
Electrocatalytic C–N coupling for urea synthesis: a critical review
Chuanju
Yang†
a,
Zhe
Li†
b,
Junpeng
Xu
c,
Yujing
Jiang
a and
Wenlei
Zhu
 *a
*a
aState Key Laboratory of Pollution Control and Resource Reuse, the Frontiers Science Center for Critical Earth Material Cycling, School of the Environment, Nanjing University, Nanjing 210023, People's Republic of China. E-mail: wenleizhu@nju.edu.cn
bNanjing Institute of Environmental Sciences, Ministry of Ecology and Environment of the People's Republic of China, Nanjing 210042, People's Republic of China
cState Key Laboratory of Pharmaceutical Biotechnology, Department of Sports Medicine and Adult Reconstructive Surgery, Nanjing Drum Tower Hospital, The Affiliated Hospital of Nanjing University Medical School; State Key Laboratory of Pharmaceutical Biotechnology, Medical School, Nanjing University, Nanjing 210093, People's Republic of China
First published on 18th March 2024
Abstract
Urea is one of the most important artificial nitrogen fertilizers in the agricultural economy and can provide essential nitrogen for plant growth. However, the industrial production of urea is very energy consumptive. In the past few years, the electrocatalytic approach has been regarded as a promising green approach for urea synthesis under mild conditions. Therefore, using carbon dioxide (CO2) and nitrogenous molecules as feedstocks, electrocatalytic urea synthesis has received extensive interest and investigation. Despite the tremendous efforts that have been made, it is still very hard to significantly increase urea synthetic efficiency due to complex mechanisms. Focusing on catalyst design, characterization, electrolytic cells chosen, urea identification/quantification, determination of intermediates, and in-depth mechanisms, the recent advances in urea electrocatalytic production are summarized and discussed. Furthermore, we analyze the current challenges and prospects for the development path of electrocatalytic urea synthesis. From an application viewpoint, we hope these insights shed light on designing efficient catalysts in future studies and bring broader application prospects for green urea synthesis.
1. Introduction
Electrosynthesis methods are appealing approaches for high-value chemical production from small molecules.1–5 By finely designing the electrocatalyst, electrochemical conversion methods can lower the energy barrier of energy-intensive processes and complete the conversions under mild conditions.6–9 For instance, various catalysts have been introduced to facilitate electrochemical CO2 reduction (CO2R).10,11 Nowadays, CO2R reaction studies are able to reach industrialization requirements with an operating current density over 1 A cm−2 towards different multi-carbon products.12,13 On the basis of CO2R, many efforts have been made including electrochemical carbon–nitrogen (C–N) coupling methods to increase the diversity of products (e.g., acetamide, urea, methylamine, formamide, etc.).14 Among these organonitrogens, urea is highly anticipated since it is an excellent nutrient carrier, and an important fertilizer in agriculture.15,16 In addition, urea is also the indispensable feedstock for urea–melamine–formaldehyde resin, urea formaldehyde, and barbiturate synthesis.17In traditional industrial manufacturing processes, urea synthesis relies on the thermal catalytic conversion of ammonium carbamate, and has made great contributions to increasing output and has driven social advancement.18,19 However, the production of feedstock ammonia and the subsequent ammonium carbamate must overcome energy-intensive processes to break the strong bonds of nitrogen gas (N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, ∼941 kJ mol−1) and carbon dioxide (C
N, ∼941 kJ mol−1) and carbon dioxide (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, ∼806 kJ mol−1), respectively.20,21 A huge amount of energy is consumed through the harsh conditions, with the emission of undesirable greenhouse gases, which is contradictory to sustainability goals.22 Therefore, the development of green and effective electrocatalytic methods for urea synthesis is crucial for the development of human society. Benefiting from the development of advanced characterization and theoretical calculation technologies, researchers can have a better understanding of the reaction mechanism and optimize the synthetic routes of catalysts.23 Thus, the electrocatalytic synthesis of urea is promising and has been widely investigated.
O, ∼806 kJ mol−1), respectively.20,21 A huge amount of energy is consumed through the harsh conditions, with the emission of undesirable greenhouse gases, which is contradictory to sustainability goals.22 Therefore, the development of green and effective electrocatalytic methods for urea synthesis is crucial for the development of human society. Benefiting from the development of advanced characterization and theoretical calculation technologies, researchers can have a better understanding of the reaction mechanism and optimize the synthetic routes of catalysts.23 Thus, the electrocatalytic synthesis of urea is promising and has been widely investigated.
Despite the great potential of resource recovery, several challenges are still impeding the industrialization of urea electrocatalytic synthesis. Although electrosynthesis methods avoid the energy penalty and direct carbon emission from natural gas reformation, fossil fuel-based energy sources are still contrary to the conception of cleaner production. Clean, safe, and renewable energy sources are urgently needed. On the other hand, opinions are divided on the mechanism of C–N bond formation, which makes it still difficult to obtain satisfactory results with the existing electrocatalytic urea synthetic systems. A decent understanding of the in-depth factors would help scientists to develop efficient urea electrocatalytic synthesis systems. In this review, the development history of urea electrocatalytic synthesis is summarized, focusing on the key steps of the common technical schemes. Focusing on catalyst design and characterization, reactor selection, product monitoring, and mechanism investigation, the advantages and shortcomings of current reaction systems are fully discussed, and possible approaches for addressing those concerns are suggested. Finally, we analyze the requirements for realizing highly efficient urea electrocatalytic synthesis, making basic studies match the requirements of practical applications.
2. Milestones in electrocatalytic urea synthesis
The formation of C–N bonds is crucial for electrocatalytic organonitrogen synthesis. For urea, with the generation of N-nucleophilic intermediates (*NH3, *NH2OH, *N2, *NO2, *NH2) and C-intermediates (*CO2, *CO, *COOH, *CH2O) on the active sites, the N-nucleophilic intermediates will attack their neighboring C-intermediates, following a configuration-stabilizing process.24,25 Currently, accompanied by greenhouse gas CO2, reactants such as N2, NO2−, NO3−, NO are employed as the nitrogen sources to produce urea during the electrocatalytic process (Fig. 1). Electrocatalytic synthesis of urea was initially reported by Shibata et al. in 1995.26 Inspired by this preliminary study, numerous reaction systems were developed and pushed electrocatalytic urea synthesis performance under ambient conditions. In 2016, Kayan et al. accomplished urea synthesis using N2 and CO2 as feedstocks on polyaniline (PANI) and poly-pyrrole (PPy)-coated platinum electrodes, which could reach 7.1% FEurea at −0.225 V vs. RHE.27 In 2020, Chen et al. proposed a Pd1Cu1/TiO2-400 catalyst that achieved a higher rate of urea production with a 8.92% FEurea at −0.4 V vs. RHE.28 However, the high bond energy of N2 leads to a large over-potential, which makes the hydrogen evolution reaction occupy a dominant position in the electrocatalytic system. In response to the competitive adsorption of N2 and CO2 on the electrode surface, developing novel reactors and catalysts with multiple active sites is gaining more attention. In 2022, Huang and coworkers prepared Zn nanobelts (Zn NBs) by electrochemical treatment of ZnO nanosheets (ZnO NSs).29 It is worth mentioning that the proposed system exhibits the urea yield rate of 15.13 mmol h−1 g−1, and the corresponding FEurea reached 11.26% at −40 mA cm−2 in a flow cell. The comprehensive utilization of nitrogen-containing molecules and CO2 points to a possible solution for dealing with pollutant recovery and carbon neutralization in a single module.30,31 According to the development trend in electrocatalytic urea synthesis, it is crucial to focus on byproduct inhibition and the maintenance of intermediates during the C–N coupling process. Hence, understanding the characteristics of different types of catalyst is beneficial for their subsequent design and modification.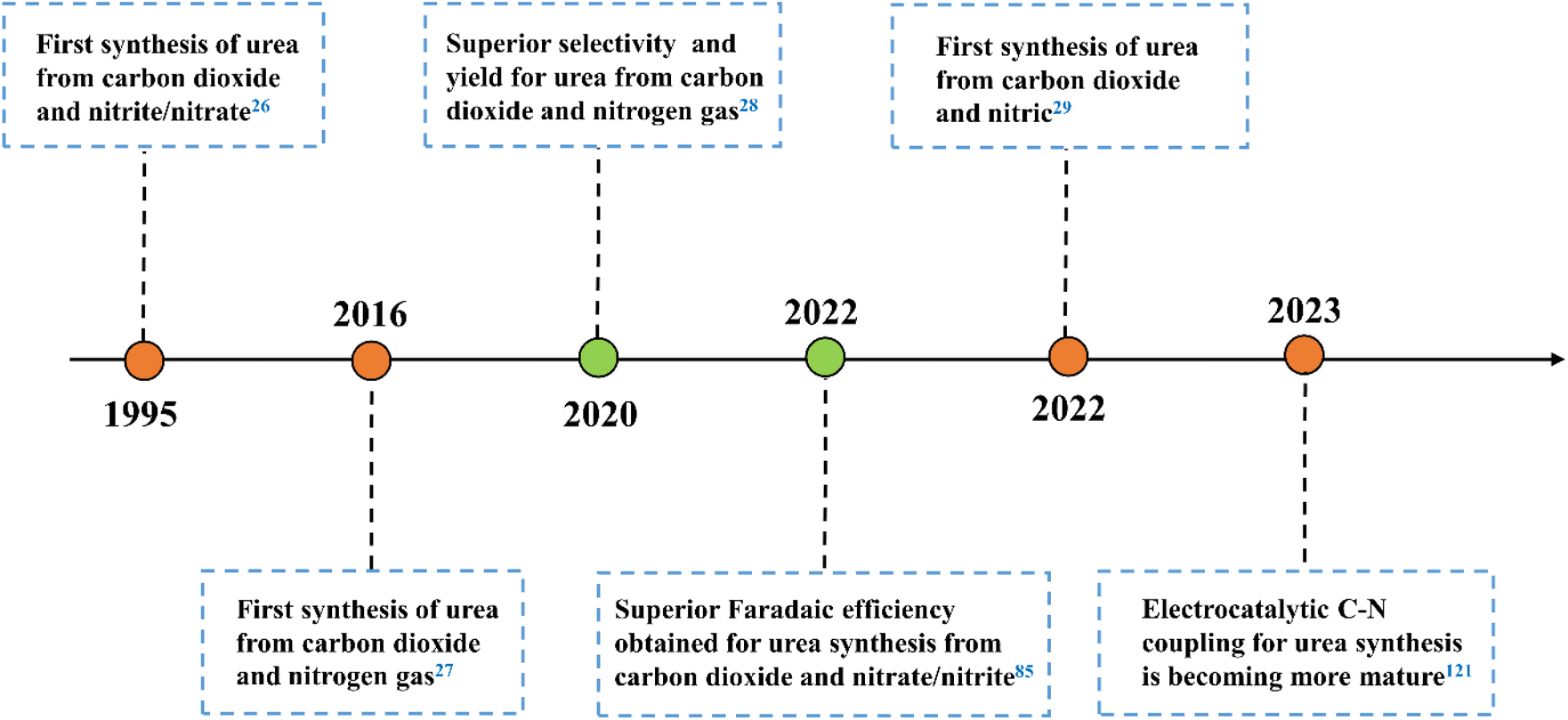 | ||
| Fig. 1 The progress of C–N coupling reaction for electrocatalytic urea synthesis. | ||
3. Catalysts for electrochemical urea synthesis
The catalyst is of great significance in building effective reaction systems with considerable yield and faradaic efficiency.32–36 The ideal catalyst should have exceptional capabilities for reactant capture, energy barrier management, bond cleavage, and coupling of intermediates.37,38 Currently, the major electrocatalytic systems for urea synthesis apply metal-based catalysts including copper, gold, indium, etc (Table 1). Aiming to enhance catalytic performances, the optimization of electronic structure, coordination environment, carrier properties, and interfacial interactions are key factors to be solved. By structural engineering, catalysts can acquire unique conformational relationships to complete the complicated reactions in an easy path.39–42| Catalyst | Media | C and N sources | Key intermediates | FE% | Detection method | Yield rate | Stability (h) | Configuration | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Pd1Cu1/TiO2-400 | 1 M KHCO3 | CO2, N2 | *NCON* | 8.92 | Urease method | 3.36 mmol g−1 h−1 | 12 | H-cell | Chen et al. (2020)28 |
| BiFeO3/BiVO4 | 0.1 M KHCO3 | CO2, N2 | *NCON* | 17 | DAMO-TSC | 4.94 mmol g−1 h−1 | 10 | H-cell | Yuan et al. (2021)95 |
| Bi-BiVO4 | 0.1 M KHCO3 | CO2, N2 | *NCON* | 12.55 | DAMO-TSC | 5.91 mmol g−1 h−1 | 10 | H-cell | Yuan et al. (2021)94 |
| CuPc NTs | 0.1 M KHCO3 | CO2, N2 | *NCON* | 12.99 | DAMO-TSC | 2.39 mmol g−1 h−1 | 10 | H-cell | Mukherjee et al. (2022)143 |
| Ni(BO3)2-150 | 0.1 M KHCO3 | CO2, N2 | *NCON* | 20 | DAMO-TSC | 9.70 mmol g−1 h−1 | 20 | H-cell | Yuan et al. (2021)116 |
| VN-Cu3N-300 | 0.1 M KHCO3 | CO2, N2 | *NCON* | 28.7 | DAMO-TSC | 2.67 mmol g−1 h−1 | 10 | H-cell | Lv et al. (2023)144 |
| VO-InOOH | 0.1 M KNO3 | CO2, NO3− | *CO2NO2 | 51 | DAMO-TSC | 9.88 mmol g−1 h−1 | 10 | H-cell | Lv et al. (2022)85 |
| Fe-Fe3O4/CNs | 0.1 M KNO3 | CO2, NO3− | *CONH2 | 16.5 | DAMO-TSC | 24.2 mmol g−1 h−1 | 10 | H-cell | Geng et al. (2023)145 |
| Ru-Cu CF | 0.1 M KNO3 | CO2, NO3− | *CONH2 | 25.4 | DAMO-TSC | 0.00253 mmol cm−2 h−1 | 10 | H-cell | Qin et al. (2022)146 |
| In(OH)3-S | 0.1 M KNO3 | CO2, NO3− | *CO2NO2 | 53.4 | DAMO-TSC | 8.89 mmol g−1 h−1 | 8 | H-cell | Lv et al. (2021)41 |
| TiO2/Nafion | 0.1 M KNO3 | CO2, NO3− | *CONH2 | 40 | Urease method | N/A | 5.2 | H-cell | Saravanakumar et al. (2017)136 |
| Cu/ZnO GDEs | 0.1 M KNO3 | CO2, NO3− | *CONH2 | 37.4 | DAMO-TSC | 16 mmol g−1 h−1 | 5 | Flow cell | Wang et al. (2023)147 |
| CuWO4 | 0.1 M KNO3 | CO2, NO3− | *CONO2 | 70.1 | Urease method | 1.64 mmol g−1 h−1 | 10 | H-cell | Zhao et al. (2023)122 |
| FeNi3 | 0.1 M KNO3 | CO2, NO3− | N/A | 16.58 | Urease method | 8.23 mmol g−1 h−1 | N/A | H-cell | Wang et al. (2023)55 |
| Co1-TiO2 | 1 M KNO3 | CO2, NO3− | *OCNO | 36.2 | Urease method | 212.8 mmol g−1 h−1 | N/A | H-cell | Tu et al. (2024)148 |
| XC72R-AuPd | 0.075 M KHCO3 + 0.025 M KNO3 | CO2, NO3− | *CONH2 | 15.6 | DAMO-TSC | 3.4 mmol g−1 h−1 | 10 | H-cell | Wang et al. (2022)40 |
| Cu-GS-800 | 0.1 M KHCO3 + 0.1 M KNO3 | CO2, NO3− | *CONH2 | 28 | DAMO-TSC | 30.63 mmol g−1 h−1 | N/A | H-cell | Leverett et al. (2022)65 |
| F-CNT-300 | 0.1 M KHCO3 + 0.1 M KNO3 | CO2, NO3− | *CONH2 | 18 | DAMO-TSC | 6.36 mmol g−1 h−1 | 8 | H-cell | Liu et al. (2022)108 |
| Bi:10%In/C NPs | 0.1 M KHCO3 + 0.1 M KNO3 | CO2, NO3− | *CONH2 | 20.31 | Urease method | 10.1 mmol g−1 h−1 | N/A | H-cell | Mao et al. (2024)125 |
| m-Cu2O | 0.1 M KHCO3 + 10 mM KNO3 | CO2, NO3− | *OCNO | 9.43 | Urease method | 29.2 mmol g−1 h−1 | N/A | H-cell | Wang et al. (2023)149 |
