
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent-free copper-catalyzed trisilylation of alkynes: a practical and atom-economical approach for accessing 1,1,1-trisilylalkanes†
Jia
Li
and
Shaozhong
Ge
 *
*
Department of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Singapore, Singapore. E-mail: chmgsh@nus.edu.sg
First published on 19th April 2024
Abstract
Organosilicon compounds are versatile reagents in chemical synthesis and materials sciences. As an important class of organosilanes, 1,1,1-trisilylalkanes can undergo various organic transformations and serve as core units for silicon-containing hyperbranched polymers. The existing catalytic approaches for accessing 1,1,1-trisilylalkanes via alkyne trisilylation not only requires pre-synthesized moisture- and air-sensitive organocalcium and organolanthanum catalysts but also suffers from limited substrate scope for both alkyne and hydrosilane reagents. For example, only alkyl-substituted alkynes can undergo organocalcium-catalyzed trisilylation with alkyl hydrosilanes to provide the desired 1,1,1-trisilylalkane products. Herein, we report a selective copper-catalyzed trisilylation reaction of both alkyl- and aryl-substituted alkynes with a readily accessible copper catalyst that is generated in situ from Cu(OAc)2 and tributylphosphine PnBu3. This copper-catalyzed trisilylation reaction features easy catalyst preparation, broad substrate scope, and mild solvent-free reaction conditions. Mechanistic studies reveal that this trisilylation reaction occurs through copper-catalyzed deprotosilylation of alkynes to form alkynylsilanes followed by double hydrosilylation of alkynylsilane.
Introduction
Organosilanes are useful building blocks in organic synthesis and materials sciences because of their diverse reactivity, non-toxicity, high stability, and ease of handling.1–3 As an important family of organosilicon compounds, 1,1,1-trisilylalkanes, particularly those containing Si–H bonds which allow their further functionalization, are widely used in the synthesis of silicon polymers.4,5 In addition, 1,1,1-trisilylalkanes can readily undergo base-induced desilylation to generate gem-disilyl-substituted carbanions.6 These carbanions are stabilized by the attached silyl groups and can react with various electrophiles.6–9 However, general approaches for preparing structurally diverse 1,1,1-trisilylalkanes from readily accessible starting materials are rather limited in scope and functional group compatibility, which in turn limits the exploration of their new reactivity. The classic synthesis of 1,1,1-trisilylalkanes has largely been based on stoichiometric reactions of trisilylmethyllithium or trisilylmethyl Grignard reagents with activated alkyl halides.10,11 However, these reactions require stoichiometric amounts of pyrophoric reagents and generate large quantities of waste when conducted on a large scale.Metal-catalyzed hydrosilylation of unsaturated hydrocarbons is a straightforward and atom-economical approach for preparing various families of organosilicon compounds,12–16 such as alkylsilanes, vinylsilanes, allylsilanes, and gem-disilylalkanes.17–24 Synthetic protocols based on lanthanum-catalyzed dihydrosilylation of silyl-substituted internal alkynes (Scheme 1A) and calcium-catalyzed trisilylation of terminal alkynes (Scheme 1B) to prepare 1,1,1-trisilylalkanes have also been developed but suffer from several significant limitations.25,26 For example, the scope of alkynes for these metal-catalyzed trisilylation reactions is limited to alkyl-substituted alkynes, and aryl-substituted alkynes only undergo dehydrogenative silylation to provide alkynylsilane products.26 In addition, the scope of hydrosilanes for these reactions is limited to alkylsilanes RSiH3 because arylsilanes ArSiH3 can readily undergo silane redistribution reactions to produce SiH4, Ar2SiH2, or Ar3SiH in the presence of alkaline earth metal or lanthanide catalysts.27–30 Furthermore, the preparation of these well-defined lanthanum and calcium pre-catalysts is rather challenging because they are highly oxophilic and moisture-sensitive.31 Lastly, the highly polar nature of metal–carbon bonds in organolanthanum and organocalcium intermediates renders these trisilylation reactions less compatible towards reactive functional groups.32 Therefore, it remains desirable to identify metal catalysts for selective alkyne trisilylation that can combine broad substrate scope, high functional group tolerance, and convenient catalyst generation.
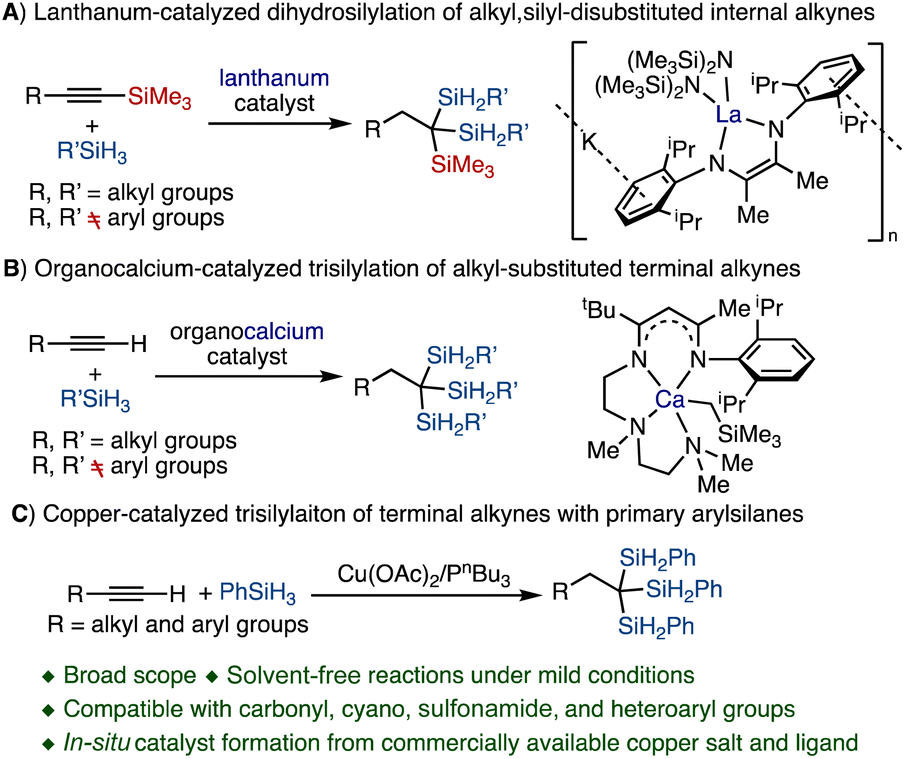 | ||
| Scheme 1 Catalytic synthesis of 1,1,1-trisilylalkanes. | ||
Deprotosilylation of terminal alkynes to form alkynylsilanes with metal acetylide species as intermediates is a key step in transition metal-catalyzed alkyne trisilylation reactions. Terminal alkynes can readily react with various copper salts under mild conditions to form stable monomeric or high-nuclearity copper acetylides.33–35 Accordingly, copper acetylide species have been proposed as reactive intermediates in a variety of alkyne functionalization reactions, such as Sonogashira coupling reactions and multi-borylation of terminal alkynes.36–43 Recently, copper complexes have been employed to catalyze hydrosilylation and deprotosilylation of terminal alkynes to generate vinylsilanes and alkynylsilanes, respectively.44,45 In these copper-catalyzed reactions between terminal alkynes and hydrosilanes, copper hydride and copper acetylide species have been proposed as key intermediates. Nevertheless, suitable conditions and copper catalysts have not been identified to integrate copper-catalyzed deprotosilylation and double hydrosilylation of alkynes into one process to produce 1,1,1-trisilylalkanes.
In continuation of our efforts in developing selective base-metal catalyzed synthesis of multi-organometallic compounds from readily accessible unsaturated hydrocarbons,46–53 we became interested in identifying selective base metal catalysts for trisilylation of alkynes to access 1,1,1-trisilylalkane compounds. We envisioned that copper complexes would be potential catalysts to promote 1,1,1-trisilylation reactions of terminal alkynes because copper acetylide and copper hydride species could be formed in the reactions of terminal alkynes with hydrosilanes. Herein, we report a copper-catalyzed 1,1,1-trisilylation reaction of terminal alkynes under mild conditions with commercially available Cu(OAc)2 and monophosphine ligand PnBu3 (Scheme 1C). Mechanistic studies suggest that alkynylcopper, alkynylsilane, and gem-disilylalkene species are key intermediates for this copper-catalyzed trisilylation reaction.
Results and discussion
Evaluation of reaction conditions
To initiate our studies on the copper-catalyzed trisilylation of alkynes, we evaluated the reaction between phenylacetylene 1a and PhSiH3 to identify selective copper catalysts and reliable conditions that promote the formation of 1,1,1-trisilylalkane 4a (Table 1). The major possible by-products of this reaction are (E)-vinylsilane 2a and gem-disilylalkane 3a from hydrosilylation and double hydrosilylation of 1a, respectively.10 The copper catalysts for this study were generated in situ by combining Cu(OAc)2 and phosphine ligands and activated by their reaction with PhSiH3. In general, the experiments were performed with alkyne 1a as a limiting reagent in the presence of 4 equivalents of PhSiH3 and 10 mol% copper catalyst at 40 °C. The results of the selected examples of these experiments are summarized in Table 1.|
|
||||
|---|---|---|---|---|
| Entry | Variation from the standard conditions | Conversion of 1a (%) | Yield of 4a (%) |
2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3a:4a :3a:4a |
|
a Reaction conditions: phenylacetylene 1a (0.300 mmol), PhSiH3 (1.20 mmol), Cu(OAc)2 (30.0 μmol), ligand (60.0 μmol for monophosphines and 30.0 μmol for bisphosphines), neat or solvent (0.3 mL) at 40 °C for 12 h; the conversion of 1a, the yield of 4a, and the ratios of 2a:3a:4a were determined by GC analysis with tridecane as the internal standard.
b CuTC = copper(I) thiophene-2-carboxylate.
c NaOtBu (20 mol%) was used.
|
||||
| 1 | None | >99 | 76 | —:12:88 |
| 2 | PnBu3 (30 mol%) | >99 | 53 | —:16:84 |
| 3 | PnBu3 (10 mol%) | 88 | 11 | 31:38:31 |
| 4 | PCy3 as the ligand | 86 | 8 | 65:23:12 |
| 5 | PtBu3 as the ligand | 90 | — | 86:14:— |
| 6 | PPh3 as the ligand | 85 | — | 66:34:— |
| 7 | Ruphos as the ligand | 89 | — | 84:16:— |
| 8 | Johnphos as the ligand | 90 | — | 80:20:— |
| 9 | xantphos as the ligand | 90 | <5 | 48:45:7 |
| 10 | binap as the ligand | 88 | — | 90:10:— |
| 11 | dppf as the ligand | 55 | — | 82:18:— |
| 12 | toluene as solvent | 70 | 5 | 21:66:13 |
| 13 | CH3CN as solvent | >99 | 44 | 8:36:56 |
| 14 | THF as solvent | 92 | 36 | 10:30:60 |
| 15 | DMA as solvent | >99 | 68 | —:12:88 |
| 16 | CuOAc as the precursor | >99 | 72 | —:24:76 |
| 17 | CuTCb as the precursor | >99 | 74 | —:15:85 |
| 18 | (iPr)CuCl as the catalystc | 30 | — | 20:80:— |
|
||||

After evaluating various phosphine ligands and solvents, we found that the neat reaction between 1a and 4 equivalents of PhSiH3 proceeded smoothly in the presence of 10 mol% Cu(OAc)2 and 20 mol% PnBu3 and afforded 1,1,1-trisilylalkane 4a in 76% GC yield with 88% selectivity (entry 1 in Table 1). The reaction with 30 mol% PnBu3 showed similar selectivity (entry 2 in Table 1). However, the reaction with 10 mol% PnBu3 proceeded with much lower selectivity (entry 3 in Table 1). The steric properties of trialkylphosphine ligands had profound influence on selectivity. For example, the reaction conducted with 20 mol% PCy3 showed only 12% selectivity toward 1,1,1-trisilylalkane 4a and the reaction with 20 mol% PtBu3 did not generate any detectable amounts of 4a (entries 4 and 5 in Table 1). When copper catalysts were generated from Cu(OAc)2 and triphenylphosphine PPh3 or bulky dialkylbiaryl phosphines, such as Johnphos and Ruphos, the reactions proceeded with high conversions of alkyne 1a, but provided (E)-vinylsilane 2a as the major product (entries 6–8 in Table 1). Similar results were obtained for the reactions conducted with copper catalysts containing bisphosphine ligands, such as xantphos, binap, and dppf (entries 9–11 in Table 1). In addition, we also tested various solvents for this reaction and found that the solvent effect on this trisilylation was noticeable (entries 12–15 in Table 1). The reactions conducted in toluene, acetonitrile, and THF proceeded with decreased chemoselectivity, and the reaction in N,N-dimethylacetamide (DMA) occurred with a similar selectivity compared to the neat reaction. Furthermore, we also found that copper catalysts generated in situ from copper(I) salts, such as CuOAc or CuTC, and PnBu3, were similarly active and selective for the copper-catalyzed alkyne trisilylation (entries 16 and 17 in Table 1). However, when the copper(I) complex (iPr)CuCl (10 mol%) together with NaOtBu (20 mol%) was used as the catalyst for the trisilylation reaction, the reaction proceeded with a low conversion and did not form a detectable amount of 1,1,1-trisilylalkane product 4a (entry 18 in Table 1).
Substrate scope of terminal alkynes
With an active copper catalyst in hand and reliable conditions identified for this Cu-catalyzed 1,1,1-trisilylation (entry 1 in Table 1), we explored the scope of terminal alkynes that undergo this trisilylation reaction, and the results are gathered in Table 2. In general, a wide range of aryl- (1a–1u), alkenyl- (1v), and alkyl-substituted alkynes (1u–1ai) reacted smoothly with PhSiH3 in the presence of 10 mol% Cu(OAc)2 and 20 mol% PnBu3 to afford the corresponding 1,1,1-trisilylalkanes (4a–4ai) in moderate to high isolated yields (up to 78%). Furthermore, several alkynes (1aj–1an) derived from commonly used drugs and bioactive molecules also underwent this Cu-catalyzed trisilylation reaction to form 1,1,1-trisilylalkane products (4aj–4an) in good yields (56–76%). The structure of 1,1,1-trisilylalkane 4r was confirmed by single-crystal X-ray diffraction analysis.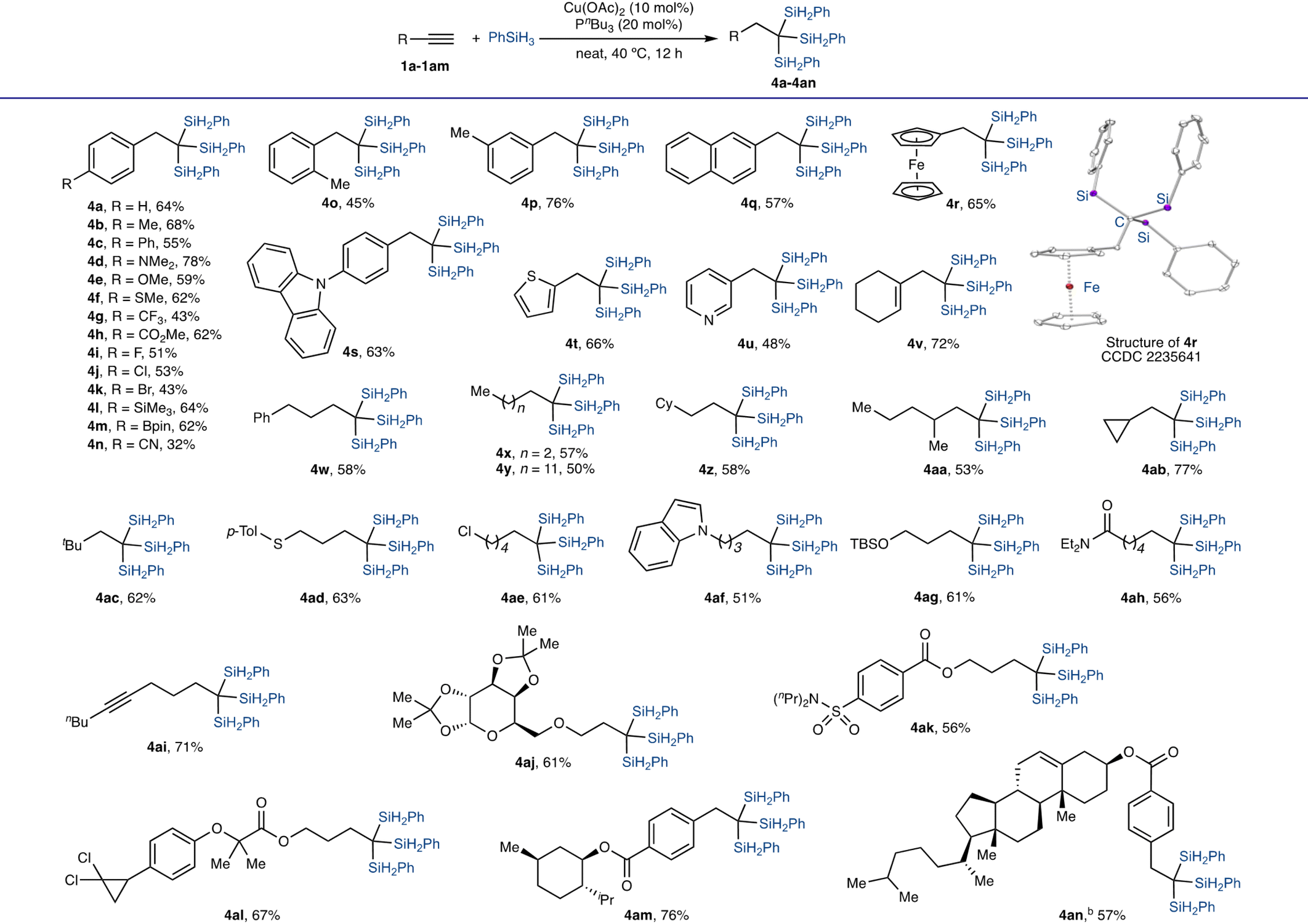
This Cu-catalyzed 1,1,1-trisilylation reaction tolerates various reactive groups. For example, alkynes containing sulfide (4f and 4ad), carboxylic ester (4h and 4ak–4an), fluoro (4i), chloro (4j, 4ae, and 4al), bromo (4k), silyl (4l), pinacol boronic ester (4m), cyano (4n), siloxy (4ag), carboxylic amide (4ah), acetal (4aj), and sulfonamide (4ak) moieties are compatible with the identified reaction conditions. In addition, alkynes containing heterocyclic aromatic groups also reacted with PhSiH3 to provide the desired 1,1,1-trisilylalkanes containing carbazole (4s), thiophene (4t), pyridine (4u), and indole (4af) in good yields.
We also conducted the copper-catalyzed 1,1,1-trisilylation of terminal alkyne 1a and 1w with nC6H13SiH3, an alkyl-substituted hydrosilane. These two reactions proceeded to form the desired 1,1,1-trisilylalkanes 4a′ and 4aw′, respectively, albeit in low isolated yields (eqn (1)).
 | (1) |
Synthetic utilities
After establishing the scope of this 1,1,1-trisilylation reaction, we subsequently showed the synthetic utility of this protocol (Scheme 2). A gram-scale reaction of phenylacetylene 1a with PhSiH3 was performed, and this reaction proceeded smoothly under standard conditions to afford 1,1,1-trisilylalkane 4a (2.46 g) in 59% isolated yield (Scheme 2A). Trisilylalkane products from these trisilylation reactions contain three primary silyl groups and they can be readily converted to other organosilicon compounds. For example, CH2 carbene formed from CH2I2 and Et2Zn could readily be inserted into all six Si–H bonds of 4a to produce trisilylalkane 5, which contains three tertiary silyl groups, in 78% isolated yield (Scheme 2B).54 Sequential alkoxylation/protodesilylation of 4a with methanol-D4 in the presence of KHMDS as a catalyst generated gem-disilylalkane 6-D13 in 80% isolated yield (Scheme 2C).55 Compound 4a could undergo arylation/desilylation and diarylation/desilylation reactions with a phenyl Grignard reagent to form gem-disilylalkanes 7 and 8 in good yields, respectively (Scheme 2D and 2E).56 The corresponding sequential alkylation/desilylation reaction with n-propylmagnesium chloride afforded gem-disilylalkane 9 in 65% isolated yield (Scheme 2F). | ||
| Scheme 2 Gram-scale synthesis of and derivatization of 4a. | ||
Mechanistic considerations
To get a preliminary understanding of this copper-catalyzed trisilylation process, we monitored the reaction of 4-ethynylanisole 1e with PhSiH3 with an attempt to identify potential intermediates. The GC-MS analysis of the reaction mixture showed that a significant amount (up to 25% GC yield) of gem-disilylalkene 10e was formed in the early stage of the reaction and then 10e was fully consumed in the late stage of the reaction (Scheme 3A). To verify the intermediacy of gem-disilylalkenes in this trisilylation reaction, we prepared gem-disilylalkene 10e and subjected it to this copper-catalyzed trisilylation reaction. As expected, 10e was converted to 1,1,1-trisilylalkane 4e in 94% GC yield (Scheme 3B). In addition, we found that alkynylsilane 11e reacted with PhSiH3 to afford 1,1,1-trisilylalkane 4e in 85% yield under standard conditions (Scheme 3C), suggesting that alkynylsilane 11e is a potential intermediate for the trisilylation reaction of alkyne 1e. Indeed, the reaction of phenylacetylene 1a with PhMe2SiH, a bulky hydrosilane, in the presence of Cu(OAc)2/PnBu3 stopped at the dehydrogenative silylation stage and afforded alkynylsilane 12 in 60% GC yield (Scheme 3D). Furthermore, we also carried out the stoichiometric reaction between copper(I) phenylacetylide 13 and 4 equivalents of PhSiH3 in the presence of 2 equivalents of PnBu3, and this reaction provided trisilylalkane 4a in 80% GC yield (Scheme 3E). However, the corresponding reaction in the absence of PnBu3 did not produce any detectable amount of 4a. | ||
| Scheme 3 Control experiments and monitoring of 1,1,1-trisilylation of alkyne 1e. | ||
Deuterium-labelling experiments were also carried out on this trisilylation reaction. For example, 4-ethynylanisole 1e-D1 reacted with PhSiH3 under standard conditions to afford trisilylalkane 4e in 61% isolated yield and no deuterium incorporation was detected by 2H NMR spectroscopic analysis (Scheme 4A). The corresponding reaction between alkyne 1e and PhSiD3 produced 4e-D8 with deuterium atoms located at the benzylic carbon and silicon atoms (Scheme 4B).
 | ||
| Scheme 4 Deuterium-labelling experiments and proposed catalytic cycles for this copper-catalyzed 1,1,1-trisilylation of terminal alkynes. | ||
Based on the results of the above mechanistic experiments and the precedent for copper-catalyzed hydrosilylation/silylation reactions of terminal alkynes,44,45 we proposed a plausible catalytic pathway for this copper-catalyzed 1,1,1-trisilylation reaction, as depicted in Scheme 4C. The activation of Cu(OAc)2 with PhSiH3 in the presence of PnBu3 (L) forms a copper hydride species LnCuH, which then reacts with an alkyne to form an alkynylcopper intermediate (I) with the concomitant release of hydrogen gas, which was detected by GC analysis. σ-Bond metathesis between copper acetylide I and PhSiH3 produces alkynylsilane 11 and regenerates LnCuH. Hydrocupration of alkynylsilane 11 with LnCuH forms an alkenylcopper species (II), which then reacts with PhSiH3 to give gem-disilylalkene intermediate 10. Subsequently, hydrocupration of gem-disilylalkene 10 with LnCuH generates an alkylcopper intermediate (III), which then reacts with PhSiH3 to afford 1,1,1-trisilylalkanes 4. Based on the reaction profile of the trisilylation of terminal alkyne 1e (Scheme 3A), accumulation of gem-disilylalkene 10e was observed, which suggests that the hydrosilylation of gem-disilylalkene 10 to generate 1,1,1-trisilylalkane 4 is the slowest reaction compared to dehydrogenative silylation of alkynes 1 and hydrosilylation of alkynylsilane 11 as shown in Scheme 4C.
Conclusions
In summary, we have developed an effective and practical protocol to access 1,1,1-trisilylalkanes by copper-catalyzed 1,1,1-trisilylation of terminal alkynes with PhSiH3. A series of alkyl- and aryl-substituted alkynes undergo this trisilylation reaction in the presence of Cu(OAc)2 and PnBu3. Mechanistic studies reveal that this trisilylation reaction proceeds through a reaction sequence combining copper-catalyzed dehydrogenative hydrosilylation and double hydrosilylation of alkynylsilane intermediates. These 1,1,1-trisilylalkane products can be readily converted to other multi-silylated compounds by manipulating their Si–H bonds. Further development of copper-catalyzed multi-functionalization reactions of unsaturated hydrocarbons and the synthetic application of 1,1,1-trisilylalkanes will be the subject of future studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ministry of Education of Singapore (A-8000984-00-00). J. L. thanks the China Scholarship Council (CSC) for providing him a PhD scholarship.References
- M. A. Brook, Silicon in Organic, Organometallic, and Polymer Chemistry, J. Wiley, New York, 2000 Search PubMed.
- I. Fleming, A. Barbero and D. Walter, Chem. Rev., 1997, 97, 2063–2192 CrossRef CAS PubMed.
- P. Gaspar and R. West, in The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport and Y. Apeloig, John Wiley and Sons, Chester, UK, 1998, vol. 2 Search PubMed.
- R. J. P. Corriu, M. Granier and G. F. Lanneau, J. Organomet. Chem., 1998, 562, 79–88 CrossRef CAS.
- K. D. Safa, S. Tofangdarzadeh and H. H. Ayenadeh, Heteroat. Chem., 2008, 19, 365–376 CrossRef CAS.
- H. Sakurai, K.-i. Nishiwaki and M. Kira, Tetrahedron Lett., 1973, 14, 4193–4196 CrossRef.
- B.-T. Gröbel and D. Seebach, Angew. Chem., Int. Ed. Engl., 1974, 13, 83–84 CrossRef.
- S. Inoue and Y. Sato, Organometallics, 1986, 5, 1197–1201 CrossRef CAS.
- K. Itami, T. Nokami and J.-i. Yoshida, Org. Lett., 2000, 2, 1299–1302 CrossRef CAS PubMed.
- C. L. Smith, L. M. James and K. L. Sibley, Organometallics, 1992, 11, 2938–2940 CrossRef CAS.
- K. D. Safa and M. Babazadeh, J. Organomet. Chem., 2005, 690, 79–83 CrossRef CAS.
- Hydrosilylation: A Comprehensive Review on Recent Advances, ed. B. Marciniec, Springer, Berlin, 2009 Search PubMed.
- Y. Nakajima and S. Shimada, RSC Adv., 2015, 5, 20603–20616 RSC.
- X. Du and Z. Huang, ACS Catal., 2017, 7, 1227–1243 CrossRef CAS.
- J. Sun and L. Deng, ACS Catal., 2016, 6, 290–300 CrossRef CAS.
- J.-W. Park, Chem. Commun., 2022, 58, 491–504 RSC.
- W. J. Teo, C. Wang, Y. W. Tan and S. Ge, Angew. Chem., Int. Ed., 2017, 56, 4328–4332 CrossRef CAS PubMed.
- C. Wang, W. J. Teo and S. Ge, Nat. Commun., 2017, 8, 2258 CrossRef PubMed.
- M.-Y. Hu, J. Lian, W. Sun, T.-Z. Qiao and S.-F. Zhu, J. Am. Chem. Soc., 2019, 141, 4579–4583 CrossRef CAS PubMed.
- X. Du, Y. Zhang, D. Peng and Z. Huang, Angew. Chem., Int. Ed., 2016, 55, 6671–6675 CrossRef CAS PubMed.
- J. Guo, H. Wang, S. Xing, X. Hong and Z. Lu, Chem, 2019, 5, 881–895 CAS.
- H. L. Sang, Y. Hu and S. Ge, Org. Lett., 2019, 21, 5234–5237 CrossRef CAS PubMed.
- S. Nishino, K. Hirano and M. Miura, Chem. – Eur. J., 2020, 26, 8725–8728 CrossRef CAS PubMed.
- J. Guo, X. Shen and Z. Lu, Angew. Chem., Int. Ed., 2017, 56, 615–618 CrossRef CAS PubMed.
- W. Chen, H. Song, J. Li and C. Cui, Angew. Chem., Int. Ed., 2020, 59, 2365–2369 CrossRef CAS PubMed.
- T. Li, R. Liu, X. Liu and Y. Chen, Org. Lett., 2023, 25, 761–765 CrossRef CAS PubMed.
- X. Liu, L. Xiang, E. Louyriac, L. Maron, X. Leng and Y. Chen, J. Am. Chem. Soc., 2019, 141, 138–142 CrossRef CAS PubMed.
- C. Guo, M. Li, J. Chen and Y. Luo, Chem. Commun., 2020, 56, 117–120 RSC.
- M. Itoh, K. Inoue, J.-i. Ishikawa and K. Iwata, J. Organomet. Chem., 2001, 629, 1–6 CrossRef CAS.
- T. Li, K. N. McCabe, L. Maron, X. Leng and Y. Chen, ACS Catal., 2021, 11, 6348–6356 CrossRef CAS.
- K. P. Kepp, Inorg. Chem., 2016, 55, 9461–9470 CrossRef CAS PubMed.
- Organometallic Chemistry and Catalysis, ed. D. Astruc, Springer Berlin Heidelberg, 2007, pp. 289–311 Search PubMed.
- A. W. Cook, Z. R. Jones, G. Wu, S. L. Scott and T. W. Hayton, J. Am. Chem. Soc., 2018, 140, 394–400 CrossRef CAS PubMed.
- G.-y. Zhao, S. Hu, S.-c. Luo, J. Lei and Y. Wei, J. Alloys Compd., 2023, 949, 169885 CrossRef CAS.
- H. Lang, A. Jakob and B. Milde, Organometallics, 2012, 31, 7661–7693 CrossRef CAS.
- C. Ghiazza, T. Billard and A. Tlili, Chem. – Eur. J., 2017, 23, 10013–10016 CrossRef CAS PubMed.
- K. Jouvin, J. Heimburger and G. Evano, Chem. Sci., 2012, 3, 756–760 RSC.
- K. Sonogashira, J. Organomet. Chem., 2002, 653, 46–49 CrossRef CAS.
- Y.-Q. Ren, C.-J. Yang, Z.-L. Li, Q.-S. Gu, L. Liu and X.-Y. Liu, J. Fluorine Chem., 2023, 267, 110107 CrossRef CAS.
- F.-L. Wang, C.-J. Yang, J.-R. Liu, N.-Y. Yang, X.-Y. Dong, R.-Q. Jiang, X.-Y. Chang, Z.-L. Li, G.-X. Xu, D.-L. Yuan, Y.-S. Zhang, Q.-S. Gu, X. Hong and X.-Y. Liu, Nat. Chem., 2022, 14, 949–957 CrossRef CAS PubMed.
- L. Liu, K.-X. Guo, Y. Tian, C.-J. Yang, Q.-S. Gu, Z.-L. Li, L. Ye and X.-Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 26710–26717 CrossRef CAS PubMed.
- X. Bai, C. Wu, S. Ge and Y. Lu, Angew. Chem., Int. Ed., 2020, 59, 2764–2768 CrossRef CAS PubMed.
- X. Liu, W. Ming, Y. Zhang, A. Friedrich and T. B. Marder, Angew. Chem., Int. Ed., 2019, 58, 18923–18927 CrossRef CAS PubMed.
- Z.-L. Wang, F.-L. Zhang, J.-L. Xu, C.-C. Shan, M. Zhao and Y.-H. Xu, Org. Lett., 2020, 22, 7735–7742 CrossRef CAS PubMed.
- Q.-C. Gan, Z.-Q. Song, C.-H. Tung and L.-Z. Wu, Org. Lett., 2022, 24, 5192–5196 CrossRef CAS PubMed.
- W. J. Teo and S. Ge, Angew. Chem., Int. Ed., 2018, 57, 12935–12939 CrossRef CAS PubMed.
- W. J. Teo, X. Yang, Y. Y. Poon and S. Ge, Nat. Commun., 2020, 11, 5193 CrossRef CAS PubMed.
- M. Hu and S. Ge, Nat. Commun., 2020, 11, 765 CrossRef CAS PubMed.
- Y. e. You and S. Ge, Angew. Chem., Int. Ed., 2021, 60, 20684–20688 CrossRef CAS PubMed.
- X. Yang and S. Ge, Organometallics, 2022, 41, 1823–1828 CrossRef CAS.
- Y. Zhao and S. Ge, Angew. Chem., Int. Ed., 2022, 61, e202116133 CrossRef CAS PubMed.
- J. Li and S. Ge, Angew. Chem., Int. Ed., 2022, 61, e202213057 CrossRef CAS PubMed.
- B. B. Tan, M. Hu and S. Ge, Angew. Chem., Int. Ed., 2023, 62, e202307176 CrossRef CAS PubMed.
- H. Wen, X. Wan and Z. Huang, Angew. Chem., Int. Ed., 2018, 57, 6319–6323 CrossRef CAS PubMed.
- J. S. Peake, W. H. Nebergall and Y. T. Chen, J. Am. Chem. Soc., 1952, 74, 1526–1528 CrossRef CAS.
- N. Hirone, H. Sanjiki, R. Tanaka, T. Hata and H. Urabe, Angew. Chem., Int. Ed., 2010, 49, 7762–7764 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2235641. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4gc00220b |
| This journal is © The Royal Society of Chemistry 2024 |