
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Turning lignin into a recyclable bioresource: transesterification vitrimers from lignins modified with ethylene carbonate†
Antoine
Duval
 *ab,
Wissam
Benali
a and
Luc
Avérous
*a
*ab,
Wissam
Benali
a and
Luc
Avérous
*a
aBioTeam/ICPEES-ECPM, UMR CNRS 7515, Université de Strasbourg, 25 rue Becquerel, 67087 Strasbourg Cedex 2, France. E-mail: antoine.duval@unistra.fr; luc.averous@unistra.fr
bSoprema, 15 rue de Saint Nazaire, 67100 Strasbourg, France
First published on 24th June 2024
Abstract
Kraft lignin (KL), an abundant and underutilized biobased aromatic resource, was valorized into polyester vitrimers showing enhanced recyclability. KL was first modified with ethylene carbonate, to expose only highly reactive primary aliphatic OH groups, allowing the direct synthesis of polyester networks by polycondensation with PEG-based dicarboxylic acid. In addition to avoiding the use of toxic acyl halides or coupling agents, this fast synthetic process yields networks in which the amount of free OH groups can be easily controlled by adjusting the stoichiometry between COOH and OH groups. The presence of free OH groups allows transesterification reactions resulting in a vitrimer behavior. Increasing the number of free OH groups promotes the stress relaxation process, but does not affect the activation energy, which is relatively low compared to typical transesterification vitrimers, owing to the specific network topology. High performance materials with improved mechanical and chemical recyclability were clearly demonstrated. Besides, the modified lignin can be recovered and isolated in high yields with only minor changes in its chemical structure, offering further upcycling opportunities. This makes lignin, the first aromatic and biobased resource, a raw material that can also be recycled, in line with the concept of a circular bioeconomy.
Introduction
Lignin is a natural polyphenol present in lignocellulosic biomass, particularly in wood. As a multifunctional oligomer with a high content of aliphatic and phenolic hydroxyl (OH) groups, lignin is considered as a valuable aromatic building block and as a major potential feedstock for the synthesis of aromatic polymers. According to the botanical origin and the fractionation process, different lignins can be obtained. Kraft lignin (KL) is for instance obtained as a by-product of cellulose in virgin paper pulp production by the Kraft process. The Kraft process is the dominant technology for paper pulp production, with more than 140 Mt of pulp produced from wood annually.1 The global production of KL is typically estimated to be between 50 and 70 Mt per year,2–5 making it by far the most abundant and economical lignin feedstock. However, only a small percent of KL is isolated from black liquor and valorized, while most of it is burnt for energy production. In this context, the recent development of new processes to recover KL from black liquor has led to a significant increase in its commercial availability,6,7 which was estimated to be 265 kt per year in 2018.8Although KL has been studied for decades, significant progress in the knowledge of its structure has been accomplished in the past few years.9–13 The Kraft pulping process generates extensive modifications on the lignin structure: ether inter-unit linkages (such as β-O-4, the most prominent one) are broken, and new bonds are formed by condensation of the reactive species created during the depolymerization. The latter are mostly carbon–carbon bonds, which are more stable than the native ether bonds. It implies that KL cannot easily be depolymerized, making it of practically no interest to produce small phenolic building blocks. In this context, the valorization of KL as a polymer in material applications could be most promising.
KL has a very high functionality in aliphatic and phenolic OH groups, which can be easily modified to introduce new chemical functions.14,15 Polymers prepared from lignin are crosslinked materials, i.e. thermoset materials. The main drawback of this class of polymers is that crosslinking prevents the material from melting, making it impossible to reshape or recycle mechanically like thermoplastics. It strongly limits the options for their end-of-life management in the context of a circular bioeconomy.
An elegant way to overcome this limitation is to prepare materials with dynamic crosslinks, so-called covalent adaptable networks (CANs).16–20 In CANs, the crosslinking bonds can change their position within the network, thus offering the possibility to reshape the materials and also achieve more sophisticated properties, such as self-healing, shape memory, or autonomous response to stimuli. Depending on the mechanisms of the exchange reactions, CANs are generally classified into dissociative and associative, although the frontier between both is sometimes difficult to draw.21 In dissociative CANs, the linkages are first broken before being formed again at another position, leading to a transition state with a reduced or even null crosslink density. The most studied dissociative linkages used in CANs are Diels–Alder adducts formed between furan and maleimide moieties.22 This strategy has been applied to lignins to develop recyclable and self-healing materials.23–26 Associative CANs, also known as vitrimers, were introduced by Leibler and co-workers.27 The first examples relied on transesterification reactions between ester linkages constituting the network and free OH groups. Since then, plenty of other dynamic linkages have been identified. Thanks to the associative nature of the exchange reactions, the crosslink density is maintained while the network topology is modified. Extensive research efforts have been devoted to the development of vitrimers lately,28–30 including the elaboration of biobased vitrimers, recently reviewed by our group.31
The development of vitrimers from lignin thus seems to be a promising approach to valorize this abundant renewable resource. Until now, only a few examples were available in the literature, mostly transesterification vitrimers synthesized from reactions between epoxy and carboxylic acid groups. To obtain them, lignin was first modified to expose carboxylic acid groups, either by ozonation32,33 or by reaction with cyclic anhydrides.34,35 Curing of the lignin-based polycarboxylic acid with a bifunctional epoxy linker led to networks containing β-hydroxy ester linkages, which can exchange bonds by transesterification. Alternatively, lignin can be modified to expose epoxy groups, and crosslinked with dicarboxylic acid linkers.36–38 Lately, lignin-based vitrimers relying on other exchangeable bonds have also been reported. Moreno et al.39 prepared lignin-based vitrimers based on acetal linkages, whereas Ma et al.40 reported lignin-based polyurethane vitrimers, where exchange reactions proceed via transcarbamoylation in the presence of an excess of OH groups. Recently, vinylogous urethane vitrimers were also reported.41,42
In this study and in a global approach towards green and efficient lignin valorization, KL was modified with ethylene carbonate to expose primary aliphatic OH groups, in the continuity of extensive research of our group on the modification of polyphenols with cyclic carbonates.43–47 This highly reactive modified KL was then crosslinked with a long chain dicarboxylic acid, to yield lignin-based polyester networks with a controlled amount of free OH groups. They have been extensively characterized by FTIR spectroscopy, TGA, DSC, swelling tests and tensile tests. The vitrimer behavior was assessed by stress relaxation experiments, and both mechanical and chemical recycling routes were tested.
