
Recycling organoiridium waste to [(1,5-cyclooctadiene)IrCl]2†
Valeriy
Cherepakhin
 and
Travis J.
Williams
*
and
Travis J.
Williams
*
Loker Hydrocarbon Research Institute, Wrigley Institute for Environment and Sustainability, and Department of Chemistry, University of Southern California, Los Angeles, California 90089-1661, USA. E-mail: travisw@usc.edu
First published on 14th February 2024
Abstract
We report the first process for iridium element recovery from organoiridium waste that is quantitative, pyrolysis-free, and generates no iridium metal. The key step is oxidative degradation of the waste by bleach to crude iridium(IV) hydroxide. Its treatment with hydrazine and then hydrogen peroxide gives synthetically important hexachloroiridic acid, which is converted to [(1,5-cyclooctadiene)IrCl]2 in 87% yield.
Iridium is a rare element with the estimated abundance in the Earth's crust of 1 ppb and the scale of worldwide production of only 7–8 tons a year,1,2 yet it is in high demand for its many unique catalytic applications.3 Since its supply is limited and demand is growing, it is crucial to have a sustainable process for its recovery from industrial and laboratory secondary sources. A few methods are known for iridium recovery,4–6 all utilize pyrolysis and calcination of the waste materials to produce the free metal, and further processing of metallic iridium is associated with a number of problems. Particularly, iridium metal is exceptionally resistant to all known “wet” oxidants, even in the powder form.7 Practically, this is surmounted by chlorination with Cl2/NaCl (625 °C),8 fusion with BaO2 (750 °C),9 or alkaline fusion with Na2O2/NaOH (600–700 °C).10,11 These suffer from incomplete conversion, reactor corrosion, and the need for a special equipment to operate at such temperatures. R&D-scale users of iridium who develop new reactivity and processes face high metal cost and can currently satisfy our desire to conserve the metal only by returning iridium-containing waste to a large-scale recycler, but only when one can be found. In this work we address these problems with a simple protocol for iridium recycling from its waste solutions that circumvents generating the metal and is suitable for implementation using common laboratory equipment.
Over several years our research group has accumulated a substantial amount of organoiridium waste, which consisted of iridium complexes mostly with carbonyl, polydentate phosphine, pyridine, amide, NHC, and η5-Cp* ligands dissolved in a mixture of common solvents and exposed to air. These resulted from development of reactions for lactate synthesis,12,13 alcohol dehydrogenation,14 H2 generation from formic acid,15,16 and ketone hydrogenation17,18 among others (Fig. S9†). The waste contained considerable amounts of sodium and potassium salts (of CO32−, HCO2−, CF3SO3−, and PF6−) but no transition metals other than iridium. Initially, all volatile components were removed to leave a semi-solid residue (1) containing 3–10 wt% of iridium (Scheme 1).
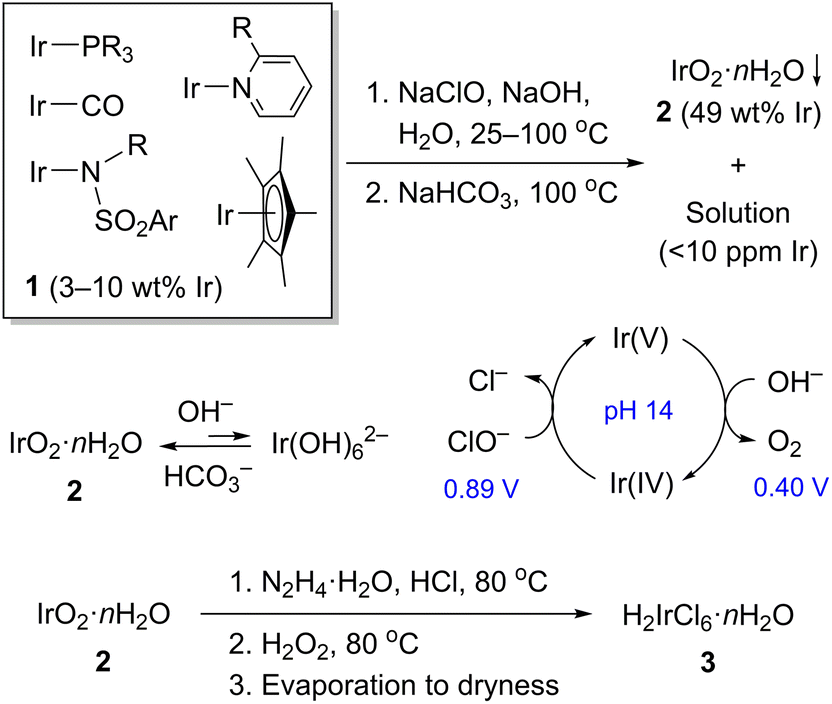 | ||
| Scheme 1 Processing organoiridium waste. | ||
As an alternative to pyrolysis, we accomplished oxidative degradation of 1 using a household solution of NaClO (7.5 wt%). This choice was justified by the facts that it is a cheap, green, and extremely powerful oxidant. Oxidation in an acidic solution was not a practical approach on our hands: these conditions give persistent organic derivatives of Ir(IV) rather than converging the system to the desired IrCl62−. By contrast, 1 is successfully oxidized in 1–3 M NaOH solution to insoluble and highly stable iridium(IV) hydroxide, IrO2·nH2O (2, Scheme 1).19,20 The waste (1) is processed as a semi-solid or a solution in CH2Cl2 to enable a good mass transfer. The reaction proceeds for one day at room