
Status and challenges for CO2 electroreduction to CH4: advanced catalysts and enhanced strategies
Bingkun
Li
a,
Lu
Liu
a,
Mingzhu
Yue
a,
Qingman
Niu
a,
Min
Li
a,
Tianyu
Zhang
a,
Wenfu
Xie
*a and
Qiang
Wang
 *ab
*ab
aBeijing Key Lab for Source Control Technology of Water Pollution, Engineering Research Center for Water Pollution Source Control & Eco-remediation, College of Environmental Science and Engineering, Beijing Forestry University, Beijing 100083, China. E-mail: wenfu2010qd@126.com; qiangwang@bjfu.edu.cn
bState Key Laboratory of Efficient Production of Forest Resources, Beijing Forestry University, Beijing 100083, China
First published on 22nd November 2023
Abstract
Sustainable energy-powered carbon dioxide (CO2) electroreduction into methane (CH4) under ambient conditions holds great promise for achieving carbon neutrality and mitigating environmental pollution. Despite the significant advancements in this field, the unsatisfactory selectivity and stability remain the major obstacles to its large-scale applications. With this background, we present a comprehensive summary of recent progress and future opportunities of CO2 electroreduction into CH4. Firstly, we delve into the elucidation of the reaction mechanism involved in CO2 electroreduction into CH4. A thorough understanding of the individual steps within this process is paramount for designing electrocatalysts and improving selectivity and efficiency. In particular, the development of advanced electrocatalysts (such as copper-based materials, carbon-based materials and other materials) for CO2 electroreduction into CH4 is elaborated. Meanwhile, a special focus has been placed on the enhanced strategies, including coordination environment manipulation, element doping, surface modification, and morphology engineering, for CO2 electroreduction into CH4. Finally, the future challenges of CO2 electroreduction into CH4 are proposed. The review aims to serves as a guiding framework for systematic exploration and optimization of electrocatalysts, thus fostering the development and advancement of CO2 electroreduction into CH4.
1. Introduction
Traditional fossil fuels have played a significant role in driving economic growth and improving living standards, whereas the increasing carbon dioxide (CO2) emissions bring about serious climate changes. CO2 electroreduction, powered by sustainable energy (solar and wind energy), provides an unprecedented possibility to alleviate anthropogenic CO2 emissions.1 Additionally, various high value-added fuels and chemicals can be obtained, further improving the feasibility in commercial applications.2,3 In the past few years, growing attention has been focused on developing efficient electrocatalysts for the CO2 reduction reaction (CO2RR) to obtain valuable chemicals like carbon monoxide (CO), formic acid (HCOOH), hydrocarbons, and alcohols.4 Among these, methane (CH4) is particularly desirable to be a promising energy carrier for generating heat and electricity due to its high energy density of 55.5 MJ kg−1.5 Moreover, CH4 plays an important role as a raw material in the production of valuable polymers and chemicals (Fig. 1).6 As the primary component of natural gas, CH4 is well-suited for the existing infrastructure storage in distribution and consumption. It should be noted that CH4 is one of the main components of natural gas, but the unsuitable mining process leads to the emission of CH4, which could exhibit a greenhouse effect 86 times that of CO2.7 Therefore, realizing the controlled production of CH4 is of great significance for eliminating the greenhouse effect. Conventional thermocatalytic CH4 production involves high temperature and pressure and is energy intensive. Alternatively, electrocatalytic CH4 production has gained widespread attention due to its advantages of environmental friendliness, a controllable reaction path, and technology compatibility. More importantly, electrocatalytic CO2 reduction can utilize clean energy, which provides a guarantee for its green development.8 Therefore, highly selective CO2 electroreduction into CH4 is an effective way to balance environmental and energy issues. | ||
| Fig. 1 Schematic illustration of advanced electrocatalysts and enhanced strategies for CO2 electroreduction into CH4. | ||
However, the selectivity and reaction kinetics of CH4 production from CO2RR are severely hindered by the thermodynamic stability and poor solubility of CO2 molecules in the aqueous system.9 Moreover, the CO2 electroreduction into CH4 suffers from the complex products via different pathways and the competition of the hydrogen evolution reaction (HER).10 To date, Cu-based materials are one of the most effective electrocatalysts for CO2 electroreduction into CH4, and tremendous research efforts have been devoted to exploring and improving the efficiency of electrocatalysts.11–13 However, the selectivity and rate of CH4 production are still far from being commercially applicable. Therefore, the development of efficient and stable electrocatalysts has become a central problem and challenge in the field of CO2 electroreduction into CH4.
Recently, there have been many reviews on CO2 electroreduction, but most of them focus on CO2 electroreduction to CO,14,15 HCOOH,16,17 or C2+ products,18–20 and few of them focus on CH4 production by CO2 electroreduction. This review provides a systematic summary of the recent progress and future opportunities for CO2 electroreduction into CH4. Firstly, the reaction mechanism of CO2 electroreduction into CH4 is briefly explained. Then, the development of advanced electrocatalysts for CO2 electroreduction into CH4 is elaborated. After that, a special perspective will be focused on the enhanced strategies, including coordination environment manipulation, elemental doping, surface modification, and morphology engineering, for CO2 electroreduction into CH4. Finally, the future challenges of CO2 electroreduction into CH4 are proposed. The review aims to provide a guideline for the systematic exploration and optimization of electrocatalysts and boost the development of CO2 electroreduction into CH4.
2. Reaction mechanism
The overall CO2 electroreduction into CH4 in aqueous solution is a multi-step process, which involves the transfer of eight electron/protons.21 Although the CO2 electroreduction into CH4 (0.17 V vs. the reversible hydrogen electrode (RHE)) is more favorable than the HER (0 V vs. RHE) or CO2 electroreduction into other products (CO of −0.10 V vs. RHE and HCOOH of −0.12 V vs. RHE) from the view of thermodynamic potential (Table 1),11 it is still encountered with sluggish kinetics because of the low solubility of CO2 and multi-electron/proton transfer process.22 Usually, the solubility of CO2 in aqueous solution is less than 34 mM, and its dissolved products are related to the pH of solution.4 The dissolved gaseous CO2 and undissociated H2CO3 molecules are the main products when the pH of solution is less than 4.3. When the pH of solution is between 4.3 and 8.3, there are mainly two forms of CO2 and HCO3− ions. When the pH of solution is greater than 8.3, CO32− begins to appear and HCO3− begins to decrease. When the pH exceeds 12, there is almost only one form of CO32− that exists in the water. Moreover, the first reduction step of CO2 into the CO2˙− radical intermediate in aqueous solution (pH = 7) is difficult with a very negative equilibrium potential of −1.9 V vs. the standard hydrogen electrode (SHE), further creating difficulties for CO2 electroreduction into CH4.23,24 The reaction pathway of CO2 electroreduction into CH4 involves two key stages: the generation of *CO and the protonation of *CO.25 Thus, *CO is a key intermediate for many products. On the one hand, the adsorbed *CO will undergo desorption into CO gas molecules when the adsorption ability is weak. On the other hand, *CO will undergo a C–C coupling process into C2+ products (e.g., C2H4 and C2H5OH) when the coverage of *CO on the surface of the electrocatalyst is high.| Reaction | E° (V vs. RHE) at pH 0 |
|---|---|
| 2H+ + 2e− → H2 | 0 |
| CO2 + 2H+ + 2e− → CO + H2O | −0.10 |
| CO2 + 2H+ + 2e− → HCOOH | −0.12 |
| CO2 + 6H+ + 6e− → CH3OH + H2O | 0.03 |
| CO2 + 8H+ + 8e− → CH4 + 2H2O | 0.17 |
| 2CO2 + 12H+ + 12e− → C2H4 + 4H2O | 0.08 |
Although CO2 electroreduction to CH4 exhibits different rate-determining steps in different catalytic systems, the transformation of *CO into *COH or *CHO is usually considered one of the most critical steps.26–28 The *CHO pathway can bring multiple products, including CH3OH and CH4, while the *COH pathway produces only CH4. The distressing thing is that challenges remain to achieve the optimal adsorption of *CO and *COH intermediates at the same time, as a result of the linear scaling relationship. During the *COH pathway, the intermediates are adsorbed on the surface of the electrocatalyst via C binding, and *C is first obtained and further reduced into *CH, *CH2, *CH3, and finally CH4. The *CHO pathway is relatively complex, which could transform into different configurations. CH4 can be produced via the protonation of *CHOH or *CH2O. It is worth noting that *CHO might transform into *OCH2via O binding, which leads to the formation of CH4 or CH3OH. The final product is determined by the break of the C–O bond in the *OCH3 intermediate. The protonation of *CO and CO desorption competed with each other, resulting in the low selectivity of CH4 production. To avoid the conversion of CO2 into CO, oxygen atoms in CO2 can be adsorbed to form *OCHO intermediate species, which can then be converted into CH4.29 But the *OCHO intermediate is easily stuck in the product of formic acid, and accompanied by the generation of CH3OH. It can be concluded that the reaction microenvironments and the adsorption ability of *CO lead to different reaction paths and different products of CO, CH4, or C2+. Meanwhile, the adsorption of key intermediates could be tuned via manipulating the C/O affinity of active sites, thus realizing the optimal formation of CH4 (Fig. 2).
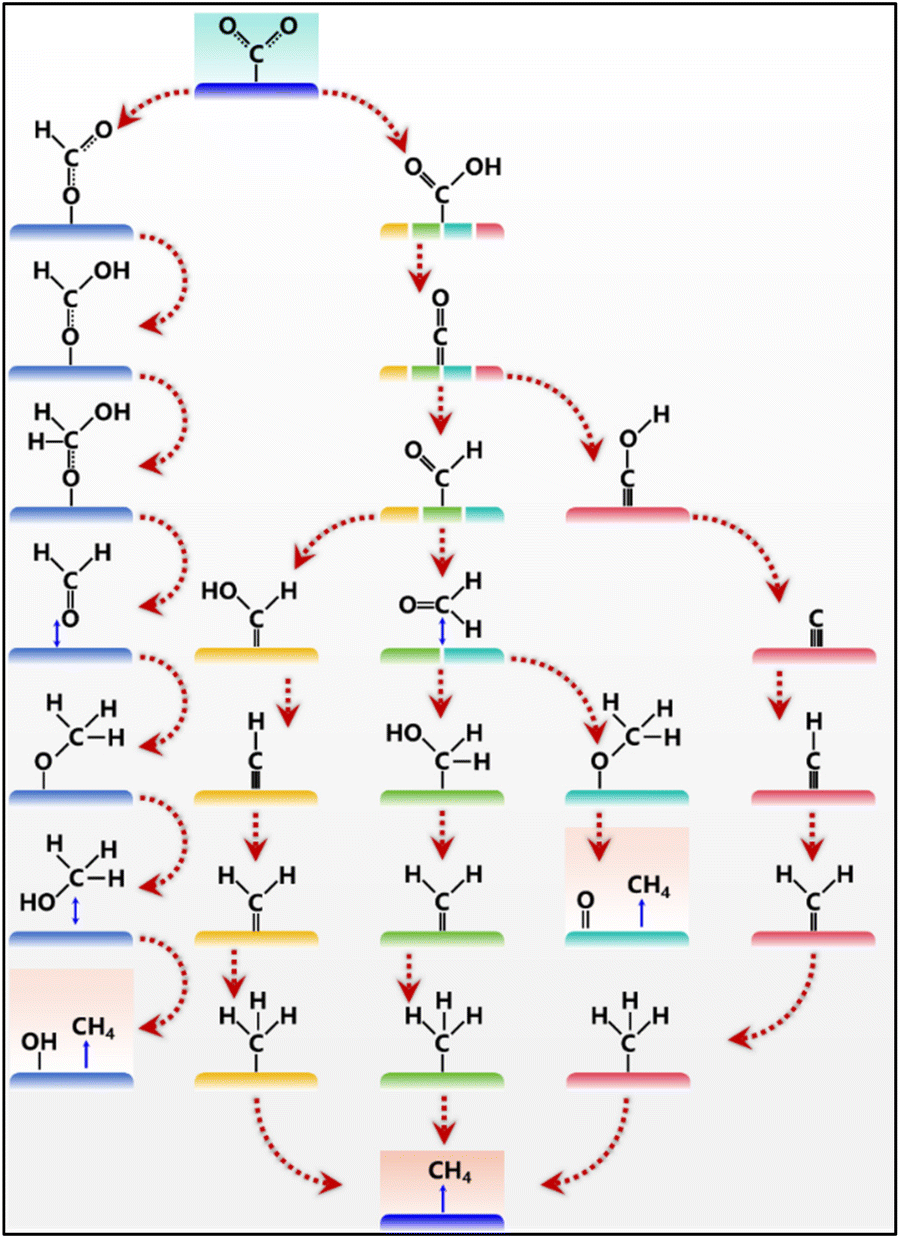 | ||
| Fig. 2 Overview of the reaction pathways for CO2 electroreduction to CH4. | ||
3. Advanced electrocatalysts
3.1. Single-atom Cu electrocatalysts
The development of single-atom catalysts (SACs) provides opportunities for elucidating the structure–activity relationships and investigating the reaction mechanisms, due to the merits of uniform active sites, adjustable coordination environments and high atom utilization.30–32 More and more SACs have been applied in the field of energy conversion and storage.33 In principle, the key step in CO2 electroreduction into CH4 is the protonation of *CO intermediates to *CHO. The main competing reaction processes are the dimerization of *CO to C2+ products and the direct desorption of *CO from the catalyst surface to produce gaseous CO.34 Among them, single-atom Cu electrocatalysts with monodisperse catalytic sites are expected to exhibit a superior performance for CO2 electroreduction into CH4, owing to the inhibited C–C coupling process. However, it is still a great challenge to achieve the controlled fabrication of Cu–SACs with the desired stability.With two-dimensional carbon nitride (CN) synthesized from melamine and alkali chlorides as support, Ajayan and co-workers realized the uniformly dispersed Cu–SAC with Cu–N2 coordination via a metal ion exchange process (Fig. 3a).35 DFT results demonstrate that the triangular 9 N pore size in poly(triazine imide) could lead to a stronger Cu–N binding energy, improving the stability of the Cu atom and optimizing the energy distribution of the reaction intermediates, thus resulting in higher catalytic activity. As a result, the prepared Cu@poly(triazine imide) electrocatalyst (Cu–PTI) exhibits a maximum FE of 68% and a partial current density of −348 mA cm−2 at −0.84 V vs. RHE. Li and co-workers synthesized a conjugated copper phthalocyanine polymer (CuPPc) via a solid-state method with CuCl2, pyromellitic dianhydride and urea (Fig. 3b).36 A high FE of 55% and a partial current density of 18 mA cm−2 are achieved on CuPPc at −1.25 V vs. RHE. The good performance stems from the spatially isolated Cu–N4 sites, which promote the generation of the *CHO intermediate through *CO protonation.
 | ||
| Fig. 3 (a) Schematic illustration of the synthesis of Cu–PHI/PTI.35 Copyright 2023, Wiley. (b) Schematic illustration of the synthesis of CuPPc.36 Copyright 2023, Wiley. (c) Schematic illustration of the synthesis of Cu SAs/GDY.37 Copyright 2018, Wiley. | ||
Although the improvement of the performance of CO2 electroreduction into CH4 can be achieved by constructing Cu–SACs, this approach is difficult to avoid the production of CO. For most Cu–SACs, the production of CO is a necessary condition for CH4 production. Mechanistic studies have proved that the *OCHO intermediate can be generated when CO2 is adsorbed on the catalyst surface through oxygen atoms, thus producing HCOOH rather than CO. This provides theoretical support for further improving CH4 selectivity while facing a huge challenge in adjusting the electronic structure of Cu–SACs to achieve regulation of intermediate adsorption. Wang and co-workers prepared a single-atom Cu electrocatalyst anchored on graphdiyne (Cu SAs/GDY) by an in situ adsorption reduction method (Fig. 3c).37 Due to the regulation of GDY, the valence state of the Cu atom is higher than zero. Moreover, the Cu–C bond enhances the charge transfer as well as modulates the intermediates, rendering an enhanced performance for CO2 electroreduction into CH4. The maximum FE of CH4 production over Cu SAs/GDY reaches 81% in the flow cell system, exceeding the most recently reported catalysts.
To date, the biggest challenge for CO2 electroreduction into CH4 on Cu-based SACs is that the single-atom Cu with an oxidized state is inevitably reduced to the metallic state under cathodic conditions. Compared with single-atom Cu with strong adsorption of the *CO intermediate, the metallic Cu shows a moderate adsorption ability for *CO. Thus, the metallic Cu or the mixture of metallic Cu and single-atom Cu can promote the dimerization of *CO, which is detrimental to the *CO hydrogenation process. Therefore, maintaining the oxidized state of single-atom Cu is an important way to facilitate the *CO hydrogenation process and the efficient production of CH4.
To achieve this goal, Qiao and co-workers incorporated Cu2+ ions into a CeO2 matrix (Cu–Ce–Ox) via a solid-solution strategy to stabilize the oxidized state of single-atom Cu during CO2 electroreduction.28 In this Cu–Ce–Ox solid solution, the accumulation of electrons around Cu2+ sites could be inhibited because of the existence of Ce3+, which can ensure the valence stability of Cu2+ during CO2 electroreduction (Fig. 4a). The electrochemical results show that Cu–Ce–Ox exhibits a maximum FE of 67.8% and a total current density of 200 mA cm−2 at −1.4 V vs. RHE for CH4 (Fig. 4b), superior to that of CuO and CuO/CeO2. DFT simulations illustrate that the rate-determining step for the CH4 pathway is the generation of *CH2O (ΔG = 1.48 eV), which is more likely to proceed than the generation of *CH–COH (ΔG = 2.50 eV) in the C2H4 pathway (Fig. 4c). The oxidized state of Cu2+ sites in Cu–Ce–Ox solid solution during CO2 electroreduction was characterized by HRTEM, XRD and XPS, suggesting that Cu2+ in Cu–Ce–Ox remains unchanged.
 | ||
| Fig. 4 (a) Schematic illustration of the self-sacrifice mechanism to protect Cu2+. (b) FEs for CO2 reduction products at different potentials over Cu–Ce–Ox. (c) Free-energy diagrams for the CH4 and C2H4 reaction pathways on the Cu–Ce–Ox model.28 Copyright 2022, American Chemical Society. (d) Schematic illustration of Cu/p-Al2O3 for the conversion of CO2 to CH4. (e) FEs for various products over Cu/p-Al2O3 SAC. (f) Free-energy diagrams for the CO2RR over Cu/p-Al2O3.29 Copyright 2021, American Chemical Society. | ||
Meanwhile, Wang and co-workers adopted the porous Al2O3 with Lewis acid sites as a substrate to support the single-atom Cu (Cu/p-Al2O3), achieving a maximum FE of 62% with a corresponding current density of 153 mA cm−2 at −1.2 V vs. RHE toward CO2 electroreduction into CH4 (Fig. 4d and e).29 The Lewis acid sites in porous Al2O3 can not only anchor the Cu atom, but also tailor the electronic structure of Cu, making its valence state close to +2. More importantly, CO2 is strongly adsorbed on Al sites with O–Al bonding through Lewis acid–base interactions, leading to the accelerated cleavage of C–O bonds. DFT calculations indicate that CH4 is generated via the *OCHO intermediate and CO2 is thermodynamically reduced into *OCHO on Cu/p-Al2O3 (ΔG = −0.25 eV). Furthermore, the key *CH3O intermediate is preferentially reduced into *CH4O rather than CH3OH owing to the low energy barrier of +0.21 eV (Fig. 4f). Therefore, Cu/p-Al2O3 is beneficial for producing CH4.
3.2. Cu-based material-supported single-atom electrocatalysts
In addition to Cu–SACs, other metal SACs have recently been reported to show high activity for CO2 electroreduction, yet the products are predominantly CO and HCOOH.32,38–41 As for CH4 production, non-Cu SACs also play very important roles in improving the performance, but more often in cooperation with Cu-based components. The introduction of non-Cu SACs into Cu-based materials can modulate the key species, such as *CO and *H, on the surface of electrocatalysts. For example, Li and co-workers presented an Ir single-atom-doped hybrid Cu3N/Cu2O electrocatalysts (Ir1–Cu3N/Cu2O) for CO2 electroreduction into CH4 (Fig. 5a).42 DFT calculations demonstrated that the coupling of Cu2O and Cu3N sites can significantly reduce the energy cost of the rate-determining step. Meanwhile, the introduced Ir could accelerate the dissociation of H2O to *H species via a nucleophilic attack, further leading to a boosted activity (Fig. 5b). As a result, Ir1–Cu3N/Cu2O preferentially catalyzes CO2 into CH4 with a maximum FE of 75% and a corresponding current density of 320 mA cm−2 at −1.3 V vs. RHE, outperforming the control electrocatalyst (Fig. 5c). | ||
| Fig. 5 (a) HAADF-STEM image of Ir1–Cu3N/Cu2O. (b) Proposed reaction mechanism for the conversion of CO2 to CH4 on Ir1–Cu3N/Cu2O. (c) FEs of various products over Ir1–Cu3N/Cu2O.42 Copyright 2022, American Chemical Society. (d) HAADF-STEM image and atomic elemental mapping of Cu–FeSA. (e) C–C coupling energy for pristine Cu vs. Cu–FeSA. (f) Comparison of the reaction products for pristine Cu and Cu–FeSA.43 Copyright 2022, Springer Nature. (g) The DFT results of CO* hydrogenation on Cu(111), Pd1Cu(111) and CO–Pd1Cu(111). (h) FEs of CH4 and C2H4 obtained using Cu and Pd1Cu SAAs.44 Copyright 2023, Springer Nature. | ||
It is also feasible to modulate the coverage of *CO on the surface electrocatalyst via the introduction of SACs, which in turn optimizes the protonation process of the intermediate products and improves the reaction efficiency. Sargent and co-workers reported a synthesis of a Cu-supported Fe-single-atom electrocatalyst (Cu–FeSA) via assembling iron phthalocyanine on the Cu(111) surface.43 The mono-dispersion of single Fe atoms in the Cu matrix was formed during the electrolysis and was confirmed by HAADF-STEM (Fig. 5d). DFT calculations were employed and demonstrated that the Fe site in Cu–FeSA shows a stronger adsorption ability for *CO than the Cu surface. Moreover, the C–C coupling process is energetically unfavorable on Cu–FeSA in comparison to Cu, indicating the suppression of C2+ products (Fig. 5e). The experimental results further validate the results of DFT calculations. At 200 mA cm−2, a maximum FE of 64% for CH4 production is achieved over Cu–FeSA (Fig. 5f). However, the main product on bare Cu is C2H4. Yang and co-workers anchored platinum group metal SACs on Cu electrocatalysts, further promoting the CO2 electroreduction performance.44 DFT calculations demonstrated that the reaction free energy and activation barriers of CO* protonation to CHO* on Cu(111) were reduced after doping a single Pd atom (Fig. 5g). Among them, octa-Pd1Cu SAA shows the highest FE (∼60%) for CO2 electroreduction into CH4 (Fig. 5h).
3.3. Cu-based oxides
Usually, Cu-based oxides (CuO and Cu2O) can catalyze CO2 into different C1 and C2+ products with uncertain selectivity because of the inevitable structure reconstruction during the reaction, which greatly limits their application in the field of CO2 electroreduction.45 Therefore, exploring advanced Cu-based oxides to produce CH4 with high selectivity is extremely desired. Recently, perovskite oxides have attracted considerable attention in CO2 electroreduction with the merits of flexible compositions and diverse structures,46 yet the production of CH4 needs further strengthening.Xia and co-workers proposed a perovskite oxide of La2CuO4, in which Cu species are partially reduced and transformed into the Cu/La2CuO4 interface, and thus deliver high activity for CO2 electroreduction into CH4 (Fig. 6a).47 At −1.4 V vs. RHE, the resultant Cu/La2CuO4 interface gives a maximum FE of 56.3% for CH4 production. Theoretical analysis indicates that the La2CuO4 can be transformed into the Cu/La2CuO4 interface, owing to the leaching of surface oxygen, thus facilitating the CO2 adsorption and electron transfer process. As a result, the Cu/La2CuO4 interface exhibits a stronger adsorption for HxCO intermediates in comparison to Cu(111), being responsible for the efficient CH4 production process (Fig. 6b). Meanwhile, Zhu and co-workers proposed a synthesis of efficient Cu-based Ruddlesden–Popper (RP) perovskite oxides for O2 electroreduction into CH4via an A-site cation deficiency engineering strategy (Fig. 6c).48 Through La-site cation deficiency engineering (La2−xCuO4−δ), a certain quantity of oxygen vacancies and CuO/RP hybrids (L2−xC) was obtained (Fig. 6d). The maximum selectivity of 22.1% and partial current density of 25.1 mA cm−2 were achieved on L1.7C electrocatalysts for CH4 (Fig. 6e), indicating that more CuO is conducive for the CO2-to-CH4 conversion. DFT simulations indicated that the pristine L2C(111) surface is preferred for C–C coupling due to the low energy of CO* dimerization (2.28 eV). However, *CO is more favorable for forming the HCO* intermediate with an energy of 1.66 eV on the CuO/L2C(111) surface compared with CO* dimerization of 2.57 eV (Fig. 5f and g). This work provides an efficient approach for manipulating the reaction pathways on perovskite oxides for CO2 electroreduction.
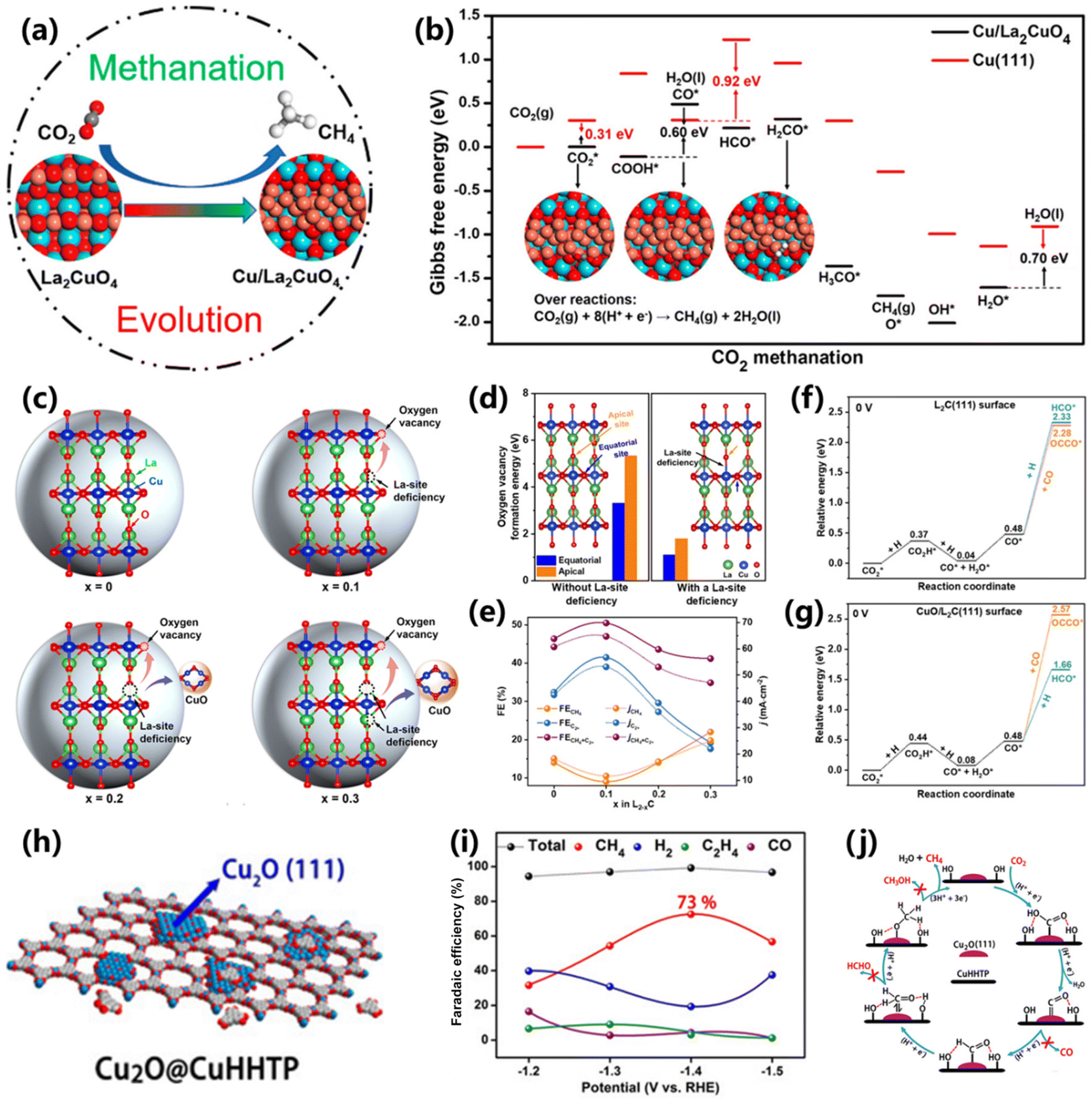 | ||
| Fig. 6 (a) Schematic of the structure-evolved copper perovskite oxide toward methane production. (b) Free-energy diagrams for the conversion of CO2 to CH4 at the Cu/La2CuO4 interface and Cu (111) surface.47 Copyright 2020, American Chemical Society. (c) Schematic of the crystal structure of La2−xCuO4−δ. (d) Oxygen vacancy formation energy on various La2−xCuO4−δ. (e) The CO2 electroreduction performance of L2−xC electrocatalysts. DFT results of CO* dimerization and protonation on the (f) L2C(111) surface and (g) CuO/L2C(111) surface.48 Copyright 2022, Wiley. (h) Schematic of the preparation of Cu2O@CuHHTP. (i) FEs of the products from the CO2RR over Cu2O@CuHHTP. (j) Reaction mechanism for CH4 production over Cu2O@CuHHTP.26 Copyright 2020, Wiley. | ||
In addition, Cao and co-workers fabricated porous conductive copper-based metal–organic framework (CuHHTP)-supported Cu2O quantum dots (Cu2O@CuHHTP) via an electrochemical treatment method (Fig. 6h).26 The Cu2O quantum dots with (111) crystalline planes were transformed from Cu2+ centers in CuHHTP under −1.2 V vs. RHE. In this composite, CuHHTP could accelerate the electron transfer, and Cu2O quantum dots surrounded with abundant hydroxyl groups facilitated the CO2 methanation process (Fig. 6j), resulting in a high FE of 73% and partial current density of 10.8 mA cm−2 for CH4 production (Fig. 6i). The crucial intermediates of *CHO, *CH2O and *OCH3 for the CH4 pathway were detected by operando ATR-FTIR. DFT calculations demonstrated that the critical protonation of *CO to *CHO needs low energy on Cu2O(111)@CuHHTP in comparison to Cu2O(111).
3.4. Cu-based chalcogenides
As mentioned before, Cu-based oxides suffer from the inevitable structure reconstruction during reactions, which is adverse to obtaining the target product with high selectivity. A self-supported copper sulfide nanosheet array (CuS@NF) was prepared by Luo and co-workers, which showed high selectivity and robust stability for CO2 electroreduction into CH4.49 The FE for CH4 production on CuS@NF reached 73% at −1.1 V vs RHE, which was higher than that of CuS nanowires and nickel foam (Fig. 7a). Moreover, the CH4 production could be maintained stable up to 60 h. The mechanism study indicates that the presence of S species can stabilize the CO2˙− intermediate during CO2 electroreduction, and the CO2˙− intermediate can couple with another CO2 molecule to form the CO2–CO2˙− intermediate, followed by the protonation to CH4 (Fig. 7b). Subsequently, Chen and co-workers proposed a vertical growth of metallic 2D Cu2Te nanosheet arrays on commercial copper foils through electrochemical etching and chemical vapor deposition methods (Fig. 7c).50 The synthetic route is facile for scalable production with an area of 500 cm2. The Cu2Te nanosheets with exposed edge-active sites demonstrate a good performance for CO2 electroreduction into CH4. Compared with the Cu2Te film (20%) and CuO film (4%), the Cu2Te nanosheets could efficiently yield CH4 with an FE of 48% at −1.0 V vs. RHE (Fig. 7d). The theoretical simulation reveals that the Cu2Te nanosheets with a highly exposed (100) facet favor CO2 adsorption and CH4 production than the Cu (111) and Cu2Te (001) facets, in which the protonation of *CO to yield *CHO needs the lowest energy (Fig. 7e). The above works bring more opportunities for exploring Cu-based electrocatalysts in the CO2RR. | ||
| Fig. 7 (a) FEs of the products from the CO2RR over CuS@NF. (b) Proposed mechanism of CuS@NF for the formation of CH4.49 Copyright 2017, Royal Society of Chemistry. (c) Schematic of the preparation of Cu2Te nanosheet arrays. (d) FEs of various products from CO2 reduction over the Cu2Te nanosheet and controlled samples. (e) The proposed mechanism of CO2 electroreduction into CH4 on edge-oriented Cu2Te.50 Copyright 2023, American Chemical Society. | ||
3.5. Cu-based composite catalysts
During CO2 reduction, the *CO protonation to *CHO is subjected to the competition of C–C coupling and the hydrogen evolution reaction. Once this competition is suppressed, the selectivity of CH4 can be improved. With this goal, Sargent and co-workers constructed an Au–Cu electrocatalyst, in which Au could not only inhibit the HER, but also change the CO2 electroreduction from the C2H4 pathway to the CH4 pathway (Fig. 8a).51 DFT calculations were first conducted and showed that low *CO coverage favors the *CO protonation vs. C–C coupling on the Au–Cu surface (Fig. 8b). Then, Au–Cu electrocatalysts with different Au contents were synthesized on the surface of polytetrafluoroethylene (PTFE) nanofibers. The CO2 concentration during the test was optimized to be 84% CO2, which could form a low *CO coverage condition and be beneficial for CH4 production. Both Au–Cu electrocatalysts exhibit good selectivity for CO2 to CH4, and 7% Au–Cu delivers the highest FE of 56% at a current density of 200 mA cm−2 (Fig. 8c). However, the continuous operation will cause a high local pH condition, which can inhibit water reduction. Under this circumstance, the available active hydrogen (*H) on the surface of catalysts will be reduced, thus preventing the *CO protonation process. To solve this problem, Sargent and co-workers further prepared a series of CoO nanoclusters on Cu electrocatalysts and achieved a desirable CO2 electroreduction into CH4.52 A volcano-like relationship between the *CO protonation and the *H binding energies is obtained on various metal–oxide-decorated Cu surfaces. Among them, CoO-decorated Cu shows the lowest energy for *CO protonation due to the moderate *H binding ability of CoO. It is concluded that the introduction of CoO could increase the density of local *H, thus enhancing the *CO protonation to *CHO rather than dimerization (Fig. 8d). CoO clusters with different sizes were synthesized and loaded on a Cu surface (CoO/Cu/PTFE) with different contents for CO2 electroreduction. The optimized CoO/Cu/PTFE with a cluster size of 2.5 nm and a loading content of 10 ng cm−2 exhibited the highest selectivity of 60% for CH4 production (Fig. 8e). | ||
| Fig. 8 (a) Schematic illustration of *CO protonation and dimerization on the Au–Cu surface. (b) Free energy difference between *CO protonation and dimerization over various Au–Cu surfaces under different *CO coverages. (c) FEs of CH4 production using various Au–Cu electrocatalysts.51 Copyright 2021, Springer Nature. (d) Schematic illustration of CO2 electroreduction into CH4 on a CoO cluster-modified Cu surface. (e) FEs of CH4 production at various sizes and loading contents on CoO/Cu/PTFE.52 Copyright 2020, Springer Nature. | ||
3.6. Carbon-based materials
Despite the excellent performance of Cu-based materials in CO2 electroreduction into CH4, the development of Cu-free electrocatalysts still attracts tremendous efforts but remains greatly challenging. Carbon-based materials have become one of the most important commonly used catalysts or supports in electrolysis due to their excellent chemical stability, high specific surface area and tunable surface properties. In addition, the electronic structure, acid/base properties, redox properties and wettability can be altered through heteroatom doping or other forms of modification, offering them the possibility of catalyzing CO2 reduction.For instance, Wu and co-workers proposed a synthesis of amine functionalized nitrogen-doped graphene quantum dots (N-aGQDs) via a molecular tuning strategy.53 The functionalization strategy is more advantageous in manipulating the type and density of functional N-containing groups on carbon substrates compared with the traditional doping strategy. The HRTEM and FTIR results confirm the successful synthesis of –NH2 functionalized GQDs (Fig. 9a and b). As a result, the prepared N-aGQDs achieve a maximum FE of 50% and CH4 partial current density of 176 mA cm−2 at −0.98 V vs. RHE (Fig. 9c), outperforming the controlled aGQD catalyst. The role of functional N-containing groups in CO2 electroreduction was further verified by tuning the types of N configurations, indicating that the –NH2 functionality has a positive effect on the selectivity and activity of CH4 production.
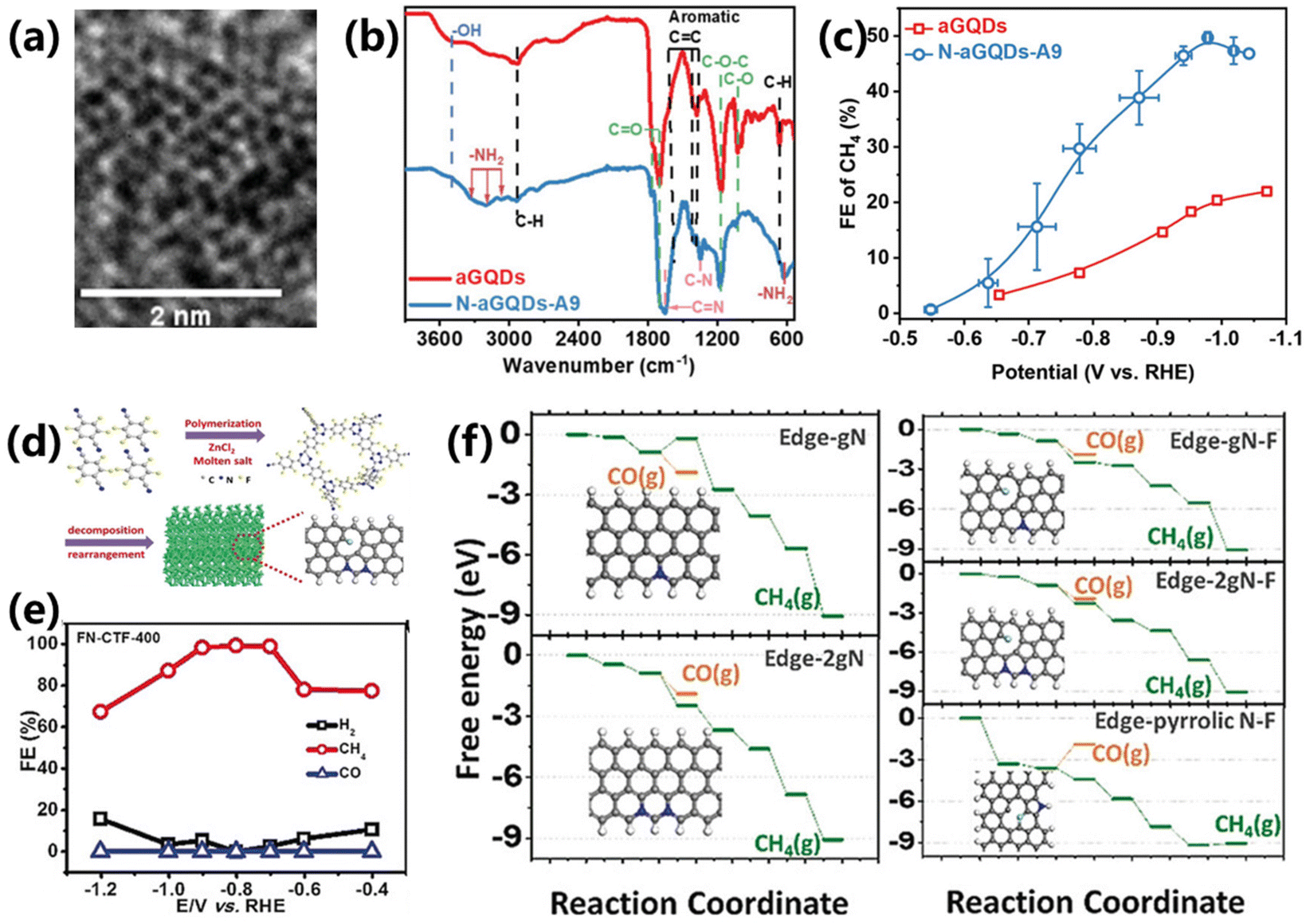 | ||
| Fig. 9 (a) HRTEM image of N-aGQDs-A9. (b) FTIR of aGQDs and N-aGQDs-A9. (c) FEs for CO2 to CH4 over aGQDs and N-aGQDs-A9.53 Copyright 2022, Wiley. (d) Schematic of the preparation of FN-CTF-400. (e) FEs for CO2 reduction over FN–CTF-400. (f) DFT results for the CO2 to CH4/CO reaction pathway and the reaction models of various doped carbons.54 Copyright 2018, Wiley. | ||
Moreover, a series of nitrogen- and fluorine-co-doped covalent triazine frameworks (FN–CTF–X) were prepared by Wen and co-workers through one-step polymerization, which shows high selectivity towards CO2 electroreduction into CH4 (Fig. 9d).54 The maximum FE for CH4 production reaches 99.3%, which is the highest value that can be retrieved so far (Fig. 9e). DFT calculations confirm that doped N is the real active center for CO2 electroreduction and fluorine doping plays a critical role in regulating the activity of N (Fig. 9f). Thus, both nitrogen and fluorine are beneficial for high CH4 selectivity. This work can facilitate research on the design of carbon-based materials for CO2 electroreduction.
3.7. Other electrocatalysts
Recently, transition-metal dichalcogenides are of particular interest in the field of electrolysis due to their unique layered structures. The planes in transition-metal dichalcogenides are usually bound through van der Waals interactions, which can be easily exfoliated to obtain the thin layer structure, rendering them promising for CO2 electroreduction. Liu and co-workers provided an electrocatalyst for the electroreduction of CO2 to CH4 based on ultrathin MoTe2 layers in an ionic liquid electrolyte system (Fig. 10a).55 The MoTe2 layers with an ultrathin structure (∼2.1 nm) and a large surface area were prepared via chemical vapor deposition and ultrasonic exfoliation methods (Fig. 10b and c). Compared with traditional bulk MoTe2 (35%), the synthesized ultrathin MoTe2 layers exhibit a FE of 83% for CO2 to CH4 at a current density of 25 mA cm−2 (Fig. 10d). The kinetic measurement demonstrates that the rate-determining step for CO2 electroreduction into CH4 is transformed from the process of CO2 to CO2˙− species (112 mV dec−1) into the process of adsorbed CO2˙− coupling with a Lewis acid CO2 molecule to form a CO2–CO2˙− adduct (68 mV dec−1), benefitting for the production of CH4 (Fig. 10e). In addition, the ultrathin MoTe2 layers can continuously produce CH4 for 45 h with an FE of 80%, indicating favorable stability.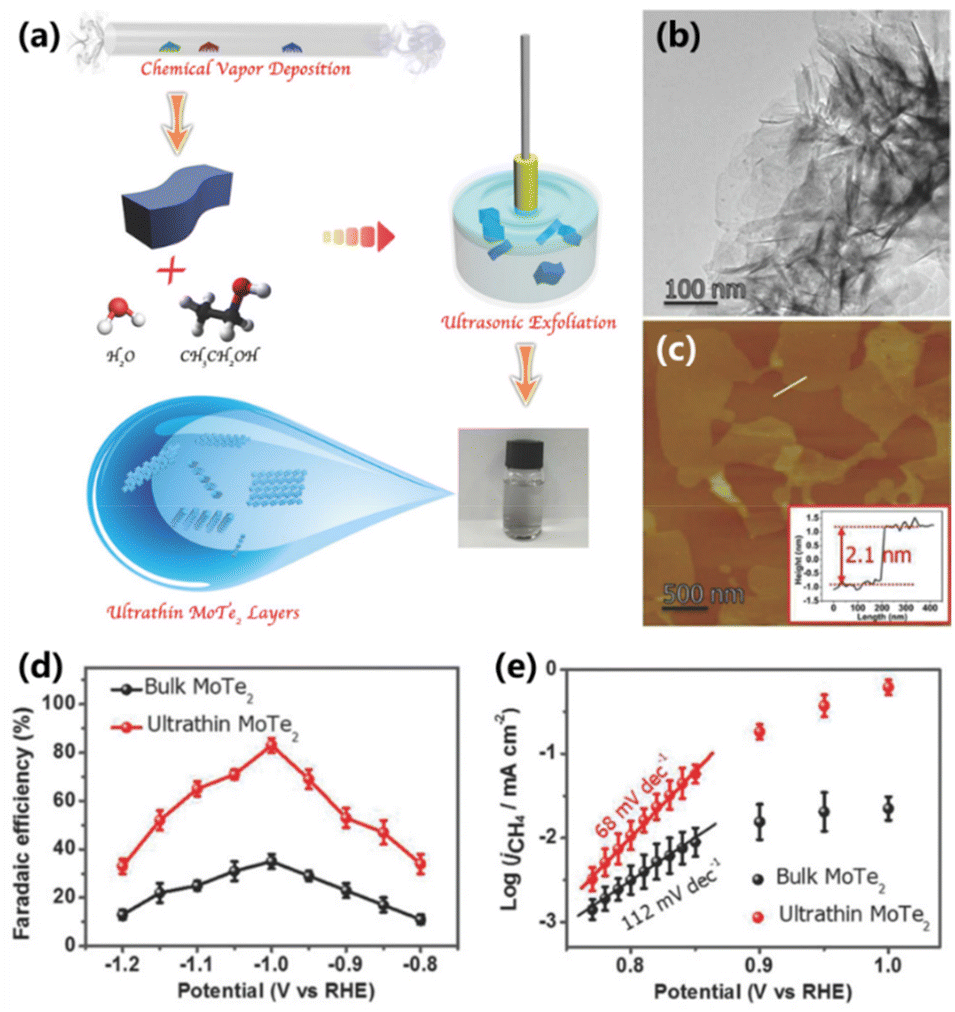 | ||
| Fig. 10 (a) Schematic for the preparation of ultrathin MoTe2 layers. (b) TEM and (c) AFM image of ultrathin MoTe2 layers. (d) FEs for the conversion of CO2 to CH4 and (e) Tafel plots over ultrathin MoTe2 and bulk MoTe2 in a CO2-saturated BmimBF4–H2O electrolyte.55 Copyright 2018, Wiley. | ||
Apart from Cu-based MOFs, Zn-1,3,5-benzenetricarboxylic acid MOFs (Zn–MOFs) were developed by Han and co-workers for CO2 electroreduction into CH4 in an ionic liquid system.56 Various Zn–MOFs with different morphologies were prepared and displayed different performances for CH4 production. Among them, sheet-like Zn–MOFs deliver the highest selectivity (∼80%) for CO2 to CH4 due to the largest electrochemical surface area. The above works provide many opportunities for exploring Cu-free electrocatalysts for CO2 electroreduction into CH4.
4. Enhanced strategy
4.1. Coordination environment manipulation
Although Cu–SACs exhibit a good performance for CO2 electroreduction into CH4, the selectivity and activity should be further improved to achieve commercialization. Through manipulating the coordination environment of SACs, the physicochemical properties can be changed, which in turn affects the adsorption behavior of reaction intermediates, and this has become a hot research topic in the current field.57 How to achieve their unique electronic structures and maximum catalytic efficiencies via coordination environment manipulation has aroused extensive research interest but great challenges remain. With the guidance of theoretical studies revealing that the introduction of boron into the Cu–N4 motif can enhance the binding of CO* and CHO* intermediates, Zeng and co-workers synthesized B, N-doped Cu–SACs (BNC–Cu) by a co-doping strategy, which delivers a superior performance towards CH4 production.34 Compared to pristine Cu–N4, the Cu–NxBy configuration can strongly stabilize *COOH and *CHO, leading to a high CH4 production (Fig. 11a). The accurate electronic structure of BNC–Cu was identified by XAS as Cu–N2B2. The synthesized BNC–Cu shows a maximum FE of 73% and partial current density of −292 mA cm−2 for CH4 production, superior to that of NC–Cu (Fig. 11b). Moreover, the FE for CH4 production was above 60% in a wide potential range from −1.23 to −1.83 V vs. RHE. Lan and co-workers further explored Cu–SACs with other coordination environments.58 A Cu-based conductive metal–organic framework (Cu–DBC) with a Cu–O4 configuration was prepared by a solvothermal method. Compared with controlled Cu single sites with other coordination, Cu–DBC exhibited the highest FEs in the potential range from −0.7 to −1.0 V vs. RHE, and achieved a maximum FE of 80% at −0.9 V vs. RHE (Fig. 11c). In addition to selectivity, Cu–DBC also showed a superior reaction efficiency with the highest current density and TOF values (Fig. 11d). DFT calculations were conducted to further study the structure–activity relationships of Cu–DBC for CO2 electroreduction into CH4. The results show that the Cu–O4 site could transform into low-valence Cu active centers easily, thus possessing the lowest energy barriers for the rate-determining step of *CO to *CHO (Fig. 11e). The above works provide foundations for designing superior SACs and understanding structure–activity relationships. | ||
| Fig. 11 (a) Thermodynamic trend for the conversion of CO2 to CH4 over various configurations as a function with Gad(CHO*). (b) FEs and partial current densities for the conversion of CO2 to CH4 over BNC–Cu and NC–Cu at various cathodic potentials.34 Copyright 2023, Springer Nature. (c) FEs for the conversion of CO2 to CH4 at various applied potentials. (d) The CO2RR performance at −0.9 V vs. RHE. (e) Free energy of the CO2 electroreduction into CH4 pathway.58 Copyright 2021, Springer Nature. | ||
4.2. Elemental doping
Elemental doping has been considered an effective method for regulating the intrinsic activity of catalytic sites in electrocatalysis.59 The introduction of heteroatoms can effectively modify the electronic structure of adjacent atoms, optimizing the interactions with key intermediates, and thereby enhancing their activity.60 Sargent and co-workers reported a porous CuAl doped with Au, Zn, or Ga via a galvanic exchange reaction, which could convert CO2 into CH4 (Fig. 12a).61 The *CO coverage on the CuAl electrocatalyst was increased after introducing the heteroatom. Meanwhile, the doped element exhibited low *CO binding energies, thus achieving the desired *CO protonation process and shifting the product from C2+ to CH4. Among them, Ga-doped CuAl delivered an FE of 30% and 53% for CH4 in 1 M KOH and 1 M KHCO3, respectively. Gong and co-workers doped various oxophilic metals, such as La, Pr, Y and Sm, into the Cu electrocatalyst via a controlled electrodeposition method.62 Owing to the high oxophilicity of the doped metal, the adsorption energy of *CHxO was increased, thus enhancing CH4 production. The best-performing La5Cu95 electrocatalyst showed a maximum FE of 64.5% with a partial current density of 193.5 mA cm−2 for CH4, much higher than that of undoped Cu (Fig. 12d). The operando ATR-SEIRAS spectra showed that the peak of *CO disappears and the peak of *CH3O appears in the range of −0.1 ∼−0.8 V vs. RHE, indicating the hydrogenation of *CO to *CH3O (Fig. 12b and c). DFT calculations demonstrated that the CO2 to *COOH process and the *CO to *CHO process on the La5Cu95 electrocatalyst are exothermic (Fig. 12e). It can be inferred that the doped La not only stabilizes *CHxO, but also accelerates the breakage of the C–O bond in *CH3O due to the stable La–O bond (Fig. 12d). The above works provide new insights into exploring efficient electrocatalysts for CO2 reduction at the atomic level.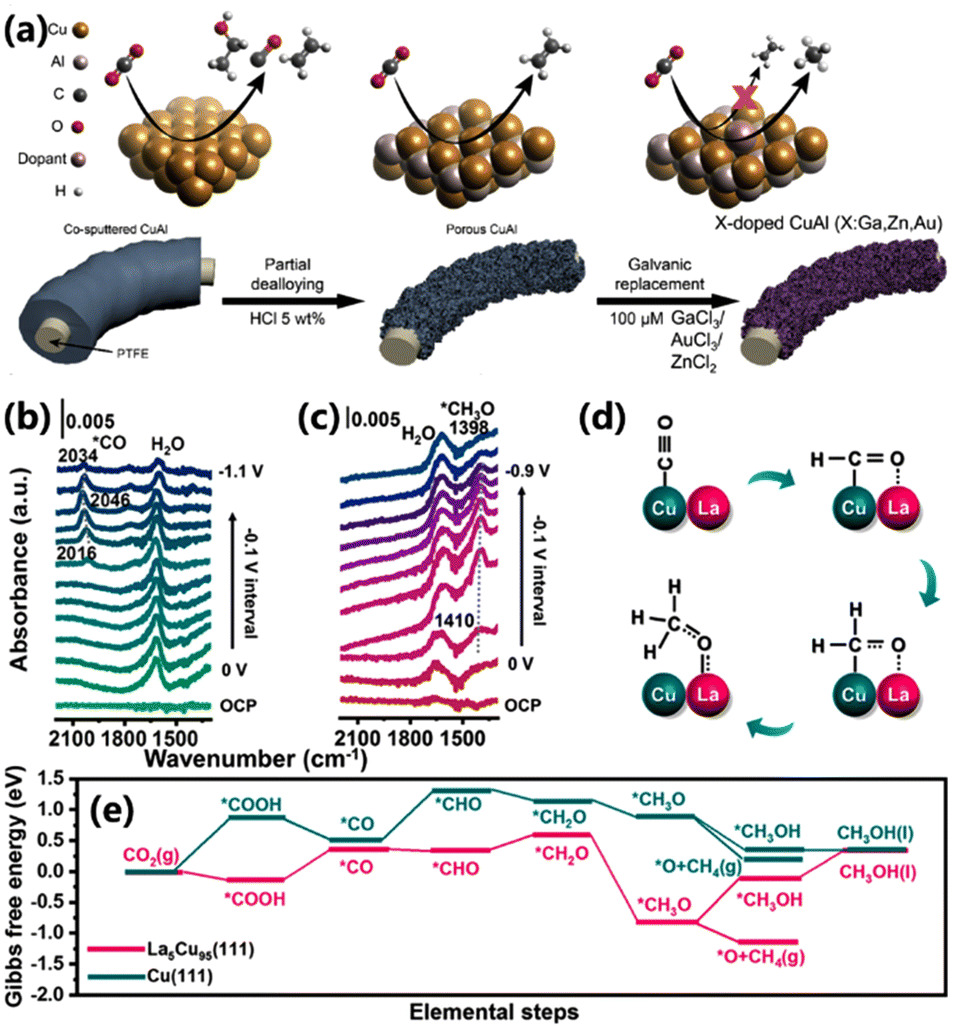 | ||
| Fig. 12 (a) Schematic of the synthesis of doped CuAl catalysts and the proposed mechanism for C–C disruption.61 Copyright 2022, Elsevier. Operando ATR-SEIRAS spectra for (b) Cu and (c) La5Cu95 electrocatalysts. (d) The reaction pathways for the conversion of CO2 to CH4 on the La5Cu95 surface. (e) The Gibbs free energy for the conversion of CO2 to CH4 on Cu(111) and La5Cu95(111).62 Copyright 2023, American Chemical Society. | ||
4.3. Heterostructure construction
Recently, heterostructure construction has been reported to be an efficient strategy to improve the intrinsic activity for CO2 electroreduction due to the unique strain effects and electronic interactions.63 Moreover, the different components in heterostructures can cause synergistic effects, thus adjusting the adsorption and conversion of critical intermediates and improving the performance. To date, the activity of CO2 electroreduction into HCOOH (In/In2O3−x,63 Bi/Bi2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 64 and Sn/SnO265), CO (Cu2O/CuO66 and Ni/Ni3ZnC0.767) and C2+ products (Cu2O@CuBTC68 and Cu2O/CeO269) has been dramatically enhanced via heterostructure construction. Therefore, exploring efficient heterostructures and revealing the underlying reaction mechanism is highly desirable.
64 and Sn/SnO265), CO (Cu2O/CuO66 and Ni/Ni3ZnC0.767) and C2+ products (Cu2O@CuBTC68 and Cu2O/CeO269) has been dramatically enhanced via heterostructure construction. Therefore, exploring efficient heterostructures and revealing the underlying reaction mechanism is highly desirable.
A heterostructure of Cu–ZnO for efficient electroreduction of CO2 to CH4 is reported by Cui and co-workers.70 The heterostructure indicated the existence of the (111) facets of metallic Cu and the (002) facets of ZnO. Moreover, the heterostructure could be modulated by tuning the feed amount of copper and zinc sources, thus exhibiting different performances. The maximum FE for CH4 production is achieved with 72.4% at −0.7 V vs. RHE when the Cu:Zn ratio was close to 1:1 (Fig. 13a). In this heterostructure, Cu is the real active site for CO2 electroreduction into CH4, and its interfacial sites are modulated by ZnO, thus accelerating the deep conversion of CO2. Compared to single Cu and ZnO, the Cu–ZnO heterostructure exhibits suitable adsorption energies of *COOH and *CHO intermediates (Fig. 13b), explaining the enhanced activity and selectivity for CH4 production.
 | ||
| Fig. 13 (a) Relationship between the FE of the CH4/CO and Cu/Zn atomic ratio of Cu–ZnO electrocatalysts. (b) DFT results for CO2 electroreduction into CH4.70 Copyright 2023, Royal Society of Chemistry. (c) Schematic of the synthesis of MOF@COF heterostructures for CO2 electroreduction into CH4. (d) DFT results for CO2 electroreduction into CH4 on COF and MCH-3, respectively.27 Copyright 2022, Wiley. | ||
One of the challenges faced by heterostructures in catalytic reactions is that the active site shows an intrinsic deficiency of buried active sites, which not only inhibits the reaction process but also creates an obstacle for synergistic mechanism study. To address the above issues, Chen and co-workers fabricated a series of honeycomb-like metal–organic frameworks@covalent organic framework (MCH–X) heterostructures via self-assembly (Fig. 13c).27 Ultrathin COF-366-OH–Cu is in situ coated on porous HMUiO-66-NH2, with tunable COF shell thickness. MCH-3 delivers a maximum FE of 76.7% for CH4 production at −1.0 V vs. RHE, much superior to that of bare COF (37.5%), MOF (15.9%) and their physical mixture (38.0%). DFT calculations indicate that the rate-determining step (generation of *COOH) required lower free energy than that of bare COF, verifying the positive effect of the MCH heterostructure (Fig. 13d).
4.4. Crystal phase engineering
For electrocatalysts, the crystal phase structure plays a critical role in the species adsorption behaviors and electron conductivity. Precise control of the crystal phase structure is of great significance for studying reaction mechanisms and improving the catalytic performance.71 For example, CO is preferentially reduced into CH4 at the (111) facets of Cu, while being reduced into C2H4 at the (111) facets.72 However, the previous works mostly conducted research in a H-type cell at a low current density, which cannot provide accurate guidance for large-scale applications.Given this, Buonsanti and co-workers further confirmed the facet-dependent selectivity of CO2 electroreduction on various shaped Cu nanoparticles in a flow cell under commercially relevant current densities.73 The electrochemical results demonstrate that CH4 is the main hydrocarbon product on the (111) phase, and the (100) phase exhibits a much higher selectivity for C2H4 (Fig. 14a). A maximum FE of 53% and a current density of 100 mA cm−2 are achieved on the (111) phase at −0.91 V vs. RHE. However, it is not accurate to conclude that the activity and selectivity of CO2 electroreduction are related to the crystal phase, especially for electrocatalysts that are prone to reconstruction during the catalytic process. Thus, the real reaction mechanism and structure–activity relationships under operating conditions still need to be confirmed through various characterization techniques. Dong and co-workers indicate that the reconstructed Cu2O/Cu interface determines the selectivity and activity of CO2 electroreduction into CH4, rather than the initial crystal phase of Cu2O.74 The authors synthesized various types of Cu2O precursors via the solvothermal method and concluded that near-surface Cu2O in all samples is reduced during CO2 electroreduction and form Cu2O/Cu interfaces (Fig. 14b and c). As a result, all three samples with different crystal phases deliver similar selectivity for CH4 production with similar Tafel slopes and long-term stability (Fig. 14d). Regardless of the initial crystal phases of Cu2O, *CO is always adsorbed at the Cu2O/Cu interface via a bridge configuration, thus exhibiting an indistinguishable performance. As a consequence, further research is needed on the true reaction mechanism of CO2 electroreduction for the materials that are prone to reconstruction.
 | ||
| Fig. 14 (a) FEs of CO2 electroreduction into CH4 over Cu nanocatalysts with various facets.73 Copyright 2020, American Chemical Society. (b) Schematic of the reconstruction of Cu2O catalysts. (c) HRTEM image of t-Cu2O after reduction at −1.6 V for 1 h. The inset shows the FFT of the corresponding t-Cu2O HRTEM. (d) The Tafel slope of c-Cu2O, t-Cu2O and o-Cu2O.74 Copyright 2022, Wiley. (e) The periodic slab model of the tw-Cu(111) surface. (f) HRTEM image of tw-Cu with a twin-boundary structure. (g) FEs of tw-Cu for CO2 electroreduction into CH4. (h) DFT results of different pathways on planar tw-Cu(111) and Cu(111).77 Copyright 2023, American Chemical Society. | ||
The construction of high-index crystal planes with rich surface structures can provide more active sites for the CO2 electroreduction, but unfortunately, high-index crystal planes on Cu-based electrocatalysts tend to reduce CO2 into C2H4 rather than CH4.75 Grain boundaries with various active sites in oxide-derived Cu have also been explored extensively for CO2 electroreduction, and C2H4 is also the main product.76 However, grain boundaries with diverse active sites pose difficulties in revealing the atomic-scale mechanism of the CO2RR. Hence, Huang and co-workers prepared a highly (111)-oriented Cu electrocatalyst with dense twin boundaries (tw-Cu) via a rotary electroplating method, showing high activity and selectivity for CO2 electroreduction into CH4 (Fig. 14e).77 A well-defined twin-boundary structure with the stacking sequence of ABC/A/CBA is confirmed by HRTEM with FFT (Fig. 14f). And the tw-Cu shows a remarkable FE of about 86% for CH4 production at −1.2 V vs. RHE (Fig. 14g), which is superior to polycrystalline Cu. DFT simulations indicate that the activation barriers of *CHO and *COH generation are lower than that of C–C coupling on tw-Cu (Fig. 14h). Compared to polycrystalline Cu with the Cu(111) surface, tw-Cu with a twin-boundary structure shows a greatly reduced reaction barrier for the CO hydrogenation process, thus explaining the high CH4 selectivity.
4.5. Surface modification
During CO2 electroreduction, the high binding of *CO is inclined to increase the coverage of *CO on the surface of the electrocatalyst, thus facilitating C–C coupling to C2+ products. Surface modification provides an efficient means to manipulate the adsorption of reaction intermediates, which is a desirable strategy to improve CH4 production yet challenging. Zeng and co-workers modified Cu nanoparticles with benzenethiol (Cu NPs–BDT) to change the electronic structure of the Cu surface and achieved high activity for CO2 to CH4.78 The modification of BDT was confirmed by FT-IR. The blue shift of C–H vibrations and disappearance of S–H bands indicate a direct interaction between Cu and BDT (Fig. 15a). DFT calculations show a strong electronic interaction in the Cu–BDT interface, resulting in a downshift of the d-band center of Cu (111) planes (Fig. 15b). The downshift of the d-band center could lead to a low *CO coverage on the surface of Cu and thus inhibit the C–C coupling process (Fig. 15c). As a result, Cu NPs–BDT exhibits a high FE of 57.7% for CH4 production, which is 1.9-fold higher than that of unmodified Cu NPs.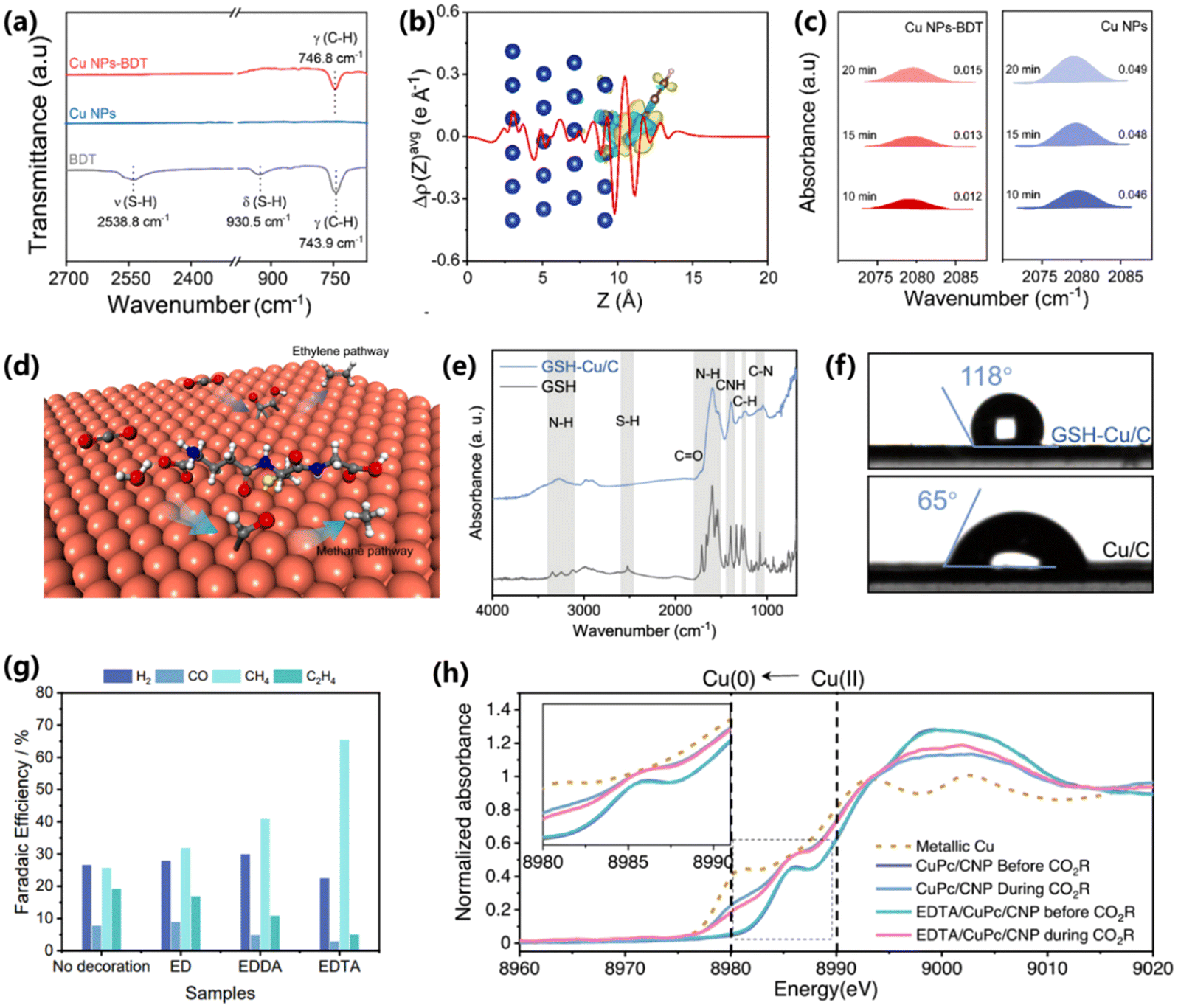 | ||
| Fig. 15 (a) FT-IR spectra of benzenethiol-modified Cu NPs, Cu NPs and benzenethiol. (b) Charge difference of the benzenethiol-modified Cu (111) plane. (c) In situ ATR-SEIRAS spectra of the *CO peak for benzenethiol-modified Cu NPs and Cu NPs.78 Copyright 2022, Royal Society of Chemistry. (d) Schematic of glutathione-modified copper electrode for CO2 electroreduction into CH4. (e) ATR-FTIR spectra of glutathione-modified Cu/C and glutathione. (f) Contact angle tests of glutathione-modified Cu/C and Cu/C.79 Copyright 2022, American Chemical Society. (g) FEs of CuPc/CNP samples with and without decoration. (h) In situ XANES spectra of CuPc/CNP and EDTA/CuPc/CNP before and during the CO2RR.80 Copyright 2023, Springer Nature. | ||
It was demonstrated in several studies that CO2 electroreduction could proceed at a high current density (>200 mA cm−2) in a flow cell configuration with an alkaline electrolyte. However, this could increase the local pH and consume the active hydrogen on the surface of the electrocatalyst. Given this, Wang and co-workers proposed a synthesis of a glutathione-modified Cu electrode (GSH–Cu/C), which exhibited a maximum FE of 61.7% and a partial current density of 153.7 mA cm−2 for CH4 production (Fig. 15d).79 The presence of glutathione on the Cu electrode is proved by the vanishing of S–H bands in the ATR-FTIR spectra (Fig. 15e). After modification of glutathione, Cu/C exhibits a hydrophobic surface with a contact angle of 118°, which could accelerate the transport of CO2 molecules (Fig. 15f). Moreover, the carboxyl and amino groups of glutathione could regulate the binding configuration of *CO and the local availability of *H, enabling a high selectivity and activity for CH4 production. Besides, Sinton and co-workers proposed a coordination method to stabilize the free Cu ions by modifying Cu with multidentate donor sites (e.g., ethylenediaminetetraacetic acid (EDTA)).80 A chelation interaction happens between Cu ions and hexadentate donor sites in EDTA, resulting Cu–N/O single sites. A high FE of 65% for CH4 production is achieved on EDTA-modified CuPc, superior to that of unmodified CuPc (Fig. 15g). Moreover, EDTA-modified CuPc exhibited good stability of 300 min at a current density of 100 mA cm−2 for CO2 electroreduction. The structure stability of EDTA-modified CuPc was further proved by in situ X-ray absorption spectroscopy. Compared to unmodified CuPc with easily reduced Cu, the oxidation state of Cu species in EDTA-modified CuPc is preserved during the CO2RR (Fig. 15h), demonstrating the positive role of surface modification.
4.6. Core–shell architecture
Constructing a hydrophobic structure via morphology manipulation would form a gas–solid–liquid interface on the surface of the electrocatalyst, which can tune the availability of local H2O and affect the CO2RR performance. For example, Yang and co-workers constructed a core–shell structure of carbon coated on a copper/cuprous oxide (H–CuOx@C) with a hydrophobic surface.81 The HAADF-STEM image and the corresponding EDS element diagram confirmed the existence of a core–shell structure with a carbon shell of 50 nm (Fig. 16a and b). The H–CuOx@C electrocatalyst possesses a maximum faradaic efficiency of 81% towards CH4 at −1.60 V vs. RHE, larger than that of wettable CuOx (12%). The activity of CO2 electroreduction into CH4 was further improved with a flow cell configuration. A maximum FE of 73.3% and conversion rate of 0.56 μmol cm−2 s−1 were achieved at current densities of 500 and 700 mAcm−2, showing a promising practical application prospect (Fig. 16c). DFT calculations suggest that the hydrophobic structure with low water coverage can improve the protonation of the *CO intermediate and the generation of CH4. Li and co-workers also synthesized a nitrogen doped carbon coated Cu electrocatalyst (Cu@NxC) via polymerization and subsequent pyrolysis methods (Fig. 16d).82 The synthesized Cu@NxC shows a well-defined core–shell structure with Cu as the core and NxC as the shell (Fig. 16e). The C and N elements are dispersed uniformly in this core–shell structure (Fig. 16f). In addition, the HRTEM image shows that the thickness of the NxC layer of Cu@NxC was about 6.4 nm (Fig. 16g). The electrochemical results demonstrate that the selectivity of CO2 electroreduction into CH4 could be adjusted by changing the temperature of pyrolysis. When the pyrolysis temperature increases from 300 °C to 400 °C, the pyrrolic N becomes the main type of nitrogen in Cu@NxC, which could adsorb *CO via bridge-bonding and favor for CH4 production (Fig. 16h).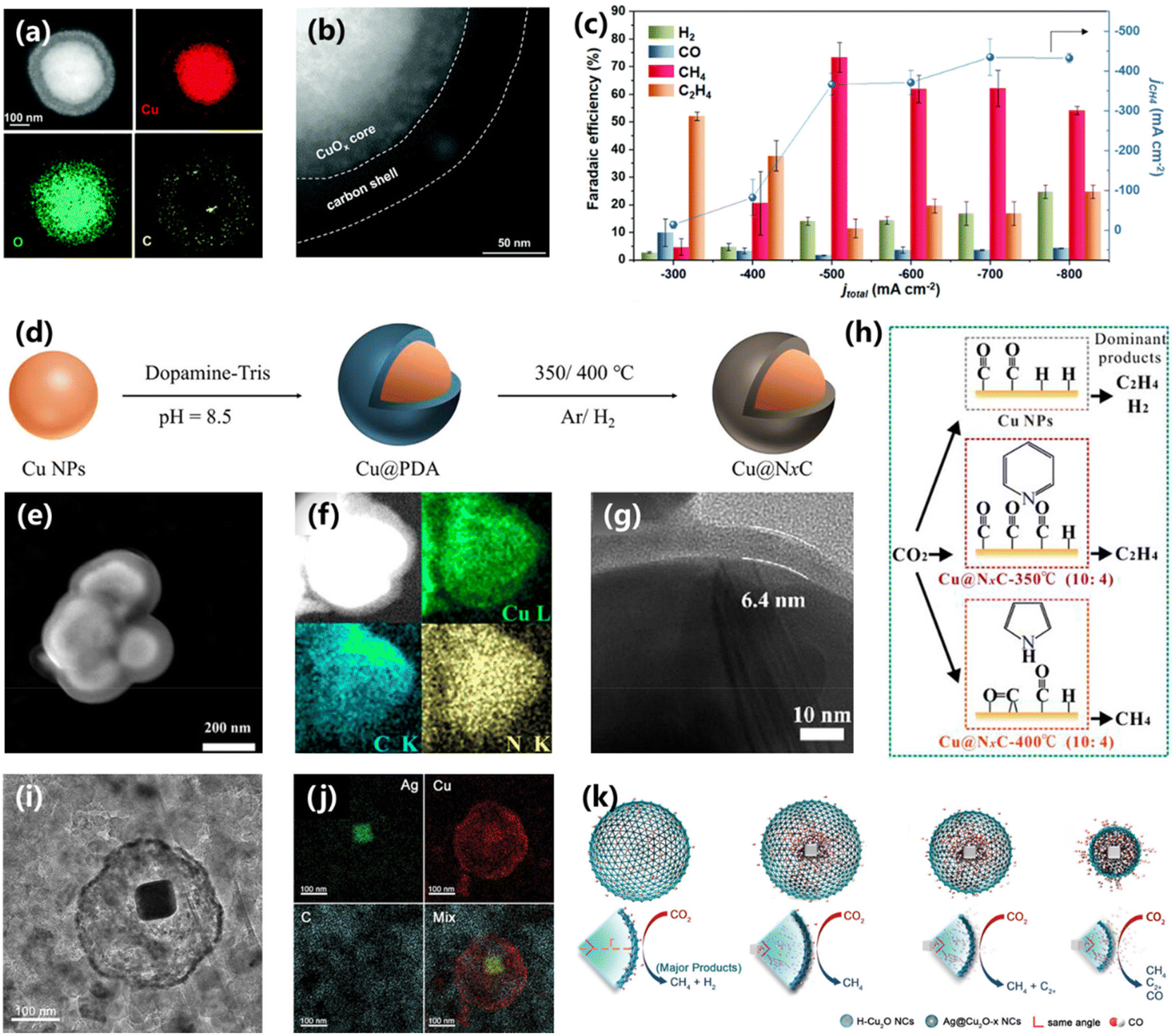 | ||
| Fig. 16 (a) HAADF-STEM image and EDS mapping images of H–CuOx@C. (b) Enlarged HAADF-STEM image of H–CuOx@C. (c) FEs and CH4 partial current density of H–CuOx@C.81 Copyright 2022, Royal Society of Chemistry. (d) Schematic of the synthesis of core–shell Cu@NxC catalysts. (e) The SEM image of Cu@NxC-350 °C and (f) the EDS mapping images. (g) TEM image of the Cu@NxC-350 °C catalyst. (h) The scheme for the proposed mechanistic pathways of CH4 and CH2CH2 over the Cu@NxC-catalysts.82 Copyright 2023, American Chemical Society. (i) TEM image and (j) the EDS mapping images of Ag@Cu2O after 2 h of electrocatalytic CO2RR. (k) Schematic of the modulation of reduction products on various Ag@Cu2O.83 Copyright 2021, Wiley. | ||
To elucidate the structure–activity relationship of CO2 electroreduction, it is essential to construct an independent catalyst model to avoid cross-talk from another catalyst, which is yet challenging. Peng and co-workers fabricated a yolk–shell nanostructure composed of an Ag core and a Cu2O shell (Ag@Cu2O-x NCs), which could avoid the above-mentioned cross-talk effect.83 The nanostructure of Ag@Cu2O-x NCs can be adjusted via changing the feeding amount of Ag. And the Cu2O shell maintained the initial cage structure despite being reduced (Fig. 16i and j). During CO2 electroreduction, the CO flux at the Cu shell is related to the size of the Cu shell. The Ag@Cu2O-6.4 NCs exhibit the most impressive FE of 74% and partial current density of 178 mA cm−2 for CH4 production at −1.2 V vs. RHE, better than that of Ag@Cu2O NCs with other sizes. The best performance is attributed to the moderate CO coverage on the Cu shell with suppressed HER and C–C coupling (Fig. 16k). DFT calculations were further employed to confirm this conclusion. Various *CO coverage ranges from 2/16 ML to 6/16 ML on the Cu(111) surface were achieved, and the formation energy of the key intermediates of *CHO and *COCOH became weaker with increasing *CO. However, as the coverage of *CO reached 2/16 or 3/16, *CHO became thermodynamically favorable with lower formation energy, demonstrating that the appropriate *CO coverage contributes to *CHO formation and CH4 production. The above works demonstrate a novel catalyst model to elucidate the structure–activity relationship.
4.7. Tandem catalysis
Since the CO2 electroreduction into CH4 requires eight proton-coupled electron transfer process in different pathways, the adsorption and desorption of key intermediates are essential for CH4 production. However, the intrinsic linear scaling relationship of intermediates binding during the CO2RR will cause low selectivity production of the target product.84 Recently, tandem catalysis has been demonstrated to be effective in breaking the limitation of linear scaling relationships, leading to high activity and selectivity. In such cases, exploring more tandem catalysts is imminent. Cobalt phthalocyanine and zinc–nitrogen–carbon were constructed into a tandem catalyst by Bao and co-workers, which exhibited a superior performance with a high CH4/CO production rate ratio to that of pure CoPc or Zn–N–C (Fig. 17a).85 A maximum FE of 18.3% and a partial current density of 44.3 mA cm−2 were achieved at −1.24 V vs. RHE. During the formation of CH4, CoPc catalyzed the reduction of CO2 into CO and Zn–N–C catalyzed the protonation of CO into CH4. DFT calculations verified that the protonation of *CO to *CHO is more likely to occur via the Langmuir–Hinshelwood mechanism, and the synergistic co-adsorption of *CO and *H on ZnN4 can enhance the CH4 production rate (Fig. 17b).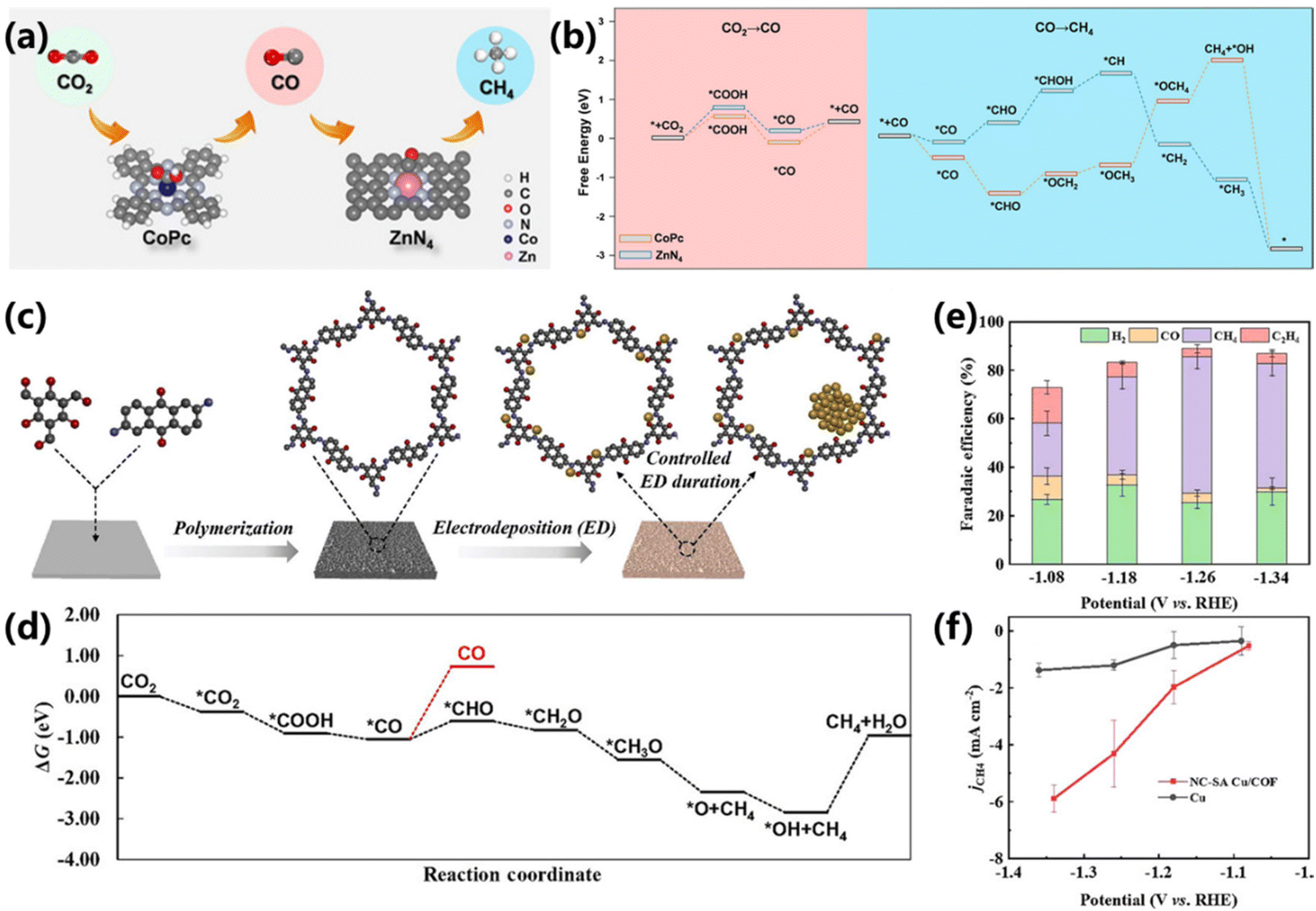 | ||
| Fig. 17 (a) Schematic of the tandem catalysis of CO2 electroreduction into CH4 over CoPc and ZnN4. (b) DFT results for CO2 electroreduction into CH4 over CoPc and ZnN4.85 Copyright 2020, Wiley. (c) Schematic synthesis of SA Cu/COF and NC–SA Cu/COF. (d) Gibbs free energy profiles of CO2RR on NC–SA Cu/COF. (e) FEs and (f) CH4 partial current densities of NC–SA Cu/COF.86 Copyright 2022, Wiley. | ||
During the formation of CH4, the activation of H2O and the generation of proton are important for the efficient conversion of CO2. Under the linear scaling relationship, there are still significant challenges in establishing the optimal balance between CO2 reduction and the generation of proton from H2O reduction. Inefficient H2O activation can lead to insufficient active hydrogen, but excessive H2O activation is bound to convert into hydrogen evolution, thereby reducing the selectivity of CO2 reduction. Therefore, Shao and co-workers reported a tandem catalyst based on covalent organic framework-supported Cu nanoclusters and isolated Cu atoms (NC–SA Cu/COF), exhibiting an enhanced performance for CO2 electroreduction into CH4 (Fig. 17c).86 DFT calculations indicate that CO2 is first adsorbed over isolated Cu atoms and then reduced into *CO, followed by moving closer to the neighboring Cu nanoclusters for further reduction into CH4. The rate-determining step for CH4 production is the reduction of *CO to *CHO (ΔG = 0.44 eV), which is easier than the desorption of *CO (Fig. 17d). Moreover, the K+ ions in COF can facilitate the adsorption and dissociation of H2O, providing *H for *CO protonation. Compared to pure COF and SA Cu/COF, the NC–SA Cu/COF delivers higher FEs (maximum FE of 56.2%) for CH4 production (Fig. 16e). In addition, the NC–SA Cu/COF also exhibits much higher CH4 partial current density (Fig. 17f), demonstrating the feasibility of tandem catalysis.
5. Summary and outlook
In this review, we have summarized the reaction mechanisms, the latest developments of advanced electrocatalysts and enhanced strategies for CO2 electroreduction into CH4. The key intermediate in the electroreduction of CO2 to CH4 is *CHO, and the reaction energy barrier can be lowered by optimizing its adsorption. Meanwhile, the generation of *CHO from *CO can be significantly influenced by modulating the coverage of *CO and *H on the surface of the electrocatalyst. Meanwhile, a significant enhancement of CH4 partial current density (>300 mA cm−2) was realized in the flow cell, but the selectivity was retained around 70% (Fig. 18). In addition, the electrocatalysts are still stuck in the realm of Cu-based materials, which is difficult to break through. Currently, only a few works have implemented a techno-economic analysis of the constructed electrocatalytic system, which is insufficient for the commercial development of CO2 electroreduction into CH4. Therefore, we present some ideas for the development of CO2 electroreduction into CH4 from the perspectives of electrocatalyst, reaction mechanism and reaction system.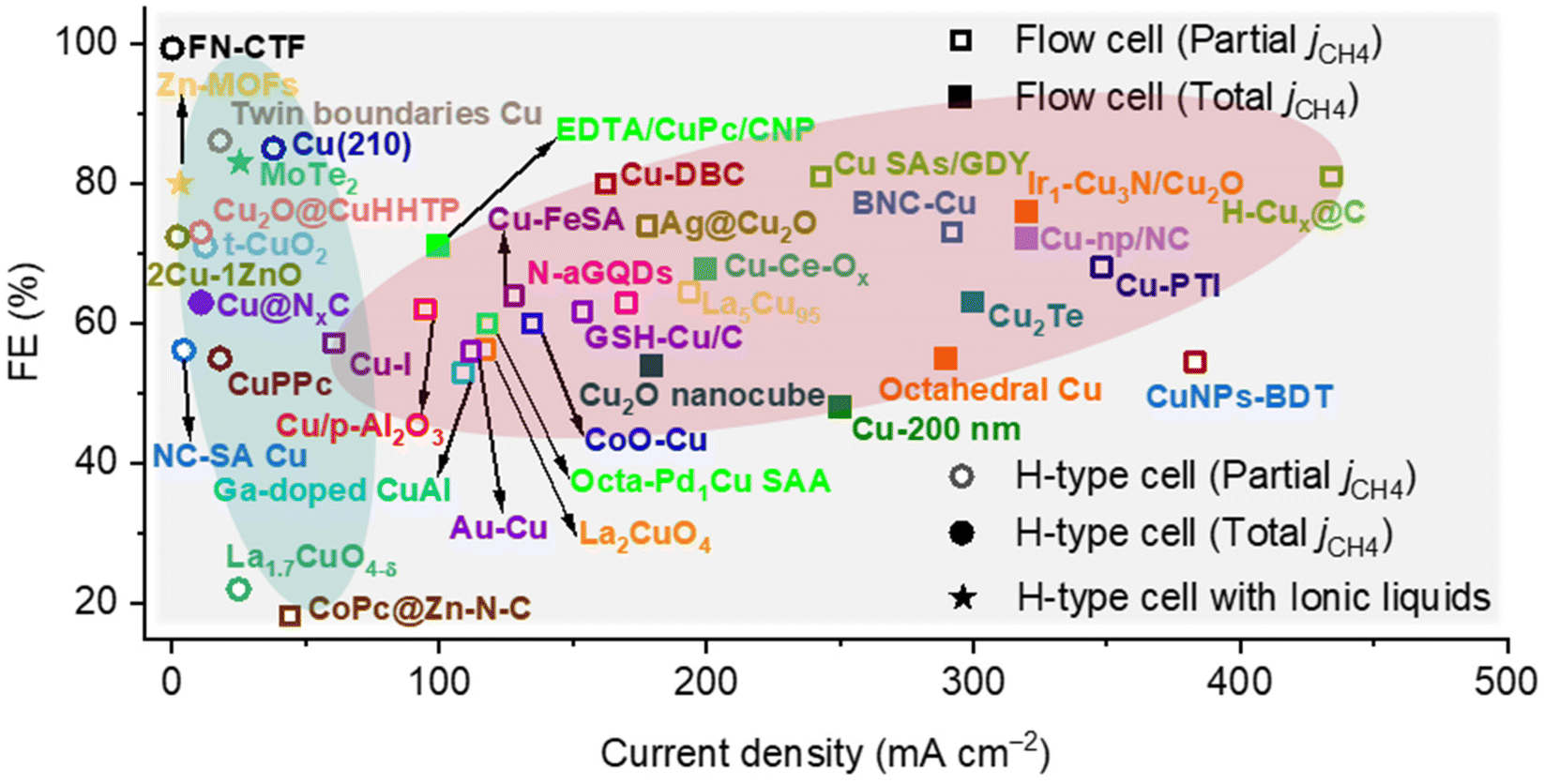 | ||
| Fig. 18 Comparison of the CO2 electroreduction into CH4 performance over electrocatalysts in recent literature. | ||
5.1. Exploring and optimizing electrocatalysts
Despite the development of various electrocatalysts and enhanced strategies, the current activity, selectivity and stability of CO2 electroreduction into CH4 are still unsatisfactory and far from being commercially available. CH4 is the deepest C1 product of CO2 electroreduction, and the CH4 pathway involves various intermediates, such as *COOH, *CO and *CHO/*COH. Under the linear scaling relationship, it is difficult but necessary to design the electrocatalyst with optimal adsorption of different intermediates. In addition to C-containing intermediates, *H is essential for the formation of CH4 but is usually overlooked. Modulating the relationship between CO2 reduction and H2O reduction by regulating the hydrophilicity and hydrophobicity of electrocatalysts is a viable strategy for improving CH4 production. Another issue is that most reported electrocatalysts are commonly designed with powdery structures, which will cause severe agglomeration of materials and decrease utilization of active sites. Powder electrocatalysts need to be attached to the surface of the current collector with the aid of a binder, which inevitably results in reduced stability. Self-supported catalysts or catalysts grown in situ on the current collector show promising prospects for CO2 electroreduction, but the existing synthetic strategy should be further upgraded to achieve the controlled surface/interface structure and large-area preparation.5.2. Monitoring the dynamic structural evolution and revealing the reaction mechanism
During the CO2 electroreduction, the morphological structure of electrocatalysts, especially for Cu-based materials, will undergo dynamic reconstruction. It is necessary to determine the real active sites and reveal the structure–activity relationships by means of in situ technologies (in situ Raman, in situ XAS, etc.). Although in situ FT-IR can capture the intermediate species produced during CO2 electroreduction, the production of CH4 involves multiple pathways with multiple intermediate species, which poses a significant obstacle to the revelation of the catalytic mechanism. In addition to the microscopic active site, the macro-structure also plays a role in the exposure of the active site and the mass transfer of CO2, and the influence mechanism still needs more advanced techniques to be determined. To date, theoretical calculations based on thermodynamic data, such as onset potential, intermediate adsorption and Gibbs free energy, have been used to propose the reaction mechanism. However, the existing experimental results show that the activity and selectivity can be affected by the morphology of the electrode, the local pH value and the type/concentration of ions at the electrode/electrolyte interface, which are usually ignored. Moreover, the DFT calculations based on the real-time surface structure and real reaction environments should be conducted to reveal the true reaction mechanisms, which will provide more guidance for the design of electrocatalysts and development of optimization strategies for CO2 electroreduction into CH4.5.3. Construction of an efficient electrolysis system
Considerable progress has been made so far in developing various types of Cu-based electrocatalysts for CO2 electroreduction into CH4, where they have demonstrated an outstanding performance at the laboratory level. Unfortunately, they are limited by the current density, selectivity, stability, etc. in practical industrial applications. Furthermore, the reactor configuration, membrane, electrolyte, pressure, temperature and many other parameters have a significant influence on the overall performance of electrochemical reactors, but have not received enough attention compared with the design of electrocatalysts at present. In order to realize the commercial application of CO2 electroreduction into CH4, techno-economic analysis and full life cycle assessment of the electrolysis system are essential. In addition, the current CO2 electroreduction is paired with an oxygen evolution reaction, which consumes 90% of the input energy. To further improve the efficiency and reduce the cost of CH4 production, the coupling system based on CO2 electroreduction and alternative oxidation reactions (e.g., organic oxidation, waste reforming) deserves more attention and exploration. Given that CO2 electroreduction into CH4 is an 8-electron/proton transfer process with sluggish kinetics, the reaction efficiency can be further improved by constructing a tandem reaction system. Of course, the coupling of CO2 electrolysis and other catalytic systems (photocatalysis, thermal catalysis) can also be constructed to realize the production of higher value-added products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was jointly supported by the Fundamental Research Funds for the Central Universities (BLX202257), the National Natural Science Foundation of China (No. 22109004 and 52300125) and the Beijing Municipal Education Commission through the Innovative Transdisciplinary Program “Ecological Restoration Engineering”.References
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- Z.-W. Yang, J.-M. Chen, L.-Q. Qiu, W.-J. Xie and L.-N. He, Angew. Chem., Int. Ed., 2022, 61, e202205301 CrossRef CAS PubMed.
- M. B. Ross, P. De Luna, Y. Li, C.-T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648–658 CrossRef CAS.
- P. Zhao, H. Jiang, H. Shen, S. Yang, R. Gao, Y. Guo, Q. Zhang and H. Zhang, Angew. Chem., Int. Ed., 2023, e202314121 CAS.
- R. Zhao, P. Ding, P. Wei, L. Zhang, Q. Liu, Y. Luo, T. Li, S. Lu, X. Shi, S. Gao, A. M. Asiri, Z. Wang and X. Sun, Adv. Funct. Mater., 2021, 31, 2009449 CrossRef CAS.
- J. Zhu, L. Lv, S. Zaman, X. Chen, Y. Dai, S. Chen, G. He, D. Wang and L. Mai, 16, 4812-4833, Energy Environ. Sci., 2023, 16, 4812–4833 RSC.
- S. Kibria Nabil, S. McCoy and M. G. Kibria, Green Chem., 2021, 23, 867–880 RSC.
- H. Zheng, Z. Yang, X. Kong, Z. Geng and J. Zeng, Chin. J. Catal., 2022, 43, 1634–1641 CrossRef CAS.
- C. Chen, J. F. K. Kotyk and S. W. J. C. Sheehan, Chem, 2018, 4, 2571–2586 CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- Z. Sun, Y. Hu, D. Zhou, M. Sun, S. Wang and W. Chen, ACS Energy Lett., 2021, 6, 3992–4022 CrossRef CAS.
- N. Han, P. Ding, L. He, Y. Li and Y. Li, Adv. Energy Mater., 2019, 10, 1902338 CrossRef.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 20627–20648 CrossRef CAS PubMed.
- S. Liang, L. Huang, Y. Gao, Q. Wang and B. Liu, Adv. Sci., 2021, 8, 2102886 CrossRef CAS PubMed.
- J. Wu, Y. Huang, W. Ye and Y. Li, Adv. Sci., 2017, 4, 1700194 CrossRef.
- P. Ding, H. Zhao, T. Li, Y. Luo, G. Fan, G. Chen, S. Gao, X. Shi, S. Lu and X. Sun, J. Mater. Chem. A, 2020, 8, 21947–21960 RSC.
- B. Chang, H. Pang, F. Raziq, S. Wang, K.-W. Huang, J. Ye and H. Zhang, Energy Environ. Sci., 2023, 16, 4714–4758 RSC.
- T. Yan, X. Chen, L. Kumari, J. Lin, M. Li, Q. Fan, H. Chi, T. J. Meyer, S. Zhang and X. Ma, Chem. Rev., 2023, 123, 10530–10583 CrossRef CAS.
- F. Pan, X. Yang, T. O'Carroll, H. Li, K.-J. Chen and G. Wu, Adv. Energy Mater., 2022, 12, 2200586 CrossRef CAS.
- R. Li, K. Xiang, Z. Peng, Y. Zou and S. Wang, Adv. Energy Mater., 2021, 11, 2102292 CrossRef CAS.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS.
- L. Sun, V. Reddu, A. C. Fisher and X. Wang, Energy Environ. Sci., 2020, 13, 374–403 RSC.
- C. Li, Z. Liu, X. Zhou, L. Zhang, Z. Fu, Y. Wu, X. Lv, G. Zheng and H. Chen, Energy Environ. Sci., 2023, 16, 3885–3898 RSC.
- L. Ou, J. Chen, Y. Chen and J. Jin, Phys. Chem. Chem. Phys., 2019, 21, 15531–15540 RSC.
- J.-D. Yi, R. Xie, Z.-L. Xie, G.-L. Chai, T.-F. Liu, R.-P. Chen, Y.-B. Huang and R. Cao, Angew. Chem., Int. Ed., 2020, 59, 23641–23648 CrossRef CAS PubMed.
- Y.-L. Yang, Y.-R. Wang, L.-Z. Dong, Q. Li, L. Zhang, J. Zhou, S.-N. Sun, H.-M. Ding, Y. Chen, S.-L. Li and Y.-Q. Lan, Adv. Mater., 2022, 34, 2206706 CrossRef CAS.