Experimental section
Materials
Softwood Kraft lignin (KL) was obtained from MeadWestvaco (Indulin AT). It was dried overnight in a vacuum oven at 40 °C and then stored in a desiccator prior to use. Ethylene carbonate (EC, ≥99%) and potassium carbonate (K2CO3, 99%) were purchased from Alfa Aesar and Fisher Chemicals, respectively. 1,5,7-Triazabicyclo[4.4.0]dec-5-ene (TBD, 98%), 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (95%) and poly(ethylene glycol) bis(carboxymethyl) ether (PEG-COOH, Mn = 600 g mol−1) were purchased from Sigma-Aldrich. PEG-COOH was characterized by 1H and 31P NMR prior to use, and the details can be found in the ESI (Fig. S1 and S2†).Chemical modification of KL with EC
KL (32.5 g, corresponding to 212.7 mmol of reactive functional groups, i.e. the sum of aliphatic OH, phenolic OH and COOH), K2CO3 (2.9 g, 21.3 mmol, 0.1 equivalent) and EC (131.0 g, 1.49 mol, 7 equivalents) were introduced into a 600 mL stainless steel reactor (Parr) equipped with a mechanical stirrer and a heating mantle. Temperature regulation was performed with a thermocouple located inside the reactor, in direct contact with the reaction mixture. The reaction temperature was set to 100 °C and continuously recorded during the synthesis. The reaction mixture was stirred under an argon flow for 3 h. It was then poured into 1 L of water previously acidified to pH 2 with a 2 M HCl solution, leading to the precipitation of the modified lignin (KL·EC), which was recovered by filtration, further washed on a filter with water, and finally dried in a vacuum oven at 40 °C overnight.Synthesis of lignin-based polyesters
KL·EC and PEG-COOH were mixed in a PTFE beaker. The amount of PEG-COOH was adjusted to obtain different COOH/OH molar ratios of 0.6, 0.8 or 1.0, between the COOH groups of PEG-COOH and the OH groups of KL·EC. TBD (0.1 molar equivalent to OH groups in KL·EC) was added, and a small volume of acetone was introduced. The mixture was stirred at room temperature until obtaining a homogeneous mixture. The beaker was then placed in a vacuum oven at 120 °C for 1 h, to allow the removal of residual acetone and water formed during the esterification reaction. The obtained paste was then compression molded in a hot press at 160 °C for 30 min, and the material was finally cured in an oven at 160 °C for 2 h.Mechanical recycling of lignin-based polyesters
Lignin-based polyester films were dipped into liquid nitrogen and immediately ground using a coffee grinder. The obtained powder was then compression molded into the desired shape in a hot press at 200 °C for 20 min.Chemical recycling of lignin-based polyesters and recovery of recycled KL·EC
Lignin-based polyester films were dipped into liquid nitrogen and immediately ground. 1 g of the obtained powder was then introduced into a 50 mL round bottom flask, to which 10 mL ethylene glycol was added. The flask was equipped with a condenser, and the mixture was stirred overnight at 150 °C or 180 °C. It was then cooled down to room temperature and centrifuged to remove the insoluble fraction. The insoluble fraction was washed with water and dried in a vacuum oven at 40 °C overnight for quantification. The solution was added dropwise to 200 mL of water, leading to the precipitation of the modified lignin (r.KL·EC). r.KL·EC was separated by centrifugation, washed with water and dried in a vacuum oven at 40 °C overnight, prior to characterization. It was then used in combination with fresh PEG-COOH to synthesize chemically recycled L-PES, following the protocol described above.Characterization
1D NMR spectra were recorded on a Bruker 400 MHz spectrometer. For 1H NMR, about 20 mg of samples were dissolved in DMSO-d6. 32 scans were recorded at 25 °C. 31P NMR of lignin samples was performed after phosphitylation with 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane in pyridine/CDCl3 (1.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v), in the presence of cholesterol as an internal standard, according to the standard protocol.48,49 128 scans were recorded at 25 °C with a 15 s delay. 2D HSQC NMR spectra were recorded on a Bruker 500 MHz spectrometer at 25 °C. About 150 mg of sample were dissolved in DMSO-d6, and 32 scans were recorded (1024 × 256 increments, 1.5 s relaxation delay, 0.2 s acquisition time).
:1 v/v), in the presence of cholesterol as an internal standard, according to the standard protocol.48,49 128 scans were recorded at 25 °C with a 15 s delay. 2D HSQC NMR spectra were recorded on a Bruker 500 MHz spectrometer at 25 °C. About 150 mg of sample were dissolved in DMSO-d6, and 32 scans were recorded (1024 × 256 increments, 1.5 s relaxation delay, 0.2 s acquisition time).
Fourier transform infrared (FTIR) spectroscopy was performed on a Nicolet iS 10 spectrometer (Thermo Scientific) in attenuated total reflectance (ATR) mode. 32 scans were collected between 500 and 4000 cm−1 at a resolution of 4 cm−1.
Size-exclusion chromatography (SEC) was performed on a Waters Acquity Advanced Polymer Chromatography (APC) system, equipped with three 150 mm APC XT columns (a 45 Å, 1.7 μm column; a 200 Å, 2.5 μm column; and a 450 Å, 2.5 μm column) regulated at 40 °C. Tetrahydrofuran (THF, HPLC grade, Fisher Scientific) was used as the eluent at a flow rate of 0.6 mL min−1. Detection was performed with an Acquity refractive index (RI) detector and an Acquity tunable UV detector operating at 280 nm. The samples were dissolved in THF at 5 mg mL−1 and filtered through 0.2 μm PTFE syringe filters prior to the injection. To ensure complete solubility in THF, lignin samples were acetylated as previously reported.50 The average molar masses and dispersities were calculated from a calibration with polystyrene standards.
Dynamic scanning calorimetry (DSC) was performed on a TA Q200 calorimeter under nitrogen. Lignin samples were first heated to 105 °C and maintained at this temperature for 10 min, to erase their thermal history. They were then cooled down to −70 °C and heated to 200 °C at 10 °C min−1. For crosslinked material samples, the thermal history was erased at 150 °C for 3 min, before applying the same cooling/heating cycle. The glass transition temperature (Tg) was taken as the midpoint of the change in the slope during the heating run. Tg values are reported as the averages and standard deviations of three replicate experiments.
Thermogravimetric analysis (TGA) was performed on a TA Q5000 instrument. The temperature was ramped at 20 °C min−1 from room temperature to 700 °C under nitrogen. The temperature where 95% of the initial weight is remaining is reported as T95%.
Dynamic mechanical analysis (DMA) was performed in torsion mode on a Discovery HR-3 rheometer from TA Instruments, using rectangular samples. The temperature was increased from −50 to 150 °C at a heating rate of 3 °C min−1. The axial force was maintained at 1 ± 0.5 N and the applied strain was 0.05% at a frequency of 1 Hz. The storage modulus (G′) and loss modulus (G′′) were recorded as a function of temperature. The relaxation temperature (Tα) was taken at the maximum of tanδ = G′′/G′. Experiments were performed in triplicate, and averages and standard deviations are reported.
Stress relaxation experiments were performed in torsion mode on a Discovery HR-3 rheometer from TA Instruments, using rectangular samples. An axial force of 1 N and a strain of 1% were applied to the material. The relaxation modulus (G) was followed over time at a constant temperature. A series of stress relaxation experiments were performed successively on the same sample from 220 to 250 °C. The relaxation modulus was normalized to the initial value, measured at 0.4 s, to obtain the normalized relaxation curves G/G0.4 s. The normalized relaxation curves were fitted with a stretched exponential decay normalized at 0.4 s,51 taking into account the potential presence of permanent linkages, as described by Li et al.52Eqn (1) was used to fit the data:
 | (1) |
For a stretched exponential decay, the average relaxation time 〈τ〉 is given by:52,53
 | (2) |
ln〈τ〉 was then plotted against 1000/T to determine the activation energy (Ea), according to the Arrhenius equation:
 | (3) |
Uniaxial tensile tests were performed on a Zwick Roll dynamometer equipped with a 5 kN load cell. Dumbbell-shaped samples with an effective length of 20 mm, a width of 4 mm and a thickness of about 1 mm were used. The tensile measurements were performed using a preload of 0.1 N and a pulling speed of 100 mm min−1 until sample failure. The elongation at break (ε), maximum tensile stress (σmax) and Young's modulus (E) are reported as averages of 5 experiments with the corresponding standard deviations.
For swelling tests, approximately 100 mg of sample (mi) were immersed in 2 ml of solvent (distilled water, acetone or THF) for 24 h at room temperature. The swollen mass (msw) was measured after carefully wiping the surface of the samples to remove excess solvent. The samples were then dried overnight in a vacuum oven at 40 °C, before measuring their final mass (mf). The experiments were performed in triplicate. The swelling ratio (SR) and gel content (GC) were calculated according to eqn (4) and (5), respectively:
 | (4) |
 | (5) |
Results and discussion
Characterization of KL modified with EC
EC can react with different functional groups present in lignin, such as aliphatic OH, phenolic OH, and COOH (Scheme 1).45,46 With phenolic OH and COOH, it leads to hydroxyethyl ethers or esters, respectively, with the release of CO2. With aliphatic OH, the reaction pathway is not clearly established, and the formation of both hydroxyethyl carbonates54 and ethers55,56 has been reported to be possible.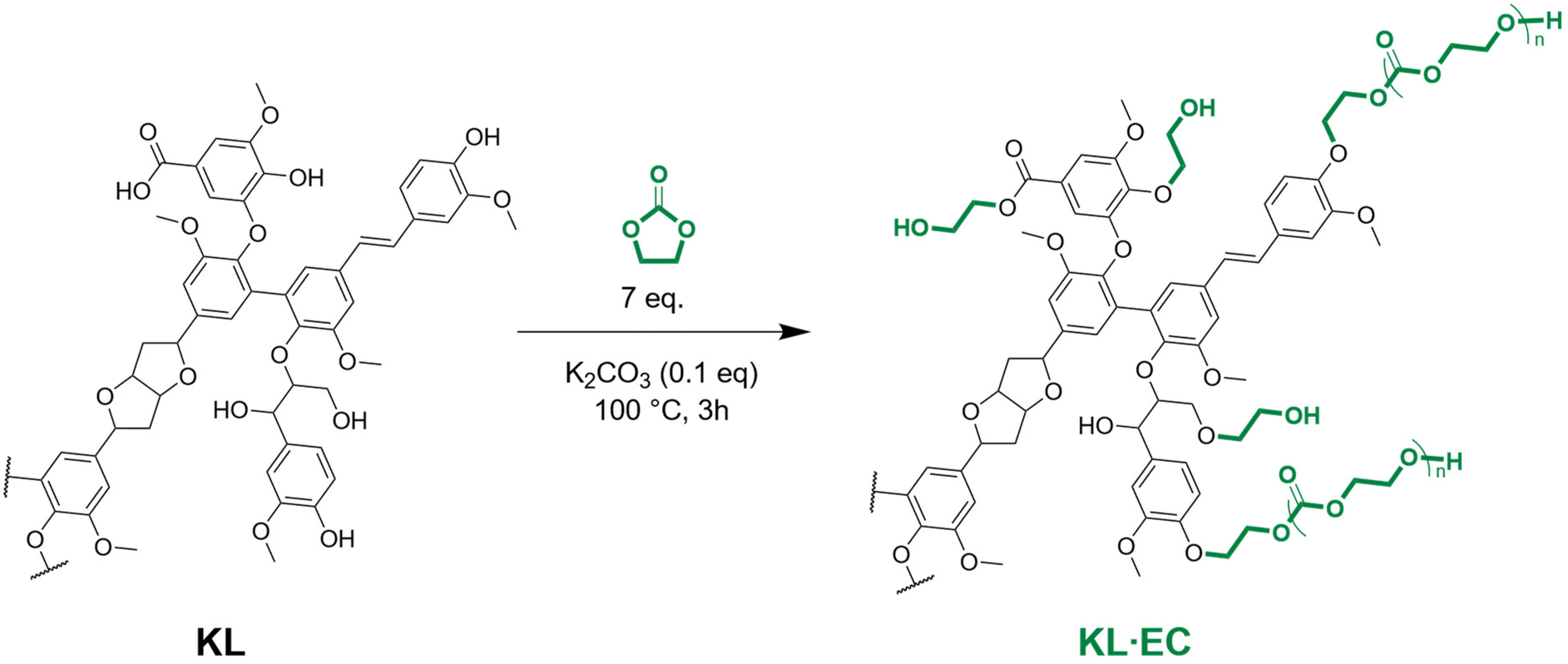 | ||
| Scheme 1 Chemical modification of KL with EC. | ||
EC is usually used in a large excess, to allow the solubilization of lignin with a sufficient liquid-to-solid ratio, without adding other solvents. In previous studies, 10 equivalents of EC per reactive group in lignin were used.45,46 To minimize the reaction E-factor,57 several attempts to reduce the amount of EC were made. Reactions with 3 or 5 equivalents of EC were unsuccessful, resulting either in low conversion of the phenolic OH groups or in the recovery of an insoluble product (Table S1, entries 1–5 and Fig. S3 in the ESI†). We then settled with 7 equivalents of EC, in good agreement with the results of Liu et al.58–60
The reaction was initially performed at 110 °C for 1 h, conditions that allowed full conversion of phenolic OH in organosolv lignins.46 However, the conversion of phenolic OH in KL, determined by 31P NMR, was only 47.9% (Table S1, entry 6 and Fig. S4 in the ESI†). Increasing the reaction time to 3 h allowed reaching a near quantitative conversion (Table S1, entry 7 and Fig. S4 in the ESI†). However, when performing the reaction at a slightly higher scale, an insoluble product was obtained, indicating that crosslinking side reactions occurred (Table S1, entry 8 in the ESI†). Other attempts with different temperatures and reaction times revealed reproducibility issues when performing the reaction at different scales. Since small scale reactions were performed in round bottom flasks immersed in an oil bath, we thought that the temperature regulation was not precise enough to allow a good reproducibility. The reaction was then performed in a stainless-steel reactor equipped with a heating mantle connected to a thermocouple placed inside the reaction mixture, which allows a more precise regulation and monitoring of the temperature of the reaction mixture (Table S1, entries 10–13 in the ESI†). The reaction performed at 110 °C led to an insoluble product, but a soluble product with full conversion of phenolic OH and COOH groups was successfully obtained after 3 h at 100 °C, as seen from the 31P NMR spectra presented in Fig. 1a. A replicate under the same conditions afforded similar results, showing that a precise control of reaction conditions ensures a high reproducibility (Fig. S5 in the ESI†).
 | ||
| Fig. 1 Comparison of KL and KL·EC: (a) 31P NMR (IS = internal standard, cholesterol), (b) details of ATR-FTIR spectra and (c) SEC (THF, PS standard calibration). | ||
These results clearly show that the control of the temperature of the reaction mixture is critical to achieve reproducible and scalable results. The crosslinking is thought to arise from transcarbonation reactions, involving OH groups and previously formed carbonate linkages (Scheme S1 in the ESI†). Crosslinking side reactions have often been reported during lignin modification with EC, whereas they appear less important when using other cyclic carbonates, such as propylene or vinyl ethylene carbonates. Indeed, the reaction with EC creates highly reactive primary OH groups, whereas the reaction with substituted CC mostly creates secondary OH groups, which are less reactive in transcarbonation reactions. It seems that reducing the temperature can help limit the crosslinking.
HSQC NMR was further used to get more precise insights into the structure of the modified lignin (Fig. 2). Interestingly, the main inter-unit linkages that are observed in KL are still present in KL·EC (β-O-4, phenylcoumaran, resinol and secoisolariciresinol, labelled in Fig. 2 as A, B, C and D, respectively). It indicates that they were not completely cleaved during the reaction. The introduction of hydroxyethyl side chains on KL·EC leads to the apparition of new signals, which appear as 4 clusters in the 60–75/3.0–4.5 ppm region (Fig. 2b). Clusters 3 and 4 correspond to the –CH2– groups in the α position to the phenol in G structures, without or with linkages at position 5, respectively. Cluster 1 regroups all the –CH2– groups next to the terminal OH groups, whereas cluster 2 corresponds to –CH2– groups close to ester or carbonate linkages.59 The aromatic region of the spectra is more difficult to interpret because of many overlapping signals, but they reveal an upfield shift of the G5 protons in the 13C dimension as a result of the grafting of hydroxyethyl chains (Fig. S6 in the ESI†), as previously reported.59
 | ||
| Fig. 2 Aliphatic side chains (a and b) and alkyl regions (c and d) of the HSQC NMR spectra of KL (a and c) and KL·EC (b and d). The corresponding chemical structures are shown in (e). | ||
HSQC NMR was further used to gain information on the reaction of lignin aliphatic OH groups during the modification with EC. The reactions of primary OH groups in A, B or D structures would lead to changes in chemical shifts of the γ-protons. However, their signals are overlapping with the protons of the grafted hydroxyethyl chains, precluding the analysis of their reactivity. Interestingly, aryl propanol side chains are clearly visible in the alkyl region of the HSQC spectra (labelled as F in Fig. 2c). After modification with EC, a new signal F′β is observed (Fig. 2d). Comparison with the predicted spectra reveals that this signal corresponds to the etherified structure F′ (Fig. 2e and Fig. S7 in the ESI†). Integration of the corresponding signals however shows that only about 50% of the OH groups were etherified. Insight into the reactivity of secondary OH groups can be gained by examining the signal of α-protons in β-O-4 structures (signal Aα in Fig. 2a). A new signal, labelled as A′α, appears in the modified lignin (Fig. 2b). Comparison with the predicted NMR spectra clearly shows that it corresponds to etherified rather than carbonated structures (structure A′ in Fig. 2e and Fig. S8 in the ESI†). However, signals Aα from unmodified OH groups are still present. Integration of both signals shows that roughly one third of the secondary OH groups have reacted.
Only a limited proportion of lignin aliphatic OH groups thus seem to react with EC under the selected reaction conditions. Unlike previously reported, etherified structures are formed rather than carbonated ones. However, the presence of carbonate linkages is clearly evidenced by HSQC NMR (cluster 2 in Fig. 2b) and confirmed by FTIR with the appearance of a new band at 1735 cm−1 on the spectrum of KL·EC (Fig. 1b). Carbonate linkages are thus rather thought to appear in the chain extension process, when hydroxyethylated structures further react with EC. Scheme 1 was drawn according to these findings.
SEC reveals a substantial increase in molar mass after modification with EC (Fig. 1c). This increase is not only due to the grafting of hydroxyethyl side chains, but also due to potential crosslinking reactions due to the formation of carbonate linkages, as previously discussed (Scheme S1 in the ESI†).46 It also leads to an increase in dispersity, from 2.51 to 3.05. Thermal stability measured by TGA increased after modification with EC (Fig. S9 in the ESI†). T95% increased from 209 to 237 °C, and the main degradation temperature increased from 368 to 412 °C. Tg is reduced from 152 to 102 °C after the modification with EC (Fig. S10 in the ESI†).
Characterization of lignin-based polyester networks (L-PES)
KL·EC possesses primary aliphatic OH groups, which are highly reactive in esterification reactions, as shown for instance by Renneckar and co-workers.58,60–62 They were exploited to synthesize lignin-based polyester networks (L-PES) by direct polycondensation with PEG-COOH (Scheme 2), in a short reaction time (1.5 h and 2 h post curing). The reaction only releases water as a safe by-product, unlike previously reported lignin-based polyesters obtained by reaction with acyl chlorides, releasing toxic HCl.63–66 It is therefore in line with the green chemistry principle of less hazardous synthesis. For comparison, an attempt was made to synthesize a material from neat KL under similar conditions, but it was still viscous during the curing step. It quickly disintegrated when immersed in a solvent, indicating a low crosslinking (Fig. S11 in the ESI†), which confirms the usefulness of the modification with EC.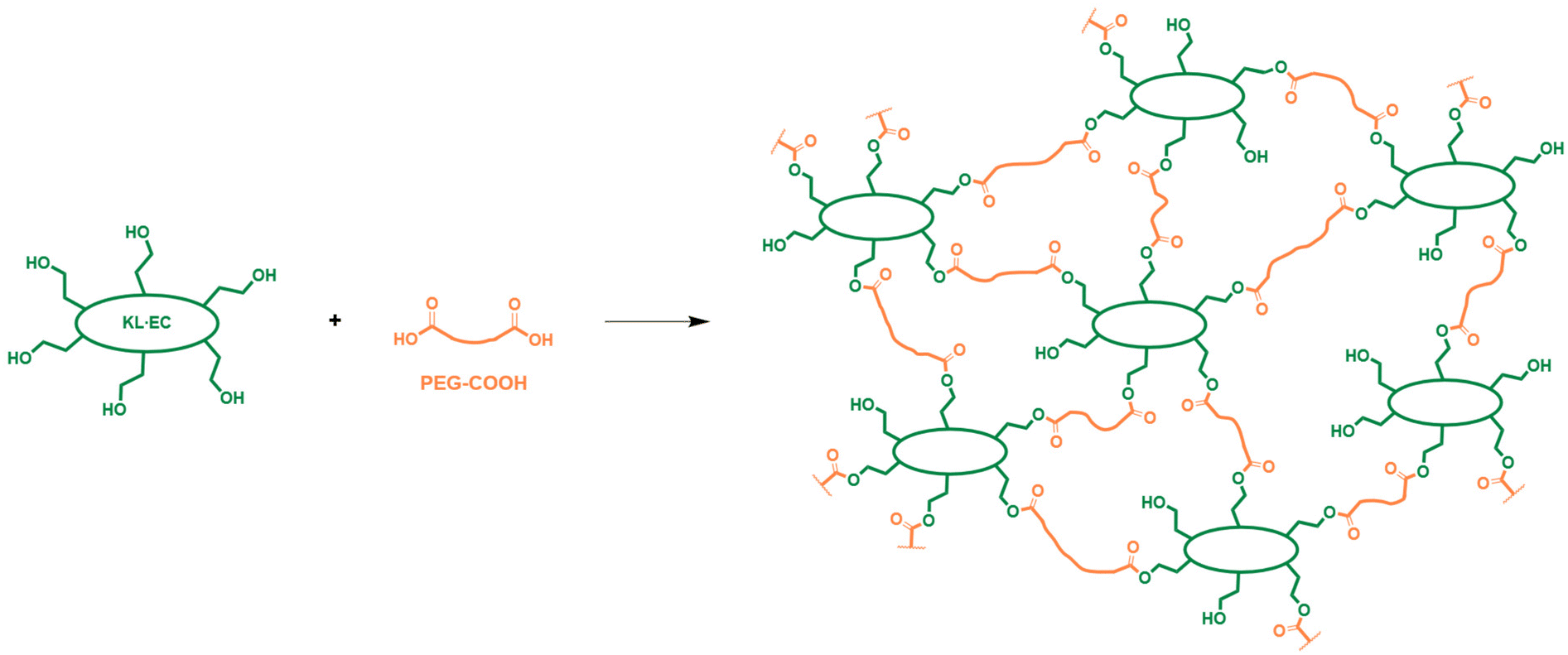 | ||
| Scheme 2 Formation of lignin-based polyester (L-PES) networks by reaction between KL·EC and PEG-COOH. | ||
The energy efficiency of the developed material synthesis was quantified using the metric developed by DeVierno Kreuder et al.,67 and compared to several other methods for the synthesis of lignin-based polyester vitrimers. The details are given in the ESI (Tables S2–S8†). Despite the additional step of KL modification with EC, the proposed methodology performs better than polycondensation with neat KL, PEG and citric acid.68,69 It also shows better or similar energy efficiency than syntheses using chemically modified lignins, either by ozonation,32 reaction with anhydride34 or epichlorohydrin.35
The COOH/OH molar ratio was adjusted to obtain various polyester networks containing different amounts of residual OH groups. The free OH groups are expected to allow transesterification reactions, as in conventional transesterification vitrimers obtained by polyaddition between carboxylic acids and epoxy groups. The composition of the different materials is given in Table 1. They have a high content of KL·EC, ranging from 47 to 59%wt. FTIR spectroscopy was performed to assess the chemical structure of the networks. Fig. 3 shows the results for L-PES-0.8, whereas similar results for the other materials are available in the ESI (Fig. S12†). A broad peak is observed between 3000 and 3600 cm−1 on the spectrum of L-PES-0.8. It shows that the material still contains free OH groups, since excess OH groups were used in the polymerization. Free OH groups would then be available for transesterification reactions. The peak at 1745 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch) confirms the formation of ester bonds in the network. In addition, a peak at 1640 cm−1 is visible on the spectra of L-PES. It is related to the presence of TBD,70 which has been added to the networks to catalyze the transesterification reactions (Table 1).
O stretch) confirms the formation of ester bonds in the network. In addition, a peak at 1640 cm−1 is visible on the spectra of L-PES. It is related to the presence of TBD,70 which has been added to the networks to catalyze the transesterification reactions (Table 1).
 | ||
| Fig. 3 FTIR spectra of KL·EC, PEG-COOH and L-PES-0.8 materials. | ||
Swelling tests were performed in water and in two different organic solvents, acetone and THF (Fig. 4). They confirm the obtention of crosslinked networks. The gel content is independent of the amount of PEG-COOH used for crosslinking. It indicates that even in the presence of a sub-stoichiometric amount of PEG-COOH, crosslinked networks with a high gel content were successfully obtained. Surprisingly, the swelling ratio of the materials follows opposite trends in water and in organic solvents. In organic solvents, the swelling ratio increases with the amount of PEG-COOH. Indeed, increasing the amount of PEG-COOH leads to a decrease in the lignin content (Table 1). Since lignin represents the crosslinking points in the networks, decreasing the proportion of lignin should lead to a decrease in crosslink density, and consequently to an increase in swelling. However, in water, the swelling ratio decreases when the amount of PEG-COOH used to crosslink KL·EC increases. This is probably related to the content of hydrophilic functions, such as free OH groups. The content of free OH groups decreases when the amount of PEG-COOH is increased, leading to a reduced hydrophilicity causing the observed decrease in swelling in water.
 | ||
| Fig. 4 Swelling ratio and gel content of L-PES materials in water, acetone and THF. | ||
Thermal and mechanical properties of L-PES
The thermal stability of the materials was evaluated by TGA (Fig. S13 in the ESI†). It shows that the materials have a high thermal stability, with T95% higher than 270 °C and main degradation temperatures of about 410 °C (Table 2). It increases with the amount of PEG-COOH, probably because it leads to networks with fewer chain ends. DSC was then performed to evaluate the Tg. A unique Tg is observed for all materials, indicating that KL·EC and PEG-COOH form a single phase (Table 2). It is indeed well known that lignins and polyethylene oxide give miscible blends with only one Tg over the whole composition range.71–75 The evolution of Tg is related to the amount of KL·EC contained in the materials (Fig. S14 in the ESI†): reducing the KL·EC content (i.e., increasing the PEG-COOH content from L-PES-0.6 to L-PES-1.0) reduces the Tg, since KL·EC has a much higher Tg than PEG-COOH (102 °C against −55 °C, respectively).| Material | T 95% (°C) | T deg (°C) |
T
g
a (°C) |
T
αb (°C) |
G′rubb (MPa) |
Ec (MPa) |
σc (MPa) |
εc (%) |
|---|---|---|---|---|---|---|---|---|
| a Average and standard deviations of 3 replicates per sample. b Average and standard deviations of 4 replicates per sample. c Average and standard deviations of 5 replicates per sample. nd = not determined. | ||||||||
| L-PES-0.6 | 271 | 407 | −9 ± 1 | 22 ± 7 | 1.9 ± 0.1 | nd | nd | nd |
| L-PES-0.8 | 285 | 405 | −12 ± 3 | 20 ± 6 | 3.4 ± 0.2 | 7.8 ± 2.8 | 1.3 ± 0.4 | 29 ± 5 |
| L-PES-1.0 | 300 | 411 | −13 ± 4 | 6 ± 2 | 1.7 ± 0.3 | 4.4 ± 0.8 | 1.0 ± 0.1 | 31 ± 5 |
DMA was then performed to evaluate the thermomechanical properties of the materials (Fig. 5). All the L-PES show typical behavior of crosslinked networks, with a constant rubbery plateau at high temperature. A relatively large variation between replicates was observed, which may be the result of the structural heterogeneity of KL. Four replicates have thus been performed (Fig. S15 in the ESI†), and the average values and standard deviations are reported in Table 2. Tα follows the same trend as Tg. It increases with the KL·EC content (Fig. S12 in the ESI†). The modulus on the rubbery plateau, G′rub, gives an indication of the crosslink density of the networks. Interestingly, the highest G′rub is obtained for L-PES-0.8. In the case of a large defect in PEG-COOH, as with L-PES-0.6, the number of connections between lignin crosslinking nodes is reduced, resulting in a low crosslink density. However, when the amount of PEG-COOH gets higher (L-PES-1.0), a lower crosslink density is also observed. It probably originates from a higher amount of network defects, such as loops, which do not contribute to the increase of the crosslink density. From the DMA results, it thus seems that L-PES-0.8 constitutes an optimum in network topology.
 | ||
| Fig. 5 DMA of L-PES materials. | ||
The mechanical properties of the materials were then evaluated by uniaxial tensile tests. The main results are given in Table 2. The stress–strain curves are available in the ESI (Fig. S16†). L-PES-0.6 was not evaluated because it was too brittle to cut adequate samples from the films. Once again, significant standard deviations were observed, confirming the heterogeneity of the materials caused by their high lignin content. The mechanical properties are relatively similar for L-PES.0.8 and L-PES-1.0. The former has a slightly higher Young's modulus, in good agreement with the DMA data. Despite the high lignin content, the materials show interesting elasticity, with an elongation at break of about 30%.
Stress relaxation
The L-PES networks contain free OH groups, which are susceptible to inducing transesterification reactions with the ester linkages constituting the networks (Scheme 3). The structure is however different from conventional transesterification vitrimers, which are mainly constituted by β-hydroxy ester linkages. Here, the networks are composed of ester linkages, and free OH groups are present at the end of dangling chains (Scheme 2). To evaluate whether this structure leads to vitrimers, the materials were subjected to stress relaxation tests between 220 and 250 °C. Normalized stress relaxation curves are presented in Fig. 6. All the materials successfully relax stress, although not completely since the normalized modulus does not tend to zero. The data were successfully fitted with a stretched exponential decay, taking into account the presence of permanent linkages, according to eqn (1).52 The results of the fits are fully detailed in the ESI (Tables S9–S11†). The parameter Gperm/G0, which corresponds to the fraction of stress that is not relaxed even after infinite time, ranges between 0.15 and 0.22. It indicates that non-dynamic linkages have been formed during material synthesis. They probably arise from lignin–lignin coupling caused by the thermal treatment at high temperature. | ||
| Scheme 3 Transesterification reaction between pendant OH groups and ester bonds constituting the L-PES networks. | ||
 | ||
| Fig. 6 Normalized relaxation modulus of L-PES materials between 220 and 250 °C: (a) L-PES-0.6, (b) L-PES-0.8, and (c) L-PES-1.0. Experimental data (solid lines) were fitted with a stretched exponential decay, which is presented as dashed lines. (d) Evolution of ln〈τ〉 with the inverse of the temperature, showing Arrhenius behavior. | ||
The corresponding non-normalized relaxation curves are available in the ESI (Fig. S17†). They show that the modulus increases with temperature for all materials, which is a typical feature of dynamic networks with associative bond exchange.76–80 A comparison of the relaxation of the different materials at the same temperature is provided in the ESI (Fig. S18†). The relaxation appears faster when the amount of free OH groups in the network increases, as expected. However, even L-PES-1.0 relaxes stress relatively fast, although it should only contain a small amount of free OH since it was synthesized with a stoichiometric ratio between COOH and OH. It indicates that transesterification vitrimers can be obtained even in the presence of a low concentration of free OH groups, whereas the classical transesterification vitrimers synthesized from epoxy and carboxylic acids usually present one free OH group per ester linkage.27
The average relaxation times 〈τ〉 were calculated from the fit parameters τ* and β (eqn (2)), and Arrhenius plots were drawn (Fig. 6d). Straight lines with R2 > 0.99 were obtained for all materials, indicating that they indeed behave as vitrimers. The activation energy Ea ranges between 55 and 66 kJ mol−1 and seems relatively independent of the amount of free OH groups. Ea is quite low as compared to classical transesterification vitrimers, for which Ea values are generally above 80 kJ mol−1.27,81 The difference probably originates from the chemical environment of the free OH groups that induce the transesterification. In these systems, the OH groups are primary and located at the end of dangling chains, whereas in the case of classical transesterification vitrimers they are secondary and included in the network in the β-position to the ester linkages.
Recycling of L-PES materials and recovery of modified lignin
Dynamic linkages within the networks offer interesting options for recycling of the materials at their end-of-life. Two alternative options for the recycling of L-PES materials have been evaluated, as illustrated in Fig. 7a. L-PES-0.8 was used for the recycling study, owing to its better properties. First, mechanical recycling was performed by simply grinding the material and reshaping it into films by compression molding for 20 min at 200 °C. The mechanically recycled material was then characterized by FTIR, DSC, DMA and swelling tests (Fig. 7, Table 3 and Fig. S19 in the ESI†). The Tg and Tα are higher than those of the virgin material, whereas its swelling ratio is lower, potentially indicating a higher crosslink density induced by the additional thermal treatment during the reprocessing step. However, its gel content and modulus on the rubbery plateau G′rub are significantly lower. This last point is surprising, since additional crosslinking should induce an increase in G′rub. It means that reprocessing at high temperature apparently induces structural changes in the network topology, resulting in significantly different properties compared to the pristine material.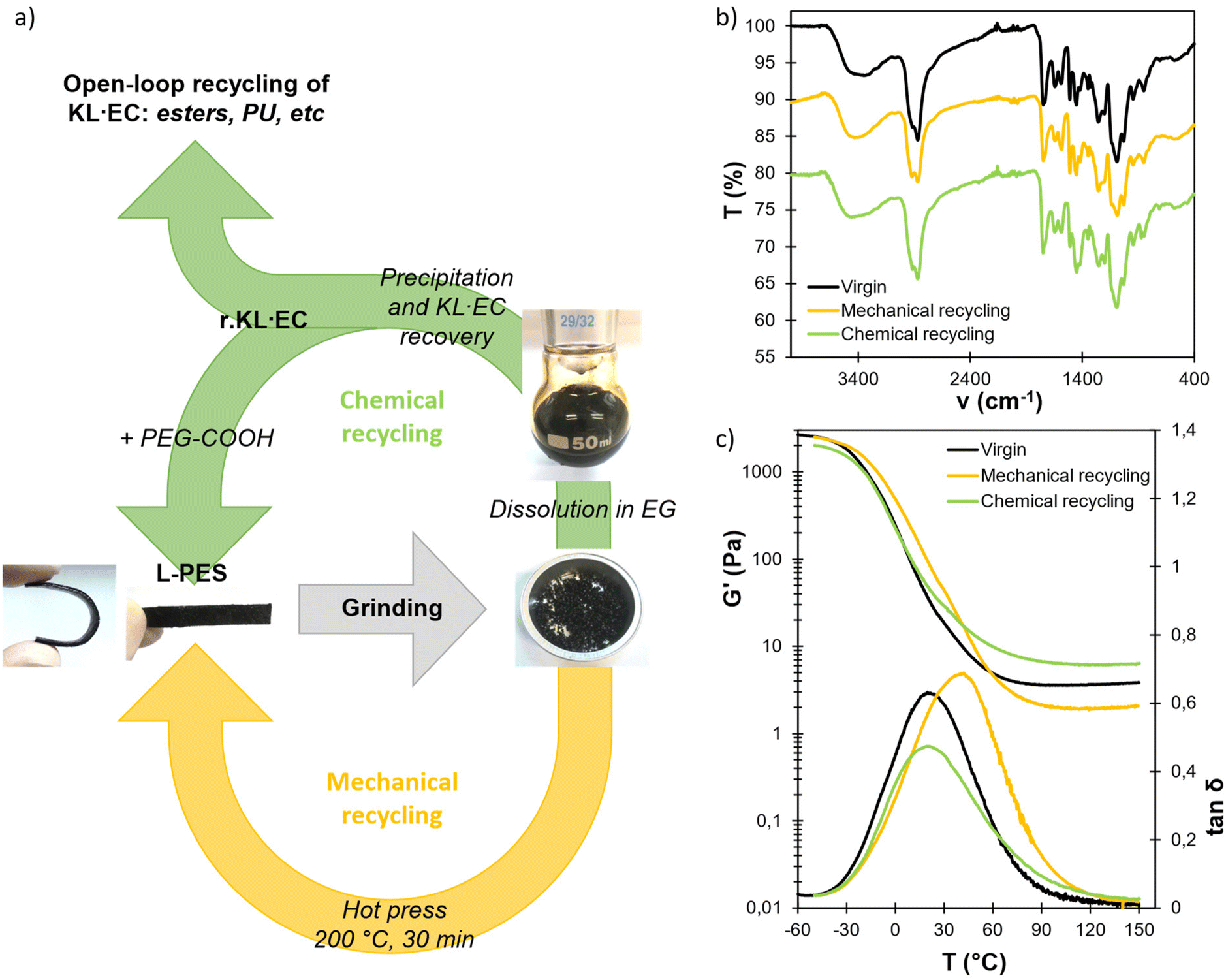 | ||
| Fig. 7 (a) Scheme of the open- and closed-loop recycling strategies of L-PES materials. Comparison of the (b) FTIR spectra and (c) DMA of virgin, mechanically and chemically recycled L-PES-0.8 materials. | ||
| T g (°C) | T α (°C) | G′rub (MPa) | SRwater (%) | SRacetone (%) | GCwater (%) | GCacetone (%) | |
|---|---|---|---|---|---|---|---|
| Virgin | −12 ± 3 | 20 ± 6 | 3.4 ± 0.2 | 73.5 ± 0.9 | 39.5 ± 1.1 | 81.2 ± 0.3 | 82.8 ± 0.6 |
| Mechanically recycled | −5 ± 3 | 41 ± 1 | 2.1 ± 0.2 | 61.0 ± 2.4 | 36.7 ± 0.1 | 76.9 ± 1.4 | 74.2 ± 1.0 |
| Chemically recycled | −8 ± 1 | 21 ± 1 | 6.5 ± 0.7 | 83.3 ± 3.9 | 39.0 ± 0.4 | 81.1 ± 0.7 | 84.6 ± 0.8 |
The material was also chemically recycled. In this context, it was first dissolved in ethylene glycol (EG) without adding an external catalyst, thanks to the disruption of the network by transesterification reactions (Fig. S20 in the ESI†). Other diols, such as propanediol or diethylene glycol, could also be used for this purpose. The modified lignin was then recovered after precipitation in water and filtration. Two different glycolysis temperatures have been evaluated, 150 and 180 °C, leading to the recovery of two recycled lignins, r.KL·EC-150 and r.KL·EC-180, respectively. The yields of the material soluble in EG and of recovered lignins are detailed in Table S12 in the ESI.† At 150 °C, only about 70% of the material was dissolved in EG, and the yield of r.KL·EC was below 30%. However, complete dissolution was observed at 180 °C, leading to a recovery yield of r.KL·EC of 84%.
The obtained recycled lignins were characterized and compared to the neat KL·EC in Fig. 8. An additional peak at 147.6 ppm is visible on the 31P NMR spectrum of r.KL·EC-150 (Fig. 8a). It does not correspond to residual EG82 and could therefore correspond to the esters of EG and PEG-COOH still bound to lignin (Fig. S21 in the ESI†), thus indicating incomplete breaking of PEG–lignin bonds by transesterification. An additional signal around 3.50 ppm in 1H NMR (Fig. 8b) and the high intensity of the C–H band around 2900 cm−1 in the FTIR spectrum are also indicative of the presence of PEG bound to r.KL·EC-150 (Fig. 8c). The incomplete cleavage of PEG–lignin bonds could explain the low recovery yield of r.KL·EC-150, because it would lead to significant water solubility. 150 °C does not appear to be a sufficient temperature for chemical recycling. However, r.KL·EC-180 does not present any additional peak in 31P and 1H NMR. The intensity of the C–H band in its FTIR spectrum is similar to that of the neat KL·EC. It indicates that efficient depolymerization is achieved at 180 °C, leading to the recovery of recycled lignin in high yield. The FTIR spectrum of r.KL·EC-180 reveals a decrease in the carbonate band at 1735 cm−1 as compared to the neat KL·EC, indicating that some hydroxyethyl carbonate groups have been removed, since carbonate bonds are labile under basic conditions.43 HSQC NMR does not reveal other significant changes in the structure of r.KL·EC-180 compared to KL·EC (Fig. S22 in the ESI†). Lignin inter-unit ether linkages are still visible in r.KL·EC-180, despite the high temperature treatment. SEC shows that the molar masses of the recycled lignins are higher than that of the neat KL·EC, and that the average molar mass increases with the dissolution temperature. It is probably related to condensation side reactions caused by the long exposure to high temperature. However, despite the increase in molar mass, r.KL·EC-180 is completely soluble in appropriate organic solvents (e.g., DMSO or THF after acetylation). The thermal stability and Tg of the recycled lignins are higher than those of the original KL·EC, because of the increase in molar mass (Fig. S23 and S24 in the ESI†). Further optimization of the temperature and reaction time for the recovery of r.KL·EC is outside the scope of the present study, but could be of interest to improve the yield and limit the changes in the recycled lignin structure.
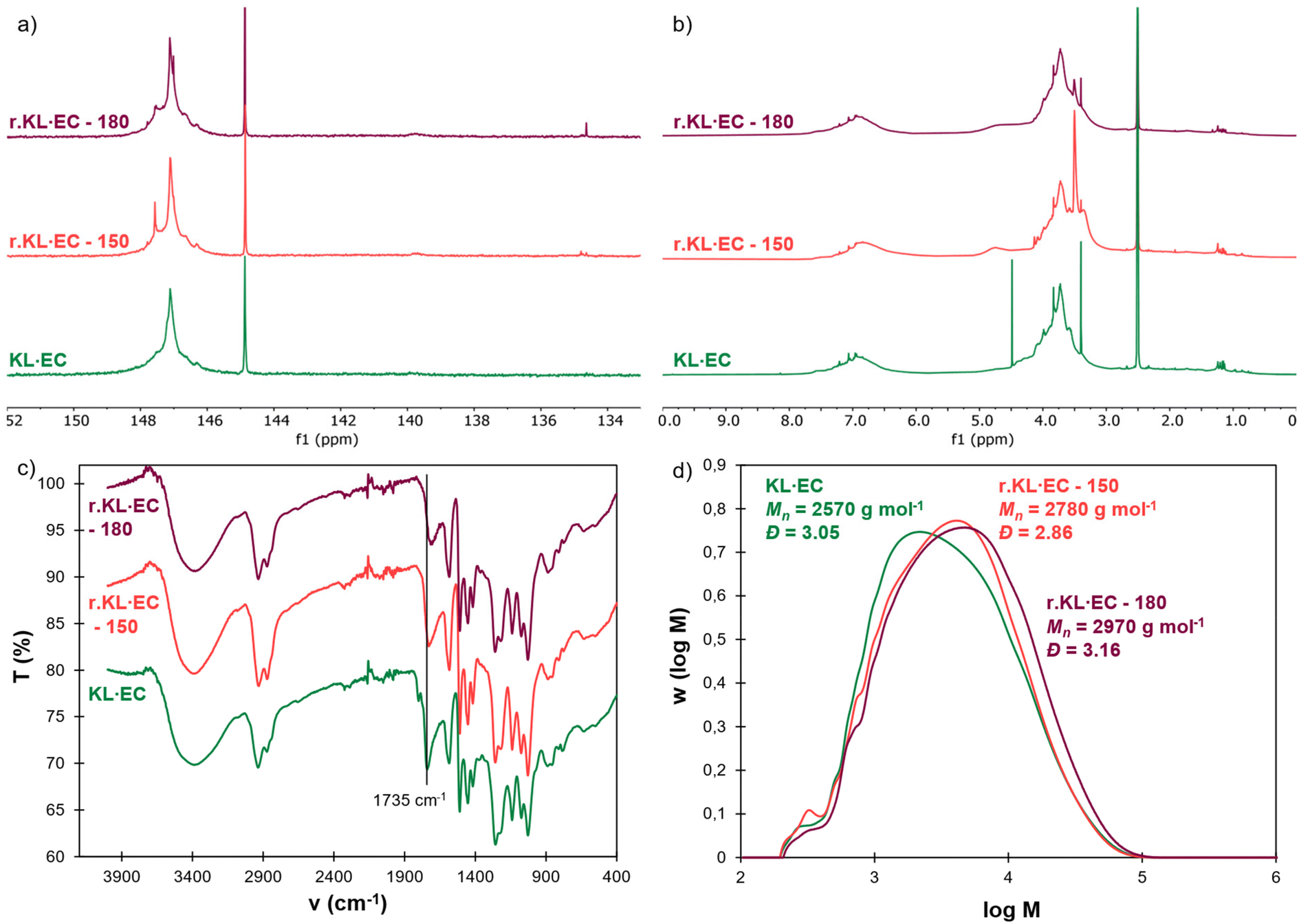 | ||
| Fig. 8 Comparison of neat KL·EC and KL·EC recycled at two different temperatures (150 or 180 °C): (a) 31P NMR, (b) 1H NMR (400 MHz, DMSO-d6), (c) ATR-FTIR and (d) SEC (THF, PS standard calibration). | ||
The recovery of r.KL·EC in high yield allows its closed-loop recycling into second generation L-PES materials (green arrow in Fig. 7a). r.KL·EC-180 was combined with fresh PEG-COOH to prepare a chemically recycled material, which has been compared to the virgin L-PES-0.8 and the mechanically recycled material (Fig. 7 and Table 3). Its Tg, Tα, gel contents and swelling ratios are similar to those of the virgin material. Interestingly, G′rub is increased, revealing that the network topology is different from that of the virgin material, as expected given the structural differences between the neat and recovered lignins. In addition to closed-loop chemical recycling, r.KL·EC could also be valorized into other polymer systems using an open-loop upcycling strategy. Indeed, its potential in the synthesis of polyurethanes47 or esters58,60–62 has been shown in various studies.
Conclusions
KL was successfully valorized into polyester vitrimers in a two-step process, which first involves its modification with EC. The entire process was solvent-free, in line with the waste prevention principle of green chemistry. After the modification with EC, lignin only contains aliphatic OH groups of high reactivity, allowing direct polycondensation with PEG-based dicarboxylic acid to synthesize polyesters. This method avoids the use of toxic acyl halides or coupling agents, in line with the green chemistry principles towards less hazardous chemistry and reduced derivatives. In addition, the fast curing reaction significantly improves the energy efficiency compared to the state-of-the-art.To achieve a vitrimer behavior, the amount of free OH groups in the networks was easily controlled by adjusting the stoichiometry between COOH and OH groups, leading to networks with a particular topology, where free primary OH groups are present as dangling chains. Increasing the amount of free OH groups logically accelerates the stress relaxation process, but does not influence the activation energy, which was quite low compared to common transesterification vitrimers (55–66 kJ mol−1).
The materials were further recycled mechanically, by grinding and compression molding, and chemically, by dissolution in ethylene glycol and lignin precipitation. Interestingly, the modified lignin could be recovered in more than 80% yield from the materials, with only limited changes in its chemical structure. It was efficiently reused to produce second generation lignin-based polyester networks and also has potential for open-loop upcycling, for instance as a building block for polyurethane or ester syntheses. It is the first example of lignin-based materials where the lignin fraction can be efficiently recovered and reused. It turns lignin, a well-known biobased aromatic precursor, into a recyclable resource, making it an even more promising feedstock for a circular bioeconomy. Further work should now be conducted to expand this successful approach to other types of lignins derived from different feedstocks and isolation processes.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Institut Carnot Mica for funding (DECOLIPO project). We acknowledge the Cronenbourg NMR Core Facility (CNRS/Université de Strasbourg, UMR 7042 LIMA, Strasbourg, France). Céline Piras and Nathan Wybo (ICPEES) are thanked for their assistance with SEC measurements.References
- D. Lachenal, in Lignocellulosic Fibers and Wood Handbook, John Wiley & Sons, Ltd, 2016, pp. 207–223 Search PubMed.
- P. C. A. Bruijnincx, R. Rinaldi and B. M. Weckhuysen, Green Chem., 2015, 17, 4860–4861 RSC.
- D. S. Bajwa, G. Pourhashem, A. H. Ullah and S. G. Bajwa, Ind. Crops Prod., 2019, 139, 111526 CrossRef CAS.
- F. Souto and V. Calado, Green Chem., 2022, 24, 8172–8192 RSC.
- M. Kienberger, S. Maitz, T. Pichler and P. Demmelmayer, Processes, 2021, 9, 804 CrossRef.
- P. Tomani, Cellul. Chem. Technol., 2010, 44, 53–58 CAS.
- L. Kouisni, A. Gagné, K. Maki, P. Holt-Hindle and M. Paleologou, ACS Sustainable Chem. Eng., 2016, 4, 5152–5159 CrossRef CAS.
- L. Dessbesell, M. Paleologou, M. Leitch, R. Pulkki and C. (Charles) Xu, Renewable Sustainable Energy Rev., 2020, 123, 109768 CrossRef CAS.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- C. Crestini, H. Lange, M. Sette and D. S. Argyropoulos, Green Chem., 2017, 19, 4104–4121 RSC.
- C. S. Lancefield, H. L. J. Wienk, R. Boelens, B. M. Weckhuysen and P. C. A. Bruijnincx, Chem. Sci., 2018, 9, 6348–6360 RSC.
- N. Giummarella, P. A. Lindén, D. Areskogh and M. Lawoko, ACS Sustainable Chem. Eng., 2020, 8, 1112–1120 CrossRef CAS.
- N. Giummarella, I. V. Pylypchuk, O. Sevastyanova and M. Lawoko, ACS Sustainable Chem. Eng., 2020, 8, 10983–10994 CAS.
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS.
- A. Duval and M. Lawoko, React. Funct. Polym., 2014, 85, 78–96 CrossRef CAS.
- C. N. Bowman and C. J. Kloxin, Angew. Chem., Int. Ed., 2012, 51, 4272–4274 CrossRef CAS PubMed.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed., 2019, 58, 9682–9695 CrossRef CAS PubMed.
- V. Zhang, B. Kang, J. V. Accardo and J. A. Kalow, J. Am. Chem. Soc., 2022, 144, 22358–22377 CrossRef CAS PubMed.
- S. V. Wanasinghe, O. J. Dodo and D. Konkolewicz, Angew. Chem., Int. Ed., 2022, 61, e202206938 CrossRef CAS PubMed.
- B. R. Elling and W. R. Dichtel, ACS Cent. Sci., 2020, 6, 1488–1496 CrossRef CAS PubMed.
- A. Gandini, Prog. Polym. Sci., 2013, 38, 1–29 CrossRef CAS.
- A. Duval, H. Lange, M. Lawoko and C. Crestini, Green Chem., 2015, 17, 4991–5000 RSC.
- P. Buono, A. Duval, L. Averous and Y. Habibi, Polymer, 2017, 133, 78–88 CrossRef CAS.
- W. Zhou, H. Zhang and F. Chen, Int. J. Biol. Macromol., 2018, 107, 790–795 CrossRef CAS PubMed.
- M. Thys, J. Brancart, G. V. Assche, R. Vendamme and N. V. den Brande, Macromolecules, 2021, 54, 9750–9760 CrossRef CAS.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2015, 7, 30–38 RSC.
- N. J. Van Zee and R. Nicolaÿ, Prog. Polym. Sci., 2020, 104, 101233 CrossRef CAS.
- M. Guerre, C. Taplan, J. M. Winne and F. E. D. Prez, Chem. Sci., 2020, 11, 4855–4870 RSC.
- M. A. Lucherelli, A. Duval and L. Avérous, Prog. Polym. Sci., 2022, 127, 101515 CrossRef CAS.
- S. Zhang, T. Liu, C. Hao, L. Wang, J. Han, H. Liu and J. Zhang, Green Chem., 2018, 20, 2995–3000 RSC.
- A. More, T. Elder, N. Pajer, D. S. Argyropoulos and Z. Jiang, ACS Omega, 2023, 8, 1097–1108 CrossRef CAS PubMed.
- C. Hao, T. Liu, S. Zhang, L. Brown, R. Li, J. Xin, T. Zhong, L. Jiang and J. Zhang, ChemSusChem, 2019, 12, 1049–1058 CrossRef CAS PubMed.
- L. Du, X. Jin, G. Qian, W. Yang, L. Su, Y. Ma, S. Ren and S. Li, Ind. Crops Prod., 2022, 187, 115439 CrossRef CAS.
- B. Xue, R. Tang, D. Xue, Y. Guan, Y. Sun, W. Zhao, J. Tan and X. Li, Ind. Crops Prod., 2021, 168, 113583 CrossRef CAS.
- R. Tang, B. Xue, J. Tan, Y. Guan, J. Wen, X. Li and W. Zhao, ACS Appl. Polym. Mater., 2022, 4, 1117–1125 CrossRef CAS.
- Y. Zheng, T. Liu, H. He, Z. Lv, J. Xu, D. Ding, L. Dai, Z. Huang and C. Si, Adv. Compos. Hybrid Mater., 2023, 6, 53 CrossRef CAS.
- A. Moreno, M. Morsali and M. H. Sipponen, ACS Appl. Mater. Interfaces, 2021, 13, 57952–57961 CrossRef CAS PubMed.
- X. Ma, S. Li, F. Wang, J. Wu, Y. Chao, X. Chen, P. Chen, J. Zhu, N. Yan and J. Chen, ChemSusChem, 2023, 16, e202202071 CrossRef CAS PubMed.
- L. Sougrati, A. Duval and L. Avérous, ChemSusChem, 2023, 16, e202300792 CrossRef CAS PubMed.
- J. Liu, A. Pich and K. V. Bernaerts, Green Chem., 2024, 26, 1414–1429 RSC.
- A. Duval and L. Avérous, ACS Sustainable Chem. Eng., 2016, 4, 3103–3112 CrossRef CAS.
- A. Duval and L. Avérous, ChemSusChem, 2017, 10, 1813–1822 CrossRef CAS PubMed.
- A. Duval and L. Avérous, ACS Sustainable Chem. Eng., 2017, 5, 7334–7343 CrossRef CAS.
- A. Duval, G. Layrac, A. van Zomeren, A. T. Smit, E. Pollet and L. Avérous, ChemSusChem, 2021, 14, 387–397 CrossRef CAS PubMed.
- A. Duval, D. Vidal, A. Sarbu, W. René and L. Avérous, Mater. Today Chem., 2022, 24, 100793 CrossRef CAS.
- A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef CAS.
- X. Meng, C. Crestini, H. Ben, N. Hao, Y. Pu, A. J. Ragauskas and D. S. Argyropoulos, Nat. Protoc., 2019, 14, 2627–2647 CrossRef CAS PubMed.
- A. Duval and L. Avérous, Green Chem., 2020, 22, 1671–1680 RSC.
- M. A. Lucherelli, A. Duval and L. Averous, ACS Sustainable Chem. Eng., 2023, 11, 2334–2344 CrossRef CAS.
- L. Li, X. Chen, K. Jin and J. M. Torkelson, Macromolecules, 2018, 51, 5537–5546 CrossRef CAS.
- L. Li, X. Chen and J. M. Torkelson, Macromolecules, 2019, 52, 8207–8216 CrossRef CAS.
- J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663–674 CrossRef CAS.
- I. Kühnel, J. Podschun, B. Saake and R. Lehnen, Holzforschung, 2015, 69, 531–538 CrossRef.
- I. Kühnel, B. Saake and R. Lehnen, React. Funct. Polym., 2017, 120, 83–91 CrossRef.
- R. A. Sheldon, M. L. Bode and S. G. Akakios, Curr. Opin. Green Sustain. Chem., 2022, 33, 100569 CrossRef CAS.
- L.-Y. Liu, Q. Hua and S. Renneckar, Green Chem., 2019, 21, 3682–3692 RSC.
- L.-Y. Liu, K. Bessler, S. Chen, M. Cho, Q. Hua and S. Renneckar, Eur. Polym. J., 2021, 142, 110082 CrossRef CAS.
- L.-Y. Liu, S. Chen, L. Ji, S.-K. Jang and S. Renneckar, Green Chem., 2021, 23, 4567–4579 RSC.
- Q. Hua, L.-Y. Liu, M. Cho, M. A. Karaaslan, H. Zhang, C. S. Kim and S. Renneckar, Biomacromolecules, 2023, 24, 592–603 CrossRef CAS PubMed.
- L.-Y. Liu, M. Cho, N. Sathitsuksanoh, S. Chowdhury and S. Renneckar, ACS Sustainable Chem. Eng., 2018, 6, 12251–12260 CrossRef CAS.
- Z. X. Guo and A. Gandini, Eur. Polym. J., 1991, 27, 1177–1180 CrossRef CAS.
- Z. X. Guo, A. Gandini and F. Pla, Polym. Int., 1992, 27, 17–22 CrossRef CAS.
- D. V. Evtugin and A. Gandini, Acta Polym., 1996, 47, 344–350 CrossRef CAS.
- C. Bonini, M. D'Auria, L. Ernanuele, R. Ferri, R. Pucciariello and A. R. Sabia, J. Appl. Polym. Sci., 2005, 98, 1451–1456 CrossRef CAS.
- A. DeVierno Kreuder, T. House-Knight, J. Whitford, E. Ponnusamy, P. Miller, N. Jesse, R. Rodenborn, S. Sayag, M. Gebel, I. Aped, I. Sharfstein, E. Manaster, I. Ergaz, A. Harris and L. Nelowet Grice, ACS Sustainable Chem. Eng., 2017, 5, 2927–2935 CrossRef CAS.
- Y. Xu, K. Odelius and M. Hakkarainen, ACS Sustainable Chem. Eng., 2019, 7, 13456–13463 CrossRef CAS.
- Y. Xu, K. Odelius and M. Hakkarainen, ACS Appl. Polym. Mater., 2020, 2, 1917–1924 CrossRef CAS.
- M. R. Islam, Z. Guo, D. Rutman and T. J. Benson, RSC Adv., 2013, 3, 24247–24255 RSC.
- J. F. Kadla and S. Kubo, Macromolecules, 2003, 36, 7803–7811 CrossRef CAS.
- S. Kubo and J. F. Kadla, Macromolecules, 2004, 37, 6904–6911 CrossRef CAS.
- J. F. Kadla and S. Kubo, Composites, Part A, 2004, 35, 395–400 CrossRef.
- S. Kubo and J. F. Kadla, J. Appl. Polym. Sci., 2005, 98, 1437–1444 CrossRef CAS.
- S. Kubo and J. F. Kadla, Holzforschung, 2006, 60, 245–252 CAS.
- A. Breuillac, A. Kassalias and R. Nicolaÿ, Macromolecules, 2019, 52, 7102–7113 CrossRef CAS.
- A. Jourdain, R. Asbai, O. Anaya, M. M. Chehimi, E. Drockenmuller and D. Montarnal, Macromolecules, 2020, 53, 1884–1900 CrossRef CAS.
- L. E. Porath and C. M. Evans, Macromolecules, 2021, 54, 4782–4791 CrossRef CAS.
- B. Soman and C. M. Evans, Soft Matter, 2021, 17, 3569–3577 RSC.
- L. Porath, B. Soman, B. B. Jing and C. M. Evans, ACS Macro Lett., 2022, 11, 475–483 CrossRef CAS PubMed.
- A. Kumar and L. A. Connal, Macromol. Rapid Commun., 2023, 44, 2200892 CrossRef CAS PubMed.
- M. Li, C. G. Yoo, Y. Pu and A. J. Ragauskas, ACS Sustainable Chem. Eng., 2018, 6, 1265–1270 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc00567h |
| This journal is © The Royal Society of Chemistry 2024 |