temperature and then is driven to completion by heating briefly with added NaClO at 90–100 °C. Reaction completion is detected visually by two key features: (1) black hydroxide 2 is suspended in a yellow solution of Ir(OH)62−. In a strongly alkaline media, iridium(IV) exists primarily as a mixture of 2 and Ir(OH)62− with the equilibrium favoring the former. Full dissolution to Ir(OH)62− requires hydrothermal conditions (20 M NaOH, 200 °C),21 while full hydrolysis to 2 happens readily at pH 7–9. (2) Adding a fresh portion of NaClO to the completed reaction actively evolves O2 at 70–100 °C. Once all reducing components are gone, an iridium-catalyzed decomposition of unreacted ClO− begins.22,23 The standard potentials of couples ClO−/Cl− (0.890 V) and O2/OH− (0.401 V vs. SHE)24 are also consistent with the spontaneous reaction: 2ClO− → 2Cl− + O2.
During the oxidation we observe reversible color changes indicative of Ir(V)/Ir(IV) redox transitions. In a control experiment, a yellow solution of Ir(OH)62− reacts with NaClO at 70 °C turning brown at first, then suddenly evolving O2, and turning back to yellow within a few seconds. The sequence can be repeated multiple times suggesting a catalytic behavior of the brown species, apparently iridium(V) (Scheme 1). Although the existence of a transient iridium(V) species has been demonstrated in 2 M HClO4,25 nothing is known about its stability at pH 14. The transient pentavalent iridium could be involved also catalytically in degradation of organic ligands associated with the metal in mixture 1. The oxidized organics seem to be completely water-soluble, thus facilitating the separation of 2.
Quantitative precipitation of 2 was achieved by buffering the reaction mixture with excess of solid NaHCO3 at 100 °C (Scheme 1). During this step, Ir(OH)62− is fully hydrolyzed to 2. Once the precipitate settles (Fig. S1-C†), a colorless supernatant contains less than 10 ppm iridium, as determined by atomic emission spectroscopy (ICP-OES, Fig. S7†). Sometimes, 2 may form intensely colored colloid solutions because of a slow and incomplete hydrolysis of Ir(OH)62− (Fig. S2†). Fortunately, these colloids can be easily converted to solid 2 by the repeated oxidation (NaClO/NaOH) and buffering steps (NaHCO3).
Material 2 is amorphous by powder X-ray diffraction. Its iridium content was established gravimetrically by means of Cs2[IrCl6] as 49.0 wt%, which corresponds to net composition of IrO2·9.3H2O. Unfortunately, the sample cannot be qualified as pure, since it contains up to 2.9 wt% of carbon revealed by the combustion analysis. The carbon content is not changed after treatment with aqueous HCl, suggesting a contamination by non-volatile organics rather than carbonate. Despite the impurity, 2 is easily used in the next steps.
Samples of 2 are resistant to mineral acids, therefore, we utilize a redox approach to convert these to a water-soluble form. A facile dissolution is possible in 6 M HCl in the presence of hydrazine (80 °C, 10 min, Fig. S3-A†). The resulting solution contains a mixture of iridium(III) aqua-chloride complexes rather than simple IrCl63−, since its UV-Vis absorption bands at 415 and 356 nm were not detected.26 Hydrogen peroxide is then added (80 °C, 10 min) to destroy the excess of hydrazine and oxidize Ir(III) to orange-black IrCl62− (Fig. S3-B†). Its formation was confirmed by UV-Vis spectroscopy: the characteristic bands were observed at 412, 428, and 486 nm (Fig. S8†).27 Subsequent careful evaporation gives a brown-black crystalline hexachloroiridic acid, H2IrCl6·nH2O (3, Fig. S3-C†), which was used without purification.28 It is worth noting that excessive heating brings a partial reduction of 3 to Ir(III) chlorides, though not impactful in our case, this process might be detrimental in alternative iridium recycling schemes.
By far, compound 3 is one of the most common precursors for synthetic chemistry involving iridium. Hence, our recycling approach can be utilized to prepare a spectrum of its derivatives. To demonstrate, we synthesized [(1,5-cyclooctadiene)IrCl]2 (4) from 3 following a modified reported procedure (Table 1).29–31 The reaction involves reduction of 3 in a boiling alcohol solvent in the presence of 1,5-cyclooctadiene ligand. Air-sensitivity of 4 in solutions is well-known,31 therefore, the synthesis and isolation were conducted under oxygen-free conditions. Clean 4 was obtained in 87% yield using isopropanol, while the balance of iridium (13%) was recovered as Cs2[IrCl6] after workup (entry 3). Surprisingly, when Cs2[IrCl6] is used as a source of iridium in this reaction, only half of it converts to 4 (Table 1, entry 4). The balance persists as beige, insoluble Cs3[IrCl6] (calc'd 26.5%, found 28.2% Cl). An analytically pure sample of 4 was obtained after slow crystallization from CH2Cl2/hexane (Fig. S4-C†). Finally, the identity of 4 was confirmed by 1H and 13C{1H} NMR spectroscopy (Fig. S5 and S6†)32 and the combustion analysis.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
||||
|---|---|---|---|---|
| Entry | Ir source | Alcohol | Yield of 4, % | Recovered Ir,b % |
| a Conditions: 3 or Cs2[IrCl6] (3.8 mmol), 1,5-cyclooctadiene (6 mL), alcohol (10 mL), and water (10 mL) were refluxed under N2 for 12 h. b Solid leftover materials were treated with hot aqua regia and then with CsCl in a minimum amount of water to form insoluble Cs2[IrCl6]. | ||||
| 1 | 2 (0.5 g) → 3 | EtOH | 87 | — |
| 2 | 2 (1.5 g) → 3 | EtOH | 83 | 17 |
| 3 | 2 (1.5 g) → 3 | i-PrOH | 87 | 13 |
| 4 | Cs2[IrCl6] (3.6 g) | i-PrOH | 49 | 50 |
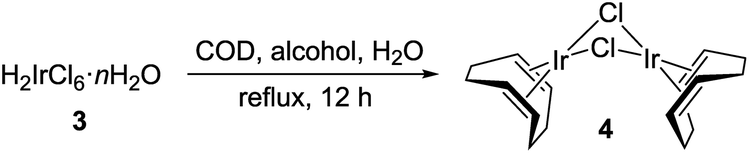
In conclusion, we developed a pyrolysis-free process for quantitative iridium recovery from laboratory-generated organoiridium waste that represents the first route available to R&D-scale iridium users. This was essential to our lab, because unlike rhodium, palladium, and ruthenium, we could find no commercial vendor who would recycle our iridium waste. The crucial steps of the process are the oxidative degradation of the waste and the redox dissolution of hydroxide 2. We show the practicality of our method by recovering iridium as synthetically important complex 4. Compounds 2, 3, and 4 require minimal to no purification. Because of its low cost and high simplicity, the process enables a very convenient way to reduce metal costs and protect this very precious natural resource. Overall, this route enables a very important option for sustainable use of iridium in laboratory practice and has potential to facilitate iridium recovery on industrial scale.
Author contributions
Conceptualization, data curation, formal analysis, investigation, methodology, project administration, validation, visualization, writing – original draft: V. C. Funding acquisition, resources, supervision, writing – review & editing: T. J. W.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is sponsored by the US Department of Energy (DE-EE0008825). We thank the NSF (CHE-2018740, DBI-0821671, CHE-0840366), the NIH (S10 RR25432), and USC Research and Innovation Instrumentation Awards for analytical tools.References
- W. M. Haynes, Abundance of Elements in the Earth's Crust and in the Sea, in CRC Handbook of Chemistry and Physics, CRC Press, 95th edn, 2014, pp. 14–19 Search PubMed.
- S. A. Singerling and R. F. Schulte, Platinum-Group Metals. In U.S. Geological Survey Minerals Yearbook–2018, USGS, 2021. https://d9-wret.s3.us-west-2.amazonaws.com/assets/palladium/production/atoms/files/myb1-2018-plati.pdf (accessed 2023-11-29).
- M. Ryan, Outlook 2023: Recycling and Thrifting – the Answer to the Iridium Question. Hydrogen Economist, November 16, 2022. https://www.pemedianetwork.com/hydrogen-economist/articles/green-hydrogen/2022/outlook-2023-recycling-and-thrifting-the-answer-to-the-iridium-question/(accessed 2023-11-29).
- C. Fan, K. Quan, Z. Han, F. Han, Z. Li, J. Liu and X. Liu, J. Sustain. Metall., 2023, 9, 909–926 CrossRef.
- G. B. Kauffman and R. D. Myers, J. Less-Common Met., 1978, 60, P1–P3 CrossRef CAS.