| B-FeNi-DASC | 0.1 M KHCO3 + 50 mM KNO3 | CO2, NO3− | *CONH2 | 17.8 | Urease method | 20.2 mmol g−1 h−1 | N/A | H-cell | Zhang et al. (2022)76 |
| Cu1-CeO2 | 0.1 M KHCO3 + 50 mM KNO3 | CO2, NO3− | *OCNO | N/A | Urease method | 52.84 mmol g−1 h−1 | 4 | H-cell | Wei et al. (2023)66 |
| Vo-CeO2-750 | 0.1 M KHCO3 + 50 mM KNO3 | CO2, NO3− | *OCNO | N/A | Urease method | 15.7 mmol g−1 h−1 | 5 | H-cell | Wei et al. (2022)86 |
| 3D Zn/Cu hybrid | 0.1 M KHCO3 + 1000 ppm KNO3 | CO2, NO3− | *CO2NO2 | 67 | DAMO-TSC | 55.3 mmol g−1 h−1 | 32 | Flow cell | Luo et al. (2023)121 |
| Cu@Zn | 0.2 M KHCO3 + 0.1 M KNO3 | CO2, NO3− | *CONH2 | 9.28 | 1H-NMR | 0.00729 mmol cm−2 h−1 | 12 | H-cell | Meng et al. (2022)151 |
| CuInS2/TF | 0.05 M KHCO3 + 0.05 M KNO3 | CO2, NO3− | *CONH2 | 19.6 | DAMO-TSC | 50.29 mmol g−1 h−1 | N/A | H-cell | Chen et al. (2024)152 |
| CuO50ZnO50 | 0.1 M Na2SO4 + 0.1 M NaNO3 | CO2, NO3− | N/A | 41 | Urease method | N/A | N/A | H-cell | Anastasiadou et al. (2023)153 |
| Te-doped Pd | 0.1 M KHCO3 + 0.01 M KNO2 | CO2, NO2− | *CONH2 | 12.2 | DAMO-TSC | N/A | 5 | H-cell | Feng et al. (2020)57 |
| ZnO-Vo | 0.2 M NaHCO3 + 0.1 M NaNO2 | CO2, NO2− | *CONH2 | 23.3 | HPLC | 16.56 mmol g−1 h−1 | 15 | H-cell | Meng et al. (2021)87 |
| Co-NiOx@GDY | 0.01 M NaNO2 | CO2, NO2− | *CONH2 | 64.3 | DAMO-TSC | 15.1 mmol g−1 h−1 | N/A | H-cell | Zhang et al. (2022)98 |
| Cu-TiO2-Vo | 0.2 M KHCO3 + 0.02 M KNO2 | CO2, NO2− | *CONH2 | 43.1 | Urease method | 20.8 mmol g−1 h−1 | N/A | H-cell | Cao et al. (2020)89 |
| Au–Cu (SANFs) | 0.5 M KHCO3 + 0.01 M KNO2 | CO2, NO2− | *CONH2 | 24.7 | DAMO-TSC | 64.8 mmol g−1 h−1 | N/A | H-cell | Liu et al. (2022)56 |
| Zn NBs | 0.2 M KHCO3 | CO2, NO | *CONH2 | 11.26 | DAMO-TSC | 15.13 mmol g−1 h−1 | N/A | Flow cell | Huang et al. (2022)29 |
3.1 Monometallic catalysts
Monometallic catalysts have been utilized in electrocatalytic urea synthesis processes due to their simplicity. With differentiated capabilities affecting electrocatalytic processes, monometallic catalysts with optimized metal types can be utilized in specific reactions, such as Pt (the most efficient catalyst for HER and ORR), Ir (high-efficiency catalyst for OER), and Cu (a good candidate for nitrate to ammonia production).43–50 As aforementioned, Shibata and colleagues indicated that carbon dioxide can simultaneously reduce with nitrite/nitrate on a Cu-loaded gas-diffusion electrode to produce urea, which sets the foundation and verifies the feasibility of electrocatalytic methods in urea synthesis.26 However, the performances of monometallic catalysts are still at a low level due to the complicated mechanisms.51 Therefore, researchers have modified monometallic catalysts by means of structural engineering in the hope of obtaining better reaction results.3.2 Alloy catalysts
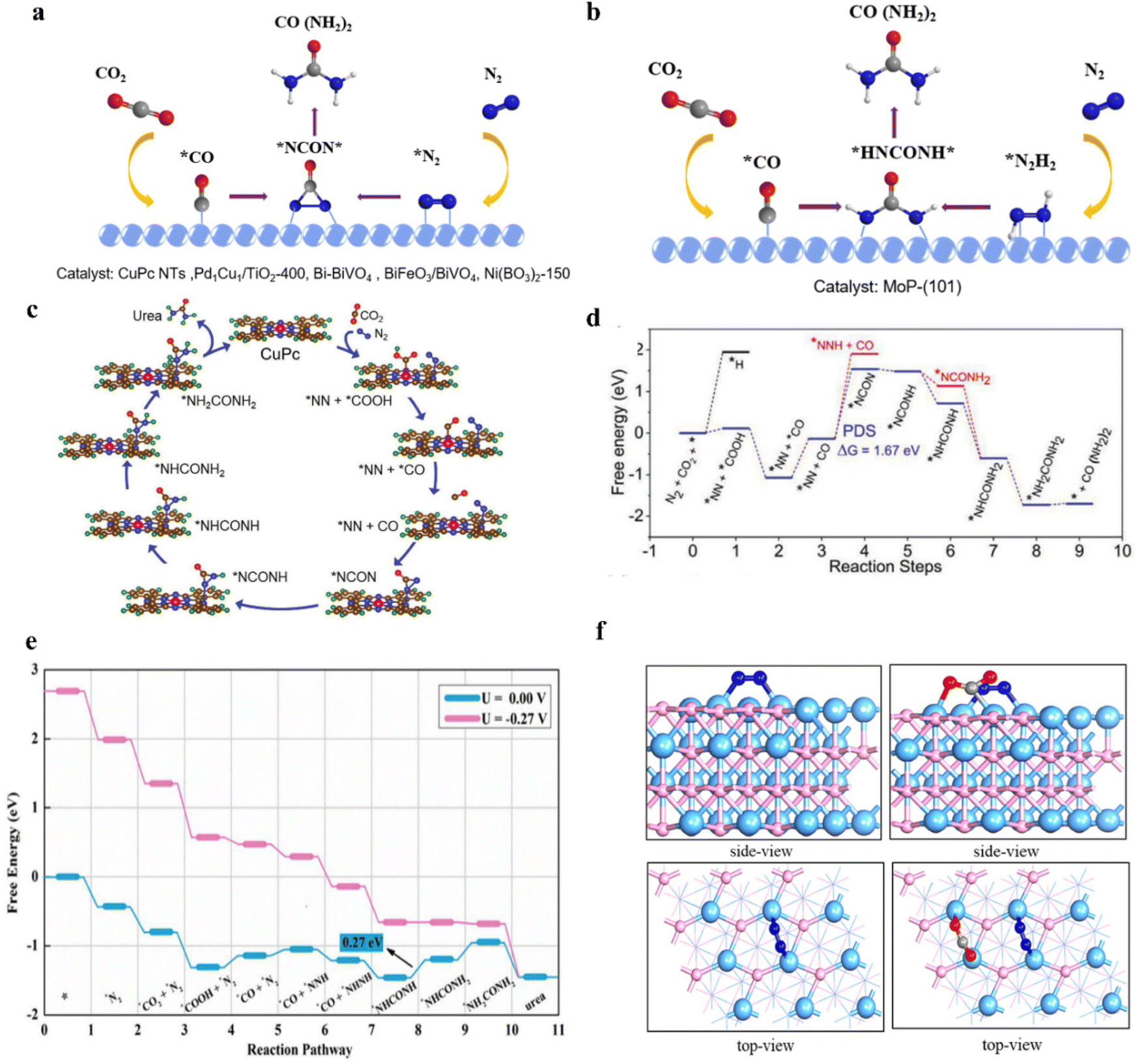
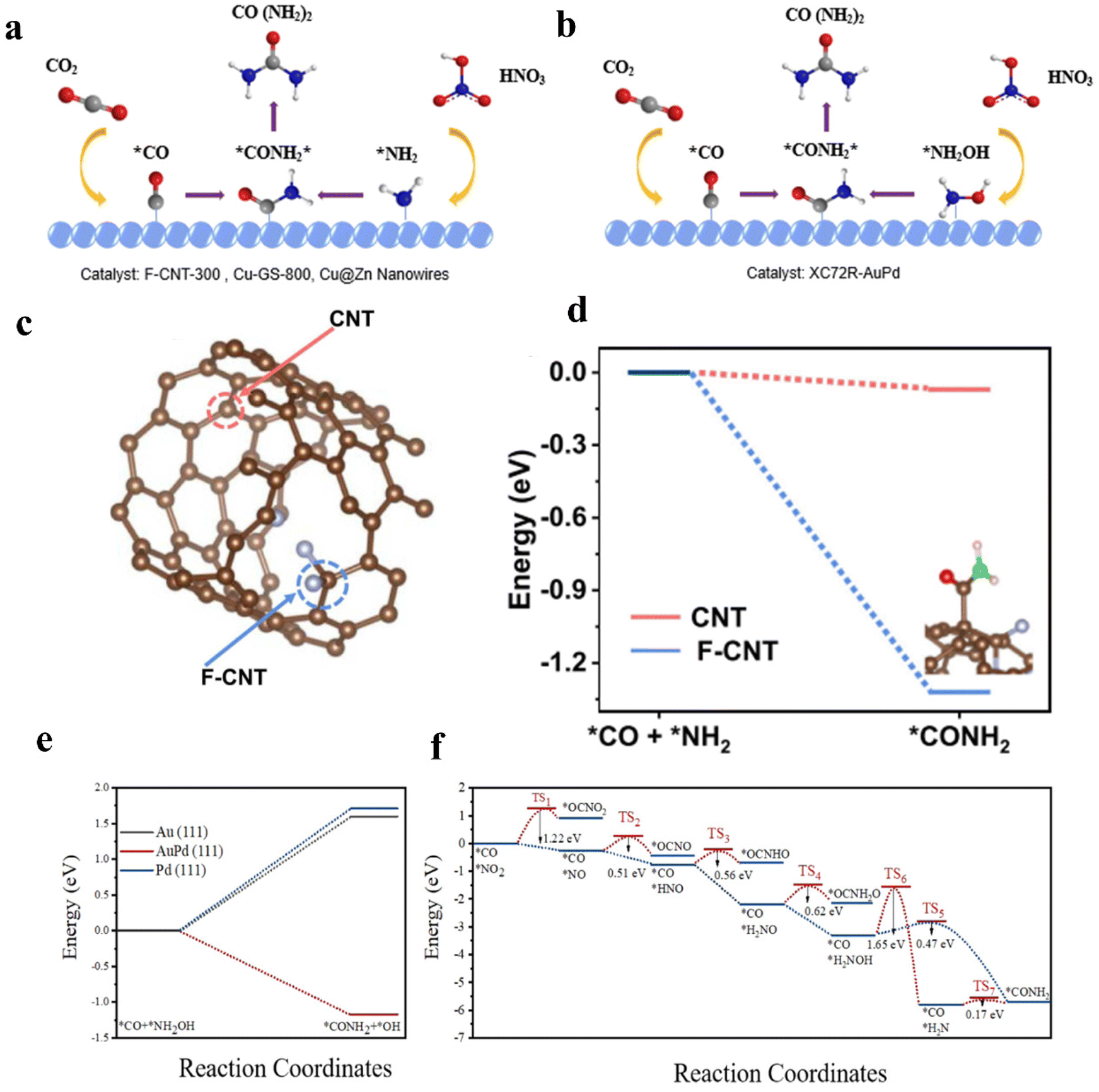
Alloying can modify the electronic and geometric structure of the active sites, which enhances the intrinsic adsorption properties of intermediates and facilitates the formation of products.52 Based on previous studies, nickel-based catalysts demonstrate excellent performance in the CO2 reduction reaction (CO2RR), while iron-based catalysts exhibit remarkable efficacy in the nitrate reduction reaction (NO3RR).53,54 In this case, Hou and his colleagues designed a FeNi/NC alloy catalyst for efficient urea synthesis.55 By effectively integrating the advantages between iron-based and nickel-based catalysts, the FeNi/NC alloy catalyst shows an excellent urea yield rate of 496.5 mg h−1 g−1 and faradaic efficiency (FE) of 16.58% at −0.9 V vs. RHE. Wang and colleagues proposed a XC72R-AuPd electrocatalyst through the one-pot reduction of metal precursors with NaBH4 (Fig. 2a).40 The catalyst exhibits exceptional catalytic properties, with a high FE of 15.6% and excellent electrochemical stability when CO2 and NO3− are used as reactants. The catalytic performance of the XC72R-AuPd outcompetes that of monometallic catalysts XC72R-Au and XC72R-Pd, which showed urea FE of 3.6% and 6.7%, respectively (Fig. 2b). They noted that a weak adsorption of CO by the catalyst would hinder the production of the active intermediate *CO, while a strong adsorption would cause catalyst poisoning. On this theoretical basis, the XC72R-AuPd electrocatalyst possesses appropriate CO adsorption capacity due to the incorporation of a small amount Pd into Au nanocrystal. Moreover, this also leads to a thermodynamically spontaneous C–N coupling between *NH2OH and *CO, which is in favor of the formation of urea (Fig. 2c). Liu and his colleagues fabricated an AuCu self-assembled nanofiber (AuCu SANF) via the hydrothermal method, coupling CO2R with nitrite reduction (NO2R) for urea electrosynthesis (Fig. 2d and e).56 The AuCu SANFs showcased excellent urea synthesis performance, achieving a high urea faradaic efficiency of 24.7% at −1.45 V vs. RHE (Fig. 2f). Also, the proportions of Au and Cu in SANF had an impact on the urea faradaic efficiency. Furthermore, the authors revealed that AuCu SANFs could modulate the coordination state and electronic structure of Au and Cu, which facilitated the adsorption and activation of reactants (CO2, NO2−). Feng and coworkers prepared the Te-doped Pd nanocrystal (denoted as Te–Pd NC) by a wet-chemical approach (Fig. 2g).57 The electrochemical synthesis of urea was achieved with nearly 12.2% faradaic efficiency at −1.1 V vs. RHE, much higher than those of Pd NCs (Fig. 2h). According to mechanism studies and theoretical calculations, doping with Te promotes the formation of *CO and eliminates active sites that strongly adsorb CO, which results in the Te-doped Pd nanocrystal exhibiting a higher stability against CO poisoning (Fig. 2i). The synergy between Te and Pd in Te–Pd NCs significantly promotes the transformation of CO2 to *CO and NO2− to *NH2, which facilitates the binding between *CO and *NH2 (Fig. 2j and k).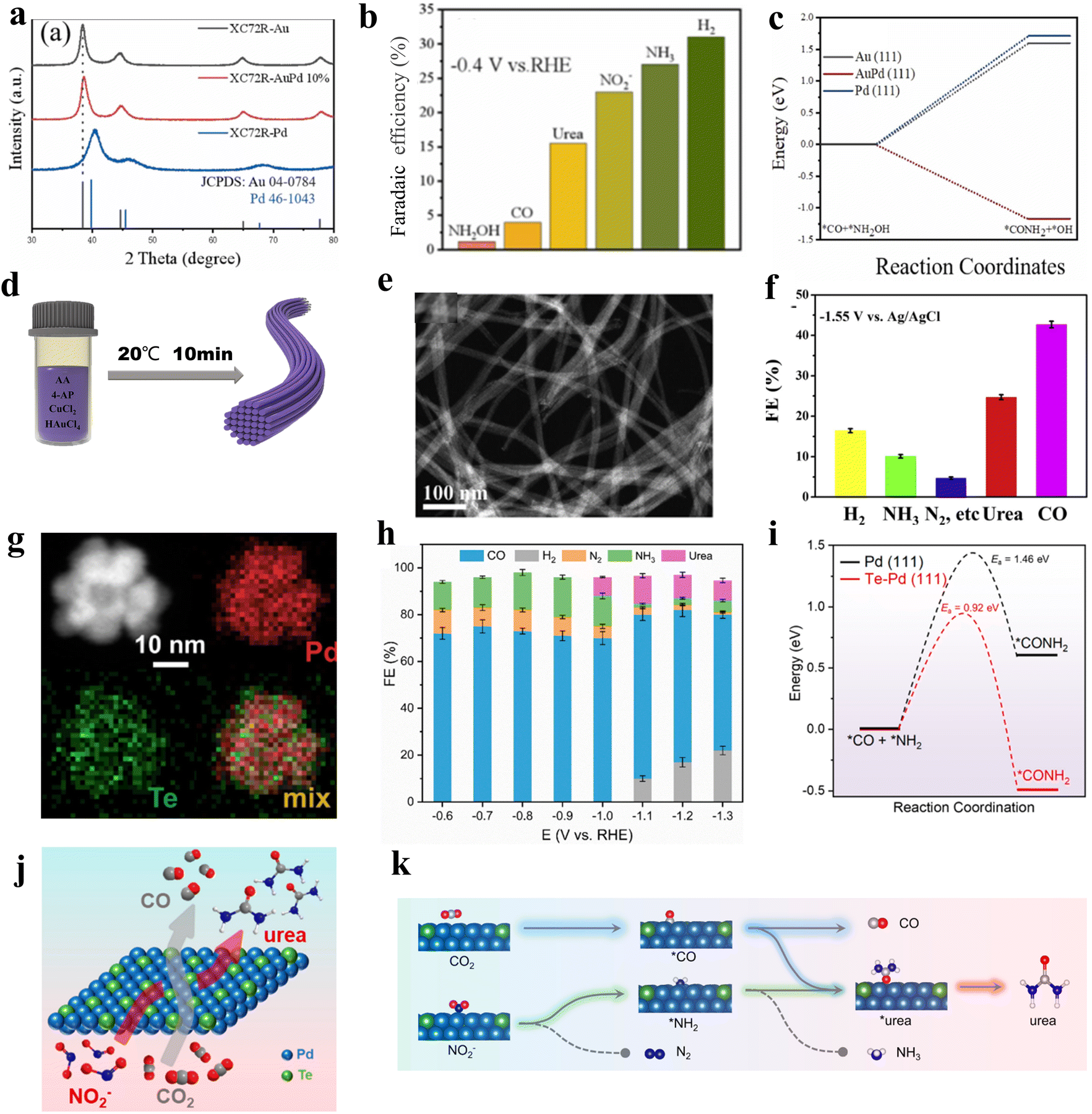 | ||
| Fig. 2 (a) XRD patterns of XC72R-AuPd samples. (b) Faradaic efficiencies for different products by using XC72R-AuPd at −0.4 V vs. RHE in H-cell. (c) Free energies for *CO + *NH2OH → *CONH2 on Au, AuPd, and Pd. Reproduced from ref. 40, with permission from Elsevier, copyright 2022. (d) The fabrication of the AuCu SANFs. (e) HAADF-STEM images of AuCu SANFs. (f) Faradaic efficiencies for different products by using AuCu SANFs at −1.55 V (vs. Ag/AgCl) in H-cell. Reproduced from ref. 56, with permission from Elsevier, copyright 2022. (g) HAADF-STEM image and corresponding elemental mappings of Te–Pd NCs. (h) Faradaic efficiencies for different products by using Te–Pd NCs/C in H-cell. (i) Free energy changes and activation barriers of *CONH2 on the Te–Pd (111) surface and pure Pd (111) surface, respectively. (j) A schematic depicts urea formation from simultaneous electroreduction of CO2 and NO2−. (k) Urea synthesis from CO2RR and NO2RR on Te–Pd NCs. Reproduced from ref. 57, with permission from American Chemical Society, copyright 2020. | ||
The study and preparation of alloy catalysts mostly rely on the interaction of two metals. Although the preparation and evaluation of alloy electrocatalysts have progressed significantly, their size, composition, and active site control still lack a reliable method to control. Furthermore, the structural relationships of alloy electrocatalysts are still not thoroughly researched. As a result, sophisticated theoretical calculations and modelling methods are necessary to achieve catalytic co-production of urea. In addition, considering the high costs of using metal precursors for metallic and alloy catalyst synthesis, functional atomic catalysts with lower precursor utilization have received much attention in recent decades.58,59
3.3 Single-atom structures
The smaller size means the existence of more low-coordinated metal atoms with catalytic activity, leading to a higher specific activity (SA).60 Owing to the low cost, minimal metal usage, large surface areas, and high activity, single-atom catalysts (SACs) have recently garnered attention in energy conversion reactions.61 In SACs, around the active center, the localized ligands impact the activity, stability, and selectivity. With a lone pair of electrons, ligands can trap isolated metal centers and regulate their electronic structure.62,63 Over the past few years, SACs have been well developed in the electrochemical carbon dioxide reduction (ECR) system and nitrogenous species reduction reaction (NRR).64 According to the existing research bases, using single-atom catalysts to realize electrocatalytic urea synthesis has been further investigated. For instance, Leverett et al. distributed Cu single atoms on a nitrogen-doped carbon substrate to form Cu–Nx–Cxvia hydrothermal methods (Fig. 3a).65 As the pyrolysis temperature increases from 800 °C to 1000 °C, the coordination structure changes from Cu–N4 sites to Cu–N3–C1 and Cu–N2–C2 sites (Fig. 3b). The configuration of Cu–Nx–Cx plays a vital role in directing CO2R and NO3R reactions. When using CO2-saturated KHCO3 as the electrolyte, the FECu–N4 for CO reaches 59% at −0.8 V vs. RHE, which is higher than that of Cu–N3–C1 and Cu–N2–C2. On the other hand, Cu–N2–C2 prefers to carry out NO3RR at similar potentials. It is noteworthy that, when introducing NO3− into the electrolyte of CO2RR, Cu–N4 reaches a 28% faradaic efficiency for urea production at −0.9 V vs. RHE, which is higher than Cu–N3–C1 and Cu–N2–C2 (Fig. 3c). DFT calculations reveal comparable energy barriers for C–N bond formation from *CO and *NH2 on each coordination structure (Fig. 3d). The key to unlocking exceptional CO2RR performance lies in the undercoordinated N atoms that surround isolated Cu species. These N atoms can strongly interact with CO2, resulting in a significant enhancement of the overall CO2RR process. Thus, CO2RR exhibits greater favorability on Cu–N4 sites, resulting in greater urea yield on those sites. However, remarkable CO2 reduction capacity does not always relate to satisfactory urea synthesis performance; the equilibrium between CO2RR and NO3RR is the key to achieving the ideal state of urea synthesis. Wei et al. fabricated a Cu single-atom catalyst on a CeO2 support (Cu-CeO2) through the wet impregnation and calcination method.66 Catalysts were labeled L-Cu1-CeO2 and H-Cu1-CeO2 based on the concentration of impregnated copper. They found copper was atomically dispersed in L-Cu1-CeO2, while the H-Cu1-CeO2 surface was distributed with numerous copper clusters and nanoparticles (Fig. 3e and f). As a result, the L-Cu1-CeO2 exhibits a urea yield rate of 52.84 mmol h−1 g−1 at −1.6 V vs. RHE, which is higher than that of CeO2 and H-Cu1-CeO2 (Fig. 3g). Based on the in situ X-ray absorption spectroscopy (XAS) results, Cu single atoms tend to form Cu clusters during the electrocatalytic process, and will recover to the single-atom state at open circuit voltage (OCV) (Fig. 3h). To identify whether the urea yield rate is related to the copper active sites or the electronic structure changes of Cu-CeO2, various metal-CeO2 single-atom structures have also been investigated. According to the experimental results, the performance of the Cu1-CeO2 structure is significantly superior to other M1-CeO2 structures. Through the projected density of states (PDOS) of adsorbed NO3− and CO2 on the M1-CeO2 catalysts, it was indicated that only Cu–O bonds offer uniquely hindered Lewis acid–base active sites that can realize efficient urea synthesis on Cu1-CeO2.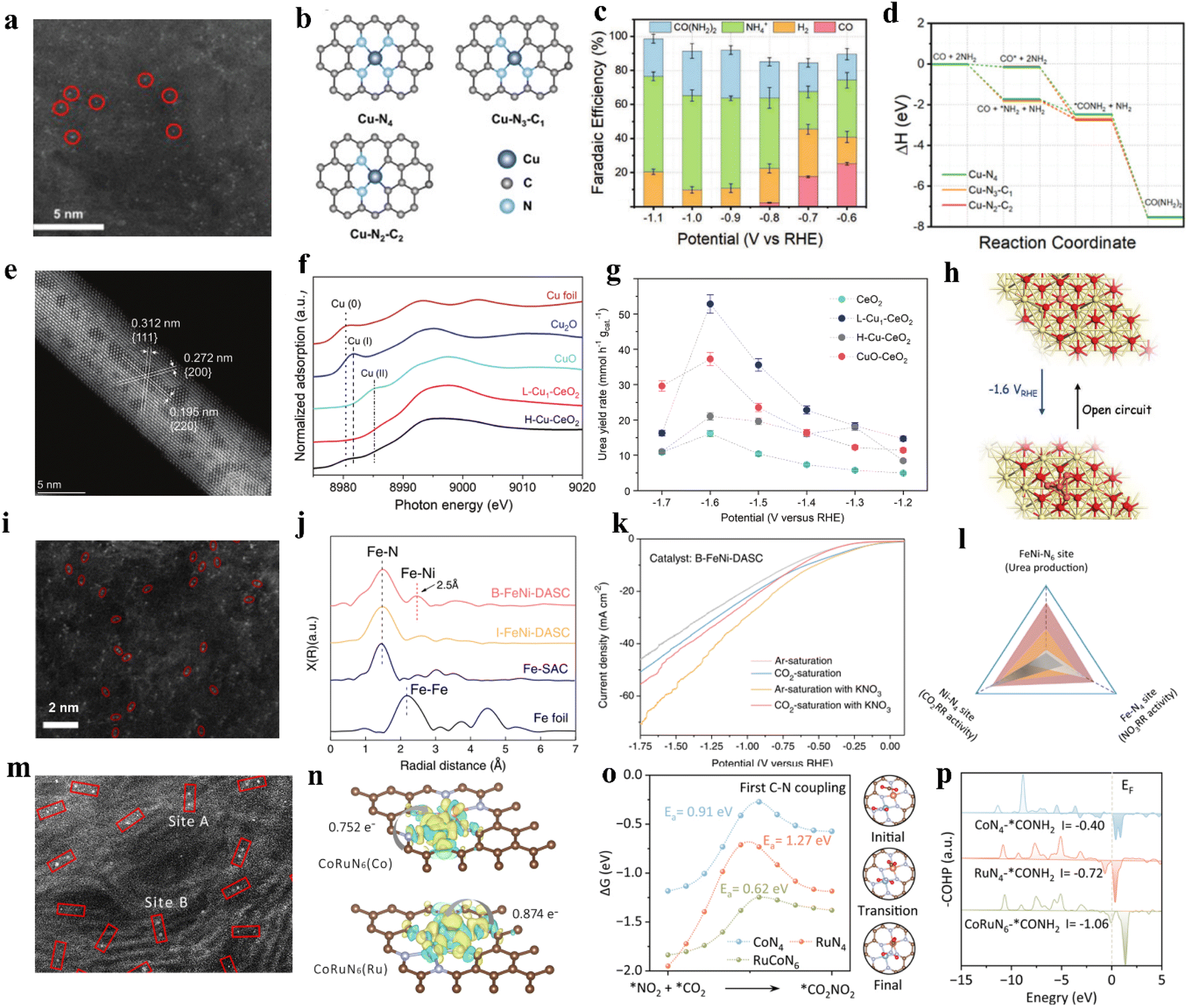 | ||
| Fig. 3 (a) HAADF-STEM image of Cu-GS-800. (b) Graphical representations of modeled Cu–Nx–Cx sites. (c) Faradaic efficiencies for different products by using Cu-GS-800 in H-cell. (d) Reaction pathway for urea production from simultaneous CO2RR and NOxRR. Reproduced from ref. 65, with permission from Wiley-VCH, copyright 2022. (e) AC-HAADF-STEM image of L-Cu1-CeO2. (f) XANES image of L-Cu1-CeO2. (g) The urea yield rates of CeO2, L-Cu1-CeO2, H-Cu-CeO2, and CuO–CeO2 catalysts at different applied potentials. (h) Schematic diagram of reconstitution of copper single-atoms to clusters suggested by the operando XAS measurements. Reproduced from ref. 66, with permission from Wiley-VCH, copyright 2023. (i) Aberration-corrected HAADF-STEM image of B-FeNi-DASC. (j) Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectra of Fe-SAC, I-FeNi-DASC, and B-FeNi-DASC. (k) LSV curves of B-FeNi-DASC. (l) Correlations between CO2RR activity, NO3RR activity, and urea production over various site configurations. Reproduced from ref. 76, with permission from Springer Nature, copyright 2022. (m) HAADF-STEM image of CoRuN6. (n) Charge density differences of Co and Ru in CoRuN6 simulated structure. (o) The reaction pathway of first C–N coupling for *CO2NO2 formation. (p) The crystal orbital Hamilton population (COHP) of *CONH2 intermediate onto CoN4, RuN4 and CoRuN6. Reproduced from ref. 77, with permission from Elsevier, copyright 2023. | ||
Unfortunately, urea synthesis through single-atom catalyst methods has received much less attention compared with the research on CO2RR and NRR. Meanwhile, competing reactions on the active sites can lead to slow rates of certain reactions and ultimately result in low urea yields, similar to monometallic catalysts. Efficient atom catalysts for urea synthesis should be developed, building upon the research on CO2RR and NRR. Zn-SACs, Fe-SACs, and Ni-SACs have been proved to be efficient CO2RR electrocatalysts.67–69 Ru SAs/N–C, Fe–N–C, and Mo/GDY have also been demonstrated as effective NRR electrocatalysts.70–72 Combining the advantages of single-atom catalysts in single reactions, the development of diatomic catalysts is gaining more and more attention as a strategy to overcome the inherent drawbacks of single-atom catalysts and enhance the reaction efficiency.
3.4 Dual-atom structures
Typically, diatomic catalysts allow two metal atoms to interact and stabilize in a restricted domain, while increased metal loading provides more active centers, meaning that catalytic performance is also improved.73 Such a catalyst usually has better activity than single-atom ones, as well as the faradaic efficiency, selectivity, and stability of target reactions. This leads to a synergy greater than the simple sum of parts, achieving “1 + 1 > 2”.74 Furthermore, the reactions on dual-atom structures are conducive to realizing simultaneous generation of C and N-intermediates on active sites. Therefore, the construction of the dual-atom structure plays a crucial role in urea synthesis.75 Wang and coworkers designed a diatomic catalyst (DAC) with bonded Fe–Ni (B-FeNi-DASC) pairs through pyrolysis of the coordination polymer (Fig. 3i and j).76 Compared with Fe-SAC, Ni-SAC, and I-FeNi-DASC electrocatalysts, B-FeNi-DASC demonstrated a better urea synthesis performance by achieving a high urea yield of 20.2 mmol h−1 g−1 with a corresponding faradaic efficiency of 17.8%. Notably, the faradaic efficiency of CO, as for NH3, showed diametrically opposed results when using Fe-SAC or Ni-SAC as electrocatalyst: the highest FECO of Fe-SAC and Ni-SAC is 86.9% and 19.2%, while the highest FENH3 of Fe-SAC and Ni-SAC is 19.9% and 65.2%, respectively. Apparently, single-atom systems present inherent disadvantages in handling intricate catalytic reactions that involve multiple reactants and intermediates. Despite the fact that I-FeNi-DASC with isolated diatomic sites Ni–N4 and Fe–N4 can activate multiple C and N species, it is unable to generate enough intermediate species without the creation of efficient C–N coupling sites. Remarkably, B-FeNi-DASC, introduced with Fe–Ni pairs in the form of FeNi-N6 configuration, can exhibit a synergistic effect and accelerate the reaction kinetics of the C–N coupling process. Furthermore, the thermodynamic and kinetic processes involved in C–N bond formation were also enhanced (Fig. 3k and l). Liu and coworkers designed Co–Ru dual-atom anchored N-doped carbon (CoRuN6) by the coordination and calcination method.77 As shown in the high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) image, numerous bright spots of Ångstrom size are evenly distributed on the carbon matrix and mainly appeared in pairs (Fig. 3m and n), which indicates the Co and Ru sites were dispersed in atomic form instead of becoming clusters. The diatomic asymmetric system successfully achieves high urea yield and selectivity by inhibiting parallel reactions (Fig. 3o and p). Consequently, the CoRuN6 electrocatalyst exhibited a urea yield rate of 8.98 mmol h−1 g−1 with a faradaic efficiency of 25.31% at −0.6 V vs. RHE. In contrast with single-atom catalysts, the selectivity of NH3 and CO byproducts was decreased on CoRuN6 (37.99% and 6.4%, respectively), which favored C and N-intermediates’ participation in the C–N coupling process.Current reports for DACs synthesis have mainly focused on atomic layer deposition (ALD), wet chemisorption, and high-temperature thermal treatment methods.78–81 However, while DACs have been intensively studied and applied, preparing highly dispersed DACs remains a significant challenge, because of the inevitable aggregation of metal atoms during the synthesis process. In addition, when using DACs in electrocatalytic urea synthesis, the accurate identification of active sites in DACs is an issue that needs further investigation.
3.5 Vacancy defects
Vacancy engineering is crucial in adjusting target reactions since the vacancies can effectively modify the electronic structure of catalysts and tune the adsorption features of CO2, nitrogen feedstocks and intermediates.75,82 For instance, oxygen vacancy (VO) is an important structural defect in metal oxides that can serve as catalytic centers with rich electron densities.83,84 Lv and colleagues fabricated VO-InOOH electrocatalyst via a facile solvothermal method. The defect-engineered catalyst delivers a 51% FEurea at −0.5 V vs. RHE.85 In contrast, InOOH electrocatalysts with no VO exhibited lower electrocatalytic performance with a 26.3% FEurea. After the long-term (24 h) chrono-amperometry test and five successive cycles of electrocatalysis, VO-InOOH displayed no obvious degradation in catalytic performance. It was also confirmed that oxygen vacancies conferred more surface electrons to In(OOH). These surface electrons are further transferred to the surface adsorbed CO2, which leads to a thicker accumulation of holes than the InOOH, and inhibits HER by repelling protons in the electrolyte. Wei and colleagues proposed a series of VO-enriched CeO2 nanorods to investigate the effects of VO concentration on the properties of catalysts (Fig. 4a).86 VO-CeO2-750 with the highest vacancy concentration exhibits a urea yield rate of 943.6 mg h−1 g−1, superior to that of partial noble-metal-based electrocatalysts. Interestingly, the active sites of VO-CeO2-750 were occupied by nitrate reduction, which was confirmed by the greatly reduced CO formation rate in co-electrolysis. However, in either nitrate reduction or C–N coupling processes, VO-CeO2-750 shows lower ammonia yield rates (Fig. 4b and c). Based on the existing data, the authors propose that VO stabilizes N-containing intermediates and exceeds their hydrogenation, thus facilitating the C–N coupling process and subsequent urea formation (Fig. 4f). Meng and coworkers prepared ZnO-VO porous nanosheets via electrodeposition.87 When using NO2− as the N-source, the ZnO-VO porous nanosheets exhibit an optimized FEurea of 23.26% at −0.79 vs. RHE, which is almost three times that of ZnO porous nanosheets. Through a series of controlled experiments, they confirmed the competitive relationship between CO2RR and NO2RR on ZnO-VO. Due to the existence of more active sites in ZnO-VO, the efficiency of urea generation is greatly promoted.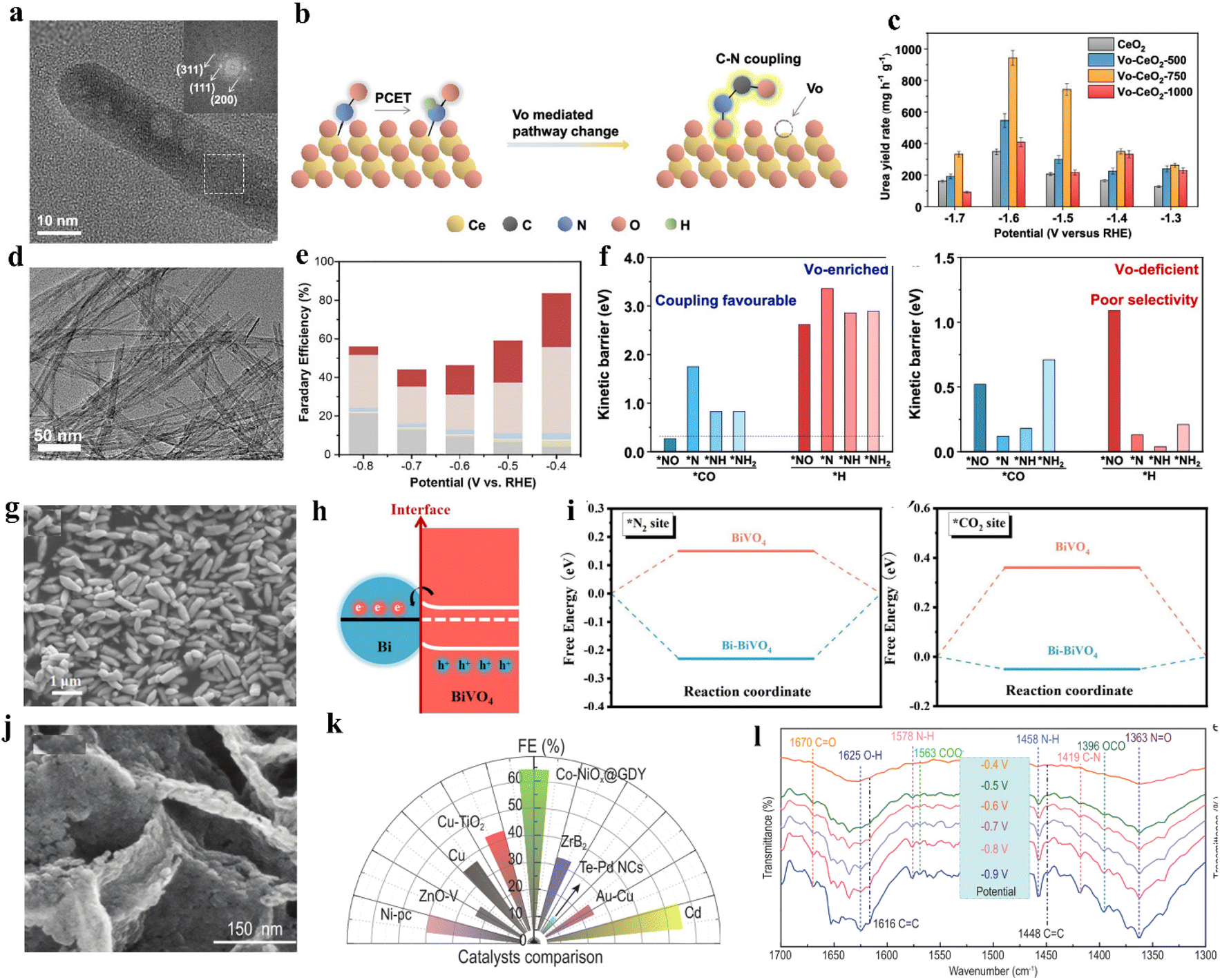 | ||
| Fig. 4 (a) TEM and HAADF-STEM images of Vo-CeO2-750. (b) Schematic diagram of Vo-mediated reaction pathway changes on CeO2. (c) Urea yield rates of CeO2, Vo-CeO2-500, Vo-CeO2-750, and Vo-CeO2-1000 at various applied potentials. Reproduced from ref. 86, with permission from American Chemical Society, copyright 2022. (d) HRTEM images of Cu-TiO2 samples. (e) Faradaic efficiencies of major reduction products for Cu-TiO2 catalysts. Reproduced from ref. 89, with permission from Elsevier, copyright 2020. (f) Comparison of the energy barriers for C–N coupling of *NO, *N, *NH, and *NH2 with *CO and protonation on Vo-enriched CeO2 and Vo-deficient CeO2. (g) SEM image of Bi-BiVO4 hybrids. (h) Schematic illustration of the charge transfer process in Bi-BiVO4 (i) N2 and CO2 adsorption on BiVO4 and Bi-BiVO4; the bottom figures are the corresponding calculation models. Reproduced from ref. 94, with permission from Wiley-VCH, copyright 2021. (j) High-magnification SEM images of Co-NiOx@GDY. (k) Comparison of the FE value of Co-NiOx@GDY with reported catalysts. (l) Infrared signals in the range of 1700–1300 cm−1. Reproduced from ref. 98, with permission from Oxford University Press, copyright 2022. | ||
It is well known that copper (Cu)-based materials have notable performances in CO2RR and NO3RR; the combination of Cu and VO-rich materials also provides an alternative catalyst for electrocatalytic urea synthesis.82,88 Cao and colleagues provided Cu-doped oxygen vacancy-rich anatase TiO2 (Cu-TiO2) nanotubes for the electrochemical co-reduction of both CO2 and NO2−, giving a FEurea of 43.1% at −0.4 V vs. RHE (Fig. 4d and e).89 Low-valence Cu doping induces abundant oxygen vacancies due to its unique d-electronic nature, favoring the exposure of bi-Ti3+ active sites. The oxygen vacancies in the proposed system provide more adsorption sites, facilitating the C–N coupling reaction.
A suitable amount of oxygen vacancies can provide more active sites and stabilize intermediates in C–N coupling reactions by modifying the energy level, conductivity, and molecular adsorption properties of the catalysts.90,91 A thorough understanding of vacancies can provide valuable insights for designing novel electrocatalysts. In future applications, with the development of preparation methods, new insights can be provided for vacancy engineering to improve the performance of urea synthesis systems.
3.6 Heterogeneous structures
Heterojunction catalysts play an important role in novel catalysts. In heterojunction catalysts, electrons can be rearranged at the heterostructure interface to modify the active sites. The synergistic effect between the active sites is conducive to promoting the reaction kinetics.92,93 Yuan and colleagues prepared Mott–Schottky heterostructure Bi-BiVO4 hybrids by the NaBH4 reduction strategy (Fig. 4g).94 When introducing N2 and CO2 as feedstocks, the proposed system delivers a urea production rate of 5.91 mmol h−1 g−1 with a FEurea of 12.55% at −0.4 V vs. RHE (Fig. 4h and i). In contrast, the electrochemical performances of pristine BiVO4 are significantly decreased. As the study mentioned, Bi-BiVO4 can form a Mott–Schottky heterostructure and a space charge region can occur due to the electron transfer from BiVO4 to metallic Bi. The unique structure greatly enhances the chemisorption capability of inert N2 and CO2 molecules, which is a prerequisite for the efficient synthesis of urea. Meanwhile, Bi-BiVO4 can effectively suppress the generation of C or N by-products, which leads to satisfactory urea production. By utilizing a similar mechanism, Yuan and colleagues put forward perovskite-structured BiFeO3/BiVO4, with FEurea up to 17.18% at −0.4 V vs. RHE in 0.1 M KHCO3, which is also higher than the pristine BiFeO3 and BiVO4.95 BiFeO3/BiVO4 hybrids endow the surfaces of BiFeO3 and BiVO4 with local nucleophilic and electrophilic regions and is beneficial for the adsorption of CO2 and N2 molecules. Graphdiyne (GDY) has several unique characteristics, including non-uniform surface charge distribution, infinite natural pores, and good stability.96,97 Zhang and colleagues indicated that the in situ growth of a multi-heterojunction interfacial structure can facilitate the intermediates’ adsorption/desorption capacity and suppress side reactions (Fig. 4j). As a result, the as-prepared Co-NiOx@GDY demonstrated a high urea yield rate of 913.2 mg h−1 g−1 and improved faradaic efficiency of 64.3% at −0.7 V vs. RHE by the co-reduction of CO2 and NO2− (Fig. 4k and l).98An increasing number of strategies have been applied for constructing heterogeneous structures, including thermal treatment, hydrothermal synthesis, electrodeposition, and ion exchange methods.99–101 However, the heterojunctions in catalysts are still not well parsed and their characterization is also limited. Relatively low current density and faradaic efficiency limit the further application of heterojunction catalysts, so their in-depth theoretical exploration needs to be further investigated.
3.7 Heteroatom doping
Heteroatom doping gives a chance to modify the electronic structure of the catalyst, optimize the intermediate binding strength, and enhance the reaction kinetics.102 In general, heteroatom doping can be divided into two categories according to atom types: metallic doping and non-metallic doping.103 Benefiting from the high electronegativity of non-metallic material, the surrounding atoms are easily polarized. Thus, non-metallic doping can change the electron density and shift the d-band center of the active sites to promote electrocatalytic reactions.104 In the study of non-metallic-based catalysts, doping carbon materials with heteroatoms (e.g., N, B, and F) is considered an efficient strategy to optimize the catalytic performance.105–107 Liu and colleagues proposed carbon nanotubes with a fluorine-rich surface (F-CNT), which achieved a urea yield rate up to 6.36 mmol h−1 g−1 with FEurea of 18% at −0.65 V vs. RHE, 3.8 times higher than the pristine CNT (1.66 mmol h−1 g−1).108 DFT calculation indicates the F-doped graphite shell provides the active sites (“C–F2 moieties”) for electrocatalytic urea synthesis. Large electronegativity of F-dopants leads to stronger charge-transfer, and effectively suppresses the activity of HER. Therefore, when F-dopants are combined with the intact CNT inner walls with high efficiency charge transfer and good conductivity, the F-CNT catalyst shows outstanding urea synthesis performance. Additionally, Mo-based materials have also emerged as promising electrocatalysts for energy storage. Jiao and colleagues put forward a catalyst containing MoP-(101) surface, which achieved the urea yield rate of 12.4 mg h−1 g−1 with 36.5% FEurea.109 They deemed that the incorporation of P will induce electron transfer from Mo to P, leading to a moderate coupling between Mo-d and P-p orbitals. Therefore, the surface Mo atoms can realize the coexistence of empty and occupied d orbitals. By means of DFT calculation, they revealed that the exposed Mo active sites in the MoP-(101) surface exhibit high reactivity towards the simultaneous activation of N2 and CO2, which facilitates the formation of characteristic C and N intermediates to generate urea in the operating system. Zhu and colleagues synthesized CuB12 monolayer to realize urea production in an electrocatalytic system.110 The empty and occupied p orbitals of non-metallic B can efficiently adsorb and activate the inert N2 molecule. On the other hand, the electron-deficient B atom can also adsorb and activate the inert CO2 by providing electrons to CO2 and breaking the inherent p bond.The degree of electronic modulation induced by the doping of heteroatoms in catalytic materials varies depending on the electronegativity and size of the dopant atoms as well as the type of dopant that ultimately forms the atoms.111 However, there are significant discrepancies between theoretical modeling simulations and practical results. The atoms’ interaction and the exact catalytic mechanism remain underexplored. A synthetic method that can provide a variety of heteroatom-doped catalysts to achieve high doping density and uniform distribution of multi-element doping is still lacking.112
3.8 Other catalysts
Facet-dependent selectivity was first proposed by Hori et al. in 1995.113 Recently, Lv and colleagues synthesized In(OH)3-S with well-defined (110) facets by a solvothermal method (Fig. 5a).41 To highlight the role played by facets on In(OH)3-S (110) in boosting urea production, the control experiments were also conducted on In(OH)3-M with mixed facets of (110) and (111). By using In(OH)3-S, the urea yield reached 533.1 mg h−1 g−1 with a 53.4% FEurea at −0.6 V vs. RHE. In contrast, In(OH)3-M reported a lower urea yield of 392.6 mg h−1 g−1 with 34.6% FEurea (Fig. 5b). The authors indicated that CO2 can capture the electrons to introduce a hole accumulation layer on the surface of the In(OH)3-S. The hole accumulation layer can restrict protons from approaching In(OH)3-S, which plays the dominant role in impeding HER on In(OH)3-S (Fig. 5c). However, the electron holes on In(OH)3-M (111) facets are combined with electrons from (110) facets, which weakens HER suppression. Besides, DFT calculation indicates that the transformation of NO3− to *NO2 intermediate is spontaneous kinetically and thermodynamically. This led to the higher coverage of *NO2 and the inhibition of CO2RR on the surface of In(OH)3-S. However, the *NO2 on In(OH)3-M is in weak adsorption, which is unfavorable to the formation of *CO2NO2. On the other hand, the (110) facets possess relatively adjacent indium atoms to endow catalysts with favorable platforms for constructing the In–O–C–O–In configuration. Such a configuration can stabilize the adsorption of *CO2, which lowers the energy barrier toward C–N coupling.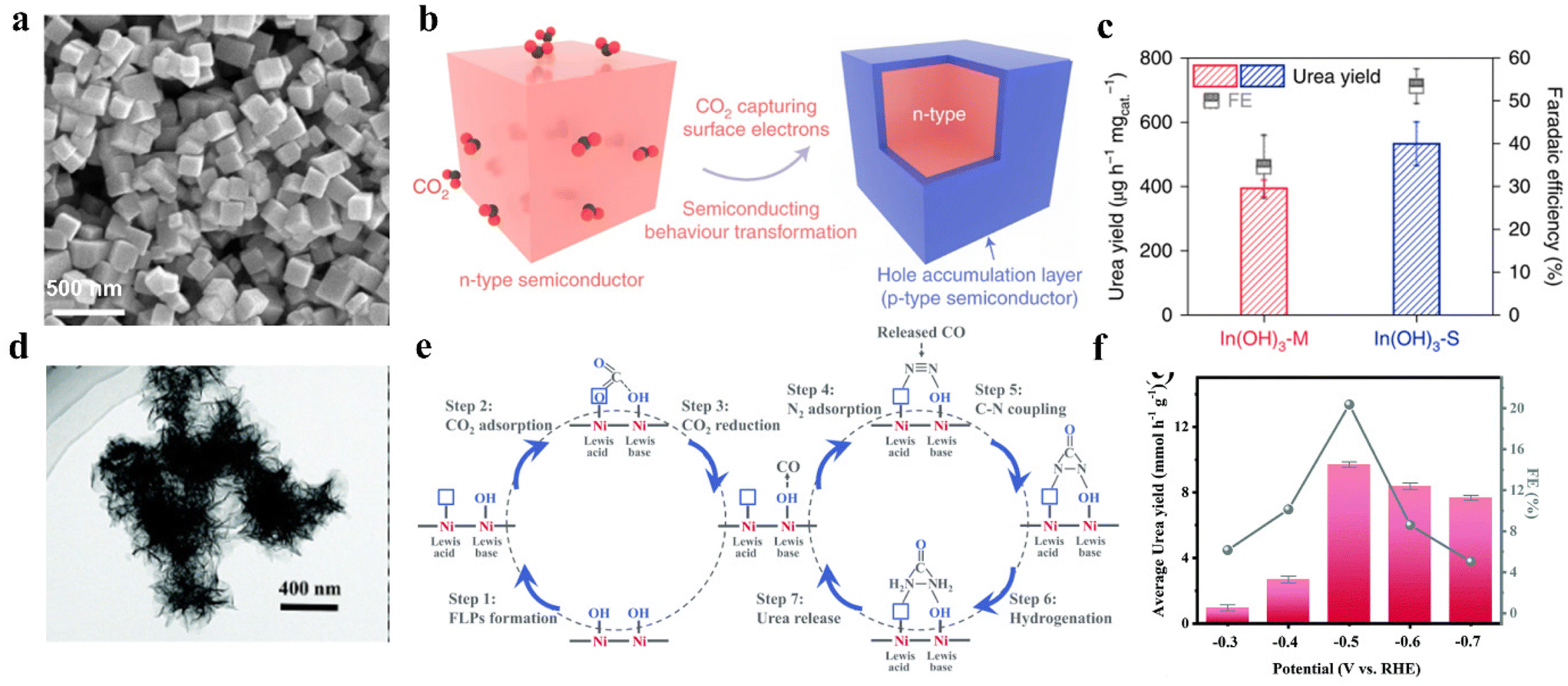 | ||
| Fig. 5 (a) SEM image of In(OH)3-S. (b) CO2 capture induced n-type In(OH)3 structure transformation. (c) Urea synthesis performance of In(OH)3-S and In(OH)3-M at −0.6 V vs. RHE. Reproduced from ref. 41, with permission from Springer Nature, copyright 2021. (d) HRTEM images of Ni3(BO3)2-150 catalysts. (e) Schematic illustration of the electrocatalytic co-activation of N2 and CO2 into urea over artificial frustrated Lewis pairs (FLPs) of Ni3(BO3)2-150 catalysts. (f) Faradaic efficiency for urea by using Ni3(BO3)2-150 catalysts in H-cell. Reproduced from ref. 116, with permission from Royal Society of Chemistry, copyright 2021. | ||
Lewis acid–base theory states that Lewis acids (LA) always have empty orbitals and the capability to form coordination bonds with Lewis bases (LB) containing lone electron pairs.114 Generally, the unsaturated metal sites act as the LA and the metal hydroxyl groups serve as the LB.115 For instance, Yuan and colleagues prepared Ni3(BO3)2-150 through a facile wet chemical strategy followed by low-temperature annealing (Fig. 5d).116 The catalyst realized an excellent urea yield rate of 9.70 mmol h−1 g−1 with a 20.36% FEurea at −0.5 V vs. RHE (Fig. 5e). In contrast with Lewis acidic Ni3(BO3)2-250 and Lewis basic Ni3(BO3)2, the flower-like Ni3(BO3)2-150 has frustrated Lewis pairs (FLPs) and unsaturated Ni sites. The existence of FLPs and unsaturated Ni sites makes Ni3(BO3)2-150 able to introduce effective absorption of CO2 and N2, and facilitate the spontaneous coupling of *NN* and *CO to generate the desired *NCON* (Fig. 5f).
The rational design and optimization of catalysts is a critical link in achieving the efficient electrocatalytic production of urea. At present, the competitive reaction on active sites and stabilization of C and N intermediates during the reaction process remain a hot spot during catalyst design. In addition to improving catalytic performance, it is also crucial to optimize the stability of the catalyst in consideration of future applications. Unstable behaviors, such as aggregation, exfoliation, and electronic structure changes of the catalyst may lead to a decrease in current density and Faraday efficiency. The visual characterization of catalysts enables us to get a better understanding of catalyst properties and facilitate the technological innovation of catalyst design.117
4. Catalyst characterization
The morphology, lattice structure, coordination environments, and surface chemical states are essential for gaining insights into the catalyst structure and reaction mechanism. In this section, characterization techniques for catalysts are summarized and classified.4.1 Microscopic methods
Electron microscopes have become an indispensable tool for characterizing the morphology, crystallography, composition and electronic properties of electrocatalysts at the nanoscale.118 In addition to scanning electron microscopy (SEM) and transmission electron microscopy (TEM), high-resolution transmission electron microscopy (HRTEM) and high-angle/medium-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) also play important roles in morphology characterization.14,119,120 The atomic-level resolution of HRTEM has enabled clear observation of crystal internal structure, atomic arrangement, and fine structures for most catalysts (Fig. 6a and b).121 HAADF-STEM is a powerful tool to study the atomic number with higher spatial resolution. A suitable microscopic method is conducive for us to gain an in-depth understanding of the fine structure of the designed catalyst, and provide ideas for catalyst modification (Fig. 6c).122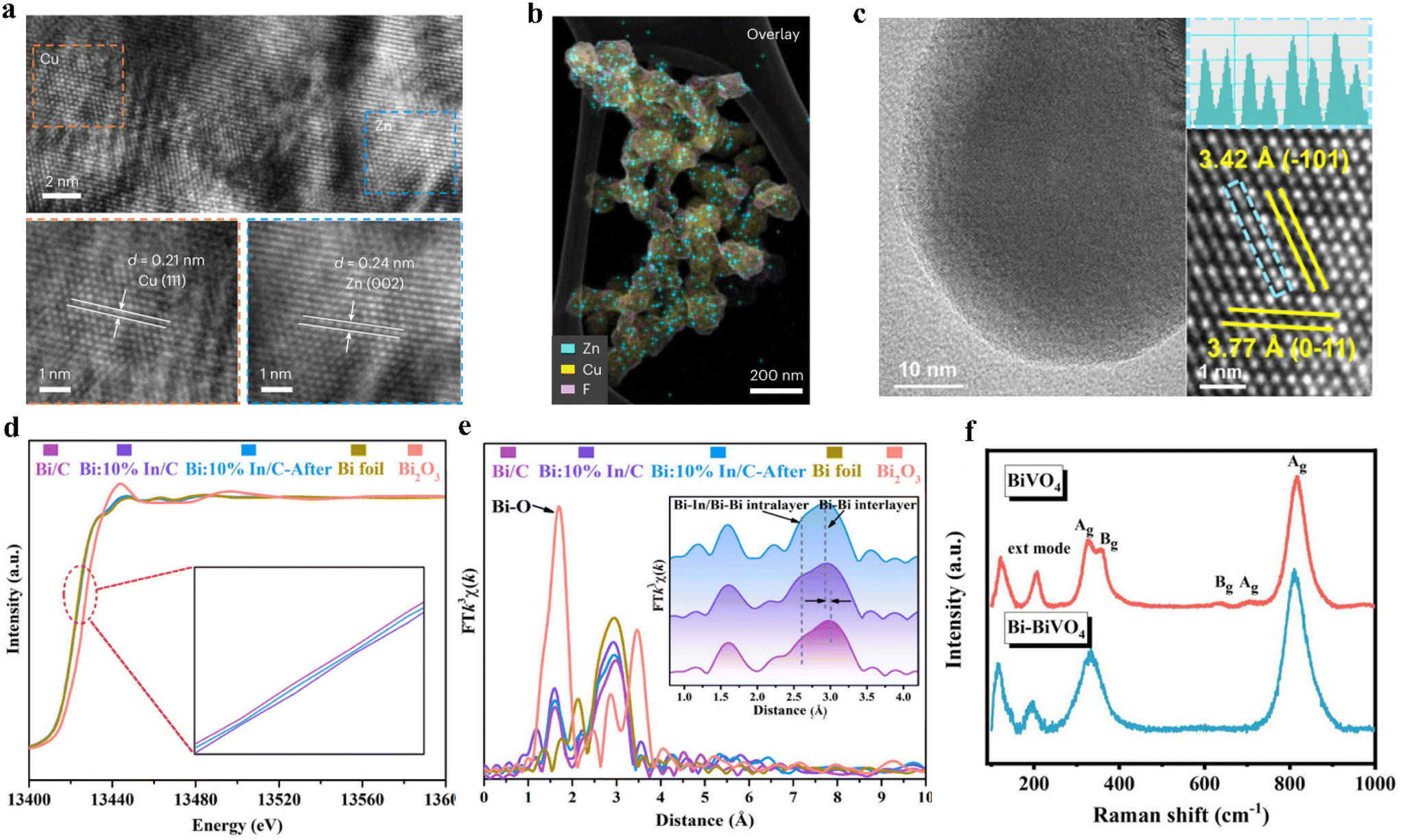 | ||
| Fig. 6 (a) HR-TEM image and the corresponding element mapping of 3D Zn/Cu hybrids. (b) The uniform deposition of Zn on Cu. Reproduced from ref. 121, with permission from Springer Nature, copyright 2023. (c) TEM and atomic-resolution HAADF-STEM image with line profile of the CuWO4. Reproduced from ref. 122, with permission from Springer Nature, copyright 2023. (d) The normalized XANES spectra at the Bi L3-edge of Bi/C, Bi:10%In/C, Bi:10%In/C after testing, Bi foil and Bi2O3. (e) R space and corresponding inverse FT-EXAFS results of Bi/C, Bi:10%In/C, Bi:10%In/C after testing, Bi foil and Bi2O3. Reproduced from ref. 125, with permission from Royal Society of Chemistry, copyright 2021. (f) Raman spectra of BiVO4 and Bi-BiVO4. Reproduced from ref. 116, with permission from Royal Society of Chemistry, copyright 2021. | ||
4.2 Spectroscopic methods
Spectroscopic characterization is crucial for identifying the coordination environment and the chemical surface states of materials. For instance, X-ray absorption spectroscopy (XAS) is a cutting-edge technique for detecting the local electronic and atomic structure of electrocatalysts, and encompasses extended X-ray absorption fine structure (EXAFS) and X-ray absorption near-edge structure (XANES).123 XANES is able to provide detailed information on catalysts including the geometry, state, and electronic structure of absorbing atoms. EXAFS is typically used to provide information on the concentration, type, and distance to the ligands of the absorbing element and neighboring atoms.124 For example, Mao and his colleagues performed XAS analysis on the Bi:10%In/C during the C–N coupling process.125 Through the XANES method, they found that the spectra of the three samples (Bi/C, Bi:10%In/C, Bi:10%In/C after testing) almost overlap with the Bi foil, indicating that the Bi species are in the Bi0 state. In comparison with Bi/C, the absorption threshold position of Bi:10%In/C slightly shifts to a higher energy, indicating that Bi is in an electron-deficient state after the introduction of In (Fig. 6d). After the introduction of In, the Fourier transforms of the Bi L3-edge EXAFS show that the interlayer Bi–Bi bond shifts towards a reduced bond length. This promoted the delocalization of Bi p-electrons and enhanced the adsorption and activation of reactive species (Fig. 6e).Raman spectroscopy provides a unique strategy for identifying the chemical characteristics of molecular features. It enables the investigation of surface adsorbates and intermediates, as well as the monitoring of structural parameters on the catalyst surface.126 It can detect diverse vibration patterns in group states and chemical bonds, resulting in the appearance, disappearance, and movement of Raman peaks (Fig. 6f).127
Although several advanced characterization techniques have been used to verify the capability of catalysts in producing urea, some limitations still remain for a single strategy.128 A combination of microscopic, spectroscopic, and other techniques is necessary to clarify the catalyst structure, which is beneficial for obtaining the practical reaction mechanism of the C–N coupling process. In addition, several in situ techniques can aid in monitoring and comprehending catalyst changes under specific reaction conditions and their effect on reaction pathways under specific reaction conditions.129 With the continuous deepening of mechanism exploration, the more favored urea production process will be revealed. At this point, a suitable reactor with considerable mass transfer and reaction stability ensures an optimal environment for the operation of catalytic systems.
5. Electrochemical cells for urea synthesis
The yield of electrocatalytic urea synthesis is affected by current density, carbon dioxide solubility, and catalytic system stability. The type of electrolyzer affects the current density and the reactant mixing degree, and the stability of the operating system determines the product yield and distribution. Therefore, electrolytic cells play a crucial role in transmuting CO2 and N-sources to value-added chemical products.39,130 Generally, the electrolytic cell is composed of two half-reactions: the reduction reaction on the cathode and the oxidation reaction on the anode. By setting an ion exchange membrane between the cathode and anode, ions can transport across the membrane and maintain the charge equilibrium of the whole system. On the other hand, a reference electrode is always added in cells to ensure the accuracy of potentials. To fulfill different experimental requirements, three types of electrochemical reactors are discussed for the electrochemical synthesis of urea, including H-type cells, flow cells, and membrane electrode assembly (MEA) cells.5.1 H-cell
The H-cell is the most widely used lab-scale electrochemical reactor, where the working and reference electrodes are placed in the cathodic compartment, while the counter electrode is in the anodic compartment. An ion-exchange membrane is used to separate the two compartments and prevent the mixing and re-oxidation of products in different phases. In general, platinum mesh and carbon rod are better candidates to be used as counter electrodes, the working electrode can utilize metal foils or electrocatalyst-loaded glassy carbon/carbon paper, while the reference electrode can be Ag/AgCl electrodes and Hg/Hg2SO4 according to the pH value of the operating system (Fig. 7a and d).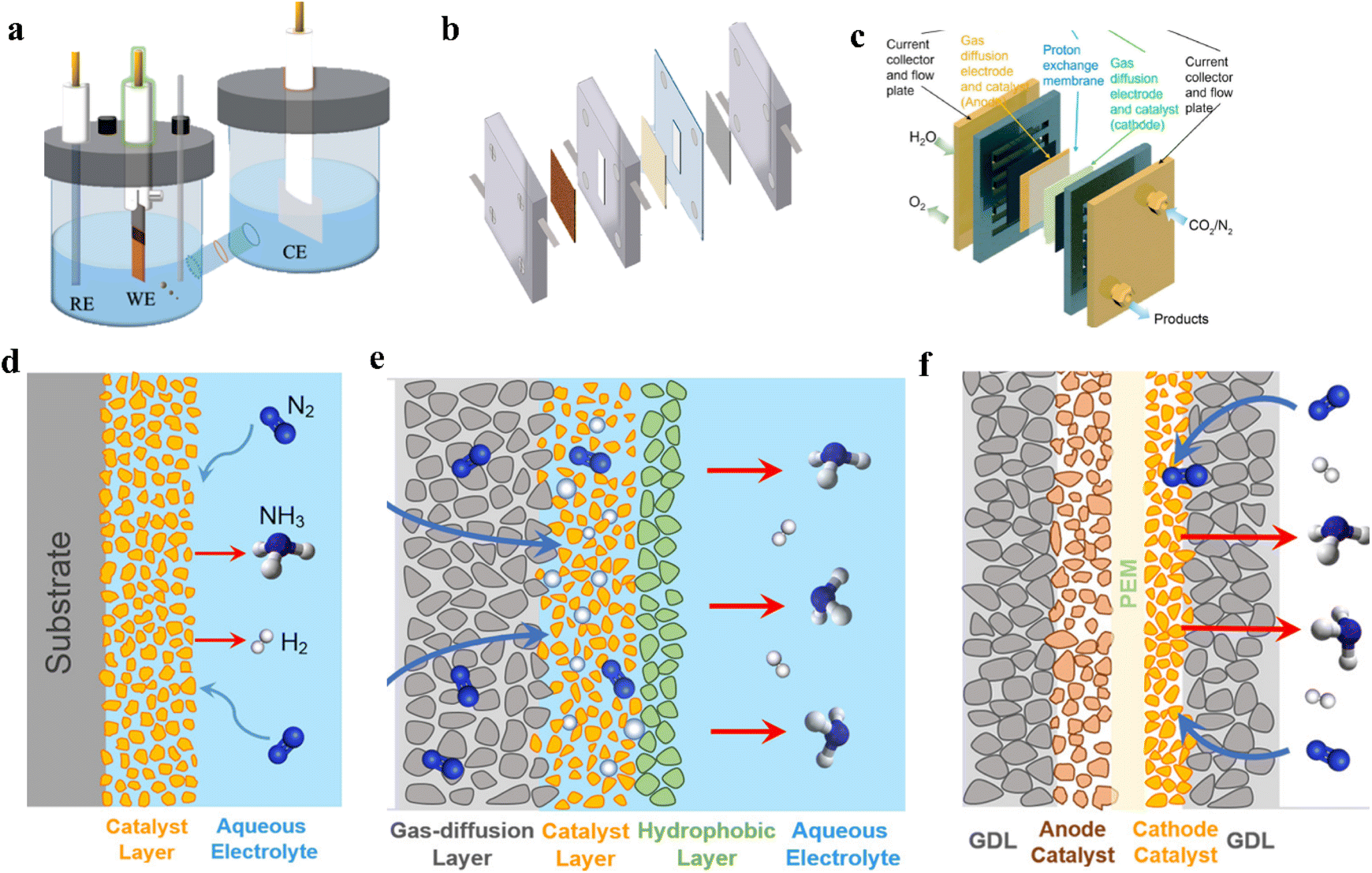 | ||
| Fig. 7 (a) H-cell for urea synthesis. (b) Flow-cell for urea synthesis. (c) MEA cell for urea synthesis. (d) Schematic illustration of solid–liquid interface in H-cell. (e) Schematic diagram of a membrane-based flow reactor. (f) Schematic illustration of solid–liquid interface in MEA cell. Reproduced from ref. 179, with permission from Royal Society of Chemistry, copyright 2023. (a and b) Reproduced from ref. 180, with permission from American Chemical Society, copyright 2022. (d, e and f) Reproduced from ref. 181, with permission from Royal Society of Chemistry, copyright 2022. | ||
During the electrocatalytic synthesis of urea, carbon dioxide (CO2) is continuously introduced into the cathode compartment to react with the nitrogen source in the electrolyte. The resulting gas products are then analyzed using a gas chromatograph (GC) and the faradaic efficiencies of gas products are determined based on the obtained data. The liquid products in the cathode chamber are collected after the electrolysis and measured using a suitable method (e.g., NMR, spectroscopic method, etc.) to determine the urea faradaic efficiency.40
However, urea synthesis in the H-cell suffers from the low solubility of CO2 and N2 (−34 mM and −0.6 mM, respectively, under ambient conditions), high resistance to electrochemical reactions, and low scalability of the device, which result in low efficiency.131 Therefore, finding a way to strengthen the interactions between feedstocks and active sites to improve the efficiency of the reactions has high priority in current research.
5.2 Flow cell reactor
Derived from the fuel cell field, the flow cell can substantially improve the mass transfer efficiency and overcome the mass transfer limitations in H-cells. In flow cells, it is feasible for the low-solubility CO2 and N2 to contact active sites through the gas phase, and the products are more easily recycled from the liquid phase. The flow cell has a similar structure to the H-cell, namely an anode compartment, a cathode compartment, and an ion exchange membrane. The cathode is a gas diffusion electrode (GDE), which has a gas diffusion layer (GDL) and a catalytic layer (CL). The GDL is hydrophobic and facilitates the transport of gases and products. It also supports the CL and allows gas to directly reach the cathode surface. The anode always employs a commercialized catalyst-loaded GDE, and the reference electrode can be Ag/AgCl or standard hydrogen electrode, depending on the experimental requirement (Fig. 7b and e). In comparison with the H-cell, lower cell resistance can be achieved in the flow cell, due to the proximity of the anode and cathode.Similar to the H-cell, the gas path of the flow cell directly connects with a gas chromatograph (GC), enabling cathode gas products from the electrolysis process to be monitored by GC. Wang and coworkers synthesized urea by electrocatalytic coupling of N2 and CO2 using Pd1Cu1/TiO2-400 as the catalyst.28 They found that the flow cell enhanced the urea formation rates compared with the H-cell. The highest rate was 3.36 mmol g−1 h−1 with a faradaic efficiency of 8.92% at −0.4 V vs. RHE in the flow cell, while the rate was 0.19 mmol g−1 h−1 with a faradaic efficiency of 1.56% at −0.4 V vs. RHE in the H-cell. However, GDE flooding is a serious problem when employing large overpotentials. In addition, the manufacturing costs of GDE and carbonate by-product formation also restrict the large-scale application of the flow cell.
5.3 Membrane electrode assemblies (MEA)
The MEA cell is a promising design for various electrochemical reduction reactions, since it possesses low ohmic loss and high energy efficiency. A MEA cell has three main components: a proton exchange membrane, a catalytic layer, and a gas diffusion layer.132,133 The proton exchange membrane conducts protons quickly and prevents the crossover of hydrogen, oxygen, and nitrogen between the cathode and anode. MEA can facilitate ion transportation and serves as a barrier against the crossover of gas and electrons. The gas, ions, and electrons can be gathered at the triple-phase boundary, which is the catalyst surface, to enhance mass transfer efficiency (Fig. 7c and f).As electrocatalytic urea synthesis is currently limited within laboratory-scale productivity, some key aspects need to be addressed to attain the standards for practical applications. For example, the yield, current density, and faradaic efficiency correspond to the mass transfer phenomenon. Therefore, reactor design plays a crucial role in eliminating the limitations of electrocatalytic urea synthesis. From the viewpoint of building a reactor, H-cells are simple to build and the products are easy to separate, but it is still difficult to meet the performance requirements owing to the impacts of concentration polarization and gas solubility. Flow cells have a relatively better mass transfer efficiency, while GDE and ion exchange membranes need to be greatly improved to meet the stability and mass transfer requirements for practical applications. MEA reactors can achieve a higher reaction efficiency, but the liquid products in the system are difficult to collect. Based on the above three common reactors, the new design of reactors should be modified accordingly to address the abovementioned concerns. To achieve a long-term electrolyzer for practical applications, the impurities of electrolytes, membrane durability, mass transfer characteristics, and electrothermal effect that causes temperature changes should be comprehensively considered.134 Particularly, the membrane is the important unit for constructing long-term stabilized electrolyzers. The further improvement of anti-degradation and anti-pollution capabilities will facilitate its practical applications in a complex reaction environment.
6 Urea identification and quantification
Product testing is an important step in electrocatalytic urea synthesis; its accuracy determines the credibility of products. Various methods have been developed for urea identification and quantification, being suitable for different reaction conditions. In order to select an optimal method for urea quantification, the experimental quantification minimum (EQM) and the potential interference of the electrocatalytic system are the key factors that need to be rated.135 So far, four main detection methods are available for urea testing: the urease method, diacetylmonoxime-thiosemicarbazide method (DAMO-TSC), 1H-NMR method, and high-performance liquid chromatography (HPLC) (Fig. 8g).28,86,87,136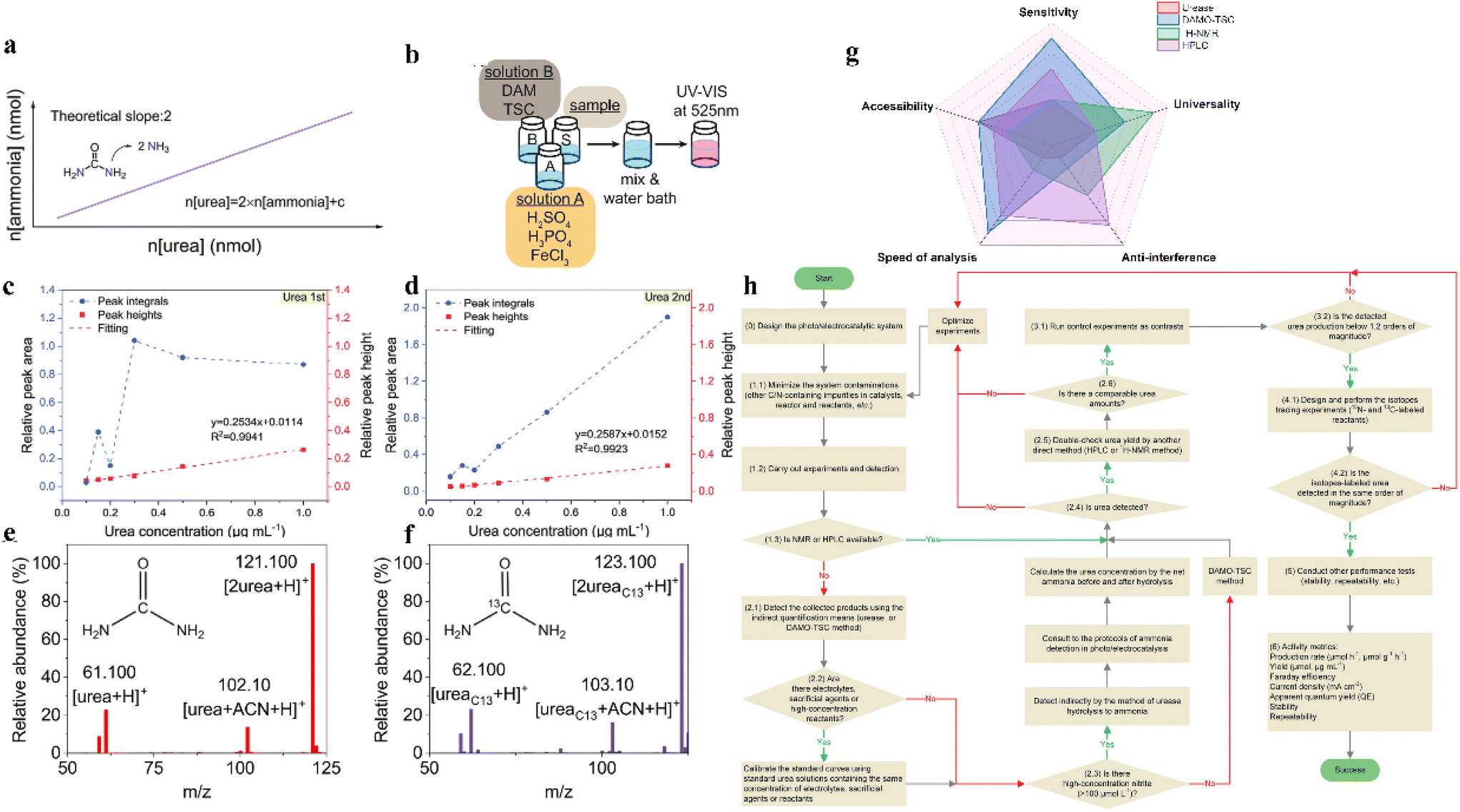 | ||
| Fig. 8 (a) The conservation principle of nitrogen atoms in the urease method. (b) Illustration of a typical urea colorimetric detection method. (c and d) Urea processed by the methods of peak heights and peak integrals at low-concentration range. Reproduced from ref. 141, with permission from Wiley-VCH, copyright 2023. (e) Mass spectrum of unlabeled (12C) urea in 0.2 M KHCO3. (f) An example of total ion chromatogram (TIC) and extracted ion chromatograms (EIC) for the 10 ppm urea standard. Reproduced from ref. 142, with permission from Elsevier, copyright 2023. (g) The comparison of four detection methods in this study. (h) The proposed photo/electrocatalytic urea synthesis and quantification protocol. Reproduced from ref. 135, with permission from Wiley-VCH, copyright 2022. | ||
6.1 Urease method
The urease method has been a commonly used method for detecting urea since 1995. In the presence of urease, the urea can be decomposed into ammonia and carbon dioxide at the optimal temperature (37 °C, 40 min).137 The amount of urea can be calculated according to the principle of conservation of nitrogen atoms (n(urea) = 2 × n(ammonia)). Then, indophenol blue method is used in determining the concentration of ammonia at λ = 655 nm (Fig. 8a).138 An appropriate dilution is essential for accurate urea quantification, since high concentrations of urea (>7.0 μg ml−1) may deviate from the linear relationship. However, the activity of urease is affected by various factors such as temperature, pH and urea concentration. Particularly, interfering metal ions from electrolyte impurities or catalyst-leaching metals cannot be ignored. The presence of Ni2+, Cu2+, Zn2+, Fe3+, and In3+ ions has a negligible effect on urea detection, while the presence of Co2+, Fe2+, and Mn2+ ions will inhibit urease activity, leading to errors in the measurement.138 The restricted reaction temperature, high cost, and long detection time also limit the operation of the urease method. Meanwhile, the NH4+ losses during the heating process may also contribute to false reporting. Therefore, adjusting the testing strategy based on the experimental details is important in the application of the urease method.6.2 Diacetylmonoxime-thiosemicarbazide method (DAMO-TSC)
The DAMO-TSC method is another widely used technique for quantifying urea. It offers several advantages over the traditional urease decomposition method, including faster detection and lower detection limit of urea.During the detection process, DAMO undergoes hydrolysis under acidic conditions to produce diacetyl. The diacetyl will react with urea to form a yellow-colored diazine product and water. Diazine can be stabilized by thiosemicarbazide and converted into a pink complex with enhanced absorbance in the presence of iron ions, which has a maximum absorption wavelength of 525 nm (Fig. 8b). Furthermore, by adding a specific concentration of antipyrine, thiourea, metal ions, and other substances, the sensitivity of the reaction between urea and diacetyl can be improved. This addition can also improve the stability of the color development.
It is noted that the effect on color development by interfering substances should be considered before detection. The colorimetric determination is affected by carbon products (such as formic acid and isopropanol), nitrogen products (such as ammonia and hydrazine), and common ionic species (such as metal cations, NO2−, and NO3−).139 Nitrite is the most problematic of these species; when the concentration of NO2− reaches 10 mmol L−1, it interferes with color development due to its strong oxidizing properties. In addition, a strongly acidic environment is necessary for color development. For most electrocatalytic urea synthesis processes, the buffering effect of KHCO3 electrolyte has a minimal tendency of pH shift to alkaline conditions. However, in higher concentrations KOH electrolytes (>1 M), the pH value of collected electrolytes should be adjusted to the optimal range before the quantification.
6.3 1H-NMR method
The 1H-NMR method is commended by its simplicity, accuracy, and specificity, and the measurements are based on the instrument selection and parameter settings.29,140 The quantification of urea by 1H-NMR is minimally affected by changing the chemical environment of hydrogen atom-influenced products and metal ions. Noteworthily, most product solutions from the electrocatalytic C–N coupling process involve huge amounts of water molecules, and the water suppression may induce baseline distortions and affect the accuracy of quantification. To avoid these disadvantages, the relative intensities can be calculated by peak integral and peak height methods. The increased variability of multiple assays is due to the proximity of the chemical shift of urea to the water peak and its increased sensitivity to interference from this peak. Therefore, the peak height method can be used to attenuate the effect of water peaks (Fig. 8c and d).1416.4 Liquid chromatography-mass spectrometry (LC-MS)
LC-MS is another method for urea quantification.87 This method simultaneously possesses the excellent separation capability of liquid chromatography with the high sensitivity and selectivity detection capability of mass spectrometry (Fig. 8e and f).142 According to the differentiated chemical composition and physical properties of individual components, products can be separated by the liquid chromatography method. The peak intensity is positively correlated to the concentration of separate products. After solvent evaporation at the interface of the LC and MS module, product ionization will occur through electrospray ionization (ESI) or atmospheric pressure chemical ionization (APCI) method. Next, ions are separated based on their charge-to-mass (m/z) ratio in the electric field.Generally, the urease method is poorly reproducible and susceptible to environmental interference. It is not suitable in the presence of low urea concentrations with high NH4+ backgrounds. The DAMO quantification method is suitable for most systems, but not for environments with high levels of NO2− (>10 mmol L−1). The 1H-NMR method is highly resistant to interference, but is strongly influenced by urea concentration, namely, experimental quantification minima are large and few studies have used the LC-MS method to quantify urea. Additionally, the environmental conditions and operating conditions should be strictly controlled during product testing to improve the accuracy, reliability, and anti-interference ability (Fig. 8h). To improve the efficiency and accuracy of product testing, a combination of two or more detection methods is usually required.
