
Electroreductive upgradation of biomass into high-value chemicals and energy-intensive biofuels
Keping
Wang
a,
Zheng
Li
b,
Zhenyan
Guo
a,
Jinshu
Huang
a,
Tengyu
Liu
a,
Min
Zhou
a,
Jinguang
Hu
 *b and
Hu
Li
*a
*b and
Hu
Li
*a
aNational Key Laboratory of Green Pesticide, Key Laboratory of Green Pesticide & Agricultural Bioengineering, Ministry of Education, State-Local Joint Laboratory for Comprehensive Utilization of Biomass, Center for R&D of Fine Chemicals, Guizhou University, Guiyang 550025, Guizhou, China. E-mail: hli13@gzu.edu.cn
bDepartment of Chemical and Petroleum Engineering, University of Calgary, 2500 University Drive, NW, Calgary, Alberta T2N 1N4, Canada. E-mail: jinguang.hu@ucalgary.ca
First published on 8th January 2024
Abstract
Biomass has always been regarded as a latent resource owing to the lack of competitive technology to convert it into an active substance. Electroreduction is considered as a burgeoning catalytic technology for upgrading biomass into a variety of high-value chemicals and energy-intensive biofuels through different transformation routes, such as hydrogenation, hydrogenolysis, deoxidation, reductive amination, and dimerization. This review summarizes recent advances in the electrocatalytic reduction of various biomass-derived molecules (e.g., levulinic acid, 5-hydroxymethylfurfural, furfural, phenol, guaiacol, benzaldehyde, acetophenone, and benzoic acid) by taking into account the particle size, morphology, and electronic structure of the catalysts as well as the applied potential, charge transfer, and acid/alkali balance of electrochemical cells. Insights into the reaction mechanisms and pathways are presented to formulate electrolytes and catalytic sites required for specific reactions. Another objective is to summarize and discuss documented catalyst modification strategies for enhancing the electrocatalytic reaction rate and selectivity. Present challenges, promising applications, and future orientations are also proposed.
1. Introduction
Vital chemicals, including but not restricted to fuels and plastics, mainly stem from unsustainable fossil subsets. The growing demand for fuels have led to the global fuel crisis; moreover, these fuels are expected to be exhausted by 2060 based on the present consumption rate.1–4 Although governments worldwide have zealously formulated viable tactics to exploit alternative renewables (e.g., wind, solar, tidal, and geothermal energies), these energy vectors are impeded by diverse regions and cannot offer carbon sources to generate high-value chemicals and energy-intensive fuels.5–7 In this view, further development and exploration of other carbon-based renewables is an imperative but challenging task.Besides non-renewable resources such as coal, natural gas, and oil, lignocellulosic biomass is identified as the most abundant and renewable organic carbon-neutral source (Fig. 1) composed of cellulose (40–50%), hemicellulose (20–35%), and lignin (15–30%).8,9 In the past, however, biomass was often discarded because of the lack of competitive techniques for transforming it into a functional material. Presently, it is known that cellulose and hemicellulose are composed of D-glucopyranose units and can be further upgraded to various platform compounds, such as levulinic acid (LA), 5-hydroxymethylfurfural (HMF), and furfural, which are ideal starting raw materials for the production of fine chemicals and fuels. In addition, lignin is a cross-linked phenolic polymer with abundant –OCH3 and alkyl groups, and a series of aromatic molecules can be obtained, including phenol, guaiacol benzaldehyde, acetophenone, and benzoic acid.10,11 Although lignocellulosic biomass is cheap and readily available, more complex pretreatment steps are required before its further conversion into desired products. Thus, increasing attention is placed on the synthesis of biomass-derived molecules.12–15 Unlike traditional fossil resources, biomass and its derivatives have high oxygen contents and low energy densities. To better utilize these renewable resources, many strategies have been concentrated on maximizing the H/C ratio (hydrogenation) and minimizing the O/C ratio (deoxygenation), among which thermoreduction and electroreduction have acquired sky-high approval.16 Thermochemical reduction of oxygenated biomass is a classic method; however, it is limited by the requirements of dense H2 pressure and elevated temperature.17,18 The involved reductive processes can be summarized into two steps. Reactants and H2 are initially adsorbed and activated on a catalyst, and then activated intermediates effectively react to produce target products. In addition, catalytic transfer hydrogenation with alcohol or acid as a hydrogen source can avoid the employment of high-pressure H2 but still cannot bypass high-temperature conditions.19–21
 | ||
| Fig. 1 Schematic diagram of the lignocellulosic biomass upgrading processes: from macromolecules to electroreductive products. | ||
In recent years, electroreductive technology has received increasing attention because it can be performed under ambient conditions, and the use of H2O as a hydrogen source follows the criterion of green chemistry. With the integration of solar, tidal, and biomass renewable energy, electricity has been indirectly endowed with the label as a sustainable source, providing sufficient background and reliance for the integration of the electrocatalysis method and “biorefinery technology”.22–24 These methods for the electroreductive upgrading of biomass to high-value chemicals or biofuels mainly focus on hydrogenation, hydrogenolysis, reductive amination, and dimerization (C–C coupling) reactions.25–27 The functional groups undergoing mutual transformations are usually hydroxyl (phenolic hydroxyl), carbonyl, furan-ring, and benzene-ring. Among them, electrochemical hydrogenation and hydrogenolysis display strong dependence and regularity on electrolytes, while reductive amination exhibits profitability for nitrogen-containing compounds. Electroreductive C–C coupling for biomass upgrading is a valuable reaction since it can extend the carbon chain. However, it has not received much attention in comparison to the well-established unimolecular conversion. Given these, it is important to gain insights into the principle of electroreductive conversion of biomass. Theoretically, if the cathodic potential is lower than the reductive potential of the reactant molecule, this reduction reaction is likely to occur.28,29 In an actual case, a more negative cathodic potential (i.e., over-potential) is usually required to realize the reduction of the substrate due to the restriction of the reaction kinetics. In comparison to the well-studied biomass electrooxidation, electroreduction is still in its infancy, and the electrocatalysts are mainly noble-metal-based and Cu-based catalysts.30–33 Adjusting the electronic structure and optimizing the surface morphology of the catalyst are typical modification strategies for enhancing the reaction rate and improving the selectivity of the desired products, including single atom,34 alloy,35 multi-site,36 doping,37 nanostructure,38 and others.
Several excellent reviews have been published on the electrocatalytic conversion of lignin components (e.g., phenol, guaiacol, and benzaldehyde).39–42 In a recent review, Akhade et al. provided a comprehensive summary of the fundamental concepts of electrochemical hydrogenation, including theoretical calculations related to electrocatalysis, evaluation of reaction parameters, reaction kinetics, and reactor design.42 To our knowledge, however, there is still no specific review that provides an overview and discussion on the progress on eletroreductive transformation of lignocellulosic biomass (including three components) into useful chemicals and biofuels. In this review, therefore, the research progress and reaction mechanisms toward the electroreductive upgrading of biomass-derived platform molecules according to its three main backbones are systematically summarized. The discussed cases include LA, HMF, and furfural originating from cellulose/hemicellulose, as well as phenol, guaiacol benzaldehyde, acetophenone, and benzoic acid stemming from lignin (Fig. 1). The developed methods for obtaining common and non-mainstream products from the electroreductive network of specific substrates are collected and discussed in detail, wherein the interaction between organic substrates and electrolytes, and the interface effect between organic substrates and electrocatalysts are highlighted. Emerging and promising reactions, like HMF ring-opening, reductive amination, C–C coupling, and ring-saturation, are also reviewed, with a focus on the electrode electroreductive behavior, preferred electrolytic parameters, and future direction. Overall, this review aims to provide a full description for formulating the electrolytes, catalytic sites, and cautions required for specific biomass conversion routes from the perspective of mechanisms.
2. The merits of biomass electroreduction relative to thermocatalytic methods
In comparison with traditional reduction routes, electrocatalytic hydrogenation (ECH) is regarded as a greener process by virtue of the following merits: (1) more sustainable hydrogen-resource supply (active hydrogen intermediates from water), (2) greener reaction processes (continuous electrons as reducing agents, and generating by-products of H2 and O2 available for fuel cells), (3) more stable operating parameters at ambient conditions (avoiding intensive pressure and temperature), and (4) capable of coupling the oxidation reaction (with the reduction reaction to promote each other). To further understand and master the ECH process, an in-depth insight into the reaction and electron transfer mechanisms is required. The electrocatalyst and electrolyte must be further optimized before conducting additional experiments, proving their vital function in ECH. As a result, the elementary concepts and general mechanisms are summarized in this part.2.1. General reaction and electrocatalysis mechanisms
In the electroreduction of organic substrates, adsorbed hydrogen (Hads) is involved in most reactions, such as hydrogenolysis and hydrogenation of the aldehyde, as well as hydrogenolysis of alcohol. These procedures are designated as the ECH route, which proceeds via the adsorption of Hads. Only a few reactions were accomplished through direct electroreduction without the participation of Hads.39 On this occasion, the carbonyl group is initially reduced to a ketyl radical intermediate via the acquisition of (e− + H+), which can gain a second (e− + H+) to form an alcohol product or couple with another radical intermediate, resulting in dimerization product formation.In a complete ECH, Hads is produced through the Volmer reaction, and then directly used for the substrate hydrogenation (Fig. 2). Understanding the whereabouts of Hads contributes to the inhibition of unwanted side-reactions and promotion of the targeted ECH. Originating from the principle of green synthesis, the use of the water-electrolyte gradually becomes dominant, which may also spark water splitting. In general, the hydrogen evolution reaction (HER) is regarded as a competing reaction with the ECH of biomass-derived platform molecules, since HER will consume Hads through the Heyrovsky and Tafel reactions. The Heyrovsky reaction proceeds in distinct pathways at different pH values, producing molecular hydrogen electrocatalytically. In contrast, the Tafel reaction is a thermochemical Hads combination and desorption process. The Heyrovsky and Tafel reactions are capable of decreasing the faradaic efficiency (FE) of ECH, and thus not conducive to reducing organic substrates. A common approach is to employ Cu- and Ag-based electrodes, featuring high HER over-potentials.42 Notably, the change of the cathodic potential to modulate the surface Hads-coverage in ECH is akin to controlling hydrogen pressure in the traditional hydrogenation procedure.40
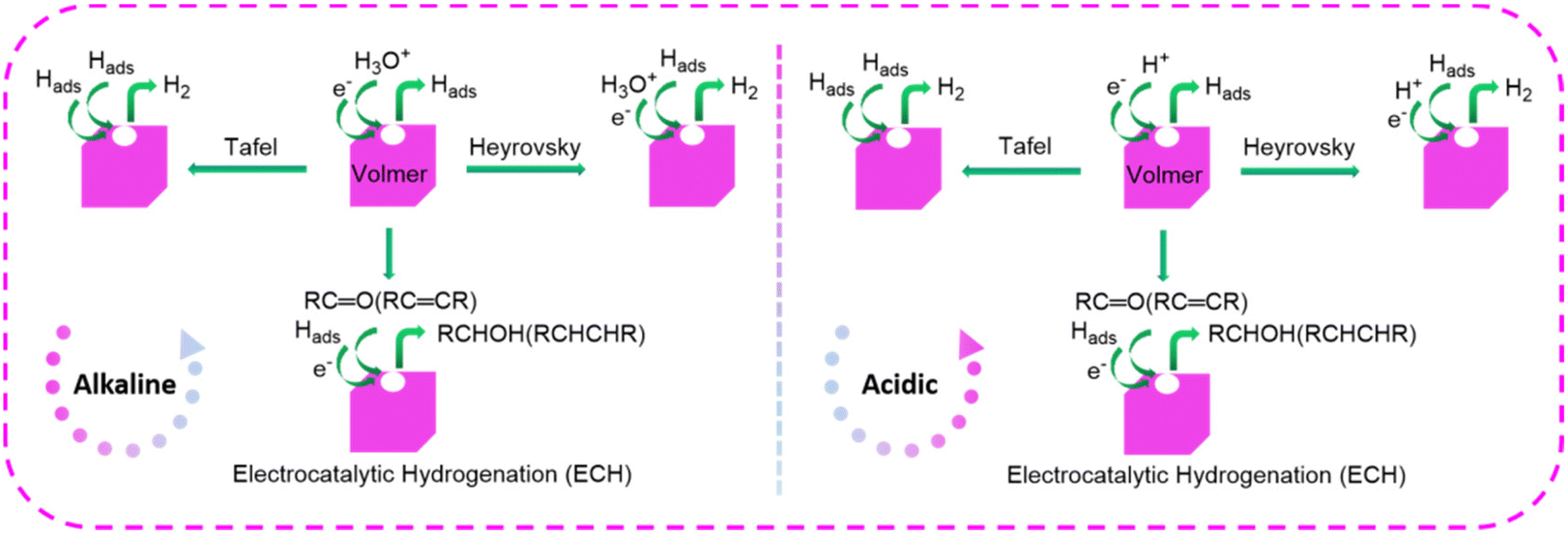 | ||
| Fig. 2 General mechanisms/pathways in the electrocatalytic hydrogenation (ECH) and hydrogen evolution reaction (HER). | ||
In the ECH route, a high-efficiency catalyst must be capable of generating the Hads through the Volmer reaction, while simultaneously suppressing the Heyrovsky and Tafel reactions effectively. To better suppress the HER, the current strategy mainly focuses on introducing auxiliary sites to passivate the Hads, or promoting the rapid interaction between Hads and substrates. For the direct electroreduction route, the employment of catalysts with high HER over-potential or metal-free sites has been reported to be the most effective strategy, because it does not carefully consider the evolution of H2.
Compared to the traditional reduction pathways, ECH is more suitable for biomass upgrading, whether from an operational or economic point of view.43–46 Along this direction, three prevalent catalytic models have been established.46 Direct utilization of inert electrodes (e.g., Pt, Pb, and carbon electrodes) is the most well-studied catalytic method, but is not efficient enough (Fig. 3A), and involves the electron being directly transferred from the electrode surface to the substrate. In such a scenario, setting a suitable potential to acquire a desirable substrate conversion and product selectivity is highly desired. The range of these inert electrode systems can be extended from the metal to the carbon electrode. Apart from the close-knit or bulk electrode materials, multi-hole or reticular electrodes are also adopted. Unlike inert electrodes, active electrodes are less dependent on the applied potential, but more determined by the active species or the type of reactions (Fig. 3B). To be precise, the selective conversion of organic molecules is achieved by loading an effective active species on the conductive supports, which are often modified electrodes.47 The modified electrode is usually designed for specific functional group conversion or similar reactions, and thus the obtained FE is higher than that of the inert electrode. Ideally, the active species could be normally regenerated in situ during the reaction process. In this aspect, massive anodic reactions (e.g., water, HMF, and glycerol oxidation) have been well-established, which provide sufficient support for cathodic reactions.48–51 Another mode is to use a redox-active medium to complete the conversion of substrates to high-value chemicals (Fig. 3C), in which electricity is exploited to directly actuate the medium oxidation–reduction cycle to fulfill the reactant conversion, thus avoiding a greater over-potential.52
 | ||
| Fig. 3 The inert electrode (A), active electrode (B), and redox-mediated system (C) for electrocatalysis. | ||
In these systems, inert electrodes can overcome the problem of the catalyst stability. However, the high cost, scarcity, and dependence on potential are unfavorable factors. For active electrode systems, abundant strategies can be used to inhibit HER, resulting in a higher FE and selectivity. However, much effort is still needed to overcome the dissolution of active sites and the identification of the active species during the reaction process. In the redox-mediated system, the transformation of substrates is determined by the property of the medium, which can bypass the inherent high over-potential of the reaction. However, some adverse reactions and unwanted wastes may be detected.
