
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn electron-donor–acceptor complex between two intermediates enables a N–N bond cleavage cascade process to access 2,3-difunctionalized pyridines†
Ya-Zhou
Liu‡
a,
Yu
Chen‡
b,
Amu
Wang
ac,
Zhongke
Shen
a,
Xueting
Zhou
d,
Jichao
Zhang
a,
Yinxiang
Jian
ac and
Xiaofeng
Ma
 *a
*a
aNatural Products Research Center, Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu, 610041, China. E-mail: maxf@cib.ac.cn
bDepartment of Chemistry and Shenzhen Grubbs Institute, Southern University of Science and Technology, Shenzhen, 518000, China
cUniversity of Chinese Academy of Sciences, Beijing, 100049, China
dSchool of Basic Medicine/State Key Laboratory of Functions and Applications of Medicinal Plants, Key Laboratory of Chemistry for Natural Products of Guizhou Province and Chinese Academy of Sciences, Guiyang, 550014, China
First published on 11th January 2024
Abstract
Chemical transformation triggered by an electron donor–acceptor (EDA) complex without the addition of an exogenous stoichiometric electron donor/acceptor is rare. Herein, we report such a process to access 2,3-difunctionalized pyridines from readily available N-aminopyridiniums (1) and activated alkenes (2) promoted by visible light. This procedure offered multi-substituted pyridines in satisfactory yields at room temperature with broad functional group tolerance. The reaction can be easily performed on a gram scale without the loss of yield. The modification of bioactive molecules including derivatives of clinical drugs and natural products was demonstrated. Mechanistic studies and DFT calculations indicated that the formal [3 + 2] cycloaddition and aza-Michael addition between 1 and 2 generated tetrahydropyrazolo[1,5-a]pyridine and a new pyridinium salt, respectively. These two intermediates formed an EDA complex, which under visible-light illumination, triggered the following single electron transfer (SET)/N–N bond cleavage/C–N bond formation cascade process with high atom economy.
Introduction
Due to exogenous photoredox catalyst-free and environmentally friendly properties, electron donor–acceptor (EDA) complex-triggered bond formation reactions have achieved huge advances in recent years, especially in the construction of challengeable C–C and C–X (N, O, S, P, etc.) bonds.1,2 Among them, N-functionalized pyridinium salts,3 including Katritzky salt,4N-amidopyridiniums (usually using N-sulfonamido-pyridiniums)5 and N-alkoxy pyridiniums,6 accomplished with their corresponding electron donors, have been extensively used as alkyl, amidyl and alkoxy radical precursors to trigger C–C, C–B or C–S bond formation and regioselective C–H functionalization of pyridines (Schemes 1A and B). However, the addition of an exogenous stoichiometric electron donor and sacrifice of additional groups are still inevitable in those series of reactions, which leaves huge room for the improvement of atom economy. Recently, Su and co-workers reported a unique example where the in situ-generated tetrahydroindolizine intermediate,7 derived from the cycloaddition reaction between N-alkylpyridinium salts and α,β-unsaturated compounds, could be used as the electron donor of the EDA complex. Upon irradiating with a 24 W LED, the tetrahydroindolizine intermediate could undergo an intramolecular SET process from the dienamine group (D part)8 to the benzoyl group (A part), leading to the formation of C2-alkylpyridines with high atom economy (Scheme 1C).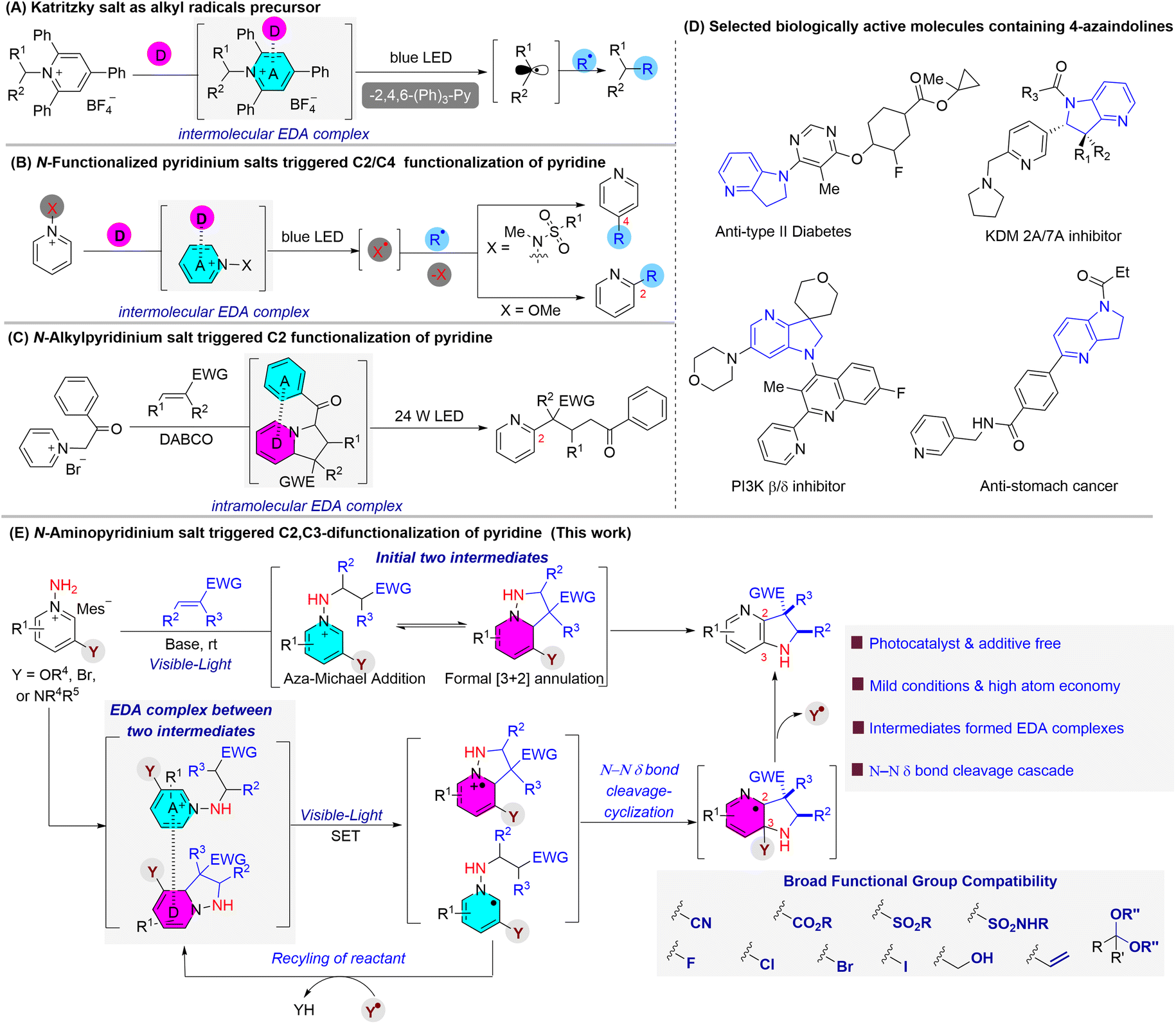 | ||
| Scheme 1 Selected examples of regioselective transformation of N-functionalized pyridines and biologically active molecules containing 4-azaindolines. | ||
Our research in this area is due to the continuing efforts to exploit mild and efficient protocols for the diversity-oriented transformation of aromatic heterocycles.9 Recently, our group revealed the regioselective formation of pyrazolo[1,5-a]pyridines from N-aminopyridinium salts.9c Two key features were observed in this protocol: (i) the formal [3 + 2] cycloadditions between N-aminopyridinium salts and α,β-unsaturated compounds are reversible, resulting in the cyclized dienamine intermediates (tetrahydro-pyrazolo[1,5-a]pyridines) detectable by NMR and HRMS; (ii) exclusive regioselectivity was observed when N-aminopyridiniums were substituted at the 3- or 5-position with a strong electron-donating substituent (such as alkoxy or amino groups). These observations plus the successful participation of enamines in EDA complexes fascinated us to explore the further possibility of using tetrahydropyrazolo-[1,5-a]pyridine intermediates to