7 Mechanism of urea synthesis process
Current studies indicate that the intermediates are critical for a coupling process. However, the exact roles played by these intermediates are still not clear. Therefore, revealing the reaction mechanism and the formation of the intermediates sheds light on enhancing the efficiency of the electrocatalytic systems.154 To date, the mechanism of the urea synthesis process during electrocatalytic urea synthesis has been investigated by temperature-programmed desorption measurements (TPD) and extensive in situ spectroscopy characterization methods, including the in situ Fourier transform infrared spectroscopy (in situ FTIR), online differential electrochemical mass spectrometry (DEMS), and operand Raman spectroscopy.7.1 Determination of intermediates
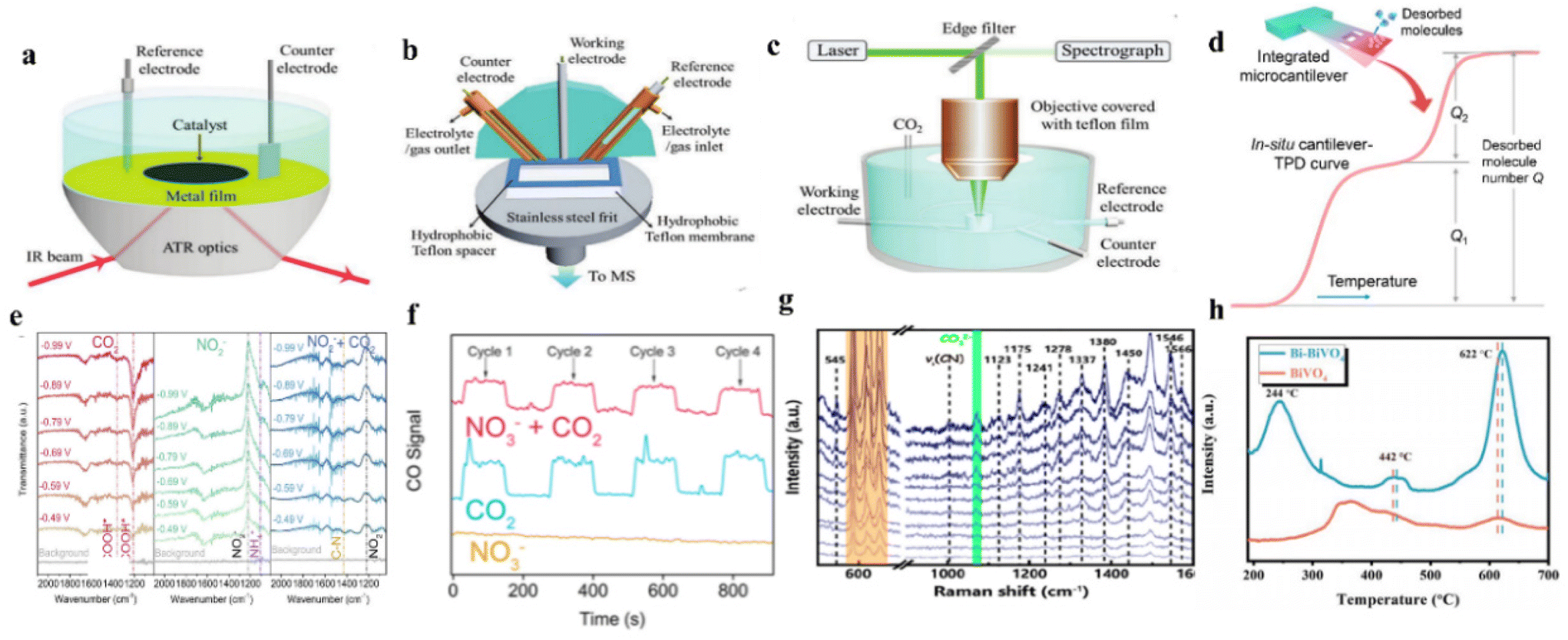 | ||
| Fig. 9 (a) Schematic of ATR-IR configurations. (b) Schematic of DEMS cell. (c) Schematic of operando Raman configurations. (d) Schematic of TPD. Reproduced from ref. 183, with permission from American Chemical Society, copyright 2023. (e) In situ ART-IR spectra of ZnO-V under CO2, NaNO2, and CO2 with NaNO2. Reproduced from ref. 182, with permission from Elsevier, copyright 2021. (f) Online DEMS spectra of CO signals over Cu@Zn. Reproduced from ref. 151, with permission from American Chemical Society, copyright 2022. (g) Operando electrocatalytic Raman spectra of Ru–Cu CF collected at −0.3 V vs. Ag/AgCl. Reproduced from ref. 146, with permission from American Chemical Society, copyright 2022. (h) (CO2-TPD) plots for BiVO4 and Bi-BiVO4. Reproduced from ref. 94, with permission from Wiley-VCH, copyright 2021. (a, b and c) Reproduced from ref. 154, with permission from Wiley-VCH, copyright 2021. | ||
In situ analyses provide solutions to monitor the generation of intermediates, bond formation, and reaction dynamics, which can provide new insights for catalyst optimization. However, in situ analyses can only detect a few key intermediate reactions, making it difficult to provide a full picture of the complex reaction system. Further exploration is needed to understand the reaction mechanism of electrochemical urea synthesis by using in situ measurements. Therefore, characterization such as in situ spectroscopic techniques (especially synchrotron radiation-Fourier transform infrared spectroscopy) and theoretical calculations will maintain their popularity in catalytic urea synthesis research.
7.2 Key intermediates in electrocatalytic urea synthesis
Generally, catalysts with the ability to simultaneously catalyze both CO2RR and NRR are more likely to carry out urea electrocatalytic production. With employing differentiated catalysts, the corresponding mechanism and pathways will be changed owing to the production of different C/N intermediates. In recent studies, theoretical calculations (density functional theory and ab initio molecular dynamics) have been used to uncover the formation steps of C–N bond. To make the internal relationship easier to understand, we have summarized the formation pathways of different intermediates with C–N bonds in this section, also displayed in Fig. 10.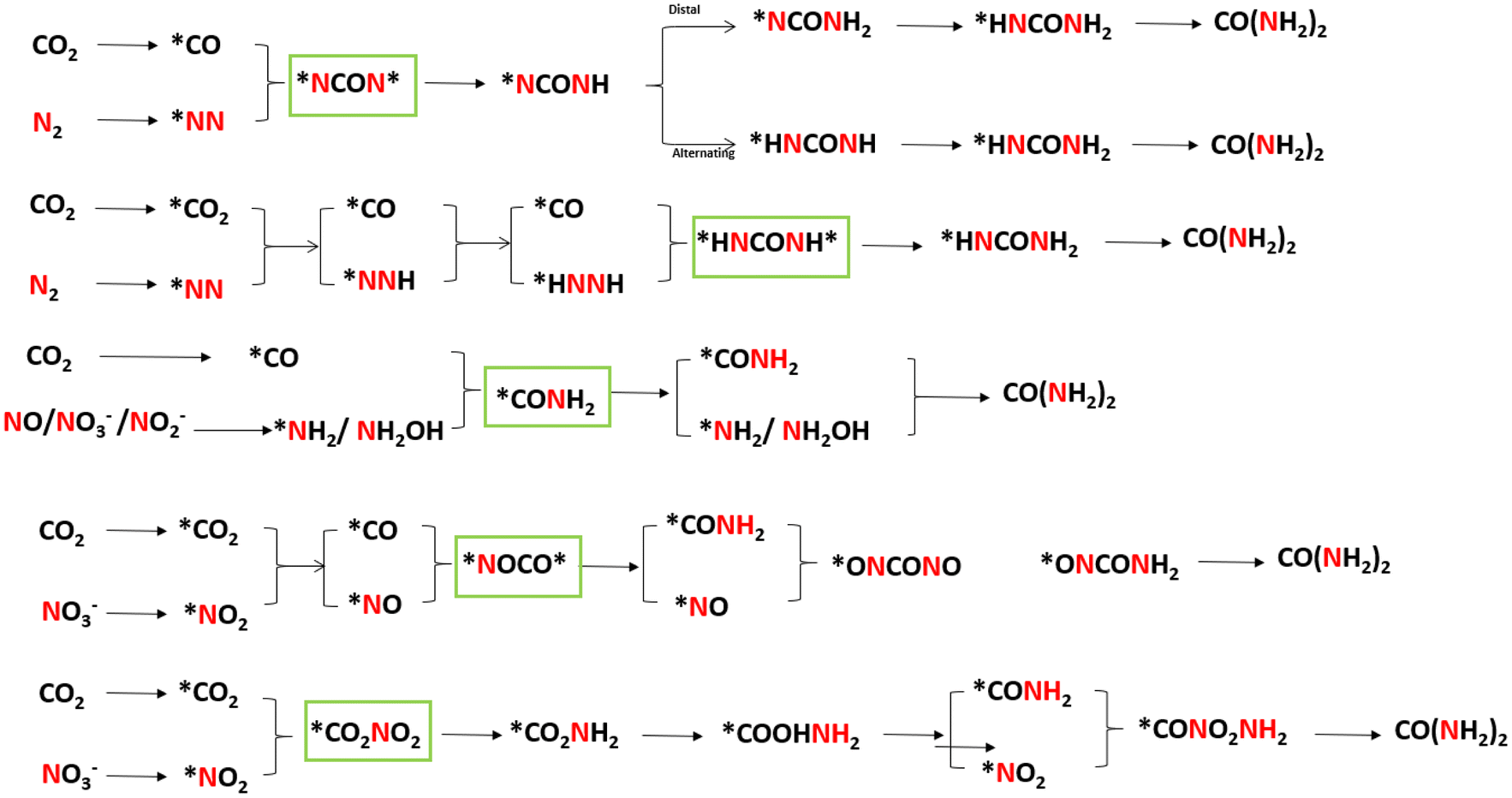 | ||
| Fig. 10 Reaction pathways for the synthesis of urea by C–N coupling and its intermediates. | ||
N* shows a strong attraction to the CO due to the matched molecular orbitals. The tower-like urea precursor *NCON* is formed after CO binding with *NN* (Fig. 11a).28 Jit Mukherjee and coworker synthesized copper phthalocyanine nanotubes (CuPc NTs) with multiple active sites, and predicted that the rate-determining step is CO* + *NN* → *NCON*.143 The *NCON* would be further hydrogenated to urea molecules (Fig. 11c and d). Another key intermediate is *NHCOHN*, which originated from the electrocatalytic process by using MoP-(101) (Fig. 11b).109 Theoretical calculation results showed that *N2H reacts with *CO and is more favorable in kinetics and thermodynamics (Fig. 11e and f). Subsequently, the *NHCOHN* intermediate can be formed by the combination of hydrogenated *N2H2 with *CO.
 | ||
| Fig. 11 (a) *CO and *N2 intermediates pathway for urea synthesis. (b) *CO and *N2H2 intermediates pathway for urea synthesis. (c) N2 and CO2 co-reduction steps for urea synthesis on CuPc. (d) The full free energy profile of urea synthesis and hydrogen adsorption on Cu center. Reproduced from ref. 143, with permission from Wiley-VCH, copyright 2022. (e) The computed free energy profiles for urea electrocatalytic on Mo-terminated MoP-(101) surface at applied potentials. (f) The optimized geometrical structures for N2 and N2 + CO2 adsorption on MoP-(101) surface. Reproduced from ref. 109, with permission from Royal Society of Chemistry, copyright 2023. | ||
 | ||
| Fig. 12 (a) *CO and *NH2 intermediates pathway for urea synthesis. (b) *CO and *NH2OH intermediates pathway for urea synthesis. (c) CNT structural model in DFT calculations. (d) C–N coupling on CNT and F-doped CNT active sites. Reproduced from ref. 108, with permission from Springer Nature, copyright 2022. (e) Free energies for *CO + *NH2OH → *CONH2 on Au, AuPd, and Pd. (f) The C–N coupling pathway on AuPd. Reproduced from ref. 40, with permission from Elsevier, copyright 2022. | ||
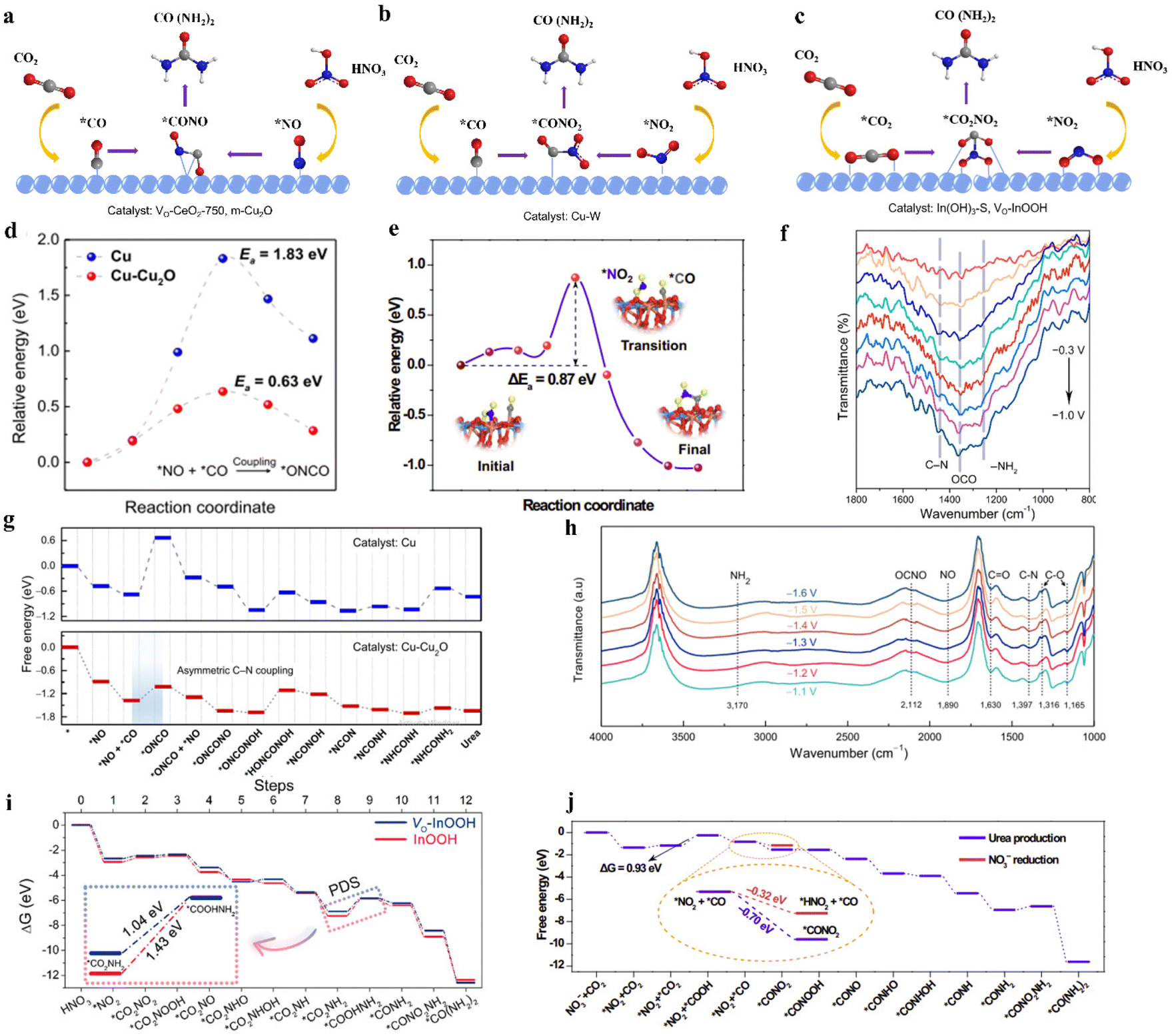 | ||
| Fig. 13 (a) *CO and *NO intermediates pathway for urea synthesis. (b) *CO and *NO2 intermediates pathway for urea synthesis. (c) *CO2 and *NO2 intermediates pathway for urea synthesis. (d) Urea formation mechanisms and corresponding free energy diagrams along the urea formation pathway on Cu and Cu-Cu2O surfaces. (e) The initial, transition, and final states during the *CONO2 formation are presented. (f) Operando SR-FTIR spectra of VO-InOOH at various potentials (−0.3 to −1.0 V vs. RHE) in the range of 800–1800 cm−1. (g) The illustration of urea formation mechanisms and corresponding free energy diagrams along the urea formation pathway on Cu and Cu-Cu2O surfaces. (h) Infrared signal in the range of 1000–4000 cm−1 under various potentials for m-Cu2O during the electrocoupling of NO3− and CO2. Reproduced from ref. 149, with permission from Chinese Chemical Society, copyright 2023. (i) Free-energy diagrams for urea production on the (110) facets of VO-InOOH and pristine InOOH at 0 V vs. RHE. Reproduced from ref. 85, with permission from American Chemical Society, copyright 2022. (j) Free-energy diagram for urea production and NO3− reduction on the CuWO4. Reproduced from ref. 122, with permission from Springer Nature, copyright 2023. | ||
In line with the experimental and theoretical results, a better catalyst should have moderate hydrogen adsorption strength to accomplish the intermediates’ hydrogenation rather than a strong hydrogen evolution. Meanwhile, a strong oxygen adsorption may induce the feedstocks and oxygenated intermediates to adsorb on the reactive sites, which dominate the direction of reactions. On the other hand, the energy barrier is an important determinant of the reaction. The low energy barrier of the C–N coupling step is more beneficial for the generation of urea and other organonitrogens. This is another determining fact that influences the effectiveness of urea production in the existing studies.
8 Summary and outlooks
Electrochemical C–N coupling presents a viable approach for the environmentally friendly and sustainable synthesis of high-value urea under mild conditions. This review aims to provide a deeper understanding of C–N coupling synthesis technology by reviewing catalyst design, characterization, electrolytic cell types, urea quantifications, and mechanistic investigations. We summarize various strategies for catalyst design, including atomic-scale modulation, vacancy engineering, heteroatom doping, alloying, crystal surface modulation, and heterostructure modification. Furthermore, the advantages and shortcomings of H-cells, flow cells, and MEAs are also concluded. Additionally, this review encapsulates four prevalent techniques for urea detection, outlining their respective application contexts.8.1 Develop superior electrocatalysts
The development of high-quality electrocatalysts with high activity and selectivity is crucial in electrocatalytic urea synthesis.161 Several factors are important in designing efficient catalysts: firstly, the ability to adsorb and activate CO2 and nitrogenous species. On catalysts, the physical adsorption of CO2 is more favorable than the chemical adsorption. For instance, Mo, Co, Rh, and Ir exhibit a more stable chemisorption of CO2, rendering them unsuitable as catalysts for urea production, while Zn, Cd, and In display a stronger physical adsorption capacity of CO2 and have been identified to possess superior ability in urea production.162,163 Additionally, the catalyst should also have sufficient active sites to enable the simultaneous reduction of various reactants. The efficiency of a catalyst can be maximized by adjusting the density of active sites on its surface, which can be achieved by alloying, defect formation, coordination, and doping methods. The modification of electrocatalysts with multiple active centers is crucial for the simultaneous reduction of carbon and nitrogen-containing species. Lastly, the stability of catalysts plays the key role in determining the feasibility of scaled-up urea synthesis. Catalyst poisoning, aggregation, sintering and dissolution always lead to the inactivation of electrocatalysts during the electrocatalytic process. From the perspective of catalyst design, a catalyst carrier with high specific surface area, chemical stability and thermal stability is beneficial for improving the stability and reactivity of proposed electrocatalysts. Meanwhile, surface modification can effectively optimize the electronic structure of electrocatalysts, which can lead to an adjustment of adsorption properties of intermediates and minimize the production of unwanted byproducts (especially CO).1648.2 Control the reaction environment
Generally, urea synthesis from carbon dioxide and nitrogenous molecules co-reduction occurs predominantly at the region of the reaction interface between the liquid electrolyte, the gaseous reactants, and the solid electrocatalysts. In this reaction environment, electrocatalyst-level, electrolyte-level, and electrochemical cells should be carefully considered. Improving the affinity of catalysts to reactants and enhancing their hydrophobicity could potentially stabilize key intermediates. The anode ions (such as OH−, HCO3−, CO32−, or NO3−) and the cathode ions (such as OH−, HCO3−, CO32−, or NO3−) have a significant impact on the local pH value, electrochemical double layer, and distribution of the electric field.150,165,166 The structure of the device influences the flow rates of mass and the pressures within the gas and liquid phases, thereby governing the stability and selectivity of the coupling reaction products.8.3 Inhibit side reactions
Side reactions lower faradaic efficiency and may affect product separation. Any irrelevant hydrogenation reactions like CO2RR, NRR, and HER should be suppressed. Meanwhile, the C–N coupling reaction should always be confined to a narrow potential window since the C/N-intermediates tend to form C–N bonds in the low overpotential region. The urea synthesis reaction requires the coupling of preferred C-intermediates/N-intermediate pairs, such as *CO/*NH2, *COOH/*NH2, and *CO2/*NO2. In addition, the over-hydrogenation of C/N sources will inhibit the necessary C–N coupling process. Moreover, the HER can have a significant impact on the formation of C/N intermediates, which will interfere with the operation of the entire system by impeding the electron transfer during urea synthesis.1678.4 Challenges for industrialized applications
To evaluate the potential for industrializing C–N coupled urea technology, a thorough techno-economic analysis (TEA) is necessary. This analysis should determine the costs of an electrochemical urea synthesis strategy, including feedstock costs, energy costs, and operation costs. In operation costs, the byproduct separation cost holds an important position in determining the comprehensive costs. The side production is a key factor affecting the feasibility of scaled-up urea electrosynthesis. A complicated product separation process is needed when various products including urea, nitrite, ammonia, and other organonitrogen products are produced in the C–N coupling system. In contrast, to develop efficient separation methods, a high-performance urea synthesis method is more suitable to reduce separation costs. Despite the increasing maturity of electrocatalytic urea synthesis technology, the electrochemical process must achieve a Faraday efficiency of at least 73.24% at a given electricity price of 0.03 kW h−1 to be competitive with current urea production.171 Therefore, it is critical to develop catalysts with excellent product yield and selectivity at industrial-scale current densities (>100 mA cm−2). It is necessary to recognize that the current density, Faraday efficiency, and yield are not solely influenced by the electrocatalyst but also by the architecture of the electrochemical reactor. The structure of the electrode has a direct implication on charge transfer, gas conversion, and mass diffusion, which can significantly affect reaction kinetics. An ideal electrocatalytic reactor should effectively facilitate electron transfer, ion transport, and controlled gas diffusion while preventing the mixing of anode and cathode electrolytes.With the maturity of electrocatalytic technology, energy sources will be the last issue to hinder large-scale application of electrosynthesis. The revolution of renewable energy is becoming essential to solve the release of greenhouse gases and fossil fuel shortages in future decades.172,173 Among the current candidates, solar-fueled photovoltaic technology is one of the most promising energy sources around the world. On the technological basis of silicon and thin-film solar cells, the photoconversion efficiencies of dye-sensitized, perovskite, organic, and quantum dot-sensitized solar cells are greatly improved.174 Particularly, the greatly developed perovskite tandem solar cell provides an opportunity to further improve the photoconversion efficiency.175,176 With the optimization of solar cell stability and operating costs, photovoltaic power supply facilities could be a strategy option for the synthesis of green urea.
8.5 Machine learning in assisting electrocatalytic urea synthesis
Electrocatalytic synthesis of urea necessitates the formation of C–N bonds, requiring catalysts that concurrently facilitate carbon dioxide reduction and nitrogen reduction reactions. However, during the urea formation process, the desorption of the intermediate *CO and further hydrogenation of *NH2 can lead to the formation of CO and NH3 by-products, interfering with the reaction. Therefore, appropriate binding energies for intermediates are also crucial. Nevertheless, traditional methods of synthesizing electrocatalysts are complex and time-consuming, which often results in a long trial-and-error process to successfully design an effective catalyst. Machine learning (ML) techniques, particularly deep learning and reinforcement learning, can be employed to predict and optimize the performance of new catalysts, addressing the bottlenecks in electrocatalytic urea synthesis.177,178 By selecting suitable descriptors and algorithms to build ML models, we can rapidly screen for catalysts rich in active sites, capable of both CO2RR and NRR, and where reaction intermediates can timely couple at active sites to form C–N bonds while suppressing the HER. With the development of “big data” and mechanistic investigation, we believe that novel designed catalysts will be increasingly applicable for developing sustainable and economically friendly urea synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
W. Z. would like to acknowledge the support from the National Natural Science Foundation of China (22176086), Natural Science Foundation of Jiangsu Province (BK20210189), State Key Laboratory of Pollution Control and Resource Reuse, the Fundamental Research Funds for the Central Universities (021114380183, 021114380189, 021114380199), the Research Funds from the Frontiers Science Center for Critical Earth Material Cycling of Nanjing University, Research Funds for Jiangsu Distinguished Professor, Carbon Peaking and Carbon Neutrality Technological Innovation Foundation of Jiangsu Province (BE2022861), Foundation of MOE Key Laboratory of Groundwater Circulation and Environmental Evolution (2023–003), China University of Geosciences (Beijing).References
- T. Kim and G. T. R. Palmore, Nat. Commun., 2020, 11, 3622 CrossRef CAS PubMed
.
- S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef PubMed
- T. Tang, Z. Wang and J. Guan, Coord. Chem. Rev., 2023, 492, 215288 CrossRef CAS
- T. Tang, Z. Wang and J. Guan, Exploration, 2023, 3, 20230011 CrossRef PubMed
- W. Wang, C. Zeng and N. Tsubaki, Green Carbon, 2023, 1, 133–145 CrossRef
- G. A. Olah, G. K. S. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS PubMed
- W. Zhu, R. Michalsky, Ö. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed
- X. Li, Y. Chen, X. Zhan, Y. Xu, L. Hao, L. Xu, X. Li, M. Umer, X. Tan, B. Han, A. W. Robertson and Z. Sun, Innov. Mater., 2023, 1, 100014 CrossRef
- X. Wang, B. Liu, Y. Zhang, T. Butburee, K. Ostrikov, S. Wang and W. Huang, EcoEnergy, 2023, 1, 108–153 CrossRef
- W. Zhu, Y.-J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS PubMed
- A. R. Woldu, Z. Huang, P. Zhao, L. Hu and D. Astruc, Coord. Chem. Rev., 2022, 454, 214340 CrossRef CAS
- D. Yang, Q. Zhu and B. Han, Innovation, 2020, 1, 100016 CAS
- P. Zhu and H. Wang, Nat. Catal., 2021, 4, 943–951 CrossRef CAS
- W. Luc, X. Fu, J. Shi, J.-J. Lv, M. Jouny, B. H. Ko, Y. Xu, Q. Tu, X. Hu, J. Wu, Q. Yue, Y. Liu, F. Jiao and Y. Kang, Nat. Catal., 2019, 2, 423–430 CrossRef CAS
- S. Verma, S. Lu and P. J. A. Kenis, Nat. Energy, 2019, 4, 466–474 CrossRef CAS
- S. Akkari, V. Vivier and C. M. Sánchez-Sánchez, Electrochim. Acta, 2024, 474, 143526 CrossRef CAS
- Y. Manaka, Y. Nagatsuka and K. Motokura, Sci. Rep., 2020, 10, 2834 CrossRef CAS PubMed
- S. Giddey, S. P. S. Badwal and A. Kulkarni, Int. J. Hydrogen Energy, 2013, 38, 14576–14594 CrossRef CAS
- S. M. Lemkowitz, J. C. Van Erp, D. M. Rekers and P. J. Van Den Berg, J. Chem. Technol. Biotechnol., 1980, 30, 85–101 CrossRef CAS
- M. Pérez-Fortes, A. Bocin-Dumitriu and E. Tzimas, Energy Procedia, 2014, 63, 7968–7975 CrossRef
- F. Barzagli, F. Mani and M. Peruzzini, Green Chem., 2011, 13, 1267–1274 RSC
- J. Zhang, C. D. Sewell, H. Huang and Z. Lin, Adv. Energy Mater., 2021, 11, 2102767 CrossRef CAS
- J. Wang, Y. Gao, H. Kong, J. Kim, S. Choi, F. Ciucci, Y. Hao, S. Yang, Z. Shao and J. Lim, Chem. Soc. Rev., 2020, 49, 9154–9196 RSC
- J. Li, Y. Zhang, K. Kuruvinashetti and N. Kornienko, Nat. Rev. Chem., 2022, 6, 303–319 CrossRef CAS PubMed
- X. Liu, Y. Jiao, Y. Zheng, M. Jaroniec and S.-Z. Qiao, Nat. Commun., 2022, 13, 5471 CrossRef CAS PubMed
- M. Shibata, K. Yoshida and N. Furuya, J. Electroanal. Chem., 1995, 387, 143–145 CrossRef
- D. B. Kayan and F. Köleli, Appl. Catal., B, 2016, 181, 88–93 CrossRef CAS
- C. Chen, X. Zhu, X. Wen, Y. Zhou, L. Zhou, H. Li, L. Tao, Q. Li, S. Du, T. Liu, D. Yan, C. Xie, Y. Zou, Y. Wang, R. Chen, J. Huo, Y. Li, J. Cheng, H. Su, X. Zhao, W. Cheng, Q. Liu, H. Lin, J. Luo, J. Chen, M. Dong, K. Cheng, C. Li and S. Wang, Nat. Chem., 2020, 12, 717–724 CrossRef CAS PubMed
- Y. Huang, R. Yang, C. Wang, N. Meng, Y. Shi, Y. Yu and B. Zhang, ACS Energy Lett., 2022, 7, 284–291 CrossRef CAS
- A. S. Dutton, J. M. Fukuto and K. N. Houk, Inorg. Chem., 2005, 44, 4024–4028 CrossRef CAS PubMed
- A. C. A. de Vooys, G. L. Beltramo, B. van Riet, J. A. R. van Veen and M. T. M. Koper, Electrochim. Acta, 2004, 49, 1307–1314 CrossRef CAS
- Z. Li, L. Wang, Y. Cai, J.-R. Zhang and W. Zhu, J. Hazard. Mater., 2022, 440, 129828 CrossRef CAS
- Z. Lyu, S. Ding, L. Fang, X. Li, T. Li, M. Xu, X. Pan, W. Zhu, Y. Zhou, D. Du and Y. Lin, Anal. Chem., 2023, 95, 4521–4528 CrossRef CAS PubMed
- H. Wang, Q.-L. Zhu, R. Zou and Q. Xu, Chem, 2017, 2, 52–80 CAS
- K. Li, Y. Cai, X. Yang, S. Wang, C. Teng, Y. Tian, Q. Min and W. Zhu, Adv. Funct. Mater., 2022, 32, 2113002 CrossRef CAS
- H. Du, J. Fu, L.-X. Liu, S. Ding, Z. Lyu, Y.-C. Chang, X. Jin, F. O. Kengara, B. Song, Q. Min, J.-J. Zhu, D. Du, C. Gu, Y. Lin, J.-S. Hu and W. Zhu, Mater. Today, 2022, 59, 182–199 CrossRef CAS
- M. Jouny, J.-J. Lv, T. Cheng, B. H. Ko, J.-J. Zhu, W. A. Goddard and F. Jiao, Nat. Chem., 2019, 11, 846–851 CrossRef CAS PubMed
- Y. Chai, Z. Lyu, H. Du, P. Li, S. Ding, Y. Jiang, H. Wang, Q. Min, D. Du, Y. Lin and W. Zhu, SusMat, 2022, 2, 392–410 CrossRef CAS
- X. Peng, L. Zeng, D. Wang, Z. Liu, Y. Li, Z. Li, B. Yang, L. Lei, L. Dai and Y. Hou, Chem. Soc. Rev., 2023, 52, 2193–2237 RSC
- H. Wang, Y. Jiang, S. Li, F. Gou, X. Liu, Y. Jiang, W. Luo, W. Shen, R. He and M. Li, Appl. Catal., B, 2022, 318, 121819 CrossRef CAS
- C. Lv, L. Zhong, H. Liu, Z. Fang, C. Yan, M. Chen, Y. Kong, C. Lee, D. Liu, S. Li, J. Liu, L. Song, G. Chen, Q. Yan and G. Yu, Nat. Sustain., 2021, 4, 868–876 CrossRef
- J.-A. Lai, Z. Zhang, X. Yang and Y. Zhang, Innov. Mater., 2023, 1, 100020 CrossRef
- Y. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529–1541 RSC
- M. Jitaru, D. A. Lowy, M. Toma, B. C. Toma and L. Oniciu, J. Appl. Electrochem., 1997, 27, 875–889 CrossRef CAS
- Y. Wang, W. Zhou, R. Jia, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 5350–5354 CrossRef CAS PubMed
- C. Ma, H.-F. Wei, M.-X. Wang, S. Wu, Y.-C. Chang, J. Zhang, L.-P. Jiang, W. Zhu, Z. Chen and Y. Lin, Nano Lett., 2020, 20, 5008–5016 CrossRef CAS PubMed
- S. Guo, X. Zhang, W. Zhu, K. He, D. Su, A. Mendoza-Garcia, S. F. Ho, G. Lu and S. Sun, J. Am. Chem. Soc., 2014, 136, 15026–15033 CrossRef CAS PubMed
- H. Tian, W. Zhu, Q. Shi, S. Ding, Z. Lyu, M. Xu, X. Pan, M. H. Engelhard, D. Dan and Y. Lin, J. Mater. Chem. A, 2022, 10, 11196–11204 RSC
- H. Lv, Z. Xi, Z. Chen, S. Guo, Y. Yu, W. Zhu, Q. Li, X. Zhang, M. Pan, G. Lu, S. Mu and S. Sun, J. Am. Chem. Soc., 2015, 137, 5859–5862 CrossRef CAS PubMed
- G. Wang, X. Li, X. Yang, L.-X. Liu, Y. Cai, Y. Wu, S. Wang, H. Li, Y. Zhou, Y. Wang and Y. Zhou, Chem. – Eur. J., 2022, 28, e202201834 CrossRef CAS PubMed
- M. Shibata and N. Furuya, J. Electroanal. Chem., 2001, 507, 177–184 CrossRef CAS
- J. He, N. J. J. Johnson, A. Huang and C. P. Berlinguette, ChemSusChem, 2018, 11, 48–57 CrossRef CAS PubMed
- K. Chen, M. Cao, G. Ni, S. Chen, H. Liao, L. Zhu, H. Li, J. Fu, J. Hu, E. Cortés and M. Liu, Appl. Catal., B, 2022, 306, 121093 CrossRef CAS
- J. Wang, Y. Wang, C. Cai, Y. Liu, D. Wu, M. Wang, M. Li, X. Wei, M. Shao and M. Gu, Nano Lett., 2023, 23, 1897–1903 CrossRef CAS PubMed
- T. Hou, J. Ding, H. Zhang, S. Chen, Q. Liu, J. Luo and X. Liu, Mater. Chem. Front., 2023, 7, 4952–4960 RSC
- S. Liu, S. Yin, Z. Wang, Y. Xu, X. Li, L. Wang and H. Wang, Cell Rep. Phys. Sci., 2022, 3, 100869 CrossRef CAS
- Y. Feng, H. Yang, Y. Zhang, X. Huang, L. Li, T. Cheng and Q. Shao, Nano Lett., 2020, 20, 8282–8289 CrossRef CAS PubMed
- S. Kim, J. Park, J. Hwang and J. Lee, EnergyChem, 2021, 3, 100054 CrossRef CAS
- Y. Liu and W. Zhang, Commun. Chem., 2023, 6, 36 CrossRef CAS PubMed
- J. Liu, Y. Cai, R. Song, S. Ding, Z. Lyu, Y.-C. Chang, H. Tian, X. Zhang, D. Du, W. Zhu, Y. Zhou and Y. Lin, Mater. Today, 2021, 48, 95–114 CrossRef CAS
- L. Wang, W. Chen, D. Zhang, Y. Du, R. Amal, S. Qiao, J. Wu and Z. Yin, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC
- Z. Li, Y. Chen, S. Ji, Y. Tang, W. Chen, A. Li, J. Zhao, Y. Xiong, Y. Wu, Y. Gong, T. Yao, W. Liu, L. Zheng, J. Dong, Y. Wang, Z. Zhuang, W. Xing, C.-T. He, C. Peng, W.-C. Cheong, Q. Li, M. Zhang, Z. Chen, N. Fu, X. Gao, W. Zhu, J. Wan, J. Zhang, L. Gu, S. Wei, P. Hu, J. Luo, J. Li, C. Chen, Q. Peng, X. Duan, Y. Huang, X.-M. Chen, D. Wang and Y. Li, Nat. Chem., 2020, 12, 764–772 CrossRef CAS PubMed
- X. Xie, C. He, B. Li, Y. He, D. A. Cullen, E. C. Wegener, A. J. Kropf, U. Martinez, Y. Cheng, M. H. Engelhard, M. E. Bowden, M. Song, T. Lemmon, X. S. Li, Z. Nie, J. Liu, D. J. Myers, P. Zelenay, G. Wang, G. Wu, V. Ramani and Y. Shao, Nat. Catal., 2020, 3, 1044–1054 CrossRef CAS
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Adv. Mater., 2020, 32, 2001848 CrossRef CAS PubMed
- J. Leverett, T. Tran-Phu, J. A. Yuwono, P. Kumar, C. Kim, Q. Zhai, C. Han, J. Qu, J. Cairney, A. N. Simonov, R. K. Hocking, L. Dai, R. Daiyan and R. Amal, Adv. Energy Mater., 2022, 12, 2201500 CrossRef CAS
- X. Wei, Y. Liu, X. Zhu, S. Bo, L. Xiao, C. Chen, T. T. T. Nga, Y. He, M. Qiu, C. Xie, D. Wang, Q. Liu, F. Dong, C.-L. Dong, X.-Z. Fu and S. Wang, Adv. Mater., 2023, 35, 2300020 CrossRef CAS PubMed
- N. Wang, Z. Liu, J. Ma, J. Liu, P. Zhou, Y. Chao, C. Ma, X. Bo, J. Liu, Y. Hei, Y. Bi, M. Sun, M. Cao, H. Zhang, F. Chang, H.-L. Wang, P. Xu, Z. Hu, J. Bai, H. Sun, G. Hu and M. Zhou, ACS Sustainable Chem. Eng., 2020, 8, 13813–13822 CrossRef CAS
- X. Liu, Z. Wang, Y. Tian and J. Zhao, J. Phys. Chem. C, 2020, 124, 3722–3730 CrossRef CAS
- R. Boppella, M. Austeria P, Y. Kim, E. Kim, I. Song, Y. Eom, D. P. Kumar, M. Balamurμgan, E. Sim, D. H. Kim and T. K. Kim, Adv. Funct. Mater., 2022, 32, 2202351 CrossRef CAS
- Z. Geng, Y. Liu, X. Kong, P. Li, K. Li, Z. Liu, J. Du, M. Shu, R. Si and J. Zeng, Adv. Mater., 2018, 30, 1870301 CrossRef
- D. Jiao, Y. Liu, Q. Cai and J. Zhao, J. Mater. Chem. A, 2021, 9, 1240–1251 RSC
- H. Zou, L. J. Arachchige, W. Rong, C. Tang, R. Wang, S. Tan, H. Chen, D. He, J. Hu, E. Hu, C. Sun and L. Duan, Adv. Funct. Mater., 2022, 32, 2200333 CrossRef CAS
- Y. Hu, Z. Li, B. Li and C. Yu, Small, 2022, 18, 2203589 CrossRef CAS PubMed
- R. Li and D. Wang, Adv. Energy Mater., 2022, 12, 2103564 CrossRef CAS
- J. Liu, S. C. Smith, Y. Gu and L. Kou, Adv. Funct. Mater., 2023, 33, 2305894 CrossRef CAS
- X. Zhang, X. Zhu, S. Bo, C. Chen, M. Qiu, X. Wei, N. He, C. Xie, W. Chen, J. Zheng, P. Chen, S. P. Jiang, Y. Li, Q. Liu and S. Wang, Nat. Commun., 2022, 13, 5337 CrossRef CAS PubMed
- C. Liu, H. Tong, P. Wang, R. Huang, P. Huang, G. Zhou and L. Liu, Appl. Catal., B, 2023, 336, 122917 CrossRef CAS
- H. Yang, Y. Chen and Y. Qin, Chin. J. Catal., 2020, 41, 227–241 CrossRef CAS
- S. Tian, Q. Fu, W. Chen, Q. Feng, Z. Chen, J. Zhang, W.-C. Cheong, R. Yu, L. Gu, J. Dong, J. Luo, C. Chen, Q. Peng, C. Draxl, D. Wang and Y. Li, Nat. Commun., 2018, 9, 2353 CrossRef PubMed
- Z. Lu, B. Wang, Y. Hu, W. Liu, Y. Zhao, R. Yang, Z. Li, J. Luo, B. Chi, Z. Jiang, M. Li, S. Mu, S. Liao, J. Zhang and X. Sun, Angew. Chem., Int. Ed., 2019, 58, 2622–2626 CrossRef CAS PubMed
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS PubMed
- H. Zhou, B. Xiong, L. Chen and J. Shi, J. Mater. Chem. A, 2020, 8, 20286–20293 RSC
- K. Zhu, F. Shi, X. Zhu and W. Yang, Nano Energy, 2020, 73, 104761 CrossRef CAS
- L. Zhang, X.-Y. Xie, H. Wang, L. Ji, Y. Zhang, H. Chen, T. Li, Y. Luo, G. Cui and X. Sun, Chem. Commun., 2019, 55, 4627–4630 RSC
- C. Lv, C. Lee, L. Zhong, H. Liu, J. Liu, L. Yang, C. Yan, W. Yu, H. H. Hng, Z. Qi, L. Song, S. Li, K. P. Loh, Q. Yan and G. Yu, ACS Nano, 2022, 16, 8213–8222 CrossRef CAS PubMed
- X. Wei, X. Wen, Y. Liu, C. Chen, C. Xie, D. Wang, M. Qiu, N. He, P. Zhou, W. Chen, J. Cheng, H. Lin, J. Jia, X.-Z. Fu and S. Wang, J. Am. Chem. Soc., 2022, 144, 11530–11535 CrossRef CAS PubMed
- N. Meng, Y. Huang, Y. Liu, Y. Yu and B. Zhang, Cell Rep. Phys. Sci., 2021, 2, 100378 CrossRef CAS
- D. Raciti and C. Wang, ACS Energy Lett., 2018, 3, 1545–1556 CrossRef CAS
- N. Cao, Y. Quan, A. Guan, C. Yang, Y. Ji, L. Zhang and G. Zheng, J. Colloid Interface Sci., 2020, 577, 109–114 CrossRef CAS PubMed
- X. Yang, K. Sun, M. Ma, C. Xu, R. Ren, J. Qiao, Z. Wang, S. Zhen, R. Hou and W. Sun, Appl. Catal., B, 2020, 272, 118968 CrossRef CAS
- Z. Chen, T. Fan, Y.-Q. Zhang, J. Xiao, M. Gao, N. Duan, J. Zhang, J. Li, Q. Liu, X. Yi and J.-L. Luo, Appl. Catal., B, 2020, 261, 118243 CrossRef CAS
- K. Wang, S. Shu, M. Chen, J. Li, K. Zhou, J. Pan, X. Wang, X. Li, J. Sheng, F. Dong and G. Jiang, Chem. Eng. J., 2020, 381, 122673 CrossRef CAS
- Q. Wu, M.-J. Mao, Q.-J. Wu, J. Liang, Y.-B. Huang and R. Cao, Small, 2021, 17, 2004933 CrossRef CAS PubMed
- M. Yuan, J. Chen, Y. Bai, Z. Liu, J. Zhang, T. Zhao, Q. Wang, S. Li, H. He and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 10910–10918 CrossRef CAS PubMed
- M. Yuan, J. Chen, Y. Bai, Z. Liu, J. Zhang, T. Zhao, Q. Shi, S. Li, X. Wang and G. Zhang, Chem. Sci., 2021, 12, 6048–6058 RSC
- G. Li, Y. Li, H. Liu, Y. Guo, Y. Li and D. Zhu, Chem. Commun., 2010, 46, 3256–3258 RSC
- Y. Fang, Y. Liu, L. Qi, Y. Xue and Y. Li, Chem. Soc. Rev., 2022, 51, 2681–2709 RSC
- D. Zhang, Y. Xue, X. Zheng, C. Zhang and Y. Li, Natl. Sci. Rev., 2023, 10, nwac209 CrossRef CAS PubMed
- M.-Y. Li, Y. Shi, C.-C. Cheng, L.-S. Lu, Y.-C. Lin, H.-L. Tang, M.-L. Tsai, C.-W. Chu, K.-H. Wei, J.-H. He, W.-H. Chang, K. Suenaga and L.-J. Li, Science, 2015, 349, 524–528 CrossRef CAS PubMed
- Z. Huang, X. Tang, J. Zhao, H. Lin, M. Nie and Q. Li, J. Mater. Chem. C, 2022, 10, 3489–3499 RSC
- B. Qiu, C. Wang, N. Zhang, L. Cai, Y. Xiong and Y. Chai, ACS Catal., 2019, 9, 6484–6490 CrossRef CAS
- W. D. Chemelewski, J. R. Rosenstock and C. B. Mullins, J. Mater. Chem. A, 2014, 2, 14957–14962 RSC
- Z. Ye, T. Li, G. Ma, Y. Dong and X. Zhou, Adv. Funct. Mater., 2017, 27, 1704083 CrossRef
- Z. Li, X. Lu, J. Teng, Y. Zhou and W. Zhuang, Nanoscale, 2021, 13, 11314–11324 RSC
- B. Zhang, L. Sun, Y. Wang, S. Chen and J. Zhang, J. Energy Chem., 2020, 41, 7–14 CrossRef
- W. Lee, G. M. Kim, S. Baik and J. W. Lee, Electrochim. Acta, 2016, 210, 743–753 CrossRef CAS
- W. Ni, Y. Xue, X. Zang, C. Li, H. Wang, Z. Yang and Y.-M. Yan, ACS Nano, 2020, 14, 2014–2023 CrossRef CAS PubMed
- X. Liu, P. V. Kumar, Q. Chen, L. Zhao, F. Ye, X. Ma, D. Liu, X. Chen, L. Dai and C. Hu, Appl. Catal., B, 2022, 316, 121618 CrossRef CAS
- D. Jiao, Y. Dong, X. Cui, Q. Cai, C. R. Cabrera, J. Zhao and Z. Chen, J. Mater. Chem. A, 2023, 11, 232–240 RSC
- C. Zhu, C. Wen, M. Wang, M. Zhang, Y. Geng and Z. Su, Chem. Sci., 2022, 13, 1342–1354 RSC
- D. Xu, S.-N. Zhang, J.-S. Chen and X.-H. Li, Chem. Rev., 2023, 123, 1–30 CrossRef CAS PubMed
- Z. Li, M. Hu, P. Wang, J. Liu, J. Yao and C. Li, Coord. Chem. Rev., 2021, 439, 213953 CrossRef CAS
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Surf. Sci., 1995, 335, 258–263 CrossRef CAS
- Z. Niu, W. D. C. B. Gunatilleke, Q. Sun, P. C. Lan, J. Perman, J.-G. Ma, Y. Cheng, B. Aguila and S. Ma, Chem, 2018, 4, 2587–2599 CAS
- J. Li, Z. Xia, Q. Xue, M. Zhang, S. Zhang, H. Xiao, Y. Ma and Y. Qu, Small, 2021, 17, 2103018 CrossRef CAS PubMed
- M. Yuan, J. Chen, Y. Xu, R. Liu, T. Zhao, J. Zhang, Z. Ren, Z. Liu, C. Streb, H. He, C. Yang, S. Zhang and G. Zhang, Energy Environ. Sci., 2021, 14, 6605–6615 RSC
- H. Hu, J. Wang, P. Tao, C. Song, W. Shang, T. Deng and J. Wu, J. Mater. Chem. A, 2022, 10, 5835–5849 RSC
- L. Zhang, W. Shi and B. Zhang, J. Energy Chem., 2017, 26, 1117–1135 CrossRef
- J. E. Kim, J. H. Jang, K. M. Lee, M. Balamurugan, Y. I. Jo, M. Y. Lee, S. Choi, S. W. Im and K. T. Nam, Angew. Chem., Int. Ed., 2021, 60, 21943–21951 CrossRef CAS PubMed
- N. Meng, J. Shao, H. Li, Y. Wang, X. Fu, C. Liu, Y. Yu and B. Zhang, Nat. Commun., 2022, 13, 5452 CrossRef CAS PubMed
- Y. Luo, K. Xie, P. Ou, C. Lavallais, T. Peng, Z. Chen, Z. Zhang, N. Wang, X.-Y. Li, I. Grigioni, B. Liu, D. Sinton, J. B. Dunn and E. H. Sargent, Nat. Catal., 2023, 6, 939–948 CrossRef CAS
- Y. Zhao, Y. Ding, W. Li, C. Liu, Y. Li, Z. Zhao, Y. Shan, F. Li, L. Sun and F. Li, Nat. Commun., 2023, 14, 4491 CrossRef CAS PubMed
- H. Sun and W. Zhou, Energy Fuels, 2021, 35, 5716–5737 CrossRef CAS
- M. Wang, L. Árnadóttir, Z. J. Xu and Z. Feng, Nano-Micro Lett., 2019, 11, 47 CrossRef CAS PubMed
- Y. Mao, Y. Jiang, Q. Gou, S. Lv, Z. Song, Y. Jiang, W. Wang, M. Li, L. Zheng, W. Su and R. He, Appl. Catal., B, 2024, 340, 123189 CrossRef CAS
- C. Chen, X. Yan, Y. Wu, S. Liu, X. Sun, Q. Zhu, R. Feng, T. Wu, Q. Qian, H. Liu, L. Zheng, J. Zhang and B. Han, Chem. Sci., 2021, 12, 5938–5943 RSC
- H. Zhang, Z. Zhou, Q. Lei and T. W. B. Lo, Curr. Opin. Electrochem., 2023, 38, 101215 CrossRef CAS
- K. Feng, H. Zhang, J. Gao, J. Xu, Y. Dong, Z. Kang and J. Zhong, Appl. Phys. Lett., 2020, 116 Search PubMed
- Z. Liang, L. Yin, H. Yin, Z. Yin and Y. Du, Nanoscale Horiz., 2022, 7, 31–40 RSC
- C. Chen, N. He and S. Wang, Small Sci., 2021, 1, 2100070 CrossRef CAS
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS
- J. Liu, L. Peng, Y. Zhou, L. Lv, J. Fu, J. Lin, D. Guay and J. Qiao, ACS Sustainable Chem. Eng., 2019, 7, 15739–15746 CrossRef CAS
- P. Senthilkumar, M. Mohapatra and S. Basu, RSC Adv., 2022, 12, 1287–1309 RSC
- F.-Y. Chen, Z.-Y. Wu, Z. Adler and H. Wang, Joule, 2021, 5, 1704–1731 CrossRef CAS
- D. Li, N. Xu, Y. Zhao, C. Zhou, L.-P. Zhang, L.-Z. Wu and T. Zhang, Small Methods, 2022, 6, 2200561 CrossRef CAS PubMed
- D. Saravanakumar, J. Song, S. Lee, N. H. Hur and W. Shin, ChemSusChem, 2017, 10, 3999–4003 CrossRef CAS PubMed
- J. J. Mccarthy, Limnol. Oceanogr., 1970, 15, 309–313 CrossRef CAS
- Y. Zhao, R. Shi, X. Bian, C. Zhou, Y. Zhao, S. Zhang, F. Wu, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Sci., 2019, 6, 1802109 CrossRef PubMed
- P. S. Francis, S. W. Lewis and K. F. Lim, TrAC, Trends Anal. Chem., 2002, 21, 389–400 CrossRef CAS
- S. Kodama and T. Suzuki, J. Food Sci., 1995, 60, 1097–1099 CrossRef CAS
- D. Li, N. Xu, Y. Zhao, R. Shi, C. Zhou, L.-P. Zhang and T. Zhang, Adv. Energy Mater., 2023, 2303885 CrossRef
- T. Yuan and O. Voznyy, Cell Rep. Phys. Sci., 2023, 4, 101521 CrossRef CAS
- J. Mukherjee, S. Paul, A. Adalder, S. Kapse, R. Thapa, S. Mandal, B. Ghorai, S. Sarkar and U. K. Ghorai, Adv. Funct. Mater., 2022, 32, 2200882 CrossRef CAS
- Z. Lv, S. Zhou, L. Zhao, Z. Liu, J. Liu, W. Xu, L. Wang and J. Lai, Adv. Energy Mater., 2023, 13, 2300946 CrossRef CAS
- J. Geng, S. Ji, M. Jin, C. Zhang, M. Xu, G. Wang, C. Liang and H. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202210958 CrossRef CAS PubMed
- J. Qin, N. Liu, L. Chen, K. Wu, Q. Zhao, B. Liu and Z. Ye, ACS Sustainable Chem. Eng., 2022, 10, 15869–15875 CrossRef CAS
- Y. Wang, S. Xia, J. Zhang, Z. Li, R. Cai, C. Yu, Y. Zhang, J. Wu and Y. Wu, ACS Energy Lett., 2023, 8, 3373–3380 CrossRef CAS
- X. Tu, X. Zhu, S. Bo, X. Zhang, R. Miao, G. Wen, C. Chen, J. Li, Y. Zhou, Q. Liu, D. Chen, H. Shao, D. Yan, Y. Li, J. Jia and S. Wang, Angew. Chem., Int. Ed., 2024, 63, e202317087 CrossRef CAS PubMed
- M. Qiu, X. Zhu, S. Bo, K. Cheng, N. He, K. Gu, D. Song, C. Chen, X. Wei, D. Wang, Y. Liu, S. Li, X. Tu, Y. Li, Q. Liu, C. Li and S. Wang, CCS Chem., 2023, 5, 2617–2627 CrossRef CAS
- J. Fu, Y. Yang and J.-S. Hu, ACS Mater. Lett., 2021, 3, 1468–1476 CrossRef CAS
- N. Meng, X. Ma, C. Wang, Y. Wang, R. Yang, J. Shao, Y. Huang, Y. Xu, B. Zhang and Y. Yu, ACS Nano, 2022, 16, 9095–9104 CrossRef CAS PubMed
- Y. Chen, Y. Liu, S. Hu, D. Wu, M. Zhang and Z. Cheng, Sci. Total Environ., 2024, 913, 169722 CrossRef CAS PubMed
- D. Anastasiadou, B. Ligt, Y. He, R. C. J. van de Poll, J. F. M. Simons and M. C. Figueiredo, Commun. Chem., 2023, 6, 199 CrossRef CAS PubMed
- X. Cao, D. Tan, B. Wulan, K. S. Hui, K. N. Hui and J. Zhang, Small Methods, 2021, 5, 2100700 CrossRef CAS PubMed
- A. M. Tripathi, W.-N. Su and B. J. Hwang, Chem. Soc. Rev., 2018, 47, 736–851 RSC
- Y. J. Wang, D. W. Chen, C. Chen and S. Y. Wang, Acc. Chem. Res., 2024, 57, 247–256 CrossRef CAS PubMed
- Y. Yu, B. Mao, A. Geller, R. Chang, K. Gaskell, Z. Liu and B. W. Eichhorn, Phys. Chem. Chem. Phys., 2014, 16, 11633–11639 RSC
- E. L. Clark, M. R. Singh, Y. Kwon and A. T. Bell, Anal. Chem., 2015, 87, 8013–8020 CrossRef CAS PubMed
- S. Jia, S. Matsuda, S. Tamura, S. Shironita and M. Umeda, Electrochim. Acta, 2018, 261, 340–345 CrossRef CAS
- P. Čičmanec, R. Bulánek and K. Frolich, J. Therm. Anal. Calorim., 2011, 105, 837–844 CrossRef
- M. Zheng, H. Ma, Z. Li, H. Yu, L. Nie, C. Ye, X. Chen and J. Wang, Appl. Catal., B, 2024, 342, 123366 CrossRef CAS
- M. Shibata, K. Yoshida and N. Furuya, J. Electrochem. Soc., 1998, 145, 595 CrossRef CAS
- M. Shibata, K. Yoshida and N. Furuya, J.
Electrochem. Soc., 1998, 145, 2348 CrossRef CAS
- Y.-G. Zhou, Y. Kang and J. Huang, CCS Chem., 2020, 2, 31–41 CrossRef CAS
- V. Hessel, N. N. Tran, M. R. Asrami, Q. D. Tran, N. V. D. Long, M. Escribà-Gelonch, J. O. Tejada, S. Linke and K. Sundmacher, Green Chem., 2022, 24, 410–437 RSC
- B. Chang, H. Pang, F. Raziq, S. Wang, K.-W. Huang, J. Ye and H. Zhang, Energy Environ. Sci., 2023, 16, 4714–4758 RSC
- Y. Ji, A. Guan and G. Zheng, Cell Rep. Phys. Sci., 2022, 3, 101072 CrossRef CAS
- M. Jiang, M. Zhu, M. Wang, Y. He, X. Luo, C. Wu, L. Zhang and Z. Jin, ACS Nano, 2023, 17, 3209–3224 CrossRef CAS PubMed
- B. Min, Q. Gao, Z. Yan, X. Han, K. Hosmer, A. Campbell and H. Zhu, Ind. Eng. Chem. Res., 2021, 60, 14635–14650 CrossRef CAS
- F. Liu, C. Shi, X. Guo, Z. He, L. Pan, Z.-F. Huang, X. Zhang and J.-J. Zou, Adv. Sci., 2022, 9, 2200307 CrossRef CAS PubMed
- Y. Huang, Y. Wang, Y. Wu, Y. Yu and B. Zhang, Sci. China: Chem., 2022, 65, 204–206 CrossRef CAS
- M. Eisa, D. Ragauskaitė, S. Adhikari, F. Bella and J. Baltrusaitis, ACS Sustainable Chem. Eng., 2022, 10, 8997–9001 CrossRef CAS
- G. Gianola, R. Speranza, F. Bella and A. Lamberti, Sol. Energy, 2023, 265, 112116 CrossRef CAS
- I. Ali, M. R. Islam, J. Yin, S. J. Eichhorn, J. Chen, N. Karim and S. Afroj, ACS Nano, 2024, 18, 3871–3915 CrossRef CAS PubMed
- H. Gao, K. Xiao, R. Lin, S. Zhao, W. Wang, S. Dayneko, C. Duan, C. Ji, H. Sun, A. D. Bui, C. Liu, J. Wen, W. Kong, H. Luo, X. Zheng, Z. Liu, H. Nguyen, J. Xie, L. Li, M. I. Saidaminov and H. Tan, Science, 2024, 383, 855–859 CrossRef CAS PubMed
- R. Lin, Y. Wang, Q. Lu, B. Tang, J. Li, H. Gao, Y. Gao, H. Li, C. Ding, J. Wen, P. Wu, C. Liu, S. Zhao, K. Xiao, Z. Liu, C. Ma, Y. Deng, L. Li, F. Fan and H. Tan, Nature, 2023, 620, 994–1000 CrossRef CAS PubMed
- X. Zhang, Y. Tian, L. Chen, X. Hu and Z. Zhou, J. Phys. Chem. Lett., 2022, 13, 7920–7930 CrossRef CAS PubMed
- S. N. Steinmann, Q. Wang and Z. W. Seh, Mater. Horiz., 2023, 10, 393–406 RSC
- S. Shin, S. Sultan, Z.-X. Chen, H. Lee, H. Choi, T.-U. Wi, C. Park, T. Kim, C. Lee, J. Jeong, H. Shin, T.-H. Kim, H. Ju, H. C. Yoon, H.-K. Song, H.-W. Lee, M.-J. Cheng and Y. Kwon, Energy Environ. Sci., 2023, 16, 2003–2013 RSC
- Z. Mei, Y. Zhou, W. Lv, S. Tong, X. Yang, L. Chen and N. Zhang, ACS Sustainable Chem. Eng., 2022, 10, 12477–12496 CrossRef CAS
- L. Hu, Z. Xing and X. Feng, ACS Energy Lett., 2020, 5, 430–436 CrossRef CAS
- Y. Zhong, H. Xiong, J. Low, R. Long and Y. Xiong, eScience, 2023, 3, 100086 CrossRef
- P. Xu, X. Li, Y. Zhou, Y. Chen, X. Wang, H. Jia, M. Li, H. Yu and X. Li, J. Phys. Chem. Lett., 2023, 14, 567–575 CrossRef CAS PubMed
Footnote |
| † These authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2024 |