X-ray crystal structure analysis of 7·(CO2)·(EtOH), it was considered that the key to CO2 release upon light irradiation was dissociation of aggregated 7·(CO2)·(EtOH) based on steric repulsion accompanying photoisomerization of the azobenzene moiety. Thus, we have developed a method for aerial CO2 capture using light energy. It is expected that by combining light at different wavelengths, various photokinetic molecules can be used to produce the driving force for light-swing CO2 capture. In this study, light-swing CO2 capture with the guanidine derivative required a large amount of solvent due to the solubility, and further improvement is necessary from a practical standpoint. However, we believe that the issue would be improved by introducing a functional group, which increases the solubility, into the substrate. In addition, this research suggests that the light-swing CO2 capture using other photoresponsive molecules would be possible under various wavelength ranges. Further study on CO2 capture based on longer wavelengths light is ongoing.Author contributions
R. M. and F. I. designed the study, the main concept ideas, and the proof outline. K. S. and A. U. performed the experiments and analysed the data. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr M. Uchiyama and Ms M. Ikurumi from Kanazawa University for elemental analysis. This work was supported by JSPS KAKENHI Grant Number JP20H03370 to F.I. R.M. acknowledges the research grant C from Kobe Gakuin University.References
- R. S. Haszeldine, Carbon capture and storage: how green can black be?, Science, 2009, 325, 1647–1652 CrossRef CAS PubMed.
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Progress in adsorption-based CO2 capture by metal-organic frameworks, Chem. Soc. Rev., 2012, 41, 2308–2322 RSC.
- M. L. Pinto, L. Mafra, J. M. Guil, J. Pires and J. Rocha, Adsorption and Activation of CO2 by Amine-Modified Nanoporous Materials Studied by Solid-State NMR and 13CO2 Adsorption, Chem. Mater., 2011, 23, 1387–1395 CrossRef CAS.
- V. Scott, S. Gilfillan, N. Markusson, H. Chalmers and R. S. Haszeldine, Last chance for carbon capture and storage, Nat. Clim. Change, 2013, 3, 105–111 CrossRef CAS.
- J. K. Moore, M. A. Sakwa-Novak, W. Chaikittisilp, A. K. Mehta, M. S. Conradi, C. W. Jones and S. E. Hayes, Characterization of a Mixture of CO2 Adsorption Products in Hyperbranched Aminosilica Adsorbents by 13CO Solid-State NMR, Environ. Sci. Technol., 2015, 49, 13684–13691 CrossRef CAS PubMed.
- D. M. Reiner, Learning through a portfolio of carbon capture and storage demonstration projects, Nat. Energy, 2016, 1, 1–7 Search PubMed.
- D. Y. C. Leung, G. Caramanna and M. M. Maroto-Valer, An Overview of Current Status of Carbon Dioxide Capture and Storage Technologies, Renewable Sustainable Energy Rev., 2014, 39, 426–443 CrossRef CAS.
- J. Gibbins and H. Chalmers, Carbon Captureand Storage, Energy Policy, 2008, 36, 4317–4322 CrossRef.
- G. Chichilnisky and P. Eisenberger, How air capture could help to promote a Copenhagen solution, Nature, 2009, 459, 1053 CrossRef CAS PubMed.
- A. Dessler, Energy for air capture, Nat. Geosci., 2009, 2, 811–811 CrossRef CAS.
- R. Pielke Jr., Air capture update, Nat. Geosci., 2009, 2, 811–811 CrossRef.
- D. W. Keith, Why capture CO2 from the atmosphere?, Science, 2009, 325, 1654–1655 CrossRef CAS PubMed.
- O. K. Farha and J. T. Hupp, Rational design, synthesis, purification, and activation of metal-organic framework materials, Acc. Chem. Res., 2010, 43, 1166–1175 CrossRef CAS PubMed.
- R. Solocow, M. Desmond, R. Aines, J. Blackstock, O. Bolland, T. Kasaarberg, N. Lewis, M. Mazzotti, A. Pfeffer, K. Sawyer, J. Siirola, B. Smit and J. Wilcox, Direct Air capture of CO2 with Chemicals, Am. Phys. Soc., 2011 Search PubMed , https://www.aps.org/policy/reports/assessments/upload/dac2011.pdf.
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kuhn, Transformation of carbon dioxide with homogeneous transition-metal catalysts: a molecular solution to a global challenge?, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed.
- A. Goeppert, M. Czaun, G. K. Surya Prakash and G. A. Olah, Air as the renewable carbon source of the future: an overview of CO2 capture from the atmosphere, Energy Environ. Sci., 2012, 5, 7833–7853 RSC.
- O. Shekhah, Y. Belmabkhout, Z. Chen, V. Guillerm, A. Cairns, K. Adil and M. Eddaoudi, Made-to-order metal-organic frameworks for trace carbon dioxide removal and air capture, Nat. Commun., 2014, 5, 4228 CrossRef CAS PubMed.
- A. Kumar, D. G. Madden, M. Lusi, K. J. Chen, E. A. Daniels, T. Curtin, J. J. Perry and M. J. Zaworotko, Direct Air Capture of CO2 by Physisorbent Materials, Angew. Chem., Int. Ed., 2015, 54, 14372–14377 CrossRef CAS PubMed.
- P. Smith, S. J. Davis, F. Creutzig, S. Fuss, J. Minx, B. Gabrielle, E. Kato, R. B. Jackson, A. Cowie and E. Kriegler, et al., Biophysical and economic limits to negative CO2 emissions, Nat. Clim. Change, 2016, 6, 42–50 CrossRef CAS.
- E. S. Sanz-Perez, C. R. Murdock, S. A. Didas and C. W. Jones, Direct Capture of CO2 from Ambient Air, Chem. Rev., 2016, 116, 11840–11876 CrossRef CAS PubMed.