realize the multi-functionalization of pyridines through an EDA complex-triggered process. Specifically, we hypothesize that, on behalf of the electron-rich properties, this kind of intermediate could potentially be activated by a visible-light-induced SET process in the presence of an electron-deficient partner, such as the N-pyridinium salt. Meanwhile, pre-installation of a readily available electron-donating group may facilitate the cleavage of the N–N bond by enol–keto tautomerization, which would be removed in the subsequent aromatization process. Herein, we report a regioselective [3 + 2] cycloaddition-initiated N–N bond cleavage/N-migration/intramolecular cyclization cascade process, between N-aminopyridiniums and α,β-unsaturated compounds, to afford medicinally important 4-azaindolines (Scheme 1D).10 We found that the initial formal [3 + 2] cycloaddition and the aza-Michael addition reaction produced two intermediates, and these intermediates formed an unprecedented EDA complex which triggered the subsequent process without the need for any additional electron acceptors or photocatalysts, and the reactions could proceed smoothly in the presence of various visible lights including sunlight, white LEDs and fluorescent lamps with similar efficiency. Remarkably, the electron-donating group not only directs regioselectivity and facilitates the cleavage of N–N bonds, but also plays a critical role in reactant recycling, preventing the sacrifice of electron acceptors (Scheme 1E). Most notably, the reaction could be easily performed on a gram scale and is compatible with an array of functional groups including halogen, cyano, ester, free alcohol, alkene, sulphone, and sulfamide. With this simple and easy handling protocol, a wide range of multi-functionalized 4-azaindolines were obtained efficiently from readily available chemical feedstocks under mild conditions and with high atom economy.
Results and discussion
Development and optimization of the reaction
Our initial attempt started with the reaction between 3-bromo-5-methoxy-N-aminopyridinium salt (1a) and acrylonitrile (2a). To our surprise, the reaction directly gave 3a in 93% yield just under the fluorescent lamp (36 W) of a laboratory fume hood, neither adding an exogenous electron-deficient substrate nor using special photochemical reaction apparatus including LED lights. A notable phenomenon of the reaction is that when a base was added to a mixture of pyridinium salt and acrylonitrile in toluene, the heterogeneous reactants simultaneously dissolved and the solution appeared to a marked yellow color, which may be due to the formation of an EDA complex.11 To prove this, we measured the UV-Vis absorption spectra of the separated reaction constituents 1a, 2a, 3a, DBU, and their mixtures, respectively (Fig. 1, see also Fig. S8 and S9† for more details). A charge-transfer (CT) band12 was observed in the visible-light region under standard conditions (orange line), while no CT band was detected in the absence of any of the reaction components. It is worth noting that the mixture of DBU and pyridinium salt formed a black suspension in toluene which is not suitable for UV-Vis absorption (see Fig. S10†). These observations suggest the existence of EDA complexes during the process of reaction.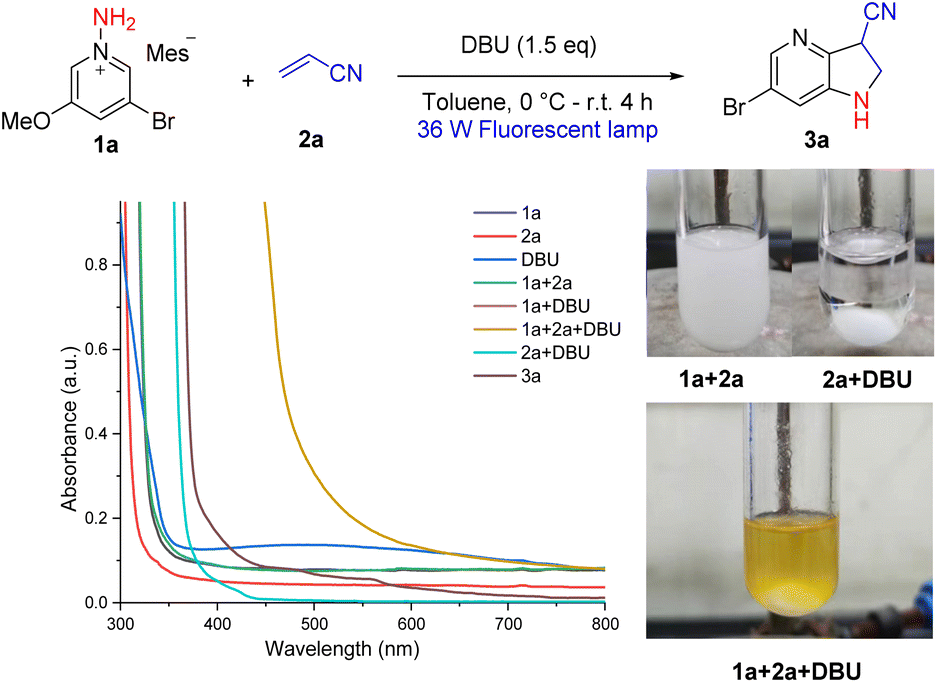 | ||
| Fig. 1 UV-Vis absorption spectra of reaction components and the picture of their reactions. Experimental conditions: 1a (0.10 mmol), 2a (0.12 mmol), and DBU (0.15 mmol) in toluene (2.0 mL). | ||
With the initial results in mind, we further explored the reaction conditions (Table 1). We found that increasing the equivalents of acrylonitrile did not reflect obvious changes in the yield (entry 2). However, changing the amount of the base reduced the yield (entry 3). Screening of different bases and solvents indicated that DBU and toluene are the most suitable for this transformation (entries 4–7). Additionally, other electron-donating groups, including ethoxy (entry 8), benzyloxy (entry 9) and dimethylamino groups (entry 10), were also investigated, which gave 3a in 88%, 91% and 82% yields, respectively. Interestingly, control reactions show that the reaction could also smoothly deliver product 3a in 89% yield under white LED irradiation (entry 11), and 87% yield under sunlight (entry 12). Meanwhile, the same reaction under the irradiation of a blue LED also led to the formation of product 3a in up to 75% yield (entry 13), while the yield of 3a was dramatically decreased (<5%) when the reaction was performed in the dark under otherwise identical conditions (entry 14).