2.2. Identifying the function of the electrocatalyst and electrolyte
The combination of reaction processes and electrocatalytic mechanisms indicate that the roles of the electrocatalyst and electrolyte are vital for ECH. Thus, the relevant discussions are given in this section. The synergy of the electrocatalyst and electrolyte aims to maximize the application of electrons.16,53,54 The former mainly offers active sites for substrate reduction, while the latter supplies conductivity to boost electron transfer and forms a circuit.The catalyst is considered as the key to a reaction, and how to select an appropriate catalyst is primary. Taking the reduction of furan-based compounds as an example, Ag- or Cu-based materials are better catalysts, while aromatic compounds are more prone to form over Pt group metals (e.g., Pt, Rh, and Pd).55,56 With the optimal catalytic sites, optimizing the morphology, constructing the interface and introducing auxiliary sites to the catalyst will help to further improve the catalytic activity.57 A catalyst or electrode with high surface area, high chemical stability, and suitable coordination numbers of the active sites are generally able to deliver better activity.43,58 The optimized catalyst in an acidic medium exhibited poor activity and stability in a basic medium, implying that a negative cathodic potential is required to offer rational current densities to reduce the substrate. For specific electrodes and substrates, even at the same potential, a change of electrolyte pH will lead to different products, giving a hint that the catalytic mechanism may be distinguishing in different electrolytes, as detailed in section 3. Considering that Hads is broadly involved in ECH, the generation of Hads is a vital step of the whole process, whose rate is directly determined by pH and over-potential, because the Volmer reaction can be promoted in a H+-abundant environment. Although the distribution of the product is significantly affected by pH, the absolute selectivity is still determined by the electrode.
3. Electrocatalytic reduction of biomass feedstocks
3.1. Electroreduction of the cellulose-derived platform molecule
 | ||
| Fig. 4 The comparison of upgrading routes in the electrochemical and thermochemical reduction of levulinic acid (LA). | ||
| Entry | Substrate | Product | Catalyst | FE (%) | Y[S] (%) | Electrochemical parameters | Ref. |
|---|---|---|---|---|---|---|---|
| Levulinic acid (LA), valeric acid (VA), gamma-valerolactone (GVL), 4-hydroxyvaleric acid (HVA), faradaic efficiency (FE), and Y[S] is yield[selectivity]. | |||||||
| 1 | 0.5 M LA | VA | Pb | 86.5 | 90[95] | 0.5 M H2SO4, E = −1.5 VRHE | 63 |
| 2 | 0.5 M LA | VA | Pb | >47 | [>90] | 0.5 M H2SO4, E = −1.5 VRHE | 64 |
| 3 | 0.5 M LA | VA | In, Zn | >44 | 96–99 | 1 M H2SO4, 50 °C | 65 |
| 4 | 0.1 M LA | VA | InPb/CF | 60.8 | 86.1[99.7] | 0.1 M H2SO4, E = −1.5 VRHE | 66 |
| 5 | 0.1 M LA | VA | PbO–In2O3 | 89.2 | 41.4[97.8] | 0.5 M H2SO4, E = −1.3 VRHE | 67 |
| 6 | 0.5 M LA | GVL | Fe | 20 | [70] | 1 M NaOH, E = −1.8 VAg/AgCl | 70 |
| 7 | 0.1 M LA | GVL | PbS-400/CP | 78.6 | — | [Bmim]BF4-MeCN–H2O, E = −2.15 VAg/Ag+, j = −13.5 mA cm−2 | 71 |
| 8 | 0.1 M LA | HVA | Pb | 81 | 93[99.9] | 0.1 M KHCO3 + 0.1 M KClO4, E = −1.9 VRHE | 72 |
| 9 | 2 M LA + methylamine | 4-(Methylamino)-pentanoic | Cu | 78 | 60 | 0.5 M KH2PO4 (pH 12), j = −0.04 A cm−2 | 73 |
In addition to the carbonyl group, the LA molecule still contains an additional –COOH group, but only the carbonyl group can react under the electroreductive conditions. From the energy densities point of view, both VA (2.84 MJ mol−1) and GVL (2.65 MJ mol−1) are higher than LA (2.42 MJ mol−1), which means that LA can be used as a benign starting feedstock to store electricity.62 Alternatively, VA is primarily used to produce biofuels and lubricants, followed by the preparation of spices and feeds, implying that the development of promising methods for the conversion of LA to VA is necessary. Clearly, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in LA must be cleaved to obtain VA, meaning that an acidic environment is required. Along with this speculation, an ECH route of LA to VA was implemented in 0.5 M H2SO4 solution over the Pb-electrode, giving a 70.8% electricity storage efficiency.63 In terms of reaction efficiency, an ideal selectivity (95%) and FE (86.5%) were also achieved at optimized conditions. In a relevant work, this electrocatalytic protocol was employed to further upgrade the acid-catalyzed downstream products of cellulose (containing FA + LA + H2SO4) in a flow cell reactor, coupled with a formic acid (FA) fuel cell.64 With this method, LA is able to be upgraded to VA as a biofuel intermediate, whereas FA is removed and fed into a fuel cell for producing electricity. The generated electricity is capable of suitably supplementing the energy cost in the electrocatalytic reduction of LA. This green catalytic scheme is expected to circumvent the complicated treatment procedures of the side-product (FA + H2SO4) isolated from the downstream cellulose hydrolysis. Although the Pb-electrode seems to be suitable for LA reduction, large-scale production may be restricted due to the incorporation of toxic Pb element. Thus, the exploration of alternative electrodes to minimize the use of Pb is of vital importance. Several valid cathodic materials that have been tested by Bisselink et al., such as Cd, In, and Zn, can efficiently produce VA from FA.65 Thereinto, both In and Zn are recognized to be more benign, with no or less by-product GVL formation. An exceptional VA selectivity (99% at In, 96% at Zn) was recorded, but the FE was unsatisfactory. This work suggests that the condition of Cd or In alone acting as a site to catalyze this reaction is untenable since the EF is negligible. As reported recently, utilization of In or Cd to construct bimetallic sites can not only provide an impressive performance for VA production, but also decrease the use of Pb.66–68 In the two-step reaction, LA is initially protonated to give HVA, followed by reduction to VA via the C–O bond cleavage. Admittedly, the conversion of LA to VA tends to occur in acidic environments, as the acidic medium is more conducive to forming Hads. The electroreduction of LA holds an extra benefit to operating in a H2SO4 medium, which can simplify the biomass upgrading process, because they (LA + H2SO4) are normally present in biomass product streams.
O bond in LA must be cleaved to obtain VA, meaning that an acidic environment is required. Along with this speculation, an ECH route of LA to VA was implemented in 0.5 M H2SO4 solution over the Pb-electrode, giving a 70.8% electricity storage efficiency.63 In terms of reaction efficiency, an ideal selectivity (95%) and FE (86.5%) were also achieved at optimized conditions. In a relevant work, this electrocatalytic protocol was employed to further upgrade the acid-catalyzed downstream products of cellulose (containing FA + LA + H2SO4) in a flow cell reactor, coupled with a formic acid (FA) fuel cell.64 With this method, LA is able to be upgraded to VA as a biofuel intermediate, whereas FA is removed and fed into a fuel cell for producing electricity. The generated electricity is capable of suitably supplementing the energy cost in the electrocatalytic reduction of LA. This green catalytic scheme is expected to circumvent the complicated treatment procedures of the side-product (FA + H2SO4) isolated from the downstream cellulose hydrolysis. Although the Pb-electrode seems to be suitable for LA reduction, large-scale production may be restricted due to the incorporation of toxic Pb element. Thus, the exploration of alternative electrodes to minimize the use of Pb is of vital importance. Several valid cathodic materials that have been tested by Bisselink et al., such as Cd, In, and Zn, can efficiently produce VA from FA.65 Thereinto, both In and Zn are recognized to be more benign, with no or less by-product GVL formation. An exceptional VA selectivity (99% at In, 96% at Zn) was recorded, but the FE was unsatisfactory. This work suggests that the condition of Cd or In alone acting as a site to catalyze this reaction is untenable since the EF is negligible. As reported recently, utilization of In or Cd to construct bimetallic sites can not only provide an impressive performance for VA production, but also decrease the use of Pb.66–68 In the two-step reaction, LA is initially protonated to give HVA, followed by reduction to VA via the C–O bond cleavage. Admittedly, the conversion of LA to VA tends to occur in acidic environments, as the acidic medium is more conducive to forming Hads. The electroreduction of LA holds an extra benefit to operating in a H2SO4 medium, which can simplify the biomass upgrading process, because they (LA + H2SO4) are normally present in biomass product streams.
Like VA, GVL is a secondary product derived from LA reduction, which can be used not only as a food and fuel additive, but also as a green solvent for biomass processing. Despite the thermocatalytic method being considered as the most mature system for reducing LA to GVL, it is still necessary to seek greener and more sustainable conversion methods to minimize energy consumption.69 From LA to GVL, two steps must be experienced (Fig. 4), namely, hydrogenation of LA to HVA and cyclization of HVA (lactonization). The former can be achieved by electrochemical approach, while the latter is a thermochemical process and sensitive to pH. Based on previous studies, the electrocatalytic reduction of LA to GVL should be feasible, but the pH optimization of electrolytes is often required. Unlike the electrocatalytic reduction of LA to VA, neutral or alkaline media appear to favor GVL production. As an example, Xin et al. employed a Pb-electrode to reduce LA to GVL in a phosphate buffer (pH 7.5), with an extraordinary selectivity (100%) and a low FE (6.2%).63 In addition, this reaction was executed at elevated pH (1 M NaOH, pH 13) with Fe-electrodes, and an integrated catalytic activity (20% FE and 70% selectivity) was obtained.70 In the electrochemical synthesis of GVL, it is difficult to simultaneously satisfy the selectivity and FE. To obtain a higher FE, the selectivity of the product is usually sacrificed. Typically, a ternary electrolyte ([Bmim]BF4–MeCN–H2O) and partially oxidized PbS-electrode were utilized for this reaction, giving a record FE (78.6%).71 Regarding the reduction of LA to GVL, the synchronous acquirement of the ideal FE and selectivity is highly anticipated, but challenging.
For LA electroreduction, HVA only appears as an intermediate whether VA or GVL is generated. It has been produced less often as a dominant product, which may be correlated with the electrolyte. As a valuable monomer, HVA can be exploited to produce biopolymers and bioplastics, apart from further production of GVL and VA. With further detailed research, an unprecedented pathway to produce HVA from LA was identified by Lucas et al., which proceeds in a co-supporting electrolyte (containing 0.1 M KHCO3 + 0.1 M KClO4).72 This electrochemical protocol could achieve 99.9% selectivity and 81% FE, with a productivity higher than 40 g L−1 h−1. More interestingly, HVA can be converted to GVL within 15 min when 0.55 M H2SO4 was used to treat the above-completed reaction. Unlike the formation of VA, HVA was produced through an outer sphere electron transfer mechanism, without the participation of Hads.
Another interesting methodology is to synthesize organic nitrogenous chemicals from LA, designated as electrocatalytic reductive amination. Mürtz et al. accomplished the preparation of 1,5-dimethyl-2-pyrrolidone in a basic electrolyte (pH 12) by combining a heating step, using LA and methylamine as the starting substrates.73 Cu was identified as a benign cathodic material to reduce LA, and pyrrolidone with a yield of 78% was acquired. In this reductive process, the current density was regarded as the primary influential factor aside from the pH. With the current density increasing from −40 mA cm−2 to −200 mA cm−2, the yield of the by-product HVA increases from 1% to 33%. Also, adjusting the electrolyte pH to 12 guarantees that a considerable part of the amine exists in alkaline form, which is beneficial to react with the aldehyde group to form an imine. The mechanism manifests that the reductive amination of LA involves three basic steps (Fig. 5): (1) formation of imine – recognized as a dynamic equilibrium process and unstable in a non-alkaline environment, (2) reduction of imine to secondary amine – deemed as an electrochemical process and dictated by the electrolyte properties and applied potential, and (3) dehydration of 4-(methylamino)-pentanoic acid – viewed as a thermochemical process and determined by temperature. Via this conversion route, C–N bonds can be introduced into biomass-derived molecules like carbonyl-containing groups to form nitrogen-containing chemicals, which extends the type of products derived from biomass.
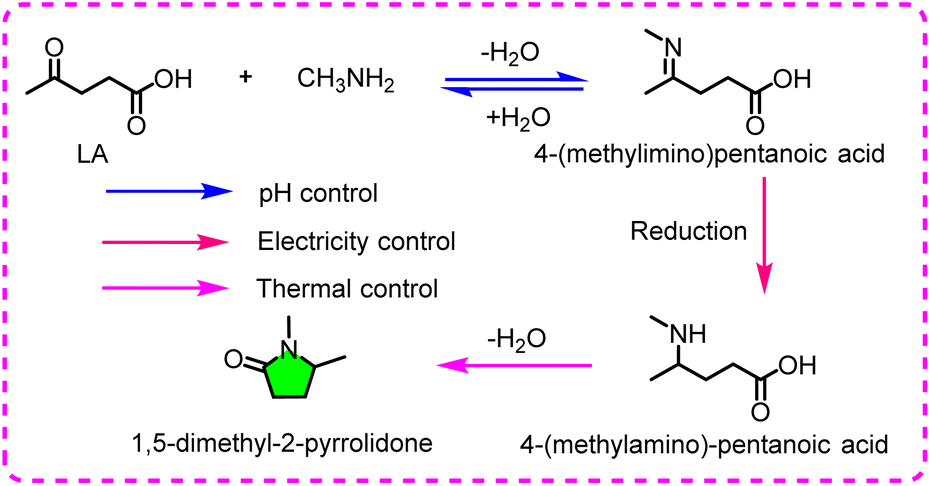 | ||
| Fig. 5 The proposed mechanism for the reductive amination of levulinic acid (LA). | ||
In all cases, the electroreductive upgrading of LA is overly dependent on planar single metal sites, making it difficult to obtain the desired current efficiency and selectivity, simultaneously. Viable strategies include optimizing the catalyst morphology, exploring new electrolytes, and constructing multiple catalytic sites, which will be discussed in detail in the section on HMF electroreduction (see below).
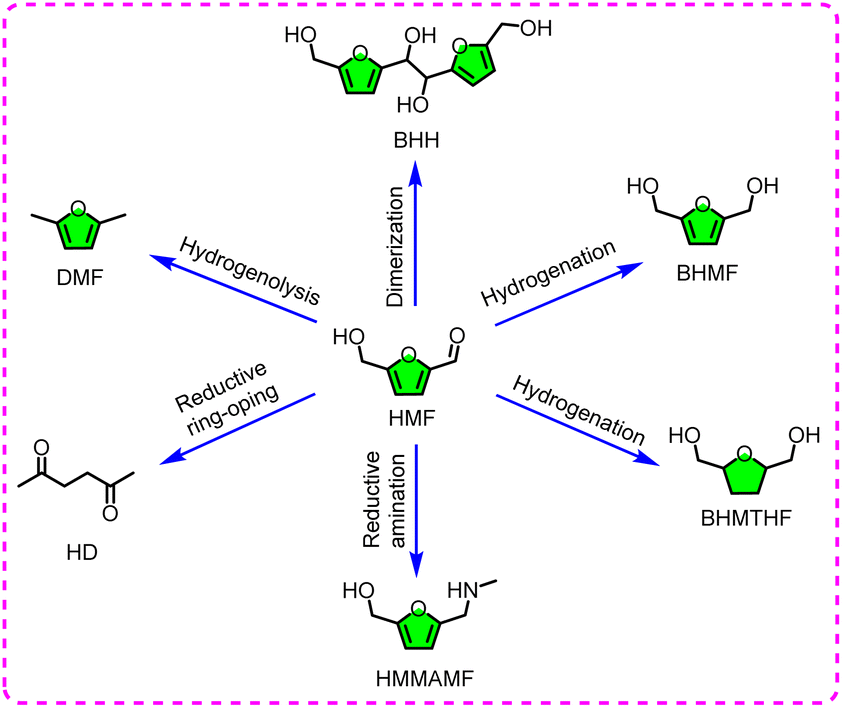 | ||
| Fig. 6 Schematic diagram of the electroreductive products obtained from HMF. | ||
| Entry | Substrate | Product | Catalyst | E.F.(%) | Y[S] (%) | Electrolysis conditions | Ref. |
|---|---|---|---|---|---|---|---|
| 5-Hydroxymethylfurfural (HMF), 2,5-bis(hydroxymethyl)furan (BHMF), 2,5-bishydroxymethyl-tetrahydrofuran (BHMTHF), 5,5′-bis(hydroxymethyl)hydrofuroin (BHH), 2,5-dimethylfuran (DMF), 2-hydroxymethyl-5-(methylaminomethyl)furan (HMMAMF), 2-hydroxymethyl-5-(ethanolaminemethyl)furan (HEMF), 2,5-hexanedione (HD), glassy carbon (GC), boron-doped diamond (BDD), faradaic efficiency (FE), and Y[S] is Yield[selectivity]. | |||||||
| 1 | 0.02 M HMF | BHMF | Ag | — | [>99.9] | Borate buffer (pH 9.2), E = −1.5 VAg/AgCl | 78 |
| 2 | 0.02 M HMF | BHMF | Ag/C | 87 | 85 | Borate buffer (pH 9.2), E = −1.3 VAg/AgCl | 79 |
| 3 | 0.02 M HMF | BHMF | Ag-TiO2 NTs | 82.2 | 0.1 mmol cm−2 h−1[89.8] | 0.5 M K2HPO4, E = −1.4 VAg/AgCl | 80 |
| 4 | 0.02 M HMF | BHMF | Ag@Cu NWAs/CF | 97.1 | [96.5] | Borate buffer (pH 9.2), E = −0.51 VRHE | 81 |
| 5 | 0.05 M HMF | BHMF | Ag/Cu | >80 | >80[80] | 0.5 M borate buffer (pH 9.2), E = −0.51 VRHE | 82 |
| 6 | 0.02 M HMF | BHMF | RuCu | 85.6 | 0.47 mmol cm−2 h−1 | 0.5 M phosphate buffer solution (PBS), E = −0.3 VRHE | 83 |
| 7 | 0.01 M HMF | BHMF | AgCu/PA | 61.8 | [94] | Borate buffer (pH 9.2), E = −1.4 VAg/AgCl | 84 |
| 8 | 0.05 M HMF | BHMF | AgCu | — | 0.567 mmol cm−2 h−1[85] | Borate buffer (pH 9.2), E = −0.51 VRHE | 85 |
| 9 | 1 mM HMF | BHMTHF | Pd/VN | >86 | 80[88] | 0.2 M HClO4, E = −0.11 VRHE | 86 |
| 10 | 0.01 M HMF | BHH | GC/BDD | 39/38 | [36]/[35] | 0.1 M carbonate buffer (pH 9.2), E = −0.7 VRHE | 87 |
| 11 | 2 g L−1 HMF | DMF | Cu | 50 | [35.6] | 500 mM H2SO4, E = −1.05 VAg/AgCl, j = −10 mA cm−2 | 88 |
| 12 | 2 g L−1 HMF | DMF | CuNi | 88 | [91.1] | 0.2 M sulfate buffer (pH 2.0), E = −0.8 VAg/AgCl | 89 |
| 13 | 0.02 M HMF | HMMAMF | Ag(Aggd) | >99 | [>99] | 0.7 M methylamine buffer (pH 11.0), E = −0.25 VRHE | 90 |
| 14 | 5 mM HMF | HEMF | TiS2 | 34.4 | [>99] | 0.7 M ethanolamine, E = −0.6 VRHE | 91 |
| 15 | 0.02 M HMF | HD | Zn | 72.4 | [81.6] | 0.2 M sulfate buffer (pH 2.0), E = −1.2 VAg/AgCl | 92 |
| 16 | 0.02 M HMF | HD | Ag-aerogel-CNx | 78 | [77] | 0.5 M H2SO4, E = −1.1 VAg/AgCl | 93 |
Among the many reductive products, DHMF is hailed as a functionalized precursor, and can be employed for producing polyethers and polyamides. To date, DHMF can be acquired through the hydrogenation of HMF, involving thermochemical and electrochemical pathways.77 The electrochemical pathway is regarded as a relatively greener strategy for HMF hydrogenation due to the use of Hads as a hydrogen source, rather than traditional high-pressure hydrogen. Ag and Cu seem to be the most advanced electrodes for HMF reduction in previous reports. In terms of electrolytes, the borate buffer (pH 9.2) offered an excellent operating environment for selectively producing BHMF and effectively restraining side reactions. As an example, Choi et al. reported that the conversion of HMF to BHMF could be achieved over an Ag electrode, with a selectivity nearing 100%, but the FE was not mentioned.78 This planar electrode only exposed limited active sites, making it extremely difficult to simultaneously obtain the expected FE and selectivity. Controlling the morphology and constructing self-supporting structures in situ are feasible methods to enhance the catalyst activity because they can increase the exposed active sites and reduce the immoderate agglomeration, together with the improvement of the electrode stability during long-term electrolysis. A self-synthesized Ag/C (Ag nanoparticles deposited on a carbon substrate) electrode was employed for electrochemical conversion of HMF to BHMF in a borate buffer (pH 9.2).79 Thanks to the high dispersion and exposure of the catalytic sites, the Ag/C catalyst exhibits high reactivity, resulting in 87% FE and 85% yield. Also, titanium dioxide (TiO2) holds a high HER over-potential, which can be recognized as a potential substrate for loading Ag to avoid excessive agglomeration of the catalytic sites, which was verified by Zheng and co-workers.80 With in-depth research, an Ag@Cu NWAs/CF (in situ growth of the Ag-decorated Cu nanowire arrays on the Cu foam) catalyst with a layered structure has been proven to be effective for reducing the aldehyde group of HMF.81 The catalyst with an efficient electron transport network and widespread nano-structures can achieve rapid ion diffusion, mass transfer, and close electrolyte contact. In addition, the combination of Cu nanoarrays and Ag-nanoparticles revealed an electronic synergy and geometric effect in terms of improving the electroreductive activity.82 The construction of bimetallic sites (including alloy) is attracting substantial attention since it can effectively reduce the consumption of precious metals with improved reactivity. For the construction of double sites, the combination of a precious metal (Ag or Ru) with Cu is found to be a suitable candidate for the hydrogenation of HMF.83–85 In the macro aspect, the introduction of Cu directly leads to the dispersion of noble metal sites and the improvement of the electrocatalytic activity. In the micro aspect, the electron transfer from the catalyst to HMF increases, resulting in the decrease in energy barrier and Gibbs free energy of the HMF hydrogenation reaction relative to the direct utilization of Ag, as certified by density functional theory (DFT) calculations. The synergistic effect of the bimetallic sites truly offers an upbeat effect for the electroreduction of HMF to BHMF, but the interaction and catalytic mechanism between the two synergic sites and individual site was not fully understood.
The electrochemical hydrogenation of HMF to BHMTHF was rarely explored, while BHMTHF is considered as the best candidate for next-generation biofuels. Therefore, developing effective catalytic systems for BHMTHF production is of great significance.94 Starting from the concept of promoting electron transport and ion diffusion, Wang et al. successfully fabricated a workable Pd/VN catalyst with a hollow nanosphere structure.86 The resulting Pd/VN catalyst was operated in an acidic medium (0.2 M HClO4) for the preparation of BHMTHF at −0.11 VRHE. The reductive products were quantified by high-performance liquid chromatography (HPLC), and an excellent electrolytic performance (86% FE and 88% selectivity) was observed. The formation of Hads was promoted. However, its desorption was inhibited on this distinct Pd/VN catalyst, hence facilitating HMF adsorption and activation.
Electroreduction of high-concentration HMF is easier to industrialize. However, most investigations have undergone severe dimerization (i.e., C–C coupling), leading to the formation of 5,5′-bis(hydroxymethyl)hydrofuroin (BHH). Dimerization is often identified as a side-reaction for HMF conversion. Therefore, BHH is rarely synthesized. In fact, dimerization can be designed as an effective strategy to extend the carbon chain, providing an interesting path to yield BHH (C12) from HMF (C6), which can be exploited to produce diesel or jet fuels after hydrogenolysis. The dimerization behavior of HMF was investigated in 0.1 M carbonate buffer (pH 9.2) using sp2-hybridized glassy carbon (GC) and sp3-hybridized boron-doped diamond (BDD) electrodes as cathodic materials.87 Both catalytic materials provided the possibility of preparing BHH, but the catalytic behavior was different. On BDD, the BHH always appeared as the target product regardless of the applied potential. When the applied potential decreased from −0.7 VRHE to −1.0 VRHE, the BHH selectivity remained almost unchanged. On GC, the product was mainly determined by the applied potential. Along with the decrease of the applied potential (from −0.7 VRHE to −1.0 VRHE), BHMF appeared as the main quantifiable product (selectivity from 0 to 31%). As a proof of concept, even an ordinary graphite foil could also be used to catalyze HMF dimerization for BHH generation. This can be attributed to the high HER over-potential of the carbon-substrate, which allows direct electron transfer, thus initiating the C–C coupling reaction.
Two major competitive reactions have been identified for the hydrogenation of HMF: (1) HER can be promoted at low HER over-potentials, leading to a decreased FE, and (2) dimerization is favored at high substrate concentrations, resulting in a low carbon balance. Considering that the hydrogenation reaction mainly occurs under non-acidic conditions, hydrogenolysis is therefore not its competitive reaction, as it mainly takes place under acidic conditions.95,96
For the hydrogenolysis of HMF, the alcohol C–O and the aldehyde CO bond in HMF are simultaneously cracked. Thus, DMF (regarded as a gasoline alternative) is the final product of this process.97 Cu seems to be the best catalyst for this reaction, exhibiting ideal reactivity under acidic conditions.88 Even when using an ordinary Cu plate as the cathodic material, 35.6% selectivity and 50% FE of DMF were achieved. In the aim to simultaneously obtain the expected productivity and high EF, a bimetallic CuNi electrode with a dendrite morphology was prepared and used to produce DMF from HMF in 0.2 M sulfate buffer (pH 2.0).89 Benefitting from the high surface area and nano-effect of the catalyst, the interfacial charge transfer of the reduction process was enhanced, leading to DMF formation in 91.1% selectivity and 88.0% FE. Also, only a slight activity decay was observed (88% to 83.5%) for the CuNi electrode after five runs under the optimal conditions. Based on the examples discussed above, a possible mechanism/pathway for the electrocatalytic reduction of HMF to DMF is summarized and depicted in Fig. 7. In this procedure, HMF is converted through the pathway of HMF → 5-methylfurfural (MFF) → DMF, rather than that of HMF → 5-methylfurfuryl alcohol (MFA) → DMF. Once MFA is formed, it is improbable for its alcohol group to be further electrochemically deoxidized to give DMF under the same conditions used for the reduction of HMF to DMF, unless advanced catalysts and/or harsh conditions are developed and/or utilized. However, in other thermodynamical reports, the pathway of HMF → MFA → DMF is admissible.98
 | ||
| Fig. 7 The electrocatalytic transformation pathway of 5-hydroxymethylfurfural (HMF) to 2,5-dimethylfuran (DMF). | ||
Considering that there is an aldehyde group in the HMF structure, it should be feasible to prepare organic nitrogen chemicals through reductive amination. Roylance and Choi performed the reductive amination of HMF in a 0.7 M methylamine buffer, for which metal electrodes like Ag, Cu, Pt, Sn, and Zn were investigated for the production of 2-hydroxymethyl-5-(methylaminomethyl)furan (HMMAMF).90 In particular, the morphology of Ag was further optimized to obtain a high-surface-area dendritic Ag electrode, giving a nearly 100% FE and selectivity at the optimal potential (−0.25 VRHE). Under identical conditions, other HMF-derived compounds can also react with methylamine to form the corresponding nitrogenous chemicals (Fig. 8). Through a vacancy construction strategy, Yan et al. fabricated a TiS2 catalyst with a high concentration of sulfur vacancy and thin nanosheet structure.91 Then, the catalytic activity of TiS2 for the reductive amination of HMF was tested in a 0.7 M ethanolamine electrolyte. The presence of sulfur vacancies is able to enhance the adsorption of HMF, and the thin nanosheets are capable of offering adequate surface active centers, resulting in a small Tafel slope and favorable kinetics.
 | ||
| Fig. 8 The electrocatalytic reductive amination of 5-hydroxymethylfurfural (HMF). | ||
Of the numerous HMF reduction reactions, the electrochemical conversion of HMF to 2,5-hexanedione (HD) is an interesting but challenging task, because this process requires six (e− + H+) to destroy the furan ring and reduce the –OH and –CHO groups, concurrently. For the thermochemical conversion, HD can be obtained by hydrogenation and hydration (DMF ring-opening) of HMF, and DMF is identified as the main intermediate in this process. On the electrochemical side, direct use of (e− + H+) as reductants can reduce HMF to HD, and no intermediates (including DMF) are observed, implying that this procedure is likely accomplished through a one-step reaction. Choi and Roylance affirmed that the direct one-step reduction of HMF to HD is viable under acidic conditions in the presence of a Zn electrocatalyst, providing 72.4% FE and 81.6% selectivity of HD.92 The electrochemical mechanism to open the furan ring is akin to that of well-established thermochemical ring-opening processes, and one experimental example is that the two carbonyl oxygen atoms of HD are from the furan ring and water, respectively. Recently, a novel Ag-aerogel-CNx (carbon-supported Ag nanoparticle aerogel) catalyst has been illustrated to be active for HD production from HMF.93 Regulating the applied potential could offer an ideal efficiency and circumvent unwanted HER, and 78% FE and 77% selectivity were recorded. The other reasons for the promotion of HMF conversion to HD are that the three-dimensional network structure exposes more catalytic sites and facilitates the dominant growth of the Ag (111) plane. Taken together, these works suggest that the occurrence of electroreduction (hydrogenolysis of alcohols and aldehydes, similar to the Clemmensen reaction) and acid-catalyzed ring-opening undergo a concerted mechanism, as displayed in Fig. 9. Here, three crucial steps must proceed for the conversion of HMF to HD: the hydrogenolysis of –OH, conversion of the –CHO group to a terminal alkane, and then ring-opening.
 | ||
| Fig. 9 Proposed mechanism for the conversion of HMF to 2,5-hexanedione (HD). | ||
The hydrogenation or hydrogenolysis of aldehyde groups in HMF is largely affected by pH, but the absolute selectivities are still determined by the electrode type. Notably, reductive amination, dimerization, and ring-opening reactions are intriguing cases for HMF electroreduction, since they create novel ways to furnish a variety of value-added bioproducts and contribute to simplifying the HMF upgrading processes.
3.2. Electroreduction of furfural
Furfural is an extremely significant biomass-derived platform molecule, originating from hemicelluloses acidolysis. Similar to HMF, furfural can also be easily reduced, and several reaction pathways are identified in the electroreductive network of furfural (Fig. 10).99 The reductive products are relatively simple in comparison with HMF reduction, mainly involving furfuryl alcohol (FFA), hydrofuroin, 2-methylfuran (MF) and tetrahydrofurfuryl alcohol (THFA), and the related parameters and performances are listed in Table 3.100–102 Thereinto, FFA has received substantial attention and efforts, owing to the bright prospects for the production of plasticizers, fuel additives, and synthetic resins. Hydrofuroin and MF are often utilized as vital precursors for producing diesel or jet fuels. | ||
| Fig. 10 Reaction pathways in the electroreduction of furfural. | ||
| Entry | Substrate | Product | Catalyst | E.F.(%) | Y[S] (%) | Electrolysis conditions | Ref. |
|---|---|---|---|---|---|---|---|
| Furfuryl alcohol (FFA), 2-methylfuran (MF), tetrahydrofurfuryl alcohol (THFA), faradaic efficiency (FE), and Y[S] is yield[selectivity]. | |||||||
| 1 | 0.05 M furan | FFA | Cu | 71 | [87] | 0.1 M Na2CO3 + NaHCO3 buffer solution (pH 10), E = −1.4 VSCE | 104 |
| 2 | 0.05 M furan | FFA | NP-Cu | 95 | 96 | PBS/MeOH = 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 25 °C, E = −1.5 VAg/AgCl :1, 25 °C, E = −1.5 VAg/AgCl |
105 |
| 3 | 0.05 M furan | FFA | Cu-NPNi/NF | — | 118.7 ± 8 μmol h−1 cm−2 | 0.5 M NaOH, E = −1.45 VAg/AgCl | 106 |
| 4 | 0.05 M furan | FFA | Cu3P/CFC | 98 | [>99.9] | 1.0 M KOH, E = −0.55 VRHE | 107 |
| 5 | 0.03 M furan | FFA | 15%-Cu/NC900 | 95 | 99[100] | 1.0 M KOH (pH 13.6), E = −1.3 VAg/AgCl | 108 |
| 6 | 0.05 M furan | FFA | Ag60Pd40 | 87 | 85 | 0.1 M potassium phosphate buffer (pH 6.9), E = −0.5 VRHE | 109 |
| 7 | 0.02 M furan | FFA | MoS2 | — | 82.5[92.1] | Borate buffer:MeCN (4:1, pH 9), j = −10 mA cm−2 |
110 |
| 8 | 0.05 M furan | Hydrofuroin | MoS2 | — | [47] | MeOH:buffer (1:4, pH 9), E = −1.2 VAg/AgCl |
111 |
| 9 | 0.01 M furan | Hydrofuroin | Carbon paper | 93 | 94 | 0.1 M KOH (pH 13), E = −1.4 VAg/AgCl | 112 |
| 10 | 0.1 M furan | MF | Cu | — | [71] | 0.5 M H2SO4 (H2O:MeCN = 4:1), E = −0.5 VRHE |
113 |
| 11 | 0.02 M furan | MF | Ru/RGO | 95 | 91 | 2.0 M H2SO4, E = −1.25 VAg/AgCl | 114 |
| 12 | 0.04 M furan | MF | Cu1/PC | >90 | — | Acetate buffer (pH 5), E = −1.0 VRHE | 115 |
| 13 | 0.05 M furan | THFA | Pd5/Pt | — | [15.3] | 0.5 M H2SO4, 15% MeCN, j = −30 mA cm−2 | 116 |
| 14 | 0.25 M furan | THFA | Pd membrane | — | [98] | 1 M H2SO4, t-BuOH, j = −75 mA cm−2 | 117 |
Unlike the electrochemical conversion of HMF to BHMF, an obvious difference was observed in the electrocatalysts and electrolytes used for the hydrogenation of furfural to FFA. The electrocatalysts are less dependent on precious metals (Ag or Pd) which are still effective, and Cu is the mainstream catalyst.103 Regarding the electrolytes, a wide pH window (6.9–13.6) can selectively produce FFA from furfural. Lin et al. tested the electrochemical behavior of several metal electrodes (Pb, Cu, Ni, and Pt) in carbonate buffer (pH 10), and the sequence for the HER over-potential is in the order of Pb > Cu > Ni > Pt, in which the Cu electrode is only a bit lower than the Pb electrode.104 Theoretically, the occurrence of HER on the Pb electrode is difficult to observe, and the current efficiency is likely to be the highest. However, the experimental results demonstrated that the highest current efficiency was observed on the Cu electrode, as Pb produced more side-products than Cu. Other reports also showed that Cu was the best catalyst for reducing furfural to FFA and close to commercial goals, while its ECH efficiency was mainly challenged by the HER and dimerization. In addition, further reducing the size of Cu to nanoscale would help to enhance the electrocatalytic activity. Taking the nanoporous Cu (NP-Cu) as an example,105 a high-surface-area and porous structure offered sufficient Hads for the hydrogenation of furfural, resulting in 95% FE and 96% to FFA. Upon retaining this nanoporous structure, the introduction of Ni could optimize the electronic structure of Cu.106 It is indicated that metal doping can clearly decrease the consumption of Cu, and is able to easily adjust its electrocatalytic performance. Besides metal doping, non-metal doping can also effectually tune the electronic structure of Cu, thereby enhancing the ECH activity of furfural. Zhang et al. fabricated Cu3P nanosheets on a carbon fiber cloth (denoted as Cu3P/CFC) through a vapor-phase hydrothermal method, which could acquire high Hads-coverage but a limited H2 desorption process, leading to high electrocatalytic performance.107 Another solution is to disperse the active sites onto the conductive substrate for gaining high ECH activity, where the conductive substrate usually has a special morphology and structure, such as pores or multilayers.108
Many studies have recognized that single metal catalysts can be used for ECH of furfural, but the effect of bimetallic sites has been less explored. In other reports, the combination of Ag and Pd can also provide good productivity for FFA without considering the economic cost.109 For instance, Zhang et al. conducted potentiostatic electrolysis experiments to systematically study the selective conversion of furfural to FFA in a neutral medium using Ag60Pd40 alloy nanoparticles.109 Considering that furfural is easily subjected to the Cannizzaro reaction in an alkaline medium, FFA and furoic acid would be formed, which can be avoided by using a neutral medium. In addition to Cu-based and noble metal catalysts, MoS2 is reported to be a potential catalyst for yielding FFA from furfural.110 MoS2 was synthesized through electrochemical deposition and solvothermal methods. The ECH behavior of MoS2 was evaluated in an alkaline solution (borate buffer:MeCN = 4:1, pH 9), affording FFA with 82.4% yield and 92.1% selectivity, while the current efficiency was not recorded due to severe HER.
As this field matures, MoS2 has been recently reported to catalyze the C–C coupling of furfural for hydrofuroin production in an alkaline environment (pH 9).111 The electrochemical dimerization of furfural could be intentionally achieved by controlling the structural phase distribution of MoS2, but the hydrofuroin selectivity (only 47%) was restricted. According to previous tips from the electrochemical dimerization of HMF, a good dimerization behavior is more easily observed using carbon-based electrodes. For example, Sun et al. conducted the electrochemical dimerization of furfural in a batch electrolyzer with carbon paper as the working electrode.112 In this typical process, a high pH (0.1 M KOH) was needed, and 94% yield of hydrofuroin and 93% FE were detected. Recent studies have reported that this reaction can be extended to the organic electrolytic systems, which can not only avoid HER but also improve the solubility of furfural in electrolytes.118,119
In short, two catalytic mechanisms for furfural conversion on metal electrodes have been generally recognized in non-acidic conditions, namely, ECH and electroreductive reduction (Fig. 11). To be specific, furfural is directly electroreduced to the FFA-radical, followed by the adsorption of Hads to obtain FFA, or direct dimerization to provide hydrofuroin.
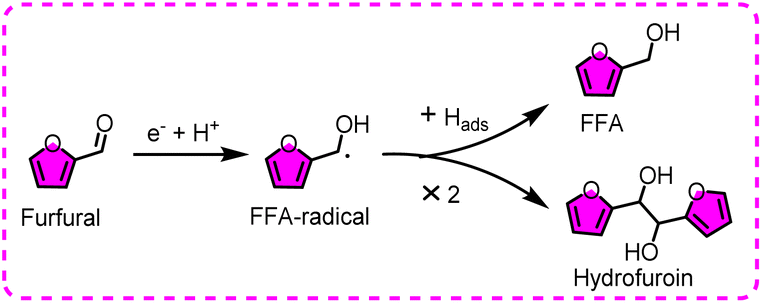 | ||
| Fig. 11 Plausible mechanisms/pathways for the ECH (upward) and dimerization (downward) of furfural. | ||
An acidic electrolyte is of crucial importance for selectively producing MF from furfural, and this tendency is consistent with the hydrogenolysis of HMF. In this case, increased selectivity toward MF would be observed at pH values below 2, as illustrated by Jung and Biddinger.113 This reaction was executed in a 0.5 M H2SO4 electrolyte using a Cu foil electrode, and 71% selectivity of MF was observed. To maximize the production efficiency of the target product, a catalyst with higher ECH performance can be considered. Bharath and Banat reported a novel tactic to fabricate Ru/RGO (reduced graphene oxide) nanocomposites using a simple microwave irradiation technique.114 The prepared Ru/RGO could catalyze the electroreduction and electrooxidation of furfural using 2.0 M H2SO4 as a cathodic electrolyte and 1.0 M KOH as an anodic electrolyte. In particular, the paired electrolyzer cell exhibited better catalytic activity than that of a half-cell. This is because it is easier for the cathode to obtain electrons from the anode, which are used for furfural reduction. In terms of reduction, MF could be obtained with 95% FE and 91% selectivity. Aside from the coupled electrooxidation strategy, a unique water/oil bi-phasic system was adopted for improving furfural hydrogenolysis. Specifically, the conversion of furfural to MF occurred in the aqueous phase, but the generated MF was more easily dissolved in the oil phase, resulting in a positive shift toward MF, which is a favorable equilibrium.120 Similar to HMF hydrogenolysis, electroreduction of furfural to MF is a one-step reaction, implying that the electrocatalytic reduction of furfural to MF and furfural to FFA are likely parallel reactions (Fig. 12). Although the conversion of FFA to MF can be possibly achieved at a highly negative potential, it does not mean that FFA is an intermediate for the electroreduction of furfural to MF. As an experimental example, when FFA was employed as the starting substrate to undergo electrolysis under the condition that furfural could be decidedly reduced to MF, only a slight MF was produced.115 In this regard, introducing ancillary sites to enhance the specific adsorption and reduction of hydroxyl groups at the catalyst surface is expected to promote the conversion of FFA to MF.
 | ||
| Fig. 12 The pathways for the eletroreduction of furfural to furfuryl alcohol (FFA) or 2-methylfuran (MF). | ||
An interesting electroreduction reaction under acidic conditions has been reported, through which furfural undergoes aldehyde hydrogenation and ring-saturation to provide tetrahydrofurfuryl alcohol (THFA).116,117 The THFA with high miscibility and biodegradability is usually used as the green solvent of industrial cleaners, epoxy resins, and dyes. However, the electrochemical route to THFA from furfural has rarely been reported due to the excellent stability of the furan ring. To handle this challenge, Lenk et al. investigated the electrochemical hydrogenation performance of furfural over the Pd5/Pt electrode, with a focus on the formation of THFA.116 The high Pd content seems beneficial to yield THFA, and the extra minor Pt content helps to increase the THFA selectivity and FE. Furfural and FFA were found to have some ring hydrogenation ability in this system, giving THFA with selectivity of 15.3% and 33%, respectively, whereas furoic acid and MF did not show this behavior. However, there is no explicit evidence to certify that this phenomenon is caused by the electronic effect of the substituent group in the furan ring, as such a reaction is in its infancy. Obviously, the Pd site is a pivotal factor for selective THFA production, and exposing more Pd sites may be beneficial to obtain higher selectivity. Excellent selectivity (98%) to THFA was observed when this reaction was carried out using a Pd-membrane reactor.117 In this unique reactor, the Pd site was electrodeposited on the Pd foil membrane to fabricate the Pd/Pd membrane catalyst, and the resultant catalyst displayed about 218-fold catalytic surface area relative to the ordinary Pd-electrode. The effects of solvent polarity and nucleophilicity on the reaction were also studied, and the results indicated that high reactivity and selectivity were detected when using a bulky and weakly nucleophilic solvent (t-BuOH). Otherwise, various by-products would be observed when using other types of solvents (e.g., EtOH, MeOH, n-BuOH, and i-PrOH). This can be attributed to the steric hindrance of these solvents that inhibit the unwanted chemical reactions. In addition, the hydrogenation of furfural to FFA was identified as the rate-determining step of the whole reaction (from furfural to THFA), and THFA might also be further reduced to yield 2-methyltetrahydrofuran (Fig. 13).
 | ||
| Fig. 13 The pathway for electrochemical hydrogenation of furfural to tetrahydrofurfuryl alcohol (MTHF). | ||
Overall, the electrochemical hydrogenation reactions of HMF and furfural are different in terms of the electrolytic conditions. For HMF, Ag and Cu are the most active elements, and the buffer electrolyte (pH 9.2) is capable of providing the most suitable electrolytic environment to generate BHMF. For furfural, Cu-based materials are reported to be the best catalysts, and the wider pH windows (6.9–13.6) are able to selectively produce FFA. It is worth noting that the preference of the electrolysis parameters for HMF and furfural hydrogenolysis is consistent, but some interesting reactions like ring-opening have not been reported in electrochemical furfural transformations.
3.3. Electroreduction of lignin-derived aromatic compounds
| Entry | Substrate | Product | Catalyst | EF (%) | C[S] (%) | Electrochemical parameters | Ref. |
|---|---|---|---|---|---|---|---|
| Cyclohexanone and cyclohexanol (KA), faradaic efficiency (FE), and C[S] is conversion[selectivity]. | |||||||
| 1 | 0.05 M phenol | Cyclohexane | 1.5% Pt/G | 44.1 | [68] | 0.2 M HClO4, j = −30 mA cm−2, 60 °C | 123 |
| 2 | 0.01 M phenol | Cyclohexanol | Pt | 85 | — | 0.05 M H2SO4, j = −5 mA cm−2 | 124 |
| 3 | 0.01 M phenol | Cyclohexanol | Pt/C | 72 | 94 | 0.5 M H2SO4, j = −15 mA cm−2 | 125 |
| 4 | 0.01 M phenol | Cyclohexanol | Pt/C | 72 | 99 | 0.5 M H2SO4, j = −5 mA cm−2 | 126 |
| 5 | 0.02 M phenol | Cyclohexanol | Pt/C | 40 | [95] | HCl (pH 5), E = −0.3 VRHE, 25 °C | 127 |
| 6 | 0.05 M phenol | KA | Pt/SSB | — | 100[98] | 0.1 M Na2SO4, j = −22.5 mA cm−2, 50 °C, 5 h | 128 |
| 7 | 0.05 M phenol | Cyclohexanol | Graphite rod, Pt/C | 98 | >99 | 0.1 M SiW12, MeOH/H2O = 1:9, j = −100 mA cm−2, 35 °C, 20 min |
130 |
| 8 | 0.018 M phenol | Cyclohexanol | Rh/C | 63 | >99 | Acetic acid (pH 5), E = −0.62 VAg/AgCl, 40 °C | 129 |
| 9 | 0.01 M phenol | Cyclohexanol | Rh/C | — | 95 | Acetate buffer (pH 5), E = −0.45 VRHE, 23 °C | 122 |
| 10 | 0.01 M phenol | Cyclohexanol | Pt1Rh1/MCN | 86 | [87] | 0.2 M HClO4, E = −0.2 VRHE, j = −3.125 mA cm−2 | 131 |
| 11 | 0.01 M phenol | Cyclohexanol | Pt3RuSn/CC | 39.5 | 91.5[96.8] | 0.2 M H2SO4, j = −20 mA cm−2, 50 °C, 100 min. | 132 |
| 12 | 1 mM phenol | Cyclohexanol | Ru/TiO2 | — | 94 | 0.2 M phosphate buffer, E = −0.9 VRHE, 50 °C | 133 |
| 13 | 0.05 M guaiacol | Cyclohexanol | Graphite rod, Pt/C | 96 | 95[56] | 0.1 M SiW12, MeOH/H2O = 1:9, j = −250 mA cm−2, 55 °C, 20 min |
130 |
| 14 | 0.01 M guaiacol | Cyclohexanol | Rh/ACC | <10 | 90[62] | 0.2 M HCl, j = −22 mA cm−2, 80 °C | 134 |
| 15 | 0.02 M guaiacol | KA | PtNiB/CMK-3 | 86.2 | 98.9[90.3] | 0.2 M HClO4, j = −10 mA cm−2, 60 °C, 60 min | 135 |
| 16 | 0.01 M guaiacol | KA | RANEY®-Ni | 26 | <100% | 0.1 M borate buffer (pH 8), j = −8 mA cm−2, 75 °C, 8 h | 136 |
| 17 | 0.1 M guaiacol | Cyclohexanol | Pt/C | 78 | 100 | 0.2 M methanesulfonic acid, j = −160 mA cm−2, 40 °C, 4 h, 240 rpm | 137 |
| 18 | 0.1 M guaiacol | Cyclohexanol | Pt/C | 80 | 98 | 0.2 M H2SO4, j = −109 mA cm−2, 50 °C, 4 h, 240 rpm | 121 |
| 19 | 0.12 M guaiacol | 2-Methoxycyclohexanol | PtRhAu | 58 | — | 0.2 M HClO4, j = −200 mA cm−2, 1 h | 138 |
| 20 | 0.02 M guaiacol | Methoxy-cyclohexanes | RhPtRu | 62.8 | [91.2] | 0.2 M HClO4, j = −50 mA cm−2, 2 h | 139 |
| 21 | 0.02 M syringol | KA | PtNiB/CMK-3 | 86.2 | 98.9[90.3] | 0.2 M HClO4, j = −10 mA cm−2, 60 °C, 90 min | 135 |
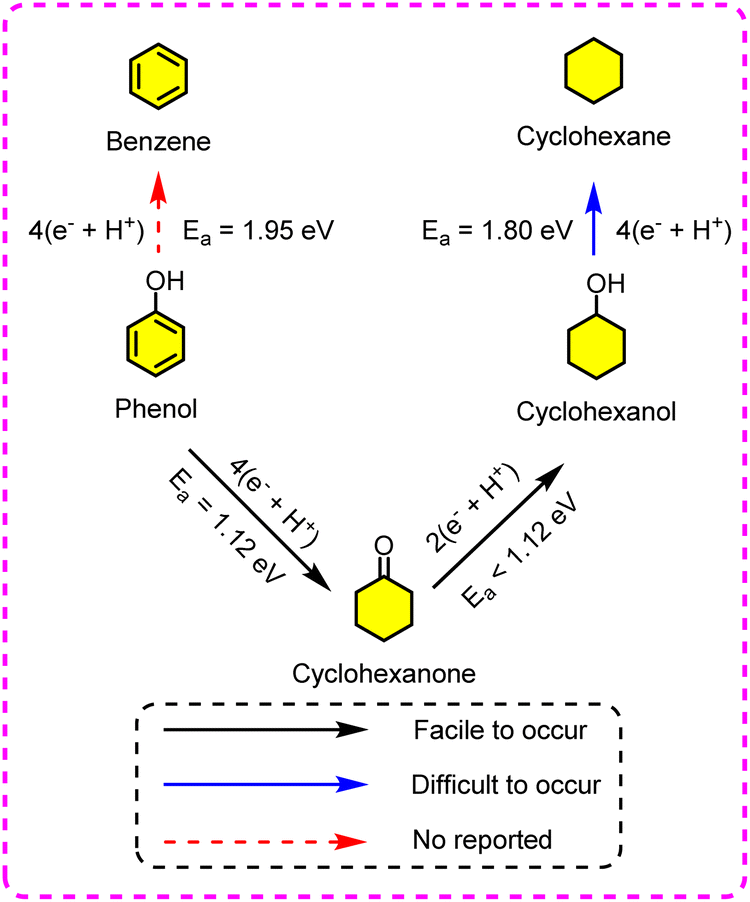
Taking ECH of phenol as an example, it is commonly accepted that Pt group metals (e.g., Pt, Rh, and Ru) are catalytic sites. Also, an appropriately elevatory temperature (5 °C to 60 °C) is beneficial to improve the electrocatalytic rate for the ECH of phenol, but it decreases beyond 60 °C.122 This means that the utilization of higher temperatures (>60 °C) to accelerate reaction rates is excluded. It allows the ECH process to combine with an acid–base-catalyzed reaction for the synthesis of hydrocarbons. Fu et al. studied the ECH of phenol in 0.2 M HClO4 electrolyte, with a 1.5% Pt/G (1.5% Pt supported on graphite) cathode.123 The temperature is the most important variable for cyclohexane production aside from current. For instance, the yield of cyclohexane increased from 44.1% to 63.7% with rising temperature from 20 °C to 60 °C, but it fell back to 50.3% at 80 °C. The difficulty was found to lie in the simultaneous hydrogenation and deoxidation (i.e., C–O bond cleavage) of phenol under mild conditions. As the C–O bond cleavage required more energy input (Ea = 1.80 eV), the reaction for the synthesis of cyclohexane was performed at an elevated temperature. As the temperature decreased, the ECH product was switched to cyclohexanol and cyclohexanone. In earlier studies, dispersed Pt was widely reported to catalyze the hydrogenation of phenol to KA, while the reactivity was not impressible to the Pt particle size.124–127 However, the current efficiency and product selectivity are affected by electrode materials. For example, the activity of the Pt/C electrode was visibly superior to that of the platinized Pt (Pt/Pt) electrode in terms of competence.126 This is due to the strong interaction between the metal Pt and the carbon support, resulting in an enhanced Pt electron density. Recently, shrimp shell biochar (SSB) was also found to serve as a Pt catalyst carrier for phenol ECH.128 After 5 h, the resultant Pt/SSB catalyst exhibited complete phenol conversion and 98% overall selectivity toward cyclohexanone and cyclohexanol. The ECH and thermocatalytic hydrogenation of phenol are demonstrated to be executed through the Langmuir–Hinshelwood mechanism, which means that the Hads of ECH is only generated by the Volmer reaction, rather than H2 re-adsorption.129 Although the electrons consumed by phenol → cyclohexanone are two times greater than that of cyclohexanone → cyclohexanol, the reaction rate of the former is faster, leading to the formation of cyclohexanone as the primary product at a low conversion. In contrast, cyclohexanone is easily converted to cyclohexanol under electrochemical conditions, suggesting that cyclohexanol is the main product after a complete reaction. Liu et al. reported that the ECH of phenol to cyclohexane was accomplished using a dual-catalyst system containing a suspended Pt/C catalyst and solvable SiW12.130 The SiW12 acted as a charge transfer catalyst to accelerate electron transfer from the cathode (graphite rod) to the Pt/C surface, and then enhance the catalytic efficiency. Almost all phenol conversions were observed at a high FE (98%), along with three quantifiable products including cyclohexanol (yield: 85%), cyclohexane (yield: 14%), and cyclohexanone (yield: 0.3%) at 35 °C within 20 min. It was manifested that more energy (Ea = 1.95 eV) was required to directly cleave the phenol C–O bond over the Pt/C electrode. Thus, phenol tends to undergo aromatic ring saturation to give cyclohexanone (Ea = 1.12 eV), as displayed in Fig. 14. Cyclohexanone can be easily converted to cyclohexanol under electrochemical conditions, while the C–O bond cleavage of cyclohexanol still requires more energy input (Ea = 1.80 eV). Hence, cyclohexanol is the dominating product in most cases. Highly dispersed nanoparticles are vastly capable of increasing the collision rate between the substrate and electrolyte, leading to a higher rate for ECH compared with that for HER.
 | ||
| Fig. 14 The proposed pathway for ECH of phenol to cyclohexanol in a dual-catalyst system. | ||
In addition to Pt sites, dispersed Rh was reported to catalyze ECH of phenol to cyclohexanol under mild conditions. Even though the conversion was able to reach a plateau (>95%), the anticipated selectivity and/or FE could not be afforded concurrently.122,129 The ECH of phenol may be a zero-order reaction, indicating that the substrate concentration is relatively independent of the reaction rate, allowing for faster and more stable adsorption on the catalyst surface.129 Given that individual Pt and Rh have a certain activity for the ECH of phenol, constructing them into alloys may provide an unexpected result. Zhou et al. reported that the highly overlapped d-orbitals in Pt and Rh facilitated phenol adsorption, and the introduction of Rh was able to generate a beneficial electronic effect to weaken the alloy-Hads interaction.131 A reliable FE (86%) and selectivity (87%) for ECH of phenol were obtained at −0.02 VRHE. Recently, a better selectivity was observed when a ternary alloy (Pt3RuSn/CC) was used as the catalyst for this reaction, but the FE was not anticipated.132 Compared with a monometallic or bimetallic catalyst, the introduction of Pt and Ru as catalytic sites for the ternary Pt3RuSn/CC catalyst synergistically could promote the hydrogen saturation of the aromatic ring, while the addition of Sn supplied extra adsorption sites for phenol ECH.
In addition, phenolic pollutants are common organic compounds in wastewater, which are mainly derived from the release of many industries like coal chemical, oil, and pharmaceutical refineries.140–142 Gu et al. developed a feasible ECH protocol to remove phenolic pollutants and recover useful cyclohexanol from wastewater, using a hierarchical Ru/TiO2 electrode.133 Remarkably, the pseudo-first-order rate constant of ECH was 0.135 min−1, which was 34 times higher relative to traditional electrochemical oxidation. Also, this method could retain good reactivity at a high phenol concentration (1000 mg L−1) and complicated water content. The substrate scope can also be extended to other substituted phenols, such as chlorophenols and methylphenols.
Considering that guaiacol bears one extra –OCH3 relative to phenol, the chemical properties should be partially overlapped. However, the presence of –OCH3 in guaiacol also leads to the diversity of ECH products, such as cyclohexanone, cyclohexanol, 2-methoxycyclohexanone, and 2-methoxycyclohexanol (Fig. 15). In the synthesis of cyclohexanol, the –OCH3 of guaiacol must be removed via demethoxylation, which is a thermodynamically favorable process. Clearly, two routes are executed to offer cyclohexanol in the existing literature, namely, demethoxylation-ring saturation and ring saturation–demethoxylation, suggesting that the selectivity and carbon balance of the target product may be reduced under conventional conditions.
 | ||
| Fig. 15 The network of the electrocatalytic transformation of guaiacol to cyclohexanol. | ||
In the dual-catalyst system discussed above, guaiacol was used as the starting substrate to investigate the ECH behavior, and 95% guaiacol conversion and 56% cyclohexanol selectivity were obtained at 96% FE, but a relatively high temperature (55 °C) was needed.130 The elevated temperature is critical for the selective production of cyclohexanol from guaiacol as it facilitates the demethoxylation step. However, a temperature over 80 °C is unfavorable for selectivity and FE,134 since hydrocarbons may be further formed via –OH hydrogenolysis, which is consistent with the phenol ECH. In contrast, the guaiacol ECH is not a zero-order reaction and is easily affected by operating parameters, such as catalyst loading, substrate adsorption, and stirring rates. In a pertinent work, Wang et al. reported a boron-doping strategy to tune the electronic structure of PtNi, and the resultant PtNiB nanoparticles were then implanted into a substrate (CMK-3) with mesoporous channels for facilitating the ECH process.135 Benefiting from this unique electronic structure (from boron-doping) and large specific surface area (from CMK-3), the as-prepared PtNiB/CMK-3 catalyst exhibited excellent catalytic performance and extensive substrate range. The related performance and parameters are depicted in Fig. 16. Specifically, the local electrons around PtNi were slightly transferred to boron atoms, giving rise to higher adsorption energy on PtNiB (−0.59 eV to −0.91 eV), thus accelerating the substrate and intermediate adsorption.135 Also, the high-surface-area and porous structure of CMK-3 can maximize the exposed active sites and supply low-resistance diffusion channels. Apart from changing the electronic structure through doping strategies, the use of a stirred slurry electrochemical reactor (SSER) also helps to improve phenol ECH. In this case, this reaction is allowed to operate at an industrial-grade current density (>|100 mA cm−2|), and the liquid–solid mass and heat transfer between reactive molecules and catalyst particles in the reaction can also be improved. Other superior properties of the SSER structure include the elimination of the requirement to deposit catalysts onto conductive substrates and the scale-up possibility using a fluidized/floating bed as an electrochemical reactor. In a classically fixed bed system, Jackson et al. studied the deficiency in carbon balance (80%) from guaiacol ECH with a RANEY® nickel electrode at a moderate temperature (75 °C), while FE was only 26% even at a low current density (−8 mA cm−2).136 Instead, an ideal carbon balance (>90%) could be obtained using this SSER configuration, of which carbon loss may be caused by transmembrane diffusion, cathodic adsorption, anodic oxidation, and evaporation.137 The adverse mass transport or diffusion limitations could be overcome at an optimal stirring rate (240 rpm), leading to improved reactive activity (full conversion of guaiacol) at a high FE (78%). Similarly, several viable noble-metal catalysts (e.g., Pt/C, Pd/C, and Ru/C) were tested for guaiacol ECH with different catholyte–anolyte pairs in SSER.121 The substrate conversion and FE were improved using NaCl catholyte and H2SO4 anolyte pairs in all cases, demonstrating that the anodic protons were successfully transported to the cathode and available for utilization for ECH. Among the three tested catalysts, Pt/C offered better activity over Ru/C and Pd/C in light of the measured reaction rates.
 | ||
| Fig. 16 The ECH of guaiacol and other phenolic compounds on the PtNiB/CMK-3 electrode. | ||
To date, many ECH studies are focused on the conversion of guaiacol to cyclohexanol, resulting in the formation of some products with –OCH3 groups being ignored. In the ECH of guaiacol, the selective production of methoxy-cyclohexanes (2-methoxycyclohexanol and 2-methoxy-cyclohexanone) is of crucial significance owing to their antimicrobial and anticancer activities.143,144 However, an unfavorable step exists when utilizing ECH to yield methoxy-cyclohexanes from guaiacol; namely, demethoxylation has thermodynamic advantages over aromatic ring hydrogenation under electroreductive conditions. Originating from the desire to promote the hydrogenation of guaiacol but inhibit its demethoxylation, some novel electrocatalysts and operating parameters have been developed. Au and Ru are often selected for regulating the electronic structure of PtRh to inhibit the adsorption of –OCH3 on the electrode surface, thus suppressing the demethoxylation and promoting the production of methoxy-cyclohexanes.138,139 In addition, the shorter running time (<2 h) and room temperature help to avoid further demethoxylation based on previous reports. For example, Peng et al. reported that a ternary alloy (PtRhAu) could catalyze the conversion of guaiacol to 2-methoxycyclohexanol in an HClO4 electrolyte.138 It was proposed that the electron of PtRh was transferred to Au, and the introduction of Au enhanced the adsorption energy of the substrate, bringing about an increased reactive activity. In terms of efficiency, 58% FE was recorded over the PtRhAu catalyst at a high current density (−200 mA cm−2), but the selectivity was unclear. Likewise, 62.8% FE and 91.2% selectivity of methoxy-cyclohexanes could be observed using the RhPtRu catalyst within 2 h at a low current density (−50 mA cm−2).139 This is caused by a synergistic effect in the RhPtRu catalyst, in which Rh and Pt bear high adsorption energies for guaiacol, thus providing a high guaiacol coverage on the RhPtRu surface. The introduction of Ru suppresses the HER, since Ru possesses the ideal ability to restrict HER as compared with that of Pt and Rh.
In contrast to phenol and guaiacol, syringol has not received much attention due to its more complex product distribution resulting from two symmetrical –OCH3 groups (at the 2-position and 6-position) in its structure. Typically, the conversion efficiency and current efficiency decrease with the increase of molecular complexity.135–139 Given that syringol also has a certain profitability in producing cyclohexanol (Fig. 16), it is usually used as an extended substrate investigation for phenol and guaiacol.135 Obviously, a longer running time (1.5-fold) is required for the conversion of syringol to cyclohexanol in comparison with that of phenol and guaiacol under the same conditions. Hence, its ECH behavior is rarely studied separately. Overall, the selective synthesis of methoxy-cyclohexanes from guaiacol or syringol via ECH is a primary bottleneck reaction since the –OCH3 group is in an electron-rich environment, which is easily cleaved to give cyclohexanol.
The ECH of benzaldehyde has been extensively investigated over various metal catalysts (e.g., Pd, Cu, Pt, Rh, and Ni), and these metals are usually loaded on conductive substrates such as Cu foam and carbon felt.145–148 Among them, Pd-based catalysts are more inclined to form benzyl alcohol, and Cu-based catalysts are more prone to produce dimer products (Fig. 17). The related parameters and performances are listed in Table 5.
 | ||
| Fig. 17 Electroreductive pathways of benzaldehyde over Pd or Cu electrode. | ||
| Entry | Substrate | Product | Catalyst | E.F. (%) | C[S] (%) | TOF (h−1) | Electrolysis conditions | Ref. |
|---|---|---|---|---|---|---|---|---|
| Cyclohexanecarboxylic acid (CCA), faradaic efficiency (FE), turnover frequency (TOF), and C[S] is conversion[selectivity]. | ||||||||
| 1 | 0.02 M benzaldehyde | Benzyl alcohol | Pd/C | 96 | — | 517 | Sodium acetate acetic acid buffer (pH 5.2), E = −0.1 VRHE | 149 |
| 2 | 0.02 M benzaldehyde | Benzyl alcohol | Pd/C | 99.6 | [99] | 3899 | Acetate buffer (pH 5), E = −0.9 VAg/AgCl | 150 |
| 3 | 0.02 M benzaldehyde | Benzyl alcohol | Pd/C | 25–100 | — | 100–900 | Alcohol:H2O, j = −(2.8–8.3) mA cm−2 |
151 |
| 4 | 0.02 M benzaldehyde | Benzyl alcohol | Pd/CF | 80–95 | — | 2762 | Acetate buffer (pH 5.2), E = −0.1 VRHE | 152 |
| 5 | 0.02 M benzaldehyde | Benzyl alcohol | Pd/Cu-CF | 92.01 | 98.51[95.46] | — | 0.1 M H2SO4, E = −0.3 VRHE | 153 |
| 6 | 0.024 M benzaldehyde | Hydrobenzoin | Cu | 37 | — | — | Phosphate buffer (pH 4.6), E = −0.5 VRHE | 154 |
| 7 | 0.04 M benzaldehyde | Hydrobenzoin | Pd/Cu | 63.2 | 85.3 | — | 0.1 M KOH, E = −0.4 VRHE | 155 |
| 8 | 0.02 M benzaldehyde and furfural | Dimer products | Cu | 80 | — | — | Phosphate buffer (pH 6.7), E = −0.5 VRHE | 156 |
| 9 | 0.5 M benzophenone | Diphenylmethanol | Pd0.2/C/T | — | [>90] | — | 0.1 M H2SO4, j = −10 mA cm−2 | 158 |
| 10 | 0.02 M acetophenone | 1-Phenylethanol | Rh/C | ∼40% | — | 1419 | Acetate buffer (pH 4.6), E = −0.8 VAg/AgCl | 159 |
| 11 | 0.02 M acetophenone | 1-Phenylethanol | Graphite | 100 | [>99.9] | — | 0.2 M NH4Cl, j = −350 mA dm−2 | 161 |
| 12 | 1 M benzoic acid | CCA | Pt1Ru1.5 | 99 | — | — | 0.05 M H2SO4, j = −1.5 mA cm−2 | 164 |
| 13 | 0.45 g L−1 benzoic acid | CCA | Pt/C/CFP | 57 | 100[100] | — | 0.05 M H2SO4, E = −0.51 VRHE | 165 |
Pd is often deemed as the most reliable electrocatalyst for converting benzaldehyde to benzyl alcohol by virtue of its low electrochemical over-potential and good anti-poisoning capacity. The ECH of phenol, furfural, and benzaldehyde were surveyed using a carbon-supported catalyst (Pd/C), where the reactivity of benzaldehyde was better than that of furfural (FE: 96% vs. 42%, TOF: 517 h−1vs. 64 h−1), while phenol was not reactive.149 Under this reaction condition, if the three substrates coexisted in the same system, benzaldehyde was preferentially hydrogenated, indicating that Pd was the most effective active site for yielding benzyl alcohol from benzaldehyde as far. A similar verdict was verified by Song et al., of which four carbon-supported catalysts (i.e., Rh/C, Pt/C, Pd/C, and Ni/C) were tested for the electroreduction of benzaldehyde to benzyl alcohol in a weakly acidic environment.150 It was discovered that the ECH of benzaldehyde underwent a one-order reaction on Rh/C and Pt/C electrodes, while a zero-order reaction on Ni/C and Pd/C electrodes, which was ascribed to high substrate coverages and low Hads-coverages on the surface, consistent with the phenol ECH. Specifically, the turnover frequency (TOF) of four catalysts for benzaldehyde ECH was in the order of Pt/C (2198 h−1) < Rh/C (2267 h−1) < Pd/C (3389 h−1) < Ni/C (14200 h−1).150 Although Ni/C bears higher intrinsic activity, a highly negative potential is needed to drive this reaction. A trend of Ni/C (35%) < Pt/C (39%) < Rh/C (64%) < Pd/C (99.6%) was observed for FE. Taken together, the Hads was consumed through ECH on Pd, leading to low Hads-coverages, while the Hads was drained via HER on Ni, resulting in low Hads-coverages, certifying that Pd is still an effective catalytic site for the electroreduction of benzaldehyde to benzyl alcohol. In a relevant work by Lopez-Ruiz et al., the effect of alcohols (including methanol, ethanol, and isopropanol) as cosolvents on the benzaldehyde ECH and HER over Pd/C in aqueous phase was investigated.151 Notably, the ECH rates and FE were lessened with the enhancement of the alcohol concentration, while the HER rates were unchanged. More importantly, the suppression of the ECH reaction became more obvious with the extension of the carbon chain of alcohol. Increasing the cathodic potential not only heightened the ECH rate, but also largely enhanced the HER rate, thereby reducing the FE of the target product. As a result, enhancement of the benzaldehyde concentration should be considered to furnish the high activity and desired FE. Koh et al. reported that acid-functionalization of the carbon support allowed for the increase in the ECH activity of the Pd catalyst, in which the introduction of Brønsted-acid sites was critical for the reduction of benzaldehyde to benzyl alcohol.152 The Brønsted-acid site near the Pd particles facilitated the proton-coupled electron transfer process to promote the ECH of benzaldehyde, which became more obvious with the decrease of the H3O+ concentration. The Brønsted-acid site is usually provided by carboxylic and phenolic groups, which can be obtained by O2 plasma treatment or oxidation with HNO3. Theoretical and experimental results indicated that the carboxylic group played an important role in the benzaldehyde ECH, since the proton transferred from the carboxylic group to electrolyte only required minimal energy (6 kJ mol−1), while the proton transfer in the phenolic groups needed more energy (168 kJ mol−1). A relatively high intrinsic activity (TOF: 2762 h−1) and 80%–95% FE were obtained for reducing benzaldehyde to benzyl alcohol. For the involved mechanism, the acid-functionalized carbon support and/or its generated H3O+ are capable of participating in the ECH of benzaldehyde, but this process must occur at the interface between Pd and support (Fig. 18). Four steps are involved in this reaction: (i) adsorption of benzaldehyde, (ii and iii) ECH of benzaldehyde and regeneration of the acidic sites, and (iv) desorption of benzyl alcohol and regeneration of the acidic sites. To retain the superior properties of the Pd-based catalyst but reduce the production cost, Wu et al. successfully fabricated a dendritic-like Pd/Cu-CF electrode via two-step electrodeposition for the ECH benzaldehyde.153 Owing to its unique dendritic-like structure, Pd/Cu-CF could expose sufficient active sites, and promote electrolyte penetration and reactant adsorption, thus resulting in 92.1% FE and 95.46% selectivity at nearly full conversion of benzaldehyde.
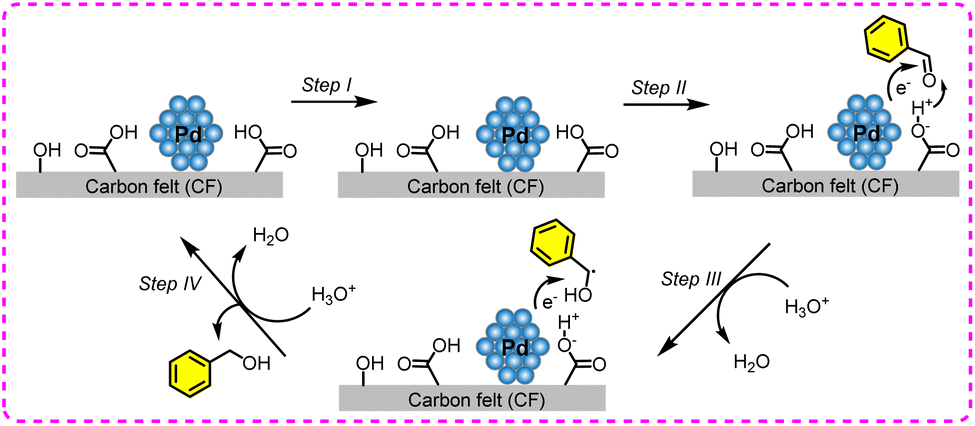 | ||
| Fig. 18 The ECH cycle of benzaldehyde over an acid-functionalized catalyst (Pd/CF). | ||
The electroreductive coupling of benzaldehyde is a valuable reaction because it can extend the carbon chain and increase the molecular weight, and the resulting hydrobenzoin can be used for antiepileptic drugs. In early studies, Anibal et al. tested the profitability of several metals (Au, Cu, Pd and Pt) to access hydrobenzoin in phosphate buffer (pH 4.6), using benzaldehyde as the starting substrate.154 Although all of the tested catalysts were active for the hydrogenation of benzaldehyde, only Cu exhibited unique C–C coupling ability (FE: 37%), attributed to the high free-radical (Ph-C˙-OH) coverage on Cu catalyst, on which free radicals were generated but not accumulated, so no product was formed. On Pd and Pt, the potential dependence and generated CO likely poisoned the catalyst, thereby limiting the accumulation of free radicals and restraining the occurrence of C–C coupling. In recent work, Yu et al. proposed that the ability of Pd sites to activate benzaldehyde was still better than that of Cu sites, and Cu was mainly responsible for the vital C–C coupling, rather than the whole reductive process.155 The experiments were carried out in an alkaline electrolyte (0.1 M KOH), with a Pd/Cu electrode. It is indicated that the direct electroreduction of benzaldehyde to benzyl alcohol is a relay process (Fig. 19), where the Pd site activates benzaldehyde to a free-radical intermediate, while Cu promotes subsequent free-radical coupling. Under the balance of these two sites, 63.2% FE and 85.3% yield towards hydrobenzoin were recorded by HPLC. More interestingly, cross-coupling and self-coupling were observed on Cu and Pd electrodes when both benzaldehyde and furfural were used as starting substrates to undergo electrolysis (Fig. 20).156 After electrolysis, hydrogenated products (benzyl alcohol and FFA) and coupled products (hydrobenzoin, hydrofuroin, and 1-(2-furyl)-2-phenyl-1,2-ethanediol) were detected. Therefore, the former is mainly yielded at a low potential (<−0.5 VRHE), and the latter is chiefly formed at a high potential (≥−0.5 VRHE), consistent with the dimerization of HMF and furfural discussed above. The reactivity tests manifested that the cross-coupling reaction was more likely to occur on the Cu electrode, while the Pd electrode tended to produce hydrofuroin. In general, the total FE of the coupled product was close to 80% on the Cu electrode at −0.5 VRHE, superior to Pd.
 | ||
| Fig. 19 The reaction mechanism for the electroreductive coupling of benzaldehyde on the Pd/Cu catalyst. | ||
 | ||
| Fig. 20 Schematic diagram of electroreductive routes in the mixed benzaldehyde-furfural system. | ||
Similar to benzaldehyde, the target functional group of acetophenone to undergo electrocatalytic conversion is carbonyl, but its reactivity is relatively lower than that of benzaldehyde, which may be caused by the electronic effect and steric hindrance of the adjacent methyl group.157 The combination of noble metal catalysts (Pd and Rh) and acidic electrolytes is still the best condition for the hydrogenation of acetophenone, and 1-phenylethanol is the end product of this reaction.158–160 In addition to Pd and Rh, graphite was reported to convert acetophenone to 1-phenylethanol with the assistance of ultrasound and achieved excellent results (FE and yield of 100%).161 The ultrasound irradiation accelerates the mass transport of acetophenone from bulk electrolyte to graphite surface, enhancing acetophenone's availability to react with the Hads, and resulting in high current efficiency and yield. Unlike benzaldehyde ECH, there is still no effective catalyst for the directional coupling of acetophenone so far. In this sense, the search and tailored design of functional electrocatalysts are crucial to realizing the electroreductive coupling of acetophenone.
Benzoic acid is composed of an electron-withdrawing group (–COOH) and an electron-donating group (phenyl), in which the –COOH group attached to the benzene-ring easily reduces the electron density of the phenyl group, making the phenyl moiety become easily adsorbed onto the electron-rich metal surface.162 Alternatively, –COOH is an extremely stabilized group under electroreductive conditions due to the presence of the p–π conjugate configuration between the carbonyl and hydroxyl groups. Accordingly, benzoic acid can be reduced to cyclohexanecarboxylic acid (CCA), which is a valuable intermediate for the synthesis of caprolactam and amrinone. Viewing that a high benzene-ring resonance energy needs to be overcome in this process, a majority of investigations were carried out under intensive pressure and/or elevatory temperature, which may lead to concurrent hydrogenation of benzene-ring and –COOH.163 Adjusting the electronic structure of the metal Pt to an electron-rich state enhances the adsorption of the benzene ring on the catalyst, thereby realizing the oriented conversion of benzoic acid to CCA. For example, Fukazawa et al. introduced Ru to optimize the electronic structure of Pt, and the electrons of the resultant Pt1Ru1.5 catalyst were transferred from Ru to Pt according to the result of X-ray photoelectron spectroscopy (XPS).164 The introduction of Ru also greatly inhibited the hydrogen evolution reaction as it had high HER over-potential, generating 99% FE towards CCA (Fig. 21). As reported by Kong et al., the use of carbon fiber paper (CFP) as a support for commercial Pt/C catalysts could adjust the selectivity of CCA and conversion of benzoic acid.165 The authors found that Pt exhibited an electron-rich state after Pt/C was deposited on CFP, while CFP existed in an electron-deficient state. More intriguingly, if the CFP was replaced with a carbon fiber cloth, it would display the opposite trend. This is due to the fact that Pt/C particles display a highly dispersive state on CFP, but a significantly agglomerating state on carbon fiber cloth. Following the results of DFT and XPS, the CFP with fewer defects was capable of inducing the intentional gathering of electrons on Pt-areas and increasing the adsorption of electron-deficient benzene-ring, thereby enhancing the reactive activity of CCA over Pt/C/CFP. In terms of electrocatalytic performance, 100% selectivity for CCA was recorded at full benzoic acid conversion within 3 h. Although the electrochemical pathways to obtain CCA from benzoic acid have been realized, the involved mechanisms remain unclear.
 | ||
| Fig. 21 Schematic diagram for the electroreduction of benzoic acid to cyclohexanecarboxylic acid over the Pt1Ru1.5 catalyst. | ||
4. Conclusions
By and large, biorefinery technology is the current and future competitive direction to produce value-added chemicals and biofuels. Although significant progress has been made in the electroreductive upgrading of biomass, it is still in its infancy due to the deficiency of mature and industrializing technology. The electroreductive system offers a green and direct transformation pathway for promising biomass valorization, but there are still many challenges to overcome before industrialization. This goal is largely limited by the type of electrodes because precious metals or toxic catalysts are widely used in these upgrading processes. The reaction mechanism study also retains enormous blanks, e.g., the electrocatalytic hydrogenation of acetophenone and benzoic acid, indicating that vast efforts need to be devoted to this direction. To overcome the current technical bottlenecks and achieve future industrialized objectives, the following aspects should be considered.(1) The rational construction of catalytic sites is crucial to improve the inherent activity of the reaction, which should be cost-effective. Electrocatalysts with high HER over-potential and strong acid/alkali resistance are highly touted for electrocatalytic hydrogenation, in which Ag, Cu, Ru, and Rh are typical examples. In addition, selecting the catalytic site according to the reaction preference is more conducive to obtaining the desired results, and the relevant cases have been summarized in the above tables.
(2) On the basis of the optimal sites, further introduction of auxiliary sites is of great significance for increasing the reactivity (activity, rate, selectivity, current density, and current efficiency). Other strategies for enhancing the intrinsic catalyst activity and increasing the number of active sites are focused on optimizing the morphology of the catalyst or adjusting the electronic structure of metals.
(3) The role of electrolytes should also be considered, as most reactions occur at high proton concentrations. These effects become more obvious in the hydrogenation and hydrogenolysis of furan-based compounds (furfural and 5-hydroxymethylfurfural). Coupling oxidation reaction to upgrade biomass is an attractive objective, but cross electrolysis is often used, meaning that substantial studies are required to optimize electrolyte composition.
(4) One of the barriers to the electroreductive upgrading of biomass is the scarcity of an in-depth understanding of reaction mechanisms and structure–activity relationship, mainly due to the difficulty in identifying those pivotal intermediates or species at the catalyst/electrolyte interface. With the rapid advances of in situ technologies and theoretical calculations, the deficiency of the involved reaction mechanisms can be supplemented, which will remarkably guide the establishment of highly efficient electrocatalytic systems.
(5) Realization of high-current-density (>|100 mA cm−2|) and continuous product separation are pivotal steps to industrialization. Most of the previous studies mainly focused on parameter optimization and feasibility analysis, which greatly limits the industrialized goals. The advent of a continuous flow reactor overcomes this limitation because it can avert product over-reduction and provide high current density, high selectivity, and high current efficiency.
In the present review, we have established a complete overview of the electroreductive upgrading of biomass-derived platform molecules into high-value chemicals (e.g., valeric acid, cyclohexanol, and hydrobenzoin) and biofuels (e.g., 2,5-dimethylfuran, 2-methylfuran, and hydrofuroin), with a focus on reactive mechanisms and feasible conversion pathways for each type of reaction. Additionally, with the outstanding materials of each catalytic system as a cornerstone, concentrating on the development of the modification tactics for these advanced catalysts and summarizing positive effects for the reactivity of target compounds are beneficial to accelerate the practical implications. Electroreduction is a prospective catalytic method for biorefinery, and an extra merit is to store electricity aside from the production of fine chemicals and biofuels. Although these biomass-derived renewable resources can relieve the pressure from traditional fossil fuels, continuous efforts are still required to address the remaining technical hurdles before designing economically viable and large-scale processes. To better integrate electrocatalysis and biorefinery, interdisciplinary cooperation is required to accelerate this progress, aiming at sustainably supplying clean and renewable chemical products to our society.
Author contributions
Keping Wang: methodology, formal analysis, writing – original draft. Zheng Li: methodology, formal analysis. Zhenyan Guo: methodology, formal analysis. Jinshu Huang: formal analysis. Tengyu Liu: formal analysis. Min Zhou: formal analysis. Jinguang Hu: conceptualization, formal analysis, writing – review & editing. Hu Li: conceptualization, formal analysis, funding acquisition, project administration, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Guizhou Provincial S&T Project (GCC[2023]011, ZK[2022]011), National Natural Science Foundation of China (22368014), and Guizhou Provincial Higher Education Institution Program (Qianjiaoji[2023]082).References
- R. Saidur, E. Abdelaziz, A. Demirbas, M. Hossain and S. Mekhilef, Renewable Sustainable Energy Rev., 2011, 15, 2262–2289 CrossRef CAS.
- N. R. Stradiotto, K. E. Toghill, L. Xiao, A. Moshar and R. G. Compton, Electroanalysis, 2009, 21, 2627–2633 CrossRef CAS.
- S. Yang, X. Xiang, Z. He, W. Zhong, C. Jia, Z. Gong, N. Zhang, S. Zhao and Y. Chen, Chem. Eng. J., 2023, 141344, DOI:10.1016/j.cej.2023.141344.
- W. H. Lie, Y. Yang, J. A. Yuwono, C. Tsounis, M. Zubair, J. Wright, L. Thomsen, P. Kumar and N. Bedford, J. Mater. Chem. A, 2023, 5527–5539, 10.1039/d2ta08306j.
- A. Kumar, N. Kumar, P. Baredar and A. Shukla, Renewable Sustainable Energy Rev., 2015, 45, 530–539 CrossRef.
- D. P. Ho, H. H. Ngo and W. Guo, Bioresour. Technol., 2014, 169, 742–749 CrossRef CAS.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS.
- Z. Fan, W. Zhang, L. Li, Y. Wang, Y. Zou, S. Wang and Z. Chen, Green Chem., 2022, 24, 7818–7868 RSC.
- H. Li, A. Riisager, S. Saravanamurugan, A. Pandey, R. S. Sangwan, S. Yang and R. Luque, ACS Catal., 2018, 8, 148–187 CrossRef CAS.
- Z. Yin, Y. Zheng, H. Wang, J. Li, Q. Zhu, Y. Wang, N. Ma, G. Hu, B. He, A. Knop-Gericke, R. Schlogl and D. Ma, ACS Nano, 2017, 11, 12365–12377 CrossRef CAS PubMed.
- D. Wang, P. Wang, S. Wang, Y. H. Chen, H. Zhang and A. Lei, Nat. Commun., 2019, 10, 2796 CrossRef PubMed.
- Z. Li, X. Li, H. Zhou, Y. Xu, S. M. Xu, Y. Ren, Y. Yan, J. Yang, K. Ji, L. Li, M. Xu, M. Shao, X. Kong, X. Sun and H. Duan, Nat. Commun., 2022, 13, 5009 CrossRef CAS PubMed.
- H. N. Wang, A. X. Guan, J. B. Zhang, Y. Y. Mi, S. Li, T. T. Yuan, C. Jing, L. J. Zhang, L. J. Zhang and G. F. Zheng, Chin. J. Catal., 2022, 43, 1478–1484 CrossRef CAS.
- H. Zhou, Z. Li, S. M. Xu, L. Lu, M. Xu, K. Ji, R. Ge, Y. Yan, L. Ma, X. Kong, L. Zheng and H. Duan, Angew. Chem., Int. Ed., 2021, 60, 8976–8982 CrossRef CAS.
- L.-R. Yang, Y.-J. Zhao, C.-J. Jiang, R. Xiong, H. Wang and J.-X. Lu, J. Catal., 2021, 401, 224–233 CrossRef CAS.
- G. Han, G. Li and Y. Sun, Nat. Catal., 2023, 6, 224–233 CrossRef CAS.
- J. Liu and B. Yang, Biofuels, Bioprod. Biorefin., 2023, 7, 696–717 Search PubMed.
- J. Böttger, T. Eckhard, C. Pflieger, O. Senneca, M. Muhler and F. Cerciello, Fuel, 2023, 344, 128025 CrossRef.
- H. Li, J. He, A. Riisager, S. Saravanamurugan, B. Song and S. Yang, ACS Catal., 2016, 6, 7722–7727 CrossRef CAS.
- H. Li, Y. Li, Z. Fang and R. L. Smith, Catal. Today, 2019, 319, 84–92 CrossRef CAS.
- W. Wu, Y. Li, H. Li, W. Zhao and S. Yang, Catalysts, 2018, 8, 264 CrossRef.
- Y. Inami, S. Iguchi, S. Nagamatsu, K. Asakura and I. Yamanaka, ACS Omega, 2020, 5, 1221–1228 CrossRef CAS PubMed.
- K. Takano, H. Tateno, Y. Matsumura, A. Fukazawa, T. Kashiwagi, K. Nakabayashi, K. Nagasawa, S. Mitsushima and M. Atobe, Bull. Chem. Soc. Jpn., 2016, 89, 1178–1183 CrossRef CAS.
- Y.-Q. Zhu, H. Zhou, J. Dong, S.-M. Xu, M. Xu, L. Zheng, Q. Xu, L. Ma, Z. Li and M. Shao, Angew. Chem., Int. Ed., 2023, 62, e202219048 CrossRef CAS PubMed.
- M. Yang, Z. Yuan, R. Peng, S. Wang and Y. Zou, Energy Environ. Mater., 2022, 5, 1117–1138 CrossRef CAS.
- Z. Jiang, Y. Zeng, D. Hu, R. Guo, K. Yan and R. Luque, Green Chem., 2023, 25, 871–892 RSC.
- M. Garedew, C. H. Lam, L. Petitjean, S. Huang, B. Song, F. Lin, J. E. Jackson, C. M. Saffron and P. T. Anastas, Green Chem., 2021, 23, 2868–2899 RSC.
- K. Li and Y. Sun, Chem. – Eur. J., 2018, 24, 18258–18270 CrossRef CAS.
- A. Fukazawa, K. Takano, Y. Matsumura, K. Nagasawa, S. Mitsushima and M. Atobe, Bull. Chem. Soc. Jpn., 2018, 91, 897–899 CrossRef CAS.
- J. Creus, M. Miola and P. P. Pescarmona, Green Chem., 2023, 25, 1658–1671 RSC.
- C. Yang, Y. Wang, L. Qian, A. M. Al-Enizi, L. Zhang and G. Zheng, ACS Appl. Energy Mater., 2021, 4, 1034–1044 CrossRef CAS.
- C. Zhang, Q. Qi, Y. Mei, J. Hu, M. Sun, Y. Zhang, B. Huang, L. Zhang and S. Yang, Adv. Mater., 2023, 35, e2208904 CrossRef.
- K. Dang, H. Dong, L. Wang, M. Jiang, S. Jiang, W. Sun, D. Wang and Y. Tian, Adv. Mater., 2022, 34, e2200302 CrossRef PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- W. Chen, S. Luo, M. Sun, X. Wu, Y. Zhou, Y. Liao, M. Tang, X. Fan, B. Huang and Z. Quan, Adv. Mater., 2022, 34, e2206276 CrossRef PubMed.
- Y. Huang, S. L. Zhang, X. F. Lu, Z. P. Wu, D. Luan and X. W. D. Lou, Angew. Chem., Int. Ed., 2021, 60, 11841–11846 CrossRef CAS PubMed.
- B. F. Rivadeneira-Mendoza, O. A. Estrela Filho, K. J. Fernandez-Andrade, F. Curbelo, F. Fred da Silva, R. Luque and J. M. Rodriguez-Diaz, Environ. Res., 2023, 216, 114424 CrossRef CAS PubMed.
- Q. Zhang and J. Guan, Adv. Funct. Mater., 2020, 30, 2000768 CrossRef CAS.
- L. Zhang, T. U. Rao, J. Wang, D. Ren, S. Sirisommboonchai, C. Choi, H. Machida, Z. Huo and K. Norinaga, Fuel Process. Technol., 2022, 226, 107097 CrossRef CAS.
- Y. P. Wijaya, K. J. Smith, C. S. Kim and E. L. Gyenge, Green Chem., 2020, 22, 7233–7264 RSC.
- F. W. S. Lucas, R. G. Grim, S. A. Tacey, C. A. Downes, J. Hasse, A. M. Roman, C. A. Farberow, J. A. Schaidle and A. Holewinski, ACS Energy Lett., 2021, 6, 1205–1270 CrossRef CAS.
- S. A. Akhade, N. Singh, O. Y. Gutierrez, J. Lopez-Ruiz, H. Wang, J. D. Holladay, Y. Liu, A. Karkamkar, R. S. Weber, A. B. Padmaperuma, M. S. Lee, G. A. Whyatt, M. Elliott, J. E. Holladay, J. L. Male, J. A. Lercher, R. Rousseau and V. A. Glezakou, Chem. Rev., 2020, 120, 11370–11419 CrossRef CAS.
- M. Rehbein, M. Guschakowski, W. Sauter, J. Kunz, U. Schröder and S. Scholl, Front. Energy Res., 2020, 8, 565570 CrossRef.
- M. Gottardo, M. Easton, V. Fabos, S. X. Guo, J. Zhang, A. Perosa, M. Selva, A. M. Bond, A. F. Masters and T. Maschmeyer, ChemSusChem, 2015, 8, 3712–3717 CrossRef CAS PubMed.
- S. K. Green, G. A. Tompsett, H. J. Kim, W. B. Kim and G. W. Huber, ChemSusChem, 2012, 5, 2410–2420 CrossRef CAS.
- S. Mohle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef PubMed.
- Y. Wang, Y.-Q. Zhu, Z. Xie, S.-M. Xu, M. Xu, Z. Li, L. Ma, R. Ge, H. Zhou, Z. Li, X. Kong, L. Zheng, J. Zhou and H. Duan, ACS Catal., 2022, 12, 12432–12443 CrossRef CAS.
- Y. Lu, C. L. Dong, Y. C. Huang, Y. Zou, Z. Liu, Y. Liu, Y. Li, N. He, J. Shi and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 19215–19221 CrossRef CAS PubMed.
- R. Ge, Y. Wang, Z. Li, M. Xu, S. M. Xu, H. Zhou, K. Ji, F. Chen, J. Zhou and H. Duan, Angew. Chem., Int. Ed., 2022, 61, e202200211 CrossRef CAS.
- S. Sun, Y. Sun, Y. Zhou, S. Xi, X. Ren, B. Huang, H. Liao, L. P. Wang, Y. Du and Z. J. Xu, Angew. Chem., Int. Ed., 2019, 58, 6042–6047 CrossRef CAS.
- L. Fan, Y. Ji, G. Wang, J. Chen, K. Chen, X. Liu and Z. Wen, J. Am. Chem. Soc., 2022, 144, 7224–7235 CrossRef CAS.
- H. Zhou, Z. Li, L. Ma and H. Duan, Chem. Commun., 2022, 58, 897–907 RSC.
- T. M. D. Alharbi, S. Elmas, A. S. Alotabi, M. R. Andersson and C. L. Raston, ACS Sustainable Chem. Eng., 2022, 10, 9325–9333 CrossRef CAS.
- X. Wang, Y. Jiao, L. Li, Y. Zheng and S. Z. Qiao, Angew. Chem., Int. Ed., 2022, 61, e202114253 CrossRef CAS PubMed.
- S. Nogami, N. Shida, S. Iguchi, K. Nagasawa, H. Inoue, I. Yamanaka, S. Mitsushima and M. Atobe, ACS Catal., 2022, 12, 5430–5440 CrossRef CAS.
- X. H. Chadderdon, D. J. Chadderdon, J. E. Matthiesen, Y. Qiu, J. M. Carraher, J. P. Tessonnier and W. Li, J. Am. Chem. Soc., 2017, 139, 14120–14128 CrossRef CAS PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- Y. Yan, H. Zhou, S. M. Xu, J. Yang, P. Hao, X. Cai, Y. Ren, M. Xu, X. Kong, M. Shao, Z. Li and H. Duan, J. Am. Chem. Soc., 2023, 145, 6144–6155 CrossRef CAS PubMed.
- P. Nilges, T. R. dos Santos, F. Harnisch and U. Schröder, Energy Environ. Sci., 2012, 5, 5231–5235 RSC.
- Y. Liu, C. Gu, L. Chen, W. Zhou, Y. Liao, C. Wang and L. Ma, ACS Appl. Mater. Interfaces, 2023, 15, 4184–4193 CrossRef CAS.
- R. Bacchiocchi, J. De Maron, T. Tabanelli, D. Bianchi and F. Cavani, Sustainable Energy Fuels, 2023, 7, 671–681 RSC.
- S. Wang, H. Huang, C. Bruneau and C. Fischmeister, Catal. Sci. Technol., 2019, 9, 4077–4082 RSC.
- L. Xin, Z. Zhang, J. Qi, D. J. Chadderdon, Y. Qiu, K. M. Warsko and W. Li, ChemSusChem, 2013, 6, 674–686 CrossRef CAS.
- Y. Qiu, L. Xin, D. J. Chadderdon, J. Qi, C. Liang and W. Li, Green Chem., 2014, 16, 1305–1315 RSC.
- R. J. M. Bisselink, M. Crockatt, M. Zijlstra, I. J. Bakker, E. Goetheer, T. M. Slaghek and D. S. van Es, ChemElectroChem, 2019, 6, 3285–3290 CrossRef CAS.
- Y. Zhang, X. Wang and Y. Shen, Fuel, 2023, 342, 127787 CrossRef CAS.
- T. Yuan, M. Chu, K. Zhang, S. Jia, S. Han, J. Zhai, H. Wang, T. Xue and H. Wu, ChemistrySelect, 2022, 7, e202201624 CrossRef CAS.
- J. Liu and S. Tao, Appl. Surf. Sci., 2023, 616, 156464 CrossRef CAS.
- S. Gyergyek, M. Grilc, B. Likozar and D. Makovec, Green Chem., 2022, 24, 2788–2794 RSC.
- T. R. dos Santos, P. Nilges, W. Sauter, F. Harnisch and U. Schröder, RSC Adv., 2015, 5, 26634–26643 RSC.
- H. Wu, J. Song, C. Xie, Y. Hu, P. Zhang, G. Yang and B. Han, Chem. Sci., 2019, 10, 1754–1759 RSC.
- F. W. S. Lucas, Y. Fishler and A. Holewinski, Green Chem., 2021, 23, 9154–9164 RSC.
- S. D. Mürtz, N. Kurig, F. J. Holzhäuser and R. Palkovits, Green Chem., 2021, 23, 8428–8433 RSC.
- Y. Kwon, E. de Jong, S. Raoufmoghaddam and M. T. Koper, ChemSusChem, 2013, 6, 1659–1667 CrossRef CAS PubMed.
- M. T. Bender, X. Yuan, M. K. Goetz and K.-S. Choi, ACS Catal., 2022, 12, 12349–12368 CrossRef CAS.
- V. G. Chandrashekhar, K. Natte, A. M. Alenad, A. S. Alshammari, C. Kreyenschulte and R. V. Jagadeesh, ChemCatChem, 2021, 14, e202101234 CrossRef.
- S. Li, M. Dong, J. Yang, X. Cheng, X. Shen, S. Liu, Z. Q. Wang, X. Q. Gong, H. Liu and B. Han, Nat. Commun., 2021, 12, 584 CrossRef CAS.
- J. J. Roylance, T. W. Kim and K.-S. Choi, ACS Catal., 2016, 6, 1840–1847 CrossRef CAS.
- X. H. Chadderdon, D. J. Chadderdon, T. Pfennig, B. H. Shanks and W. Li, Green Chem., 2019, 21, 6210–6219 RSC.
- Z. Zhao, X. Luo, J. Peng, S. Wang, T. Guo and H. Zheng, Sustainable Energy Fuels, 2022, 6, 5281–5289 RSC.
- Y. Zhong, R. Ren, Y. Peng, J. Wang, X. Ren, Q. Li and Y. Fan, Mol. Catal., 2022, 528, 112487 CrossRef CAS.
- G. Sanghez de Luna, P. H. Ho, A. Sacco, S. Hernandez, J. J. Velasco-Velez, F. Ospitali, A. Paglianti, S. Albonetti, G. Fornasari and P. Benito, ACS Appl. Mater. Interfaces, 2021, 13, 23675–23688 CrossRef CAS.
- K. Ji, M. Xu, S. M. Xu, Y. Wang, R. Ge, X. Hu, X. Sun and H. Duan, Angew. Chem., Int. Ed., 2022, 61, e202209849 CrossRef CAS PubMed.
- H. Li, T. Zhang, M. Peng, Q. Zhang, J. Liu, J. Zhang, Y. Fu and W. Li, Int. J. Hydrogen Energy, 2022, 47, 28904–28914 CrossRef CAS.
- G. S. de Luna, A. Sacco, S. Hernandez, F. Ospitali, S. Albonetti, G. Fornasari and P. Benito, ChemSusChem, 2022, 15, e202102504 CrossRef CAS PubMed.
- S. Li, X. Sun, Z. Yao, X. Zhong, Y. Cao, Y. Liang, Z. Wei, S. Deng, G. Zhuang, X. Li and J. Wang, Adv. Funct. Mater., 2019, 29, 1904780 CrossRef CAS.
- R. Kloth, D. V. Vasilyev, K. J. J. Mayrhofer and I. Katsounaros, ChemSusChem, 2021, 14, 5245–5253 CrossRef CAS PubMed.
- P. Nilges and U. Schröder, Energy Environ. Sci., 2013, 6, 2925–2931 RSC.
- Y.-R. Zhang, B.-X. Wang, L. Qin, Q. Li and Y.-M. Fan, Green Chem., 2019, 21, 1108–1113 RSC.
- J. J. Roylance and K.-S. Choi, Green Chem., 2016, 18, 5412–5417 RSC.
- M. Zhang, S. Xu, M. Boubeche, D. Decarolis, Y. Huang, B. Liu, E. K. Gibson, X. Li, Y. Wang, H. Luo, C. R. A. Catlow and K. Yan, Green Chem., 2022, 24, 9570–9578 RSC.
- J. J. Roylance and K.-S. Choi, Green Chem., 2016, 18, 2956–2960 RSC.
- S. Panigrahy, R. Mishra, P. Panda, M. Kempasiddaiah and S. Barman, ACS Appl. Nano Mater., 2022, 5, 8314–8323 CrossRef CAS.
- Y. Kwon, Y. Y. Birdja, S. Raoufmoghaddam and M. T. Koper, ChemSusChem, 2015, 8, 1745–1751 CrossRef CAS PubMed.
- D. K. Lee, S. R. Kubota, A. N. Janes, M. T. Bender, J. Woo, J. R. Schmidt and K. S. Choi, ChemSusChem, 2021, 14, 4563–4572 CrossRef CAS PubMed.
- X. Yuan, K. Lee, M. T. Bender, J. R. Schmidt and K. S. Choi, ChemSusChem, 2022, 15, e202200952 CrossRef CAS PubMed.
- K. Wang, M. Wu, Y. Liu, Y. Yang and H. Li, New J. Chem., 2022, 46, 5312–5320 RSC.
- M.-Y. Chen, C.-B. Chen, B. Zada and Y. Fu, Green Chem., 2016, 18, 3858–3866 RSC.
- B. Munirathinam, L. Lerch, D. Hüne, L. Lentz, T. Lenk, M. Görke, G. Garnweitner, N. Schlüter, F. Kubannek, D. Schröder and T. Gimpel, ChemElectroChem, 2022, 9, e202200885 CrossRef CAS.
- S. Jung and E. J. Biddinger, ACS Sustainable Chem. Eng., 2016, 4, 6500–6508 CrossRef CAS.
- S. Jung, A. N. Karaiskakis and E. J. Biddinger, Catal. Today, 2019, 323, 26–34 CrossRef CAS.
- A. S. May and E. J. Biddinger, ACS Catal., 2020, 10, 3212–3221 CrossRef CAS.
- Y. Cao and T. Noel, Org. Process Res. Dev., 2019, 23, 403–408 CrossRef CAS PubMed.
- L. Liu, H. Liu, W. Huang, Y. He, W. Zhang, C. Wang and H. Lin, J. Electroanal. Chem., 2017, 804, 248–253 CrossRef CAS.
- Z. Yang, X. Chou, H. Kan, Z. Xiao and Y. Ding, ACS Sustainable Chem. Eng., 2022, 10, 7418–7425 CrossRef CAS.
- R. J. Dixit, K. Bhattacharyya, V. K. Ramani and S. Basu, Green Chem., 2021, 23, 4201–4212 RSC.
- X. Zhang, M. Han, G. Liu, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Appl. Catal., B, 2019, 244, 899–908 CrossRef CAS.
- W. Xu, C. Yu, J. Chen and Z. Liu, Appl. Catal., B, 2022, 305, 121062 CrossRef CAS.
- J. T. Brosnahan, Z. Zhang, Z. Yin and S. Zhang, Nanoscale, 2021, 13, 2312–2316 RSC.
- S. Huang, Y. Jin, M. Zhang, K. Yan, S.-P. Feng and J. C.-H. Lam, Green Chem., 2022, 24, 7974–7987 RSC.
- S. Huang, B. Gong, Y. Jin, P. H. L. Sit and J. C.-H. Lam, ACS Catal., 2022, 12, 11340–11354 CrossRef CAS.
- X. Shang, Y. Yang and Y. Sun, Green Chem., 2020, 22, 5395–5401 RSC.
- S. Jung and E. J. Biddinger, Energy Technol., 2018, 6, 1370–1379 CrossRef CAS.
- G. Bharath and F. Banat, ACS Appl. Mater. Interfaces, 2021, 13, 24643–24653 CrossRef CAS PubMed.
- P. Zhou, Y. Chen, P. Luan, X. Zhang, Z. Yuan, S.-X. Guo, Q. Gu, B. Johannessen, M. Mollah, A. L. Chaffee, D. R. Turner and J. Zhang, Green Chem., 2021, 23, 3028–3038 RSC.
- T. Lenk, V. Rueß, J. Gresch and U. Schröder, Green Chem., 2023, 25, 3077–3085 RSC.
- R. S. Delima, M. D. Stankovic, B. P. MacLeod, A. G. Fink, M. B. Rooney, A. Huang, R. P. Jansonius, D. J. Dvorak and C. P. Berlinguette, Energy Environ. Sci., 2022, 15, 215–224 RSC.
- M. Temnikova, J. Medvedev, X. Medvedeva, N. H. Delva, E. Khairullina, E. Krivoshapkina and A. Klinkova, ChemElectroChem, 2022, 10, e202200865 CrossRef.
- K. Yan, M. L. Huddleston, B. A. Gerdes and Y. Sun, Green Chem., 2022, 24, 5320–5325 RSC.
- M. Jiang, J. Tan, Y. Chen, W. Zhang, P. Chen, Y. Tang and Q. Gao, Chem. Commun., 2023, 59, 3103–3106 RSC.
- Y. P. Wijaya, K. J. Smith, C. S. Kim and E. L. Gyenge, J. Appl. Electrochem., 2020, 51, 51–63 CrossRef.
- N. Singh, Y. Song, O. Y. Gutiérrez, D. M. Camaioni, C. T. Campbell and J. A. Lercher, ACS Catal., 2016, 6, 7466–7470 CrossRef CAS.
- B. Zhao, Q. Guo and Y. Fu, Electrochemistry, 2014, 82, 954–959 CrossRef CAS.
- K. Amouzegar and O. Savadogo, Electrochim. Acta, 1994, 39, 557–559 CrossRef CAS.
- A. Martel, B. Mahdavi, J. Lessard, H. Ménard and L. Brossard, Can. J. Chem., 1997, 75, 1862–1867 CrossRef CAS.
- K. Amouzegar and O. Savadogo, Electrochim. Acta, 1998, 43, 503–508 CrossRef CAS.
- N. Singh, U. Sanyal, G. Ruehl, K. A. Stoerzinger, O. Y. Gutiérrez, D. M. Camaioni, J. L. Fulton, J. A. Lercher and C. T. Campbell, J. Catal., 2020, 382, 372–384 CrossRef CAS.
- X. Lu, J. Wang, W. Peng, N. Li, L. Liang, Z. Cheng, B. Yan, G. Yang and G. Chen, Fuel, 2023, 331, 125845 CrossRef CAS.
- Y. Song, O. Y. Gutiérrez, J. Herranz and J. A. Lercher, Appl. Catal., B, 2016, 182, 236–246 CrossRef CAS.
- W. Liu, W. You, Y. Gong and Y. Deng, Energy Environ. Sci., 2020, 13, 917–927 RSC.
- L. Zhou, X. Zhu, H. Su, H. Lin, Y. Lyu, X. Zhao, C. Chen, N. Zhang, C. Xie, Y. Li, Y. Lu, J. Zheng, B. Johannessen, S. P. Jiang, Q. Liu, Y. Li, Y. Zou and S. Wang, Sci. China: Chem., 2021, 64, 1586–1595 CrossRef CAS.
- Y. Du, X. Chen and C. Liang, Mol. Catal., 2023, 535, 112831 CrossRef CAS.
- Z. Gu, Z. Zhang, N. Ni, C. Hu and J. Qu, Environ. Sci. Technol., 2022, 56, 4356–4366 CrossRef CAS PubMed.
- M. Garedew, D. Young-Farhat, J. E. Jackson and C. M. Saffron, ACS Sustainable Chem. Eng., 2019, 7, 8375–8386 CrossRef CAS.
- Y. Zhou, Y. Gao, X. Zhong, W. Jiang, Y. Liang, P. Niu, M. Li, G. Zhuang, X. Li and J. Wang, Adv. Funct. Mater., 2019, 29, 1807651 CrossRef.
- C. H. Lam, C. B. Lowe, Z. Li, K. N. Longe, J. T. Rayburn, M. A. Caldwell, C. E. Houdek, J. B. Maguire, C. M. Saffron, D. J. Miller and J. E. Jackson, Green Chem., 2015, 17, 601–609 RSC.
- Y. P. Wijaya, R. D. D. Putra, K. J. Smith, C. S. Kim and E. L. Gyenge, ACS Sustainable Chem. Eng., 2021, 9, 13164–13175 CrossRef CAS.
- T. Peng, T. Zhuang, Y. Yan, J. Qian, G. R. Dick, J. Behaghel de Bueren, S. F. Hung, Y. Zhang, Z. Wang, J. Wicks, F. P. Garcia de Arquer, J. Abed, N. Wang, A. S. Rasouli, G. Lee, M. Wang, D. He, Z. Wang, Z. Liang, L. Song, X. Wang, B. Chen, A. Ozden, Y. Lum, W. R. Leow, M. Luo, D. M. Meira, A. H. Ip, J. S. Luterbacher, W. Zhao and E. H. Sargent, J. Am. Chem. Soc., 2021, 143, 17226–17235 CrossRef CAS PubMed.
- M. Wang, T. Peng, C. Yang, B. Liang, H. Chen, M. Kumar, Y. Zhang and W. Zhao, Green Chem., 2022, 24, 142–146 RSC.
- J. Chen, Y. Qi, X. Pan, N. Wu, J. Zuo, C. Li, R. Qu, Z. Wang and Z. Chen, Water Res., 2019, 158, 338–349 CrossRef CAS PubMed.
- C. Fan, A. Lu, Y. Li and C. Wang, Chem. Eng. J., 2010, 160, 20–26 CrossRef CAS.
- C. Guo, Q. Cao, B. Chen, S. Yang and Y. Qian, J. Cleaner Prod., 2019, 211, 380–386 CrossRef CAS.
- T. Felicetti, R. Cannalire, D. Pietrella, G. Latacz, A. Lubelska, G. Manfroni, M. L. Barreca, S. Massari, O. Tabarrini, K. Kieć-Kononowicz, B. D. Schindler, G. W. Kaatz, V. Cecchetti and S. Sabatini, J. Med. Chem., 2018, 61, 7827–7848 CrossRef CAS PubMed.
- G. Valdameri, C. Gauthier, R. Terreux, R. Kachadourian, B. J. Day, S. M. B. Winnischofer, M. E. M. Rocha, V. Frachet, X. Ronot, A. Di Pietro and A. Boumendjel, J. Med. Chem., 2012, 55, 3193–3200 CrossRef CAS PubMed.
- D. C. Cantu, A. B. Padmaperuma, M.-T. Nguyen, S. A. Akhade, Y. Yoon, Y.-G. Wang, M.-S. Lee, V.-A. Glezakou, R. Rousseau and M. A. Lilga, ACS Catal., 2018, 8, 7645–7658 CrossRef CAS.
- S. Han, X. Zhang, R. Wang, K. Wang, J. Jiang and J. Xu, Chem. Eng. J., 2023, 452, 139299 CrossRef CAS.
- Q. Zhai, S. Han, C.-Y. Hse, J. Jiang and J. Xu, Fuel Process. Technol., 2022, 227, 107109 CrossRef CAS.
- N. Sarki, R. Kumar, B. Singh, A. Ray, G. Naik, K. Natte and A. Narani, ACS Omega, 2022, 7, 19804–19815 CrossRef CAS PubMed.
- U. Sanyal, K. Koh, L. C. Meyer, A. Karkamkar and O. Y. Gutiérrez, J. Appl. Electrochem., 2020, 51, 27–36 CrossRef.
- Y. Song, U. Sanyal, D. Pangotra, J. D. Holladay, D. M. Camaioni, O. Y. Gutiérrez and J. A. Lercher, J. Catal., 2018, 359, 68–75 CrossRef CAS.
- J. A. Lopez-Ruiz, U. Sanyal, J. Egbert, O. Y. Gutiérrez and J. Holladay, ACS Sustainable Chem. Eng., 2018, 6, 16073–16085 CrossRef CAS.
- K. Koh, U. Sanyal, M. S. Lee, G. Cheng, M. Song, V. A. Glezakou, Y. Liu, D. Li, R. Rousseau, O. Y. Gutierrez, A. Karkamkar, M. Derewinski and J. A. Lercher, Angew. Chem., Int. Ed., 2020, 59, 1501–1505 CrossRef CAS PubMed.
- Y. Wu, Z. Guo, C. Sun, X. Ren and Q. Li, Fuel Process. Technol., 2022, 237, 107436 CrossRef CAS.
- J. Anibal, A. Malkani and B. Xu, Catal. Sci. Technol., 2020, 10, 3181–3194 RSC.
- J. Yu, P. Zhang, L. Li, K. Li, G. Zhang, J. Liu, T. Wang, Z. J. Zhao and J. Gong, Nat. Commun., 2022, 13, 7909 CrossRef CAS PubMed.
- J. Anibal and B. Xu, ACS Catal., 2020, 10, 11643–11653 CrossRef CAS.
- L. C. Meyer, U. Sanyal, K. A. Stoerzinger, K. Koh, J. L. Fulton, D. M. Camaioni, O. Y. Gutiérrez and J. A. Lercher, ACS Catal., 2022, 12, 11910–11917 CrossRef CAS.
- C. M. Mulero, A. Saez, J. Iniesta and V. Montiel, Beilstein J. Org. Chem., 2018, 14, 537–546 CrossRef CAS PubMed.
- U. Sanyal, J. Lopez-Ruiz, A. B. Padmaperuma, J. Holladay and O. Y. Gutiérrez, Org. Process Res. Dev., 2018, 22, 1590–1598 CrossRef CAS.
- M. Villalba, M. L. Bossi and E. J. Calvo, Chem. Chem. Phys., 2015, 17, 10086–10092 RSC.
- S. Calado, G. de Melo, M. E. P. da Silva, M. E. B. da Silva, J. A. da Paz, C. M. B. de Menezes Barbosa, F. D. de Menezes, R. N. A. Loureiro, M. Navarro, J. Â. P. da Costa, G. F. da Silva, A. A. O. Villa and M. Vilar, Int. J. Hydrogen Energy, 2020, 45, 22855–22872 CrossRef.
- H. Zhang, X. Gao, Y. Ma, X. Han, L. Niu and G. Bai, Catal. Sci. Technol., 2017, 7, 5993–5999 RSC.
- X. H. Lu, Y. Shen, J. He, R. Jing, P. P. Tao, A. Hu, R. F. Nie, D. Zhou and Q. H. Xia, Mol. Catal., 2018, 444, 53–61 CrossRef CAS.
- A. Fukazawa, Y. Shimizu, N. Shida and M. Atobe, Org. Biomol. Chem., 2021, 19, 7363–7368 RSC.
- A. Kong, M. Liu, H. Zhang, Z. Cao, J. Zhang, W. Li, Y. Han and Y. Fu, Chem. Eng. J., 2022, 445, 13671 Search PubMed.
| This journal is © The Royal Society of Chemistry 2024 |