
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile and sustainable recovery of spent LiFePO4 battery cathode materials in a Ca(ClO)2 system†
Gongqi
Liu
ab,
Zejian
Liu
abc,
Jing
Gu
abc,
Shujia
Wang
a,
Yufeng
Wu
*d,
Haoran
Yuan
*abc and
Yong
Chen
ab
aGuangzhou Institute of Energy Conversion, Chinese Academy of Sciences (CAS), Guangzhou 510640, PR China. E-mail: yuanhr@ms.giec.ac.cn
bGuangdong Provincial Key Laboratory of New and Renewable Energy Research and Development, Guangzhou 510640, PR China
cSchool of Engineering Science, University of Science and Technology of China, Hefei, 230026, PR China
dInstitute of Circular Economy, Beijing University of Technology, Beijing 100124, China. E-mail: wuyufeng3r@126.com
First published on 31st January 2024
Abstract
Spent LiFePO4 batteries are gradually increasing in popularity and interest, and their stable and insoluble olive-shaped structure poses a great challenge for the sustainable recycling of Li. In this study, a simple and sustainable Ca(ClO)2 system was proposed for the recovery of spent LiFePO4 battery cathode materials. The effects of both mechanochemical activation and hydrometallurgical enhanced leaching on the deconstruction of LiFePO4 and the leaching rates of Li and Fe in the Ca(ClO)2 system were studied. Compared with mechanochemical activation, Ca(ClO)2-assisted hydrometallurgical enhanced leaching can simultaneously achieve the separation and enrichment of the target components Li+ and Fe3+ and the Ca2+ impurities. The transformation path and reaction mechanism of LiFePO4 in the Ca(ClO)2 system were proposed based on the phase composition and micromorphology of the reaction products. In addition, the economic evaluation results show that Ca(ClO)2-assisted hydrometallurgical enhanced leaching has a high recovery economy. The developed Ca(ClO)2 system realizes the strong dissolution of spent LiFePO4 battery cathode materials and the sustainable comprehensive recovery of valuable components.
1. Introduction
Lithium-ion batteries are a valuable energy storage technology.1,2 Lithium iron phosphate (LiFePO4) battery cathode materials are considered to be one of the main choices for lithium-ion batteries in new energy electric vehicles because of their low cost, high energy density, good reversibility, and stable thermodynamic properties.3–5 The manufacturing capacity of LiFePO4 battery cathode materials has been increasing in tandem with the rise in the electric vehicle market demand and the ongoing enhancement of lithium-ion battery electrode material performance.6,7 According to the 2023 white paper published by EV Tank, China Battery Industry Research Institute and China YiWei Institute of Economics, active materials for LiFePO4 battery cathode materials in China accounted for 58.65% of the lithium battery market share in 2022.8The necessity for safe disposal and comprehensive recovery of spent LiFePO4 battery cathode materials (SLFPB-Ms) is growing along with the production of LiFePO4 battery cathode materials.9 Although SLFPB-Ms are thought to be environmentally benign, improper handling can result in significant environmental problems, such as the leakage of fluorine-containing electrolytes, which can lead to organic and fluorine pollution.10,11 Moreover, SLFPB-Ms have a very high recovery value and a concentration of the valuable metal lithium that is significantly higher than that of the source mineral.9,12 As a result, recycling SLFPB-Ms can help the lithium battery industry grow sustainably while also preventing pollution.13,14 Compared with the recycling of spent LiNixCoyMn1−x−yO2 and LiCoO2 batteries, which are enriched with large amounts of precious metals, the economic driving force for recycling SLFPB-Ms is weak.15,16 Developing low-carbon, efficient, and sustainable extraction technologies suitable for SLFPB-Ms recycling has proven to be a daunting challenge for scientists and technologists.17,18
Typically, direct regeneration and hydrometallurgical processes are conventional methods for recovering SLFPB-Ms.19–21 The direct regeneration approach has the advantages of a short process flow and low consumption of acid and alkali reagents, but the regenerated LiFePO4 usually contains impurity components, resulting in poor electrochemical performance.11,22 The hydrometallurgical method is widely used for the comprehensive recovery of SLFPB-Ms due to its good selectivity and high recovery rate for target elements.23 In hydrometallurgical systems, a range of inorganic acids (such as H2SO4, HCl, HNO3 and H3PO4)24,25 and organic acids (such as citric acid, oxalic acid, acetic acid, etc.) are employed to recover precious metals from SLFPB-Ms. To improve the leaching rates and recovery rates of important metals, the hydrometallurgical recovery of SLFPB-Ms usually uses microwaves, field strength, or powerful oxidants, which leads to further increases in energy consumption and secondary pollution risk.26,27
A simplified recovery strategy using direct oxidation has been developed by scientists and technicians in response to the challenging dissociation of the SLFPB-Ms in recent years. This strategy has the advantage of achieving the efficient release of lithium and the directed conversion of iron phosphate without destroying the crystal structure of lithium iron phosphate.28 It has been reported that Na2S2O8,29 (NH4)2S2O8,30 H2O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31and NaClO32,33 as oxidation additives can achieve the directed decomposition of lithium iron phosphate and the efficient recovery of lithium under mild conditions. However, it is challenging to encourage the widespread use of these compounds in industrial production due to their highly oxidizing, poisonous, and costly chemical characteristics. Therefore, it is urgent and necessary to develop a green, inexpensive and sustainable hydrometallurgical system to realize the efficient recovery of SLFPB-Ms. Ca(ClO)2 is one of the most widely used disinfectants in the water treatment industry and is also used extensively as an oxidant in industrial production because of its high efficiency, low toxicity, and low cost.34–36 An innovative and interesting experiment is the use of Ca(ClO)2 as a clean, green oxidant in the recycling of SLFPB-Ms.
31and NaClO32,33 as oxidation additives can achieve the directed decomposition of lithium iron phosphate and the efficient recovery of lithium under mild conditions. However, it is challenging to encourage the widespread use of these compounds in industrial production due to their highly oxidizing, poisonous, and costly chemical characteristics. Therefore, it is urgent and necessary to develop a green, inexpensive and sustainable hydrometallurgical system to realize the efficient recovery of SLFPB-Ms. Ca(ClO)2 is one of the most widely used disinfectants in the water treatment industry and is also used extensively as an oxidant in industrial production because of its high efficiency, low toxicity, and low cost.34–36 An innovative and interesting experiment is the use of Ca(ClO)2 as a clean, green oxidant in the recycling of SLFPB-Ms.
This paper proposes a sustainable approach for the recovery of SLFPB-Ms based on a Ca(ClO)2-enhanced dissociation system. Compared with the traditional enhanced leaching system, Ca(ClO)2 acts as both an oxidizing agent and a purifying agent in the recovery process of SLFPB-Ms. This allows for the simultaneous release of Li+ ions and the precipitation of impurity ions, thereby cutting down on the processing workflow time. We examined the effects of Ca(ClO)2-assisted mechanochemical activation and hydrometallurgical enhanced leaching on the phase conversion of the lithium iron phosphate ore. A single-factor experiment was used to determine the directed conversion efficiency of the impurity as well as the recoveries of the target elements Li and Fe. By combining characterization techniques such as phase and microscopic morphology, the reaction mechanism of LiFePO4 in the Ca(ClO)2 system has been proposed. Furthermore, the proposed technology was analyzed from both an economic and technological standpoint, yielding positive results. The results of this research may provide insight for the directional transformation of target elements and high-efficiency recovery of SLFPB-Ms.
2. Experimental
2.1 Materials and reagents
The spent LiFePO4 battery used in this work came from a new energy end-of-life vehicle dismantling enterprise in Huzhou, China. The initial predischarge of the SLFPB-Ms was conducted in a saturated sodium chloride solution. After manual dismantling, crushing, and sorting, the organic separator, copper foil, aluminum foil, steel shell, and electrode powder were obtained. The obtained electrode powder was finely ground and sieved to obtain 200 mesh SLFPB-Ms as the raw material for this study. SLFPB-Ms (0.2 g) was dissolved in aqua regia, and the solution was measured for each metal element with an inductively coupled plasma-optical emission spectrometer (ICP-OES), and the results are shown in Table S1.† All reagents used in the experimental procedures were of analytical grade and were purchased from MACKLIN (Shanghai, China). All solutions were prepared with deionized water obtained from a Dura Pro 24FV (Shanghai, China).2.2 Experimental procedures
Fig. 1 illustrates the process flow for the enhanced dissociation of SLFPB-Ms and the directed conversion of Li, Fe, and P in the Ca(ClO)2 system.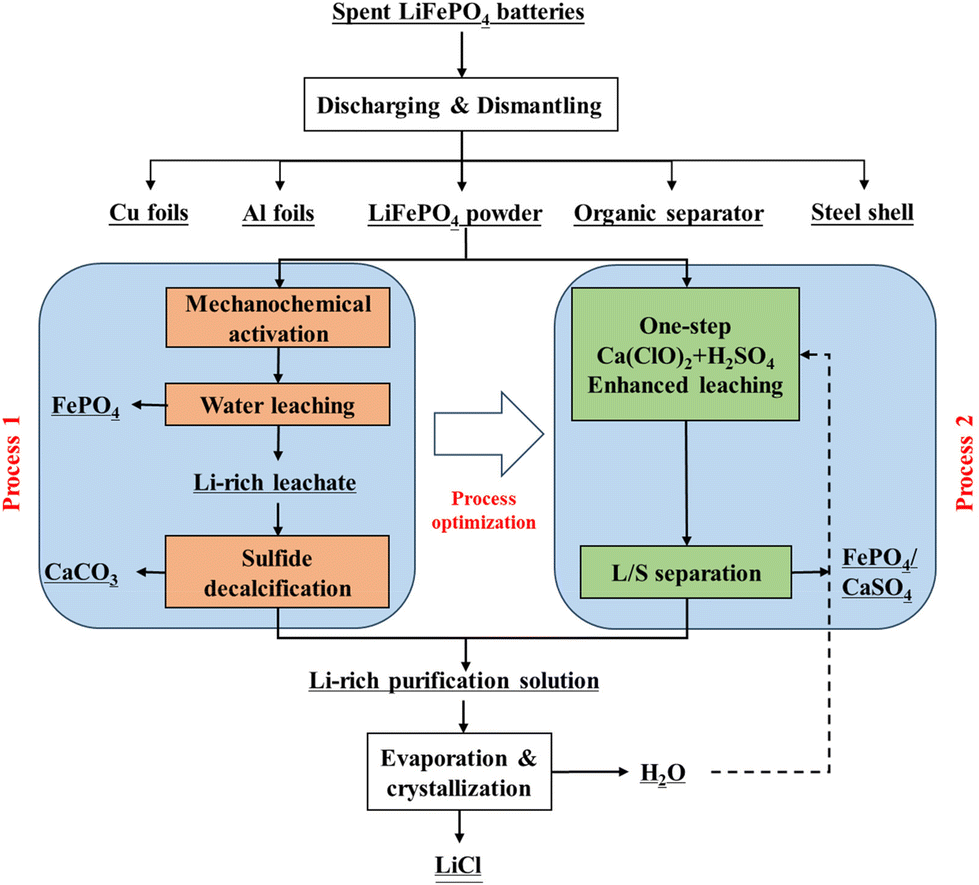 | ||
| Fig. 1 Schematic diagram of the enhanced deconstruction pathway and process optimization of SLFPB-Ms in the Ca(ClO)2 system. | ||
:1 g mL−1 to dissolve it. The slurry was filtered through a Brinell funnel to obtain the Li-rich leachate and leached residue. The leached residue was washed with deionized water three times, and the washing solution was mixed with the Li-rich leachate, and the concentration of elements was measured by ICP-OES after achieving a constant volume with 5% dilute nitric acid. The average value of three test results was taken. The leaching rates of Li, Fe and P were calculated using the following equation: | (1) |
:1 mL g−1. The slurry was stirred at a speed of 500 rpm, and the pH of the system was adjusted with H2SO4 at a reaction temperature of 25–75 °C for 10–110 min. Following the reaction, the slurry was filtered through a Brinell funnel to obtain the Li-rich leachate and leached residue. The leached residue was washed three times with deionized water, and the washing solution was mixed with the Li-rich leachate, and the concentration of elements was measured by ICP-OES after achieving a constant volume with 5% dilute nitric acid. The average value of the three test results was calculated. The leaching rates of Li, Fe, and P were calculated by eqn (1).
2.3 Analysis and characterization
The contents of Li, Fe and P in the raw materials and other solid phase-products were determined by inductively coupled plasma emission spectrometry (ICP-OES, Perkin Optima 8000, America), and the crystal structures of the target components were analyzed by X-ray diffractometry (XRD, X′ Pert PRO MPD, Netherlands). The surface morphology of the products was observed using a scanning electron microscope (SEM, Hitachi/SU-70, Japan), and the surface chemical composition was analyzed using energy dispersive X-ray spectroscopy (EDS). In addition, X-ray photoelectron spectroscopy (XPS, ESCALAB 250Xi, Thermo Fischer, America) was used to characterize the valence states of the elements in the solid phase-products.3. Results and discussion
3.1 Ca(ClO)2-assisted mechanochemical activation
Fig. 2 illustrates the effect of various factors on the leaching rates of Li, Fe, and P in Ca(ClO)2-assisted mechanochemical activation. Fig. 2a shows that the mass ratio of Ca(ClO)2/LiFePO4 has a significant effect on the leaching rate of valuable elements after mechanochemical activation. When the mass ratio of Ca(ClO)2/LiFePO4 is less than 1.0 g g−1, the leaching rate of Li increases linearly. The leaching rate of Li is maintained at approximately 98.32% when the mass ratio of Ca(ClO)2/LiFePO4 is more than 1.0 g g−1. The introduction of oxidizing agents facilitates the transition of Fe2+ to Fe3+, thereby enhancing the disintegration of LFP and the formation of FePO4. This process ultimately promotes the release of Li+ ions.37 Conversely, the leaching effect of Fe and P is inversely proportional to the leaching rate of Li.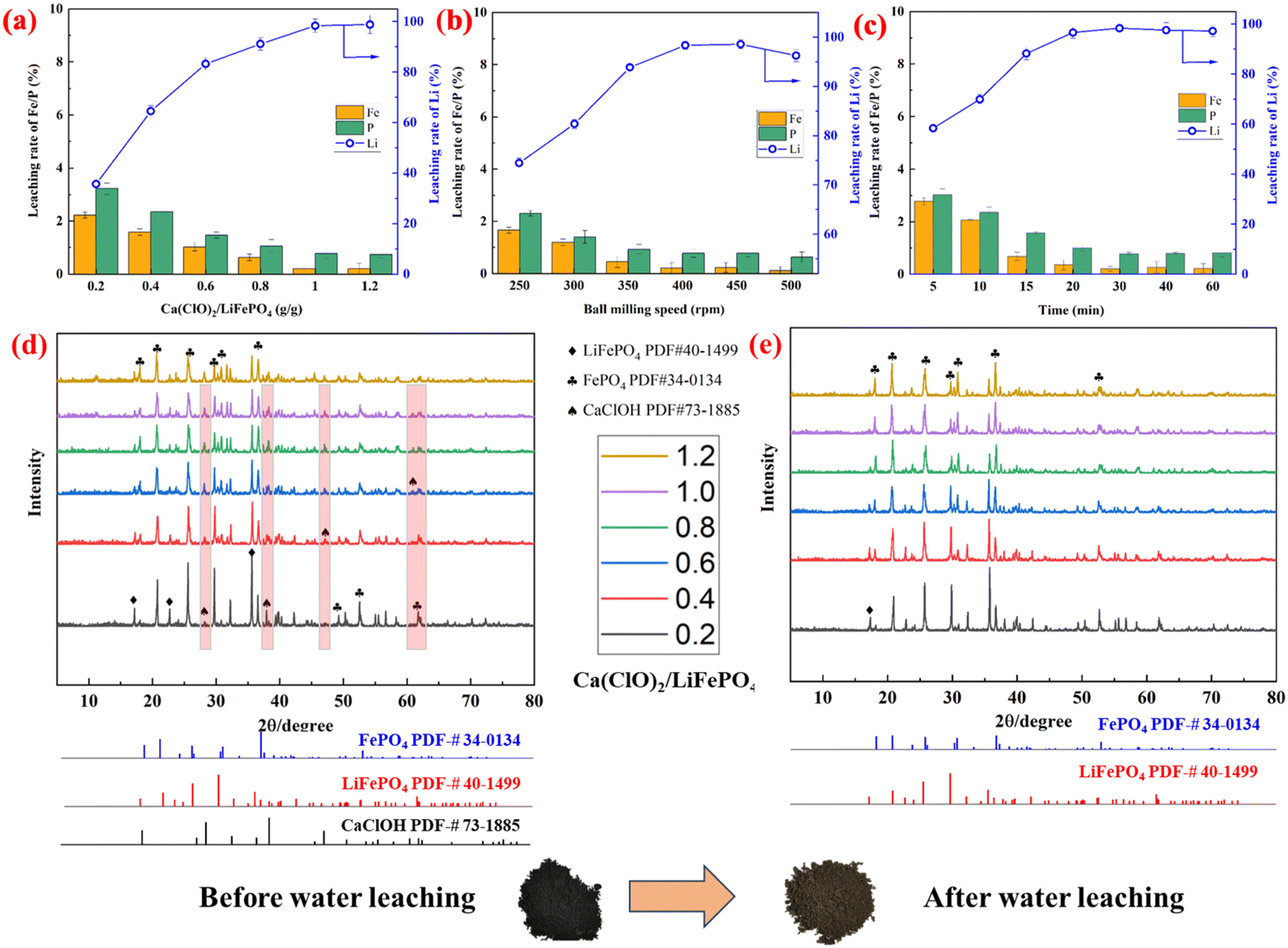 | ||
| Fig. 2 Leaching rates of Li, Fe and P at different (a) mass ratios of Ca(ClO)2/LiFePO4 (400 rpm, 30 min), (b) ball milling speeds (Ca(ClO)2/LiFePO4 = 1.0 g g−1, 30 min), and (c) milling times (Ca(ClO)2/LiFePO4 = 1.0 g g−1, 400 rpm), and the XRD patterns of activated materials (d) before and (e) after water leaching. | ||
Fig. 2b shows the effect of ball milling speed on the leaching rate of valuable elements under other fixed experimental conditions. It is evident that the leaching rate of Li displays a gradual increase when the ball milling speed is less than 400 rpm and a declining trend when the speed is larger than 400 rpm. The potential explanation lies in the fact that the elevated ball milling speed induces a higher shear force, expediting the deconstruction of LFP and facilitating the release of Li+ ions.28,38 However, the high speed of ball milling can also lead to material agglomeration, impeding the dissolution of Li and causing a continuous decline in the leaching rates of Fe and P. The effect of ball milling time on the leaching rate of Li, Fe and P is displayed in Fig. 2c. The leaching reaction of Li can approach equilibrium in a shorter time (<30 min), and Fe and P can be optimally enriched in the solid phase at this time.
As seen in Fig. 2d, the physical phases of the activated material following mechanochemical activation under varying mass ratios of Ca(ClO)2/LiFePO4 can be compared to observe that LiFePO4 oxidizes to FePO4 as the Ca(ClO)2 addition ratio increases. This could be explained by the oxidant Ca(ClO)2 linking several mechanical forces, including impact, shear, and friction, to facilitate the release of Li and the conversion of FePO4. In the LiFePO4 ore phase, the addition of the oxidant Ca(ClO)2 changes Fe2+ to Fe3+, which facilitates the release of Li+ while preserving its original crystal structure. Fe2+ oxidized during the ball milling process, causing Ca(ClO)2 to eventually break down. During the ball milling process, Ca(ClO)2 was gradually decomposed as Fe2+ was oxidized. A small number of CaClOH (PDF#73-1885) characteristic diffraction peaks appeared in the XRD patterns of the activated materials, which could be attributed to the decomposition of Ca(ClO)2 with moisture in the air during mechanochemical activation to produce the intermediate product CaClOH, as shown in the following equation:
| Ca2+ + Cl− + OH− ⇌ CaClOH | (2) |
It was discovered by comparing the leached residue obtained after the activated materials were leached by water that the distinctive peak of FePO4 was more notable in the leached residue with a mass ratio of Ca(ClO)2/LiFePO4 of 1.0 g g−1 than with other mass ratios (Fig. 2e). Additionally, a faint LiFePO4 signal was observed, which can be attributed to the inadequate oxidant addition of the mechanochemical activation reaction.39 The fact that there was no calcium-containing chemical phase in the leached residue as opposed to before the leaching could be the cause of the compounds’ adequate solubility in the aqueous solution of CaClOH and other compounds.
The activated materials and the residue after water leaching were characterized by SEM and XPS, and the results are shown in Fig. 3. It is evident that following Ca(ClO)2-assisted mechanochemical activation, the material's surface is comparatively smooth (Fig. 3a), which favors the formation of favorable reaction conditions for Li+ release. However, after Li+ leaching, the crystal structure shifted from LiFePO4 to FePO4, but the shape of the residue following water leaching did not significantly alter (Fig. 3b), suggesting that lithium iron phosphate has a high level of structural stability.
 | ||
| Fig. 3 SEM of (a) activated materials and (b) leached residue, XPS image of (c) XPS spectra, (d) Fe 2p, (e) Ca 2p and (f) Li 1s for activated materials and leached residue (mass ratio of Ca(ClO)2/LiFePO4 = 1.0 g g−1, 400 rpm and 30 min). | ||
Furthermore, XPS analysis was carried out to identify the surface chemical composition and valence states of the target elements in the solid phase. The Li, Fe, P, Ca, Cl, and O signals are clearly visible in the activated material (before leaching), while the Cl signal is clearly diminished and the Li signal is obviously weakened in the leached residue (after leaching), according to the XPS spectra of Fig. 3c. This indicates that Cl and Li are dissolved in the solution as soluble matter. The Fe 2p high-resolution XPS spectra (Fig. 3d) confirm the presence of the FePO4 phase in the XRD results by demonstrating that the Fe3+ peak strength and peak width of Fe 2p in the solid phase increase during the leaching reaction. After leaching, a noticeable shift to the left was observed in the Fe 2p characteristic binding energy of the leached residue. This phenomenon can be attributed to the reduction of peaks associated with interfering elements coupled with the increased prominence of Fe3+ characteristic peaks after leaching.40 These observations are consistent with the results of the XRD analyses. The high-resolution XPS spectrum of Ca 2p is shown in Fig. 3e. Calcium salts from the activated material enter the solution as soluble matter because the binding energy signal of Ca 2p in the leached residue is weaker than it was prior to the leaching reaction.
Notably, as shown in Fig. S2a,† the characteristic peak of Ca 2p in the leached residue was more noticeable when Ca(ClO)2 was in excess (mass ratio of Ca(ClO)2/LiFePO4 = 1.4). We deduced the existence of CaCO3 in the solid phase based on the combination of the high-resolution XPS spectra of C 1s produced under these reaction conditions (Fig. S2b†). This may be caused by the reaction of Ca(ClO)2 with CO2, and H2O in air during the mechanochemical activation to produce insoluble CaCO3, and the reaction is given in eqn (3).
| Ca(ClO)2 + H2O + CO2 = CaCO3↓ + 2HClO | (3) |
Furthermore, the high-resolution XPS spectrum of Li 1s (Fig. 3f) demonstrates that the activated material possesses a distinct characteristic peak of Li 1s prior to water leaching, whereas the intensity of the same peak in the leached residue is noticeably weakened.41 This provides evidence in favor of the successful separation of Li in SLFPB-M after mechanochemical activation and water leaching.
FePO4 is also present in the leached residue and the activated materials, as evidenced by the high-resolution XPS spectra of P 2p and O 1s in Fig. S3.† The P 2p spectra at 133.25 eV and 134.15 eV were decomposed into P 2p3/2 and P 2p1/2, indicating the presence of P in the solid phase as phosphate.42 In the O 1s spectrum, the characteristic peak at 531.12 eV was attributed to P-O.43
3.2 Hydrometallurgical enhanced leaching
The aforementioned experiments on Ca(ClO)2-assisted mechanochemical activation and water leaching demonstrate that Ca, Li, and Cl are combined as soluble ions in Li-rich leachate. To produce a pure lithium salt product, Ca ions must be eliminated since the target elements and contaminant components coexist in the Li-rich leachate. It may simplify the recycling process and reduce the loss of Li during the decalcification process to apply a one-step method to achieve both efficient leaching of Li and selective separation of the impurity component Ca. To achieve this, a hydrometallurgical enhanced leaching experiment for SLFPB-Ms in a Ca(ClO)2 system was designed. The effects of the mass ratio of Ca(ClO)2/LiFePO4, pH, reaction temperature and time on the leaching rate of Li were examined in the hydrometallurgical enhanced leaching experiment. Furthermore, the concentration variations in impurity Ca ions within the system were analyzed, and the results are shown in Fig. 4. | ||
| Fig. 4 The leaching rate of Li and concentration of Ca ions at different (a) mass ratios of Ca(ClO)2/LiFePO4 (pH 6.0, 55 °C, 50 min), (b) pH values (Ca(ClO)2/LiFePO4 = 1.2 g g−1, 55 °C, 50 min), and (c) temperatures (Ca(ClO)2/LiFePO4 = 1.2 g g−1, pH 6.0, 50 min) and (d) times (Ca(ClO)2/LiFePO4 = 1.2 g g−1, pH 6.0, 55 °C), and (e) the XRD patterns of the leached residue. | ||
Under the experimental conditions of pH 6.0, 50 min and 55 °C, Fig. 4a shows the effect of the mass ratio of Ca(ClO)2/LiFePO4 on the leaching rate of Li and the concentration of Ca ions. The leaching rate of Li increased steadily to 96.6%, and the concentration of Ca ions in the slurry was 145.0 mg L−1 when the mass ratio of Ca(ClO)2/LiFePO4 was 1.0 g g−1. The leaching rate of Li exceeded 98.5%, and the concentration of Ca ions increased to 302 mg L−1 when the mass ratio of Ca(ClO)2/LiFePO4 increased to 1.2 g g−1. The above results show that the Li-rich leachate is not only enriched in Li ions but also mixed with a large number of impurity Ca ions, which is not conducive to the enrichment of Li.
Fig. 4b shows the effect of pH on the leaching rate of Li and the concentration of Ca ions at a Ca(ClO)2/LiFePO4 mass ratio of 1.0 g g−1, 55 °C and 50 min. The leaching rate of Li was not significantly affected by pH, and a high leaching rate of Li (>98.3%) was achievable in the pH range of 2–7. When the pH was low, the concentration of Ca ions was kept at a high level (>4230.0 mg L−1), but when the pH was higher than 6.0, the concentration of Ca ions rapidly dropped to less than 330 mg L−1. As demonstrated by eqn (4), this is mostly caused by the precipitation interaction of Ca2+ with SO4.2–44,45
| Ca2+(aq) + SO42−(aq) → CaSO4(s) | (4) |
The effect of temperature on the leaching rate of Li and the concentration of Ca ions is depicted in Fig. 4c. It is evident that the leaching rate of Li increases with temperature in the 25–55 °C range and subsequently somewhat decreases after reaching 98.74%. It is possible that raising the temperature will help in increasing the Ca(ClO)2 activity, speeding up the rate of reaction, and thus encouraging the leaching reaction to proceed forward. On the other hand, a high temperature reduced the oxidative characteristic of Ca(ClO)2 and promoted its breakdown. Temperature had less of an impact on the Ca ion concentration, which stayed between 302 and 312 mg L−1.
Fig. 4d shows the effect of time on the leaching rate of Li and the concentration of Ca ions. The trend of the Li leaching rate was inversely linked with the Ca ion concentration when the reaction time was less than 50 minutes. As the reaction time was further increased, both the leaching rate of Li and the concentration of Ca tended to stabilize. After 50 min, the maximum leaching rate of Li was 98.74%, while the concentration of Ca ions was kept at less than 310 mg L−1.
In summary, the optimal conditions for the selective leaching of Li from SLFPB-Ms through hydrometallurgical enhanced leaching were determined as follows: a mass ratio of Ca(ClO)2/LiFePO4 at 1.2 g g−1, pH maintained at 6.0, leaching time set at 50 minutes, and leaching temperature held at 55 °C. Under these optimized leaching conditions, the Li leaching rate reached 98.74%, with the concentration of Ca ions remaining below 310 mg L−1.
XRD analyses of the leached residue obtained under different mass ratios of Ca(ClO)2/LiFePO4 were performed, and the results are shown in Fig. 4e. As the mass ratios of Ca(ClO)2/LiFePO4 increases, the characteristic peaks of LiFePO4 (PDF#40-1499) gradually disappear, whereas the diffraction peaks of CaSO4 (PDF#43-0606) and FePO4 (PDF#34-0134) emerge in the leached residue. Local enlargement of the plots shows that when the mass ratio of Ca(ClO)2/LiFePO4 is 0.8 g g−1, the characteristic peaks of LiFePO4 at 17.3°, 32.3° and 49.3° disappear, whereas the characteristic peaks of the FePO4 signal appear at 18.1° and 30.8°. When the Ca(ClO)2/LiFePO4 mass ratio is further increased to 1.2 g g−1, the characteristic peaks of CaSO4 appear at 31.9° and 49.2°.
The micromorphology of the leached residue was analyzed by SEM, and the results are shown in Fig. 5a. The surface boundary of the leached residue is clear, and the particle size is uniform. The EDS analysis of the leached residue surface elements is depicted in Fig. 5b. The findings indicate that the major components are C, Fe, O, P, Ca, and S, of which C may originate from the amorphous carbon coating on the surface of the particles. Fig. 5c–f shows the distribution analysis of the major elements. The distribution of each element is uniform, and there is no elemental segregation, which further proves that the solid-phase products formed by the hydrometallurgical enhanced leaching process are rich in CaSO4 and FePO4.
 | ||
| Fig. 5 Leached residue of the hydrometallurgical enhanced leaching process: (a) SEM image, (b) EDS spectra, and element mapping images of (c) Ca, (d) S, (e) O, (f) Fe, (g) P and (h) C. | ||
3.3 Purification of Li from lithium-rich leachate
It is necessary to eliminate the Ca ion impurities from the Li-rich leachate produced by Ca(ClO)2-assisted mechanochemical activation-water leaching to further enrich the lithium salt product. After heating 50 mL of the Li-rich leachate to 60 °C in a water bath, the pH was adjusted by adding saturated (NH4)2CO3 solution dropwise until no precipitate was formed while the mixture was stirred with a magnetic device. Following 30 min of reaction, the slurry was divided into liquid and solid phases using a Brinell funnel, and the phase and microscopic morphology of the precipitated slag obtained are shown in Fig. 5 and 6.| (NH4)2CO3(aq) ⇌ 2NH4+(aq) + CO32−(aq) | (5) |
| Ca2+(aq) + CO32−(aq) = CaCO3(s) | (6) |
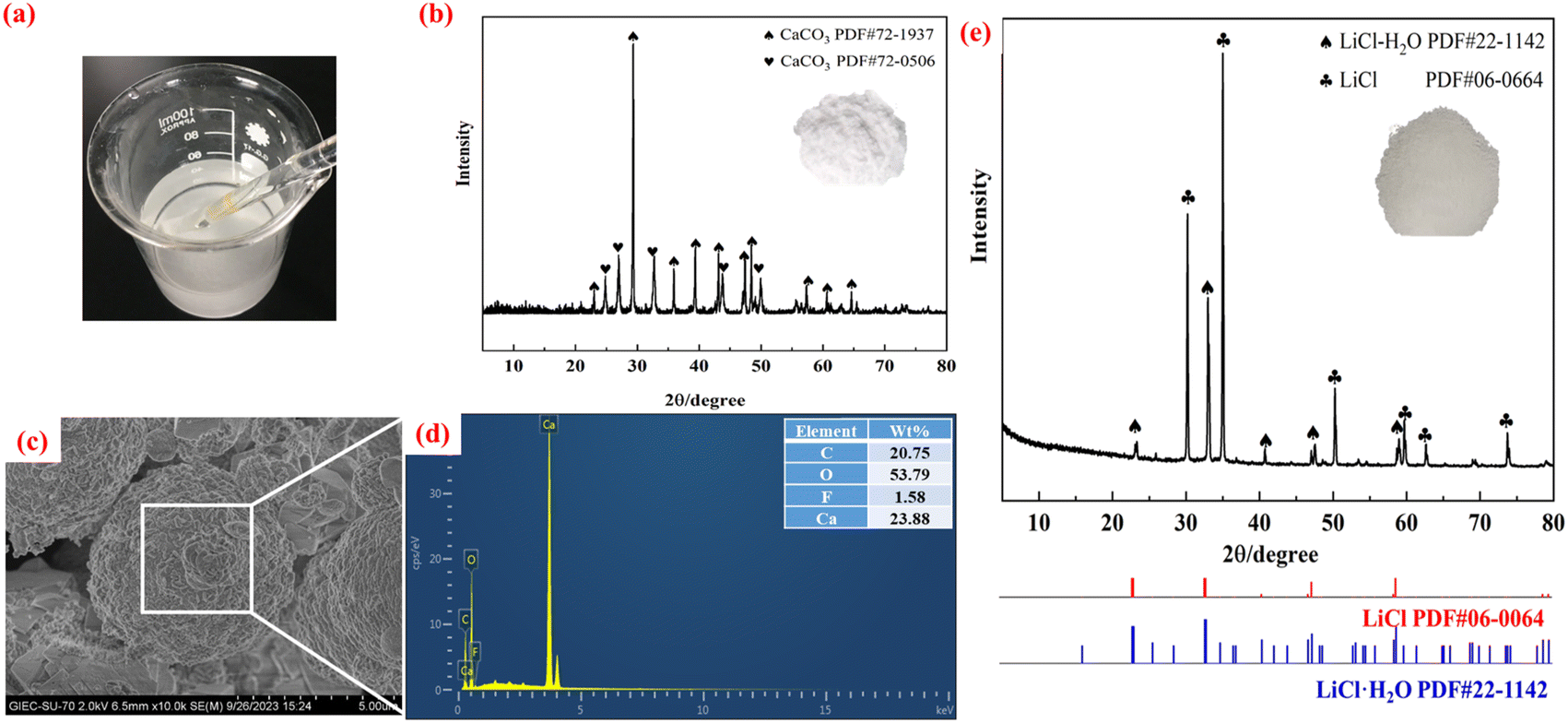 | ||
| Fig. 6 (a) Precipitation reaction process, (b) XRD, (c) SEM, and (d) energy spectrum of the precipitated slag, (e) XRD images of the LiCl products. | ||
From the precipitation reaction (Fig. 6a), it can be seen that milky white sediments are produced. The white precipitates are made up of two crystalline phases of CaCO3, as can be observed from the precipitates’ XRD patterns (Fig. 6b), which match the standard maps of CaCO3 (PDF#72-1937) and CaCO3 (PDF#72-0506). This may be related to external factors such as pH, the heating technique and the stirring speed, which require more discussion in terms of microscopic thermodynamics and macroscopic dynamics. The microscopic morphology of the precipitates is shown in Fig. 6c, and its appearance is spherical with a dense and homogeneous structure. The energy spectrum analysis (Fig. 6d) reveals that the main components are C (20.75%), O (53.79%) and Ca (23.88%), which is in agreement with the results of the XRD phase analysis. A trace amount of impurity F ions (<1.58%) was also detected, which may be caused by the small amount of F ions doped into the active material powder during the leaching process, which is enriched in the precipitated slag in the form of insoluble CaF2.
To enrich the lithium salt by evaporation crystallization, the solution after removal of calcium ions was mixed with the solution after acid leaching. The crystallization reaction takes place in an evaporating dish. 50 mL of the mixture solution was heated in an electric oven at 150 °C. The solution was continuously stirred with a glass rod until crystallites formed. The crystallites were washed three times with ultrapure water and then dried in a drying oven at 105 °C for 24 h to obtain lithium salt products, and their morphology and phase components are shown in Fig. 6e. The XRD pattern of the LiCl crystal is in good agreement with that of the standard PDF card. The characteristics of the produced LiCl product and industrial grade LiCl are compared in Table S2.†46 LiCl has a higher moisture content and purity than industrial grade LiCl. However, more research and testing are needed to fully understand its electrochemical performance.
3.4 Transformation pathways/mechanism of target elements in the Ca(ClO)2 system
Based on the aforementioned investigations, we hypothesize that Fig. 7 depicts the LiFePO4 conversion process and reaction path in the Ca(ClO)2 system. | ||
| Fig. 7 Transformation mechanism of LiFePO4 in the Ca(ClO)2 system. | ||
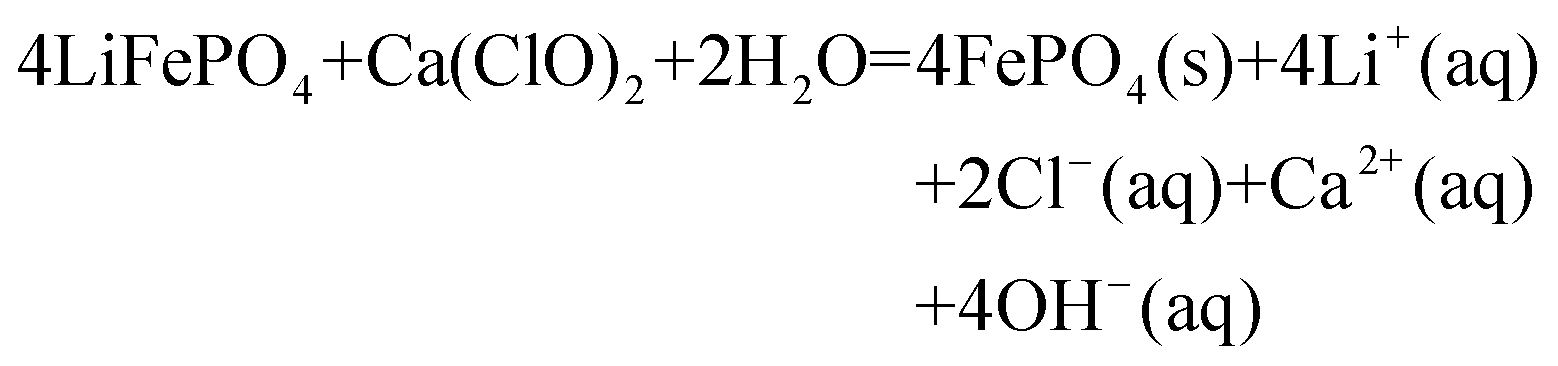 | (7) |
| Li+(aq) + Cl−(aq) = LiCl(s) | (8) |
 | (9) |
3.5 Techno-economic evaluation
To demonstrate the advantages of the proposed technology for the recovery of SLFPB-Ms in the Ca(ClO)2 system, an economic benefit assessment analysis was performed.30,48 As the recovery method has not yet reached industrialization, the evaluation is predicated on appropriate parameters derived from the 1.0 kg spent LiFePO4 battery recovery conducted in a laboratory. For more details, see text S1. Utilizing the economic analysis approach, the flow, material balance, reagent usage, energy consumption, and input–output costs of the recycling process were meticulously documented. The input costs of energy and chemical reagents are labeled in red font and the output benefits of the recycled products are labeled in green font, as shown in Fig. 8. | ||
| Fig. 8 Techno-economic evaluation (a) input and output for disposal of 1.0 kg of spent LiFePO4 batteries, (b) economic comparison of two processes in the Ca(ClO)2 system, and (c) a graph of the inputs and outputs for our work versus the persulfate system. | ||
Fig. 8a and b show that process 1 (mechanochemical activation) and process 2 (hydrometallurgical enhanced leaching) have the same pretreatment step and therefore have the same input ($1.13) and output ($0.58) costs. Because process 1 takes longer than process 2, it uses more energy and costs more reagents during the separation step. Process 2 requires $4.744 while process 1 requires $3.914 in the purification step.
Furthermore, the benefits of recovering 1.0 kg of spent LiFePO4 batteries in the persulfate system proposed in our group's previous research were compared (Fig. S3†).40 The profits of the three processes are as shown in Fig. 8c: process 2 (5.037$) > persulfate system (4.768$) > process 1 (4.04$). This demonstrates that it is economically possible to recover spent LiFePO4 batteries through hydrometallurgical enhanced leaching in the Ca(ClO)2 system.
However, it is worth noting that the above data are from small-scale laboratory experiments. It is only a guideline analysis for its industrial feasibility and has not been put into large-scale commercial recovery, so it is not an accurate economic assessment.
4. Conclusion
A sustainable process to achieve enhanced dissociation of spent LiFePO4 cathode materials by both mechanochemical activation and hydrometallurgical enhanced leaching in a Ca(ClO)2 system is proposed. Compared with the traditional enhanced leaching system, Ca(ClO)2 acts as both an oxidizing agent and a purifying agent in the recovery process of SLFPB-Ms. This allows for the simultaneous release of Li+ ions and the precipitation of impurity ions, thereby cutting down on the processing workflow time. Compared with mechanochemical activation, Ca(ClO)2-assisted hydrometallurgical enhanced leaching can simultaneously achieve the separation and enrichment of the target components Li+ and Fe3+ and the Ca2+ impurities. LiCl products can be obtained by further evaporation and crystallization.On the basis of XRD, XPS and SEM characterization, a reaction pathway and mechanism of strong dissociation of LiFePO4 in the Ca(ClO)2 system were proposed. Furthermore, the technical-economic evaluation showed that Ca(ClO)2-assisted hydrometallurgical enhanced leaching has higher economic advantages. In summary, this recycling approach shows great promise for industrialization planning and is a low-cost, sustainable process that meets the needs for economic, environmental and efficient recycling of spent lithium-ion batteries.
Author contributions
Gongqi Liu: investigation, data curation, and writing – original draft preparation; Zejian Liu: visualization and data curation; Jing Gu: writing – reviewing and editing; Shujia Wang: writing – reviewing and editing; Haoran Yuan: supervision, formal analysis, project administration, and funding acquisition; Yufeng Wu: writing – reviewing and editing; Yong Chen: validation and supervision.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Key R&D Program of China, China (2022YFC3902600), the CAS Project for Young Scientists in Basic Research, China (YSBR-044), and the China Postdoctoral Science Foundation (2023M733510).We would like to thank AJE (https://www.aje.cn/services/editing) for its linguistic assistance during the preparation of this manuscript.
References
- L. Yue, C. Ma, S. Yan, Z. Wu, W. Zhao, Q. Liu, Y. Luo, B. Zhong, F. Zhang, Y. Liu, A. A. Alshehri, K. A. Alzahrani, X. Guo and X. Sun, Nano Res., 2022, 15, 186–194 CrossRef CAS.
- L. Yue, D. Wang, Z. Wu, W. Zhao, Y. Ren, L. Zhang, B. Zhong, N. Li, B. Tang, Q. Liu, Y. Luo, A. M. Asiri, X. Guo and X. Sun, Chem. Eng. J., 2022, 433, 134477 CrossRef CAS.
- C. M. Costa, J. C. Barbosa, R. Gonçalves, H. Castro, F. Del Campo and S. Lanceros-Méndez, Energy Storage Mater., 2021, 37, 433–465 CrossRef.
- X. Jin, P. Zhang, L. Teng, S. Rohani, M. He, F. Meng, Q. Liu and W. Liu, Waste Manage., 2023, 165, 189–198 CrossRef CAS PubMed.
- Y. Tong, C. Qin, X. Zhu, Z. Lv, X. Huang and J. Chen, ACS Sustainable Chem. Eng., 2023, 11, 6722–6730 CrossRef CAS.
- K. Yan, Q. Chen, Z. Zhang, H. Nie, R. Wang and Z. Xu, Green Chem., 2023, 25, 9156–9166 RSC.
- C. Li, B. Liu, N. Jiang and Y. Ding, Nano Res. Energy, 2022, 1, 9120031 CrossRef.
- H. Zhou, Y. Zhang, L. i. Li and Z. Cao, Green Chem., 2023, 25, 7696–7706 RSC.
- F. Meng, J. McNeice, S. S. Zadeh and A. Ghahreman, Miner. Process. Extr. Metall., 2021, 42, 123–141 CrossRef CAS.
- X. Yang, T. Liu and C. Wang, Nat. Energy, 2021, 6, 176–185 CrossRef CAS.
- Q. Liang, H. Yue, S. Wang, S. Yang, K.-h. Lam and X. Hou, Electrochim. Acta, 2020, 330, 135323 CrossRef CAS.
- Y. Miao, L. Liu, Y. Zhang, Q. Tan and J. Li, J. Hazard. Mater., 2022, 425, 127900 CrossRef CAS PubMed.
- B. Makuza, Q. Tian, X. Guo, K. Chattopadhyay and D. Yu, J. Power Sources, 2021, 491, 229622 CrossRef CAS.
- W. Wang and Y. Wu, Resour., Conserv. Recycl., 2017, 127, 233–243 CrossRef.
- M. Fan, X. Chang, Y. Guo, W. Chen, Y. Yin, X. Yang, Q. Meng, L.-J. Wan and Y. Guo, Energy Environ. Sci., 2021, 14, 1461–1468 RSC.
- X. Zhou, W. Yang, X. Liu, J. Tang, F. Su, Z. Li, J. Yang and Y. Ma, Waste Manage., 2023, 155, 53–64 CrossRef CAS PubMed.
- G. Ji, J. Wang, Z. Liang, K. Jia, J. Ma, Z. Zhuang, G. Zhou and H. Cheng, Nat. Commun., 2023, 14, 584 CrossRef CAS PubMed.
- K. Liu, J. Wang, M. Wang, Q. Zhang, Y. Cao, L. Huang, M. Valix and D. C. Tsang, Green Chem., 2023, 25, 6642–6651 RSC.
- S. Jiang, X. Li, Q. Gao, X. Lyu, S. N. Akanyange, T. Jiao and X. Zhu, Sep. Purif. Technol., 2023, 324, 124630 CrossRef CAS.
- X. Wang, X. Wang, R. Zhang, Y. Wang and H. Shu, Waste Manage., 2018, 78, 208–216 CrossRef CAS PubMed.
- X. Li, Q. Zhou, X. Zhang, M. Ge, H. Zhang, Y. Yin and S.-T. Yang, ACS Sustainable Chem. Eng., 2023, 11, 14457–14466 CrossRef CAS.
- X. Li, S. Gao, F. Zhou, X. Qu, Y. Cai, D. Wang and H. Yin, ACS Sustainable Chem. Eng., 2023, 11, 12695–12703 CrossRef CAS.
- J. Kumar, R. R. Neiber, J. Park, R. A. Soomro, G. W. Greene, S. A. Mazari, H. Y. Seo, J. H. Lee, M. Shon and D. W. Chang, Chem. Eng. J., 2022, 431, 133993 CrossRef CAS.
- H. Li, S. Xing, Y. Liu, F. Li, H. Guo and G. Kuang, ACS Sustainable Chem. Eng., 2017, 5, 8017–8024 CrossRef CAS.
- X. Chen, C. Luo, J. Zhang, J. Kong and T. Zhou, ACS Sustainable Chem. Eng., 2015, 3, 3104–3113 CrossRef CAS.
- S. Yang, Y. Shi, Q. Li, K. Liu, H. Wang, Q. Pan, X. Zhang, G. Yang and Y. Su, Chem. Eng. J., 2023, 476, 146554 CrossRef CAS.
- Z. Jiang, P. Zhu, Y. Yang, W. Jin, G. Zou, H. Hou, J. Hu, W. Sun and X. Ji, Chem. Eng. J., 2023, 466, 143186 CrossRef CAS.
- E. Fan, L. Li, X. Zhang, Y. Bian, Q. Xue, J. Wu, F. Wu and R. Chen, ACS Sustainable Chem. Eng., 2018, 6, 11029–11035 CrossRef CAS.
- J. Zhang, J. Hu, Y. Liu, Q. Jing, C. Yang, Y. Chen and C. Wang, ACS Sustainable Chem. Eng., 2019, 7, 5626–5631 CrossRef CAS.
- D. Peng, J. Zhang, J. Zou, G. Ji, L. Ye, D. Li, B. Zhang and X. Ou, J. Cleaner Prod., 2021, 316, 128098 CrossRef CAS.
- Y. Dai, Z. Xu, D. Hua, H. Gu and N. Wang, J. Hazard. Mater., 2020, 396, 122707 CrossRef CAS PubMed.
- K. Liu, M. Wang, Q. Zhang, Z. Xu, C. Labianca, M. Komárek, B. Gao and D. C. Tsang, J. Hazard. Mater., 2023, 445, 130502 CrossRef CAS PubMed.
- Q. Jing, J. Zhang, Y. Liu, C. Yang, B. Ma, Y. Chen and C. Wang, J. Phys. Chem. C, 2019, 123, 14207–14215 CrossRef CAS.
- Y. Zhou, C. Li, C. Fan, M. Fu, L. Tao, M. Yu and M. Zhang, Environ. Prog. Sustainable, 2015, 34, 1586–1595 CrossRef CAS.
- Q. Wen, M. Ma, H. Hou, W. Yu, G. Gui, Q. Wu, K. Xiao, Y. Zhu, S. Tao and S. Liang, Environ. Res., 2022, 203, 111825 CrossRef CAS PubMed.
- X. Ma, Z. Cheng, W. Bai, R. Tang, G. Wu, X. Zhan and Z.-H. Hu, Sci. Total Environ., 2023, 856, 159080 CrossRef CAS PubMed.
- Q. Zhang, E. Fan, J. Lin, S. Sun, X. Zhang, R. Chen, F. Wu and L. Li, J. Hazard. Mater., 2023, 443, 130160 CrossRef CAS PubMed.
- J. Guan, Y. G. Li, Y. G. Guo, R. J. Su, G. L. Gao, H. X. Song, H. Yuan, B. Liang and Z. H. Guo, ACS Sustainable Chem. Eng., 2017, 5, 1026–1032 CrossRef CAS.
- K. Liu, Q. Tan, L. Liu and J. Li, Environ. Sci. Technol., 2019, 53, 9781–9788 CrossRef CAS PubMed.
- G. Liu, Z. Liu, J. Gu, H. Yuan, Y. Wu and Y. Chen, Chem. Eng. J., 2023, 471, 144265 CrossRef CAS.
- Z. Ruan, S. Wang, S. Zhang, X. Duan, J. Chen and X. Ren, J. Environ. Chem. Eng., 2023, 11, 110798 CrossRef CAS.
- R. Dedryvère, M. Maccario and L. Croguennec, Chem. Mater., 2008, 20, 7146–7170 CrossRef.
- Y. Jie, S. Yang, P. Shi, D. Chang, G. Fang, C. Mo, J. Ding, Z. Liu, Y. Lai and Y. Chen, Metals, 2021, 11, 1641 CrossRef CAS.
- W. Ma, P. W. Brown and D. Shi, Cem. Concr. Res., 1992, 22, 531–540 CrossRef CAS.
- Y. Wang, X. Peng, L. Chai and Y. Shu, Chin. J. Process Eng., 2009, 9, 84–87 CAS.
- N. Linneen, R. Bhave and D. Woerner, Sep. Purif. Technol., 2019, 214, 168–173 CrossRef CAS.
- B. Zhang, L. He, J. Wang, Y. Liu, X. Xue, S. He, C. Zhang, Z. Zhao, L. Zhou and J. Wang, Energy Environ. Sci., 2023, 16, 3873–3884 RSC.
- J. Zhang, W. Hu, J. Zou, X. Wang, P. Li, D. Peng, Y. Li, R. Zhao and D. He, ACS Sustainable Chem. Eng., 2022, 10, 13424–13434 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc04418a |
| This journal is © The Royal Society of Chemistry 2024 |