
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent-driven isomerization of muconates in DMSO: reaction mechanism and process sustainability†
Ibrahim
Khalil
 *a,
Fatima
Rammal
a,
Lisa
De Vriendt
a,
An Sofie
Narmon
a,
Bert F.
Sels
a,
Sebastian
Meier
b and
Michiel
Dusselier
*a
*a,
Fatima
Rammal
a,
Lisa
De Vriendt
a,
An Sofie
Narmon
a,
Bert F.
Sels
a,
Sebastian
Meier
b and
Michiel
Dusselier
*a
aCenter for Sustainable Catalysis and Engineering (CSCE), KU Leuven, Celestijnenlaan 200F, B-3001 Leuven, Belgium. E-mail: khalil.ibrahim@kuleuven.be; michiel.dusselier@kuleuven.be
bDepartment of Chemistry, Technical University of Denmark, Kemitorvet 207, 2800 Kgs. Lyngby, Denmark
First published on 26th March 2024
Abstract
The production of renewable chemicals and monomers is fundamental for transitioning to a future circular economy. Currently, cis,cis-muconic acid (ccMA) is a bio-sourced platform chemical with great potential for added-value chemicals, monomers, and specialty polymers. Among the three isomers, the trans,trans (tt-isomer) stands out due to its reactivity for polymerization and unique ability as a substrate for the Diels–Alder cycloaddition reaction. Whereas earlier research has focused on producing this isomer, the most promising solvent-driven isomerization in DMSO-containing water yields moderate ttMA due to a competitive ring-closing lactonization reaction, especially in highly concentrated systems. This work highlights the unique ability of DMSO, among several other solvents, to produce ttMA. In addition, we report the effect of the acidity of the initial MA concentration and the amount of water on the lactonization reaction. Control of reaction conditions and use of muconates (diethyl muconates = DEM) countered the competitive lactonization, reaching >90% tt-isomer selectivity. The involvement of water and DMSO in the isomerization mechanism was investigated in detail by probing the reaction mechanism with in situ NMR. Identifying the reaction products and several intermediates led us to propose a plausible mechanism. Based on this knowledge, condition optimization led to a significant thirty-fold ttDEM productivity improvement, viz. from 10 to 328 mM h−1. The DEM can be isolated almost quantitatively from the DMSO solvent system by extraction.
Introduction
The migration from a petroleum-based chemical industry can be assisted by the development of processes that produce and convert platform molecules derived from renewable carbon sources.1–5 Biomass serves as one of the most promising renewable sources for high-value chemicals.6–10 An example of such a high-value chemical is 2,4-hexenedioic acid, known as muconic acid (MA).11,12 Due to its conjugated double bonds and carboxyl groups at both ends, MA serves as a precursor for valuable monomers, such as adipic acid (AA), ε-caprolactam, and terephthalic acid (TPA).13–15 It is also used to prepare the novel hexenedioic acid (HDA) and cyclohexane-1,4-dicarboxylic acid (CHDA) monomers.15–17 These monomers are mainly used for the production of polyamides (PA), polyesters, and polyurethanes.18–20 Note that MA itself can be used as a monomer for homo- and co-polymerization reactions.15,21MA is present in three geometric isomeric forms, two of which can be obtained from bio-based sources: cis,cis-MA and cis,trans-MA (ccMA and ctMA).17,22–25ctMA can also be obtained from the spontaneous isomerization of ccMA under acidic conditions.15,26 However, the preferred isomer for polymerization and certain chemical transformations (e.g., Diels–Alder cycloaddition), trans,trans-MA (ttMA), can only be obtained chemically by synthesis or isomerization.12,26,27 The most prominent catalytic isomerization protocol for the production of ttMA and muconates (muconic esters) is the homogenous I2 catalyzed isomerization,28,29 besides two heterogeneously catalyzed methods using 5%Pd/C and Ru-hydride/zeolite (0.2%Ru) have been developed.30,31 Research on non-catalytic isomerization routes showed that dimethyl sulfoxide (DMSO) could promote the formation of ttMA from ctMA, albeit at moderate yield and in low concentrated solutions.26 Other solvents, such as triethylamine, toluene, acetone, acetonitrile, 2-propanol, methanol, hexanol, tetrahydrofuran, and ethyl acetate, showed no isomerization activity. The main side product observed in all solvents was mono-muconolactone (Mlac), as a result of a competitive irreversible intramolecular lactonization, in essence, a ring closing of MA.15,26,32 This reaction can be reduced, providing higher ttMA yields, by controlling the water amounts in DMSO.32 The role of water was only effective until a concentration of 0.5 M, yielding 39% ttMA at 50% ctMA conversion. Above this concentration, the DMSO-driven system suffers from low yields and selectivity, where at 2 M concentration, ttMA was hardly formed with <1% yield at 50% conversion.32 Besides the moderate results, the mechanism of DMSO-driven isomerization is not fully understood, therefore missing handles to improve ttMA yields in concentrated solutions.
In this work, we first investigated the isomerization of ccMA into ctMA in various solvents (including DMSO), where the lactones formation was also followed experimentally as a function of the concentration of MA and the used solvent. We effectively countered the competitive lactonization by protecting the carboxylic groups by esterification. Additionally, the dynamics of isomerization of both MA and muconates were followed with in situ NMR measurements. Although we find that the role of water was less pronounced when using muconates, its presence was always required for high tt-isomer production. Further, careful identification and real-time tracking of intermediates and their byproducts clarified the role of DMSO in the isomerization mechanism. This insight led us to achieve outstanding process isomerization efficiency, outperforming known catalyst-free MA isomerizations. As opposed to current systems, we can isomerize >0.5 M muconates solutions in high productivity and selectivity > 95%. The recovery of the DEM isomers from DMSO was successfully performed, with recovery levels surpassing 90%.
Experimental
Materials
cis,cis-Muconic acid (ccMA) was purchased from Fluorochem with a purity >99%. Ethanol (99.8%) and anhydrous magnesium sulfate MgSO4 were supplied by Fisher. Ethyl acetate (99.5%), DMSO (99.5%), acetonitrile (99.9%), N,N-dimethylformamide (DMF) (>99%), acetone (>99%), toluene (99.5%), and 96% sulfuric acid in water H2SO4/H2O were supplied by Acros. Sodium chloride (>99.5%) was supplied by Carl Roth. Anhydrous DMSO (99.9%), triethylamine (>99.5%), and the 9 mL glass pressure tubes were supplied from Merck. The length of the glass pressure tubes was optionally reduced (10.2 to around 6 cm) at a glassblower workshop for performing low-volume reactions (1–2 mL).Preparation of ctMA and ctDEM
The reaction conditions for the isomerization (towards tt-isomer and Mlac products)
The formation of tt-isomer from the ct-form was performed in 9 mL pressure tubes (or modified pressure tubes with a total volume of 6 mL). Solutions of 3–5 mL of different solvents (H2O, ethanol, acetonitrile, DMSO, DMF, acetone, triethylamine, or toluene) containing ccMA, ctMA, or ctDEM in a concentration range from 30 to 3200 mM were prepared. Optionally, other solvents and substrates were added to the mixture such as H2O, DMSO2, sulfolane, and methylphenyl sulfoxide. For the experiments with dry DMSO, a commercial anhydrous DMSO was used, where the liquid was extracted under a flow of argon. After filling all the components, the glass pressure tube was sealed at room temperature, and reactions were conducted at the desired temperature (a range of 80 to 150 °C was investigated) in an oil bath. The conversion and product distribution were determined using 1H-NMR and DMSO2 as an external standard for the experiments studying the isomerization of MA. For analyzing the isomerization results of DEM, gas phase chromatography (GC) was used with n-heptane as an external standard.Recovery of ctDEM and ttDEM from DMSO
The mixture containing DEM isomers (ctDEM and ttDEM) was extracted from DMSO by first dissolving 10 mL of the reaction mixture in 10 mL of distilled water. 5 mL of EtOAc was then added, and ttDEM and ctDEM were extracted after vigorous stirring, followed by degassing.33 This step was repeated three times to collect 15 mL EtOAc phase, which was then washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The recovery level r was calculated as:| r = [DEM]EtOAc/[DEM]initial. |
Analysis methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The gas mixture was further driven to the column (initially at 80 °C) with a flow of 2.6 mL min−1. After 5 min at 80 °C, the temperature was increased to 200 °C (10 °C min−1) and held for 3 min, and then to 280 °C (20 °C min−1) and held there for 6 min. The FID detector used was maintained at 320 °C. n-Heptane was used as an external standard. The calibration curve was derived, yielding a response ratio for ctDEM of ActDEM/Aheptane = 1.408 [ctDEM]/[heptane]. The same response factor was used for the quantification of ttDEM.
:1. The gas mixture was further driven to the column (initially at 80 °C) with a flow of 2.6 mL min−1. After 5 min at 80 °C, the temperature was increased to 200 °C (10 °C min−1) and held for 3 min, and then to 280 °C (20 °C min−1) and held there for 6 min. The FID detector used was maintained at 320 °C. n-Heptane was used as an external standard. The calibration curve was derived, yielding a response ratio for ctDEM of ActDEM/Aheptane = 1.408 [ctDEM]/[heptane]. The same response factor was used for the quantification of ttDEM.
All the quantified products were identified based on retention times, using pure components or mixtures with various ratios as references, with the relevant retention times being n-heptane (∼1.5 min)/ttDEM (∼15.0 min)/ctDEM (∼15.3 min).
Time series of 1H-NMR spectra (using the zgcppr pulse sequence) were acquired by accumulating 64 transients with an interscan relaxation delay of 3 seconds and sampling the FID for 1.7 s (16384 complex data points), yielding a time resolution of 5 min per spectrum. All real-time kinetic NMR tracking was implemented in the form of pseudo-2D experiments. With these experiments, the conversion of MA acids and esters was tracked at the indicated temperatures, and approximately 250 time points were acquired in the pseudo-2D experiments. The kinetic data were processed using zero filling to 32768 complex data points and an exponential window function with 3 Hz line broadening (LB) in Bruker Topspin 4.1.3. Reaction progress data from the pseudo-2D experiments were integrated in the same software. Signal areas were plotted using software pro Fit 7 (QuantumSoft).
Products in post-reaction mixtures were identified at 25 °C using a suite of homo- and heteronuclear 2D NMR assignment spectra including 1H–1H COSY, 1H–1H TOCSY, 1H–13C HSQC, 1H–13C HSQC-TOCSY and 1H–13C HMBC spectra to annotate the assignments, provided herein.
Assignment spectra were acquired using Topspin 3.5 pl6. These spectra were processed with zero filling to at least twice the number of acquired complex data points in Bruker Topspin 4.1.3, and analyzed in the same software.
Results and discussion
Isomerization vs. lactonization of muconic acid: solvent dependency
The first isomerization of ccMA to ctMA is known to occur under conditions as mild as 25 °C in water. However, this isomerization step is pH-dependent and should preferentially be performed at a pH below 4. The isomerization is also temperature-dependent, and can be accelerated from a few days to less than an hour by increasing the temperature from 25 to 75 °C.18,26,34,35 Fig. S1† summarizes the isomerization products obtained over the course of 4 hours for a solution of 30 mM ccMA in water at 75 °C. The complete conversion of ccMA occurs within 40 minutes, leading to ctMA initially, but just before ccMA conversion is complete, Mlac starts to be generated due to the intramolecular lactonization of the ctMA intermediate. This reaction is known to be assisted by water, as previously reported by Tessonnier et al. (Fig. S2 and S3†).26 To investigate the solvent effect on the isomerization of ccMA and the competitive lactonization towards Mlac, we studied the reaction in a selection of solvents with different polarity and proticity, namely water, ethanol, acetone, toluene, acetonitrile (MeCN), N,N-dimethylformamide (DMF), triethylamine (Et3N), and dimethyl sulfoxide (DMSO) (Fig. S4†). The reactions were conducted at 90 °C for 4 hours (30 mM ccMA), and the results are presented in Fig. 1.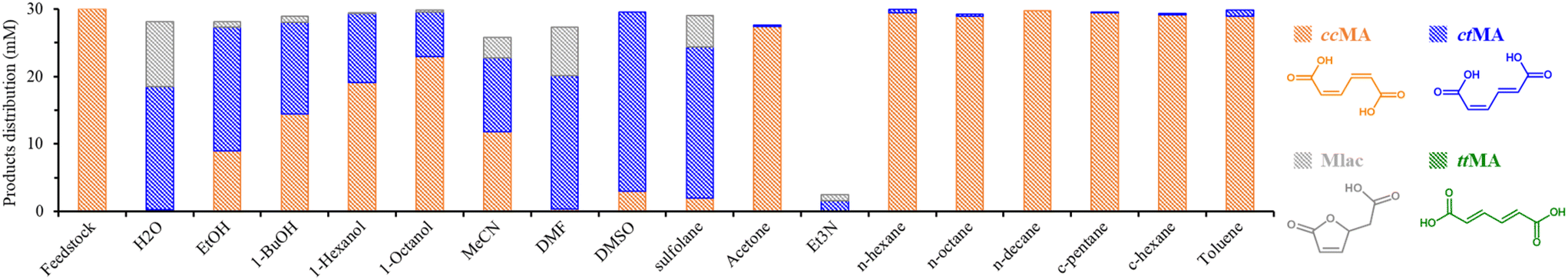 | ||
| Fig. 1 The product distribution (in mM) for the conversion of 30 mM ccMA into ctMA and mono-muconolactone (Mlac) in various solvents (3 mL) at 90 °C for 4 hours. The reaction was performed in a glass pressure tube sealed under static conditions at room temperature. Abbreviations for solvents: EtOH = ethanol, 1-BuOH = 1-butanol, MeCN = acetonitrile, DMF = N,N-dimethylformamide, DMSO = dimethyl sulfoxide, Et3N = triethylamine, c-pentane = cyclopentane, and c-hexane = cyclohexane. The product distribution was determined using 1H-NMR in DMSO-d6 using DMSO2 as an external standard. | ||
A good carbon balance was observed overall (80–98%) except for Et3N, in which the degradation of MA was noticed. The degradation may be linked to the basic nature of Et3N since, correspondingly, partial loss in the carbon balance was also noticed when using MeCN, which shares basic characteristics. In addition to the basicity, the polarity of the solvents seems to influence the conversion of ccMA, since minimal conversion (1 to 5%) was noticed in acetone, cycloalkanes, alkanes, and toluene (least polar solvents in our range) in comparison to complete conversion in H2O (polar) and DMF (basic nature and semi-polar). We note that the solubility of ccMA in the least polar solvents was very limited. EtOH and DMSO afforded partial conversion of ccMA with 70% and 90%, respectively. For EtOH, the high conversion of ccMA can be due to its protic properties next to its polarity. Interestingly, when testing alcohols with longer alkyl chains (1-butanol, 1-hexanol, and 1-octanol), we notice a decrease in the ccMA isomerization, which is due to the decrease in the proticity of the alcohols (less acidic proton) when the size of the alkyl group increases. For DMSO, a higher conversion of ccMA was observed besides its lower polarity and aprotic properties. Similar activity was obtained with sulfolane solvent (owing sulfone groups). From the solvent screening, it is clear that solvent properties play an essential but multifaceted role in assisting the isomerization, given its effect cannot be ascribed to only basicity, polarity, or proticity.
Under the studied conditions, the formation of ttMA was not noticed, while Mlac was mainly formed in solvents promoting the first isomerization, except for DMSO, albeit in very little amounts in ethanol. These results suggest that lactonization becomes more favorable than the second isomerization, viz. ctMA to ttMA, in these solvents.15 Due to their high ctMA yields in the first screening, water, ethanol, DMSO, and DMF solvents were selected to further investigate the tendency for lactonization vs. isomerization.
The parallel (Mlac ← ctMA → ttMA) reaction network in the selected solvents was investigated kinetically into more detail by varying the initial concentration of ctMA from 30 to 300 mM. ctMA was used as feedstock instead of ccMA to eliminate complexity due to the contribution of the first isomerization step. Interestingly, the formation of ttMA was not noticed in any of the conducted reactions, instead, Mlac was the only observed product (Fig. 2).
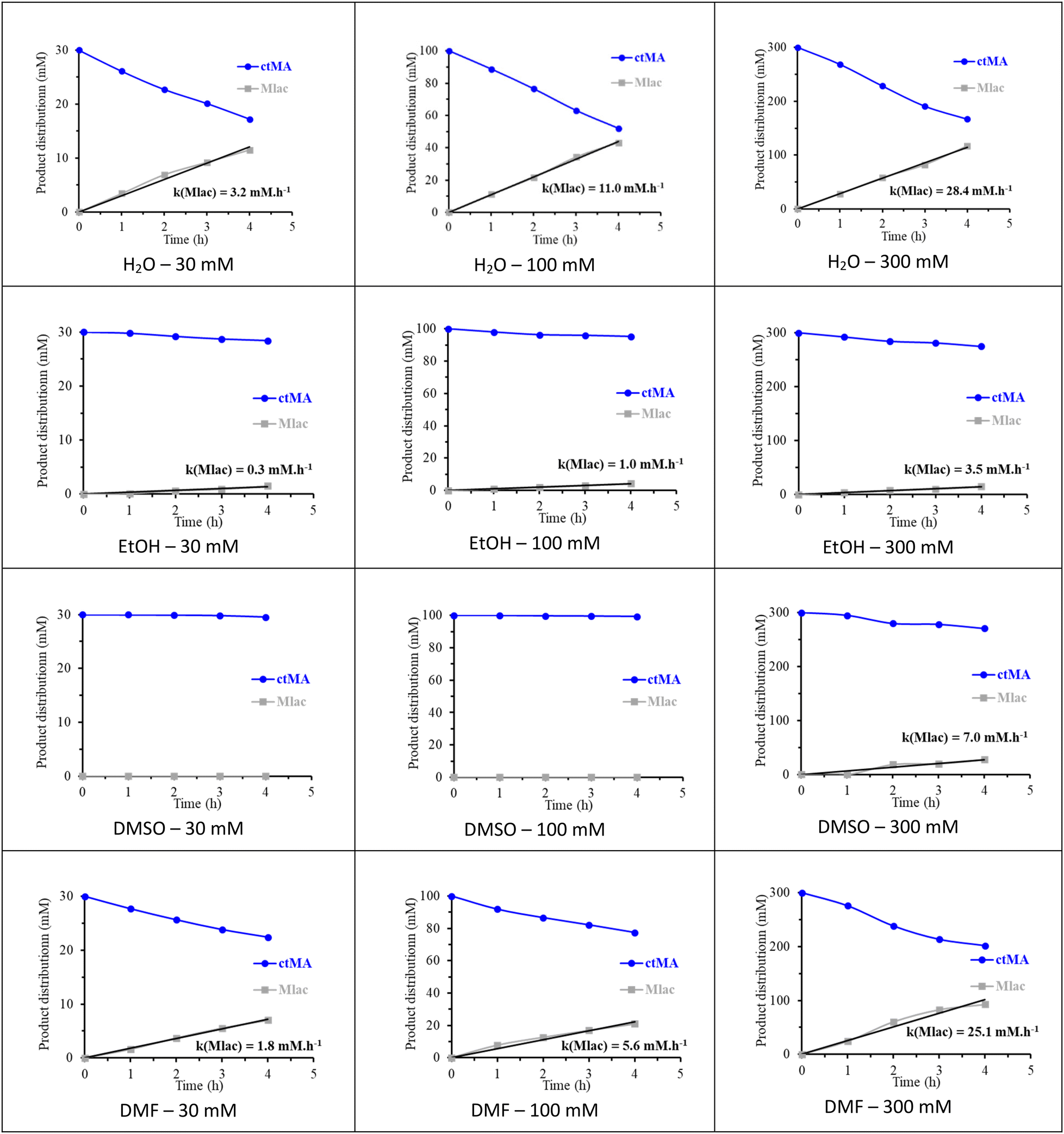 | ||
| Fig. 2 The time profile of the conversion of ctMA (30, 100, and 300 mM) solutions in H2O, EtOH, DMSO, and DMF solvents at 90 °C. The amount of remaining ctMA and produced Mlac is reported in mM. The formation of ttMA was- not noticed. The product distribution was determined using 1H-NMR in DMSO-d6 using DMSO2 as an external standard. | ||
At 30 mM concentration, the amount of produced Mlac after 4 hours was higher in H2O and DMF (11.5 mM and 7.0 mM, respectively) in comparison to EtOH (1.5 mM) and DMSO (no formation of Mlac). The possible involvement of acidic hydrogen from the solvent in favoring the formation of Mlac from ctMA was proposed by Tessonnier et al. according to the in water reported mechanism (Fig. S3†).26 We believe that the basic nature of DMF can assist the formation of Mlac according to the same mechanism. Increasing the initial concentration of ctMA from 30 mM to 100 mM was followed by an increase in the formation rate of Mlac in H2O (from 3.2 to 11.0 mM h−1), DMF (from 1.8 to 5.6 mM h−1), and EtOH (from 0.3 to 1.0 mM h−1), pointing to the occurrence of an autocatalyzed lactonization mechanism of ctMA, as presented in Fig. S5.†32 Only in DMSO, Mlac was not formed in the 100 mM solution, which can be explained by the effect of DMSO in increasing the pKa of carboxylic acid groups, disfavoring Brønsted acid catalyzed mechanisms such as the acid-catalyzed intramolecular lactonization of ctMA in this case.36 In addition, DMSO can stabilize ctMA by interacting with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, as shown later in the proposed reaction mechanism (Fig. 5B), which will likely lead to less Mlac formation. A further increase in the initial concentration of ctMA to 300 mM increases the formation rate of Mlac even more, as 28.4, 25.1, and 3.4 mM h−1 were measured in H2O, DMF, and EtOH, respectively. At this concentration, around 9% of Mlac was formed after 4 hours in DMSO (7.0 mM h−1). Enhanced Mlac formation at high concentrations of MA confirms that lactonization follows an acid-catalyzed pathway with MA, both reactant and catalyst, as reported in literature and illustrated in Fig. S5.†15,32 Interestingly, by following the evolution of the Mlac productivity as a function of the initial concentration of ctMA, one may conclude a positive order of the lactonization reaction in ctMA (Fig. S6†). Remarkably, Mlac formation was only visible in DMSO in the 300 mM ctMA solution.
C bonds, as shown later in the proposed reaction mechanism (Fig. 5B), which will likely lead to less Mlac formation. A further increase in the initial concentration of ctMA to 300 mM increases the formation rate of Mlac even more, as 28.4, 25.1, and 3.4 mM h−1 were measured in H2O, DMF, and EtOH, respectively. At this concentration, around 9% of Mlac was formed after 4 hours in DMSO (7.0 mM h−1). Enhanced Mlac formation at high concentrations of MA confirms that lactonization follows an acid-catalyzed pathway with MA, both reactant and catalyst, as reported in literature and illustrated in Fig. S5.†15,32 Interestingly, by following the evolution of the Mlac productivity as a function of the initial concentration of ctMA, one may conclude a positive order of the lactonization reaction in ctMA (Fig. S6†). Remarkably, Mlac formation was only visible in DMSO in the 300 mM ctMA solution.
Upon an increase of the reaction temperature to 120 °C, the production of Mlac increased for all solvents (Fig. 3, 100 mM solution). The maximal Mlac yield was again obtained in H2O and DMF, at 73 and 52 mM, respectively. The formation of di-muconolactone (Dlac) was also noticed in H2O at 120 °C. Less Mlac formation was found in EtOH, while ttMA was only formed in DMSO, with a ttMA to Mlac ratio of 6.7. The absence of ttMA at 90 °C can be explained by the relatively short reaction time (4 hours), where Tessonnier et al. reported the need for around 50–60 hours to produce 20% at 87 °C, respectively.32 This unique ability shows that DMSO selectively promotes the ctMA to ttMA isomerization, whereas other solvents (H2O and DMF) are more efficient for the primary ccMA to ctMA isomerization.
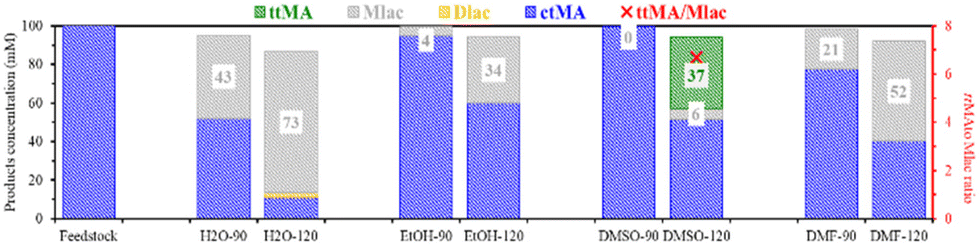 | ||
| Fig. 3 The temperature effect on the product distribution (in mM) of the conversion of 100 mM ctMA in H2O, EtOH, DMSO, and DMF at 90 and 120 °C after 4 hours. The product distribution was analyzed with quantitative 1H-NMR analysis in DMSO-d6 using DMSO2 as an external standard. | ||
In conclusion, this first screening clarified that the competitive lactonization reaction is boosted by several factors, including temperature, initial MA concentration (or acidity), and the solvent properties. The observations align with earlier reports, studying isomerization/lactonization of MA.15,26,28 The second isomerization forming ttMA is kinetically hindered in all solvents, except in DMSO. The role of DMSO in ttMA formation may be ascribed either to an indirect (stabilization) effect, such as the stabilization of MA to prohibit the acid-catalyzed intramolecular lactonization, or to its direct involvement in the reaction mechanism. However, this role is limited to low substrate concentration, given that Mlac formation is higher for more concentrated MA solutions.
Role of water on the reaction pathways
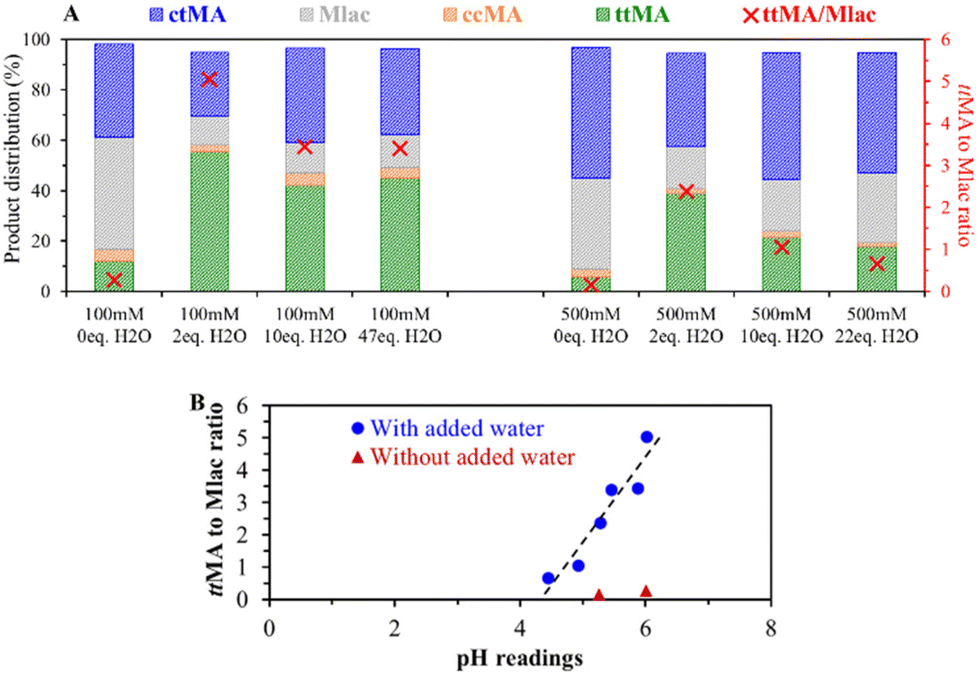 | ||
| Fig. 4 (A) The product distribution of the isomerization of 100 and 500 mM ctMA in DMSO as a function of the amount of added water equivalents to MA. The reactions were performed in glass pressure tubes at 150 °C for 1 hour. The values of the ttMA to Mlac ratio are plotted on the right (red) y-axis. (B) Effect of the initial pH reading value on the obtained ttMA to Mlac molar ratio at the end of the isomerization reaction. The points in blue correspond to 6 reactions performed at initial 100 and 500 mM concentrations with added H2O equivalents (see Fig. 4A). The red triangles show experiments without added water (0 eq. H2O) at 100 and 500 mM initial concentration of ctMA. | ||
Mlac was the major side product and the only one quantified in this study due to its predominance over other side products. However, other side products still arose at high enough concentrations to allow their identification and they were formed in competition to isomerization. Due to their minor amounts, these products were not identified and are represented in the graphs as the loss in carbon balance (product distribution not reaching 100%). Their chemical structures, along with their chemical shifts for 1H-NMR and 13C-NMR, are shown in Fig. S8.† Beyond Mlac, the di-muconolactone form of MA (Dlac) was analyzed. In addition, a hydroxylated version of Mlac and some aldehyde species were present, likely a result of redox chemistry on the MA chain, as shown in Fig. S8.†
In our attempts to reduce the acid-catalyzed side reactions, including lactonization, we studied the effect of adding a base. Controlled additions of base, such as triethylamine, to reach pH around 6 (MA to triethylamine molar ratio of 17), lead to various degradation products. Similar results were obtained after the addition of alkaline salts, e.g., KHCO3. Also, no beneficial effect was noticed with weak organic bases such as pyridine and potassium thiocyanate, in contrast, their presence rather slows the ctMA to ttMA isomerization. Thus, higher pH does not necessarily recover a lost isomerization activity. We also noted that the trend between pH and Mlac formation does not extrapolate to the tests without added water (red triangles in Fig. 4B) and thus fails to explain the low ttMA to Mlac ratio in dry DMSO. The low ttMA to Mlac ratio in the absence of water, therefore, suggests the direct involvement of water in the isomerization.
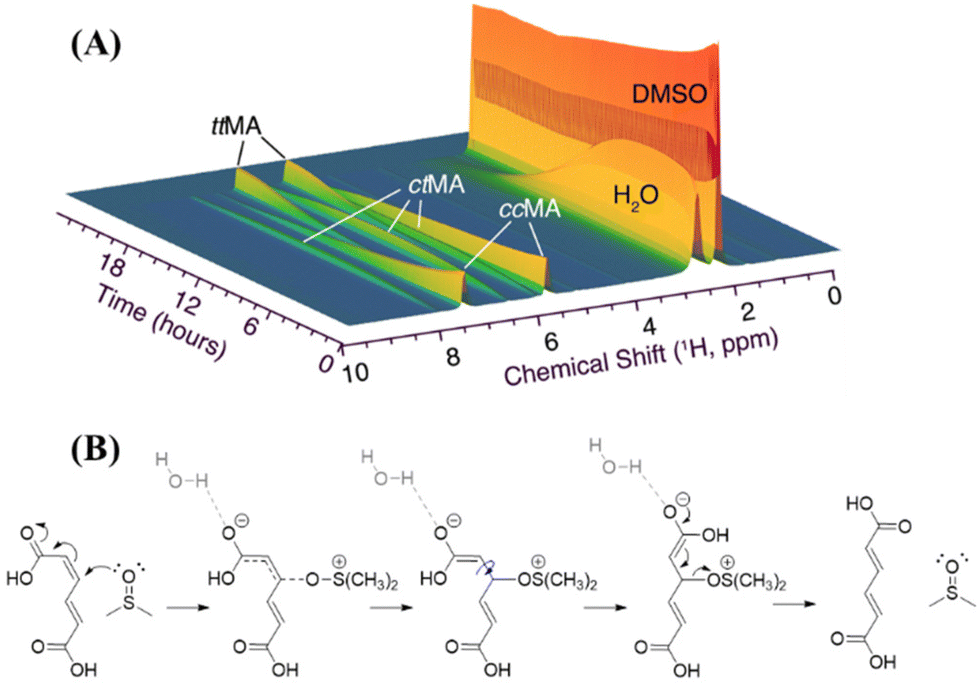 | ||
| Fig. 5 (A) The evolution of the isomerization products of ccMA in DMSO as well as the water signal from in situ monitored 1H-NMR measurements. (B) Plausible transient catalytic attack of DMSO on the CC of MA followed by the rearrangement of the conjugated CO functionality. | ||
In different contexts, a mechanism for the isomerization of double bonds, that are conjugated with carboxylic acid groups, and involves a Michael-type addition of a nucleophile has been proposed.37,38 While elaborating on this mechanism, we propose a plausible mechanism for the isomerization of MA in DMSO (Fig. 5B). This proposal is based on the unique molecular geometry of DMSO, which is characterized by a trigonal pyramidal-like structure with a highly polarized S–O single bond, in which the oxygen carries a partial negative and S a partial positive charge (Fig. S9†).39 Therefore, DMSO can initiate a transient nucleophilic Michael addition via its nucleophilic oxygen atom on the CC of MA, forming the corresponding enolate intermediate. Interestingly, in the conversion of ccMA to ttMA, some olefinic intermediates containing an alcoholic group next to the double bonds were observed in the real-time NMR spectra after the accumulation of ctMA. The signals of this intermediate in real-time NMR are marked with asterisks in Fig. S10.† Given the signals of the olefinic and sp3 hybridized alcoholic groups evolve synchronically, they belong to the same intermediate molecule. This intermediate may be attributed to an adduct of a nucleophilic solvent addition on a double bond in MA. This enolate is stabilized by the presence of water. After the rotation of the C–C bond, ttMA is generated after the elimination of DMSO. The presence of the hydroxylated Mlac (Fig. S8†) indicates that the coordination of DMSO and the further hydrolysis of this coordination can occur. Among the side products formed in the reaction, enol forms that are stabilized through cyclization were identified. This indicates that DMSO/H2O mixtures elicit a combined effect by increasing the pKa value of MA while reducing lactonization and affording transient nucleophilic addition while stabilizing enol forms during isomerization.
Towards a selective and highly productive isomerization method
The rate of the isomerization for both ctMA and ctDEM was first followed in DMSO at 120 °C, as shown in Fig. 6A and B. The two ct-isomers seem to react very analogously, judging from the reaction progress curves. This observation suggests a minor influence of deprotonation of the carboxylic groups in ct to tt isomerization, which is in line with our proposed mechanism (Fig. 5B). Importantly, as lactonization was successfully counteracted with DEM, high selectivity to its tt-isomer was achieved, e.g., from 88% (ctMA → ttMA) to 100% (ctDEM → ttDEM) as shown in Fig. 5. In situ1H-NMR in DMSO-d6 at 100 °C showed similar findings (Fig. S12†). The isomerization of ctDEM was further tested in the initial selection of solvents, including EtOH, DMSO, and DMF, but not H2O due to the insolubility of DEM in H2O. Even though the use of esters avoided Mlac formation in all solvents, ttDEM was only observed in DMSO, confirming its direct involvement in the isomerization mechanism.
 | ||
| Fig. 6 The time profile of the isomerization of 100 mM ctMA and ctDEM in DMSO (5 mL) and 2eq. H2O at 120 °C. The rate plots for the conversion of ct-isomer and the formation of the tt-isomer are obtained for the slope of the data points plotted between 0 and 4 hours. The reactions were performed in glass pressure tubes sealed under static conditions. The product distribution was determined by 1H-NMR for the reaction in A and by GC for the reaction in B. | ||
At 100 mM ctDEM, the ttDEM yield increases rapidly with reaction temperature, from 5.8% in 24 hours at 80 °C to 76.4% in an hour at 150 °C (Table S2 entries 1–4† and Fig. 7), while maintaining a high selectivity towards ttDEM (>5%). All reactions were performed in the presence of 2 equivalents H2O relative to ctDEM in DMSO. Due to the instability of DMSO with possible decomposition at higher temperatures, the temperature range was limited to 150 °C. For the same concentration, the absence of water reduces the formed ttDEM to 18.3% (entry 5), while the presence of water between 2 and 20 equivalents left ttDEM yield unaltered (between 74% and 77%). Higher water equivalents retarded ttDEM formation to 47.1%, however, Mlac was not observed.
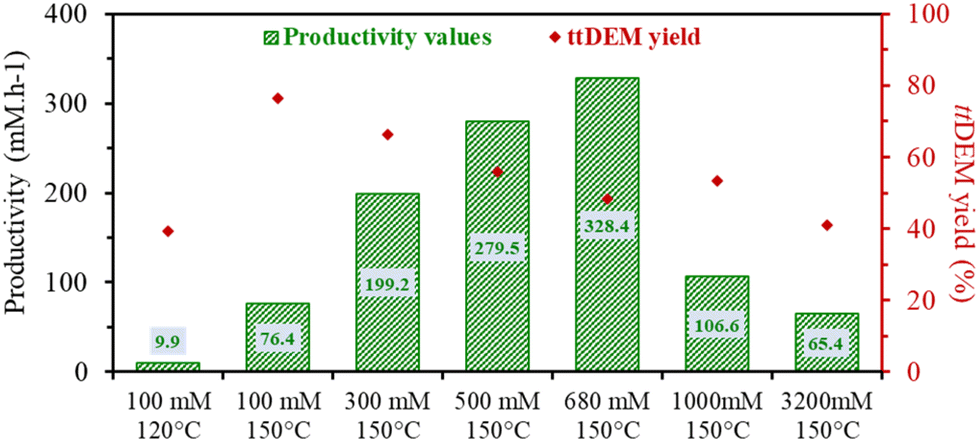 | ||
| Fig. 7 A comparison of the increase in the productivity values as a function of the optimized reaction conditions. The values were selected for systems producing 40% or more of ttDEM. The yields of ttDEM corresponding to every productivity value are shown in red (right-hand y-axis). | ||
The addition of other reagents was also tested. As mentioned before the addition of organic base or alkaline salt was not beneficial to the isomerization, while degradation of muconates occurred. Methylphenyl sulfoxide (MPSO) was used as a sulfoxide-type solvent and co-solvent with DMSO, but ttDEM formation was not influenced, even slowed down for high MPSO contents (Table S2 – entries 10–15†). The isomerization activity vanished in pure MPSO solvent. We believe that this lower isomerization capacity of MPSO may result on the one hand from the lower polarization of its oxygen atom in comparison to DMSO, and the steric effect on the other hand. The addition of sulfones into DMSO was further tested, specifically sulfolane and dimethyl sulfone (DMSO2). Sulfolane behaves like MPSO at low and high loadings (Table S2 – entries 16–20†). Interestingly, DMSO2 beneficially affected the production of ttDEM (Table S2 – entries 21–23†). For a better visualization of the effect of DMSO2, the isomerization was conducted at a lower temperature (120 °C) in the presence of 10 eq. DMSO2 (Fig. S14†), and the results were compared to the ones obtained in the absence of DMSO2 (Fig. 6B), showing a noticeable 10.3 to 13.7 mM h−1 increase in the ttDEM formation rate. Similar results were obtained with in situ NMR monitoring measurement for a solution of ccMA (Fig. S15†). The presence of DMSO2 was also found to increase the conversion rate of ccMA in the first place by a factor of 1.2–1.3 (from 37% to 45% at 2.5 hours). The formation rate of ttMA, obtained from the second isomerization, increased by three-fold at the 5 hours point.
Finally, the effect of increasing the initial concentration of ctDEM was tested (in presence of DMSO and 2 eq. H2O at 150 °C). The progressive increase in the concentration led to higher productivity values of ttDEM where a maximum of 328.4 mM h−1 is obtained for an initial concentration of 680 mM h−1 (Table S3 – entry 6† and Fig. 7). The productivity drops at higher concentrations. A total selectivity towards ttDEM was noted up to 100 mM concentrated solutions and starts to drop at higher concentrations to reach around 95% between 500 and 2000 mM (Table S3†). The decrease in selectivity is mostly due to the reactivity of the tt-isomer in oligomerization reactions. However, contrary to ctMA, the increase in the amount of ctDEM does not reduce pH; pH varied only from 10 to 9.7 in the presence of 1000 mM ctDEM and 2000 mM H2O. The non-acidic conditions enabled the tt-isomer formation, consistent with the absence of Mlac formation, even at concentrations as high as 1000 to 3200 mM (Table S3 – entries 7–9†).
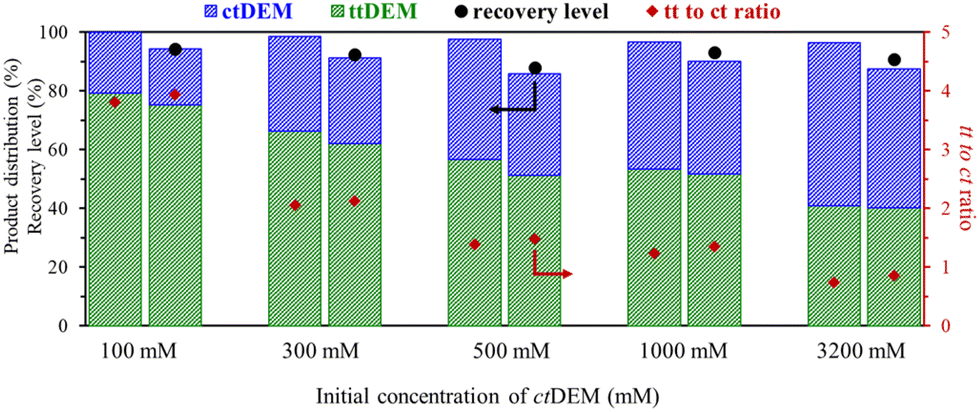 | ||
| Fig. 8 The product distribution after isomerization and after extraction from DMSO. The recovery level (r) after extraction is plotted in black circle on a scale of 0 to 100% (left-hand y-axis). The ratio of tt to ct isomers was also calculated before and after the recovery and is plotted in red diamonds on the right-hand y-axis. | ||
Sustainability of the isomerization methods: Green Chemistry principles
To assess the sustainable character of our isomerization method, we scored it along the principles of Green Chemistry and compared it to the best available isomerization practices in terms of tt-isomer productivity (Table S4†).40–42 The solvent-driven isomerization method was compared to the most common reported isomerization method using homogeneous iodine catalysts. Seven of the twelve principles of green chemistry were selected and compared in Table S5.† The remaining five principles (designing safer chemicals, design for degradation, real-time analysis for pollution prevention, reduce derivatives, and inherently safer chemistry for accident prevention) are not considered relevant at this status of research. The details of the comparison and considered parameters can be found in the ESI (pages S12 and S13†).The comparison between the isomerization methods revealed that iodine catalysis yields the highest ttMA (86%) and tt-muconates (95%) amounts. All the reactions show a high atom economy (AE), and they generally exhibit a low level of hazards, except for iodine catalysis, which uses toxic tetrahydrofuran (THF) and methanol (MeOH) as solvents. Also, iodine itself is a hazardous chemical and harmful to steel reactors. The need for the esterification of MA will affect both methods by increasing the waste production (E′-factor) and decreasing the atom economy (AE′), due to water formation from the esterification. However, this step was not considered due to its need either before or after the isomerization to access the high potential of these muconates, known for their high solubility and their use in polymerization as well as other chemical transformations (towards terephthalates and adipates).15,21 Thus, with this consideration, all methods show a moderate E-factor (between 14.1 and 20.6), except for the isomerization of MA in DMSO (40.8) and the isomerization of DMM with iodine (41.8) (Table S5†). The main contributor to the high E-factor of the MA-in-DMSO method is the low yield of produced ttMA (40%), while the low initial concentration of di-methyl muconate (DMM = 118 mM) was the main contributor for DMM with iodine (Table S4†). Our new method (DEM-in-DMSO) resulted in an E-factor of 20.6, which is nearly half the E-factor of the same method using MA (40.8). Since the amount of solvent accounts for a very large part of the waste production, increasing the initial concentration of DEM will allow reducing the E-factor. By doubling the concentration of DEM (500 to 1000 mM), the E-factor will drop to nearly 11.3 at a production yield of 53.3% of ttDEM. This drop in waste production clearly stresses the high potential of the solvent-driven isomerization of muconic esters.
Conclusions
This work presents interesting solvent effects on the selective thermal isomerization of MA isomers and their esters (muconates). To avoid the competitive acid-catalyzed lactonization, use of esters in this isomerization chemistry is highly advised to be able to reach the highest isomer products selectivity. Whereas the primary cc to ct isomerization is less sensitive to the solvent properties, the tt isomer formation is only successful in DMSO with minor equivalents of water. A mechanism that explains the beneficial role of DMSO is proposed based on dedicated intermediate and side product analysis with real-time NMR. This mechanism considers direct nucleophilic addition chemistry, that promotes the subsequent ct to tt isomerization, while water plays a crucial role in stabilizing the intermediate adduct. High 328 mM h−1tt isomer productivity with >95% selectivity can be achieved with this DMSO/H2O solvent isomerization method after optimization of temperature and reagent concentration based on these mechanistic understandings. After isomerization of the muconates, the product isomers can be recovered almost quantitatively by extraction in ethyl acetate. A Green Chemistry principles analysis reveals a few promising metrics of the DMSO/H2O method especially at high substrate/solvent ratios in highly concentrated reactions.Author contributions
Ibrahim Khalil: conceptualization, investigation, methodology, funding acquisition, writing, review and editing. Fatima Rammal: investigation, methodology, review and editing. Lisa De Vriendt: investigation, review and editing. An Sofie Narmon: investigation, writing, review and editing. Bert Sels: resources, review and editing. Sebastian Meier: investigation, methodology, funding acquisition, resources, writing, review and editing. Michiel Dusselier: supervision, conceptualization, visualization, resources, funding acquisition, review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
I. Khalil acknowledges the FWO foundation for postdoctoral grants number 1260321N and 12A3M24N. Part of this work was performed in the framework of the Catalisti cluster SBO project SPICY (“Sugar-based chemicals and Polymers through Innovative Chemocatalysis and engineered Yeast”), with the financial support of VLAIO (Flemish Agency for Innovation and Entrepreneurship) (HBC.2017.0597). S. M acknowledges funding by the Independent Research Fund Denmark (grants 0217-00277A and 2035-00119B). The 800 MHz NMR spectra were recorded at the NMR Center DTU, supported by the Villum Foundation.References
- J.-P. Lange, Nat. Catal., 2021, 4, 186–192 CrossRef CAS.
- C. Zhou, I. Khalil, F. Rammal, M. Dusselier, P. Kumar, M. Lacroix, E. Makshina, Y. Liao and B. F. Sels, ACS Catal., 2022, 12, 11485–11493 CrossRef CAS.
- I. Bechthold, K. Bretz, S. Kabasci, R. Kopitzky and A. Springer, Chem. Eng. Technol., 2008, 31, 647–654 CrossRef CAS.
- F. Brandi, I. Khalil, M. Antonietti and M. Al-Naji, ACS Sustainable Chem. Eng., 2021, 9, 927–935 CrossRef CAS.
- M. A. Shah, I. Khalil, S. Tallarico, T. Donckels, P. Eloy, D. P. Debecker, M. Oliverio and M. Dusselier, Dalton Trans., 2022, 51, 10773–10778 RSC.
- W. Schutyser, T. Renders, S. Van den Bosch, S.-F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- T. D. J. te Molder, S. R. A. Kersten, J.-P. Lange and M. P. Ruiz, Ind. Eng. Chem. Res., 2021, 60, 13515–13522 CrossRef CAS.
- F. Rammal, J. Matthijssen, I. Khalil, S. Calderon-Ardila, E. Makshina and B. F. Sels, ACS Catal., 2023, 13, 15811–15823 CrossRef CAS.
- Y. Liao, S.-F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. Van Aelst, M. Maesen, H. Matsushima, J. M. Thevelein, K. Van Acker, B. Lagrain, D. Verboekend and B. F. Sels, Science, 2020, 367, 1385–1390 CrossRef CAS PubMed.
- M. Dusselier, P. Van Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415 RSC.
- N. A. Rorrer, J. R. Dorgan, D. R. Vardon, C. R. Martinez, Y. Yang and G. T. Beckham, ACS Sustainable Chem. Eng., 2016, 4, 6867–6876 CrossRef CAS.
- N. Z. Xie, H. Liang, R. B. Huang and P. Xu, Biotechnol. Adv., 2014, 32, 615–622 CrossRef CAS PubMed.
- B. Briou, B. Améduri and B. Boutevin, Chem. Soc. Rev., 2021, 50, 11055–11097 RSC.
- B. H. Shanks and P. L. Keeling, Green Chem., 2017, 19, 3177–3185 RSC.
- I. Khalil, G. Quintens, T. Junkers and M. Dusselier, Green Chem., 2020, 22, 1517–1541 RSC.
- P. Carter, J. L. Trettin, T.-H. Lee, N. L. Chalgren, M. J. Forrester, B. H. Shanks, J.-P. Tessonnier and E. W. Cochran, J. Am. Chem. Soc., 2022, 22, 9548–9553 CrossRef PubMed.
- C. Ling, G. L. Peabody, D. Salvachúa, Y.-M. Kim, C. M. Kneucker, C. H. Calvey, M. A. Monninger, N. M. Munoz, B. C. Poirier, K. J. Ramirez, P. C. St. John, S. P. Woodworth, J. K. Magnuson, K. E. Burnum-Johnson, A. M. Guss, C. W. Johnson and G. T. Beckham, Nat. Commun., 2022, 13, 4925 CrossRef CAS PubMed.
- J. E. Matthiesen, J. M. Carraher, M. Vasiliu, D. A. Dixon and J. P. Tessonnier, ACS Sustainable Chem. Eng., 2016, 4, 3575–3585 CrossRef CAS.
- S. Capelli, A. Rosengart, A. Villa, A. Citterio, A. Di Michele, C. L. Bianchi, L. Prati and C. Pirola, Appl. Catal., B, 2017, 218, 220–229 CrossRef CAS.
- S. Capelli, D. Motta, C. Evangelisti, N. Dimitratos, L. Prati, C. Pirola and A. Villa, ChemCatChem, 2019, 11, 3075–3084 CrossRef CAS.
- G. Quintens, J. Vrijsen, P. Adriaensens, D. Vanderzande and T. Junkers, Polym. Chem., 2019, 40, 5555–5563 RSC.
- K. A. Curran, J. M. Leavitt, A. S. Karim and H. S. Alper, Metab. Eng., 2013, 15, 55–66 CrossRef CAS PubMed.
- C. W. Johnson, D. Salvachúa, P. Khanna, H. Smith, D. J. Peterson and G. T. Beckham, Metab. Eng. Commun., 2016, 3, 111–119 CrossRef PubMed.
- D. R. Vardon, N. A. Rorrer, D. Salvachúa, A. E. Settle, C. W. Johnson, M. J. Menart, N. S. Cleveland, P. N. Ciesielski, K. X. Steirer, J. R. Dorgan and G. T. Beckham, Green Chem., 2016, 18, 3397–3413 RSC.
- T. Nicolaï, Q. Deparis, M. R. Foulquié-Moreno and J. M. Thevelein, Microb. Cell Fact., 2021, 20, 114 CrossRef PubMed.
- J. M. Carraher, T. Pfennig, R. G. Rao, B. H. Shanks and J. P. Tessonnier, Green Chem., 2017, 19, 3042–3050 RSC.
- M. Shiramizu and F. D. Toste, Angew. Chem., Int. Ed., 2013, 52, 12905–12909 CrossRef CAS PubMed.
- A. E. Settle, L. Berstis, S. Zhang, N. A. Rorrer, H. Hu, R. M. Richards, G. T. Beckham, M. F. Crowley and D. R. Vardon, ChemSusChem, 2018, 11, 1768–1780 CrossRef CAS PubMed.
- J. W. Frost, A. Miermont, D. Schweitzer and V. Bui, US Patent, US8426639B2, 2013 Search PubMed.
- V. Bui, D. MacRae and D. Schweitzer, US Patent, US8809583, 2014 Search PubMed.
- I. Khalil, M. G. Rigamonti, K. Janssens, A. Bugaev, T. Donckels, S. Robijns, D. De Vos and M. Dusselier, Single-Atom Ru-zeolite isomerization catalysis for a bio-based route towards terephthalates from muconic acid, 16 August 2022, PREPRINT (Version 1) available at Research Square.
- J. M. Carraher, P. Carter, R. G. Rao, M. J. Forrester, T. Pfennig, B. H. Shanks, E. W. Cochran and J.-P. Tessonnier, Green Chem., 2020, 22, 6444–6454 RSC.
- A. A. Turkin, F. Rammal, S. Eyley, W. Thielemans, E. V. Makshina and B. F. Sels, ACS Sustainable Chem. Eng., 2023, 11, 8968–8977 CrossRef CAS.
- J. M. Carraher, J. E. Matthiesen and J. P. Tessonnier, J. Mol. Liq., 2016, 224, 420–422 CrossRef CAS.
- J. W. Frost and K. M. Draths, US Patent, US5616496, 1997 Search PubMed.
- E. Rossini, A. D. Bochevarov and E. W. Knapp, ACS Omega, 2018, 3, 1653–1662 CrossRef CAS PubMed.
- N. Hayama, Y. Kobayashi and Y. Takemoto, Tetrahedron, 2021, 89, 132089 CrossRef CAS.
- M. F. Baruch, J. E. Pander, J. L. White and A. B. Bocarsly, ACS Catal., 2015, 5, 3148–3156 CrossRef CAS.
- Y.-C. Wen, H.-C. Kuo and H.-W. Jia, J. Mol. Struct., 2016, 1109, 154–160 CrossRef CAS.
- A. S. Narmon, E. Leys, I. Khalil, G. Ivanushkin and M. Dusselier, Green Chem., 2022, 24, 9709–9720 RSC.
- S. Y. Tang, R. A. Bourne, R. L. Smith and M. Poliakoff, Green Chem., 2008, 10, 268–269 RSC.
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc00395k |
| This journal is © The Royal Society of Chemistry 2024 |