- M. Fan, S. Li, H. Deng, X. Zhang, G. Luo, Z. Huang and M. Chen, Sep. Purif. Technol., 2022, 289, 120765 CrossRef CAS.
- H. Renner, G. Schlamp, I. Kleinwächter, E. Drost, H. M. Lüschow, P. Tews, P. Panster, M. Diehl, J. Lang, T. Kreuzer, A. Knödler, K. A. Starz, K. Dermann, J. Rothaut, R. Drieselmann, C. Peter, R. Schiele, J. Coombes, M. Hosford and D. F. Lupton, Platinum Group Metals and Compounds, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2018, pp. 1–73 Search PubMed.
- G. B. Kauffmann, L. A. Teter, J. W. Hogarth and F. P. Dwyer, Inorg. Synth., 1966, 8, 223–227 CrossRef.
- A. K. Keep and S. Collard, Improvements to Processes for the Preparation of Iridium Acetate, GB2413323A, 2005.
- R. Gilchrist, Chem. Rev., 1943, 32, 277–372 CrossRef CAS.
- M. Stettner, M. Günther, S. F. Alameda, V. Thiel and S. Voss, Process for Digestion of a Metallic Iridium- and/or Iridium Oxide-Comprising Mixture of Solid Particles, US10590511B2, 2020.
- Z. Lu, I. Demianets, R. Hamze, N. J. Terrile and T. J. Williams, ACS Catal., 2016, 6, 2014–2017 CrossRef CAS.
- Z. Lu, V. Cherepakhin, T. Kapenstein and T. J. Williams, ACS Sustainable Chem. Eng., 2018, 6, 5749–5753 CrossRef CAS PubMed.
- V. Cherepakhin and T. J. Williams, ACS Catal., 2018, 8, 3754–3763 CrossRef CAS PubMed.
- J. J. A. Celaje, Z. Lu, E. A. Kedzie, N. J. Terrile, J. N. Lo and T. J. Williams, Nat. Commun., 2016, 7, 11308 CrossRef CAS PubMed.
- V. K. Do, N. A. Vargas, A. J. Chavez, L. Zhang, V. Cherepakhin, Z. Lu, R. P. Currier, P. A. Dub, J. C. Gordon and T. J. Williams, Catal. Sci. Technol., 2022, 12, 7182–7189 RSC.
- I. Demianets, V. Cherepakhin, A. Maertens, P. J. Lauridsen, S. M. Sharada and T. J. Williams, Polyhedron, 2020, 182, 114508 CrossRef CAS PubMed.
- L. Zhang, Z. Lu, A. R. Rander and T. J. Williams, Chem. Commun., 2023, 59, 8107–8110 RSC.
- R. Gilchrist and E. Wichers, J. Am. Chem. Soc., 1935, 57, 2565–2573 CrossRef CAS.
- F. Krauss and H. Gerlach, Z. Anorg. Allg. Chem., 1925, 143, 125–128 CrossRef CAS.
- R. Albrecht, H. Poddig, J. Hunger, M. Ruck, P. Benrath, A. Möller and T. Doert, Z. Anorg. Allg. Chem., 2021, 647, 667–672 CrossRef CAS.
- G. H. Ayres and M. H. Booth, J. Am. Chem. Soc., 1955, 77, 825–827 CrossRef CAS.
- G. H. Ayres and M. H. Booth, J. Am. Chem. Soc., 1955, 77, 828–833 CrossRef CAS.
- S. G. Bratsch, J. Phys. Chem. Ref. Data, 1989, 18, 1–21 CrossRef CAS.
- S. E. Castillo-Blum, D. T. Richens and A. G. Sykes, Inorg. Chem., 1989, 28, 954–960 CrossRef CAS.
- C. K. Jørgensen, Acta Chem. Scand., 1956, 10, 500–517 CrossRef.
- E. Blasius and W. Preetz, Z. Anorg. Allg. Chem., 1965, 335, 16–35 CrossRef CAS.
- E. Pietsch, Gmelins Handbuch der Anorganischen Chemie, Verlag Chemie, 1939, vol. 67 Search PubMed.
- G. Winkhaus and H. Singer, Chem. Ber., 1966, 99, 3610–3618 CrossRef CAS.
- J. L. Herde, J. C. Lambert and C. V. Senoff, Inorg. Synth., 1974, 15, 18–20 CrossRef CAS.
- F. A. Cotton, P. Lahuerta, M. Sanau and W. Schwotzer, Inorg. Chim. Acta, 1986, 120, 153–157 CrossRef CAS.
- L. Jürgensen, M. Frank, M. Pyeon, L. Czympiel and S. Mathur, Organometallics, 2017, 36, 2331–2337 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data (PDF). See DOI: https://doi.org/10.1039/d4gc00151f |
| This journal is © The Royal Society of Chemistry 2024 |