- X. L. Zhou, J. Q. Shan, L. Chen, B. Y. Xia, T. Ling, J. J. Duan, Y. Jiao, Y. Zheng and S. Z. Qiao, J. Am. Chem. Soc., 2022, 144, 2079–2084 CrossRef CAS.
- S. Chen, B. Wang, J. Zhu, L. Wang, H. Ou, Z. Zhang, X. Liang, L. Zheng, L. Zhou, Y.-Q. Su, D. Wang and Y. Li, Nano Lett., 2021, 21, 7325–7331 CrossRef CAS.
- X. Liang, N. Fu, S. Yao, Z. Li and Y. Li, J. Am. Chem. Soc., 2022, 144, 18155–18174 CrossRef CAS PubMed.
- H. Zhang, J. Wang, Z. Zhao, H. Zhao, M. Cheng, A. Li, C. Wang, J. Wang and J. Wang, Green Chem., 2018, 20, 3521–3529 RSC.
- W. Xie, H. Li, G. Cui, J. Li, Y. Song, S. Li, X. Zhang, J. Y. Lee, M. Shao and M. Wei, Angew. Chem., Int. Ed., 2021, 60, 7382–7388 CrossRef CAS.
- S. Ji, Y. Chen, X. Wang, Z. Zhang, D. Wang and Y. Li, Chem. Rev., 2020, 120, 11900–11955 CrossRef CAS.
- Y. Dai, H. Li, C. Wang, W. Xue, M. Zhang, D. Zhao, J. Xue, J. Li, L. Luo, C. Liu, X. Li, P. Cui, Q. Jiang, T. Zheng, S. Gu, Y. Zhang, J. Xiao, C. Xia and J. Zeng, Nat. Commun., 2023, 14, 3382 CrossRef CAS PubMed.
- S. Roy, Z. Li, Z. Chen, A. C. Mata, P. Kumar, S. C. Sarma, I. F. Teixeira, I. F. Silva, G. Gao, N. V. Tarakina, M. G. Kibria, C. V. Singh, J. Wu and P. M. Ajayan, Adv. Mater., 2023, 2300713 CrossRef PubMed.
- K. Zhang, J. Xu, T. Yan, L. Jia, J. Zhang, C. Shao, L. Zhang, N. Han and Y. Li, Adv. Funct. Mater., 2023, 33, 2214062 CrossRef CAS.
- G. Shi, Y. Xie, L. Du, X. Fu, X. Chen, W. Xie, T.-B. Lu, M. Yuan and M. Wang, Angew. Chem., Int. Ed., 2022, 61, e202203569 CrossRef CAS PubMed.
- Q. Wang, K. Liu, K. Hu, C. Cai, H. Li, H. Li, M. Herran, Y.-R. Lu, T.-S. Chan, C. Ma, J. Fu, S. Zhang, Y. Liang, E. Cortés and M. Liu, Nat. Commun., 2022, 13, 6082 CrossRef CAS.
- K. Li, S. Zhang, X. Zhang, S. Liu, H. Jiang, T. Jiang, C. Shen, Y. Yu and W. Chen, Nano Lett., 2022, 22, 1557–1565 CrossRef CAS PubMed.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS PubMed.
- X. Zu, X. Li, W. Liu, Y. Sun, J. Xu, T. Yao, W. Yan, S. Gao, C. Wang, S. Wei and Y. Xie, Adv. Mater., 2019, 31, 1808135–1808142 CrossRef PubMed.
- S. Chen, Z. Zhang, W. Jiang, S. Zhang, J. Zhu, L. Wang, H. Ou, S. Zaman, L. Tan, P. Zhu, E. Zhang, P. Jiang, Y. Su, D. Wang and Y. Li, J. Am. Chem. Soc., 2022, 144, 12807–12815 CrossRef CAS PubMed.
- S.-F. Hung, A. Xu, X. Wang, F. Li, S.-H. Hsu, Y. Li, J. Wicks, E. G. Cervantes, A. S. Rasouli, Y. C. Li, M. Luo, D.-H. Nam, N. Wang, T. Peng, Y. Yan, G. Lee and E. H. Sargent, Nat. Commun., 2022, 13, 819 CrossRef CAS.
- M. Chhetri, M. Wan, Z. Jin, J. Yeager, C. Sandor, C. Rapp, H. Wang, S. Lee, C. J. Bodenschatz, M. J. Zachman, F. Che and M. Yang, Nat. Commun., 2023, 14, 3075 CrossRef CAS.
- X. Wang, Q. Hu, G. Li, H. Yang and C. He, Electrochem. Energy Rev., 2022, 5, 28 CrossRef CAS.
- J. Wang, C. Cheng, B. Huang, J. Cao, L. Li, Q. Shao, L. Zhang and X. Huang, Nano Lett., 2021, 21, 980–987 CrossRef CAS PubMed.
- S. Chen, Y. Su, P. Deng, R. Qi, J. Zhu, J. Chen, Z. Wang, L. Zhou, X. Guo and B. Y. Xia, ACS Catal., 2020, 10, 4640–4646 CrossRef CAS.
- J. Zhu, Y. Wang, A. Zhi, Z. Chen, L. Shi, Z. Zhang, Y. Zhang, Y. Zhu, X. Qiu, X. Tian, X. Bai, Y. Zhang and Y. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202111670 CrossRef CAS.
- Z. Zhao, X. Peng, X. Liu, X. Sun, J. Shi, L. Han, G. Li and J. Luo, J. Mater. Chem. A, 2017, 5, 20239–20243 RSC.
- H. Wang, G. Zhan, C. Tang, D. Yang, W. Liu, D. Wang, Y. Wu, H. Wang, K. Liu, J. Li, M. Huang and K. Chen, ACS Nano, 2023, 17, 4790–4799 CrossRef CAS.
- X. Wang, P. Ou, J. Wicks, Y. Xie, Y. Wang, J. Li, J. Tam, D. Ren, J. Y. Howe, Z. Wang, A. Ozden, Y. Z. Finfrock, Y. Xu, Y. Li, A. S. Rasouli, K. Bertens, A. H. Ip, M. Graetzel, D. Sinton and E. H. Sargent, Nat. Commun., 2021, 12, 3387 CrossRef CAS PubMed.
- Y. Li, A. Xu, Y. Lum, X. Wang, S. F. Hung, B. Chen, Z. Wang, Y. Xu, F. Li, J. Abed, J. E. Huang, A. S. Rasouli, J. Wicks, L. K. Sagar, T. Peng, A. H. Ip, D. Sinton, H. Jiang, C. Li and E. H. Sargent, Nat. Commun., 2020, 11, 6190 CrossRef CAS.
- R. M. Yadav, Z. Li, T. Zhang, O. Sahin, S. Roy, G. Gao, H. Guo, R. Vajtai, L. Wang, P. M. Ajayan and J. Wu, Adv. Mater., 2022, 34, 2105690 CrossRef CAS.
- Y. Wang, J. Chen, G. Wang, Y. Li and Z. Wen, Angew. Chem., Int. Ed., 2018, 57, 13120–13124 CrossRef CAS PubMed.
- X. Liu, H. Yang, J. He, H. Liu, L. Song, L. Li and J. Luo, Small, 2018, 14, 1704049 CrossRef PubMed.
- X. Kang, Q. Zhu, X. Sun, J. Hu, J. Zhang, Z. Liu and B. Han, Chem. Sci., 2016, 7, 266–273 RSC.
- H. Chen, X. Guo, X. Kong, Y. Xing, Y. Liu, B. Yu, Q.-X. Li, Z. Geng, R. Si and J. Zeng, Green Chem., 2020, 22, 7529–7536 RSC.
- Y. Zhang, L.-Z. Dong, S. Li, X. Huang, J.-N. Chang, J.-H. Wang, J. Zhou, S.-L. Li and Y.-Q. Lan, Nat. Commun., 2021, 12, 6390 CrossRef CAS.
- X. Yan, C. Chen, Y. Wu, Y. Chen, J. Zhang, R. Feng, J. Zhang and B. Han, Green Chem., 2022, 24, 1989–1994 RSC.
- A. Zhang, Y. Liang, H. Zhang, Z. Geng and J. Zeng, Chem. Soc. Rev., 2021, 50, 9817–9844 RSC.
- A. Sedighian Rasouli, X. Wang, J. Wicks, C.-T. Dinh, J. Abed, F.-Y. Wu, S.-F. Hung, K. Bertens, J. E. Huang and E. H. Sargent, Chem. Catal., 2022, 2, 908–916 CrossRef CAS.
- J. Zhao, P. Zhang, T. Yuan, D. Cheng, S. Zhen, H. Gao, T. Wang, Z.-J. Zhao and J. Gong, J. Am. Chem. Soc., 2023, 145, 6622–6627 CrossRef CAS PubMed.
- Y. Liang, W. Zhou, Y. Shi, C. Liu and B. Zhang, Sci. Bull., 2020, 65, 1547–1554 CrossRef CAS.
- W. Wang, G. Ruan, X. Wang, C. Wu and Q. Wang, New J. Chem., 2023, 47, 8894–8905 RSC.
- H. Hu, L. Gui, W. Zhou, J. Sun, J. Xu, Q. Wang, B. He and L. Zhao, Electrochim. Acta, 2018, 285, 70–77 CrossRef CAS.
- H. Yang, Y.-W. Hu, J.-J. Chen, M. S. Balogun, P.-P. Fang, S. Zhang, J. Chen and Y. Tong, Adv. Energy Mater., 2019, 9, 1901396 CrossRef.
- X. Wei, S. Xiao, R. Wu, Z. Zhu, L. Zhao, Z. Li, J. Wang, J. S. Chen and Z. Wei, Appl. Catal., B, 2022, 302, 120861 CrossRef CAS.
- Z. Zhou, S. Liang, J. Xiao, T. Zhang, M. Li, W. Xie and Q. Wang, J. Energy Chem., 2023, 84, 277–285 CrossRef CAS.
- H. Wang, H. Zhang, Y. Huang, H. Wang, A. Ozden, K. Yao, H. Li, Q. Guo, Y. Liu, A. Vomiero, Y. Wang, Z. Qian, J. Li, Z. Wang, X. Sun and H. Liang, ACS Nano, 2023, 17, 346–354 CrossRef CAS PubMed.
- X. Ma, D. Jiao, J. Fan, Y. Dong and X. Cui, Inorg. Chem. Front., 2023, 10, 168–173 RSC.
- C. Chen, X. Sun, X. Yan, Y. Wu, M. Liu, S. Liu, Z. Zhao and B. Han, Green Chem., 2020, 22, 1572–1576 RSC.
- K. J. P. Schouten, Z. Qin, E. Pérez Gallent and M. T. M. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS.
- G. L. De Gregorio, T. Burdyny, A. Loiudice, P. Iyengar, W. A. Smith and R. Buonsanti, ACS Catal., 2020, 10, 4854–4862 CrossRef CAS PubMed.
- B. Deng, M. Huang, K. Li, X. Zhao, Q. Geng, S. Chen, H. Xie, X. A. Dong, H. Wang and F. Dong, Angew. Chem., Int. Ed., 2022, 61, e202114080 CrossRef CAS PubMed.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Phys. Chem. B, 2002, 106, 15–17 CrossRef CAS.
- Z. Chen, T. Wang, B. Liu, D. Cheng, C. Hu, G. Zhang, W. Zhu, H. Wang, Z.-J. Zhao and J. Gong, J. Am. Chem. Soc., 2020, 142, 6878–6883 CrossRef CAS.
- J. Cai, Q. Zhao, W.-Y. Hsu, C. Choi, Y. Liu, J. M. P. Martirez, C. Chen, J. Huang, E. A. Carter and Y. Huang, J. Am. Chem. Soc., 2023, 145, 9136–9143 CrossRef CAS PubMed.
- C. Wang, X. Kong, J. Huang, Y. Yang, H. Zheng, H. Wang, S. Dai, S. Zhang, Y. Liang, Z. Geng, F. Li and J. Zeng, J. Mater. Chem. A, 2022, 10, 25725–25729 RSC.
- Y. Shi, K. Sun, J. Shan, H. Li, J. Gao, Z. Chen, C. Sun, Y. Shuai and Z. Wang, ACS Catal., 2022, 12, 8252–8258 CrossRef CAS.
- M. Fan, R. K. Miao, P. Ou, Y. Xu, Z.-Y. Lin, T.-J. Lee, S.-F. Hung, K. Xie, J. E. Huang, W. Ni, J. Li, Y. Zhao, A. Ozden, C. P. O'Brien, Y. Chen, Y. C. Xiao, S. Liu, J. Wicks, X. Wang, J. Abed, E. Shirzadi, E. H. Sargent and D. Sinton, Nat. Commun., 2023, 14, 3314 CrossRef CAS PubMed.
- X. Y. Zhang, W. J. Li, X. F. Wu, Y. W. Liu, J. Chen, M. Zhu, H. Y. Yuan, S. Dai, H. F. Wang, Z. Jiang, P. F. Liu and H. G. Yang, Energy Environ. Sci., 2022, 15, 234–243 RSC.
- Y. Cui, C. Kong, C. Yang, Y. Su, Y. Cheng, D. Yao, G. Chen, K. Song, Z. Zhong, Y. Song, G. Wang, Z. Li and L. Zhuang, ACS Catal., 2023, 13, 11625–11633 CrossRef CAS.
- L. Xiong, X. Zhang, L. Chen, Z. Deng, S. Han, Y. Chen, J. Zhong, H. Sun, Y. Lian, B. Yang, X. Yuan, H. Yu, Y. Liu, X. Yang, J. Guo, M. H. Rümmeli, Y. Jiao and Y. Peng, Adv. Mater., 2021, 33, 2101741 CrossRef CAS PubMed.
- M. Xie, Y. Shen, W. Ma, D. Wei, B. Zhang, Z. Wang, Y. Wang, Q. Zhang, S. Xie, C. Wang and Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202213423 CrossRef CAS.
- L. Lin, T. Liu, J. Xiao, H. Li, P. Wei, D. Gao, B. Nan, R. Si, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2020, 59, 22408–22413 CrossRef CAS PubMed.
- Q. Zhao, Y. Wang, M. Li, S. Zhu, T. Li, J. Yang, T. Lin, E. P. Delmo, Y. Wang, J. Jang, M. Gu and M. Shao, SmartMat, 2022, 3, 183–193 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |