
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirectional glycolysis of waste PET using deep eutectic solvents for preparation of aromatic-based polyurethane elastomers†
Fei
Li
ab,
Xiaoqian
Yao
 a,
Rong
Ding
ab,
Yinan
Bao
ac,
Qing
Zhou
*ab,
Dongxia
Yan
a,
Yi
Li
a,
Junli
Xu
a,
Jiayu
Xin
ab and
Xingmei
Lu
*ab
a,
Rong
Ding
ab,
Yinan
Bao
ac,
Qing
Zhou
*ab,
Dongxia
Yan
a,
Yi
Li
a,
Junli
Xu
a,
Jiayu
Xin
ab and
Xingmei
Lu
*ab
aBeijing Key Laboratory of Ionic Liquids Clean Process, CAS Key Laboratory of Green Process and Engineering, State Key Laboratory of Mesoscience and Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China. E-mail: qzhou@ipe.ac.cn; xmlu@ipe.ac.cn
bSchool of Chemical and Engineering, University of Chinese Academy of Sciences, Beijing 100049, PR China
cCollege of Chemical Engineering, Beijing Institute of Petrochemical Technology, Beijing 102617, China
First published on 14th August 2024
Abstract
Glycolysis of waste polyethylene terephthalate (PET) is a green and high-value PET recovery approach, the product of which can be used as raw materials to prepare polyurethanes. Enhancing the conversion rate and monomer selectivity of PET glycolysis through the development of efficient catalysts is crucial for reducing both the energy consumption and operational costs associated with the process. In this study, a series of deep eutectic solvents (DESs) were synthesised based on L-proline (Pro) and its derivatives and used as catalysts for the glycolysis of PET using 1,4-butanediol (BDO). Under the optimized reaction conditions (5.0 g PET, 25.0 g BDO, 2 wt% Pro/Zn(Ac)2 catalyst, 210 °C, 15 min and atmospheric pressure), PET was degraded completely, and the yield of monomeric bis(4-hydroxybutyl)terephthalate (BHBT) reached 67.1%. Based on the results of in situ-IR, 1H NMR and density functional theory (DFT), the possible catalytic mechanism of DESs synergistically promoting PET depolymerization was proposed. Finally, aromatic-based polyurethane elastomers (PUEs) were prepared with PET glycolysis products bis(2-hydroxyethyl)terephthalate (BHET) and BHBT as chain extenders. Compared with the control group, the thermal and mechanical properties of aromatic-based PUEs were improved, which proved the feasibility of high value utilization of PET glycolysis products.
1 Introduction
Polyurethanes (PUs) are highly adaptable polymers, whose characteristics can be readily adjusted by varying the types and functionalities of the polyol and isocyanate components. Thus, PUs have excellent physical and chemical properties and are widely used in foams,1 coatings,2 adhesives, elastomers,3,4 and medical materials.5 As the main raw material of PUs, the development of the polyol industry relies heavily on petroleum-based resources. With the consumption of petroleum resources and the prominence of environmental problems, the replacement of raw petroleum-based resources with recycled or environmentally friendly resources is currently a common global trend.6PET as one of the most important thermoplastic polyesters has exploded in the marketplace over the past decade, increasingly used for bottles and containers as it is light weight, resealable and shatter-resistant.7 In 2020, the global demand for PET bottles was 27 million metric tons and it is expected to reach 42 million metric tons by 2030.8 The dramatically increasing consumption of PET leads to great environmental pressure, including the demand for non-renewable fossil fuel precursors, the resulting waste accumulation, and the CO2 emissions associated with both the production and disposal of PET products.9,10 Therefore, the recycling of waste PET is a top priority. However, the cost of producing PET from petroleum raw materials is lower than the cost of recycling waste PET, which poses a big challenge for the recycling of PET. With this in mind, it is a solution to improve the economics of PET recycling by using waste PET as the raw material to prepare high-value PU products.11 As one of the main recycling methods of waste PET, chemical methods offer the possibility of reintroducing waste PET into the material cycle without downgrading of quality.12 To date, the commercially available chemical recycling technologies include hydrolysis,13 glycolysis,14 aminolysis,15,16 and alcoholysis17 reactions. In the glycolysis process, diverse glycols are used to depolymerize PET in the presence of catalysts to obtain various oligomers and/or monomers with different structures under relatively mild conditions. These glycolysis products of PET can be introduced into PU by two approaches. The first is to directly introduce the products into the soft segment of PU as polyester polyols, and the second is to introduce them into the hard segment of PU as chain extenders. The above two approaches can not only partially replace petroleum resources, but also retain the aromatic ring structure of PET to prepare aromatic-based PU products.18
PUs incorporating benzene rings typically exhibit enhanced heat resistance and higher hardness. However, an excessive content of rigid benzene rings can result in reduced flexibility of the PU. Aiming at adjusting the proportion of the rigid benzene ring and enriching the structure of products obtained by PET degradation, thereby tailoring properties of PUs, we prefer to make PET more efficiently degraded by different kinds of diol compounds. This is achieved by promoting more efficient degradation of PET using various diol compounds. The efficiency of PET degradation reactions, however, is influenced by the choice of the diol. The reactivity of diol compounds in PET glycolysis is primarily governed by their chemical properties. The nucleophilic activity of diols is influenced by both the intermolecular electronic effect and steric hindrance. Consequently, the relative reactivity of diols follows the order: 1,2-propanediol > ethylene glycol (EG) > 1,4-butanediol (BDO). To mitigate the potential reduction in reactivity associated with diols, the development of efficient catalysts is a suitable strategy to enhance the reactivity and overall process efficiency.
In the past decade, most catalysts have been developed focusing on the process of PET glycolysis with EG, such as traditional metal salts19 and metal oxides, new solid acid catalysts,20 polyoxometalates,21 metal-doped graphene hybrid materials,22 as well as ionic liquids (ILs),23,24 DESs,25,26etc. The special properties of ionic liquids make it easy to separate the catalyst from the solid glycolysis products and also provide an excellent catalytic effect on PET glycolysis. Wang et al. applied [3a-C3P(C4)3][Gly] to PET glycolysis for the first time, achieving 100% PET conversion and proposing a detailed reaction mechanism of the glycolysis of PET.27 But the yield for monomeric BHET was not high at the time. With the progress of research, a series of new ionic liquids have been continuously synthesized, such as [Bmim]ZnCl3, [Amim]ZnCl3, etc., which not only ensured the complete degradation of PET, but also achieved more than 80% of monomer yield. However, ionic liquids are expensive, which limits their large-scale application. As a novel catalyst with an adjustable structure, low cost, and easy preparation,28 DESs initially showed excellent catalytic effects in PET degradation.29 The excellent catalytic effect of DESs is due to the synergistic catalysis between the hydrogen bond donor (HBD) and the hydrogen bond acceptor (HBA), which eventually leads to the cleavage of PET chains. Wang et al. conducted glycolysis of PET using urea/metal salts DESs and EG, and PET completely degraded after only 30 min.30 However, urea is easy to decompose in the reaction and the catalyst's cycle performance is poor. In order to improve the stability of the catalyst, Liu et al. prepared DESs with 1,3-dimethylurea instead of urea. Under the optimized conditions, PET degraded completely and the yield of BHET was 82%.31 The above studies indicate that it is feasible to design DESs with high stability for efficient degradation of PET. Proline and its derivatives have relatively high thermal stability. In addition, their amino and carboxyl groups can form hydrogen bonds with the hydroxyl group of diols, which is expected to improve the electronegativity of oxygen in diols, enhance its reactivity and accelerate PET depolymerization. Therefore, proline and its derivatives were used as DES components for PET glycolysis in this study.
As mentioned above, long chain diols such as BDO have lower reactivity and more demanding reaction conditions. Accordingly, high requirements for catalysts are put forward. At the same time, considering that 1,2-propanediol and diethylene glycol are more difficult to separate from the alcoholysis products, they are suitable for utilization in the form of whole components. Therefore, the PET depolymerization monomer products of EG and 1,4-butanediol are more suitable for direct industrial application. And 1,4-butanediol has a moderate carbon–carbon chain length, which can make the soft and hard segments produce microphase separation, so that the crystallinity of the urethane hard chain segments is better. Thus, the isocyanate-1,4-butanediol hard chain can be better oriented, so that the crystallization and the oriented arrangement make it easier for the polymer molecules to form hydrogen bonds between the polymer molecules, which also means that it can produce a better ordered crystallization, which ultimately manifests itself in the polymer with excellent toughness and hardness. Therefore, the process of polymerizing PU using the butanediol glycolysis product of PET is of great importance. However, the current studies on butanediol glycolysis PET are quite few, and the studies on the preparation of PUs from its glycolytic products are even fewer. Most of them are the study of mixing butanediol with other diols,32 and all of them use traditional Zn(Ac)2 as the catalyst,32,33 which is prone to heavy metal residue and makes it difficult to separate and purify the glycolysis products. Thus, in this study, an alkaloid-containing DES was used for the first time to achieve efficient butanediol glycolysis of PET and to optimize the glycolysis process.
In summary, considering the low activity of long-chain alcohols for PET alcoholysis and the structure-regulating role of 1,4-butanediol in polyurethane synthesis, in this work, a series of DESs were synthesized by using the reaction of PET glycolysis with BDO as the probe reaction, and the PET conversion rate and the monomer yield rate obtained by the process were used as indicators to screen the catalyst. The performance of the selected catalysts was evaluated once more, by the process of PET glycolysis with EG. Finally, aromatic-based polyurethane elastomers (PUEs) with application prospects were prepared from monomers obtained by PET glycolysis, and the thermal and mechanical properties of the polyurethane samples were comparatively analysed using the aliphatic chain extender 1,4-butanediol as a control. This work can improve the economy of the green recovery route of PET and provide preliminary theoretical guidance and experimental support for the industrialization of the process.
2 Experimental
2.1 Materials
PET pellets were provided by Jindong Commercial Co. Ltd (Jiangsu, China), and ground into 40–60 mesh with a small grinding mill (ZN-02). Zinc acetate (Zn(Ac)2), manganese acetate (Mn(AC)2), cobalt acetate (Co(AC)2) and anhydrous lead acetate (Pb(AC)2) were purchased from Xilong Chemical Co. Ltd (Guangdong, China). Ethylene glycol (EG), 1,4-butanediol (BDO), 2-pyrrolidinone, N-ethyl-2-pyrrolidinone (NEP), L-proline (Pro), isophorone diisocyanate (IPDI), N,N-dimethylformamide (DMF) and the other reagents were purchased from Aladdin Reagent Co., Ltd (Shanghai, China). All reagents were used as provided without further purification.2.2 Synthesis of DESs
Six DESs: 2-pyrrolidinone/Zn(Ac)2, NEP/Zn(Ac)2, Pro/Zn(Ac)2, Pro/Co(Ac)2, Pro/Pb(AC)2 and Pro/Mn(Ac)2 were synthesized by a heating and stirring method.34 In a typical procedure, 2-pyrrolidinone, Pro, and NEP were mixed with metal salts respectively at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at the temperature of 80 °C–110 °C until a clear and homogeneous liquid formed. In this process, nitrogen should be introduced in advance to avoid side reactions such as oxidation during heating. Finally, the resulting liquid was put into a vacuum drying oven and dried at 70 °C for 48 h to remove water.
:1 at the temperature of 80 °C–110 °C until a clear and homogeneous liquid formed. In this process, nitrogen should be introduced in advance to avoid side reactions such as oxidation during heating. Finally, the resulting liquid was put into a vacuum drying oven and dried at 70 °C for 48 h to remove water.
2.3 General process of PET glycolysis
For the glycolysis of PET with BDO, 5.0 g PET and a certain amount of BDO were added to a 50 mL three-necked flask equipped with a thermometer, magnetic stirring device, nitrogen inlet and reflux condensing device. After the oil bath is heated to the specified reaction temperature, a specified amount of catalyst is added to the flask. The PET glycolysis reactions were carried out at temperatures ranging from 170 to 220 °C for reaction times from 5 min to 60 min, under atmospheric pressure. In the experiments of PET glycolysis with EG, 5.0 g PET waste, 25.0 g EG and 5 wt% Pro/Zn(Ac)2 (based on weight of PET as the catalyst) were added to a glass reactor at 190 °C for 60 min.After the reaction was completed, the reaction solution was added to the right amount of acetonitrile. Undegraded PET powder and crude product were separated by filtration, respectively. The undegraded PET was dried at 70 °C for 12 h and weighed. The monomer products BHBT and BHET are obtained by PET glycolysis with BDO and EG, respectively. The conversion of PET (CPET) and the yield of BHBT (YBHBT) and BHET (YBHET) are calculated using eqn (1) and (2) as follows:
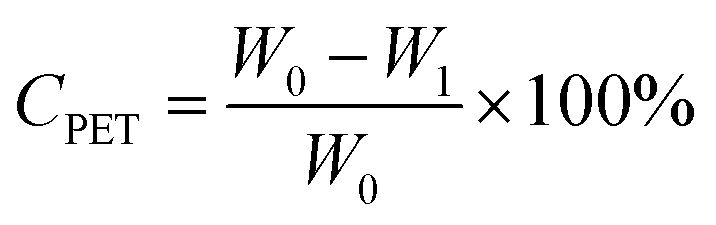 | (1) |
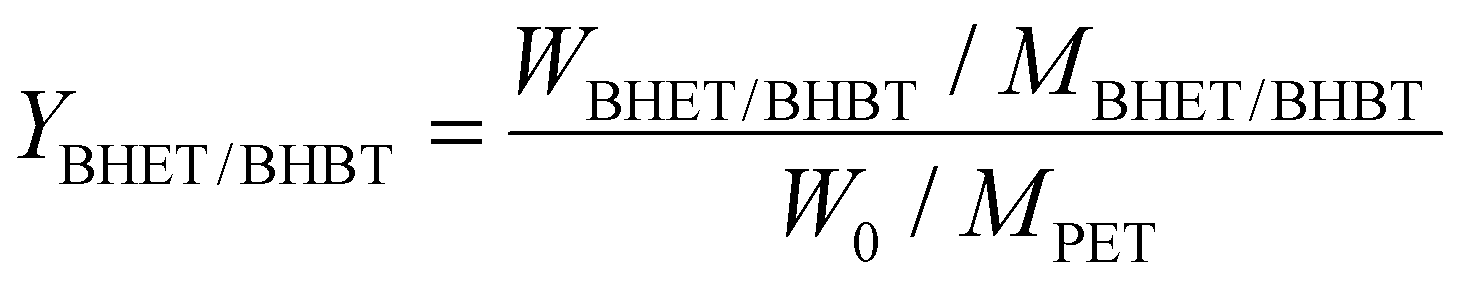 | (2) |
2.4 Synthesis of aromatic-based PU
Three groups of PUEs were synthesized to analyse the impact of glycolysis monomers on final PU properties. All the samples were synthesized according to the following process: 0.02 mol of IPDI, 0.01 mol of PTMEG-2000 and DMF (70 g) were added to the reactor. The reaction mixture was stirred at 60 °C under a nitrogen atmosphere for 4 h. Then stoichiometric amounts of the chain extenders (0.01 mol) were added to the homogenized mixture at 70 °C for about 2 h and cast in a polytetra fluoroethylene (PTFE) mold and cured at 70 °C for 48 h. The differences among the three samples are as follows: (1) in the synthesis of PUE-1, the chain extender of polyurethane was BHET; (2) in the synthesis of PUE-2, the chain extender of polyurethane was BHBT; (3) in the synthesis of PUE-3, the chain extender of polyurethane was BDO. Overall, the synthesis process is shown in Fig. 1.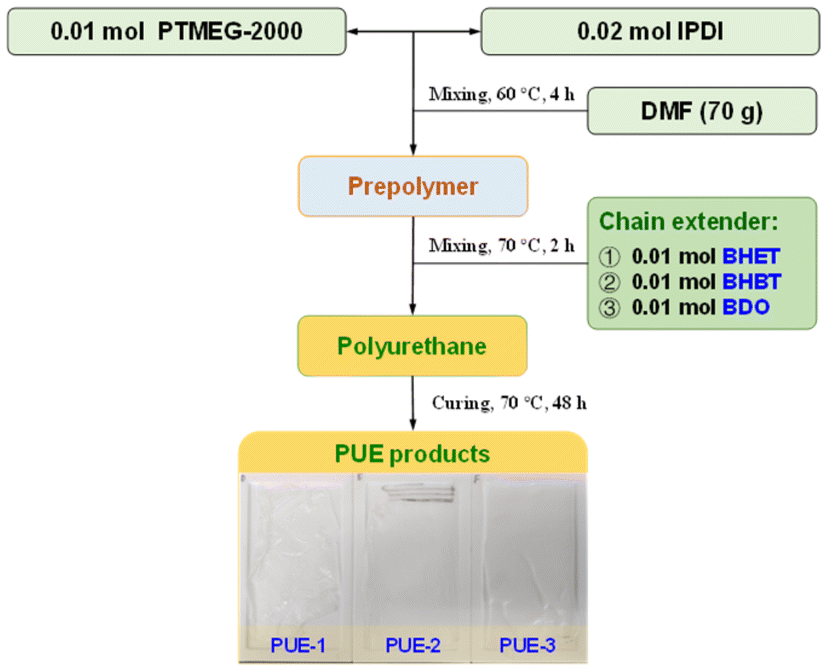 | ||
| Fig. 1 Process flowchart of aromatic-based PU synthesis. | ||
2.5 Characterization
The analysis for catalysts and main products was conducted by ESI-MS, 1H NMR, 13C NMR, HPLC, FT-IR, DSC and TGA. Mass spectra (MS) were obtained on a Bruker micro-TOF instrument with electrospray ionization (ESI). Nuclear magnetic resonance (NMR) spectra were obtained using a Bruker Avance-III600 with DMSO-d6 as the solvent. High performance liquid chromatography (HPLC) spectra were obtained on a BET-C18 column with the flow phase being acetonitrile/water (7:3). The flow rate was 0.5 mL min−1 using an injection volume of 20 μL, at the column temperature of 35 °C. The FT-IR analysis was performed using a Thermo Nicolet 380 (USA). Solid samples were tested by the potassium bromide tablet method; liquid samples are directly coated on smooth KBr wafers and thin film samples can be tested directly. The thermal performance was tested by using a DTG-60H (Shimadzu, Japan) and DSC1 (Mettler-Toledo, Switzerland) at a heating rate of 10 °C min−1 under a N2 atmosphere. The melting point and thermal stability of the samples were measured by DSC1 and TGA, respectively. TGA controlled temperature ranges from 25 °C to 600 °C. The mechanical properties of the samples were determined using a universal testing machine INSTRON.3365 (USA). The test sample size was 80 mm, width was 5 mm, and fixture distance was 30 mm. Data for all mechanical properties were collected under the same environmental conditions.
2.6 DFT calculations
All calculations were carried out using the Gaussian 09 program. The B3LYP/6-311+G(d,p) method was used to optimize the structure, and subsequent frequency calculations at the same level verified that the optimized structure was the ground state without any virtual frequency (NImag = 0).3 Results and discussion
3.1 Investigation of the catalytic activity
DESs synthesized with metal salts and L-proline (Pro) and its derivatives were used as catalysts for the glycolysis of PET, respectively. The catalytic performances of these catalysts were investigated based on the conversion of PET, the yield of BHBT and BHET, which is shown in Table 1.| Entry | Catalyst | Temp. (°C) | Time (min) | C PET (%) | Y BHBT (%) | Y BHET (%) |
|---|---|---|---|---|---|---|
| Reaction conditions (entries 1–12): PET (5.0 g), BDO (25.0 g), catalysts (0.10 g), atmospheric pressure; reaction conditions (entry 13): PET (5.0 g), EG (25.0 g), catalysts (0.10 g), atmospheric pressure. | ||||||
| 1 | — | 210 | 20 | 0.1 | — | — |
| 2 | Pro | 210 | 20 | 14.3 | 2.0 | — |
| 3 | NEP/Zn(Ac)2 | 210 | 20 | 100.0 | 62.7 | — |
| 4 | 2-Pyrrolidinone/Zn(Ac)2 | 210 | 20 | 100.0 | 63.9 | — |
| 5 | Pro/Zn(Ac)2 | 210 | 20 | 100.0 | 66.4 | — |
| 6 | Pro/Co(Ac)2 | 210 | 20 | 99.8 | 60.7 | — |
| 7 | Pro/Pb(Ac)2 | 210 | 20 | 97.5 | 48.1 | — |
| 8 | Pro/Mn(Ac)2 | 210 | 20 | 97.0 | 47.0 | — |
| 9 | Zn(Ac)2 | 210 | 20 | 98.4 | 54.9 | — |
| 10 | Co(Ac)2 | 210 | 20 | 97.1 | 47.1 | — |
| 11 | Pb(Ac)2 | 210 | 20 | 96.7 | 40.3 | — |
| 12 | Mn(Ac)2 | 210 | 20 | 96.1 | 39.2 | — |
| 13 | Pro/Zn(Ac)2 | 190 | 60 | 100 | — | 87.3 |
According to the blank experiment (entry 1), PET glycolysis reactions, without a catalyst, show almost no degradation of PET even at high temperatures such as 210 °C. As a common kind of catalyst for PET glycolysis research, when metal salts were used alone for PET glycolysis with BDO (entries 9–12), the results of PET conversion are almost the same under the same reaction conditions. But, in the yield of BHBT, it is obvious that Zn2+ is superior to other metals, including Co2+, Pb2+ and Mn2+. This is consistent with the results of PET glycolysis with EG using these metal salts as catalysts. Pro and derivatives, another component of the synthesised DESs, their amino and carboxyl groups can form hydrogen bonds with the hydroxyl group of diols, which is expected to improve the electronegativity of oxygen in the diol and accelerate PET depolymerization. Thus, Pro was used alone as the catalyst for PET glycolysis with BDO (entry 2). The result showed that the conversion of PET was only 14.3%, and the yield of BHBT was even lower, only 2%. However, when Pro and derivatives and metal salts with good catalytic performance were used to prepare DESs for PET glycolysis with BDO (entries 3–8), both the PET conversion rate and BHBT yield were significantly improved. The catalytic activity order of the DESs containing Pro and derivatives (entries 3–5) is Pro/Zn(Ac)2 > 2-pyrrolidinone/Zn(Ac)2 > NEP/Zn(Ac)2. When Pro was used as a component of the synthesised DESs (entries 5–8), the catalytic effect follows the rule of metal salt order: Pro/Zn(Ac)2 > Pro/Co(Ac)2 > Pro/Pb(Ac)2 > Pro/Mn(Ac)2. Under the presence of Pro/Zn(Ac)2 (entry 3), PET can be completely degraded and the yield of BHBT reached up to 66.4%. It suggests that the high catalytic activity of the DESs can be attributed to the synergetic catalysis between Pro and derivatives and metal salt. That is to say, the metal ion may play a major catalytic role as Lewis acids, while the basicity of Pro and its derivatives are enhanced by forming hydrogen bonds with metal salts, which improves the overall catalytic performance of acid–base synergistic DES catalysts. Also, the screened Pro/Zn(Ac)2 catalyst was applied and showed an excellent catalytic effect for PET glycolysis with EG (entry 13), where the conversion of PET reached 100%, and the yield of BHET reached 87.3% at 190 °C within 60 min, which is higher than that catalysed by the other catalysts.27,32–36 The detailed comparison results of degradation conditions and catalytic activities are shown in Table 2.
| Source | Catalyst | Solvent | Temp. (°C) | Time (min) | C PET (%) | Yield (%) |
|---|---|---|---|---|---|---|
| This work | Pro/Zn(Ac)2 | BDO | 210 | 20 | 100 | 66.4 |
| Pro/Zn(Ac)2 | EG | 190 | 60 | 100 | 87.3 | |
| Ref. 32 | Zn(Ac)2 | BDO | 180–210 | — | 100 | — |
| Ref. 33 | Zn(Ac)2 | BDO & TEG | 220 | 600 | — | — |
| Ref. 34 | Zn(Ac)2 | EG | 197 | 100 | 98.78 | — |
| Ref. 30 | Urea/ZnCl2 | EG | 170 | 30 | 100 | 82.8 |
| Ref. 27 | [3a-C3P(C4)3][Gly] | EG | 180 | 480 | 100 | — |
| Ref. 35 | [Bmim][FeCl4] | EG | 178 | 240 | 100 | 59.2 |
| Ref. 36 | MAF-6 | EG | 180 | 240 | 92.4 | 81.7 |
In order to better represent the reaction efficiency of this process of PET butanediol glycolysis catalysed by this catalyst, quantitative typical metrics were calculated according to the CHEM21 Metrics Toolkit,37 as shown in Table 3. The selectivity was 67.1%, the atom economy (AE) was 83.3%, the reaction mass efficiency (RME) was 25.0%, the optimum efficiency (OE) was 30.0%, and the total process mass intensity (PMI) was 5.7 g g−1. (The details of the calculations are shown in section 5 of the ESI.†)
| Route | Selectivity (%) | AE (%) | RME (%) | OE (%) | PMI (g g−1) |
|---|---|---|---|---|---|
| BDO | 67.1 | 83.3 | 25.0 | 30.0 | 5.7 |
In summary, the catalyst, consisting of proline and Zn(Ac)2, is biodegradable and has an excellent catalytic effect on both butanediol and ethylene glycol glycolysis. And the metal contents of the DESs are considerably less than those using traditional metal salts, which reduces the difficulty of subsequent product purification. This kind of DESs is both environment-friendly and energy-efficient. Due to the advantages of easy preparation, high activities and relatively low cost, this kind of catalyst showed a promising industrial prospect in the efficient recycling of waste PET.
3.2 Characterization of the optimized DES
The structure of the prepared DES (Pro/Zn(Ac)2 (1:1)) was analysed by FT-IR and NMR. The thermal properties were checked by DSC and TGA.
In order to prove the interaction in the eutectic solvent, 1H NMR spectra of Pro, Zn(Ac)2 and Pro/Zn(Ac)2 DES catalysts synthesized from Pro and Zn(Ac)2 were comparatively studied (Fig. 2(a–c)). It can be seen from Fig. 2(a–c) that DES contains the basic displacement peaks of the two raw materials. δ = 2.078 ppm in DES and δ = 2.088 ppm in Pro represent the chemical shift of hydrogen directly linked to nitrogen, while the peaks of δ = 1.823 ppm in DES and δ = 1.808 ppm in Zn(Ac)2 represent the chemical shift of methyl hydrogen in the acetic acid root, respectively. In addition, in Zn(Ac)2, the peak of water normally appears at δ = 3.300 ppm, while in DES and Pro, the chemical shift of the water peak is at δ = 4.330 ppm and δ = 4.205 ppm, respectively. This increase of chemical shift is mainly caused by the formation of a hydrogen bond between the carboxyl group in Pro and water. It is noteworthy that the movement degree of water peak to low field in the DES is significantly higher than that in Pro. From this result, it can be reasonably inferred that in the synthesis process of the DES, Pro acted as the HBD and its carboxyl group reacted with Zn(Ac)2 to form a stable hydrogen bond, and the target DES catalyst Pro/Zn(Ac)2 was synthesized.
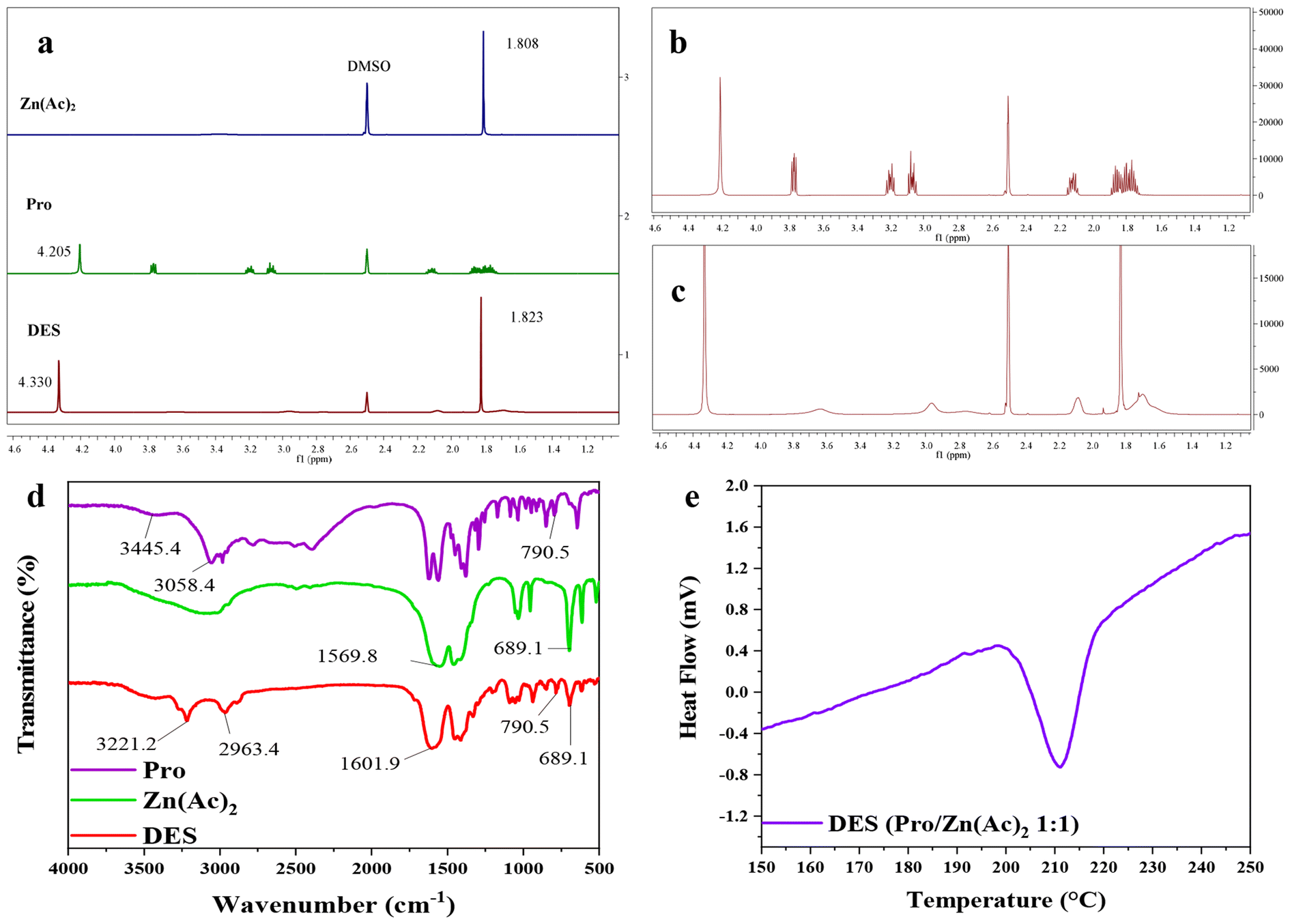 | ||
| Fig. 2 (a–c) 1H NMR spectra of (a: Zn(Ac)2, Pro and DES), (b: Pro) and (c: DES); (d) FT-IR spectra of Pro, Zn(Ac)2 and DES; and (e) DSC curve of DES. | ||
FT-IR spectra are shown in Fig. 2(d). It shows the spectra of the DES and the two raw materials, respectively. The similarity between the DES and raw materials suggests that the DES contains basic functional groups of two kinds of raw materials, such as peaks at 790.5 and 689.1 cm−1. But there are some obvious differences between them. It can be seen that the peaks of the O–H bond and N–H bonds in Pro showed obvious blue shifts from 3445.4 cm−1 to 3221.2 cm−1 and from 3058.4 cm−1 to 2963.4 cm−1 in the DES, which also indicated that new intermolecular hydrogen bonds were formed by the coordination between Pro and Zn(Ac)2.
The DSC curve of the DES is shown in Fig. 2(e). According to the figure, the melting endothermic peak of the DSC curve at 211 °C corresponds to the melting point of DES synthesis, which is lower than the melting point of Zn(Ac)2 (253 °C) and Pro (228 °C) under the same test conditions. It is due to the formation of hydrogen bonds between Pro and Zn(Ac)2. This also proves that the catalyst for the preparation is the DES rather than a simple mixture.
3.3 Characterization of the main product BHBT
NMR, ESI-MS and DSC were carried out to identify the structure of the main product obtained by PET glycolysis with BDO.It can be seen from the ESI-MS spectra in Fig. 3(a) that the peak at m/z 311.1593 was obtained, which is consistent with the molecular weight of BHBT after being ionized by H+ (in ESI, the fraction could be ionized by H+, Na+, or K+). Therefore, it can be initially inferred that the main product is BHBT.
 | ||
| Fig. 3 (a) ESI-MS spectrum, (b) DSC curve, (c) 1H NMR spectrum and (d) 13C NMR spectrum of the main product. | ||
The DSC curve of the main product is shown in Fig. 3(b), a sharp melting endothermic peak appears at 76.3 °C, corresponding to the melting point of the product. This value is consistent with the melting point of the BHBT reported by Lyoo,38 and no other peaks appear on the spectrum, so it can be determined that the product is BHBT with high purity.
1H NMR and 13C NMR spectra are shown in Fig. 3(c) and (d), respectively. The signals are shown as follows. 1H NMR: δ = 8.08, 4.47, 4.32, 3.46, 1.76, 1.55; 13C NMR: δ = 165.52, 134.23, 129.92, 65.66, 60.73, 29.37, 25.45. The signal at δ 8.08 ppm represents the four aromatic protons of the benzene ring. δ = 4.47 ppm is characteristic of the protons of the two –OH. Signals at 4.32, 1.76, 1.55 and 3.46 ppm indicate the presence of the methylene protons from COO-CH2 to CH2-OH in turns. In the spectrum of 13C NMR (Fig. 3d), the characteristic signal at δ 165.52 ppm is of the carbon atom of –COO–. The signal at δ 134.23 ppm is the benzene ring carbon directly connected with esteryl carbon, and δ = 129.92 ppm is the carbon on the benzene ring that has not been replaced by substituents. δ = 65.66, 60.73, 29.37 and 25.45 ppm were the four methylene carbon atoms of –CH2-CH2-CH2-CH2–, respectively. The results of 1H NMR and 13C NMR spectra were consistent with the predicted results of BHBT. Thus, the main product of PET glycolysis with BDO is the monomer BHBT.
3.4 Optimization of reaction conditions
In order to evaluate the influence of process parameters on PET glycolysis by BDO, four factors including reaction temperature, reaction time, amount of catalyst and glycolysis agent BDO were specifically investigated, as shown in Fig. 4. (Specific glycolysis data are shown in Tables S1–S4.†)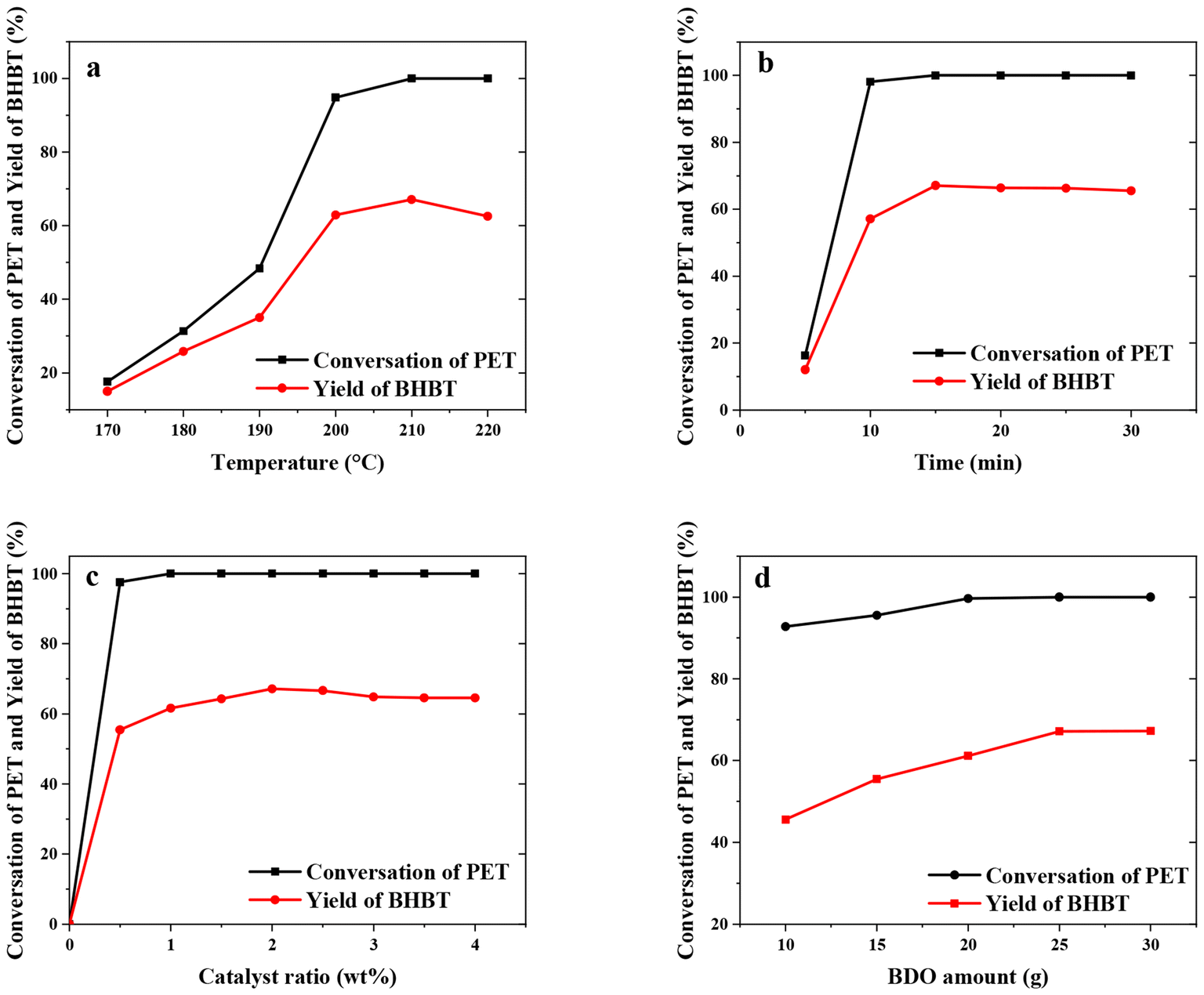 | ||
| Fig. 4 Effects of different reaction parameters on PEF glycolysis: (a) reaction temperature (b) reaction time (c) catalyst ratio and (d) m(BDO)/m(PET). | ||
Reaction temperature is the main factor to determine the energy consumption and reaction time, so we first studied the effect of reaction temperature on the reaction. The scope of investigation was as follows: temperature in the range of 170 °C to 220 °C, reaction time 30 min, 2% catalyst ratio, 5 g PET and 25 g BDO, as shown in Fig. 4(a). The results showed that the reaction of PET glycolysis was greatly affected by temperature. PET reacted completely at 210 °C, and the yield of BHBT reached 65.4%. Glycolysis is a typical endothermic reaction, so rising temperature is beneficial for the forward reaction. However, when the temperature further increases to 220 °C, the yield of BHBT decreases to a certain extent, which is about 62.6%, because there is a chemical reaction balance between monomer BHBT and oligomer in the reaction process. Increasing the temperature will promote the balance to move towards oligomer generation, resulting in a decrease in the yield of monomer BHBT.31 Therefore, 210 °C is the relatively suitable reaction temperature in the range of reaction conditions.
Fig. 4(b) reflects the effect of reaction time on PET conversion rate and BHBT yield. At the initial reaction stage, that is, within 10 min, the PET conversion rate and BHBT yield increased rapidly with time, reaching 98.1% and 57.1% at 10 min, respectively. When the reaction time was 15 min, the PET conversion rate and BHBT yield reached the maximum value of 100% and 67.1%, respectively, and remained unchanged with the further extension of reaction time. This reaction trend is due to the low concentration of BHBT, the surface product of PET, and the low mass transfer resistance. Because the reaction reached equilibrium at 15 min, continuing the reaction for a longer period of time not only increased the reaction energy consumption, but also led to more side effects, such as the increase of product EG content, which would cause ester exchange reaction with the monomer BHBT.39 In addition, the proportion of by-products at 5.8 min in HPLC analysis increased from 2.5% to 4.2% when reaction time was changed from 15 min to 30 min, which could prove the above speculation. Although it will not affect the degradation of PET, the yield of the main product BHBT monomer will be reduced due to the influence of the side reaction mentioned above, so 15 min is determined as the best reaction time.
The amount of catalyst is one of the considerations related to the process economy in the process of industrialization. Considering this, the optimization of catalyst dosage is also of great significance. Using the above optimized reaction temperature of 210 °C and time of 15 min, the influence of catalyst dosage was investigated as shown in Fig. 4(c). As can be seen from Fig. 4(c), PET could hardly degrade even at 210 °C in the absence of a catalyst. When the amount of the catalyst is only 0.5% of the mass of PET, the degradation rate of PET can reach 97.6% after 15 min, and the yield of BHBT can reach 55.5%, which directly shows that the synthesized DES catalyst has excellent catalytic performance. On this basis, the PET conversion rate and BHBT yield can be improved by increasing the amount of catalyst. When the amount of catalyst is 2%, the catalytic effect is the best. When the amount of catalyst is increased, the index data remain basically stable. Thus, a 2% catalyst dosage is relatively suitable for PET glycolysis with BDO in the range of reaction conditions.
In the PET glycolysis reaction system, BDO is not only the substrate raw material for the reaction with PET, but also as the solvent of the reaction to make catalyst DES and PET powder full contact, and promote the reaction. The influence of the amount of BDO on PET glycolysis reaction was investigated as shown in Fig. 4(d). When other conditions remain the same, the PET conversion rate and the yield of BHBT increase gradually with the increasing amount of BDO, and finally remain basically unchanged. When the mass ratio of PET:BDO = 1:5, the conversion of PET was 100%, and the yield of BHBT was 67.1%. When PET:BDO = 1:6, the conversion of PET remained 100%, while the yield of BHBT was 67.2%. Therefore, through the comprehensive consideration of economy and production efficiency, when the mass ratio of PET:BDO = 1:5, the basic requirements of the reaction can be met at the same time, and the economy of the process can be maximized.
3.5 Reaction mechanism
The process of PET glycolysis with BDO was monitored continuously for 6 h by in situ infrared spectroscopy. The monitoring temperature of 170 °C was lower than the optimal reaction temperature of 210 °C. The reaction time at this temperature was longer and it was easier to capture the changes in the reaction system. As shown in Fig. 5, the characteristic peaks of 1720 cm−1, 1271 cm−1 and 737 cm−1 are the characteristic peaks of carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, C–O single bond and benzene ring in the product BHBT and soluble oligomer ester group respectively. These three characteristic peaks appear and increase rapidly in the initial 20 min of the reaction. The peak intensity increases gently from 2.5 h to 6 h, indicating that the alcoholysis rate of PET is fast at the initial stage of the reaction, and the apparent reaction rate decreases with the extension of time. When the reaction approaches 6 h, the reaction basically reaches equilibrium. In the reaction process, the carbonyl peak of the characteristic group in the infrared spectrum shows an obvious red shift from 1717 cm−1 to 1723 cm−1. This is due to the existence of a strong electron-sucking effect, which reduces the electron cloud density near the carbonyl CO of PET and low polyester, thus resulting in a blue shift. This phenomenon is mainly related to the strong electron-absorbing effect of Zn2+ in Pro/Zn(Ac)2 hydrogen bond receptor.40
O bond, C–O single bond and benzene ring in the product BHBT and soluble oligomer ester group respectively. These three characteristic peaks appear and increase rapidly in the initial 20 min of the reaction. The peak intensity increases gently from 2.5 h to 6 h, indicating that the alcoholysis rate of PET is fast at the initial stage of the reaction, and the apparent reaction rate decreases with the extension of time. When the reaction approaches 6 h, the reaction basically reaches equilibrium. In the reaction process, the carbonyl peak of the characteristic group in the infrared spectrum shows an obvious red shift from 1717 cm−1 to 1723 cm−1. This is due to the existence of a strong electron-sucking effect, which reduces the electron cloud density near the carbonyl CO of PET and low polyester, thus resulting in a blue shift. This phenomenon is mainly related to the strong electron-absorbing effect of Zn2+ in Pro/Zn(Ac)2 hydrogen bond receptor.40
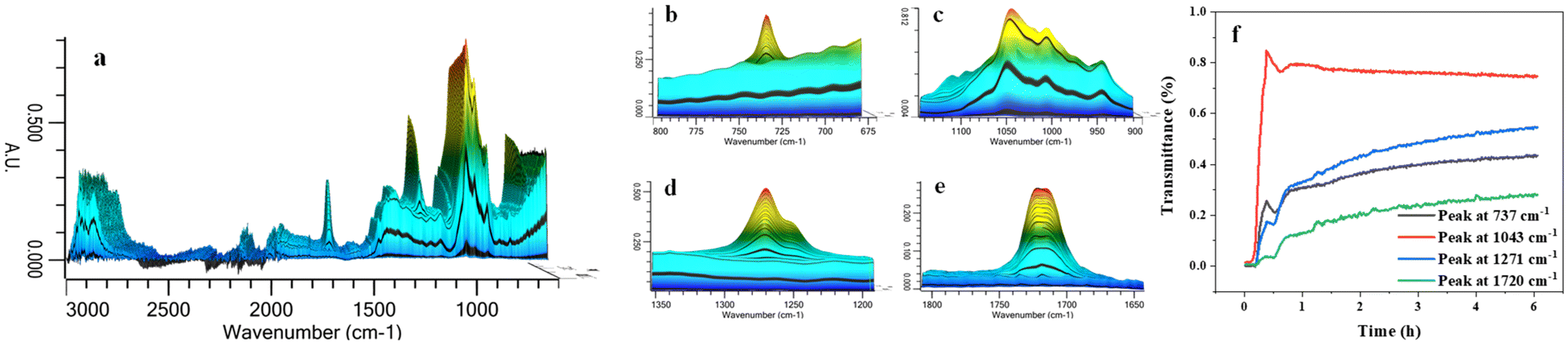 | ||
| Fig. 5 (a) In situ IR spectra of PET glycolysis, (b) peak at 737 cm−1, (c) peak at 1043 cm−1, (d) peak at 1271 cm−1, (e) peak at 1720 cm−1 and (f) the changes of four main characteristic peaks. | ||
Previous studies have shown that DES catalysts can activate the hydroxyl group of alcohol, thus improving the activity of the PET glycolysis reaction. To confirm this prediction, 1H NMR was used to study the interaction between Pro/Zn(Ac)2 and BDO. As shown in Fig. 6, in the 1H NMR spectrum of pure BDO, the chemical shift of its hydroxyl hydrogen (marked with “↓” in Fig. 6) was 3.92 ppm. When the ratio of butanediol to Pro/Zn(Ac)2 was 3, 1 and 1/3, respectively, the chemical shift of hydroxyl hydrogen in butanediol gradually shifted to 4.27 ppm, 4.38 ppm and 4.49 ppm. That is, as the amount of Pro/Zn(Ac)2 increases, the chemical shift of hydroxyl hydrogen of butanediol gradually increases. This displacement change is mainly due to the strong hydrogen bond between the –OH of BDO and the catalyst. In other words, hydrogen bonding between the Pro/Zn(Ac)2 catalyst and the glycolysis agent BDO enhances the electronegativity of hydroxyl oxygen in the glycolysis agent and its reactivity, thus accelerating the depolymerization of the PET chain.
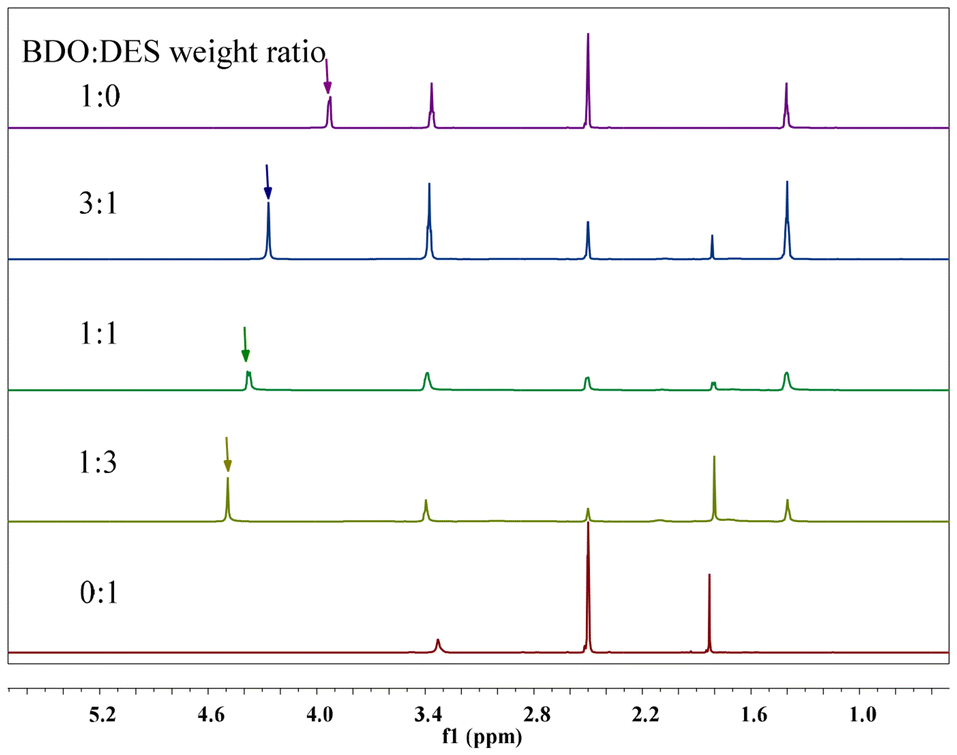 | ||
| Fig. 6 1H NMR spectra of BDO and Pro/Zn(Ac)2 mixture with different ratio. Symbol in the 1H NMR spectra identify the O–H (↓) of BDO. | ||
In order to explore the specific interaction sites between the catalyst and the alcohol solvent butanediol even further, we used density-functional theory (DFT) to investigate the optimal interaction structure between the low-eutectic solvent catalysts with different hydrogen bond donors and the alcohol solvent butanediol, and finally obtained the stable structure with the lowest energy as shown in Fig. 7. Meanwhile, the interaction energies between the catalyst and butanediol in the optimal interaction structure were obtained as shown in Table 2. From Table 3, it is easy to find that the interaction energy between the low eutectic solvent with Pro as the hydrogen bond donor and butanediol is the largest at 23.42 kcal mol−1, followed by pyrrolidinone/Zn(Ac)2 and NEP/Zn(Ac)2, which gives some theoretical support to the experimental results of the order of the catalytic activity for catalyst performance evaluation in section 3.1. Also, this finding confirms the activation of alcohol hydroxyl groups by hydrogen bond donors during PET glycolysis.
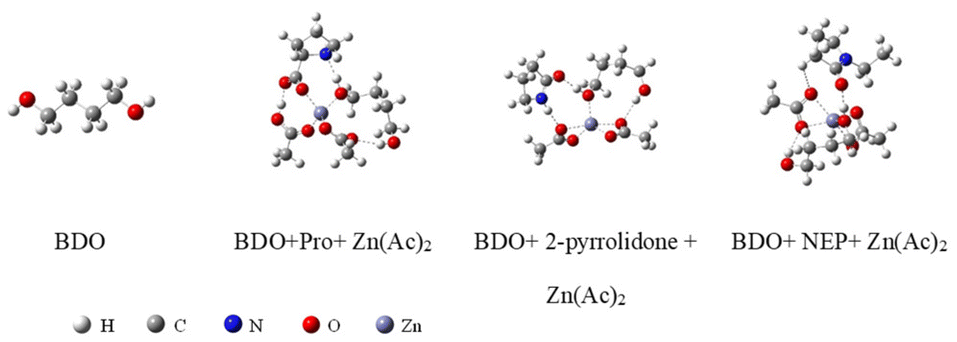 | ||
| Fig. 7 Optimum structure of different DESs and BDO. | ||
Based on the above studies, the catalytic mechanism of Lewis acid-hydrogen bond synergistically promoting PET depolymerization during PET glycolysis was proposed, as shown in Fig. 8. As the hydrogen bond acceptor of Lewis acid, the strong electron-absorbing effect reduces the density of electron cloud around carbonyl group, which enhances the carbon positivity of carbonyl group. The hydrogen bond donor forms strong hydrogen bond with the glycolysis agent BDO, and elongates the hydroxy-oxygen hydrogen bond of BDO, thus increasing the electronegativity of hydroxy-oxygen in butanediol. The synergistic effect of hydrogen bond donor and acceptor components promoted the transesterification reaction, and finally accelerated the rupture of long-chain PET (Table 4).
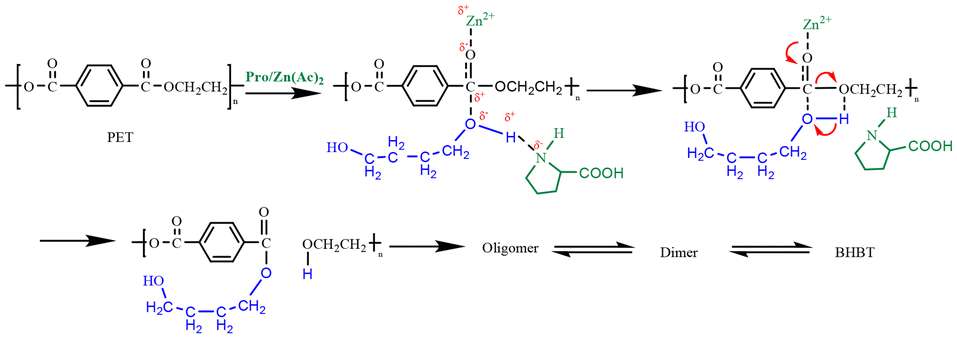 | ||
| Fig. 8 Possible mechanism of PET glycolysis. | ||
| DES component | The interaction energy ΔE (kcal mol−1) |
|---|---|
| Pro + Zn(Ac)2 | 23.42 |
| 2-Pyrrolidone + Zn(Ac)2 | 16.96 |
| NEP + Zn(Ac)2 | 12.98 |
3.6 Characterization of aromatic-based polyurethane
The structure and thermal properties of aromatic-based polyurethane elastomers (PUE-1 and PUE-2) prepared with BHET and BHBT as chain extenders and the control group of polyurethane elastomers (PUE-3) prepared with BDO as chain extenders were characterized by FT-IR, TGA and DSC. Mechanical properties were measured with a universal testing machine.FT-IR spectra of PUE-1, PUE-2 and PUE-3 are shown in Fig. 9(a). It can be seen from the figure that the structures of the three synthetic materials are very similar. The absorption peak of the amide group –CO-NH– is at 3325 cm−1. The wider absorption peak at 2900 cm−1 is due to the stretching vibration of alkyl C–H.
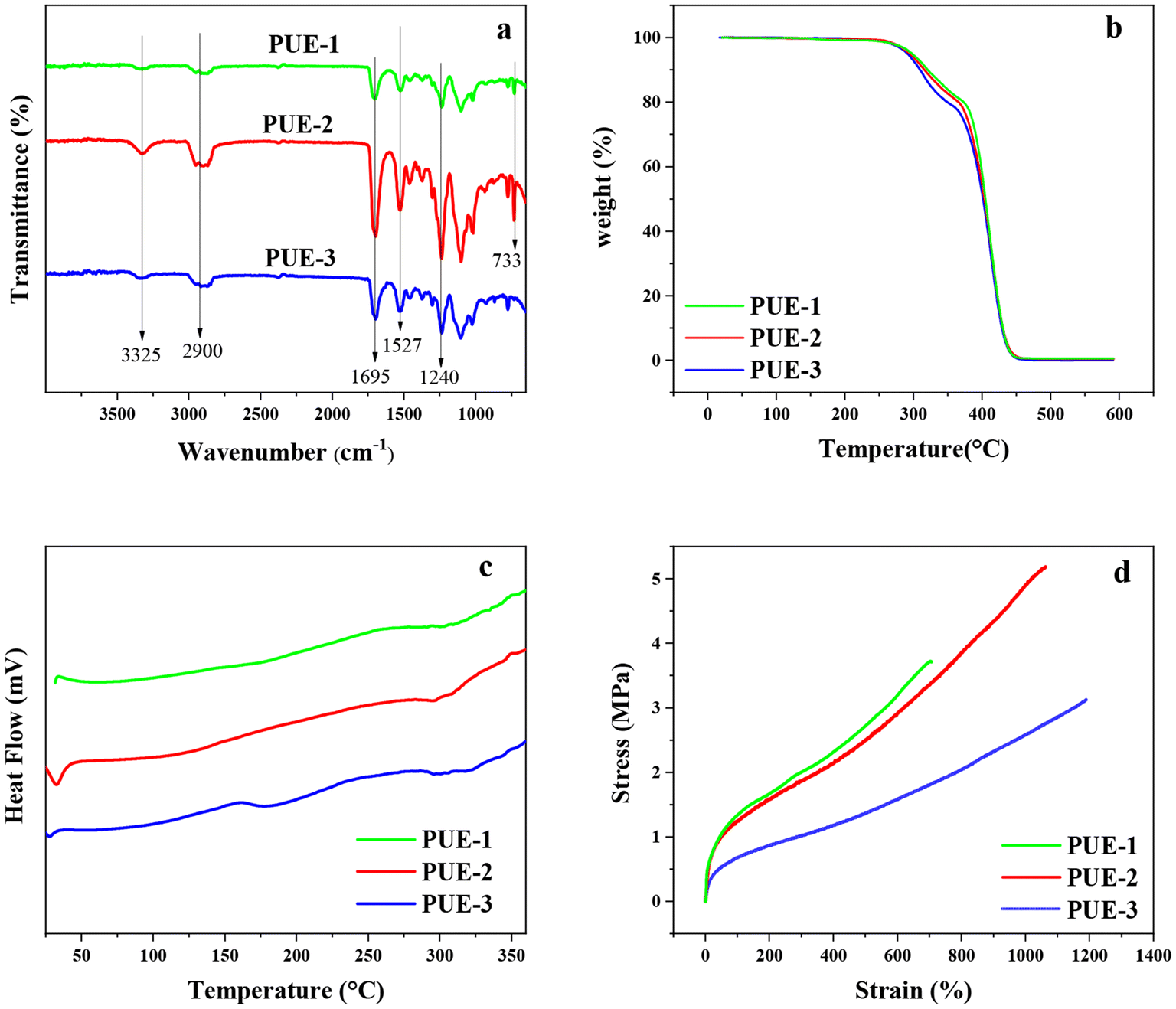 | ||
| Fig. 9 (a) FT-IR spectra, (b) TGA curve, (c) DSC curve and (d) stress–strain curve of synthetic PUEs by different chain extender. | ||
The characteristic absorption peaks of carbonyl CO bond and C–O bond are located at 1695 cm−1 and 1240 cm−1, respectively.
Because the benzene rings of samples PUE-1 and PUE-2 are symmetrically substituted substituents with the same structure, the absorption peaks of 1600 cm−1 and 1580 cm−1 do not appear. However, the bending vibration absorption of benzene ring C–H exists in samples PUE-1 and PUE-2 at 733 cm−1. However, the PUE-3 sample showed no peak at 733 cm−1, indicating the presence of a benzene ring in PUE-1 and PUE-2 structures, and BHET and BHBT were successfully introduced into the sample. The formation of the carbamate group (–NHCOO–) can be determined by combining the wave numbers of 3325 cm−1, 1695 cm−1 and 1240 cm−1. In addition, there is no absorption peak near 2250 cm−1 of the isocyanate group (–NCO), indicating that the reaction of –NCO is basically complete.41 FT-IR spectra showed that polyurethane was successfully prepared, and PET glycolysis monomers were successfully introduced into PUE-1 and PUE-2.
TGA was used to measure the thermal stability of three kinds of synthetic polyurethane elastomer materials. As shown in Fig. 9(b), the samples with BHET and BHBT as chain extenders have better thermal stability, while the samples with BDO as chain extenders have slightly lower thermal stability, which may be due to the stronger heat resistance of benzene rings compared with aliphatic carbon chains. Therefore, the thermal stability of the products has been improved. In the process of heating up, all three samples showed thermal weight losses twice. The first loss occurred at about 270 °C, which was caused by the decomposition of the carbamate group in the hard segment of the polyurethane material. And the temperature range was 270–370 °C. When the temperature is higher than 370 °C, the dissociated soft and hard segment decompose gradually.42 Compared with aliphatic-based PUEs, this kind of aromatic-based PUEs has a relative advantage in heat-resistant products.
The glass transition temperature of the synthetic polyurethane elastomer was measured by DSC, as shown in Fig. 9(c). The glass transition temperature of the soft and hard segments of the three samples is shown by Tgs and Tgh respectively. As can be seen from the figure, the Tgs and Tgh values of the three samples are not significantly different, but the Tgs and Tgh values of the samples using BHET and BHBT as chain extenders are higher, while the Tgs and Tgh values of the samples using BDO as chain extenders are lower. Crystallinity and crosslinking degree are two important parameters affecting the glass transition temperature Tg of polymers. In general, increasing crystallinity and/or crosslinking in the polymer structure results in an increase in Tg. When the polymer chains are stacked together in a regular manner as in linear polyurethanes, the crystallinity is high and the structure is more regular. Due to the rigid structure of the benzene ring, the crystallinity of the sample is improved. Therefore, the regularity of PUE-1 and PUE-2 prepared with BHET and BHBT as chain extenders is higher than that of PUE-3 prepared with flexible aliphatic chain BDO as the chain extender.43,44
The stress–strain curve of the polyurethane elastomer sample is shown in Fig. 9(d). Under the same elongation, the samples prepared with a chain extender containing a benzene ring have higher tensile stress, that is, higher tensile strength. The order of tensile strength is PUE-2 > PUE-1 > PUE-3, because the rigid benzene ring structure enhances its tensile strength.3 However, the elongation at break of the PUE-1 sample using BHET as the chain extender was 707%, and its tensile strength was 3.7 MPa. The PUE-3 sample with BDO as the chain extender has the largest elongation at break, but its tensile strength is low, its elongation at break is 1193%, and its tensile strength is 3.1 MPa. PUE-2 samples using BHBT as the chain extender have moderate tensile strength and high elongation at break. Compared with PUE-1, it has higher elongation because BDO can better neutralize the rigid structure of the benzene ring and obtain moderate flexibility. In addition, the rigidity conferred by the benzene ring makes PUE-2 have higher tensile strength than PUE-3. The elongation at break of PUE-2 is 1063%, and its tensile strength is 5.2 MPa. The results of tensile property studies show that the aromatic-based polyurethane elastomers have relatively high tensile strength. The tensile strength and elongation at break of PUE-2 are greater than those of PUE-1, because BHBT has a longer chain length than BHET, which produces a more suitable microphase separation. And the isocyanate-BHBT chain can be better arranged in the curl orientation so that the polymer molecules are more likely to form hydrogen bonding between the polymer molecules, which exhibits excellent tensile strength elongation at break.
The above results comprehensively show that compared with aliphatic BDO, the thermal and mechanical properties, including thermal stability and tensile properties, of the polyurethane materials prepared by using the alcoholysis product monomers of PET, BHET and BHBT, as chain extenders are improved.
4 Conclusions
In this study, we successfully synthesized six deep eutectic solvents (DESs) with high catalytic efficacy, using a combination of metal salts and L-proline (Pro) along with its derivatives. These DESs demonstrated superior catalytic performance compared to their respective metal salt counterparts, with Pro/Zn(Ac)2 identified as the most effective catalyst. The synergistic effect of the HBD and HBA in the DES was confirmed by in situ IR, 1H NMR and DFT calculation. This effect makes the hydroxyl oxygen in the glycolysis agent BDO attack carbonyl carbon in PET more easily, thus accelerating the depolymerization of the PET chain. The benzene ring, being rigid, offers improved heat resistance compared to aliphatic small molecules. Consequently, incorporating the glycolysis products of PET into polyurethane formulations is expected to enhance the heat resistance and tensile strength of the resulting materials. Indeed, aromatic-based polyurethane elastomers synthesized from bis(2-hydroxyethyl) terephthalate (BHET) and bis(4-hydroxybutyl) terephthalate (BHBT) exhibited superior thermal stability and tensile strength when compared to their aliphatic-based counterparts. The results show that it is feasible to prepare polyurethane products from waste PET.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported financially by the National Natural Scientific Fund of China (no. 22238011, 22178364, U23A20135), the Strategic Priority Research Program of Chinese Academy of Science (no. XDA29040700), the Youth Innovation Promotion Association of the Chinese Academy of Sciences (no. 2022048) and the Beijing Natural Science Foundation (no. 2232030).References
- N. Mahmood, Z. S. Yuan, J. Schmidt, M. Tymchyshyn and C. B. Xu, Green Chem., 2016, 18, 2385–2398 RSC.
- Z. X. Lin, Z. Sun, C. P. Xu, A. Q. Zhang, J. Xiang and H. J. Fan, RSC Adv., 2021, 11, 27620–27626 RSC.
- H. Gang, D. Lee, K. Y. Choi, H. N. Kim, H. Ryu, D. S. Lee and B. G. Kim, ACS Sustainable Chem. Eng., 2017, 5, 4582–4588 CrossRef CAS.
- K. Seshimo, H. Sakai, T. Watabe, D. Aoki, H. Sugita, K. Mikami, Y. Mao, A. Ishigami, S. Nishitsuji, T. Kurose, H. Ito and H. Otsuka, Angew. Chem., Int. Ed., 2021, 60, 8406–8409 CrossRef.
- C. Xu, Y. Huang, L. Tang and Y. Hong, ACS Appl. Mater. Interfaces, 2017, 9, 2169–2180 CrossRef CAS.
- Y. Tian, H. Zhao and X. Wang, Green Chem. Eng., 2020, 1, 3–4 CrossRef.
- H. Wang, Z. X. Li, Y. Q. Liu, X. P. Zhang and S. J. Zhang, Green Chem., 2009, 11, 1568–1575 RSC.
- I. Tiseo, Global demand for polyethylene terephthalate 2010–2030, https://www.statista.com/statistics/1128658/polyethylene-terephthalate-demand-worldwide/.
- Y. C. Liu, X. Q. Yao, H. Y. Yao, Q. Zhou, J. Y. Xin, X. M. Lu and S. J. Zhang, Green Chem., 2020, 22, 3122–3131 RSC.
- C. Song, B. Zhang, L. Hao, J. Min, N. Liu, R. Niu, J. Gong and T. Tang, Green Energy Environ., 2022, 7, 411–422 CrossRef CAS.
- V. Jamdar, M. Kathalewar, K. A. Dubey and A. Sabnis, Prog. Org. Coat., 2017, 107, 54–63 CrossRef CAS.
- B. Geyer, G. Lorenz and A. Kandelbauer, eXPRESS Polym. Lett., 2016, 10, 559–586 CrossRef CAS.
- S. Ghosh, M. A. Makeev, Z. M. Qi, H. Y. Wang, N. N. Rajput, S. K. Martha and V. G. Pol, ACS Sustainable Chem. Eng., 2020, 8, 6252–6262 CrossRef CAS.
- L. H. Deng, R. H. Li, Y. Chen, J. H. Wang and H. Song, J. Mol. Liq., 2021, 334, 116419 CrossRef CAS.
- J. P. K. Tan, J. Tan, N. Park, K. J. Xu, E. D. Chan, C. Yang, V. A. Piunova, Z. K. Ji, A. Lim, J. D. Shao, A. Bai, X. Y. Bai, D. Mantione, H. Sardon, Y. Y. Yang and J. L. Hedrick, Macromolecules, 2019, 52, 7878–7885 CrossRef CAS.
- C. N. Hoang, Y. H. Dang, C. T. Pham and D. Hoang, ACS Omega, 2020, 5, 7044–7050 CrossRef CAS PubMed.
- T. Sako, I. Okajima, T. Sugeta, K. Otake, S. Yoda, Y. Takebayashi and C. Kamizawa, Polym. J., 2000, 32, 178–181 CrossRef CAS.
- P. M. Spasojević, V. V. Panić, J. V. Džunuzović, A. D. Marinković, A. J. J. Woortman, K. Loos and I. G. Popović, RSC Adv., 2015, 5, 62273–62283 RSC.
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, S. Arnaiz and J. I. Gutiérrez-Ortiz, Polym. Degrad. Stab., 2010, 95, 1022–1028 CrossRef.
- M. L. Zhu, Z. X. Li, Q. Wang, X. Y. Zhou and X. M. Lu, Ind. Eng. Chem. Res., 2012, 51, 11659–11666 CrossRef CAS.
- P. T. Fang, B. Liu, J. L. Xu, Q. Zhou, S. J. Zhang, J. Y. Ma and X. M. Lu, Polym. Degrad. Stab., 2018, 156, 22–31 CrossRef CAS.
- A. M. Al-Sabagh, F. Z. Yehia, D. R. K. Harding, G. Eshaq and A. E. ElMetwally, Green Chem., 2016, 18, 3997–4003 RSC.
- J. Sun, D. Liu, R. P. Young, A. G. Cruz, N. G. Isern, T. Schuerg, J. R. Cort, B. A. Simmons and S. Singh, ChemSusChem, 2018, 11, 781–792 CrossRef CAS.
- Z. Jiang, D. Yan, J. Xin, F. Li, M. Guo, Q. Zhou, J. Xu, Y. Hu and X. Lu, Polym. Degrad. Stab., 2022, 199, 109905–109913 CrossRef CAS.
- Y. Wang, S. Wang and L. Liu, Green Chem. Eng., 2022, 3, 5–14 CrossRef.
- Y. Chen and T. Mu, Green Energy Environ., 2019, 4, 95–115 CrossRef.
- H. Wang, Y. Q. Liu, Z. X. Li, X. P. Zhang, S. J. Zhang and Y. Q. Zhang, Eur. Polym. J., 2009, 45, 1535–1544 CrossRef CAS.
- J. Wang, S. Zhang, Z. Ma and L. Yan, Green Chem. Eng., 2021, 2, 359–367 CrossRef.
- H. Qin, X. T. Hu, J. W. Wang, H. Y. Cheng, L. F. Chen and Z. W. Qi, Green Energy Environ., 2020, 5, 8–21 CrossRef.
- Q. Wang, X. Q. Yao, Y. R. Geng, Q. Zhou, X. M. Lu and S. J. Zhang, Green Chem., 2015, 17, 2473–2479 RSC.
- B. Liu, W. Fu, X. Lu, Q. Zhou and S. Zhang, ACS Sustainable Chem. Eng., 2018, 7, 3292–3300 CrossRef.
- S. H. Mansour and N. E. Ikladious, Polym. Test., 2002, 21, 497–505 CrossRef CAS.
- M. R. Patel, J. V. Patel, D. Mishra and V. K. Sinha, J. Polym. Environ., 2007, 15, 97–105 CrossRef CAS.
- A. S. Goje and S. Mishra, Macromol. Mater. Eng., 2003, 288, 326 CrossRef CAS.
- H. Wang, R. Y. Yan, Z. X. Li, X. P. Zhang and S. J. Zhang, Catal. Commun., 2010, 11, 763 CrossRef CAS.
- R.-X. Yang, Y.-T. Bieh, C. H. Chen, C.-Y. Hsu, Y. Kato, H. Yamamoto, C.-K. Tsung and K. C.-W. Wu, ACS Sustainable Chem. Eng., 2021, 9, 6541–6550 CrossRef CAS.
- C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–3121 RSC.
- W. S. Lyoo, J. H. Kim and W. S. Ha, J. Appl. Polym. Sci., 1996, 62, 473–480 CrossRef CAS.
- L. Zhou, X. M. Lu, Z. Y. Ju, B. Liu, H. Y. Yao, J. L. Xu, Q. Zhou, Y. F. Hu and S. J. Zhang, Green Chem., 2019, 21, 897–906 RSC.
- P. Chakraborty, J. Adhikary, R. Sanyal, A. Khan, K. Manna, S. Dey, E. Zangrando, A. Bauza, A. Frontera and D. Das, Inorg. Chim. Acta, 2014, 421, 364–373 CrossRef CAS.
- C. Q. Fang and R. Yang, Polym. Eng. Sci., 2018, 58, 2149–2155 CrossRef CAS.
- H. Y. Yao, X. M. Lu, L. Ji, X. Tan and S. J. Zhang, Ind. Eng. Chem. Res., 2021, 60, 4180–4188 CrossRef CAS.
- S. Balcioglu, H. Parlakpinar, N. Vardi, E. B. Denkbas, M. G. Karaaslan, S. Gulgen, E. Taslidere, S. Koytepe and B. Ates, ACS Appl. Mater. Interfaces, 2016, 8, 4456–4466 CrossRef CAS.
- S. M. Cakić, I. S. Ristić, M. M. Cincović, D. T. Stojiljković and J. B. Simendić, Int. J. Adhes. Adhes., 2016, 70, 329–341 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc00292j |
| This journal is © The Royal Society of Chemistry 2024 |