|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yieldb |
| a Reaction conditions: 1 (0.10 mmol), 2 (0.12 mmol), and base (0.15 mmol) in solvent (2.0 mL) at 0 °C – rt for 4 h. b Isolated yield. | ||
| 1 | — | 93% |
| 2 | 1.5 eq. of 2a | 91% |
| 3 | 1.0 eq. of DBU | 62% |
| 4 | Cs2CO3 instead of DBU | 82% |
| 5 | DIPEA instead of DBU | 85% |
| 6 | DCM instead of toluene | 41% |
| 7 | 1,4-Dioxane instead of toluene | 79% |
| 8 | 1b instead of 1a | 88% |
| 9 | 1c instead of 1a | 91% |
| 10 | 1d instead of 1a | 82% |
| 11 | White LED | 89% |
| 12 | Sunlight | 87% |
| 13 | Blue LED | 75% |
| 14 | In the dark | <5% |
Evaluation of the substrate scope
With the optimal reaction conditions in hand, we then evaluated the substrate scope of this protocol (Scheme 2). 2-Benzylacrylonitrile and 4-(2-cyanoallyl) benzonitrile reacted smoothly and the N-tosyl-protected products 3b and 3c were isolated in 73% and 78% yield respectively over two steps, due to the stability of the free amine product during purification. Notably, a nitrile derivative from a Morita–Baylis–Hillman reaction was also successfully applied in this protocol and 3d was generated in 65% yield with excellent diastereoselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr). Next, the scope was extended to vinyl-sulfone derivatives under the standard conditions. Phenyl vinyl sulfones, with or without substituent groups, were successfully employed in the protocol and gave the corresponding N-tosyl-protected products (4a–4c) in satisfactory yields (69%–75%), whose structure was unambiguously confirmed by X-ray analysis of 4b. To our delight, this strategy also exhibited good tolerance for sulfanylamides, which are found in various on-market medicines, three N-tosyl protected products (4d–4f) were obtained in moderate to good yields (59%–81% over two steps).
:1 dr). Next, the scope was extended to vinyl-sulfone derivatives under the standard conditions. Phenyl vinyl sulfones, with or without substituent groups, were successfully employed in the protocol and gave the corresponding N-tosyl-protected products (4a–4c) in satisfactory yields (69%–75%), whose structure was unambiguously confirmed by X-ray analysis of 4b. To our delight, this strategy also exhibited good tolerance for sulfanylamides, which are found in various on-market medicines, three N-tosyl protected products (4d–4f) were obtained in moderate to good yields (59%–81% over two steps).
 | ||
| Scheme 2 Substrate scopes. aStandard conditions: 0.1 mmol of 1, 0.12 mmol of 2, and 0.15 mmol of DBU in 2 mL of toluene, 0 °C – rt, under the irradiation of a 36 W fluorescent lamp, 4 h; isolated yields are reported; bat 50 °C, in 2 mL of 1,4-dioxane, 12 h; isolated yields are reported. | ||
The reaction scope was further examined by employing a wide range of α,β-unsaturated esters. n-Hexyl methacrylate worked well under the reaction conditions and gave the product 5a in 81% yield. Additionally, α,β-unsaturated esters bearing benzyl groups regardless of their electronic properties were also successfully applied in this protocol and produced the corresponding products (5b–5h) in 72%–85% yields. A methyl methacrylate derivative of citric acid delivered the product 5i in high yield after two steps of transformation. Most notably, substituted α-methylene-γ-butyrolactone, a subunit present in a host of important bioactive molecules,13 was also reacted well under slightly modified reaction conditions (at 50 °C in 1,4-dioxane), giving the corresponding N-tosyl-protected product 5j in 40% yield as a single diastereoisomer. Besides, Morita–Baylis–Hillman ester was also successfully applied in this protocol and produced 5k in 52% yield with 5:1 diastereoselectivity.
In addition to the 3-bromo-5-methoxypyridinium salts, the scope of the reaction with other pyridinium salts was also evaluated. As expected, 1-amino-3-methoxypyridinium exhibited good tolerance to a variety of α,β-unsaturated compounds, giving products 6a–6c in satisfactory yields. Additionally, 2,3-disubstituted pyridinium was also suitable for this method and generated 6d in 56% yield. The modified or unmodified aromatic groups at the C5 or C6 positions of 3-methoxypyridine did not show obvious influence on the yield in our protocol and all test examples delivered the products (6e–6l) in satisfactory yields no matter the reaction was with acrylonitrile or methacrylate esters. 3-Methoxy-6-benzyl-N-amino-pyridinium was also reacted well with α-methylene-γ-butyrolactone, and N-tosylated product 6m was achieved in 51% yield over two steps, which further verified the generality of this transformation. Besides, 3-bromo-5-methoxy-1-(methylamino)-pyridinium (1n) was also suitable for this protocol and reacted smoothly with various activated alkenes including α,β-unsaturated nitriles, esters and sulphones, offering the corresponding 1-N-Me-4-azaindolines 6n–6q in satisfactory to good yields.
To further demonstrate the versatility of our methodology, we explored the late-stage modification of bioactive molecules and carried out the reaction on a gram scale. A vinyl-sulfamide derivative from the first-line medicine Amantadine was encompassed into this strategy, which gave product 7a in 61% yield. Also, methacrylate ester derivatives from the natural product diosgenin, galactose and the on-market medicine Abiraterone were subjected to the standard reaction conditions, producing the corresponding products (after N-tosyl protection) 7b–7d in around 45% yield. Additionally, gram-scale reactions were also performed, starting from 2.5 mmol of 3-bromo-5-methoxy-1-aminopyridinium salt and 3.0 mmol of acrylonitrile, and 3a was collected in 89% yield, which would be beneficial for further investigation of biological functions of these compounds.
Notably, in our previous experiments, we have noticed that the regioselective formal [3 + 2] cycloaddition between 3-substituted pyridinium salts and α,β-unsaturated compounds could be switched if a 2,3-di-substituted pyridinium salt was employed.9c At the present stage, we were curious if the cyclization was still possible when the 2-position of pyridine was occupied with a substituent. Therefore, 2-chloro-3-ethoxy-5-bromopyridinium salt (8a) was subjected to the standard reaction to couple with acrylonitrile and methacrylonitrile. As shown in Scheme 3, the reactions went smoothly and offered products 9a and 9b in 62% and 75% isolated yield, respectively. The structure of 9a was confirmed by single crystal X-ray diffraction analysis of the N-tosyl protected derivative (9a-Ts). Additionally, phenyl and n-hexyl methacrylate were also employed in this transformation, delivering the products 9c and 9d in 68% and 78% yield, respectively. Impressively, the alkenyl and hydroxyl groups were also tolerated in this reaction and the corresponding products 9f–9h were isolated in satisfactory yields (49%–65% yields). Moreover, α-methylene-γ-butyrolactone was also reacted well and produced the product (9i) in 51% yield after tosylation. A similar process with β-phenyl-substituted α-methylene-γ-butyrolactone gave the product 9j in 45% yield and higher than 20:1 d.r. Besides 8a, 2-chloro-3-methoxy-5-bromopyridinium salt (8b) and 2-chloro-3-benzyloxy-5-bromopyridinium salt (8c) were also reacted well and gave tosyl-protected products 9k and 9l in 63% and 59% yields, respectively, over two steps.
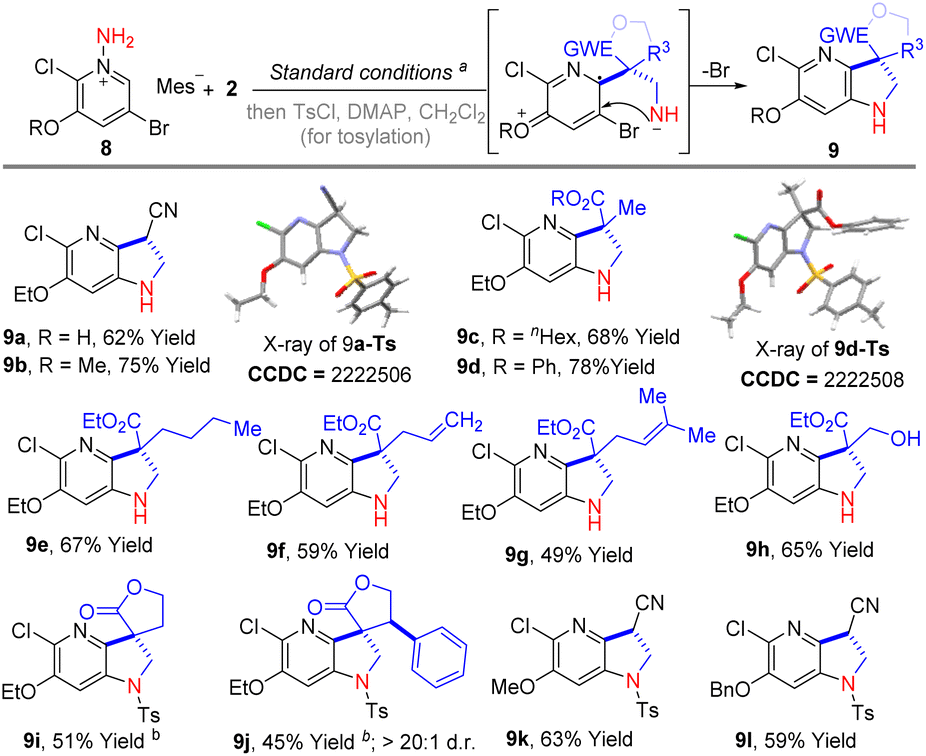 | ||
| Scheme 3 Substrate scope of 2-chloride-3-ethoxyl-5-bromopyridinium salt 8. aStandard conditions: 0.1 mmol of 8, 0.12 mmol of 2, 0.20 mmol of DBU in 2 mL of toluene, 0 °C – rt, under the irradiation of a 36 W fluorescent lamp, 4 h; isolated yields are reported. Temperature: b at 50 °C, in 2 mL of 1,4-dioxane, 12 h; isolated yields are reported. | ||
Mechanistic investigations
N–N bond cleavage of N-amidopyridiniums has been used extensively for C4 selective pyridine C–H functionalizations.2,3,14 In all cases, it has been well established that a strong electron-withdrawing group such as N-sulfonamide or N-benzoyl amide is essential to weaken the N–N bond. In certain cases, unselective C3-functionalized pyridines via 1,3-migration of N-iminopyridiniums were observed as a byproduct.15,16 Therefore, it appears that the electron-donating group such as OR or NR2 is critical to promote the N–N bond cleavage/N-migration cascade process of N-aminopyridiniums in our case. To further understand the insight of the reaction mechanism, a series of control experiments were performed (Fig. 2). First, classical radical inhibitors TEMPO and BHT were added respectively to the reaction between 1a and hexyl methacrylate under the standard reaction conditions. The reactions only resulted in the decomposition of the starting materials and product 5a was not detected in the reaction mixture, which suggests that the reaction may proceed through a radical pathway (Fig. 2A). To prove the presence of the methoxy radical, methyl phenyl sulfone, its radical trapper,6 was added to the reaction of 1a with acrylonitrile under standard conditions, which gave product 3a in dramatically decreased yield (37%) along with 7% yield of methyl benzenesulfinate 10a, indicating that the methoxy radical is crucial for the transformations (Fig. 2B). It is worth noting that during the reaction between 2-chloride-3-ethoxy-5-bromopyridinium salt 8a and phenyl methacrylate, we also detected the presence of phenyl 3-bromo-2-methylacrylate by LC-MS (see Fig. S18† for more details), which suggests that phenyl methacrylate may capture the bromine radical produced during the reaction.17 This could also explain why the reaction between 8 and activated alkenes delivered the 4-azaindoline products in lower yields (40%–79%).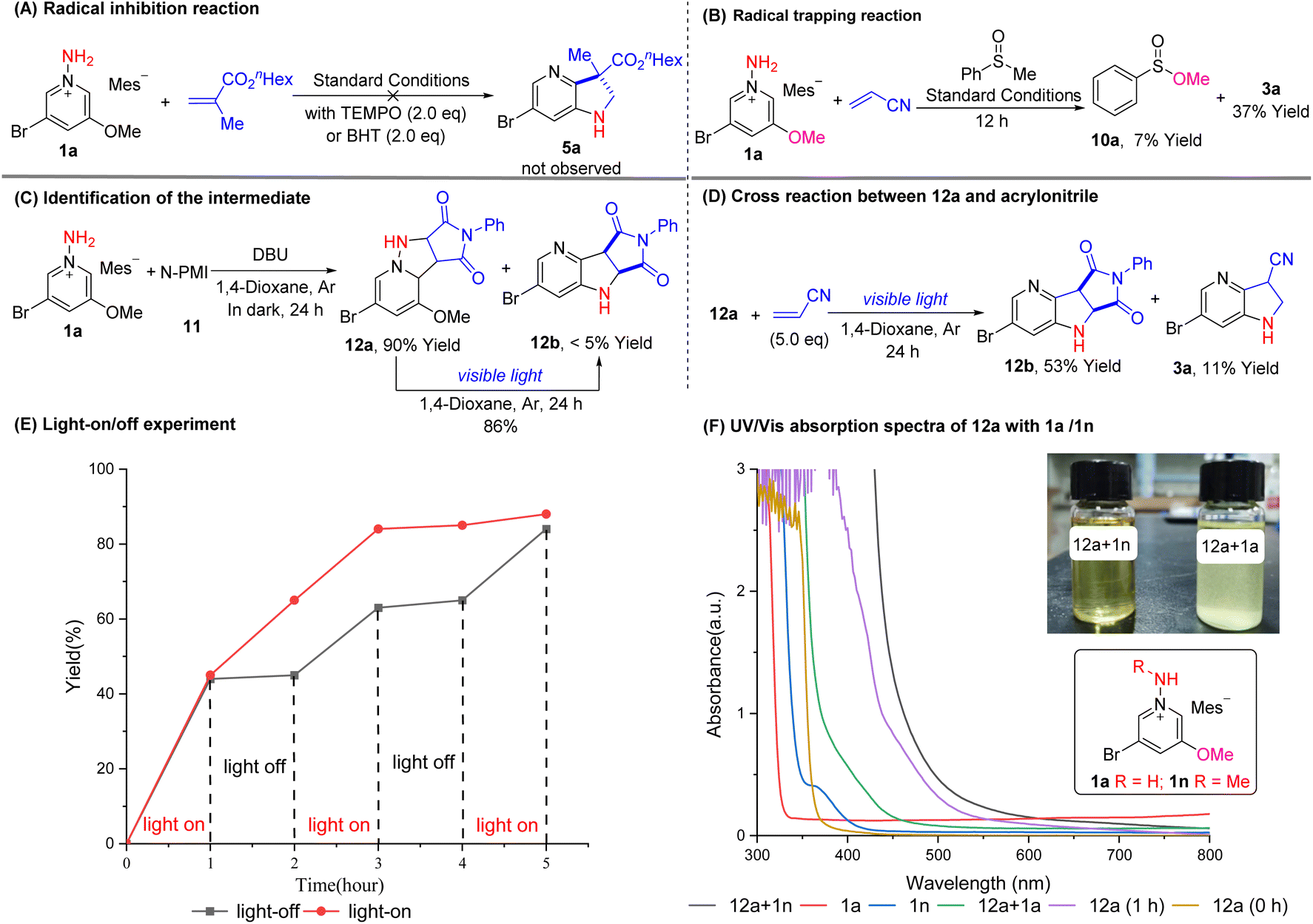 | ||
| Fig. 2 Mechanistic study. (A) Radical inhibition reaction. (B) Radical trap reaction. (C) Identification of the intermediate. (D) Cross reaction of 12a with acrylonitrile. (E) Light-on/off experiment of 1a (0.05 M), 2a (0.06 M) and DBU (0.075 M) in toluene. The red line represents the yield of the reaction continuously irradiated with visible-light. The black line represents the yield of reaction under irradiation of visible light in the first, third and fifth hour. (F) UV-VIS absorption spectra of 12a (0.05 M), 1a (0.05 M), 1n (0.05 M) and their mixture (12a + 1a or 12a + 1n) in acetonitrile. 1a was partially dissolved. | ||
Additionally, during the development of the reaction, we discovered that the reaction of pyridinium 1a and N-PMI (11) could give a room temperature stable intermediate 12a (90% isolated yield) under identical conditions but in the dark, with less than 5% yield of 4-azaindoline product 12b (Fig. 2C). This indicates that the visible light is not essential for the formal [3 + 2] cycloaddition, while it may be critical for the rearrangement from the intermediate to the final product. Furthermore, irradiation of 12a with visible light in dioxane produced the rearrangement product 12b in 86% yield, highlighting the necessity of light during the N–N bond cleavage and rearrangement. Also, at the beginning of the conversion of 12a into 12b, no obvious CT band was observed, while a clear CT band appeared after the reaction was stirred for 1 h (see Fig. S19†). Besides, re-subjected intermediate 12a in the presence of acrylonitrile under visible light gave 53% yield of 12b and 11% yield of 3a (Fig. 2D), suggesting the reversibility of formal [3 + 2] cycloaddition. To determine whether the reaction involves a radical chain process, a light on–off experiment was performed between 1a and 2a under standard conditions (Fig. 2E). The absence of light resulted in no conversion indicating the necessity of continuous light irradiation and a radical chain mechanism is unlikely for this transformation.18 At this stage, two key information could be obtained from these experiments: (i) the intramolecular EDA complex as in Su's case was not involved in our reactions.7 (ii) In the solution, 12a (as the electron donor) and another electron-poor species (as the electron acceptor), generated from a retro-Mannich reaction19 of 12a or 3-bromo-5-methoxy-1-aminopyridinium salt (1a), may form an EDA complex to trigger the following reaction.
To figure out the potential electron acceptor, we used the 3-bromo-5-methoxy-1-(methylamino)pyridinium salt 1n to mimic the aza-Michael adduct intermediate, which should have a similar N-alkylated structure (such as intermediate 15 in Scheme 4A, vide infra) to the species from the retro-Mannich reaction of 12a, to launch the UV-Vis experiments with 12a. As shown in Fig. 2F, it was found that when 12a was mixed with 1n, a clear yellow solution was formed immediately along with a significant redshift in the UV-Vis spectrum (black line), which is similar to the reaction in which 12a was stirred alone in dioxane for 1 hour (purple line). In contrast, there was no noticeable color change and redshift appeared for the mixture of 12a and 1a (green line). These results highly suggested that in our case, the entity from aza-Michael addition (or the retro-Mannich reaction after formal [3 + 2] addition) may work as the electron acceptor rather than N-aminopyridinium salt itself. Moreover, the Cyclic Voltammetry (CV) experiments of 3-bromo-5-methoxy-1-aminopyridinium salt (1a) and 3-bromo-5-methoxy-1-(methylamino)-pyridinium salt (1n), respectively, showed that the irreversible reduction of 1a and 1n (see Fig. S16 and S17† for more details). We also detected the conversion of 1n into 3-bromo-5-methoxypyridine after 2 cycles of CV experiments, which indicated that, after accepting an electron, it would more likely decompose to the corresponding pyridine rather than being oxidized back to the corresponding pyridinium salt 1n. All taken together, these results suggested that the formal [3 + 2] cycloaddition of N-aminopyridiniums and activated alkenes produced tetrahydropyrazolo[1,5-a]pyridine derivatives such as 12a, meanwhile, the retro-Mannich reaction of 12a or aza-Michael addition of aminopyridiniums and activated alkenes may generate a new N-alkylated-amidopyridinium salt which would form an EDA complex with 12a. In the presence of light, this EDA complex triggered the following process of N–N bond cleavage, which, in turn, led to the formation of an alkoxyl radical (or dimethyl amino radical or bromine radical) keeping the reaction going smoothly.20
 | ||
| Scheme 4 (A) Proposed mechanism. (B) DFT calculation. The energies are given in kcal mol−1. All the structures were calculated at the (U)B3LYP functional with Grimme's D3(BJ) dispersion correction. | ||
Based on the above observations, a plausible mechanism for the EDA complexes enabling the intermolecular synthesis of 4-azaindolines was rationalized as shown in Scheme 4A. Initially, N-aminopyridinium salt 1 when reacted with the base could form a highly active intermediate N-aminopyridine ylide 13, which would be subsequently trapped by α,β-unsaturated compounds via a formal [3 + 2] cycloaddition process to generate tetrahydropyrazolo[1,5-a]pyridine 14 (or 12a). The new pyridinium salt 15 could be obtained by the aza-Michael addition or retro-Mannich reaction of 14. Then, intermediates 14 and 15 would form an EDA complex (16), which upon irradiation by visible-light generated 16*. Radical cation 14′ and radical 15′ were obtained through a SET process. Deprotonation of 14′ gave a new radical 17, which would then transform into 18 by imine–enamine tautomerism. Intramolecular re-cyclization of 18 could generate a radical intermediate 19. The homolytic fission of the C–O bond and aromatization in 19 gave the 4-azaindoline product, along with the release of the methoxy radical.21 Meanwhile, the methoxy radical would abstract a hydrogen atom from the radical 15′,22 which was followed by intramolecular cyclization to form intermediate 14 to re-enter the reaction process. In the whole process, intermediate 14 serves as a formal catalyst promoting the reaction to deliver the desired product. A similar process could also be used to explain the reactions starting from 3-bromo-5-(dimethylamino)pyridinium salt 1d and 2-chloride-3-ethoxy-5-bromopyridinium salt 8.
A computational study was then carried out to excavate deeper insight into the mechanism (Scheme 4B). The energy barrier for the formation of tetrahydropyrazolo[1,5-a]pyridine 14via formal [3 + 2] annulation, and for the generation of 15 either through Michael addition between 1 and 2 promoted by DBU or through a retro-Mannich reaction of 14 is lower than 20 kcal mol−1 supported the ready formation of 14 and 15 at room temperature. Subsequently, the intermediate 15 can be captured by 14 to give the complex 16, which is exothermic by 1.7 kcal mol−1. The excitation energy of complex 16 was also calculated using TD-DFT, and the obtained absorption peak of 478 nm with the oscillator strength of 0.02 is consistent with our experimental result, in which visible-light is used to promote the reaction. The calculation result of complex 16 provides important evidence to support that 16 is the key EDA complex. The SET process then occurs at the excited state from intermediate 14 to 15 of complex 16 to give the radical cation 14′ and radical 15′. Subsequently deprotonation of 14′ gives radical intermediate 17, which is exothermic by 17.2 kcal mol−1. For 17, the N–N bond cleavage can occur easily via a transition state TS2 with an energy barrier of 10.9 kcal mol−1, to generate intermediate 18, and this is exothermic by 10.3 kcal mol−1. Next, the radical intermediate 19 can be obtained from 18 through a slight exergonic process by overcoming an energy barrier of 18.9 kcal mol−1viaTS3. Finally, product 3 could be formed by homolytic fission of C–O along with the release of the methoxy radical, and this process requires an energy barrier of 8.7 kcal mol−1. The dissociative methoxy radical will be captured by 15′ to give MeOH and adduct 14, which is exothermic by 52 kcal mol−1. The computational studies showed that once MeO radical gets close to radical 15′, the cyclization to form 14 occurred immediately; unfortunately, we were unable to identify a feasible transient state structure for such a process. In addition, the other situations including the intramolecular atom transfer (HAT) in 15′ and the intermolecular single electron transfer process between the MeO radical and 15′, have been proven unlikely to happen by calculation (see Fig. S21† for details). The computational result shows that before forming the key EDA complex, the overall energy barrier (from 13 to TS1 in Fig. S20†) is 19.5 kcal mol−1, and after SET, the overall energy barrier (from 18 to TS3 in Scheme 4B) is 18.9 kcal mol−1. This is also in good accordance with our experimental result, in which all the reactions proceeded smoothly at room temperature.
Conclusions
In summary, we have developed a visible-light induced intermolecular protocol to access 2,3-difunctionalized pyridines from N-aminopyridinium salts and activated alkenes promoted by an in situ formed EDA complex under mild conditions. Mechanistic studies and DFT calculations suggested that the two intermediates from the reactants via formal [3 + 2] cycloaddition and its ring-opening product (or aza-Michael addition from the reactants) are responsible for EDA complex formation. Besides, the in situ generated methoxy or bromine radical plays an important role in the regeneration of the intermediate 14 to maintain the continuous formation of the EDA complex. This unique and straightforward procedure has many advantages such as high atom economy, broad functional group compatibility, mild reaction conditions, easy handling, and photocatalyst- and photoreactor–free conditions, and can be readily performed on a gram scale. The synthetic application was also demonstrated by late-stage modifications of a series of on-market drugs and natural products. Besides, this study also demonstrated how intermediates produced transiently during the reaction can contribute to the formation of photoactive EDA complexes and initiate the subsequent radical process and may open up new avenues for the application of EDA complex photochemistry in synthetic chemistry.Author contributions
X. M. conceived the idea and guided the project. Y.-Z. L. carried out reaction condition optimization, substrate screening experiments and mechanistic studies. A. W., Z. S., X. Z., J. Z. and Y. J. performed some of the substrate screening experiments. Y. C. performed the computational studies. X. M. and Y.-Z. L. wrote the manuscript with feedback from other authors.Conflicts of interest
The authors declare the following competing interests that one patent has been registered (CN202310032248.5).Acknowledgements
We thank the National Natural Science Foundation of China (22001246), the Sichuan Science and Technology Program (2022JDRC0132 and 2022ZYD0047), the Biological Resources Program (KFJ-BRP-008) from the Chinese Academy of Sciences (CAS), the CAS Pioneer Hundred Talents Program and the Thousand Talents Program of Sichuan Province, for the financial support.References
- (a) A. K. Wortman and C. R. J. Stephenson, Chem, 2023, 9, 2390 CrossRef CAS; (b) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461 CrossRef CAS PubMed; (c) C. G. S. Lima, T. de M. Lima, M. Duart, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389 CrossRef CAS; (d) Z. Yang, Y. Liu, K. Cao, X. Zhang, H. Jiang and J. Li, Beilstein J. Org. Chem., 2021, 17, 771 CrossRef CAS PubMed; (e) T. Tasnim, M. J. Ayodele and S. P. Pitre, J. Org. Chem., 2022, 87, 10555 CrossRef CAS PubMed.
- M. Tavakolian and M. Hosseini-Sarvari, ACS Sustainable Chem. Eng., 2021, 9, 4296 CrossRef CAS.
- (a) M. Kim, Y. Koo and S. Hong, Acc. Chem. Res., 2022, 55, 3043 CrossRef CAS PubMed; (b) F.-S. He, S. Ye and J. Wu, ACS Catal., 2019, 9, 8943 CrossRef CAS.
- Y. Gao, S. Jiang, N.-D. Mao, H. Xiang, J.-L. Duan, X.-Y. Ye, L.-W. Wang, Y. Ye and T. Xie, Top. Curr. Chem., 2022, 380, 25 CrossRef CAS.
- (a) S. Jung, S. Shin, S. Park and S. Hong, J. Am. Chem. Soc., 2020, 142, 11370 CrossRef CAS PubMed; (b) I. Kim, S. Park and S. Hong, Org. Lett., 2020, 22, 8730 CrossRef CAS PubMed; (c) H. Choi, G. R. Mathi, S. Hong and S. Hong, Nat. Commun., 2022, 13, 1776 CrossRef CAS PubMed; (d) S. Shin, S. Lee, W. Choi, N. Kim and S. Hong, Angew. Chem., Int. Ed., 2021, 60, 7873 CrossRef CAS PubMed.
- D. Xie, Y. Wang, X. Zhang, Z. Fu and D. Niu, Angew. Chem., Int. Ed., 2022, 61, e20220492 Search PubMed.
- R.-B. Hu, S. Sun and Y. Su, Angew. Chem., Int. Ed., 2017, 56, 10877 CrossRef CAS PubMed.
- (a) E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750 CrossRef CAS PubMed; (b) E. Arceo, A. Bahamonde, G. Bergonzini and P. Melchiorre, Chem. Sci., 2014, 5, 2438 RSC; (c) M. Silvi, E. Arceo, I. D. Jurberg, C. Cassani and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 6120 CrossRef CAS PubMed; (d) A. Bahamonde and P. Melchiorre, J. Am. Chem. Soc., 2016, 138, 8019 CrossRef CAS PubMed.
- (a) X. Ma, J. J. Farndon, T. A. Young, N. Fey and J. F. Bower, Angew. Chem., Int. Ed., 2017, 56, 14531 CrossRef CAS; (b) X. Ma, Y. Liu, L. Du, J. Zhou and I. E. Marko, Nat. Commun., 2020, 11, 914 CrossRef CAS PubMed; (c) A. Wang, Y.-Z. Liu, Z. Shen, Z. Qiao and X. Ma, Org. Lett., 2022, 24, 1454 CrossRef CAS PubMed.
- (a) P. A. Gerken, J. R. Wolstenhulme, A. Tumber, S. B. Hatch, Y. Zhang, S. Muller, S. A. Chandler, B. Mair, F. Li, S. M. B. Nijman, R. Konietzny, T. Szommer, C. Yapp, O. Fedorov, J. L. P. Benesch, M. Vedadi, B. M. Kessler, A. Kawamura, P. E. Brennan and M. D. Smith, Angew. Chem., Int. Ed., 2017, 56, 15555 CrossRef CAS; (b) F. G.-L. de Turiso, Y. Shin, M. Brown, M. Cardozo, Y. Chen, D. Fong, X. Hao, X. He, K. Henne, Y.-L. Hu, M. G. Johnson, T. Kohn, J. Lohman, H. J. McBride, L. R. McGee, J. C. Medina, D. Metz, K. Miner, D. Mohn, V. Pattaropong, J. Seganish, J. L. Simard, S. Wannberg, D. A. Whittington, G. Yu and T. D. Cushing, J. Med. Chem., 2012, 55, 7667 CrossRef; (c) K. Takai, Y. Inous, Y. Konishi, A. Suwa, Y. Uruno, H. Matsuda, T. Nakako, M. Sakai, H. Nishikawa, G. Hashimoto, T. Enomoto, A. Kiamura, Y. Uematsu, A. Kiyoshi and T. Sumiyoshi, Bioorg. Med. Chem. Lett., 2014, 24, 3189 CrossRef CAS; (d) M. Badland, I. Devillers, C. Durand, V. Fasquelle, B. Gaudillière, H. Jacobelli, A. C. Manage, I. Pevet, J. Puaud, A. J. Shorter and R. Wrigglesworth, Tetrahedron Lett., 2011, 52, 5292 CrossRef CAS.
- E. de Pedro Beato, D. Spinnato, W. Zhou and P. Melchiorre, J. Am. Chem. Soc., 2021, 143, 12304 CrossRef CAS PubMed.
- J. K. Kochi, Pure Appl. Chem., 1991, 63, 255 CrossRef CAS.
- H.-W. He, F.-Y. Wang, D. Zhang, D. C.-Y. Chen, D. Xu, H. Zhou, X. Liu and G. Xu, J. Agric. Food Chem., 2022, 70, 10316 CrossRef CAS.
- (a) W. Lee, Y. Koo, H. Jung, S. Chang and S. Hong, Nat. Chem., 2023, 15, 1091 CrossRef CAS PubMed; (b) Y. Moon, B. Park, I. Kim, G. Kang, S. Shin, D. Kang, M.-H. Baik and S. Hong, Nat. Commun., 2019, 10, 4117 CrossRef PubMed; (c) N. Kim, C. Lee, T. Kim and S. Hong, Org. Lett., 2019, 21, 9719 CrossRef CAS; (d) J. Kim, M. Kim, J. Jeong and S. Hong, J. Am. Chem. Soc., 2023, 145, 14510 CrossRef CAS PubMed; (e) S. Hong, B. Kweon, W. Lee, S. Chang and S. Hong, Org. Lett., 2023, 25, 2722 CrossRef CAS; (f) W. Choi, M. Kim, K. Lee, S. Park and S. Hong, Org. Lett., 2022, 24, 9452 CrossRef CAS; (g) C. Kim, J. Jeong, M. Vellakkaran and S. Hong, ACS Catal., 2022, 12, 13225 CrossRef CAS; (h) C.-Y. Tan, M. Kim, I. Park, S. Kim and S. Hong, Angew. Chem., Int. Ed., 2021, 61, e202213857 CrossRef.
- For examples of N–N bond cleavage/N-migration of N-alkoxycarbonyliminopyridinium and N-sulfonylimino pyridinium ylides with symmetrical pyridines, see: (a) T. Sasaki, K. Kanematsu and A. Kakehi, J. Org. Chem., 1971, 36, 2978 CrossRef CAS; (b) R. A. Abramovitch and T. Takaya, J. Org. Chem., 1972, 37, 2022 CrossRef CAS; (c) M. W. Barker and W. E. McHenry, J. Org. Chem., 1979, 44, 1175 CrossRef CAS.
- Shi and coworkers found that when N-tosyl-3-bromo-5-methoxypyridiniumimide ylide was reacted with 2-trimethylsilylphenyl triflate in the presence of 3 equiv. of CsF at 70 °C for 24 h, 3-MeO-5-Ts-pyrido[3,2-b]indole and 3-Br-5-Ts-pyrido[3,2-b]indole were formed unselectively in almost the same yield. J. Zhao, P. Li, C. Wu, H. Chen, W. Ai, R. Sun, H. Ren, R. C. Larock and F. Shi, Org. Biomol. Chem., 2012, 10, 1922 RSC.
- P. Jia, Q. Li, W. C. Poh, H. Jiang, H. Liu, H. Deng and J. Wu, Chem, 2020, 6, 1766 CAS.
- M. Ociepa, J. Turkowska and D. Gryko, ACS Catal., 2018, 8, 11362 CrossRef CAS.
- (a) X.-L. Jiang, S.-F. Wu, J.-R. Wang, H. Lu, G.-J. Mei and F. Shi, Org. Biomol. Chem., 2018, 16, 8395 RSC; (b) X.-P. Mu, Y.-H. Li, N. Zheng, J.-Y. Long, S.-J. Chen, B.-Y. Liu, C.-B. Zhao and Z. Yang, Angew. Chem., Int. Ed., 2021, 60, 11211 CrossRef CAS PubMed.
- J. C. Walton and A. Studer, Acc. Chem. Res., 2005, 38, 794 CrossRef CAS.
- F. Parsaee, M. C. Senarathna, P. B. Kannangara, S. N. Alexander, P. D. E. Arche and E. R. Welin, Nat. Rev. Chem., 2021, 5, 486 CrossRef CAS.
- (a) L. Chang, Q. An, L. Duan, K. Feng and Z. Zuo, Chem. Rev., 2022, 122, 2429 CrossRef CAS; (b) A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668 CrossRef CAS PubMed; (c) Q. An, Z. Wang, Y. Chen, X. Wang, K. Zhang, H. Pan, W. Liu and Z. Zuo, J. Am. Chem. Soc., 2020, 142, 6216 CrossRef CAS PubMed; (d) Q. An, Y.-Y. Xing, R.-H. Pu, M. Jia, Y. Chen, A. Hu, S.-Q. Zhang, N. Yu, J. Du, Y. Zhang, J. Chen, W. Liu, X. Hong and Z. Zuo, J. Am. Chem. Soc., 2023, 145, 359 CrossRef CAS PubMed; (e) F. Liu, S. Ma, Z. Lu, A. Nangia, M. Duan, Y. Yu, G. Xu, Y. Mei, M. Bietti and N. Houk, J. Am. Chem. Soc., 2022, 144, 6802–6812 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2222506, 2222507 and 2222508. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3gc04425d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |