
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA highly efficient and sustainable catalyst system for terminal epoxy-carboxylic acid ring opening reactions†
Tizian-Frank
Ramspoth
 *a,
Jitte
Flapper
b,
Keimpe J.
van den Berg
b,
Ben L.
Feringa
*a and
Syuzanna R.
Harutyunyan
*a
*a,
Jitte
Flapper
b,
Keimpe J.
van den Berg
b,
Ben L.
Feringa
*a and
Syuzanna R.
Harutyunyan
*a
aStratingh Institute for Chemistry, Advanced Research Center Chemical Building Blocks Consortium (ARC CBBC), University of Groningen, Nijenborgh 7, 9747 AG Groningen, The Netherlands. E-mail: s.harutyunyan@rug.nl; b.l.feringa@rug.nl; t.ramspoth@rug.nl
bDepartment Resin Technology, AkzoNobel Car Refinishes BV, 2171 AJ Sassenheim, The Netherlands
First published on 22nd January 2024
Abstract
The nucleophilic ring opening of epoxides by carboxylic acids is an indispensable transformation for materials science and coating technologies. Due to this industrial significance, improvements in operational energy consumption and catalyst sustainability are highly desirable for this transformation. Herein, an efficient, environmentally benign and non-toxic halide free cooperative catalyst system based on an iron(III) benzoate complex and guanidinium carbonate is reported. The novel catalyst system shows improved activity over onium halide catalysts under neat conditions and in several solvents, including anisole and nBuOAc. Detailed mechanistic studies using FeCl3/DMAP as a catalyst revealed the importance of a carboxylate bridged cationic trinuclear μ3-oxo iron cluster and guanidinium carbonate or DMAP as a carboxylate reservoir due to its superior activity.
Introduction
The formation of β-hydroxyesters derived from the nucleophilic ring opening of epoxides by carboxylic acids is a reaction of high value. Apart from its relevance for accessing drug precursors1,2 and its occurrence in biologically relevant processes,3 the transformation is industrially important for the synthesis of monomers for photocurable resins and adhesives4 as well as for crosslinking reactions.5,6 Due to this industrial significance for large scale applications, the reaction demands exhaustive optimization towards sustainability. In this regard, research has mainly focussed on replacement of reactants with biofeedstock derived materials.7–12 However, the design of a broadly applicable and sustainable catalyst system with high activity and compatibility with environmentally benign solvent is necessary to fully exhaust the optimization potential for this invaluable transformation. The origin and environmental fate of the catalyst material from cradle to grave can have a significant impact on a large scale and its activity directly influences the energy consumption of the process.13,14Industrial applications largely employ catalysts such as triethylamine,15–24n-tetrabutylammoniumbromide25–34 or triphenylphosphine,35–44 which can have adverse effects on health,45–47 aquatic life46 or tropospheric ozone concentrations48,49 and often rely on halogenated petrochemical feedstock for their synthesis.50–53 More benign alternatives such as triethylbenzylammoniumchloride54–56 are likewise accessed through halogenated precursors57 and alkali hydroxides,58,59 alkali carbonates,60,61 metallic Lewis acids62,63 or heterogeneous catalysts64 often have limitations of catalytic activity or chemoselectivity with respect to polyether bond formation.
The need for a sustainable and highly active catalyst system for industrially relevant epoxy-carboxylic acid reactions, which can operate under neat conditions and in environmentally benign solvents, is therefore high.
Cooperative approaches utilizing a Lewis acid (LA) such as FeCl3 or CrBr3 in conjunction with an amine65 or specifically with pyridine66 have been reported to achieve enhanced catalytic activity and improved chemoselectivity for this reaction (Scheme 1a and b). The effect is rationalized by simultaneous activation of the epoxide electrophile and carboxylic acid nucleophile through Lewis acidic and basic interactions with the metal salt and the amine, respectively. However, these catalyst systems have not been further explored, as the design of an LA/(L)B cooperative catalyst system is challenging, due to potentially inhibiting interactions between both catalyst components. Furthermore, concerns regarding the environmental fate and the toxicological profile of catalyst components like pyridine67–69 and triethylamine48 remain for these systems as well.
 | ||
| Scheme 1 Reported LA/LB cooperative catalyst systems for the nucleophilic ring opening of epoxides by carboxylic acids; (a) iron or chromium salts in conjunction with amines,65 (b) FeCl3/pyridine66 and (c) this work: the ([Fe3O(benzoate)6(H2O)3]NO3)/guanidinium carbonate catalyst system. | ||
In this work, we have developed an effective, sustainable and easily accessible cooperative catalyst system, composed of [Fe3O(Benzoate)6(H2O)3]NO3 and guanidinium carbonate (Scheme 1c). Its constituents are naturally abundant, considered environmentally friendly and are already accessible via industrially established routes without the involvement of halogenated compounds. Additionally, this catalyst system outcompetes state-of-the-art catalysts in industrially common solvents, in green solvent alternatives and under neat conditions, which further contributes to a more sustainable reaction profile through reduction of the reaction time and temperature.
Results and discussion
In our quest to find a sustainable catalyst system for epoxy-carboxylic acid ring opening reactions we decided to focus on LA/LB cooperative systems as the most promising candidates to catalyse the target transformation. We started by screening metal-based Lewis acids in a model reaction of benzoic acid (1a) with 1,2-epoxyhexane (2) in toluene at reflux (Table 1). Under these conditions, Zr(acac)4 (Table 1, entry 2) and FeCl3 (Table 1, entry 3) showed the highest catalytic activity. The performance of iron(III) acetylacetonate and iron(III) nitrate salts was inferior compared to that of the chloride salt (Table 1, entries 4 and 5). Other metal salts, such as AlCl3, also showed lower catalytic activity and were limited by solubility (Table 1, entry 7 and ESI 3†), while triflate salts promoted epoxide homopolymerization instead (Table 1, entries 8 and 9).|
|
||
|---|---|---|
| Entry | Catalyst [2 mol%] | Conv., 3a + 4aa [%] |
Reaction conditions: 1a (1.5 mmol), 2 (1.5 mmol), reflux, 3 h.a Determined by 1H-NMR spectroscopy of 1a and 3a + 4a, regioisomer ratios (3a/4a) range from 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :33 to 74:26.b DMAN = 1,8-bis(dimethylamino)naphthalene. :33 to 74:26.b DMAN = 1,8-bis(dimethylamino)naphthalene. |
||
| 1 | None | 3 |
| 2 | Zr(acac)4 | 66 |
| 3 | FeCl3 | 64 |
| 4 | FeCl3 (1 mol%) | 32 |
| 5 | Fe(NO3)3·9 H2O | 52 |
| 6 | Fe(acac)3 | 37 |
| 7 | AlCl3 | 7 |
| 8 | Fe(OTf)3 | n.d. |
| 9 | Sc(OTf)3 | n.d. |
| 10 | DMAP | 63 |
| 11 | DMANb | 13 |
| 12 | Pyridine | 42 |
| 13 | 2,6-Lutidine | 11 |
| 14 | FeCl3/DMAP [1/1] | 82 |
| 15 | FeCl3/pyridine [1/1] | 78 |
| 16 | FeCl3/2-picolinic acid [1/1] | 40 |
| 17 | FeCl3 + L-proline [1/1] | 22 |
| 18 | FeCl3/2,2′-bipyridyne [1/1] | 42 |
| 19 | FeCl3/4,4′-bipyridyne [1/1] | 58 |
| 20 | FeCl3/DIPEA [1/1] | 71 |
| 21 | FeCl3/DMAN [1/1] | 70 |
| 22 | FeCl3/Barton's base [1/1] | 83 |
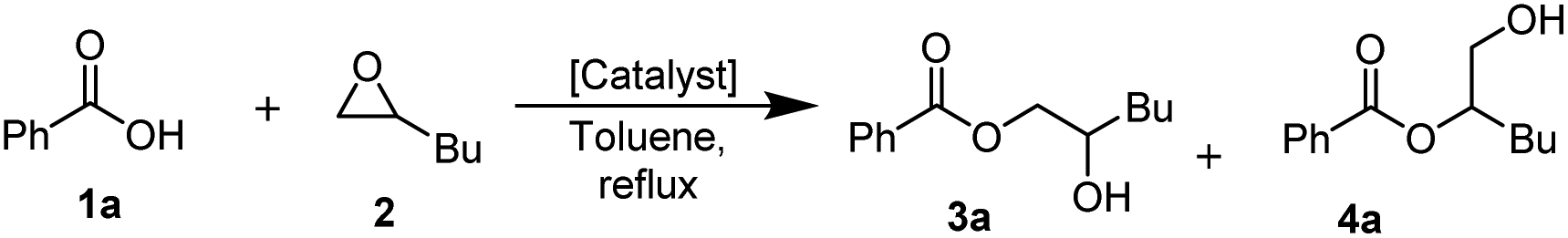
Following LA catalyst experiments, the screening of nitrogen-based Lewis bases was performed as well under the same reaction conditions (Table 1, entries 10–13). This led to the identification of 4-dimethylaminopyridine (DMAP) as the most potent structure (Table 1, entry 10).
We combined equal amounts of the best performing stand-alone LA and LB catalysts (FeCl3 and DMAP) and confirmed that the duo catalytic system surpasses the catalytic activity of the 2-fold amount of either of its components as stand-alone catalysts (Table 1, entry 14). Toluene was identified as the most optimal solvent for this transformation. Running the reaction in more polar solvents resulted in either lower catalytic activity or decreased chemoselectivity (ESI, 4†). The effect of the LA/LB ratio on the catalytic performance of the DMAP/FeCl3 combination was studied and it was found that there is a marginal increase in conversion for higher LA/LB ratios (ESI, 5†).
Next, we decided to re-evaluate the nature of the LB combined with FeCl3 (Table 1, entries 15–22). The results revealed that the DMAP/FeCl3 catalyst system performs slightly better than the previously reported pyridine/FeCl3 catalyst, while bidentate ligands such as picolinic acid, L-proline and 2,2′-bipyridine performed worse than 4,4′-bipyridine, and sterically hindered bases such as N,N-diisopropylethylamine (DIPEA), DMAN, and Barton's base performed well in conjunction with FeCl3 (for the full set of screening data, see ESI 3†).
To understand the precise role of DMAP, we performed the reaction using higher loadings of it as a standalone catalyst (Table 2). For 25% and 50 mol% of DMAP loading (Table 2, entries 1 and 2), the conversion towards the β-hydroxyester products (3a + 4a) reached 74% and 51% respectively. With 50 mol% of DMAP, 53% of β-hydroxyester products (3a + 4a) were isolated. Additionally, an ionic ring opening product (ROP), which formed via the reaction of DMAP with 1,2-epoxyhexane (2), was isolated as a formate salt (ROP-FA, 5b) after reverse phase column chromatography. Employing 75% of DMAP led to a conversion of 49% of (3a + 4a) (Table 2, entry 3).
|
|
|||
|---|---|---|---|
| Entry | DMAP [mol%] | (3a + 4a)a [%] | Yield, 5bb [%] |
| Reaction conditions: 1a (1.5 mmol), 2 (1.5 mmol), reflux, 20 h.a Determined by GC-FID; the value in parentheses corresponds to the isolated yield.b Isolated yield based on DMAP. Product 5 was also observed in entries 1 and 3 but the yield was not determined. | |||
| 1 | [25] | 74 | n.d. |
| 2 | [50] | 51 (53) | 52 |
| 3 | [75] | 49 | n.d. |
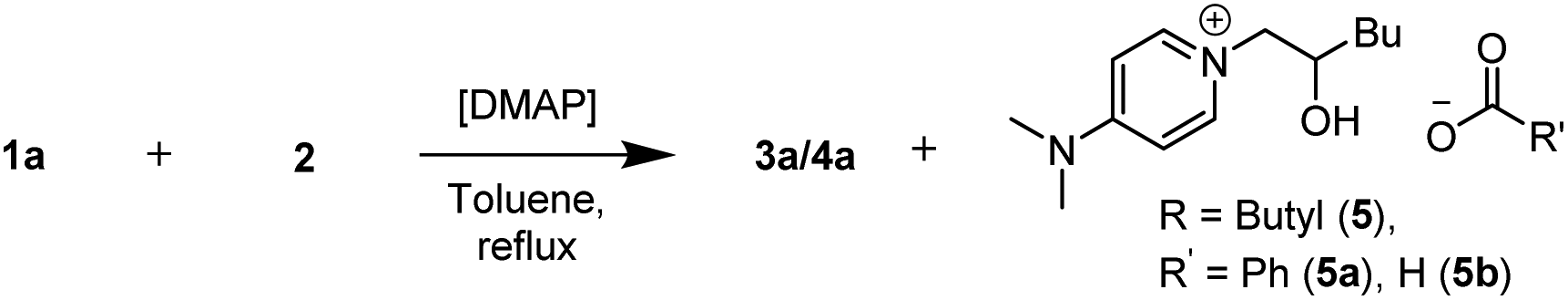
No formation of ROP (5) was observed in the absence of benzoic acid. Next, we screened ROP-FA (5b) as a stand-alone catalyst in combination with FeCl3 and achieved comparable catalytic activity to DMAP (ESI, 8†). Similar structures, resulting from the nucleophilic addition of an amine to an epoxide, have been previously proposed to be mechanistically relevant for the amine catalysed epoxide ring opening with carboxylic acids.7 Employing ROP-FA (5b) as a substrate with benzoic acid (1a) did not lead to product formation in the presence of DMAP, DMAP/FeCl3 or sodium benzoate (ESI, 8†).
Upon leaving a reaction mixture containing FeCl3/DMAP at room temperature, dark red, rod-shaped crystals were formed. We were unable to obtain a high-resolution structure by X-ray crystallography, but core structural features (ESI, 14†) have been identified by NMR-spectroscopy and HRMS. The obtained material consists of a cationic μ3-oxo trinuclear iron cluster, bridged by six benzoates, similar to the structures reported in the literature.70–721H- and 13C-NMR spectroscopy and HRMS confirmed the presence of ROP species in the crystal structure. The crystals exhibited catalytic activity similar to the FeCl3/DMAP system (Table 3, entry 1). To understand the role of the cationic μ3-oxo trinuclear benzoate type cluster, we independently prepared clusters 771 and 8 and tested them in our reaction. Cluster 7 showed almost identical catalytic activity to FeCl3 (Table 3, entry 2 versusTable 1 entry 4). Combination of 7 with DMAP and cluster 8 alone exhibited comparable catalytic performance to the FeCl3/DMAP system (Table 3, entries 2–4).
|
|
|||
|---|---|---|---|
| Entry | Catalyst [0.33 mol%] | Conv., 2a [%] | 3a/4ab |
| Reaction conditions: 1a (1.5 mmol), 2 (1.5 mmol), reflux, 3 h.a Determined by GC-FID, with mesitylene as the external standard.b Determined by 1H-NMR signals of both regioisomers of the product.c Assumed structure.d The ratio 7/DMAP = 0.33 mol%/1 mol%. | |||
| 1 | Fe3O(benzoate)6(ROP)3Clc | 73 | 2.7 |
| 2 | 7 | 33 | 2.4 |
| 3 | 7/DMAPd | 87 | 2.6 |
| 4 | 8 | 83 | 2.6 |

Next, we confirmed the presence of an equilibrium between the benzoate ligands coordinated to the cluster and free benzoic acid in solution, by observing a scrambled β-hydroxyester product when using 2,3,4,5,6-deuterobenzoic acid as a substrate in the presence of the [Fe3O(Benzoate)6(H2O)3]NO3 (7)/DMAP catalyst system (ESI, 13†). Following this, the electronic and steric effects of the benzoic acid substrate have been investigated (Scheme 2).
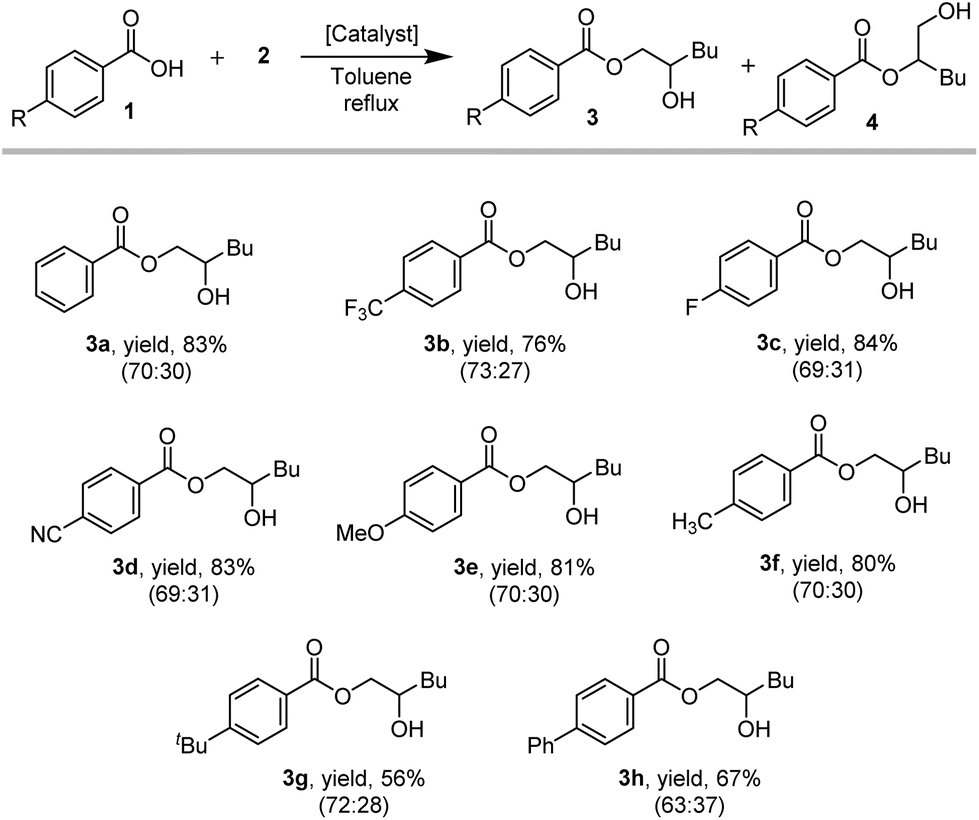 | ||
| Scheme 2 Substrate scope of para-substituted benzoic acids with 1,2-epoxyhexane. Reaction conditions: 1 (1.5 mmol), 2 (1.5 mmol), reflux, overnight; 1 mol% FeCl3 + 1 mol% DMAP. Combined isolated yields of both regioisomers (3 + 4) is given; only the major isomer is depicted for clarity, ratio of 3:4 depicted in parentheses. | ||
The products derived from p-CN (3d/4d; 69:31) as well as p-methoxy (3e/4e; 70:30) substituted benzoic acids were isolated in comparable yields to 3a/4a (70:30), indicating that the electronic properties of the carboxylic acid have a minor effect on the reaction outcome. However, significantly lower yields were obtained for the products with bulkier aromatic substituents, such as p-tbutyl (3g/4g; 72:28) or p-phenyl (3h/4h; 63:37) (Scheme 2). Furthermore, comparable yields and regioisomer ratios have been obtained for an extended substrate scope including aliphatic carboxylic acids and an aromatic epoxide for FeCl3/DMAP and TBAB as catalysts (ESI, 14†).
Next, we compared the conversions of different para-substituted benzoic acids (p-H, p-CF3, p-CH3) after 3 h of reaction time. From the assessed benzoic acids, the most nucleophilic p-toluic acid (1f) reacted fastest, followed by benzoic acid (1a) and p-CF3 benzoic acid (1b). Although the trend is marginal, it suggests an equal or slightly higher reactivity for more nucleophilic carboxylic acids (ESI, 9†).
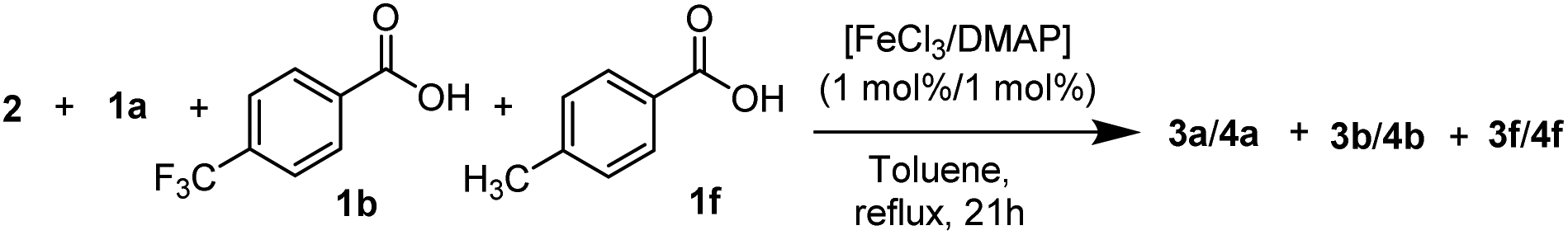
However, in a direct competition experiment, employing p-CF3, p-CH3 and p-H benzoic acid in equal amounts as substrates (totalling one equivalent of acid substrate w.r.t. 2), a different result was observed (Table 4). The p-CF3 benzoic acid (1b) reacted preferably in this scenario: after 20 minutes, 39% of 1b was consumed, while more than 90% of benzoic-(1a) and p-toluic acid (1f) were still present in the reaction medium (Table 4, entry 1). After 3 h, 1b was completely consumed, while 27% and 42% of 1a and 1f were still present, respectively (Table 4, entry 2). After 21 h, all three carboxylic acid substrates were completely consumed (Table 4, entry 3), although a decrease in 1H-NMR yield was observed, likely due to transesterification or other side processes.
|
|
||||||
|---|---|---|---|---|---|---|
| Time |
1aa |
(3a + 4a)b | 1ba | (3b + 4b)b | 1ga | (3g + 4g)b |
| a Conversions and formation of 1 in [%] determined by 1H-NMR of the crude. b Conversions and formation of (3 + 4) in [%] determined by 1H-NMR of the crude. 2/1a/1b/1f = 1/0.33/0.33/0.33 eq. in toluene, reflux. | ||||||
| 0.33 h | 7 | 7 | 39 | 30 | 5 | 6 |
| 3 h | 73 | 75 | 100 | 100 | 58 | 59 |
| 21 h | 100 | 99 | 100 | 93 | 100 | 92 |
In order to gain a conclusive mechanistic insight, we studied the reaction kinetics. We determined the reaction orders in catalysts and substrates for the FeCl3/DMAP system and DMAP as a standalone catalyst (for the results with FeCl3 as a standalone catalyst see ESI 18–21†) using the visual kinetics analysis method described by Burés et al.73 Catalyst degradation and product inhibition effects were ruled out by performing "same excess” experiments at two different starting concentrations for each of the catalyst systems (ESI, 18†). For the FeCl3/DMAP system, the reaction is zero order in benzoic acid (Table 5), while for the DMAP catalysed reaction, a half order in benzoic acid (1a) was found. For both the DMAP catalysed reaction and the cooperative FeCl3/DMAP system, a first order in epoxide and the catalyst was determined.
| Entry | Catalyst | Reaction order in | ||
|---|---|---|---|---|
| 1a | 2a | Catalyst | ||
| Reaction conditions: toluene, reflux, oN. | ||||
| 1 | FeCl3/DMAP (1:1) |
0 | 1 | 1 |
| 2 | DMAP | 0.5 | 1 | 1 |
The results indicate a correlation between the reactivity and acidity of the nucleophiles. In a direct competition experiment, the observed selectivity across the three acids is contrary to the observed conversion tendencies of the p-substituted acids as standalone substrates (ESI, 9†). The slower reacting p-CF3 benzoic acid (1b) appears to be consumed faster than its more electron rich analogues (1a, 1f), which suggests that the ionic ROP and potentially unreacted DMAP are acting predominantly as a base. In alignment with the change in the order of the acid from 0.5 in the DMAP catalysed reaction to 0 when using the FeCl3/DMAP catalyst system, we propose that the main purpose of the Lewis basic species is to create a reservoir of carboxylates via deprotonation of the carboxylic acid.
The carboxylate nucleophile is consumed from within this reservoir, whereas the selectivity in a direct competition experiment between different carboxylic acid substrates is determined by the relative concentrations of the respective carboxylates present in the reaction medium. As the amount of base is limited, the acid with the highest pKa is the dominant carboxylate in the reservoir, leading to the observed preference in acid consumption during the ring opening of 2.
Based on the acquired data, a mechanistic proposal for this cooperative catalytic transformation is presented in Scheme 3.
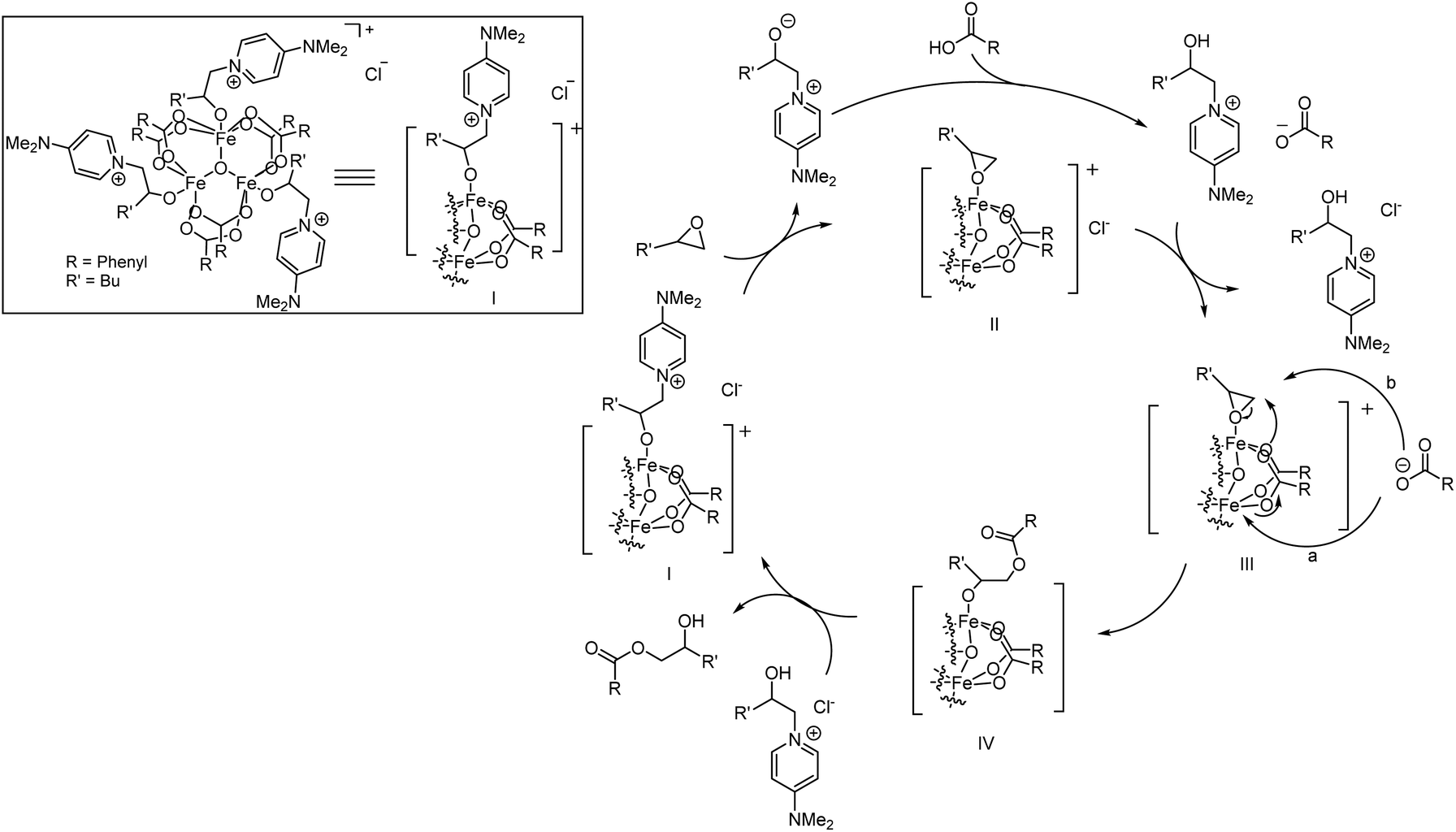 | ||
| Scheme 3 Proposed catalytic cycle for the FeCl3/DMAP catalyzed nucleophilic ring opening reaction of epoxides with carboxylic acids. | ||
Formation of I can proceed via coordination of carboxylic acid in the presence of ROP/DMAP as a suitable ligand to occupy the remaining coordination vacancies. Ligand exchange in favour of the epoxide gives rise to species II, where both substrates coordinate to the catalyst. The released ROP/DMAP deprotonates a carboxylic acid molecule outside of the coordination sphere of the catalyst, followed by exchange of the counter ion, leading to species III. In the following step, either an intramolecular nucleophilic attack of a carboxylate ligand on the coordinated epoxide, accompanied by a concerted regeneration of the catalytically active cluster structure from the carboxylate which is present as a counter ion (Scheme 3, a) or a direct outer sphere type attack of the carboxylate counterion (Scheme 3, b) occurs. This leads to the formation of neutral species IV, with the product coordinated to the cluster via its generated alcoholate moiety. Upon protonation, the β-hydroxyester product is released and species I is regenerated.
Steps III and IV are expected to be rate determining, as no product inhibition is observed and the previous steps are characterized by zero order behaviour of the carboxylic acid and its acidity dependent selectivity.
Considering drawbacks of the FeCl3/DMAP catalyst system, such as the corrosiveness of the iron source and the toxicity, the detrimental biodegradability and the negative impact of DMAP on aquatic life,74 we decided to explore sustainable alternatives based on the above mechanistic proposal. Based on our results, FeCl3 can be readily replaced by the [Fe3O(Benzoate)6(H2O)3]NO3 cluster (7), which avoids potential HCl formation. As DMAP is proposed to increase the reactivity of the nucleophile via formation of a carboxylate based ionic species, a suitable surrogate would have to combine the ability to irreversibly deprotonate the carboxylic acid with high stability of the formed cation, in order to promote the reaction of the carboxylate. We hypothesised that the naturally ubiquitous guanidine75 would be a proficient substitute to achieve high catalytic activity in conjunction with the iron(III) benzoate cluster (7), due to its high basicity and the stability of the corresponding cation.76 As guanidine reacts with atmospheric water and CO2,77,78 guanidinium carbonate (GC) was used. We were delighted to see that GC exhibited equally high activity in conjunction with [Fe3O(Benzoate)6(H2O)3]NO3 (7) as the initial FeCl3/DMAP system under model conditions (Table 6, entries 1 and 2). Interestingly, unlike DMAP, GC has no significant effect on the reaction progress when employed as a standalone catalyst (Table 6, entry 3). The regioisomeric ratios observed for both cooperative catalyst systems are in alignment with the findings reported for the FeCl3/DMAP system (Scheme 2).
| Entry | Catalyst | [mol%] | Conv. [%], 1 h | Conv. [%], 3h | ||
|---|---|---|---|---|---|---|
| 1a | 2 | 1a | 2 | |||
| Reaction conditions: 1a (1.5 mmol), 2 (1.5 mmol), reflux in toluene.a Determined by 1H-NMR spectroscopy.b Determined by GC-FID.c GC = guanidinium carbonate. Regioisomeric ratios for entries 1 and 2 are 69:31 after 20 h. |
||||||
| 1 | 7/GCc | [0.33/1] | 57 (69:31) |
53 | 86 | 84 |
| 2 | FeCl3/GC | [1/1] | 57 (69:31) |
58 | 83 | 82 |
| 3 | GCc | [2] | n.d. | n.d | 9 | 9 |
To render the system more environmentally friendly, we explored anisole and n-butylacetate (nBuOAc)79 as alternative solvents (Table 7). The catalytic activity of our 7/GC catalyst system in anisole at 115 °C was comparable to that of its performance in toluene at reflux and it slightly improved in nBuOAc (Table 7, entry 1). In contrast, the activity of the FeCl3/DMAP system, other common ammonium salts and PPh3 was worse in both solvents (Table 7, entries 2–5). Tetraphenylphosphoniumbromide (PPh4Br), on the other hand, showed similar activity to 7/GC, both in anisole and nBuOAc (Table 7, entry 6); however, it shares sustainability disadvantages with the other onium salts, due to its synthesis pathway.80 The regioisomeric ratios observed after reaction completion were comparable across all tested catalyst systems. In addition, the activity results obtained in (1-methoxy-2-propyl) acetate as a solvent were comparable to the catalyst performances observed in anisole (ESI, 23†).
| Entry | Catalyst | [mol%] | Conv. [%], 1 h in Anisole | Conv. [%], 1 h, in nBuOAc | ||
|---|---|---|---|---|---|---|
|
1aa |
2b |
1aa |
2b |
|||
| Reaction conditions: 1a (1.5 mmol), 2 (1.5 mmol), 115 °C in anisole or nBuOAc, [a], [b], and [c] are the same as in Table 6; regioisomeric ratios after 20 h are within the range of 68–32 to 73–27 (for details see the ESI†). | ||||||
| 1 | 7/GCc | 0.33/1 | 51 | 52 | 66 | 63 |
| 2 | FeCl3/DMAP | 1/1 | 30 | 30 | 41 | 43 |
| 3 | TBAB | 2 | 19 | 17 | 26 | 27 |
| 4 | Et3NBn | 2 | 19 | 20 | 24 | 27 |
| 5 | PPh3 | 2 | 20 | 19 | 16 | 11 |
| 6 | PPh4Br | 2 | 47 | 52 | 55 | 53 |
Next, we looked into the recyclability of the 7/GC catalyst system (Fig. 1). Re-addition of substrates 1a and 2 to a reaction performed in anisole in the presence of the 7/GC catalyst system after 16 h showed catalytic activity similar to a standard run (79% vs. 82% conversion of 2, 3 h). The recycled catalyst material was isolated in 72% yield through precipitation (assuming guanidinium benzoate formed) from reaction mixtures after 16 h. The recycled material showed a slight drop in the activity (74% vs. 82% conversion of 2, 3 h). In terms of recyclability, 7/GC is advantageous over the FeCl3/DMAP and FeCl3/pyridine system, both of which contain a liquid catalytic component (DMAP forms the liquid ionic species 5 under reaction conditions). This non-volatile character has also operational benefits for large scale industrial applications.
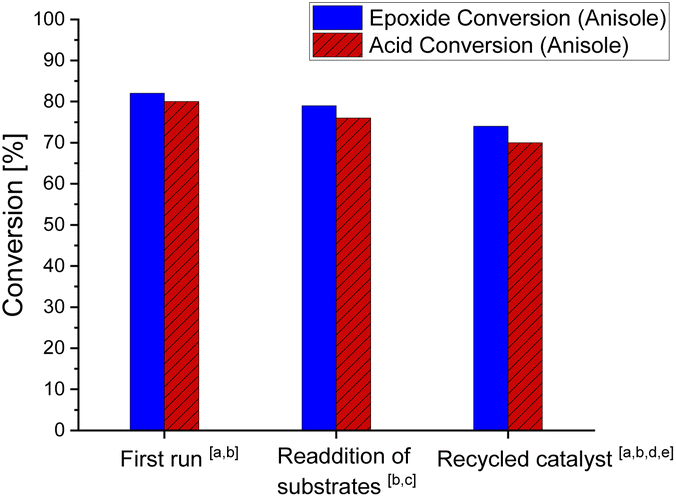 | ||
| Fig. 1 Reaction conditions: 1a (4.5 mmol), 2 (4.5 mmol), 0.33/1 mol% 7/GC, reflux in toluene, and v = 9 mL, adetermined by 1H-NMR spectroscopy, bdetermined by GC-FID, cre-addition to reaction after 16 h dequal isolated mol ratio (0.33/1) of [Fe3O(Benzoate)6(H2O)3]NO3 and guanidinium benzoate assumed, and e1a (1.5 mmol), 2 (1.5 mmol), and v = 3 mL. | ||
To demonstrate the industrial applicability further, the catalyst system was tested in the synthesis of 2-hydroxy-3-phenoxypropyl methacrylate (11), a common structural motif in dental composites81 and photosensitive resins.82 When the reaction was performed neat at 65 °C, the [Fe3O(Benzoate)6(H2O)3]NO3(7)/GC system was superior to currently employed catalysts such as TBAB83 or triethylamine (Et3N).81 Importantly, it performed significantly better than the other common alternative onium halide catalysts tested, including PPh4Br (Table 8, entries 1–6). After 3 hours, the acid was almost completely consumed by the 7/GC catalyst, whereas TBAB led to 47% of product formation and PPh4Br reached 60% conversion to the product in the same time period.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Amount [mol%] | 11 [%] | |
| 1 h | 3 h | |||
| Reaction conditions: 9 (1.5 mmol), 10 (1.5 mmol), 65 °C.a Determined by 1H-NMR spectroscopy.b GC = guanidinium carbonate. For all tested catalysts, only traces of side products have been observed. | ||||
| 1 | 7/GCb | [0.33/1] | 59 | 88 |
| 2 | TBAB | [2] | 24 | 47 |
| 3 | Et3N | [2] | 3 | 8 |
| 4 | Et3NBn | [2] | 22 | 48 |
| 5 | PPh3 | [2] | 26 | 51 |
| 6 | PPh4Br | [2] | 36 | 60 |
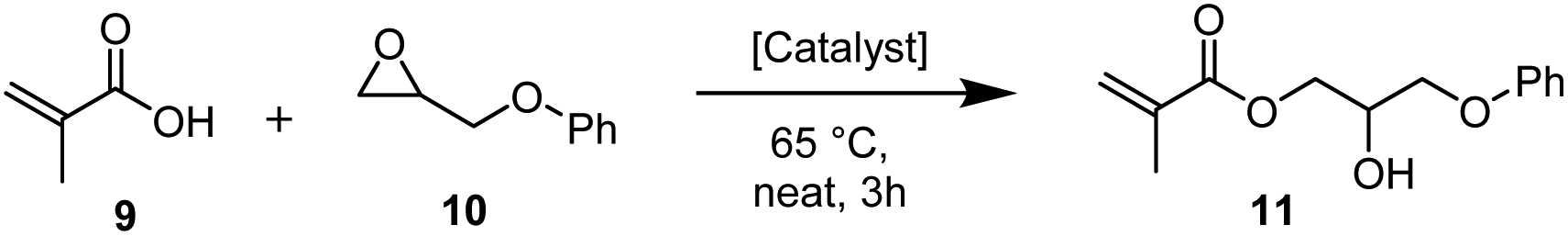
For all tested catalysts, the depicted isomer is the major product and only traces of side product formation have been observed.
We opted to conclude this study with an estimation of the sustainability benefit of the [Fe3O(Benzoate)6(H2O)3]NO3/guanidinium carbonate system. The main advantage of the introduced catalyst system comes from its higher activity under neat conditions and in green solvent alternatives, compared to the largely utilized onium halide and amine catalysts. Additionally, current information suggests that 7/GC is more environmentally benign than other common catalysts, including DMAP and pyridine, considering the toxicological profiles, synthesis pathways and environmental fates. The iron catalyst component is readily synthesized by ligation of Fe(NO3)3·9 H2O as a halogen free iron precursor, with sodium benzoate in water. The synthesis was successfully conducted on a gram scale. Benzoic acid, industrially produced via oxidation of toluene,84,85 which has in itself still limited accessibility through renewable resources,86–88 is abundantly present in biomass (e.g. plants, fruits, styrax tree).89 It is, like sodium benzoate, used as a food additive.90 The iron complex can be isolated almost quantitatively via precipitation and washing with water. The expected side products are sodium nitrate, used as a fertilizer,91 and nitric acid, a reactant for the synthesis of the iron precursor.92 The carbonate salt of guanidine, an abundant motif in nature93,94 that can be accessed photosynthetically,95 is generated as a side product in the pyrolysis of the metabolic waste product urea in the presence of water and CO2 in the synthesis of melamine.96–98 It is already used as an ingredient in hair relaxer formulations.99 Based on the current assessment of regulatory needs conducted by the European Chemicals Agency (ECHA), guanidine carbonate is unlikely to pose risks of toxicity or bioaccumulation and does not require further regulatory risk management100 in contrast to various issues associated with the use of TBAB46,49,50 and other common catalysts (see ESI 27†).
Conclusions
In this work, we have identified [Fe3O(Benzoate)6(H2O)3]NO3(7)/guanidinium carbonate as a highly active, non-volatile, sustainable and non-toxic, halide free cooperative catalyst system for the ring opening reaction of terminal epoxides with carboxylic acids. This system has been demonstrated to be superior to currently employed catalysts in the synthesis of 2-hydroxy-3-phenoxypropyl methacrylate at 65 °C under solvent free conditions. In addition, [Fe3O(benzoate)6(H2O)3]NO3/guanidinium carbonate proved to be highly active in toluene and 1-methoxy-2-propylacetate and no significant loss of activity was observed when switched to anisole or nBuOAc as a greener solvent alternative. Furthermore, its recyclability has been demonstrated in anisole. The development resulted from detailed mechanistic studies of the FeCl3/DMAP catalyst system, which led to identification of a cationic carboxylate bridged μ3-oxo trinuclear iron cluster and a basic ionic species formed via nucleophilic ring opening of the epoxide by DMAP as catalytically active structures. The presented data support a mechanistic pathway operating via a cooperative interaction of the DMAP/ionic species and the iron(III) benzoate cluster, allowing the substitution of FeCl3 with [Fe3O(Benzoate)6(H2O)3]NO3 and replacement of DMAP by guanidinium carbonate.Author contributions
T.-F. R. designed the project, carried out the research, visualised and analysed the data and prepared the original manuscript draft. S. R. H. conceptualized and supervised the research project with contributions from B. L. F, K. v. d. Berg. and J. F. All authors commented on the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is part of the Advanced Research Center for Chemical Building Blocks Consortium,ARC CBBC, which is co-founded and co-financed by the Dutch Research Council (NWO) and the Netherlands Ministry of Economic Affairs and Climate Policy. The authors thank Renze Sneep for high resolution mass spectrometry measurements.References
- Y. Luo, Y. Zhang, P. Zhou, Y. He, H. Zhu, X. Li, X. Wu and H. Liu, Preparation method for 3-chloro-2-hydroxy propyl aromatic acid ester, CN108623461A, 2018, A China National Intellectual Property Administration.
- M. R. Monaco, S. Prévost and B. List, Angew. Chem., Int. Ed., 2014, 53, 8142–8135 CrossRef CAS PubMed.
- M. Arand, H. Wagner and F. Oesch, J. Biol. Chem., 1996, 8, 4223–4229 CrossRef PubMed.
- L. Zhou, Y. Song, P. Wang and H. Chen, Efficient synthesis method of hydroxypropyl methacrylate, CN114634414A, 2022, A China National Intellectual Property Administration.
- S. Thomas, C. Sinturel and R. Thomas, Micro and Nanostructured Epoxy/Rubber Blends, Wiley-VCH, 2014 Search PubMed.
- M. J. Gadman, J. J. Florio and M. C. Salvi, CoatingsTech, 2023, 18–31 CAS.
- S. P. Amaud, N. M. Malitowski, K. M. Casamayor and T. Robert, ACS Sustainable Chem. Eng., 2021, 9, 17142–17151 CrossRef.
- S. Wang, J. Dai, N. Teng, J. Hu, W. Zhao and X. Liu, ACS Sustainable Chem. Eng., 2020, 8, 16842–16852 CrossRef CAS.
- T. P. Kainulainen, P. Erkkilä, T. I. Hukka, J. A. Sirviö and J. P. Heiskanen, ACS Appl. Polym. Mater., 2020, 2, 3215–3225 CrossRef CAS.
- Y. Sun, Z. Zhou, H. Jiang, Y. Duan, J. Li, X. Liu, L. Hong and C. Zhao, Dent. Mater., 2022, 38, 281–293 CrossRef CAS PubMed.
- S. Wang, Y. Wu, J. Dai, N. Teng, Y. Peng, L. Cao and X. Liu, Eur. Polym. J., 2020, 123, 109439 CrossRef CAS.
- J.-T. Miao, S. Peng, M. Ge, Y. Li, J. Zhong, Z. Weng, L. Wu and L. Zheng, ACS Sustainable Chem. Eng., 2020, 8, 9415–9424 CrossRef CAS.
- K. Schröder, K. Matyjaszewski, K. J. T. Noonan and R. T. Mathers, Green Chem., 2014, 16, 1673–1686 RSC.
- A. Kokel and C. Schäfer, Application of Green Chemistry in Homogeneous Catalysis, Green Chem., 2018, 375–414 Search PubMed . Elsevier, ISBN 9780128092705.
- Z. Wang, Y. Hu and J. Yu, Anaerobic sealant and preparation method thereof, CN115160962A, 2022, A China National Intellectual Property Administration.
- I. S. Sirotin, E. A. Gorbunova, V. Xuan-Son, D. V. Onichin and V. V. Kireev, Phosphase-containing oligoester acrylate and method for its production, RU2743697C1, 2019, Federal Service for Intellectual Property (Rospatent).
- J. Hao, Silane coupling agent containing hydroxyl group and (methyl) acryloyloxy, CN113968879A, 2021, A China National Intellectual Property Administration.
- L. Wang, J. Xiao and Y. Wu, High-weather-resistance water-based modified polyacrylic acid bridge concrete protective coating, CN116463022A, 2023, A China National Intellectual Property Administration.
- W. Shan, W. Li, H. Wang, Q. Zhang, Z. Hua and X. Han, Hexa-functional group polyurethane acrylate containing triazine ring, and preparation method and application thereof, CN110872372A, 2020, A China National Intellectual Property Administration.
- H. Irie and O. Shibata, Active energy ray-curable resin composition and concrete protection material, JP2023000195A, 2023, Japan Patent Office.
- Y. Chen, Resin composition, glue dripping material, glue pressing material and preparation methods and application of glue dripping material and glue pressing material, CN113004855A, 2021, A China National Intellectual Property Administration.
- P. J. Homnick, T. P. Klun, B. R. Pietz, C. S. Lyons and K. K. Stensvad, Fluorinated coupling agents and fluorinated (co)polymer layers made using the same, WO2021229338A1, 2021, World Intellectual Property Organization.
- S. Wang, X. Liu, J. Lu, L. Chen, T. Sun and W. Yao, Photosensitive polypropylene material and preparation method thereof, CN114085457A, 2022, A China National Intellectual Property Administration.
- B. Ren and T. Guan, Polymerizable polyurethane associative thickener with side group at tail end as well as preparation method and application of polymerizable polyurethane associative thickener, CN111944112A, 2020, A China National Intellectual Property Administration.
- H. Fan, J. Zheng, Y. Cai, X. Liang and J. Wei, Ultraviolet light-thermocuring composition for 3D printing and application of ultraviolet light-thermocuring composition, 112794943A, A China National Intellectual Property Administration, 2021 Search PubMed.
- F. Xu, S. Wang, Y. Li, X. Ding and Y. Dong, Cationic softening agent and preparation method and application thereof, CN113373690A, 2021, A China National Intellectual Property Administration.
- P. Desai and R. N. Jagtap, Photo-curable multifunction acrylated/methacrylated epoxy resin and one-pot preparation thereof, 2022153095A1, World Intellectual Property Organization, 2022 Search PubMed.
- S. H. Koo, C. Y. Boo, H. I. Kim, J. H. Bang, S. H. Yang, S. H. Han and H. S. Yeo, Novel glyceride compound and preparation method thereof, WO2022260401A1, 2022, World Intellectual Property Organization.
- Y. Miyamoto, M. Kamiya, M. Aburano and J. M. Yatvin, Lithographic printing plate precursor and method of use, WO2022051095A1, 2022, World Intellectual Property Organization.
- X. Zhang, M. Han and J. Wang, Preparation of cationic surfactants, US2022033348A1, 2022, United States Patent and Trademark Office.
- L. Ning and D. Peng, Electronic material and use thereof, CN110713616A, 2020, A China National Intellectual Property Administration.
- A. Gu, L. Ning, G. Liang and L. Yuang, Remoldable bismaleimide resin and application thereof, WO2021018158A1, 2021, World Intellectual Property Organization.
- J. Xie, Preparation method of ultraviolet-cured waterborne polyurethane containing HMMM resin type diol, CN112375204A, 2021, A China National Intellectual Property Administration.
- K. Satake, K. Asai, K. Sunahara and Y. Satake, Polymer material, WO2020175382A1, 2020, World Intellectual Property Organization.
- S. Yamada and Y. Kameyama, Epoxy (meth)acrylate resin composition, curable resin composition, cured product, and article, JP2021055005A, 2021, Japan Patent Office.
- M. Kiguchi, T. Harada and R. Ogiwara, Two-liquid curable coating agent and multilayer film, JP2022136407A, 2022, Japan Patent Office.
- Y. Sawai, K. Mishima, N. Miyashita and T. Yamakawa, Photosensitive colored composition, cured object, banks, organic electroluminescent element, and image display device, WO2022176976A1, 2022, World Intellectual Property Organization.
- M. Liu, J. Xu, G. Lin, W. Tu and Q. Ding, UV-curable water-based cathode electrophoretic coating as well as preparation method and application thereof, CN113801565A, 2021, A China National Intellectual Property Administration.
- J. Zhang, Y. Long, J. Chen and Y. Wang, Curable resin and liquid crystal sealant prepared from same, CN116217892A, 2023, A China National Intellectual Property Administration.
- T. Robert and S. A. Pérocheau, Process for producing free-radically curable compositions based on novel reactive diluents and reactive diluents, WO2022058473A1, 2022, World Intellectual Property Organization.
- T. Yuan, H. Wu, J. Huang, J. Zhang and Z. Yang, Bio-based multifunctional flame-retardant epoxy acrylate as well as preparation method and application thereof, CN113292910A, 2021, A China National Intellectual Property Administration.
- J. Miao, L. Wu, M. Ge, S. Peng and L. Zheng, Bio-based 3D printing resin and preparation method thereof, CN113583165A, 2021, A China National Intellectual Property Administration.
- J. Dai, S. Wang, X. Liu and J. Zhu, Light-cured resin based on organic polyatomic acid as well as preparation method and application of light-cured resin, CN111978444A, 2020, A China National Intellectual Property Administration.
- X. Xia, D. Lin, X. Wan, Y. Hong, Y. Chen, J. Han, J. Cai and T. Chen, Isocyanate-based epoxy resin rust-inhibitive primer, CN113637388A, 2021, A China National Intellectual Property Administration.
- (a) https://echa.europa.eu/registration-dossier/-/registered-dossier/14938/2/1 , accessed 14.10.23; (b) https://www.sigmaaldrich.com/NL/en/sds/vetec/v000751 , accessed 29.10.23.
- (a) https://echa.europa.eu/registration-dossier/-/registered-dossier/15982/2/1 , accessed 14.10.23; (b) https://www.sigmaaldrich.com/NL/en/sds/mm/8.18839 , accessed 29.10.23.
- (a) https://echa.europa.eu/registration-dossier/-/registered-dossier/13659/2/1 , accessed 14.10.23; (b) https://www.sigmaaldrich.com/NL/en/sds/mm/8.08270 , accessed 29.10.23.
- (a) D. Tong, J. Chen, D. Qin, Y. Ji, G. Li and T. An, Sci. Total Environ., 2020, 737, 139830 CrossRef CAS PubMed; (b) https://echa.europa.eu/en/registration-dossier/-/registered-dossier/14938/2/1 , accessed 03.12.2023; (c) https://www.sigmaaldrich.com/NL/en/sds/mm/8.08352?userType=anonymous , accessed 03.12.2023.
- S. Chen, L. Artiglia, F. Orlando, J. Edebeli, X. Kong, H. Yang, A. Boucly, P. C. Arroyo, N. Prisle and M. Ammann, ACS Earth Space Chem., 2021, 11, 3008–3021 CrossRef PubMed.
- M. A. Johnson, J. D. Reedy and K. Yang, Method for preparing tetrabutylammonium bromide, US3965178A, 1976, United States Patent and Trademark Office.
- X. Gong, J. Li, H. Li, W. Wang and Y. Cui, Synthesis method of tetrabutylammonium bromide, CN116023269A, 2023, A China National Intellectual Property Administration.
- A. Zhu, H. Zhou and L. Jin, Triphenylphosphine, CN108178773A, 2018, A China National Intellectual Property Administration.
- S. Guangri, B. Caifeng and S. Youmei, Synthesis of triphenyl phosphine, CN1069273A, 1993, A China National Intellectual Property Administration.
- W. Liu, P. Lu and C. Chen, Positive working photosensitive material, WO2020048957A1, 2020, World Intellectual Property Organization.
- M. Wang, X. Jian, J. Wang, Y. Zou, L. Zong, Z. Bi and C. Tang, Preparation method of high-refractive-index anti-blue-light modified epoxy acrylate material and optical filter, CN116023813A, 2022, A China National Intellectual Property Administration.
- Y. Zhang, X. Fan, X. Fan and K. Lu, High-toughness glass fiber and preparation method thereof, CN114606771A, 2022, A China National Intellectual Property Administration.
- Y. Yu, C. Zhou, H. Ying, J. Liu and P. Chen, Synthesis method of benzyltriethylammonium chloride, CN115521209A, 2022, A China National Intellectual Property Administration.
- L. Sun, T. Yang, H. Li and Y. Lou, Preparation method of glycidyl tertiary carboxylic ester, WO2013097724A1, 2013, World Intellectual Property Organization.
- M. Imai, K. Sugimoto, K. Fuji, A. Otsuji, T. Okhuma, M. Takagi, R. Suzuki and K. Takuma, Sulfur-containing unsaturated carboxylate compound and its cured products, US6458908B1, 2000, United States Patent and Trademark Office.
- M. Paradas-Palomo, S. Flores-Penalba, J. Garcia-Miralles, H.-G. Kinzelmann, R. M. P. Sebastián, J. C. Marquet, J. C. Aguilera, F. Ariolo and T. Hemery, Method for producing functionalized polyesters, WO2018206181A1, 2018, World Intellectual Property Organization.
- D. Yea, L. Yejin, K. Park and J. Lim, J. Ind. Eng. Chem., 2021, 97, 287–298 CrossRef CAS.
- G. Merfeld, C. Molaison, R. Koeniger, A. Ersin Acar, S. Mordhorst, J. Suriano, P. Irwin, R. S. Warner, K. Gray, M. Smith, K. Kovaleski, G. Garrett, S. Finley, D. Meredith, M. Spicer and T. Naguy, Prog. Org. Coat., 2005, 52, 98–109 CrossRef CAS.
- W. J. Blank, Z. A. He and M. Picci, J. Coat. Technol., 2002, 74, 33–41 CrossRef CAS.
- S. Behrouz, M. N. S. Rad, M. A. Piltan and M. M. Doroodmand, Helv. Chim. Acta, 2017, 100, e1700144 CrossRef.
- A. E. Gurgiolo, Process for preparation of β-hydroxy esters by reaction of organic carboxylic acids and vicinal epoxides, US4069242A, 1978, United States Patent and Trademark Office.
- Y. Zhao, W. Wang, J. Li, F. Wang, X. Zheng, H. Yun, W. Zhao and X. Dong, Tetrahedron Lett., 2013, 54, 5849–5852 CrossRef CAS.
- (a) https://echa.europa.eu/registration-dossier/-/registered-dossier/13681/2/1 , accessed, 29.10.23; (b) https://www.sigmaaldrich.com/NL/en/sds/sial/270970 , 29.10.23.
- Y. Grosse, D. Loomis, K. Z. Guyton, F. E. Ghissassi, V. Bouvard, L. Benbrahim-Tallaa, H. Mattock and K. Straif, Lancet Oncol., 2017, 18, 1003–1004 CrossRef PubMed.
- D. H. Stuermer, D. J. Ng and C. J. Morris, Environ. Sci., 1982, 16, 582–587 CrossRef CAS PubMed.
- A. M. Bond, R. J. H. Clark, D. G. Humphrey, P. Panayiotopoulos, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 1998, 11, 1845–1852 RSC.
- F. F. Moreira, T. Hasegawa, D. J. Evans and F. S. Nunes, J. Coord. Chem., 2007, 60, 185–191 CrossRef CAS.
- Y. Li, J. J. Wilson, L. H. Do, U.-P. Apfel and S. J. Lippard, Dalton Trans., 2012, 41, 9272–9275 RSC.
- C. D.-T. Nielsen and J. Burés, Chem. Sci., 2019, 10, 348–353 RSC.
- (a) https://echa.europa.eu/registration-dossier/-/registered-dossier/17217/2/1 , accessed 24.02.2023; (b) https://www.sigmaaldrich.com/NL/en/sds/mm/8.51055 , accessed, 29.10.23.
- B. Marescau, D. R. Deshmukh, M. Kockx, I. Possemiers, I. A. Qureshi, P. Wiechert and P. P. De Deyn, Metabolism, 1992, 41, 526–532 CrossRef CAS PubMed.
- A. Gobbi and G. Frenking, J. Am. Chem. Soc., 1993, 115, 2362–2372 CrossRef CAS.
- C. A. Seipp, M. J. Williams, M. K. Kidder and R. Custelcean, Angew. Chem., Int. Ed., 2016, 56, 1042–1045 CrossRef PubMed.
- S. Li and C. Lanzhen, Process for synthesizing guanidine carbonate, CN1560031A, 2005, A China National Intellectual Property Administration.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- S. Guancheng and H. Guoyu, Method for preparing tetraphenyl phosphoric bromide, CN101418014A, 2009, A China National Intellectual Property Administration.
- M. Trujillo-Lemon, K. L. Wall and K. Esquibel, Carbamate-methacrylate monomers and their use in dental applications, US20100307378A1, 2010, United States Patent and Trademark Office.
- S. Matsumoto, Photosensitive resin composition, JP2021117442A, 2021, Japan Patent Office.
- Y. Gao, Y. Wen, C. Zeng, F. Wang, J. Wang, M. Liu, Y. Bi, Q. Liu, S. Yan and J. Luo, Phenolic aldehyde modified mud-resistant polycarboxylate superplasticizer and preparation method thereof, CN111560092A, 2020, A China National Intellectual Property Administration.
- L. G. Wade, Organic Chemistry, Pearson new international ed., 2013, Pearson Education Limited, Harlow, p. 985 Search PubMed.
- A. Schoeters, Loop oxidation of toluene to benzoic acid, WO2017017524A1, 2017, European Patent Office.
- Y. P. Wijaya, I. Kristianto, H. Lee and J. Jae, Fuel, 2016, 182, 588–596 CrossRef CAS.
- T. Dai, C. Li, B. Zhang, H. Guo, X. Pan, L. Li, A. Wang and T. Zhang, ChemSusChem, 2016, 9, 3434–3440 CrossRef CAS PubMed.
- A. M. Niziolek, O. Onel, Y. A. Guzman and C. A. Floudas, Energy Fuels, 2016, 30, 4970–4998 CrossRef CAS.
- A. del Olmo, J. Calzada and M. Nuñez, Crit. Rev. Food Sci. Nutr., 2017, 57, 3084–3103 CrossRef CAS PubMed.
- https://www.food.gov.uk/business-guidance/approved-additives-and-e-numbers , accessed 10.07.23.
- J. Davidson and J. A. Le Clerc, J. Agron, 1917, 9, 145 CrossRef CAS.
- E. Wildermuth, H. Stark, G. Friedrich, F. L. Ebenhöch, B. Kühborth, J. Silver and R. Rituper, Iron Compounds, in Ullmann's Encyclopedia of Industrial Chemistry, 2000, Wiley-VCH, Weinheim Search PubMed.
- T. Walligmann, M. Tokarska-Schlattner, D. Neumann, R. M. Epand, R. F. Epand, R. H. Andres, H. R. Widmer, T. Hornemann and V. Saks, Molecular System Bioenergetics: Energy for Life, Wiley-VCH, 2007 Search PubMed.
- E. Schulze and E. Steiger, Biol. Chem., 1887, 11, 43–65 CrossRef.
- B. Wang, T. Dong, A. Myrlie, L. Gu, H. Zhu, W. Xiong, P. Maness, R. Zhou and J. Yu, Green Chem., 2019, 21, 2928–2937 RSC.
- A. Schmidt, K. Wegleitner, J. H. Hatzl, R. Sykora and F. Weinrotter, Process for the preparation of guanidine carbonate, US3952057A, 1972, United States Patent and Trademark Office.
- J. M. Lee, Process for manufacturing melamine from urea, US5384404A, 1993, United States Patent and Trademark Office.
- H. Gabriel and R. Dicke, Method for increasing the guanidine carbonate yield in a combined melamine process and guanidine carbonate treatment process, EP3626706A1, 2020, European Patent Office.
- P. T. Browning, Hair Relaxer, WO2003039291A2, 2003, World Intellectual Property Organization.
- https://echa.europa.eu/assessment-regulatory-needs/-/dislist/details/0b0236e1873b78fe , accessed, 24.02.2023.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc04301k |
| This journal is © The Royal Society of Chemistry 2024 |