- C. A. Seipp, N. J. Williams, M. K. Kidder and R. Custelcean, CO2 Capture from Ambient Air by Crystallization with a Guanidine Sorbent, Angew. Chem., Int. Ed., 2017, 56, 1042–1045 CrossRef CAS PubMed.
- R. Hanna, A. Abdulla, Y. Xu and D. G. Victor, Emergency Deployment of Direct Air Capture as a Response to the climate Crisis, Nat. Commun., 2021, 12, 368 CrossRef CAS PubMed.
- F. Kraxner, S. Nilsson and M. Obersteiner, Negative emissions from BioEnergy use, carbon capture and sequestration (BECS)-the case of biomass production by sustainable forest management from semi-natural temperate forests, Biomass Bioenergy, 2003, 24, 285–296 CrossRef CAS.
- M. Fajardy and N. M. Dowell, Can BECCS deliver sustainable and resource efficient negative emissions?, Energy Environ. Sci., 2017, 10, 1389–1426 RSC.
- G. T. Rochelle, Amine Scrubbing for CO2 Capture, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- F. Inagaki, Y. Okada, C. Matsumoto, M. Yamada, K. Nakazawa and C. Mukai, Energyless CO2 Absorption, Generation, and Fixation Using Atmospheric CO2, Chem. Pharm. Bull., 2016, 64, 8–13 CrossRef PubMed.
- F. Inagaki, C. Matsumoto, T. Iwata and C. Mukai, CO2-Selective Absorbents in Air: Reverse Lipid Bilayer Structure Forming Neutral Carbamic Acid in Water without Hydration, J. Am. Chem. Soc., 2017, 139, 4639–4642 CrossRef CAS PubMed.
- R. Murakami, H. Kawamitsu, R. Otsuka, H. Tanishima, C. Matsumoto and F. Inagaki, Adv. Mater. Interfaces, 2024, 11, 230881 Search PubMed.
- S. A. Mazari, B. Si Ali, B. M. Jan, I. M. Saeed and S. Nizamuddin, An Overview of Solvent Management and Emissions of Amine-Based CO2 Capture Technology, Int. J. Greenhouse Gas Control, 2015, 34, 129–140 CrossRef CAS.
- M. T. Ho, G. W. Allinson and D. E. Wiley, Reducing the Cost of CO2 Capture from Fuel Gases Using Pressure Swing Adsorption, Ind. Eng. Chem. Res., 2008, 47, 4883–4890 CrossRef CAS.
- R. M. Siqueira, G. R. Freitas, H. R. Peixoto, J. F. D. Nascimento, A. P. S. Musse, A. E. B. Torres, D. C. S. Azevedo and M. Bastos-Neto, Carbon Dioxide Capture by Pressure Swing Adsorption, Energy Procedia, 2017, 114, 2182–2192 CrossRef CAS.
- S. Voskian and T. A. Hatton, Faradaic Electro-Swing Reactive Adsorption for CO2 Capture, Energy Environ. Sci., 2019, 12, 3530–3547 RSC.
- S. Jin, M. Wu, R. G. Gordon, M. J. Aziz and D. G. Kwabi, PH Swing Cycle for CO2 Capture Electrochemically Driven through Proton-Coupled Electron Transfer, Energy Environ. Sci., 2020, 13, 3706–3722 RSC.
- R. Sharifian, R. M. Wagterveld, I. A. Digdaya, C. Xiang and D. A. Vermaas, Electrochemical Carbon Dioxide Capture to Close the Carbon Cycle, Energy Environ. Sci., 2021, 14, 781–814 RSC.
- H. Seo, M. Rahimi and T. A. Hatton, Electrochemical Carbon Dioxide Capture and Release with a Redox-Active Amine, J. Am. Chem. Soc., 2022, 144, 2164–2170 CrossRef CAS PubMed.
- A. M. Afaradi, B. Kudisch, N. Ni, J. Thomas, T. Y. George, K. Ragabimoghadam, H. J. Jiang, D. G. Nocera, M. J. Aziz and R. Y. Liu, Reversible CO2 Capture and On-Demand Release by an Acidity-Matched Organic Photoswitch, J. Am. Chem. Soc., 2023, 145, 26720–26727 CrossRef PubMed.
- G. Hartley, The Cis-form of azobenzene, Nature, 1937, 140, 281 CrossRef CAS.
- J. Griffiths, Photochemistry of azobenzene and its derivatives, Chem. Soc. Rev., 1972, 1, 481–493 RSC.
- A. A. Beharry and G. A. Woolley, Azobenzene photoswitches for biomolecules, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- H. M. D. Bandara and S. C. Burdette, Photoisomerization in different classes of azobenzene, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- F. A. Jerca, V. V. Jerca and R. Hoogenboom, Advances and opportunities in the exciting world of azobenzenes, Nat. Rev. Chem., 2022, 6, 51–69 CrossRef PubMed.
- Y. Zhu and W. Zhang, Reversible tuning of pore size and CO2 adsorption in azobenzene functionalized porous organic polymers, Chem. Sci., 2014, 5, 4957–4961 RSC.
- R. Huang, M. R. Hill, R. Babarao and N. V. Medhekar, CO2 Adsorption in Azobenzene Functionalized Stimuli Responsive Metal–Organic Frameworks, J. Phys. Chem. C, 2016, 120(30), 16658–16667 CrossRef CAS.
- D. Xue, W. Zhang, X. Liu, S. Qi and L. Sun, Fabrication of azobenzene-functionalized porous polymers for selective CO2 capture, Chin. J. Chem. Eng., 2022, 43, 24–30 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2312097. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4gc00736k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |