
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot mechanochemical hydrogenation and acetylation of 4-nitrophenol to 4-aminophenol and paracetamol†
Jimin
Park
 abe,
Jacob S.
Maier
a,
Caria
Evans
bce,
Marta
Hatzell
ad,
Stefan
France
bce,
Carsten
Sievers
*abe and
Andreas S.
Bommarius
*abe
abe,
Jacob S.
Maier
a,
Caria
Evans
bce,
Marta
Hatzell
ad,
Stefan
France
bce,
Carsten
Sievers
*abe and
Andreas S.
Bommarius
*abe
aSchool of Chemical and Biomolecular Engineering, Georgia Institute of Technology, 311 Ferst Dr. NW, Atlanta, GA 30332, USA. E-mail: andreas.bommarius@chbe.gatech.edu; carsten.sievers@chbe.gatech.edu
bRenewable Bioproducts Institute, Georgia Institute of Technology, 500 10th St. NW, Atlanta, GA 30332, USA
cSchool of Chemistry and Biochemistry, Georgia Institute of Technology, 901 Atlantic Dr NW, Atlanta, GA 30332, USA
dGeorge W. Woodruff School of Mechanical Engineering, Georgia Institute of Technology, 801 Ferst Dr., Atlanta, GA 30332, USA
eCenter for a Renewables-based Economy from WOOD (ReWOOD), 500 10th St. NW, Atlanta, GA 30332, USA
First published on 29th February 2024
Abstract
The mechanochemical hydrogenation of 4-nitrophenol was examined in a ball mill reactor. Hydrogen gas flows through the reactor at ambient temperature and pressure in the presence of Pd/C catalyst and acetic anhydride. Under optimized conditions, simultaneous hydrogenation and acetylation in one pot increased paracetamol selectivity by avoiding undesirable side reactions of 4-aminophenol. Side product formation was further reduced through a novel bubbler configuration allowing for the delivery of acetic anhydride vapor to the reaction vessel. Ball mill results were compared to traditional stirred tank hydrogenations in a Parr reactor and shown to have superior yield and process mass intensity.
Introduction
Paracetamol, also known as acetaminophen or acetyl-para-aminophenol (APAP), is one of the most widely used over-the-counter (OTC) active pharmaceutical ingredients (APIs) in the world, with a global production rate of 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 metric tons per year.1 An analgesic used to alleviate fevers and pain, paracetamol, commonly prescribed, is listed on the WHO List of Essential Medicines.2 Despite its widespread use and importance, various countries are experiencing paracetamol shortages in part due to supply chain issues and highly localized production.1,3,4 Because of rising costs of processing pharmaceuticals as well as strict environmental regulations, paracetamol production has shifted away from developed countries. Fixing paracetamol supply shortages is not as simple as transferring existing processes back to developed nations. For the reshoring of drug supplies to be viable, new routes that mitigate existing environmental issues are important. As a result, there is an increased interest in developing new processes for paracetamol production with a focus on Green Chemistry. Such developments may reduce the dependence of many countries on pharmaceutical imports, improve supply chain stability, and provide an alternative to traditional paracetamol synthesis methods which suffer from poor environmental metrics.
000 metric tons per year.1 An analgesic used to alleviate fevers and pain, paracetamol, commonly prescribed, is listed on the WHO List of Essential Medicines.2 Despite its widespread use and importance, various countries are experiencing paracetamol shortages in part due to supply chain issues and highly localized production.1,3,4 Because of rising costs of processing pharmaceuticals as well as strict environmental regulations, paracetamol production has shifted away from developed countries. Fixing paracetamol supply shortages is not as simple as transferring existing processes back to developed nations. For the reshoring of drug supplies to be viable, new routes that mitigate existing environmental issues are important. As a result, there is an increased interest in developing new processes for paracetamol production with a focus on Green Chemistry. Such developments may reduce the dependence of many countries on pharmaceutical imports, improve supply chain stability, and provide an alternative to traditional paracetamol synthesis methods which suffer from poor environmental metrics.
State-of-the-art methods to produce paracetamol are dependent on petrochemical feedstocks, produce large excesses of waste, and often employ hazardous reagents.5 For example, the Hoechst-Celanese route (Beckmann rearrangement) employs hazardous compounds such as hydrofluoric acid and thionyl chloride.6 In addition, this route sources its phenol from the cumene process using the petrochemical benzene as feedstock. Another route begins from the nitration of benzene and features a subsequent Bamberger rearrangement which requires stoichiometric amounts of iron-acid reducing agents, forming equivalent amounts of non-reusable iron and iron–oxide sludge.7,8 Routes from nitrobenzene also suffer from poor selectivity through unavoidable aniline formation.9 New routes should start from renewable raw materials and mitigate harsh chemistries that fail to meet environmental regulations in developed countries. Lignin is a promising renewable source of hydroxylated aromatics that may be used as building blocks for various APIs because of its abundance, renewability, and carbon neutrality (Fig. 1).10
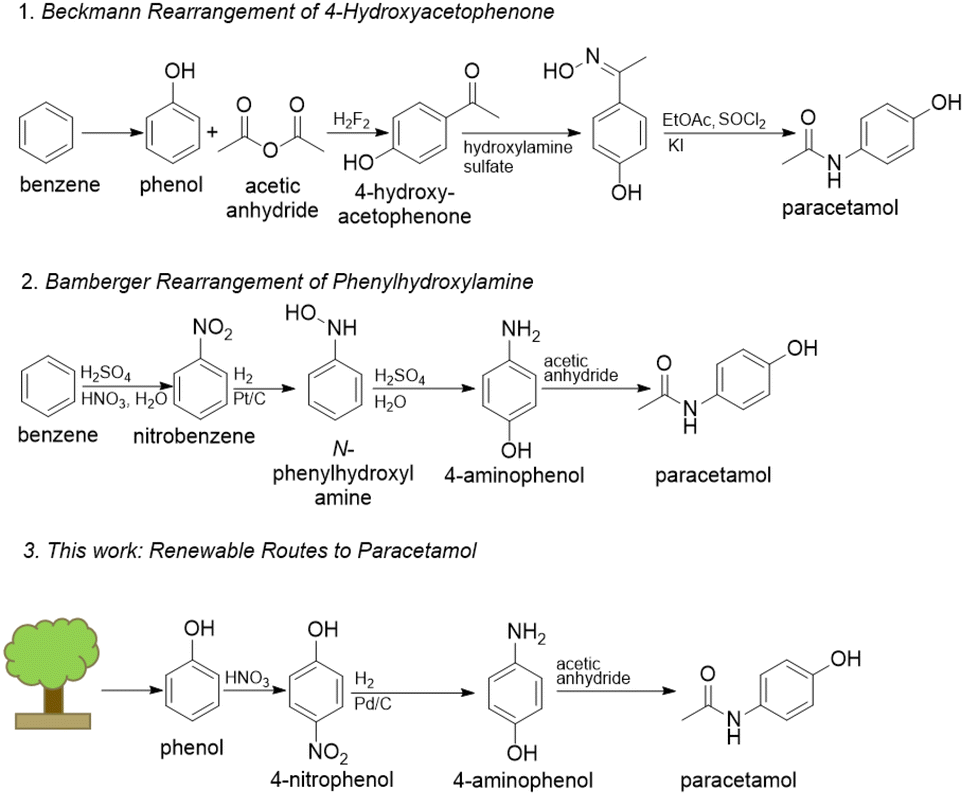 | ||
| Fig. 1 Current industrial routes to paracetamol as well as the proposed route.5,6,9 | ||
To that end, 4-nitrophenol may be a possible alternative feedstock to the commonly used nitrobenzene. 4-Nitrophenol can be obtained through the nitration of phenol, which can be obtained by lignin depolymerization followed by upgrading of the building blocks as an alternative to the non-renewable cumene process.10–12 Furthermore, the hydrogenation of 4-nitrophenol to 4-aminophenol catalysed by noble metals is more environmentally friendly than routes from nitrobenzene because of the elimination of the need for strong mineral acids and improved selectivity.13
Traditional methods of hydrogenating 4-nitrophenol are conducted in a batch configuration with high hydrogen pressures and solvent loads under RANEY®-Nickel or noble metal catalysts.8,13 These conditions introduce safety issues associated with the presence of high pressures of flammable hydrogen as well as poor Green Chemistry metrics from the high amount of required solvent. One method of avoiding pressurized hydrogen is to perform liquid phase transfer hydrogenation of 4-nitrophenol using reagents such as formates or alcohols.14 However, these processes still tend to require high solvent loads in batch phase.
Mechanochemistry provides a method to perform synthesis reactions in an environmentally friendly manner. It can quickly drive reactions between solid reactants with little to no solvents, often resulting in less energy use, reduced waste generation, and improved Green Chemistry metrics. Because of this, organic synthesis via ball-milling of reactants has been gaining increasing attention in recent years because of its often-advantageous Green Chemistry aspects.15–17 Paracetamol has been synthesized mechanochemically before, either through transfer hydrogenation of 4-nitrophenol or through the Beckmann rearrangement.18,19 However, current mechano-chemical methods to paracetamol remain sparse and use expensive reagents or significant amounts of additives. Additionally, mechanochemically conducted hydrogenation reactions, which traditionally require gas-phase reactants, are less well studied in the literature. To our knowledge, only one case of the mechanochemical transfer hydrogenation of 4-nitrophenol has been published so far, with none using hydrogen gas.18
The immediately obvious method of mechanochemical hydrogenation involves the use of catalytic transfer hydrogenation (CTH) agents such as formates and alcohols with noble metal catalysts, as demonstrated in a study by Portada et al., where 4-nitrophenol was milled with Pd/C in the presence of ammonium formate to obtain 4-aminophenol and paracetamol in high yields.18 Transfer hydrogenations of various reactants, such as carbonyls, ketones, and nitro derivatives have been increasingly discussed in the literature.18,20–22 However, a key question remains whether transfer hydrogenation is truly a preferred Green method of hydrogenation when compared to using elemental hydrogen. Indeed, transfer hydrogenation reagents suffer from pitfalls such as high cost, high molecular weight (compared to H2), and the fact that elemental hydrogen tends to be involved in their main synthesis routes. As such, transfer hydrogenation can result in poorer Green Chemistry metrics such as atom economy and E-factor.
Thus, the potential of gas-phase ball mill reactions for the direct catalytic hydrogenation of 4-nitrophenol with elemental hydrogen should be explored. In fact, gas-phase ball mill reactions have been well documented in the literature and provide the key advantage of strong solid–gas interactions under ambient pressures and minimal solvent loads.23–26 Furthermore, under ambient pressure gas flow conditions, the dangers of elemental hydrogen gas can be safely mitigated.
In this study, the catalytic transfer hydrogenation of 4-nitrophenol with formic acid is compared to the catalytic hydrogenation of 4-nitrophenol with hydrogen gas flow in a ball mill. Several reaction configurations were considered, including one-pot direct synthesis of paracetamol from 4-nitrophenol through the addition of acetic anhydride as well as different methods of acetic anhydride delivery to the ball mill reactor. These configurations are compared with respect to important Green Chemistry metrics such as conversion, selectivity, reaction mass efficiency (RME), and process mass intensity (PMI) with the goal of providing a clear recommendation for the most viable process.
Experimental
Materials
Paracetamol (APAP) (99%), Pd/C (5 wt%), Pt/C (5 wt%), and Ru/C (5 wt%) were obtained from Sigma Aldrich. 4-Nitrophenol (4-NP) (99%) was obtained from Acros Organics. 4-Aminophenol (4-AP) (98%) was obtained from Alfa Aesar. Formic acid (99%) and acetic anhydride (99%) were obtained from Fisher Scientific. Hydrogen, nitrogen, and argon were obtained from Airgas. All chemicals were used without further purification.Synthesis of side products
4-N-Hydroxyphenylformamide (4-NHPF) was synthesized by adding 4-aminophenol to an excess of formic acid and stirring at 120 °C for 2 h. Formic acid was evaporated off and the resulting white solid was washed three times with water, redissolved in methanol and dried with sodium sulfate. The methanol was evaporated off resulting in white solid 4-NHPF.4-Acetamidophenol acetate (4-APA) was synthesized by adding paracetamol to an excess of acetic anhydride and stirring at 80 °C for 2 h. Acetic anhydride was evaporated, and the solid products were purified by reverse phase column chromatography (C8 column) with a 30% acetonitrile/water mobile phase, resulting in an off-white solid.
4,4′-[(2-Amino-5-hydroxy-2,5-cyclohexadiene-1,4-diylidene)dinitrolo] bis-phenol (CAS 71082-02-5), hereafter called the “trimer”, was synthesized by reacting 4-aminophenol and Pd/C in isopropanol at 120 °C for 5 h. The resulting solid was purified by reverse phase column chromatography (C8 column) with a 30% acetonitrile/water mobile phase and used for quantification and identification.
Mechanochemical hydrogenation procedure
Retsch MM 200 and 400 shaker mills were used for transfer hydrogenations and gas flow hydrogenations, respectively. In a typical hydrogenation reaction, 1 mmol 4-nitrophenol, 25 mg of 5 wt% Pd/C, 2.2 eq. acetic anhydride (if applicable), and a 10 mm stainless steel ball were added to a 25 mL custom made steel milling vessel with gas inlet and outlet ports. Gas tubing was connected to the vessel, which was purged for 15 min with nitrogen (60 SCCM). Gas flow was then switched to hydrogen (20–60 SCCM) and the contents of the vessel were milled at 20 Hz for 30 min with one 27 g steel ball. A typical bubbler reaction used a 50 mL bubbler filled with acetic anhydride heated to its boiling point of 140 °C with a stirred oil bath. The gas lines were heated to 120 °C. Purging procedures were the same as before.Pressurized stirred tank hydrogenation procedure
Pressured stirred tank hydrogenation experiments were run using a 100 mL pressurized steel autoclave from Parr Instruments Co. In a typical reaction, 2 mmol of 4-nitrophenol, 50 mg of 5 wt% Pd/C, and 25 mL of isopropanol were charged into the reactor. The reactor was then closed and sealed, purged to 6.9 bar three times with nitrogen, then purged to 6.9 bar three times with hydrogen. The reactor was then further pressurized to 10 bar with hydrogen, heated to 80 °C, and stirred for 30 min. For stirred tank hydrogenation reactions under hydrogen flow, the reactor gas inlet was connected to a hydrogen line with a mass flow controller. The reactor was purged similarly to the batch hydrogenation conditions. After purging, hydrogen was bubbled through the bottom of the reactor (20–60 SCCM) with stirring and heating to 80 °C for 30 min.Quantification of reaction products
After reaction, the inside of each reactor was washed with methanol, and the wash solution was transferred quantitatively to a volumetric flask. Samples were then taken and analysed using HPLC with 0.1% formic acid in water and acetonitrile mobile phase and C18 stationary phase. The method used a gradient of 5% acetonitrile to 35% acetonitrile by 15 minutes.Side products were identified by HPLC retention time, LC-MS, and NMR. A Bruker AV3 HD 500 was used for NMR analysis. An Agilent LC-MS (1260 LC with 6120BMS MS) was used for mass spectrometry analysis. LC-MS methods were identical to HPLC methods.
Conversion, yield and selectivity calculations
Conversion was defined as: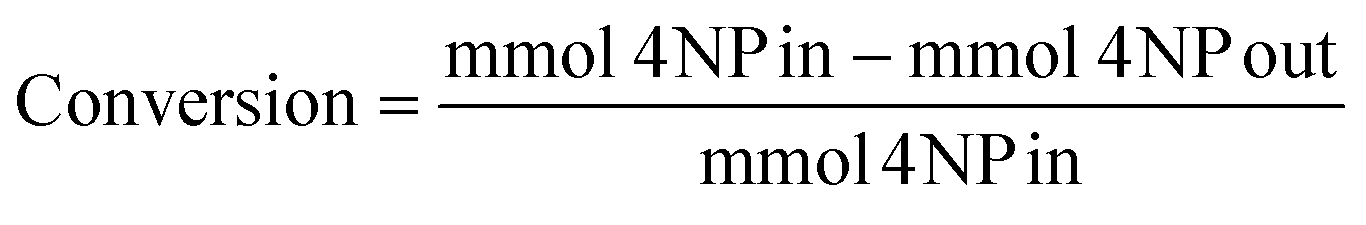
About 3–10% of the phenolic reagents used could adsorb to the Pd/C catalyst and were unquantifiable. Yield was defined as mmol of 4-AP or APAP obtained divided by mmol 4-NP input. However, as all detectable reactants and products for the one pot reactions were quantified with standards, “effective” yield for the one-pot reactions was defined using the mols of quantifiable species only, thus excluding any adsorbed species. Selectivity was defined as yield divided by conversion.
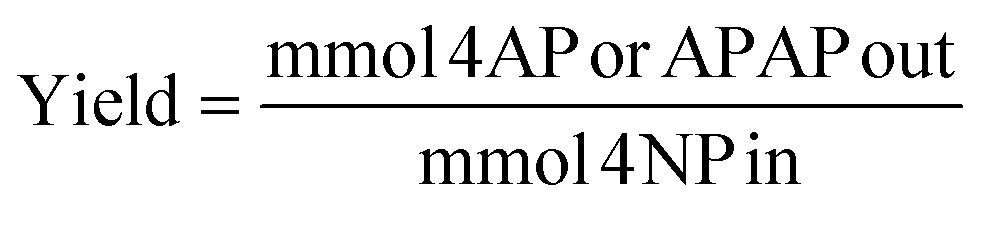
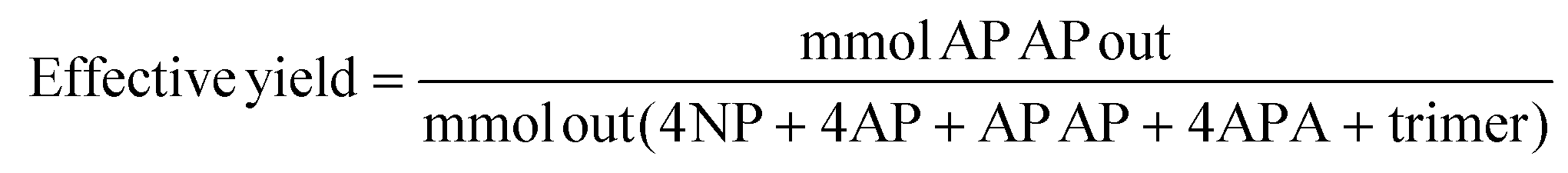
Catalyst characterization
TEM images were obtained with a FEI Tecnai F30. The instrument was operated at 300 kV with a thermally assisted field emission (TFE) gun. A CompuStage single-tilt holder was used to hold samples. Images were shown on a scale ranging from 50 to 200 nm. Images were converted and analyzed using a Gatan GIF system (Tridiem 863 UHS). Samples were prepared by dissolving 5 mg of sample in methanol and sonicated for 1 h. The sample was then dispersed onto a holey carbon–copper 200 mesh grid. SEM/EDS was conducted using a Thermo Axia Variable Pressure Model. Samples were prepared on carbon tape and analysed under high vacuum at 10.00 kV and a working distance of 0.6 mm. An Everhard-Thornley detector was used for SEM imaging, ranging from 30–100 μm. EDS mapping acquisition time was 60 seconds. X-ray photoelectron spectra (XPS) were taken with a Thermo Scientific Scanning X-ray photoelectron spectrometer using monochromatic Al Kα X-ray (1.486 keV) source. High-energy-resolution spectra were recorded for Pd 3d, O 1s, C 1s, and Fe 2p. A pass-energy of 50.0 eV and step size of 0.10 eV was used for acquisition. 200 eV and a step size of 1.0 eV was used for the survey.Results and discussion
The viability of three transfer hydrogenation agents, ammonium formate, formic acid, and sodium borohydride, was tested in the presence of Pd/C in similar conditions to those in the original synthesis by Portada et al. Formic acid was found to be the best performing agent with 99% conversion and 83% selectivity to 4-aminophenol (ESI Fig. 1†).4-Aminophenol side reaction routes
Despite the satisfying initial results of the transfer hydrogenation of 4-NP with formic acid and ammonium formate, 17% of the reactants formed detectable side products, which conflicted with the reports of Portada et al., who claimed quantitative yields.18Unfortunately, the desired intermediate product 4-AP was unstable even under ambient conditions and formed a condensation product in the presence of Pd/C (Fig. 2). This product (hereafter called the “trimer”) was identified through LC-MS and NMR and compared with literature,27,28 and likely occurs through the oxidation of 4-AP to form reactive quinone intermediates. The ease of 4-AP degradation through oxidation is well described in literature.29 Interestingly, the trimer is formed even in oxygen-free environments (such as a typical hydrogenation reaction). This may occur due to the presence of oxygen atoms in 4-NP: certain hydrogenation intermediates such as nitroso compounds or nitric oxide free radicals may act as oxidating agents and attack 4-AP throughout the reaction. The oxygen atoms may also allow Pd/C to act as a Lewis-acid to catalyse the condensation reaction. In general, the oxidation state of the catalyst could be important for further optimization.
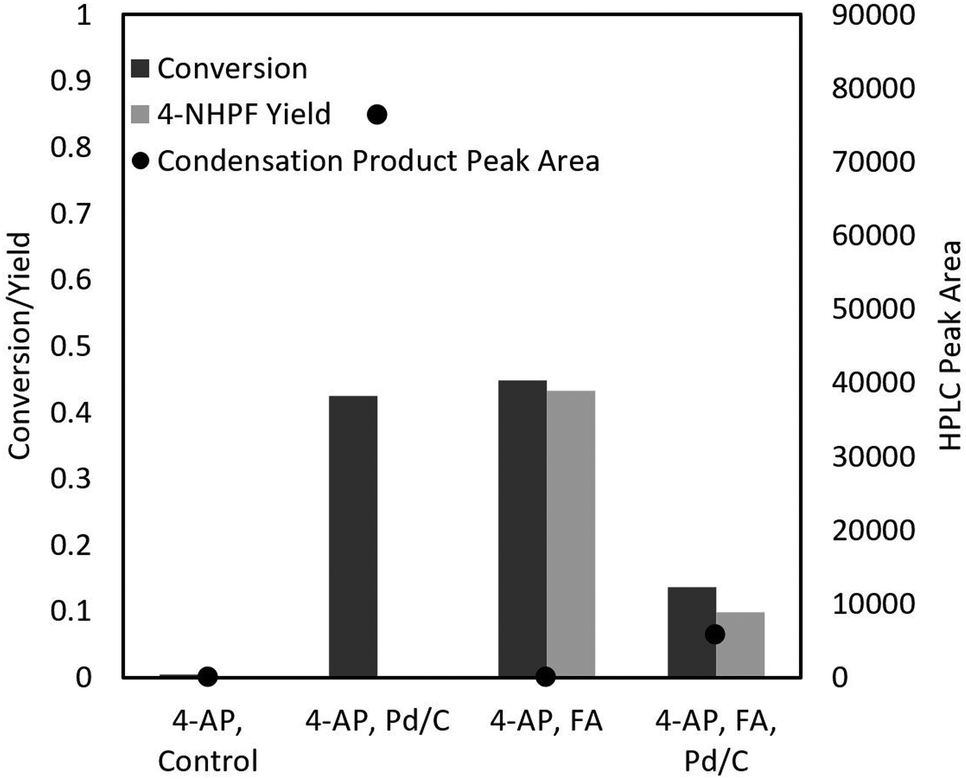 | ||
| Fig. 2 Side product formation experiments. 1 mmol of 4-NP was milled with 50 mg of Pd/C (if applicable) and 3.3 mmol of formic acid (FA, if applicable) at 30 Hz for 30 min in a closed 15 mL stainless steel milling vessel. | ||
Furthermore, 4-AP readily attacks the formate groups used during the transfer hydrogenation, selectively forming 4-NHPF (4-N-hydroxyphenyl formamide) as the byproduct (Fig. 2). This is demonstrated through the milling of 4-AP with and without the presence of formic acid and Pd/C. Fig. 3 shows possible routes to the side products.
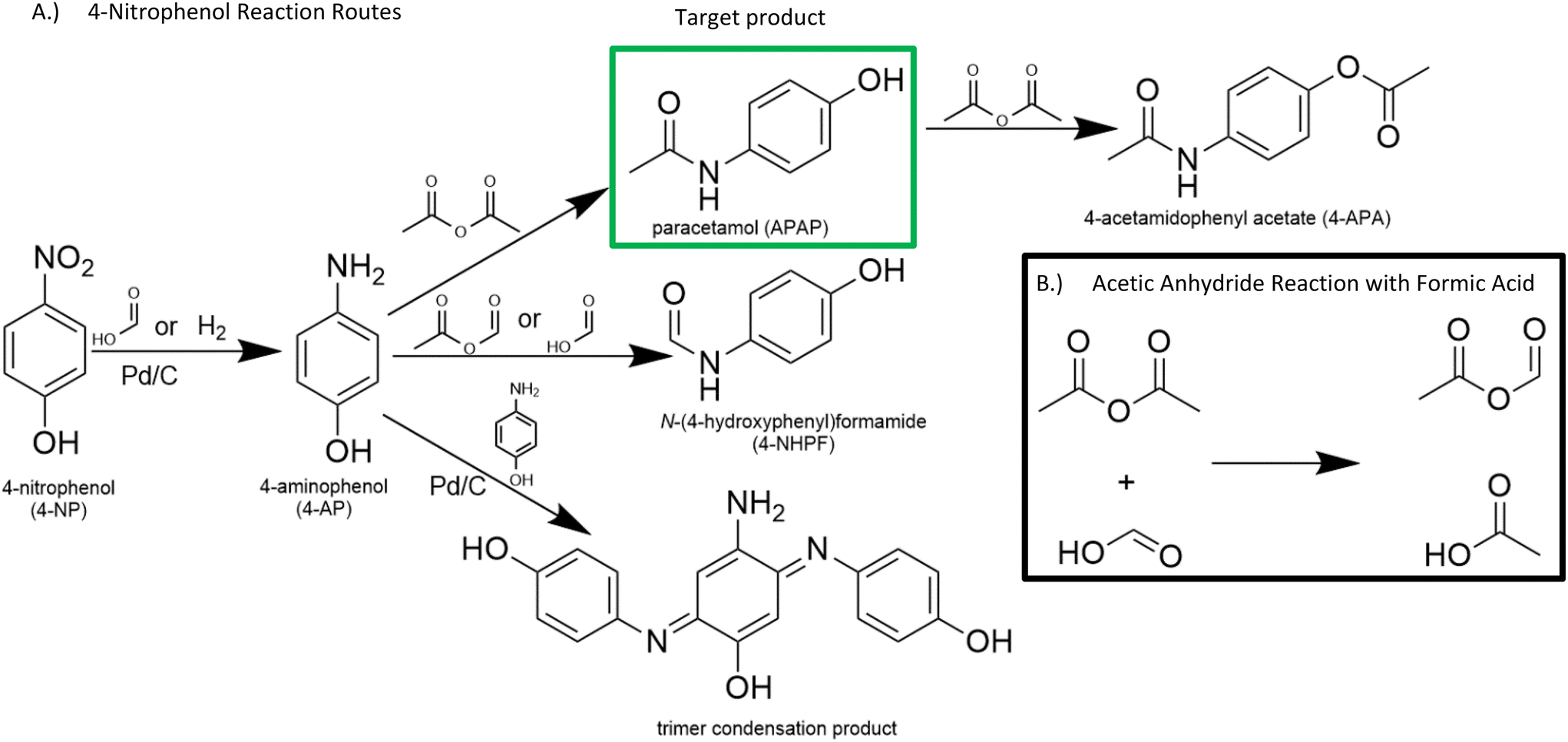 | ||
| Fig. 3 Reaction diagram. 4-Nitrophenol (4-NP) is hydrogenated to form 4-aminophenol (4-AP) which is then acetylated to form paracetamol (APAP). 4-AP undergoes side reactions with formic acid to form 4-nhydroxyphenylformamide (4-NHPF) or with itself in the presence of Pd to form trimer side products. Furthermore, acetic anhydride and formic acid may react to form a mixed anhydride. | ||
A clear difficulty with the transfer hydrogenation reaction is that unreacted or excess formic acid can react with 4-aminophenol as it is accumulated, producing the undesired 4-NHPF side product. Furthermore, the Pd/C catalyst also facilitated the formation of the trimer side product. Because of these factors, the need for strategies to improve selectivity became clear. Two possible techniques to achieve this are liquid assisted grinding and the prevention of 4-AP accumulation.
Strategies to reduce side product formation: LAG
Liquid assisted grinding (LAG), which entails the addition of a small amount of solvent to the grinding jar, is one possible method of increasing selectivity.30 LAG agents were added with the same liquid volume/reactant mass ratio for all conditions (η = 2.85). Promisingly, the addition of solvent significantly reduced the production of the main side products in all cases (Fig. 4). Polar protic solvents were the most effective, eliminating the presence of 4-NHPF and significantly reducing trimer formation, resulting in an overall increase in selectivity to 4-AP from 0.77 to 0.85 under optimal conditions. The advantages of protic solvents in hydrogenation processes have been well described in literature31,32 and may explain their superior performance in this reaction. Specifically, protic solvents can facilitate the heterolysis of H2 through H-shuttling effects: an H from an activated H2 molecule can be accepted by solvent molecules which can then donate a proton to the organic reactants.31 Another possibility is the stronger interactions of the polar reactants with protic solvents through H-bonding, which could lower the activation energy barrier of the reaction by stabilizing a polar transition state.32 Certain solvents may also provide better mixing in the ball mill: as the experiments used only a single ball, certain parts of the milling jar, such as the sides, were prone to caking of the solid reactants and thus “dead space” where the reactants were not well-mixed was formed. Despite the selectivity improvements with the addition of solvent, the formation of the trimer could not be solved through LAG alone, as it remained detectable.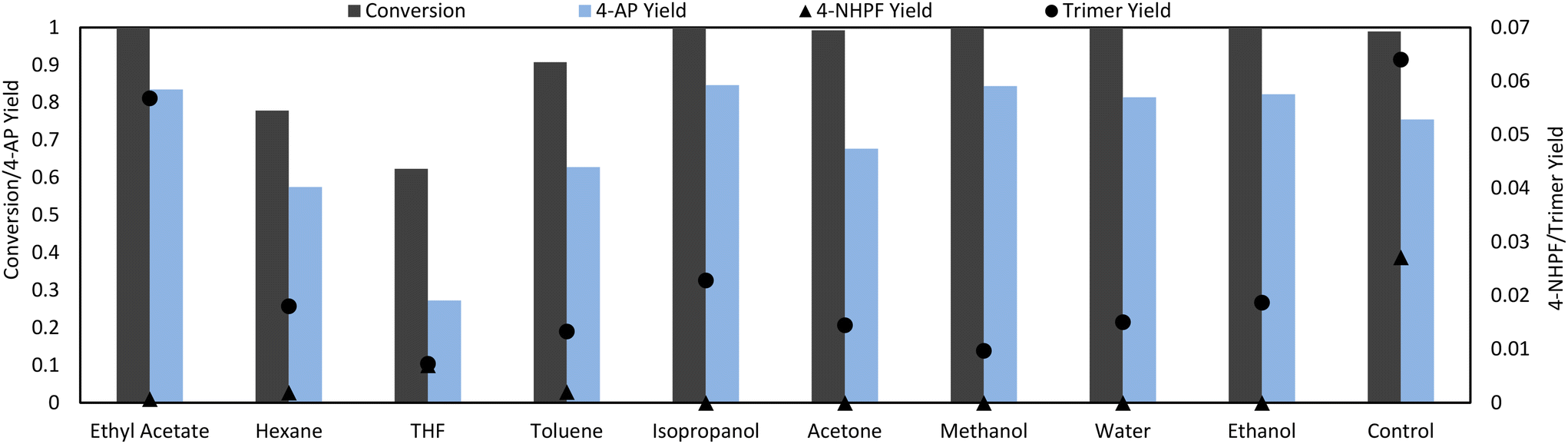 | ||
| Fig. 4 LAG solvent screen. 1 mmol 4-NP was milled at 30 Hz with 3.3 eq. formic acid, 50 mg Pd/C (5 wt%) and 0.5 mL of solvent at 30 Hz, 5 min in a 15 mL closed steel vessel. | ||
Strategies to reduce side product formation: one-pot transfer hydrogenation and acetylation
A key parameter in the prevalence of the 4-AP condensation reaction is the accumulation of 4-AP as the hydrogenation reaction proceeds, which allows for the self-reaction of 4-AP to proceed. As such, another strategy to reduce condensation is to run the hydrogenation and acetylation steps simultaneously, such that any 4-AP formed is immediately reacted to the more stable and desired APAP. A simple method of achieving this is to add the hydrogen source and acetic anhydride at the same time. Disappointingly, when 4-NP was reacted in one pot with formic acid and acetic anhydride, 4-NHPF formation increased greatly: APAP selectivity was 64% and the balance formed 4-NHPF (Fig. 5). As such, the selectivity of the one pot reaction was considered inferior to the selectivity of the previous LAG reactions, which were about 80% selective to 4-AP (Fig. 4).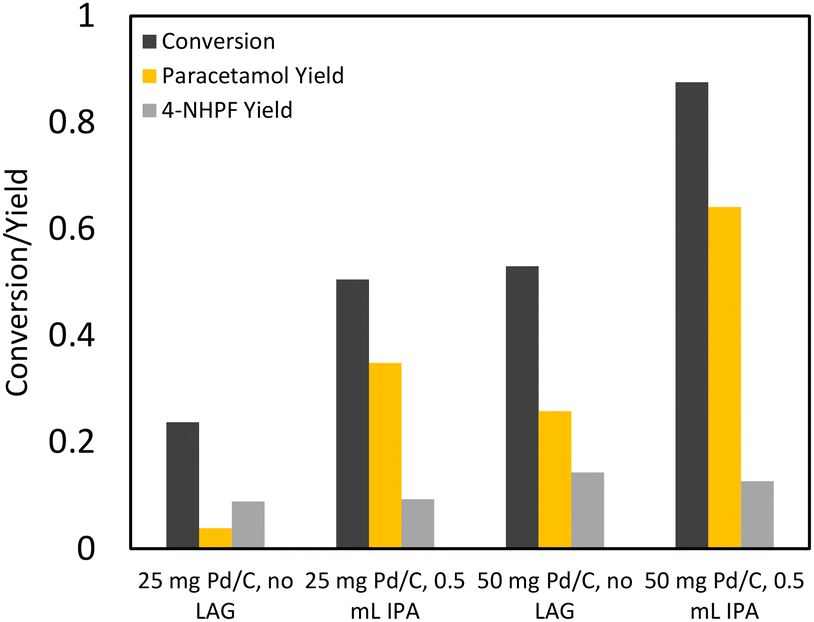 | ||
| Fig. 5 One-pot transfer hydrogenation and acetylation. 1 mmol 4-NP was milled at 30 Hz with 3.3 eq. formic acid, 50 mg Pd/C (5 wt%) and 2.2 eq. acetic anhydride for 15 min. | ||
The low selectivity of the one pot transfer hydrogenation and acetylation was caused by the reaction of acetic anhydride and formic acid to form acetyl formic anhydride (Fig. 3B). This reaction is highly favourable at room temperature, and the resulting mixed anhydride can react with 4-aminophenol to form either APAP or 4-NHPF. In addition, the mixed anhydride formation removes formic acid required for the hydrogenation step. Because of these two factors, a one pot transfer hydrogenation and acetylation reaction using formic acid as the hydrogen donor is not viable. Alternate transfer hydrogenation agents that avoid reactions with acetic anhydride such as isopropanol or sodium borohydride were considered but ultimately discarded due to factors such as low reactivity, high cost, and low selectivity to 4-AP (ESI section 2†). Furthermore, a simple two-step process where the hydrogenation and acetylation are separated was discarded because of the insufficient selectivity of the transfer hydrogenation to 4-AP.
Interestingly, the addition of LAG significantly improved the conversion and yield of the one-pot reaction. This may be due to superior mass transfer and proton-shuttle effects provided by the solvent, or to the dilution of formic acid and acetic anhydride in the reactor, making mixed anhydride formation less favourable.
Despite unavoidable 4-NHPF formation, the one pot reaction formed undetectable or trace amounts of the trimer. This was likely because the concurrent reaction of 4-AP to APAP and 4-NHPF reduced the 4-AP self-condensation reaction. As such, these results validated the hypothesis that immediately reacting 4-AP to APAP as it is formed and preventing 4-AP accumulation can reduce trimer formation. To develop a viable reaction, side reactions with acetic anhydride must be avoided.
Strategies to reduce side product formation: one-pot elemental hydrogenation and acetylation
As conventional transfer hydrogenation agents were deemed unviable for a one-pot process, the possibility of using elemental hydrogen as the hydrogenating agent was considered. Hydrogen does not react with acetic anhydride unless at high temperatures and over specific catalysts so that undesired side reactions are avoided.33 A key concern with using elemental hydrogen is the safety of process configurations with high hydrogen pressures. Ball milling, through its superb solid–solid and solid–gas mixing capabilities, provides a method to run hydrogenation reactions using hydrogen gas flow at ambient temperatures and pressures. Through using continuous hydrogen flow, build-up of large reservoirs of hydrogen gas is avoided, increasing process safety. Furthermore, the atmosphere of the ball mill is devoid of oxygen, reducing the possibility of ignition.Initial reactions of 4-NP with hydrogen gas in the presence of different Pd/C loadings showed similar results in terms of 4-AP yield to the experiments with formic acid (Fig. 6A). However, 4-NHPF formation did not occur as formic acid was no longer used in this reaction. The only detectable side product was the trimer, which formed in significant yields of up to 10%. Importantly, the addition of isopropanol to the milling jar improved conversion and reduced trimer formation. 25 mg Pd/C loading was used for subsequent reactions as it had similar results to 50 mg.
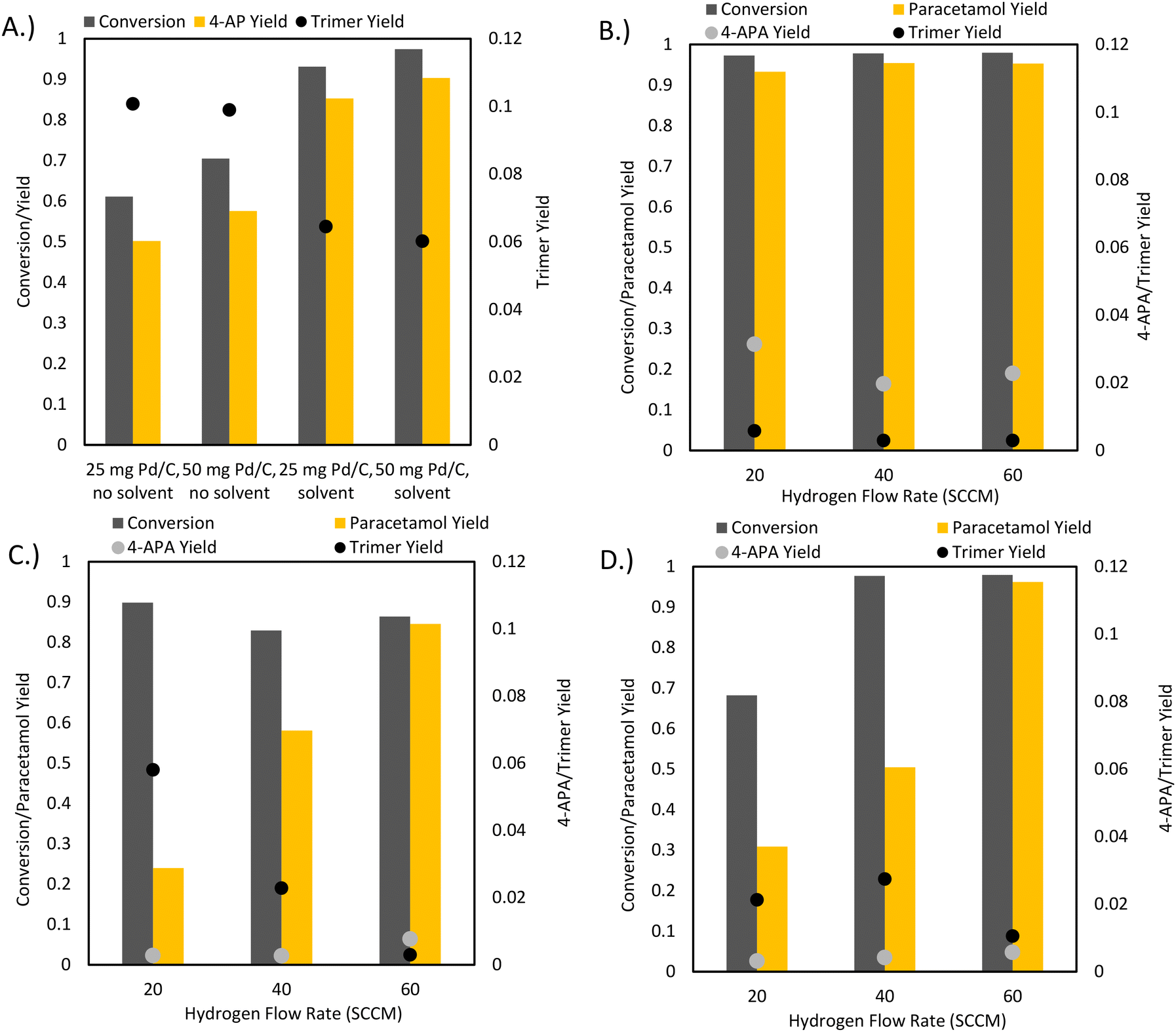 | ||
| Fig. 6 Hydrogen flow reactions. 1 mmol of 4-NP was milled under hydrogen flow at 20 Hz for 30 min with 5 wt% Pd/C in a 15 mL custom steel milling vessel with gas inlet and outlet ports. (A) 4-NP was milled in 20 SCCM hydrogen flow in the absence of acetic anhydride to form 4-AP. (B) 4-NP was milled in hydrogen flow with 0.5 mL of acetic anhydride to form APAP. (C) 4-NP was milled in neat grinding conditions with acetic anhydride bubbler. (D) 4-NP was milled in 0.5 mL isopropanol with an acetic anhydride bubbler which delivered two equivalents of acetic anhydride over 30 min at 60 SCCM H2. | ||
To reduce the formation of the trimer, a one pot reaction was run where acetic anhydride was charged to the reactor as a LAG agent (Fig. 6B). This reaction produced paracetamol with up to 97% selectivity, a clear improvement over previous conditions that were 85–90% selective at best. This increase in selectivity coincided with a significant reduction in the abundance of the trimer, supporting the idea of reducing side product formation through preventing 4-AP accumulation. In fact, the formation of the undesired trimer was almost eliminated, with only about 0.5% final yield to the side product, a clear improvement compared to the original conditions where it was produced with more than 6% yield. However, the large excess of acetic anhydride resulted in the acetylation of the hydroxyl group of APAP to form 4-APA with up to 3% final yield. Because of this undesired reaction, it was concluded that using an excess of acetic anhydride as the reaction solvent was suboptimal. In addition, the reaction suffered from evaporation due to the gas flow. Tuning the relative rates of the condensation and acetylation reactions is vital for further improvements to selectivity.
The immediately obvious method of manipulating the rate of the acetylation reaction is through fed batch addition of acetic anhydride. Ideally, acetic anhydride would be delivered at a molar rate equivalent to the formation rate of 4-AP, such that any 4-AP is reacted as it is formed. Similarly, any acetic anhydride reaching the reactor is also reacted immediately as 4-AP is a far superior nucleophile to APAP, reducing possible interactions with the APAP product.
However, fed batch addition of liquid reagents to a rapidly vibrating ball mill reactor is not trivial. For a controlled flow of liquid through the milling vessel to be possible, the consistency of the reaction slurry is important. Too little liquid results in a mud-like consistency that does not flow freely; as such, a high liquid level is required in the milling vessel. However, this defeats the purpose of ball milling, as a high liquid level results in reactor conditions very similar to a traditional well-mixed stirred tank reactor. Other issues also arise, such as clogging, the fact that one of the reactants (hydrogen) is gas-phase, and a large acetic anhydride excess. Thus, a fed batch addition of acetic anhydride in the liquid phase to the ball mill reactor is not feasible. As such, the reactor setup was modified such that hydrogen could be flowed through a bubbler filled with acetic anhydride (ESI Fig. 3†). Through adjusting the bubbler temperature (∼139.5 °C, the boiling point of acetic anhydride) and hydrogen flow rate, the amount of gas phase acetic anhydride delivered to the reactor can be tuned accordingly. This vastly reduces the complexity of the reaction conditions through delivering both reagents in the same phase as well as eliminating issues with the flow delivery.
Expectedly, the mass of acetic anhydride delivered to the reactor was linearly correlated with the hydrogen flow rate: higher hydrogen flow rates resulted in more delivered acetic anhydride and thus higher paracetamol yields. The optimal hydrogen flow rate through the bubbler was set as 60 SCCM. Initial solvent-free bubbler conditions displayed incomplete conversion of 4-nitrophenol. This was likely due to reactant caking along the walls of the reactor and thus isopropanol was added as a grinding aid, resulting in improved conversion (Fig. 6C and D).
Promisingly, the prevalence of 4-APA decreased by three-fold compared to the initial LAG one-pot conditions using acetic anhydride. This indicates that under the bubbler conditions, the acetylation of paracetamol was suppressed. The suppression of the over-acetylation reaction coincided with a final paracetamol selectivity of 99% for both the neat and LAG bubbler conditions. Given that the acetic anhydride LAG conditions contained a large acetic anhydride excess compared to the bubbler reactions, these results were expected: the high excess of acetic anhydride in LAG conditions would favour the acetylation of APAP. In the bubbler setup, acetic anhydride was delivered to the milling vessel at roughly the rate of 4-AP conversion. Because of the ease of the over acetylation of APAP, the advantage of a fed-batch addition of acetic anhydride is clear, with our optimized conditions (Fig. 6D, 60 SCCM H2) achieving 97% conversion with 99% selectivity to APAP. The bubbler provides a simple method of such a fed batch addition approach in a mechanochemical reactor, as the simultaneous pumping of liquid and gas into a rapidly shaking vessel introduces design difficulties, such as the need for separate gas and liquid inlet ports. Finally, acetic anhydride is a poor hydrogenation solvent: polar protic solvents were shown to have the best performance during the initial solvent screening. The bubbler allows the use of other solvents such as isopropanol that can improve the performance of the hydrogenation reaction.
An important safety consideration is the possibility of the ignition of flammable acetic anhydride vapor under bubbler conditions. The possibility of ignition is mitigated by remaining well under the auto-ignition temperature (316 °C) of acetic anhydride in the bubbler as well as maintaining an oxygen free atmosphere throughout the reactor lines. Furthermore, mild milling frequencies are recommended to eliminate the chance of sparking during the milling process. As this reaction is run with hydrogen flow under a fume hood, the concentration of hydrogen or acetic anhydride in the air in the event of a leak will remain under ignition limits. Under normal circumstances, ignition is impossible as the temperature and concentrations remain far under ignition limits and the reactor system is devoid of oxygen. However, robust temperature control and monitoring is recommended for safe operation.
Catalyst surface species, performance, and deactivation
Despite the high final yield of APAP, up to 5% of the input mols of reactant could not be attributed to one of the identified reaction components (4-NP, 4-AP, APAP, 4-APA, or a trimer product). As no other side product peaks could be detected by HPLC, it is likely that the missing species remained adsorbed on the catalyst (Fig. 7). When the compounds 4-AP, 4-NP, and APAP are milled with Pd/C, more than 10% of the mass cannot be quantified. In the case of 4-AP, this is because of the aforementioned side reaction. However, both 4-NP and APAP showed no traces of side product peaks in HPLC with the mass simply disappearing, which is most easily explained by catalyst sticking. Indeed, the ability of activated carbon to adsorb phenolic compounds is well described in the literature.34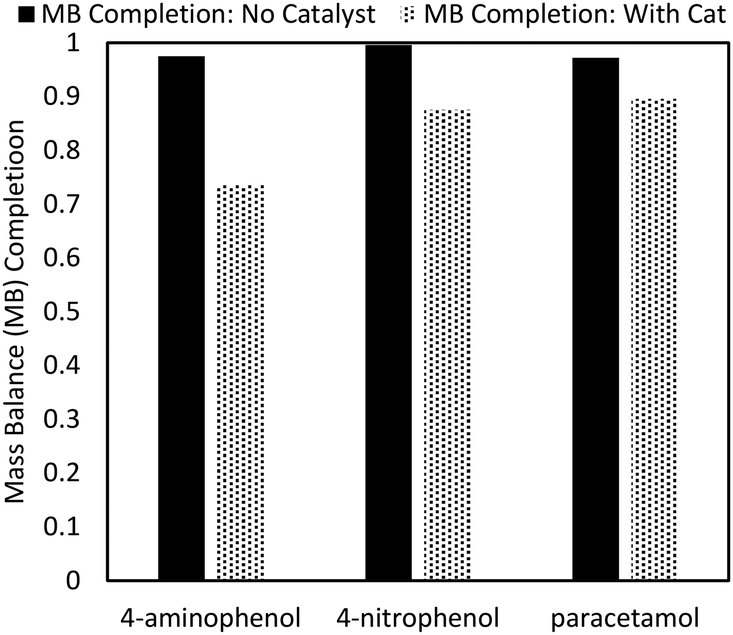 | ||
| Fig. 7 Mass balance (MB) examination. 1 mmol of feedstock was milled with 50 mg Pd/C at 30 Hz for 30 min. The resulting wash was quantified for mass completion. | ||
As such, the “effective yield” was used for quantification: the adsorbed species were not included in the denominator of the yield calculation. All products of the one-pot reaction were quantified with standards, allowing for a complete mass balance using quantifiable species only.
The performance of Pd/C as catalyst was also compared to that of two other commonly used noble metals, Pt/C and Ru/C, at different loadings. Palladium was the best performing metal in terms of final paracetamol yield, followed by platinum and ruthenium (ESI Fig. 2†). All three metals formed the trimer. Importantly, the catalyst also affected selectivity, with a 5% selectivity decrease when the catalyst loading was doubled as well as an increase in the presence of the trimer. This was unsurprising considering that palladium also catalyses the condensation reaction. As the reaction rate also directly depends on catalyst loading, there is a clear trade-off between reaction rate and selectivity that must be considered for future studies. Palladium was ultimately chosen as the most desirable catalyst because of its superior performance as well as the lower activity and environmental and/or safety concerns of other hydrogenation catalysts such as nickel or iron-acid systems reported in the literature.35
The recyclability of the catalyst over multiple reactions was also considered (Fig. 8). A typical reaction cycle involved 70 mg 4-NP and 25 mg 5 wt% Pd/C milled for 30 minutes with one 27 g steel ball. After each reaction, the milling vessel was opened to air while additional feedstock (4-NP) was added. The reaction was then run again using the same catalyst from the previous run with no further regeneration. These experiments showed significant catalyst deactivation over the reaction time scale, with conversion decreasing from 99% to ∼70% by the third recycle.
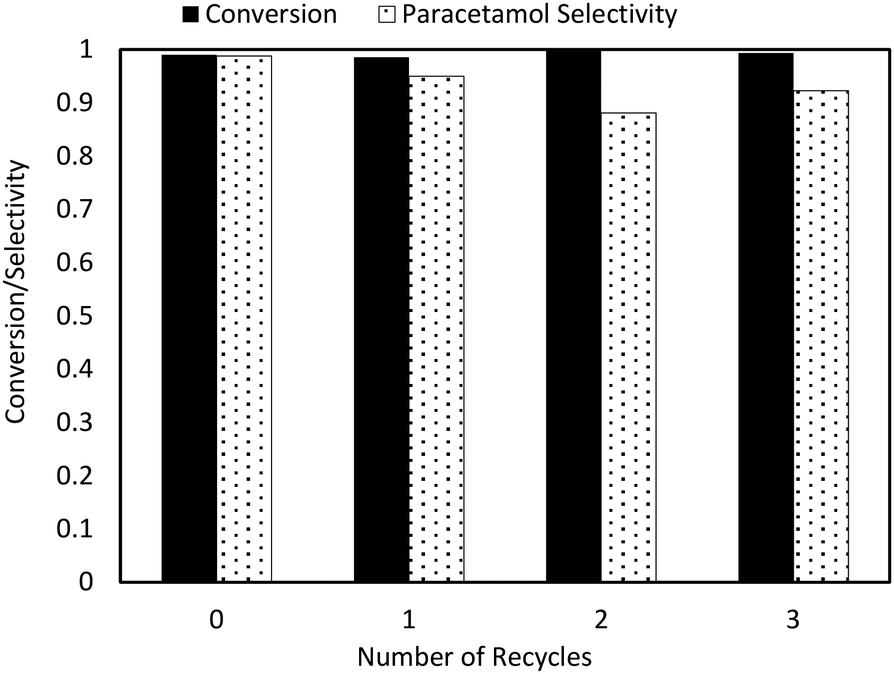 | ||
| Fig. 8 Catalyst reusability studies. First cycle: 1 g steel balls (×27), 70 mg 4-NP, 25 mg 5 wt% Pd/C, and 0.25 mL isopropanol were milled at 15 Hz for 30 min under 60 SCCM hydrogen flow with acetic anhydride vapor. Subsequent cycles added 70 mg 4-NP, 0.1 ml isopropanol (to prevent caking), and milled under the same conditions as the first cycle with no further addition or regeneration of catalyst. | ||
The instability of the catalyst under the above conditions was a cause for concern. One possible cause of catalyst deactivation was exposure to air during the addition of more feedstock. The palladium catalyst could easily oxidize when exposed to air, resulting in lowered activity. Another more likely cause could be the significant impact forces when milling at the relatively high frequency of 20 Hz with one heavy (27 g) steel ball. The kinetic energy imparted from the impacts could cause sintering of Pd particles as well as destroy the carbon support, which could adversely affect both the dispersion of active catalyst particles and the surface area of the support. Indeed, the possibility of activity loss of Pd based catalysts from sintering is well described in the literature.36,37
To address these issues, the recycle procedure was modified to avoid oxygen exposure as well as use more mild milling conditions. To eliminate the possibility of oxidation, the outlet gas port of the ball mill vessel was opened, and the additional 4-NP was fed into the port while hydrogen flowed through the vessel. To reduce the possibility of catalyst destruction, twenty-seven 1 g stainless steel balls were used instead of one 27 g steel ball. In addition, the milling frequency was lowered from 20 Hz to 15 Hz. The use of many smaller balls not only reduced impact energy of each individual collision but also improved mixing inside the ball mill.
The implementation of the above changes to the recycle procedure resulted in much more stable catalyst performance, with no activity loss noted after four reaction cycles (three recycles). There was a slight decrease in selectivity from optimized bubbler conditions resulting from the addition of additional feedstock to the reactor. As more feedstock is added per reaction cycle, paracetamol is formed and accumulates, increasing the prevalence of its side reaction with acetic anhydride. However, this phenomenon is unrelated to the catalyst and solely due to reactant feed rates, which can be easily solved with further optimization. In general, the catalyst remains highly reusable under mild milling conditions.
The spent catalyst was then characterized to confirm why recyclability was superior with mild milling conditions. TEM imaging showed sintered Pd particles after milling under harsh conditions. The palladium particles in the fresh catalyst (Fig. 9A) were relatively well dispersed and had small particle sizes of 3–3.5 nm. After milling under harsh conditions (20 Hz, 1 × 27 g ball, 30 min), significant sintering of the palladium was observed, with agglomeration of the particles on the order of >200 nm being visible in TEM images (Fig. 9B). On the other hand, the catalyst milled under milder conditions (Fig. 9C) maintained its dispersion, with an average particle size range of 3.5–4.0 nm. While milling can cause sintering, the effects of ball milling on noble–metal catalysts are not yet well understood and should be further examined in the future.
 | ||
| Fig. 9 TEM of the effects of ball milling on the palladium catalyst. Figure A represents the fresh catalyst examined without further preparation. Figure B represents the catalyst after milling at 20 Hz with 1 × 27 g stainless steel ball for 30 min under 60 SCCM hydrogen. Figure C represents the catalyst after milling at 15 Hz with 27 × 1 g steel balls for 30 min under 60 SCCM hydrogen. | ||
In addition, milling caused changes in both the structure of the carbon support as well as the elemental composition of the catalyst surface. With milling, the carbon support structure became less ordered as seen in SEM (ESI Fig. 4†). Harsher milling conditions seemed to cause more severe transformation of the catalyst support. Palladium also migrated away from the surface after milling according to XPS (ESI Table 1†), likely caused by both sintering and restructuring of the support. The migration of Pd away from the surface was more pronounced under harsh milling conditions. Finally, the oxygen content decreased during milling. According to XPS (ESI Fig. 5†), the fresh catalyst contained peaks corresponding to palladium metal (335.2 and 340.5 eV) as well as PdO (337.2 and 342.5 eV).38,39 PdO was likely fully reduced after milling the catalyst in hydrogen atmosphere, as peaks at 337.2 and 342.5 eV that were present in the fresh catalyst sample disappeared in the milled samples. This suggests that reactivating spent catalyst through a pre-treatment step involving reduction may not be necessary.
In general, catalyst deactivation could occur through several factors, including sintering, support destruction, and migration of palladium away from the surface. These phenomena are all more prevalent in harsh milling conditions, and thus mild milling conditions are recommended for better catalyst recyclability. Additionally, while out-of-scope for this investigation, the catalyst itself may be optimized, such as through using more robust supports that can withstand impacts in a ball mill.
Comparison to pressurized stirred tank process
Stirred tank hydrogenations were also run for a direct comparison of traditional pressurized hydrogenation procedures to the mechanochemical process. Stirred tank hydrogenation using high pressure as well as hydrogen flow (where hydrogen was bubbled through the solvent in the reactor) in ambient pressure were examined. In addition, the performances of the hydrogenation to 4-AP and the one-pot reaction to APAP in the Parr reactor were compared.Expectedly, the reactions with ambient hydrogen flow in the stirred tank reactor were inferior in terms of yield and selectivity to both the ball mill reaction configurations (Fig. 10). This is likely in part due to the poorer mass transfer in the stirred tank reactor: high hydrogen pressures result in higher hydrogen saturation concentration. At ambient pressures, the dissolved hydrogen concentration is too low for high hydrogenation rates. Because ball mill conditions at ambient hydrogen pressures demonstrated good reaction rates, the stirred tank flow conditions are most likely kinetically limited, with poor hydrogen mass transfer to the surface of the catalyst. The low rate of hydrogenation may also explain the poor selectivity of the stirred tank flow conditions, as the formation of 4-aminophenol is slower. As less 4-aminophenol is available for acetylation, the acetylation of paracetamol may become more favourable. This is further exacerbated by the high reaction temperatures in the stirred reactor compared to the ambient temperatures of the ball mill, which increases the speed of the acetylation of the hydroxyl group. This can be seen through the high production of 4-APA in the one-pot flow reaction.
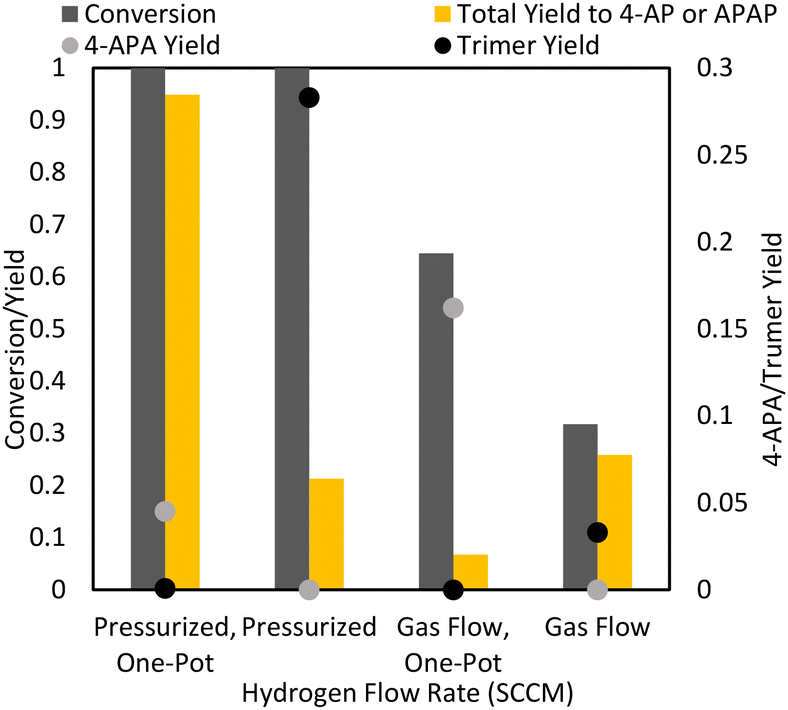 | ||
| Fig. 10 Stirred tank reactions. 2 mmol 4-NP, 50 mg Pd/C, 2.2 eq. acetic anhydride (when applicable), and 25 mL isopropanol was reacted under 10 bar or 60 SCCM hydrogen at 80 °C for 30 min. One-pot reactions targeted APAP. | ||
Overall, the best stirred-tank reaction involved the one-pot hydrogenation and acetylation under high hydrogen pressure, which obtained APAP with 95% yield and selectivity. Without the presence of acetic anhydride, various side products were formed, including the trimer described for previous reactions. One possibility for the low selectivity of the reaction to 4-AP may be the higher temperatures involved in the stirred tank reaction: the ball mill is run at room temperature (at 20 Hz, there is no significant heat accumulation in the milling vessel) while the stirred tank reactor is run at 80 °C. 4-AP is susceptible to degradation at higher temperatures. Importantly, in the presence of acetic anhydride, APAP was obtained in high yields, i.e., almost no trimer side product was formed through reducing 4-AP accumulation.
Green chemistry analysis
The best performing conditions using every reactor configuration described so far were compared regarding common Green Chemistry metrics. As expected, all ball mill conditions have vastly superior process mass intensity (PMI) to the stirred tank reaction because of their massive reduction of solvent loads (Table 1). As solvent loading is usually the main driver of PMI in pharmaceutical processes, this is a large improvement reducing waste generation. It is important to note that the calculated PMI values describe the yield of paracetamol straight from the reaction vessel without any separation or purification steps. Furthermore, it is possible to further reduce the PMI of the stirred tank conditions by increasing the 4-NP concentration. In addition, the ball mill conditions have certain disadvantages to the stirred tank process. The most prevalent is the high amount of hydrogen used in gas flow conditions: almost three times as much hydrogen is used in the ball mill with hydrogen flow as in the pressurized stirred tank reactor, resulting in a lower reaction mass efficiency (RME). Optimization of the acetic anhydride delivery rate through the bubbler as well as using hydrogen recycle streams can mitigate this problem and perhaps even make the flow conditions superior in terms of hydrogen mass used. Furthermore, the use of continuous hydrogen flow reduces the possibility and severity of accidents.40–42 Another problem is the incomplete conversion of 4-nitrophenol in the ball mill, with about 2–3% unconverted material remaining. Optimization with the number and size of balls to induce better mixing as well as modifying the vessel dimensions to avoid dead space and caking may solve this issue. As such, the ball mill conditions are superior in terms of Green Chemistry to the traditional stirred tank process.| Hydrogenating agent | Reactor | Configuration | Paracetamol yield | RME | PMI |
|---|---|---|---|---|---|
| Formic acid | Ball mill | One-pot CTH wet | 0.641 | 0.241 | 8.18 |
| Hydrogen | Ball mill | One-pot wet | 0.953 | 0.166 | 6.01 |
| Hydrogen | Ball mill | One-pot bubbler | 0.846 | 0.243 | 4.12 |
| Hydrogen | Ball mill | One-pot bubbler wet | 0.962 | 0.276 | 6.40 |
| Hydrogen | Stirred tank | One-pot pressurized | 0.949 | 0.325 | 20–80 |
In addition to traditional Green Chemistry metrics such as RME and PMI, the energy expenditure of mechanochemical processes should not be ignored. As the energy expenditure of mechanochemical processes does not scale linearly with the size of the reaction, it would be misleading to use energy expenditure calculations from bench scale conditions.43 However, large scale mechanochemical processes have been shown to benefit from economies of scale: energy expenditure decreases with scale-up.43 Furthermore, the continuous mechanochemical scale-up of various processes is feasible through the use of techniques such as twin-screw extrusion.17
The potential of mechanochemical pharmaceutical synthesis for reducing carbon emissions, ecotoxicity, and even operating costs has been demonstrated: for example, Galant et al. have shown that twin screw extrusion synthesis of nitrofurantoin results in order-of-magnitude improvements in the aforementioned metrics compared to solvent based synthesis.44 These improvements are largely driven by mechanochemical reaction intensification as well as reduction of solvent evaporation.44,45 As the extent of such improvements can vary from process to process, a rigorous examination of these considerations at scale, while out of the scope of this work, can be beneficial. Regarding the APAP synthesis process described here, the 5–20-fold PMI decrease afforded by mechanochemical synthesis can be translated to a proportional reduction in the energy required for solvent separations. In addition, as the mass of acetic anhydride delivered during the reaction remains orders of magnitude lower than the solvent loading of a stirred tank reaction, the energy required to deliver gas-phase acetic anhydride can be mitigated by the reduction of solvent usage.
Among the ball mill conditions, the formic acid conditions were discarded because of their low paracetamol yield (owing to side product formation) and higher PMI. The key decision point is the viability of the LAG conditions: while they result in noticeably higher final product yield through improving conversion and selectivity, they also significantly increase the PMI. Given the possibility of side products and unreacted feedstock interfering in downstream separations processes, the use of LAG may be warranted despite the higher waste generation. Another consideration is that optimization may reduce the need for LAG. Conditions such as different catalyst loadings, supports, milling temperature and frequency, number of balls, etc. may all be leveraged to improve the conversion and selectivity of the solvent-free reactions to meet or exceed the best conditions described currently.
Finally, the fed-batch addition of acetic anhydride to the ball mill is likely more desirable than the addition of an excess, not only for improving the RME but also for preventing the over-acetylation of paracetamol to 4-APA. Further optimization of the process should match the rate of acetic anhydride addition to the rate of formation of 4-aminophenol in the reactor.
Conclusions
The reaction network and side products of the hydrogenation of 4-nitrophenol with formic acid and elemental hydrogen followed by acetylation with acetic anhydride in a ball mill were elucidated, revealing three main side products: 4-N-hydroxyphenyl formamide (4-NHPF), 4-acetamidophenyl acetate (4-APA), and a trimer condensation product.LAG was examined as a method of providing superior hydrogenation conditions by inducing proton transfer, stabilizing transition states, and improving mixing.
Formates were discarded as a viable hydrogenation agent because the formation of 4-NHPF was unavoidable when it was used. Hydrogen gas was instead continuously flowed into the ball mill at ambient pressures as a safe method of hydrogenation while avoiding 4-NHPF formation.
The trimer, which formed from the condensation of the unstable intermediate product 4-AP, was avoided through one-pot hydrogenation. Avoiding 4-AP accumulation was key to achieving high selectivity and was accomplished through simultaneous hydrogenation and acetylation such that 4-AP would be immediately acetylated to the more stable and desired paracetamol instead of undergoing side reactions.
4-APA formation was favoured in one-pot conditions requiring excess acetic anhydride and was mitigated through a novel acetic anhydride bubbler setup, which allowed for tuneable fed-batch acetic anhydride delivery to the milling vessel and the avoidance of excess reactants. Under optimized conditions, paracetamol was obtained in high yields of 96% with a corresponding selectivity of 99%.
Overall, the aforementioned conditions improved upon the original synthesis by Portada et al. through eliminating the use of formates as hydrogen donors, improving selectivity, and simplifying the typical two step hydrogenation and acetylation to a one step process.18
Finally, the various ball mill reactor configurations were tested and compared with the traditional stirred tank hydrogenation process in a pressurized Parr reactor regarding Green Chemistry metrics. Ball mill conditions were superior to the stirred tank process: PMI was reduced by a factor of 5–20 with comparable yield and superior selectivity (95% stirred vs. 99% ball mill), although conversion was slightly lower in the ball mill.
Further studies will focus on scale-up of the one-pot reaction configurations.
Author contributions
Jimin Park (conceptualization, data curation, formal analysis, investigation, methodology, project administration, resources, software, supervision, validation, visualization, writing – original draft, writing – review and editing), Jacob Maier (investigation, formal analysis, methodology, visualization), Caria Evans (formal analysis, investigation, methodology), Marta Hatzell (project administration, writing – review and editing), Stefan France (methodology, resources, writing – review and editing), Carsten Sievers (conceptualization, methodology, project administration, resources, supervision, validation, writing – review and editing), Andreas Bommarius (conceptualization, methodology, project administration, resources, supervision, validation, writing – review and editing).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the Renewable Bioproducts Institute at Georgia Institute of Technology for providing funding for this project through the Paper Science and Engineering Fellowship. We would also like to thank Jacob Dewitt, Erin Phillips, and Karoline Hebisch from the Sievers group for assistance with building the gas flow ball mill set up and catalyst characterization.References
- N. Tsolakis and J. S. Srai, Oper. Manage. Res., 2018, 11, 83–104 CrossRef.
- WHO Model List of Essential Medicines, https://list.essentialmeds.org/.
- H. Ellis-Petersen, India limits medicine exports after supplies hit by coronavirus, The Guardian, 2020 Search PubMed.
- S. Kinkartz, Germany sees alarming shortage in essential medicine, DW, 2022 Search PubMed.
- R. Joncour, N. Duguet, E. Metay, A. Ferreira and M. Lemaire, Green Chem., 2014, 16, 2997–3002 RSC.
- J. Park, M. Kelly, J. Kang, S. Seemarkurti, J. Ramirez, M. C. Hatzell, C. Sievers and A. S. Bommarius, Green Chem., 2021, 23, 7499–7498 RSC.
- R. Joncour, A. O. Ferreira, N. Duguet and M. Lemaire, Org. Process Res. Dev., 2018, 22, 312–320 CrossRef CAS.
- M. J. Vaidya, S. M. Kulkarni and R. V. Chaudhari, Org. Process Res. Dev., 2003, 7, 202–208 CrossRef CAS.
- C. Rode, M. Vaidya and R. Chaudhari, Org. Process Res. Dev., 1999, 3, 465–470 CrossRef CAS.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- L. Ducry and D. M. Roberge, Angew. Chem., Int. Ed., 2005, 44, 7972–7975 CrossRef CAS.
- L. L. Silva, M. J. Stellato, M. V. Rodrigues, B. J. Hare, J. C. Kenvin, A. S. Bommarius, L. Martins and C. Sievers, J. Catal., 2022, 411, 187–192 CrossRef CAS.
- H. Lu, H. Yin, Y. Liu, T. Jiang and L. Yu, Catal. Commun., 2008, 10, 313–316 CrossRef CAS.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS.
- G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. Harris, G. Hyett and W. Jones, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- J. Andersen and J. Mack, Green Chem., 2018, 20, 1435–1443 RSC.
- T. Portada, D. Margetić and V. Štrukil, Molecules, 2018, 23, 3163 CrossRef.
- R. Mocci, E. Colacino, L. D. Luca, C. Fattuoni, A. Porcheddu and F. Delogu, ACS Sustainable Chem. Eng., 2021, 9, 2100–2114 CrossRef CAS.
- A. Y. Li, A. Segalla, C.-J. Li and A. Moores, ACS Sustainable Chem. Eng., 2017, 5, 11752–11760 CrossRef CAS.
- C. Wang, S. Deng, R. Chen, G. Liu, T. Cheng and R. Liu, Synlett, 2022, 33, 1858–1862 CrossRef CAS.
- V. J. Kolcsár and G. Szőllősi, ChemCatChem, 2022, 14, e202101501 CrossRef.
- A. W. Tricker, K. L. Hebisch, M. Buchmann, Y.-H. Liu, M. Rose, E. Stavitski, A. J. Medford, M. C. Hatzell and C. Sievers, ACS Energy Lett., 2020, 5, 3362–3367 CrossRef CAS.
- J. A. DeWitt, E. Phillips, K. L. Hebisch, A. Tricker and C. Sievers, Faraday Discuss., 2022, 243, 65–76 RSC.
- C. Bolm and J. G. Hernández, Angew. Chem., Int. Ed., 2019, 58, 3285–3299 CrossRef CAS.
- S. Immohr, M. Felderhoff, C. Weidenthaler and F. Schüth, Angew. Chem., Int. Ed., 2013, 52, 12688–12691 CrossRef CAS PubMed.
- L. Lerner, J. Phys. Chem. A, 2011, 115, 9901–9910 CrossRef CAS.
- A. C. Sousa, L. O. Martins and M. P. Robalo, Adv. Synth. Catal., 2013, 355, 2908–2917 CrossRef CAS.
- S. Mitchell and R. Waring, Kirk–Othmer Encyclopedia of Chemical Technology, 2000 Search PubMed.
- P. Ying, J. Yu and W. Su, Adv. Synth. Catal., 2021, 363, 1246–1271 CrossRef CAS.
- M. Li, C. Zhang, Y. Tang, Q. Chen, W. Li, Z. Han, S. Chen, C. Lv, Y. Yan and Y. Zhang, ACS Catal., 2022, 12, 11960–11973 CrossRef CAS.
- H. Wan, A. Vitter, R. V. Chaudhari and B. Subramaniam, J. Catal., 2014, 309, 174–184 CrossRef CAS.
- T. H. L and S. W. Polichnowski, Process for hydrogenating acetic anhydride using a catalyst containing a noble metal, European Patent Office Pat, EP0070860B1, 1981 Search PubMed.
- A. Dąbrowski, P. Podkościelny, Z. Hubicki and M. Barczak, Chemosphere, 2005, 58, 1049–1070 CrossRef PubMed.
- Y.-G. Wu, M. Wen, Q.-S. Wu and H. Fang, J. Phys. Chem. C, 2014, 118, 6307–6313 CrossRef CAS.
- L. Yang, C. Fan, L. Luo, Y. Chen, Z. Wu, Z. Qin, M. Dong, W. Fan and J. Wang, Catalysts, 2021, 11, 725 CrossRef CAS.
- M. Danielis, S. Colussi, C. de Leitenburg, L. Soler, J. Llorca and A. Trovarelli, Angew. Chem., 2018, 130, 10369–10373 CrossRef.
- M. C. Militello and S. J. Simko, Surf. Sci. Spectra, 1994, 3, 387–394 CrossRef CAS.
- M. C. Militello and S. J. Simko, Surf. Sci. Spectra, 1994, 3, 395–401 CrossRef CAS.
- M. D. Johnson, S. A. May, J. R. Calvin, J. Remacle, J. R. Stout, W. D. Diseroad, N. Zaborenko, B. D. Haeberle, W.-M. Sun and M. T. Miller, Org. Process Res. Dev., 2012, 16, 1017–1038 CrossRef CAS.
- P. J. Cossar, L. Hizartzidis, M. I. Simone, A. McCluskey and C. P. Gordon, Org. Biomol. Chem., 2015, 13, 7119–7130 RSC.
- T. Yu, J. Jiao, P. Song, W. Nie, C. Yi, Q. Zhang and P. Li, ChemSusChem, 2020, 13, 2876–2893 CrossRef CAS PubMed.
- M. D. Kaufman Rechulski, M. Käldström, U. Richter, F. Schüth and R. Rinaldi, Ind. Eng. Chem. Res., 2015, 54, 4581–4592 CrossRef CAS.
- O. Galant, G. Cerfeda, A. S. McCalmont, S. L. James, A. Porcheddu, F. Delogu, D. E. Crawford, E. Colacino and S. Spatari, ACS Sustainable Chem. Eng., 2022, 10, 1430–1439 CrossRef CAS.
- K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145–2162 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc04281b |
| This journal is © The Royal Society of Chemistry 2024 |