
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioderived silicon nano-quills: synthesis, structure and performance in lithium-ion battery anodes†
Nancy
Chen‡
a,
Morteza
Sabet‡
 *ab,
Nawraj
Sapkota
bc,
Mihir
Parekh
bc,
Shailendra
Chiluwal
bc,
Kelliann
Koehler
d,
Craig M.
Clemons
e,
Yi
Ding
f,
Apparao M.
Rao
*bc and
Srikanth
Pilla
*aghij
*ab,
Nawraj
Sapkota
bc,
Mihir
Parekh
bc,
Shailendra
Chiluwal
bc,
Kelliann
Koehler
d,
Craig M.
Clemons
e,
Yi
Ding
f,
Apparao M.
Rao
*bc and
Srikanth
Pilla
*aghij
aDepartment of Automotive Engineering, Clemson University, Greenville, SC, USA
bClemson Nanomaterials Institute, Clemson University, Anderson, SC, USA
cDepartment of Physics and Astronomy, Clemson University, Clemson, SC, USA
dElectron Microscopy Laboratory, Clemson University, Anderson, SC, USA
eUSDA Forest Service, Forest Products Laboratory, Madison, WI, USA
fU.S. Army DEVCOM GVSC, Warren, MI, USA
gCenter for Composite Materials, University of Delaware, Newark, DE, USA
hDepartment of Mechanical Engineering, University of Delaware, Newark, DE, USA
iDepartment of Materials Science and Engineering, University of Delaware, Newark, DE, USA
jDepartment of Chemical and Biomolecular Engineering, University of Delaware, Newark, DE, USA
First published on 12th March 2024
Abstract
Cellulose nanocrystals (CNCs) are bioderived one-dimensional species with versatile surface chemistry and unique self-assembling behavior in aqueous solutions. This work presents a scientific approach to leverage these characteristics for creating CNC network templates and processing them to engineer a novel silicon (Si)-based material called silicon nano-quill (SiNQ) for energy storage applications. The SiNQ structure possesses a porous, tubular morphology with a substantial ability to store lithium ions while maintaining its structural integrity. The presence of Si suboxides in the SiNQ structure is demonstrated to be crucial for realizing a stable cycling performance. One of the defining attributes of SiNQ is its water dispersibility due to Si–H surface bonds, promoting water-based Si-graphite electrode manufacturing with environmental and economic benefits. The incorporation of only 17 wt% SiNQ enhances the capacity of graphitic anodes by ∼2.5 times. An initial coulombic efficiency of 97.5% is achieved by employing a versatile pre-lithiation. The SiNQ-graphite anodes with high active loading, when subjected to accelerated charging/discharging conditions at 5.4 mA cm−2, exhibit stable cycling stability up to 500 cycles and average coulombic efficiency of >99%. A generalized physics-based cyclic voltammetry model is presented to explain the remarkable behavior of SiNQs under fast-charging conditions.
Introduction
Cellulose nanocrystals (CNCs) are one-dimensional (1D), non-toxic cellulosic materials with exceptional mechanical properties and versatile surface chemistry.1–3 They can be processed through diverse chemical treatment methods to enable their use for targeted applications.4 CNCs exhibit a remarkable self-assembling behavior in aqueous solutions, leading to the formation of 3D networks consisting of individual CNCs.5 The CNC networks can be utilized as a template in sol–gel processing6–8 to synthesize distinct silicon (Si)-containing structures for efficient energy storage in lithium-ion batteries (LIBs).Silicon, possessing high theoretical gravimetric capacity (i.e., 4200 mA h g−1 for Li22Si5 at 415 °C, and 3579 mA h g−1 for Li15Si4 at room temperature),9–11 has emerged as an alternative anode active material for advancing high-energy LIBs.12–15 Using commercial Si products in LIB anodes poses several technical challenges, particularly due to the large volume changes of Si during (de)lithiation,16 which can result in loss of electrical contact and capacity fade over the operational lifetime. Silicon nanostructures with several morphologies have been proposed to mitigate the volume change issue, among which 1D tubular structure is proven to effectively mitigate mechanical failure by relieving mechanical stresses in the material.17–20 Moreover, pore generation can enhance Li-ion (Li+) diffusion through Si structure and improve its charge storage capabilities.21,22 However, the synthesis methods commonly employed for fabricating porous tubular Si nanostructures frequently use hazardous chemicals, demand considerable financial investment, and give rise to safety issues.23,24
In the current study, we exploit the self-assembling properties of CNCs to fabricate Si-based materials with considerable porosity via a template-assisted sol–gel synthesis and subsequent metallothermic reduction. The devised process yields a novel Si-based material comprising interconnected 1D tubular porous structures resembling porcupine quills, leading us to call our products silicon nano-quills (SiNQs). One of the defining attributes of SiNQs is their water dispersibility, enabling the use of water-based slurries in lieu of the standard N-methyl-2-pyrrolidone (NMP)-based mixtures, which are necessary for conventional Si-based slurry compositions due to the hydrophobic nature of commercial Si (c-Si). Notably, NMP introduces undesirable occupational and environmental challenges, encompassing risks of human exposure, reproductive toxicity, and consequentially rigorous EU regulations.25 Leveraging water-dispersible materials mitigates these risks and presents potential economic advantages by lowering production costs due to diminished hazardous waste management requirements.
A significant aspect of our study centers on developing SiNQ-graphite composite anodes using aqueous slurries. Particularly, electric vehicle (EV) battery manufacturers tend to move toward Si-graphite composite anodes to benefit from the high Li+ storage capability of Si and the ultrahigh stability of the graphite.26 We successfully develop SiNQ-graphite composite anodes containing 17 wt% SiNQ in the active material and evaluate their electrochemical performance. A benchmark study of Si-graphite anodes, where a similar amount of graphite is replaced with Si-based materials, confirms that our SiNQ-graphite anodes exhibit superior capacity retention at higher active mass loadings. The cyclic voltammetry (CV) results of our SiNQ-graphite anodes reveal a previously unexplained shift of the dominant delithiation peak at high scan rates from graphite to Si. This observation and superior capacity retention indicate that SiNQ may be suitable for Si-graphite composite electrodes for next-generation batteries with fast charging/discharging capabilities. We present a generalized physics-based model to explain this novel observation, which the battery community could adopt to explain cyclic voltammetry data for other appropriate battery electrodes. The derived model is a fundamental contribution to the battery literature because the widely used CV model27 has assumptions (e.g., semi-infinite diffusion, and both species of redox couple freely diffusing with the same phase), which make it unsuitable for use in rechargeable metal-ion batteries.
Results and discussion
Three-dimensional porous nano-quills
The synthesis process of Si-containing materials in this study is schematically shown in Fig. 1. In the first step, CNCs (Fig. 1a) were utilized to fabricate a silicate gel through a versatile sol–gel process (Fig. 1b). Tetraethyl orthosilicate (TEOS) was used as the silicate precursor. The silicate gel, which contains templates of hexadecyltrimethylammonium bromide (CTAB)-modified CNCs (which we term CNC templates for brevity), was freeze-dried and then calcined in air at 600 °C to remove the templates and produce porous SilicaNQs (Fig. 1c). The SilicaNQ powder was pre-mixed with magnesium powder and thermally reduced at 650 °C under an inert atmosphere. Lastly, the thermoreduction product was purified using a mild hydrochloric acid solution to realize the final SiNQ material (Fig. 1d).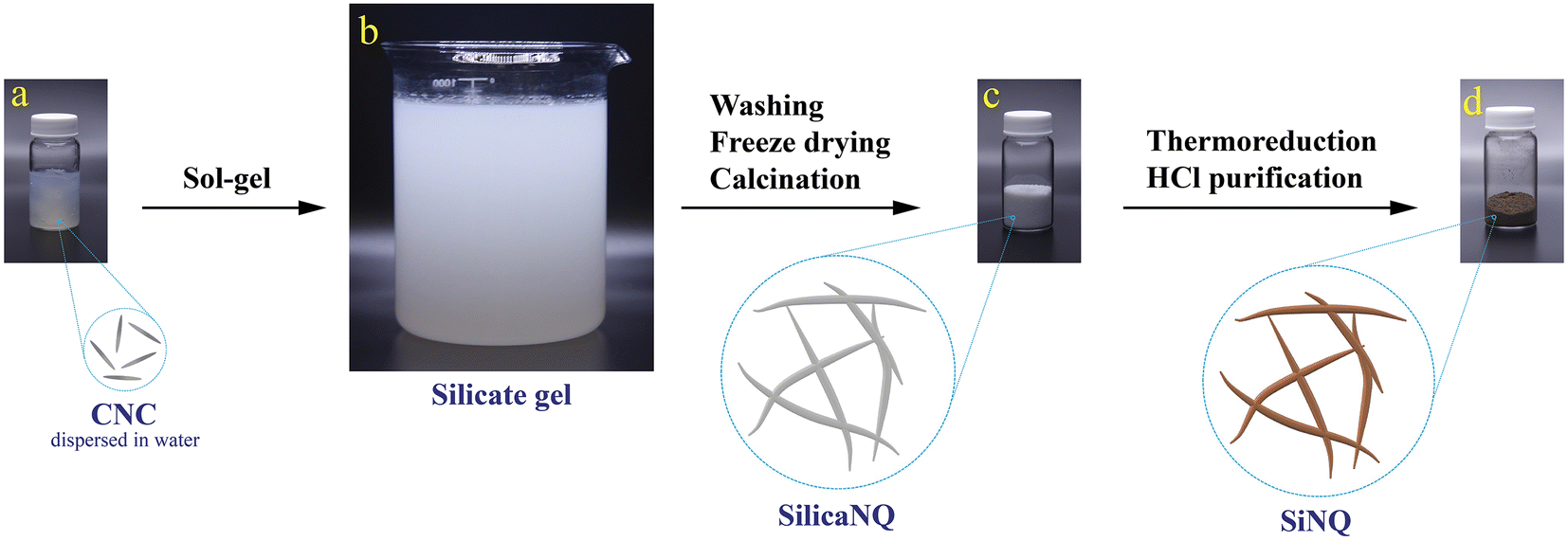 | ||
| Fig. 1 The process roadmap for producing porous silicon nano-quills from wood-derived CNCs; (a) uniform suspension of 10 wt% CNCs in water, (b) synthesis of silicate gel via a basic sol–gel reaction, (c) highly porous silica nano-quills derived from calcination and removal of CNC templates, and (d) silicon nano-quills prepared by thermoreduction at 650 °C and mild acid purification. | ||
We performed detailed microstructural studies to explore the formation of SilicaNQ nanoparticles through the templating approach. The as-received CNCs are needle-like particles with an average length of 144 nm and width of 12.5 nm, as shown in Fig. 2a. The CNCs and CTAB were added to the sol–gel medium for preparing a silicate gel. Silicate gels are often synthesized by hydrolyzing monomeric, tetrafunctional alkoxide precursors such as TEOS, employing a base (e.g., NH3) as a catalyst. The hydrolysis reaction replaces alkoxide groups of TEOS with hydroxyl groups. Subsequent condensation reactions involving the silanol groups (Si–O–H) produce siloxane bonds (Si–O–Si), plus the alcohol or water as by-products.28 The morphology of CNC-templated silicate gel (Fig. 2b) is characterized by interconnected 1D arms resembling the porcupine quills. After air calcination, SilicaNQ particles with an average length of 252 nm and an average width of 40.2 nm were obtained (Fig. 2c). However, their length distribution does not follow a normal distribution, and SilicaNQ particles with a length of up to 700 nm were observed. Comparing the length of as-received CNCs and final SilicaNQ particles indicates CNCs form a network during the sol–gel process. Adding CTAB to the CNC suspension causes an ion exchange reaction between the sodium counter ion of the CNCs and the cetyltrimethylammonium ion of CTAB, destabilizing the CNC dispersion and resulting in a network of modified CNCs.29 The silicate gel was then formed on the network of CNCs, resulting in longer SilicaNQ particles up to 700 nm.
 | ||
| Fig. 2 Electron microscopic studies of CNCs, SilicaNQs, and SiNQs; (a) needle-like CNC particles with an average length of 144 nm and width of 12.5 nm, (b) porcupine quill-like silicate gel product from sol–gel reaction, (c) SilicaNQ particles from calcination process showing average length of 252 nm and width of 40.2 nm, (d) BF S-TEM image of SilicaNQs reveals the hollow tunnels within 1D silica arms, (e) ultrahigh porous surface of SilicaNQs with an ordered arrangement of hexagonal pores, (f) a single SilicaNQ arm possessing hexagonal surface pores and a central hollow core, (g) TEM and S-TEM micrographs of SiNQ-a, (h) TEM and S-TEM micrographs of SiNQ-g, and (i) high-resolution TEM micrograph of SiNQ arms, revealing crystalline pockets distributed in an amorphous matrix. Electron diffraction patterns show the presence of major crystal systems of elemental Si in the structure of SiNQ material. | ||
The bright-field scanning transmission electron microscopic (BF S-TEM) study of SilicaNQ revealed the existence of tunnels at the core of 1D silica arms (Fig. 2d), verifying the removal of CNC templates and generation of hollow cores. Furthermore, the surface of SilicaNQ arms is covered with hexagonal pores uniformly distributed over the surface (secondary electron S-TEM image, Fig. 2e and f). The presence of hexagonal pores with diameters in the 3–5 nm range indicates the formation of cylindrical CTAB micelles on CNC surfaces during the sol–gel process. The CTAB molecules can arrange themselves to form cylindrical micelles, where the positively charged quaternary ammonium ions of the CTAB molecules form the outer surface of the cylinder.30 An electrostatic attraction between the positively charged micelle surface and anionic silanol species (–Si–O−) creates a stable surfactant-silicate composite,31 which eventually forms silicate gel on the CNC templates. By heat treatment of silicate gel at 600 °C, CTAB molecules were removed to generate hexagonal-shaped pores on the surface of SilicaNQ particles. A hollow core is also visible in Fig. 2f, indicating the removal of CNCs during the calcination treatment.
Magnesiothermic reduction is commonly used to convert silica materials into Si. The following chemical reaction describes the main reduction process as
| SiO2(s) + 2Mg(g) → Si(s) + 2MgO(s) | (1) |
Upon exposure to gaseous magnesium (Mg) and diffusion of Mg molecules into the surface of silica particles, elemental Si and Mg byproducts are formed. Subsequently, the solid-state diffusion process enables the transportation of Mg deeper into the material, thus leading to the following chemical reactions throughout the material:32,33
| Si(s) + 2Mg(g) → Mg2Si(s) | (2) |
| Mg2Si(s) + SiO2(s) → 2MgO(s) + 2Si(s) | (3) |
In general, magnesiothermic reduction is an exothermic process with the progressive formation of Si from the surface toward the core.34 The accumulation of generated heat can cause a substantial temperature rise, leading to local melting and distortion of the Si product.33 A common practice in the literature is to conduct a magnesiothermic reduction in ceramic crucibles, which usually exhibit low thermal diffusivity at high temperatures. For example, the thermal diffusivity of alumina at 650 °C is ∼0.02 cm2 s−1.35 At the same temperature, the thermal diffusivity of graphite is ∼0.2 cm2 s−1.36 Graphite's significantly higher thermal diffusivity makes it a better crucible for dissipating the heat generated during reduction reactions. To verify this, we conducted SilicaNQ reductions in both alumina and graphite crucibles, schematically shown in Fig. S1.† The as-reduced powders were treated with a mild hydrochloric acid solution to remove Mg-containing byproducts and obtain the final SiNQ material. Hereafter, the SiNQ material prepared using alumina crucible and purified using HCl solution is referred to as SiNQ-a, while the SiNQ material from graphite crucible followed by HCl treatment is referred to as SiNQ-g.
The microstructural characteristics of SiNQ materials were studied to identify the effects of heat accumulation during the reduction process. The TEM micrographs of SiNQ-a and SiNQ-g are presented in Fig. 2. The 1D morphology of SilicaNQ arms was conserved after reduction in both crucibles. A closer view of individual arms (S-TEM micrographs are presented in the inset, Fig. 2g and h) reveals that both materials are porous. However, SiNQ-a arms seem flattened and more distorted, while SiNQ-g arms exhibit a tubular geometry with less distortion. Gas physisorption measurements were performed on SilicaNQ and both SiNQ materials, and the results are presented in Fig. S2.† The SilicaNQ material has a Brunauer–Emmett–Teller (BET) specific surface area of 1266 m2 g−1, with a Barrett–Joyner–Halenda (BJH) total pore volume of 1.25 cm3 g−1. The adsorption–desorption isotherms show an apparent hysteresis (Fig. S2a†), indicating the existence of mesopores in the material structure.37 The pore size distribution of SilicaNQ (Fig. S2b†) is characterized by two peaks: a sharp peak with an average pore diameter of ∼4.0 nm, which corresponds to hexagonal surface pores (as presented in Fig. 2e and f) originated from CTAB micelles; and a broad peak with an average pore diameter of 18.9 nm, which is related to open tunnels from CNC template removal (see Fig. 2d). After reduction to SiNQ-a, the surface area and total pore volume decreased to 232 m2 g−1 and 0.42 cm3 g−1, respectively. While the surface area and total pore volume of SiNQ-g material were measured to be 400 m2 g−1 and 0.64 cm3 g−1, respectively. The lower surface area and pore volume of SiNQ-a, compared to SiNQ-g, implies a considerable fusion and distortion of the material during reduction in the alumina crucible. The S-TEM observations also verified a greater extent of distortion in SiNQ-a particles (see Fig. 2g and h), confirming that the graphite crucible improves heat dissipation from the reacting materials and preserves the original nano-quill structure.
Hysteresis is present in isotherms of SiNQ-a (Fig. S2c†) and SiNQ-g (Fig. S2e†), implying that mesopores are preserved during the reduction of SilicaNQ using either crucible. However, multiple peaks with pore diameter of <10 nm appear in their pore size distribution graph (see Fig. S2d and f†). For example, SiNQ-a shows mesopores with diameters of 3.5, 4.4, 6.3, and 7.5 nm, and SiNQ-g has mesopores of 3.7, 4.5, 5.8, and 9.0 nm. Although SiNQs maintain surface mesopores, these results show that the geometry of surface pores was altered during SilicaNQ reduction. In addition, we identified a peak with a pore diameter of >20 nm from BJH pore size analysis of SiNQs. This peak, with an average diameter of ∼25 nm, corresponds to the presence of a hollow core within SiNQ arms. This hollow space, together with surface mesopores, is expected to accommodate volume changes during Li+ insertion and extraction processes. However, we anticipate that clear morphological differences between SiNQ-a and SiNQ-g materials affect their charge storage performance while interacting with Li ions.
High-resolution TEM of SiNQs was performed to reveal the material's phase composition after thermal reduction. The resulting structure is characterized by randomly distributed crystalline pockets in an amorphous matrix, as shown in Fig. 2i. The majority of crystalline pockets show interplanar spacing of 0.31 nm and 0.19 nm. The former corresponds to Si(111) planes, and the latter corresponds to Si(220) planes.38 Moreover, electron diffraction patterns of SiNQs (inset, Fig. 2i) reveal the presence of other Si crystal planes, including (311), (400), (331), and (422). The phase evolution from SilicaNQ to SiNQ-a or SiNQ-g was also evaluated through X-ray diffraction (XRD) analysis, and the results are shown in Fig. 3a. The XRD spectrum of SilicaNQ displays a broad band centered at around 2θ = 23°, corresponding to amorphous silicon oxide.39 Upon reduction of SilicaNQ using both crucibles, the characteristic peaks of crystalline Si appear in XRD patterns of SiNQs, supporting the results from electron diffraction patterns (Fig. 2i). In addition, both SiNQ-a and SiNQ-g materials show the band for amorphous silicon oxide. This indicates a partial conversion from silica to Si and the presence of Si oxides in the as-reduced materials. However, the band for the amorphous phase is more noticeable for SiNQ-g material, implying a lower degree of conversion when using a graphite crucible. It is noted that characteristic peaks of magnesium orthosilicate (Mg2SiO4) are detected in the XRD spectrum of SiNQ-a, although SiNQ materials were subjected to the same purification procedure using hydrochloric acid solution. We repeated acid treatment of SiNQ-a, but Mg2SiO4 peaks persisted. This can be due to structural collapse/fusing of as-reduced material in alumina crucible because of local temperature rise and trapping of Mg2SiO4 byproduct within the SiNQ-a structure.
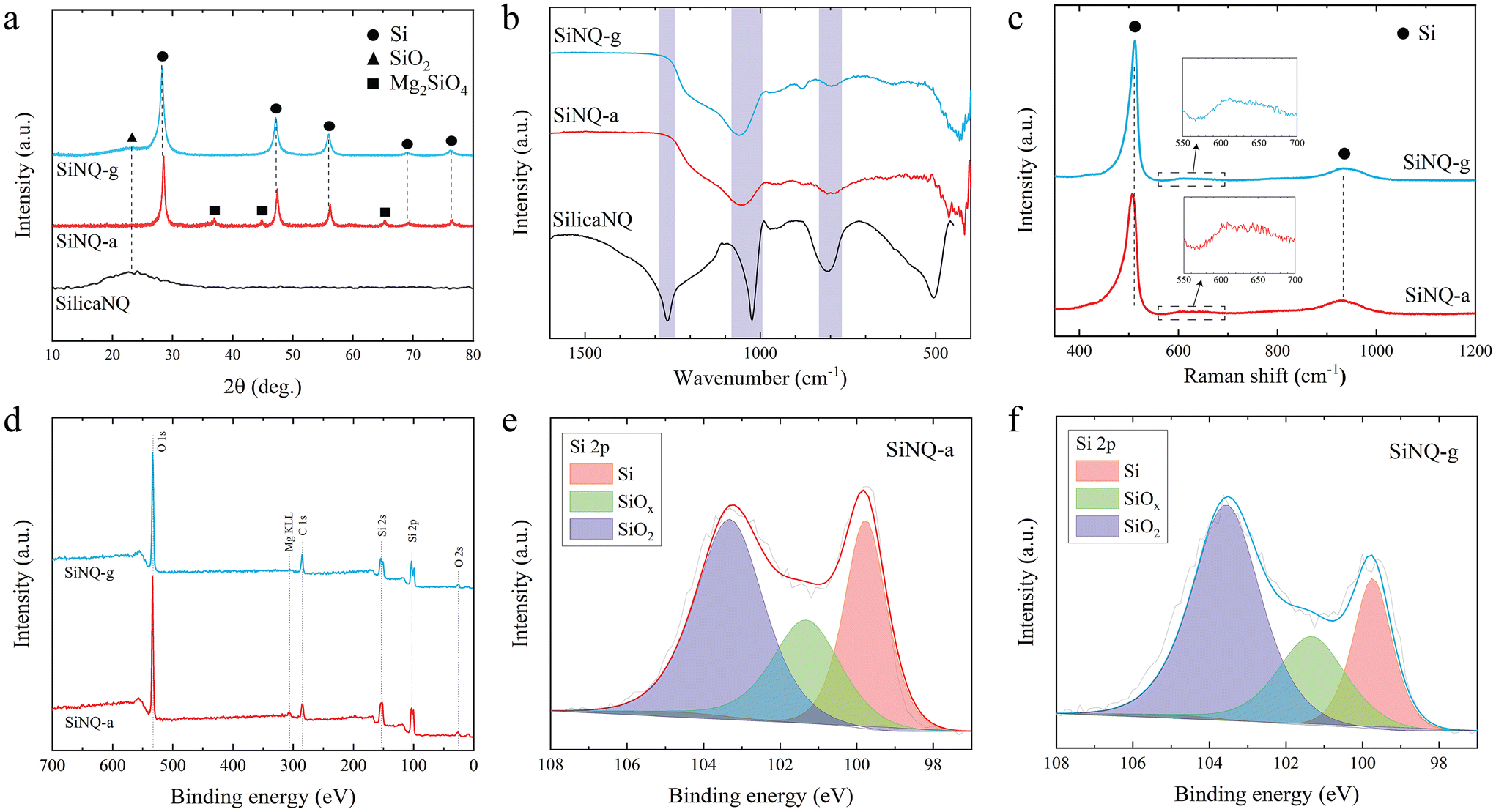 | ||
| Fig. 3 Characterization of Si-based materials in this study; (a) XRD spectra of SilicaNQ and both SiNQs, (b) FTIR spectra of SilicaNQ and both SiNQs, (c) Raman spectra of both SiNQs, (d) XPS survey spectra of both SiNQs and high-resolution Si spectra of (e) SiNQ-a and (f) SiNQ-g materials after 936 seconds of etching. Peak fitting is used to calculate the distribution of Si, SiOx, and SiO2 species. | ||
Fourier-transform infrared (FTIR) spectra of Si-based materials in this study are shown in Fig. 3b. For comparison, the FTIR spectrum of commercial Si (c-Si, crystalline Si nanoparticles with spherical shape and average size of 100 nm) is illustrated in Fig. S3a.† A distinctive broad peak, appearing at 1250 cm−1, in the spectrum of SilicaNQ is indicative of amorphous silica.40 SilicaNQ also exhibits the 1090 cm−1 peak for asymmetric Si–O–Si vibrations, while the symmetric counterpart of these vibrations is present at 795 cm−1.41 These two peaks are also visible but at a lower intensity for SiNQ-a and SiNQ-g materials. The presence of O in thermally reduced materials is evident from S-TEM elemental maps (Fig. S4†). For SiNQs, the O–H deformation vibration band at 870–820 cm−1 is due to the hydroxyl group linked to a Si atom.42 The c-Si material also shows 1090 cm−1 peak for its native silica surface layer. Raman spectra of SiNQs are presented in Fig. 3c. The ∼520 cm−1 peak is the characteristic feature of crystalline Si. However, when compared to c-Si, the Si peak in SiNQs exhibits a slight shift towards lower wavenumbers and an increased peak width (see Fig. S3b†). These peak shift and peak broadening effects are attributed to the inclusion of silicon oxides in the material composition.43 The appearance of a peak at approximately 920 cm−1 in the second-order Raman spectrum of silicon (Fig. 3b and S3b†) typically indicates a two-phonon scattering process, which is inherent to Si-based materials.44 Additionally, a peak at 630 cm−1, attributed to the wagging vibrations of Si–H bonds,45,46 is observed for both SiNQ-a and SiNQ-g materials. This peak is absent for c-Si material. The presence of Si–H bonds in SiNQs can promote their dispersibility in water-based slurries. We dispersed a similar amount of SiNQ-g and c-Si powders in DI water using magnetic stirring and ultrasonication methods to assess this. SiNQ-g material can be easily dispersed in DI water and create a stable suspension for up to one week, as shown in Fig. S3,† while c-Si is hardly dispersed in DI water. Even after ultrasonication, a large portion of c-Si gets separated and floats on top of the water. The zeta potential measurements of dispersions of SiNQ-g and c-Si prepared using ultrasonication also verify the stability of SiNQ-g dispersion, with a zeta potential of less than −30 mV (Fig. S3c†). We attribute such excellent dispersibility of SiNQ-g in aqueous solution to the presence of Si–H bonds.
XPS spectra were analyzed to identify and quantify the presence of different silicon species in SiNQ-a and SiNQ-g materials. The XPS survey spectra in Fig. 3d show C, O, and Si as expected. The SiNQ-a material contained trace amounts of aluminum and magnesium (0.34 and 0.22 atomic percent, respectively). The presence of aluminum can be attributed to residual contamination originating from the alumina crucible employed during reduction. The detected magnesium by XPS corresponds to Mg2SiO4 byproduct trapped within the SiNQ-a structure (as revealed by XRD results, Fig. 3a). On the other hand, SiNQ-g material demonstrated no such traces, confirming the absence of thermoreduction byproducts. High-resolution Si 2p spectra of SiNQs are presented in Fig. 3e and f. Peak fitting of Si 2p spectra provides further insight into the distribution of Si (red), SiOx suboxides (x < 2, green), and SiO2 (purple) species. Argon etching was also used to collect depth profiles for each material to understand how the ratio of these species changed from the surface to the bulk. As seen in Fig. S5,† SiNQ-a has a very inconsistent ratio of SiO2 and SiOx, while SiNQ-g is consistent throughout the depth profile. This consistency in Si-based species for SiNQ-g is likely a result of more homogenous reduction, which implies a more effective heat dissipation because of using graphite crucible instead of alumina during exothermic reduction reactions. The Si![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SiOx:SiO2 atomic ratio was estimated over 936 seconds of argon etching, and average atomic ratios of 27:25:48 (for SiNQ-a) and 27:16:57 (for SiNQ-g) were obtained. The higher SiOx:SiO2 atomic ratio of SiNQ-a material can be attributed to the local temperature rise in the alumina crucible during reduction, leading to a higher degree of conversion from SiO2 to SiOx.
:SiOx:SiO2 atomic ratio was estimated over 936 seconds of argon etching, and average atomic ratios of 27:25:48 (for SiNQ-a) and 27:16:57 (for SiNQ-g) were obtained. The higher SiOx:SiO2 atomic ratio of SiNQ-a material can be attributed to the local temperature rise in the alumina crucible during reduction, leading to a higher degree of conversion from SiO2 to SiOx.
Hydrofluoric acid treatment of SiNQs
We attempted to remove silicon oxides from the HCl-treated SiNQ product to obtain a material based on pure Si and assess its electrochemical performance. To this end, we chose SiNQ-a and subjected it to hydrofluoric acid (HF) etching. The S-TEM elemental mapping of HF-etched product (i.e., SiNQ-a-HF) verified the removal of O-containing species (see Fig. S6a and b†). However, HF treatment clearly damaged the original tubular structure of nano-quills. As revealed earlier, the nano-quill arms are composed of crystalline Si pockets distributed in an amorphous silicon oxide matrix. The HF treatment removed the amorphous phase and left behind a chain of elemental Si.The electrochemical cycling performance of anodes containing SiNQ-a with and without HF treatment is presented in Fig. S6c.† At a current density of 420 mA g−1, initial reversible capacities of 1003 mA h g−1 (for SiNQ-a) and 910 mA h g−1 (for SiNQ-a-HF) were acquired. We attribute the higher reversible capacity of SiNQ-a anode to the active material's unique morphology. It is noted that the surface area and porosity of SiNQ-a significantly decrease after HF etching. The higher surface area/porosity and high aspect ratio of SiNQ-a allow for more extensive Li–Si alloying due to the shortened diffusion length,47 resulting in a more effective Li+ storage. On the other hand, SiNQ-a-HF presumably undergoes severe pulverization and loss of active material during the first lithiation, leading to a lower initial reversible capacity. Both anodes exhibited a similar cycling performance up to about 30 cycles. Afterwards, the anode with SiNQ-a-HF showed a capacity decay, and its specific capacity reached 550 mA h g−1 at the 150th cycle, while the delivered capacity of SiNQ-a anode remained stable at around 875 mA h g−1. The SiNQ-a battery shows a gradual capacity rise after about 90 cycles. A similar behavior, known as the ‘capacity climbing’ phenomenon, is reported in SiOX batteries. The lithiation reaction of silicon oxide triggers the formation of electrochemically active Si out of the original material through the following reactions:48
| SiO2 + 4Li → Si + 2Li2O | (4) |
| 2SiO2 + 4Li → Si + Li4SiO4 | (5) |
This phenomenon has been supported in the literature and observed for other SiOX-based materials.49 Therefore, we attribute the slight capacity climb behavior observed from the SiNQ-a anode to the contribution from SiOx. We disassembled the cycled batteries and investigated the active materials using S-TEM. The acquired elemental mapping for Si, O, and C in cycled active materials is presented in Fig. S6d and e.† The cycled SiNQ-a maintained its 1D morphology without an apparent pulverization or swelling. On the other hand, the cycled SiNQ-a-HF is characterized by swollen particles and clear pulverization. This indicates that the unique composition of SiNQs with a silicon oxide matrix can play a crucial role in buffering the volumetric changes and maintaining the particle's integrity during cycling, leading to a very stable cycling performance. Thus, in further work, we utilized SiNQs without HF treatment to prepare Si-graphite composite anodes.
Water-based SiNQ-graphite anodes
Recent developments in commercial LIB anodes for electric vehicles (EVs) have demonstrated that Si-graphite composite materials are promising candidates for efficient lithium storage, taking advantage of the high capacity of Si-based materials and ultrahigh stability of graphite.50,51 The content of Si-based materials in commercial Si-graphite electrodes for EV batteries is typically low (<10 wt%).52,53 The Si content may vary depending on the targeted balance between increased energy density and cycling stability. In the current study, we adhered to the slurry composition reported by researchers at the Oak Ridge National Laboratory and Argonne National Laboratory,54,55 where 17 wt% of the graphite is replaced with Si-based materials. We combined SiNQ-a or SiNQ-g with conventional mesocarbon microbead (MCMB) graphite to explore the potential performance enhancement derived from our newly developed Si-based nano-quills. As such, we prepared 17% SiNQ-a/MCMB and 17% SiNQ-g/MCMB electrodes on a copper foil current collector. Commercial Si (100 nm dia.) was also used to make a 17% c-Si/MCMB electrode. An MCMB electrode (without Si addition) was also prepared for comparison. The active mass loading in each of the four electrodes was ∼4.5 mg cm−2.Utilizing cyclic voltammetry, we investigated the behavior of 17% c-Si/MCMB and 17% SiNQ-g/MCMB electrodes through cathodic and anodic scans, and the results are presented in Fig. S7.† Within the cathodic scan (Fig. S7a and b†), characteristic peaks around 0.03, 0.10, and 0.17 V represent graphite lithiation to form intercalated LiCx phases.56 These peaks coalesce with the cathodic signature of LixSi alloying observed at 0.18 V.57,58 In the anodic scan, subtle peaks at 0.25 and 0.29 V denote the progressive delithiation mechanism transitioning from the LiC6 phase to intermediary species.56 An additional anodic peak, discernible at approximately 0.5 V, is attributable to the dealloying process of LixSi.58
Cyclic voltammetry measurements were performed over a spectrum of scan rates spanning 0.1 to 2.0 mV s−1 to elucidate the battery reaction kinetics. Notably, for the 17% SiNQ-g/MCMB anode, the anodic peaks synonymous with LiCx delithiation (0.25 and 0.29 V) are pronounced at lower scan rates. However, at higher scan rates (greater than 0.8 mV s−1), the graphite peaks are subsequently eclipsed by the anodic peak of LixSi (see Fig. S7d†). In Si-graphite composite electrodes comprising porous Si, the shift of dominant delithiation peak from carbon allotrope to Si at higher scan rates has also been observed but unexplained.59 On the other hand, for 17% c-Si/MCMB anode, the anodic peak associated with graphite delithiation remains pronounced up to 2 mV s−1, where the peak currents for graphitic and Si delithiation are roughly at the same level (see Fig. S7c†). The ratio of graphitic peak current to Si peak current decreases with increasing scan rate, as shown in Fig. S7e and f.† Therefore, at scan rates higher than 2 mV s−1, we expect the Si delithiation peak current to be higher than the graphite delithiation peak current, even for c-Si. Thus, although the scan rates at which the dominant peak shifts from graphite to Si might differ for c-Si and SiNQ-g, the dominant peak shifting at higher scan rates is a common phenomenon observed in both cases. This behavior is counter-intuitive because the (de)intercalation of Li from graphite is expected to be faster than the phase-change governed delithiation of Si-based materials.
We developed a detailed model for this counter-intuitive phenomenon, as explained in the ESI section (see Fig. S8 and eqn (S1)–(S14)†). Our derivation clearly indicates that researchers should exercise caution while using the standard power-law correlation with the scan rate, v, where b = 0.5 indicates a diffusion-controlled process and b = 1 indicates a capacitive process, as defined by eqn (6) for analysis of CV results as
| i = avb. | (6) |
Additionally, we would like to guide the researchers to ref. 27, which shows that eqn (6) is derived for redox couples (with both reduced and oxidized species present within the electrolyte) under the conditions of semi-infinite diffusion, none of which are valid in current LIBs. It is noteworthy that when a redox reaction takes place at any given electrode–electrolyte interface for metal-ion batteries (viz., lithium-ion batteries, sodium-ion batteries, potassium-ion batteries, calcium-ion batteries, etc.), three processes govern the reaction rate: (a) electronic conduction through the electrode, (b) metal-ion diffusion within the electrolyte, and (c) metal (ion) diffusion through the solid electrode. Our analysis (see ESI section†) may be used to interpret CV data obtained in metal-ion batteries as long as the metal (ion) diffusion through the solid electrode is the rate-limiting process. For cases where either process (a) or (b) is the rate-limiting process (e.g., in highly concentrated electrolytes) or in cases with metallic anodes with plating reactions (e.g., lithium metal anodes), we intend to present a thorough analysis to help the battery community with the interpretation of CV data in future.
All graphite-based anodes were cycled at 0.1C for 200 cycles, with a formation cycle at 0.05C. A current density of 900 mA g−1 was chosen as 1C. The charge–discharge profiles of the anodes and their corresponding coulombic efficiency values for the first 20 cycles are plotted in Fig. S9.† The MCMB anode shows an initial coulombic efficiency (ICE) of 88%. Incorporating SiNQ-g, SiNQ-a, and c-Si shifts the ICE to 81.3, 82.8, and 84.4%, respectively. At 0.1C, the MCMB anode exhibits a stable capacity of around 0.9 mA h cm−2 over 200 cycles (Fig. 4a). Both 17% SiNQ-g/MCMB and 17% c-Si/MCMB anodes deliver an initial reversible capacity of 2.2 mA h cm−2, which is around 2.5 times the capacity of pure MCMB anode. Notably, this achievement is particularly impressive for anodes containing SiNQ-g, given its composition is comprised primarily of silicon oxides, yet manages to deliver a capacity on par with commercial Si products, which are almost entirely made of elemental Si. After 200 cycles, a distinct divergence occurs, where the 17% c-Si/MCMB anode shows a capacity drop to 0.9 mA h cm−2, corresponding to a capacity retention of 36%. Meanwhile, the 17% SiNQ-g/MCMB anode exhibits a capacity retention of 75%. The 17% SiNQ-a/MCMB anode delivers an initial reversible capacity of 1.65 mA h cm−2 with a capacity retention of 81% after 200 cycles.
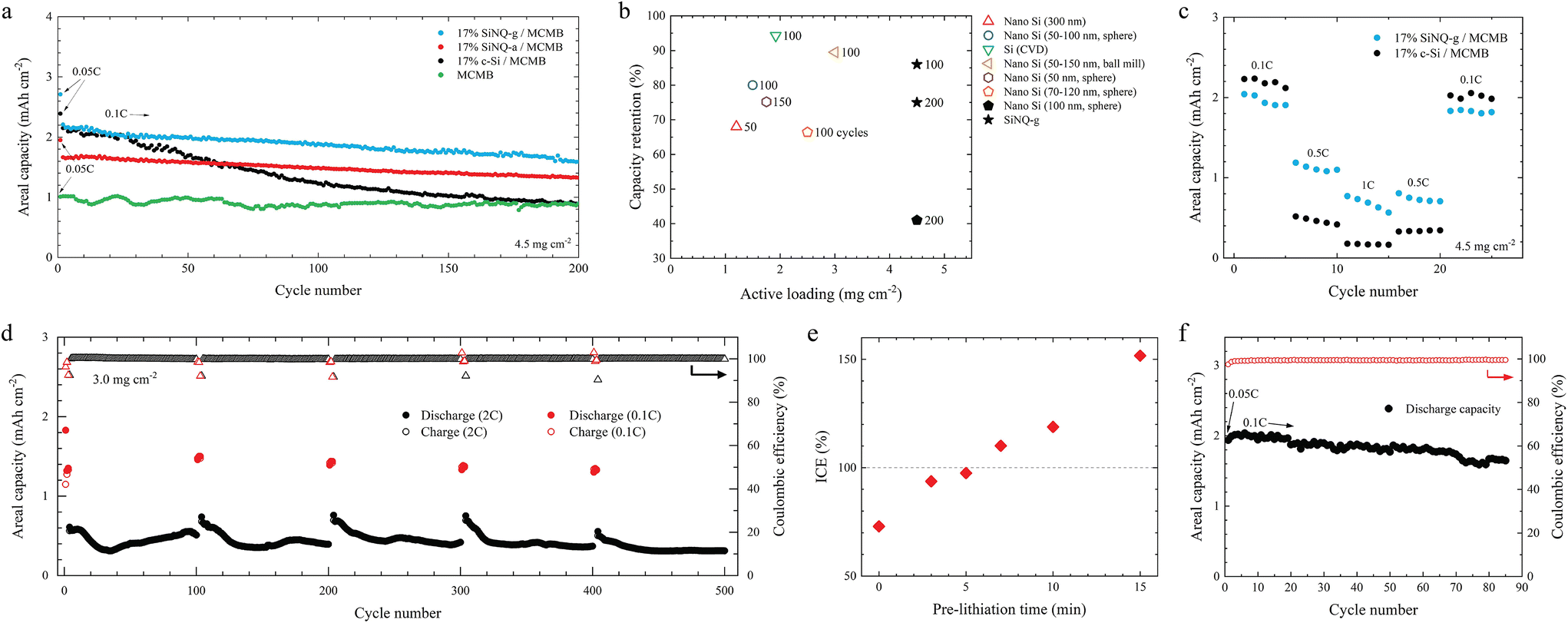 | ||
| Fig. 4 Electrochemical performance of Si-graphite electrodes in this study. (a) Cycling performance of composite electrodes containing 17% c-Si, 17% SiNQ-a, or 17% SiNQ-g, (b) a literature survey of capacity retention vs. active mass loading for water-based Si-graphite anodes presented in Table S1,† (c) rate performance comparison of composite electrodes containing 17% SiNQ-g and 17% c-Si, (d) cycling performance of 17% SiNQ-g/MCMB electrode under alternative cycling in which the anode underwent 3 cycles at 0.1C followed by 97 cycles at 2C, and this process was repeated 5 times, (e) ICE of 17% SiNQ-g/MCMB electrode as a function of pre-lithiation time, (f) cycling performance of 17% SiNQ-g/MCMB electrode pre-lithiated for 5 minutes. All graphite-based half cells in this study were cycled between 1.5 V and 0.005 V (1C = 900 mA g−1). The formation cycle was performed at 0.05C. | ||
To better analyze the responsible mechanisms for capacity decay (Fig. 4a), we presented the differential capacity curves corresponding to the formation, 1st, 2nd, 3rd, 100th, and 200th cycles in Fig. S10.† The differential capacity plots exhibit key peaks in alignment with the observations drawn from the charge–discharge curves. The MCMB anode exhibits characteristic delithiation peaks at approximately 0.11, 0.16, and 0.24 V, as well as lithiation peaks around 0.08, 0.11, and 0.20 V, which correspond to the voltage plateaus resulting from lithium-graphite (de)intercalation.60,61 The evolution of peaks with cycling (Fig. S10a†) indicates a peak shift towards lower voltages during lithiation and a peak shift towards higher voltages during delithiation. In addition, the peak intensity dramatically decreases when the battery undergoes more cycles. All electrodes with Si-based materials consistently showcase a peak at the lithiation endpoint of <0.05 V. This peak signifies a phase transition from an amorphous to a crystalline structure, corresponding with a stoichiometry of Li15Si4.62 A broad delithiation peak (between 0.25–0.4 V) indicates the formation of amorphous Si.63 Moreover, a prominent delithiation peak surfaces around 0.5 V denotes a two-phase reaction, marking a transition from the crystalline Li15Si4 to amorphous LixSi species.63,64 This peak levels off for the 17% c-Si/MCMB anode after 200 cycles; however, its strength remains unchanged in the case of SiNQ-based anodes. This observation highlights the possibility of gradual pulverization of crystalline phases from c-Si and a loss of active material during long-term cycling, which can cause severe capacity decay over 200 cycles. For SiNQs, the crystal pockets are embedded in a matrix of amorphous silicon oxides, which can act as a protecting medium and preserve the two-phase reaction over cycling. After the formation cycle, a small peak appears at around 0.25 V during lithiation of the 17% c-Si/MCMB anode (inset, Fig. S10b†). This peak intensifies in subsequent cycles. However, no distinct peak at this voltage level is observed for MCMB, 17% SiNQ-a/MCMB, or 17% SiNQ-g/MCMB anodes. The appearance of an extra lithiation peak at 0.25 V for the 17% c-Si/MCMB electrode can be attributed to the electrolyte decomposition. To investigate this further, we performed electrochemical impedance spectroscopy (EIS) on 17% SiNQ-g/MCMB and 17% c-Si/MCMB electrodes at the 5th and 25th cycles. The Nyquist plots (presented in Fig. S11a and b) were modeled using the Randles circuit shown in Fig. S11c,† and the series resistance (R1), the SEI resistance (R2), and the charge-transfer resistance (R3) values were estimated. The R1, R2, and R3 variations are depicted in Fig. S11d.† A higher series resistance is detected for the 17% SiNQ-g/MCMB anode. This can be due to the higher porosity of SiNQ-g, compared to c-Si, which leads to a higher internal impedance. R1 remains almost stable for both anodes, cycling up to 25 cycles. However, there is a significant increase in R2 and R3 when cycling the 17% c-Si/MCMB anode from the 5th to 25th cycle, while R2 dropped for the 17% SiNQ-g/MCMB anode and R3 remained unchanged. The increase of R2 and R3 over cycling for the 17% c-Si/MCMB electrode agrees with the advent of an extra lithiation peak (inset, Fig. S10b†) and implies the occurrence of side reactions. This is due to the pulverization of c-Si particles over the charging–discharging process and the electrolyte decomposition over newly formed Si surfaces,65 which eventually affect the cycling stability of the 17% c-Si/MCMB anode (see Fig. 4a).
Among explored anodes with Si-graphite active material, the anode containing SiNQ-g presents the best cycling stability. We conducted a literature survey for water-based graphitic anodes in which 14–17 wt% of the active material consists of Si-based materials (see Table S1†) and presented a comparative graph in Fig. 4b. The 17% SiNQ-g/MCMB anode outperforms the others by combining a high active mass loading (4.5 mg cm−2) and decent capacity retention (75%, 200 cycles). In the context of rate performance, the 17% SiNQ-g/MCMB anode outperforms the 17% c-Si/MCMB anode at higher examined current rates, as illustrated in Fig. 4c. The anode with c-Si exhibits a huge capacity drop from 0.1C to 1C, while the 17% SiNQ-g/MCMB anode delivers a high capacity when subjected to fast charging–discharging at 1C.
SiNQs for fast charging–discharging batteries
Batteries with fast charging and discharging capabilities are attractive for EVs, making the latter suitable for various applications, from long-distance travel to urban commuting. Such capabilities would reduce the time required to replenish the vehicle's energy and enable regenerative braking and rapid acceleration, improving the overall performance of EVs. To enable fast charging/discharging batteries, Si-graphite anodes must demonstrate enhanced kinetics during both lithiation and delithiation at high current densities. To examine this, we cycled 17% SiNQ-g/MCMB electrodes (with active mass loading of 3.0 mg cm−2) under three different conditions: (i) continuous cycling with lithiation at 0.1C (0.27 mA cm−2) and delithiation at 2C (5.4 mA cm−2) in each cycle, (ii) continuous cycling with lithiation at 2C and delithiation at 0.1C in each cycle, and (iii) alternative cycling in which the anode underwent 3 cycles at 0.1C followed by 97 cycles at 2C, and this process was repeated 5 times (i.e., a total of 500 cycles). The results from these three testing conditions are presented in Fig. S12† and Fig. 4d.The battery subjected to lithiation at 0.1C and delithiation at 2C (case i) demonstrated a reversible discharge capacity of 1.21 mA h cm−2, maintaining stability in subsequent cycles and achieving a CE of >99% after three cycles (Fig. S12a†). The battery cycled at 2C lithiation, and 0.1C delithiation (case ii) delivered a stable capacity of 0.64 mA h cm−2, with a CE of >99% after the second cycle (Fig. S12b†). To assess the anode's recuperative performance following periods of rapid cycling, we subjected the anode to the cycling conditions referenced in case iii. As presented in Fig. 4d, the battery demonstrated an initial reversible discharge capacity of 1.32 mA h cm−2 at 0.1C. Following cycles of high current rate exposure (at 2C), the battery's capacity exhibited a notable recovery, achieving values of 1.46, 1.39, 1.33, and 1.30 mA h cm−2, respectively, during each subsequent cycling interval (at 0.1C) interspersed within the test regimen. Our developed CV model (ESI section†) explains the superior performance of SiNQ-g material at high rates. The unique porous morphology and composition of SiNQ-g material enable it to effectively interact with Li+ ions at high current densities, thus making SiNQ-g suitable anode materials for advancing next-generation fast charging/discharging batteries.
Pre-lithiation of SiNQ-based electrodes
As previously demonstrated, the 17% SiNQ-g/MCMB anode exhibits an ICE of 81.3% (Fig. S9e†). The active Li loss in the first cycle would alter the specific energy density of full cells, where this anode is paired with a cathode containing a limited Li inventory. As such, we conducted a systematic pre-lithiation study to efficiently preset Li+ ions in the anode active materials and compensate for the initial Li loss. The utilized pre-lithiation process entails direct contact between the 17% SiNQ-g/MCMB electrode and a Li chip under pressure while both of them are immersed in an electrolyte for a specified duration. The charge–discharge profiles of 17% SiNQ-g/MCMB electrodes pre-lithiated for 0, 3, 5, 7, 10, and 15 minutes are shown in Fig. S13.† We noticed that the ICE increases monotonically with the pre-lithiation time (Fig. 4e). An ICE of 97.5% is achieved following a 5-minute pre-lithiation; thus, it is considered the optimal pre-lithiation condition. The optimally pre-lithiated electrode was subjected to cycling at 0.1C. As seen in Fig. 4f, a stable cycling with an average CE of 99.4% was achieved.Conclusions
A scalable process was introduced to prepare water-dispersible porous SiNQs. Microstructural characterizations revealed that SiNQs are composed of crystalline Si pockets distributed in an amorphous Si suboxide matrix. We showed that utilizing a high-thermal-diffusivity crucible for the thermoreduction process results in a Si-based product that better retains the morphology of original silica particles. High-loading robust SiNQ-graphite composite anodes with 17 wt% SiNQ in the active material and an active mass loading of 4.5 mg cm−2 were prepared using an aqueous slurry. The unique 1D hollow morphology of SiNQ arms enables them to withstand the volume changes during Li+ insertion–extraction processes, leading to high-capacity anodes with substantial cycling performance. Notably, SiNQ-g materials, predominantly composed of Si suboxides, demonstrate a capacity on par with commercially obtained pure Si nanostructures, underscoring their efficiency. Additionally, a general physics-based cyclic voltammetry model was presented to explain the unusual shift in the dominant delithiation peak from graphite to Si at high scan rates. In agreement with cyclic voltammetry observations, SiNQ-based anodes demonstrated high capacity and outstanding cycling stability when cycled at a high current density of 5.4 mA cm−2. We employed a simple pre-lithiation technique and produced anodes with nearly 100% ICE.Experimental
Materials and chemicals
Cellulose nanocrystals (CNCs) in the form of 10 wt% aqueous dispersion were kindly provided by the USDA Forest Products Laboratory (Madison, WI, USA). Absolute ethanol (200 proof, CAS Number 64-17-5, Fisher Chemical) was purchased from Fisher Scientific (Hampton, NH, USA). Hexadecyltrimethylammonium bromide (CTAB, CAS Number 57-09-0, ≥98%), tetraethyl orthosilicate (TEOS, CAS Number 78-10-4, ≥99%), lithium hexafluorophosphate (LiPF6, CAS Number 21324-40-3, battery grade, ≥99.99%), lithium hydroxide (LiOH, CAS Number 1310-65-2, 98%), N-methyl-2-pyrrolidone (NMP, CAS Number 872-50-4, ≥99.5%), fluoroethylene carbonate (FEC, CAS Number 114435-02-8, 99%), poly(acrylic acid) (PAA, CAS Number 9003-01-4, average Mw ∼450000), poly(vinylidene fluoride) (PVDF, CAS Number 24937-79-9), ethylene carbonate (EC, CAS Number 96-49-1, >99%), dimethyl carbonate (DMC, CAS Number 616-38-6, >99.9%), and a solution of 1M LiPF6 in EC/diethyl carbonate (DEC) with EC:DEC volume ratio of 50:50 were purchased from Sigma Aldrich (St Louis, MO, USA). Ammonium hydroxide (NH3, CAS Number 1336-21-6, 28–30% ACS) was acquired from VWR International (Radnor, PA, USA). Magnesium powder (CAS Number 7439-95-4, 99+%, pure) was obtained from Acros Organics. Commercial Si (spherical, 100 nm, >97%), Carbon black (SuperP, CAS Number 1333-86-4) and Li chip (99.9% purity, 16 mm diameter, and 600-micron thickness) were acquired from MTI Corporation (Richmond, CA, USA). Mesocarbon microbeads (MCMB) synthetic graphite (spherical, 9–14 μm) was purchased from MSE Supplies (Tucson, AZ, USA).
Synthesis of SilicaNQs
The synthesis process commenced with the formulation of an aqueous solution consisting of deionized (DI) water (800 mL), ethanol (200 mL), ammonium hydroxide (4 mL), CNCs (200 mg, dry content), and CTAB (200 mg). This mixture underwent ultrasonication for ten minutes to ensure a homogeneous dispersion. The resulting solution, containing CNCs, was subjected to continuous stirring at 350 rpm, during which TEOS (5.6 mL) was added dropwise using a syringe pump at a rate of 0.22 mL min−1. The mixture was reacted for two hours under mild stirring. Then, the silicate gel was separated from the aqueous medium via vacuum filtration, washed with DI water to eliminate any residual chemicals, and freeze-dried using a FreeZone freeze-dry system (Labconco, MO, USA). The freeze-dried powder was calcined in a box furnace overnight at 600 °C at a ramp rate of 1 °C min−1. This process resulted in ∼200 mg of SilicaNQ product.Fabrication of SiNQs
Within an argon-filled glove box, SilicaNQ was mixed with Mg powder at a weight ratio of 1:0.8, corresponding to silica:Mg molar ratio of 1:2. Two types of crucibles were considered to explore the role of heat accumulation during the reduction reactions. To prepare SiNQ-a, the mixture was transferred to an alumina crucible placed inside a rectangular air-tight steel reactor. To prepare SiNQ-g, the mixture was transferred to a cylindrical graphite crucible equipped with a screw-on lid. The reactors were sealed to inhibit oxygen ingress into the system before positioning them in the middle of a tube furnace. The tube was purged with argon at a rate of 200 sccm for 30 minutes prior to heating. The mixture was then subjected to the following thermal cycle: (i) heating from room temperature to 450 °C with a ramp rate of 5 °C min−1, (ii) heating from 450 °C to 650 °C with a ramp rate of 1 °C min−1, (iii) isothermal heating at 650 °C for 6 hours, and (iv) natural cooling to room temperature inside the tube. Due to the formation of Mg-based byproducts, a chemical etching step was imperative. A 2 M hydrochloric acid solution was employed to dissolve magnesium oxide and silicide by-products. The as-reduced material from alumina or graphite crucibles was dispersed in the HCl solution under constant agitation for 48 hours. This was succeeded by multiple washes with DI water until a neutral pH was achieved. In addition, to remove silicon suboxides and obtain a highly pure Si-based product, SiNQ-a material (i.e., reduced in alumina crucible and HCl treated) was subjected to HF etching. This was performed by soaking the SiNQ-a powder in a 5% HF solution for 15 minutes, followed by thorough washing with DI water.
Electrochemical measurements
The effects of HF etching on the electrochemical cycling performance of SiNQs were explored. A slurry of SiNQ-a (with or without HF etching) was cast onto the conductive Bucky Paper (60 g m−2, NanoTechLabs, NC, USA) current collector. The mass ratio of active material, binder (PVDF), and conductive additive (Super P) in the NMP-based slurry was 60:20:20. Electrodes with an average active mass loading of ∼1.1 mg cm−2 were achieved.
Graphite-based electrodes were prepared by casting an aqueous slurry onto copper foil and subsequent calendaring. The active materials were 100% MCMB graphite (as the control electrode) or a combination of Si-based materials (i.e., c-Si, SiNQ-a, or SiNQ-a) and MCMB graphite with a mass ratio of 17:83. Lithiated PAA (LiPAA) was prepared by adding LiOH to an aqueous solution of PAA (until pH 7) and was used as the binder. The mass ratio of active material, LiPAA binder, and Super P in aqueous slurries was 88:10:2. The average active mass loading was ∼4.5 mg cm−2.
All half cells were assembled in CR2032-type coin cells with a Celgard 2324 separator. Electrodes with a diameter of 12 mm were punched and paired with Li counter electrodes in an argon-filled glovebox (water and oxygen content <0.1 ppm). For Bucky Paper electrodes with SiNQ-a (with or without HF etching) active material, the electrolyte was composed of 1 M LiPF6 in EC/DMC solvents with EC:DMC volume ratio of 50:50. These anode half cells were tested over a voltage range of 1.0–0.01 V. All graphite-based anodes were tested using an electrolyte composed of 1 M LiPF6 in EC/DEC (EC:DEC volume ratio of 50:50), with 10% FEC additive, over a voltage range of 1.5–0.005 V.
Galvanostatic charge/discharge experiments were performed using an Arbin battery analyzer (LBT Series). Cyclic voltammetry measurements were performed on 17% c-Si/MCMB and 17% SiNQ-g/MCMB anodes at scan rates ranging from 0.1 to 2.0 mV s−1. Electrochemical impedance spectroscopy measurements were conducted on 17% c-Si/MCMB and 17% SiNQ-g/MCMB anodes at room temperature over a frequency range of 100 kHz to 0.01 Hz. Cells were measured in the fully lithiated state (i.e., 0.005 V) after the 5th and 25th cycles to ensure identical conditions. All CV and EIS experiments were carried out using a Gamry Reference 3000 potentiostat.
Pre-lithiation was performed on 17% SiNQ-g/MCMB electrodes following the short-circuit electrochemical method reported by Bai et al.66 Briefly, the 17% SiNQ-g/MCMB electrode was put under direct contact with a Li chip and soaked in 50 μL of electrolyte (1 M LiPF6 EC/DEC (50:50) and 10% FEC additive). The assembly was placed under ∼5.5 kPa compressive pressure to ensure close contact between the electrode and the Li chip. A range of pre-lithiation durations (i.e., 0, 3, 5, 7, 10, and 15 min) were considered. After pre-lithiation, each electrode was paired with a fresh Li chip in the CR2032 coin cell with 50 μL of the same electrolyte and cycled between 1.5–0.005 V to evaluate the initial coulombic efficiency.
Characterizations
Transmission electron microscopy studies were carried out using a Hitachi T9500 high-resolution TEM at 300 kV with the selected area electron diffraction (SAED). Scanning TEM examinations were conducted using a Hitachi SU9000 ultra-high-resolution SEM at 30 kV with energy-dispersive X-ray spectroscopy (EDS). Dimensional analysis of microstructural features in TEM or S-TEM images was performed using ImageJ software.Fourier-transform infrared spectroscopic analysis was conducted directly on solid samples with a ThermoFisher Nicolet iS50 spectrometer equipped with a diamond ATR. Raman spectroscopy was performed on powders using a Renishaw inVia Raman microscope, with 532 nm laser excitation and 10× objective beam focusing. A minimum of 5 spectra were collected from different points on each sample with 30 s data accumulation per point and then averaged. The specific surface area and total pore volume of materials were evaluated using a Quantachrome Autosorb iQ gas sorption analyzer using N2 gas.
XPS data was collected using a PHI VersaProbe III with a monochromatic Al Kα X-ray source (hν = 1486.6 eV) and an Al anode powered at 25 W and 15 kV. The system was calibrated to both Ag and Au metallic binding energy. All data was calibrated to adventitious C 284.8 eV. The system base pressure was maintained at 1 × 10−6 Torr or better. Samples were prepared by pressing powders into a disc and then adhering samples to the platen using 3M double-sided tape. The analysis area was set to 500 × 500 μm2 and a beam diameter of 100 μm. XPS survey data was collected using 3 sweeps, a pass energy of 224 eV, and a step size of 0.8 eV. High-resolution spectra for C, O, and Si were collected at 45 degrees with 3 sweeps, a pass energy of 69 eV, and a step size of 0.125 eV. Argon etching was used for depth profiling of the samples. Peak fitting parameters were set for Si and Si oxides using a Shirley baseline with an FWHM of 1.2 and 2.1 eV, respectively. All peaks were fit with a G/L 60. All Si binding energies were constrained to 99.8 ± 0.25 eV. Silicon suboxide (SiOx) peaks were identified at 101.2 ± 0.5 eV. Silicon dioxide (SiO2) was constrained to 103.93 ± 0.25 eV. All peak fitting was done in Casa XPS.
Author contributions
M.S., A.M.R., and S.P. conceived the project. N.C. and M.S. developed the concept. N.C., M.S., N.S., M.P., S.C., and K.K. conducted the experiments, performed data analysis, and developed the model. N.C. and M.S. drafted the manuscript. S.P., A.M.R., Y.D., and C.M.C. reviewed and edited the manuscript. All authors discussed and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the US Endowment for Forestry and Communities [Contract No. 20-00082] to synthesize and characterize silicon nano-quills (SiNQs) described in this study. The authors also acknowledge financial support through Clemson University's Virtual Prototyping of Autonomy Enabled Ground Systems (VIPR-GS), under Cooperative Agreement W56HZV-21-2-0001 with the US Army DEVCOM Ground Vehicle Systems Center (GVSC), to develop and test batteries described in this study. The battery data presented herein was obtained at the Clemson Nanomaterials Institute, which Clemson University operates.References
- D. Trache, M. H. Hussin, M. K. M. Haafiz and V. K. Thakur, Nanoscale, 2017, 9, 1763–1786 RSC.
- W. Chen, H. Yu, S. Y. Lee, T. Wei, J. Li and Z. Fan, Chem. Soc. Rev., 2018, 47, 2837–2872 RSC.
- C. Chen and L. Hu, Acc. Chem. Res., 2018, 51, 3154–3165 CrossRef CAS PubMed.
- U.S. Endowment for Forestry and Communities, P3Nano – Advancing Commercialization of Cellulosic Nanomaterials, https://www.usendowment.org/what-we-do/future-markets/p3nano-advancing-commercialization-of-cellulosic-nanomaterials/.
- Y. Habibi, L. A. Lucia and O. J. Rojas, Chem. Rev., 2010, 110, 3479–3500 CrossRef CAS PubMed.
- Y. Wan, Z. Yang, G. Xiong, S. R. Raman and H. Luo, RSC Adv., 2015, 5, 48875–48880 RSC.
- A. Korpi and M. Kostianinen, ChemNanoMat, 2022, 8(4), e20210045 Search PubMed.
- K. Zheng, J. A. Bortuzzo, Y. Liu, W. Li, M. Pischetsrieder, J. Roether, M. Lu, A. R. Boccaccini, B. Glaser, J. Lehmann and W. Zech, Biol. Fertil. Soils, 2015, 35, 219–230 Search PubMed.
- Y. Cui, Nat. Energy, 2021, 6, 995–996 CrossRef CAS.
- C. J. Wen and R. A. Huggins, J. Solid State Chem., 1981, 37, 271–278 CrossRef CAS.
- M. N. Obrovac and L. Christensen, Electrochem. Solid-State Lett., 2004, 7, 5–9 CrossRef.
- C. Z. Ke, F. Liu, Z. M. Zheng, H. H. Zhang, M. T. Cai, M. Li, Q. Z. Yan, H. X. Chen and Q. B. Zhang, Rare Met., 2021, 40, 1347–1356 CrossRef CAS.
- W. An, P. He, Z. Che, C. Xiao, E. Guo, C. Pang, X. He, J. Ren, G. Yuan, N. Du, D. Yang, D. L. Peng and Q. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 10308–10318 CrossRef CAS PubMed.
- X. Han, L. Gu, Z. Sun, M. Chen, Y. Zhang, L. Luo, M. Xu, S. Chen, H. Liu, J. Wan, Y. B. He, J. Chen and Q. Zhang, Energy Environ. Sci., 2023, 16, 5395–5408 RSC.
- Z. Sun, Q. Yin, H. Chen, M. Li, S. Zhou, S. Wen, J. Pan, Q. Zheng, B. Jiang, H. Liu, K. Kim, J. Li, X. Han, Y. He, L. Zhang, M. Li and Q. Zhang, Interdiscip. Mater., 2023, 2, 635–663 CrossRef.
- X. H. Liu, J. W. Wang, S. Huang, F. Fan, X. Huang, Y. Liu, S. Krylyuk, J. Yoo, S. A. Dayeh, A. V. Davydov, S. X. Mao, S. T. Picraux, S. Zhang, J. Li, T. Zhu and J. Y. Huang, Nat. Nanotechnol., 2012, 7, 749–756 CrossRef CAS PubMed.
- R. Liu, C. Shen, Y. Dong, J. Qin, Q. Wang, J. Iocozzia, S. Zhao, K. Yuan, C. Han, B. Li and Z. Lin, J. Mater. Chem. A, 2018, 6, 14797–14804 RSC.
- A. W. Nemaga, J. Mallet, J. Michel, C. Guery, M. Molinari and M. Morcrette, J. Power Sources, 2018, 393, 43–53 CrossRef CAS.
- C. Wang, C. Yang and Z. Zheng, Adv. Sci., 2022, 9, 2105213 CrossRef CAS PubMed.
- M. T. Mcdowell, S. W. Lee, W. D. Nix and Y. Cui, Adv. Mater., 2013, 25, 4966–4985 CrossRef CAS PubMed.
- J. Sakabe, N. Ohta, T. Ohnishi, K. Mitsuishi and K. Takada, Commun. Chem., 2018, 1, 1–9 CrossRef CAS.
- Z. Chen, D. L. Danilov, R. A. Eichel and P. H. L. Notten, Adv. Energy Mater., 2022, 12, 2201506 CrossRef CAS.
- T. Zhao, D. Zhu, W. Li, A. Li and J. Zhang, J. Power Sources, 2019, 439, 227027 CrossRef CAS.
- F. Xi, Z. Zhang, X. Wan, S. Li, W. Ma, X. Chen, R. Chen, B. Luo and L. Wang, ACS Appl. Mater. Interfaces, 2020, 12(43), 49080–49089 CrossRef CAS PubMed.
- J. Sherwood, T. J. Farmer and J. H. Clark, Chem, 2018, 4, 2010–2012 CAS.
- G. Zhu, D. Chao, W. Xu, M. Wu and H. Zhang, ACS Nano, 2021, 15, 15567–15593 CrossRef CAS PubMed.
- v A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, Ltd, 3rd edn, 2022 Search PubMed.
- C. J. Brinker and G. W. Scherer, Sol-Gel Science: The Physics and Chemistry of Sol-Gel Processing, Academic Press, 1990 Search PubMed.
- T. Abitbol, H. Marway and E. Cranston, Nord. Pulp Pap. Res. J., 2014, 29, 46–57 CrossRef CAS.
- N. C. Das, H. Cao, H. Kaiser, G. T. Warren, J. R. Gladden and P. E. Sokol, Langmuir, 2012, 28, 11962–11968 CrossRef CAS PubMed.
- S. H. Wu and H. P. Lin, Chem. Soc. Rev., 2013, 42, 3862–3875 RSC.
- J.-K. Yoo, J. Kim, M.-J. Choi, Y.-U. Park, J. Hong, K. M. Baek, K. Kang, Y. S. Jung, J. Yoo, M. Choi, K. M. Baek, Y. S. Jung, J. Kim, Y. Park, J. Hong and K. Kang, Adv. Energy Mater., 2014, 4, 1400622 CrossRef.
- J. Entwistle, A. Rennie and S. Patwardhan, J. Mater. Chem. A, 2018, 6, 18344–18356 RSC.
- Y. Tsuboi, S. Ura, K. Takahiro, T. Henmi, A. Okada, T. Wakasugi and K. Kadono, J. Asian Ceram. Soc., 2017, 5, 341–349 CrossRef.
- Aluminium oxide Al2O3 – Thermal diffusivity/conductivity, https://www.linseis.com/en/applications/ceramics-and-glass-industry/lfa-1000-aluminum-oxide-al2o3-thermal-diffusivity-conductivity/.
- Graphite – Thermal conductivity/thermal diffusivity, https://www.linseis.com/en/applications/research-development-academia/lfa-1000-graphite-thermal-conductivity/.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57(4), 603–619 CrossRef CAS.
- G. Viera, M. Mikikian, E. Bertran, P. R. I. Cabarrocas and L. Boufendi, J. Appl. Phys., 2002, 92, 4684–4694 CrossRef CAS.
- T. D. Hatchard and J. R. Dahn, J. Electrochem. Soc., 2004, 151, A838 CrossRef CAS.
- D. B. Mawhinney, J. A. Glass and J. T. Yates, J. Phys. Chem. B, 1997, 101, 1202–1206 CrossRef CAS.
- A. Beganskienė, V. Sirutkaitis, R. Juškėnas, A. Kareiva and M. Kurtinaitienė, Mater. Sci., 2004, 10(4), 287–290 Search PubMed.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies, John Wiley & Sons, Ltd, 3rd edn, 2001 Search PubMed.
- I. S. Curtis, R. J. Wills and M. Dasog, Nanoscale, 2021, 13, 2685–2692 RSC.
- C. Meier, S. Lüttjohann, V. G. Kravets, H. Nienhaus, A. Lorke and H. Wiggers, Phys. E, 2006, 32, 155–158 CrossRef CAS.
- V. A. Volodin and D. I. Koshelev, J. Raman Spectrosc., 2013, 44, 1760–1764 CrossRef CAS.
- Y. Huang, Y. Zeng, Z. Zhang, X. Guo, M. Liao, C. Shou, S. Huang, B. Yan and J. Ye, Sol. Energy Mater. Sol. Cells, 2019, 192, 154–160 CrossRef CAS.
- M. Ko, S. Chae and J. Cho, ChemElectroChem, 2015, 2, 1645–1651 CrossRef CAS PubMed.
- S. Xun, X. Song, L. Wang, M. E. Grass, Z. Liu, V. S. Battaglia and G. Liu, J. Electrochem. Soc., 2011, 158, A1260 CrossRef CAS.
- Z. Gu, X. Xia, C. Liu, X. Hu, Y. Chen, Z. Wang and H. Liu, J. Alloys Compd., 2018, 757, 265–272 CrossRef CAS.
- P. Li, H. Kim, S. T. Myung and Y. K. Sun, Energy Storage Mater., 2021, 35, 550–576 CrossRef.
- S. He, S. Huang, S. Wang, I. Mizota, X. Liu and X. Hou, Energy Fuels, 2021, 35, 944–964 CrossRef CAS.
- J. Moon, H. C. Lee, H. Jung, S. Wakita, S. Cho, J. Yoon, J. Lee, A. Ueda, B. Choi, S. Lee, K. Ito, Y. Kubo, A. C. Lim, J. G. Seo, J. Yoo, S. Lee, Y. Ham, W. Baek, Y. G. Ryu and I. T. Han, Nat. Commun., 2021, 12, 2714 CrossRef CAS PubMed.
- R. Schmuch, R. Wagner, G. Hörpel, T. Placke and M. Winter, Nat. Energy, 2018, 3, 267–278 CrossRef CAS.
- P. F. Cao, M. Naguib, Z. Du, E. Stacy, B. Li, T. Hong, K. Xing, D. N. Voylov, J. Li, D. L. Wood, A. P. Sokolov, J. Nanda and T. Saito, ACS Appl. Mater. Interfaces, 2018, 10, 3470–3478 CrossRef CAS PubMed.
- B. Hu, S. Jiang, I. A. Shkrob, J. Zhang, S. E. Trask, B. J. Polzin, A. Jansen, W. Chen, C. Liao, Z. Zhang and L. Zhang, J. Power Sources, 2019, 416, 125–131 CrossRef CAS.
- J. Xu, X. Wang, N. Yuan, B. Hu, J. Ding and S. Ge, J. Power Sources, 2019, 430, 74–79 CrossRef CAS.
- Z. Yi, W. Wang, Y. Qian, X. Liu, N. Lin and Y. Qian, ACS Sustainable Chem. Eng., 2018, 6, 14230–14238 CrossRef CAS.
- L. Zhang, C. Wang, Y. Dou, N. Cheng, D. Cui, Y. Du, P. Liu, M. Al-Mamun, S. Zhang and H. Zhao, Angew. Chem., Int. Ed., 2019, 58, 8824–8828 CrossRef CAS PubMed.
- P. Li, J.-Y. Hwang and Y.-K. Sun, ACS Nano, 2019, 13, 2624–2633 CAS.
- L. J. Krause, T. Brandt, V. L. Chevrier and L. D. Jensen, J. Electrochem. Soc., 2017, 164, A2277–A2282 CrossRef CAS.
- A. Missyul, I. Bolshakov and R. Shpanchenko, Powder Diffr., 2017, 32, S56–S62 CrossRef CAS.
- L. Luo, J. Wu, J. Luo, J. Huang and V. P. Dravid, Sci. Rep., 2014, 4, 1–7 Search PubMed.
- M. Wetjen, D. Pritzl, R. Jung, S. Solchenbach, R. Ghadimi and H. A. Gasteiger, J. Electrochem. Soc., 2017, 164, A2840–A2852 CrossRef CAS.
- T. Yoon, C. C. Nguyen, D. M. Seo and B. L. Lucht, J. Electrochem. Soc., 2015, 162, A2325–A2330 CrossRef CAS.
- Y. W. Cheng, C. H. Chen, S. A. Wang, Y. C. Li, B. L. Peng, J. H. Huang and C. P. Liu, Nano Energy, 2022, 102, 107688 CrossRef CAS.
- S. Bai, W. Bao, K. Qian, W. Li, B. Sayahpour, B. Sreenarayanan, D. Tan, S. Ham and Y. S. Meng, Adv. Energy Mater., 2023, 13, 2301041 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Thermoreduction setups; structural and physical characteristics of Si-based materials; effects of HF etching; cyclic voltammetry results and the corresponding model; charge–discharge profiles and differential capacity plots of Si-graphite anodes used in this study; electrochemical impedance spectroscopy and analysis; electrochemical cycling of SiNQ-graphite anodes under fast charging and discharging conditions; optimization of the pre-lithiation process; comparisons of the electrochemical performance of Si-graphite electrodes from aqueous slurries. See DOI: https://doi.org/10.1039/d4gc00498a |
| ‡ Author contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |